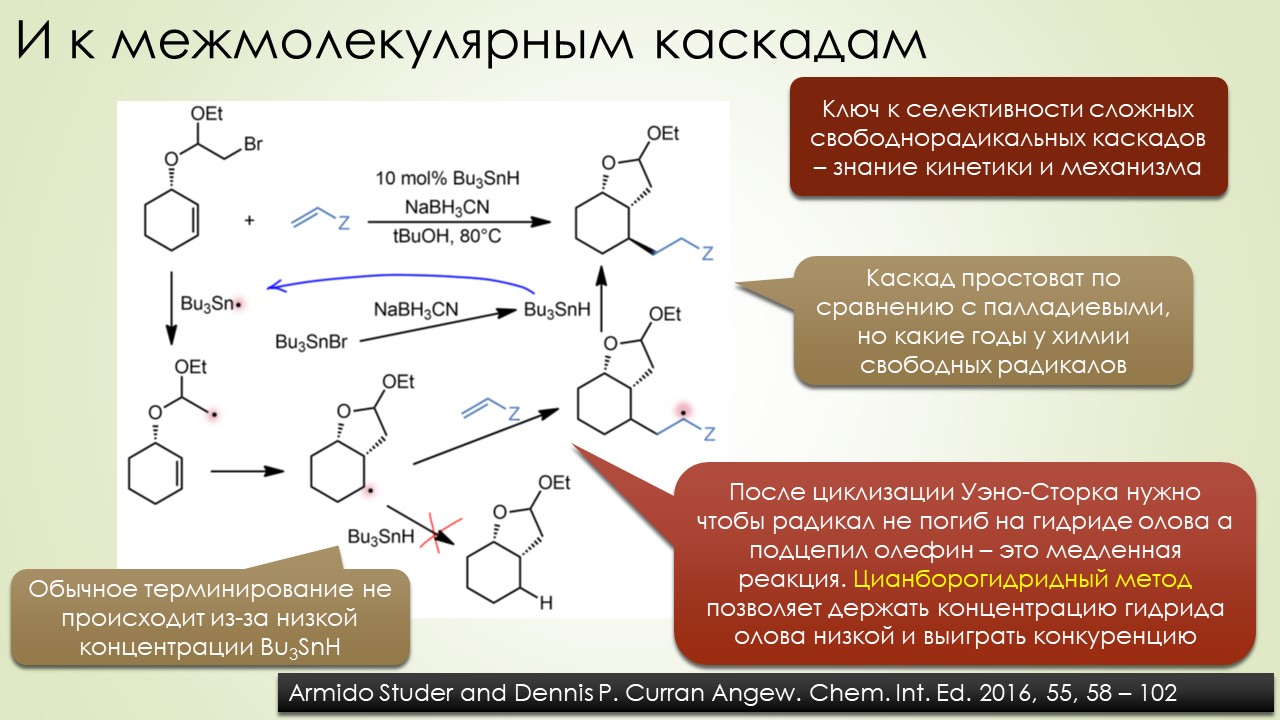
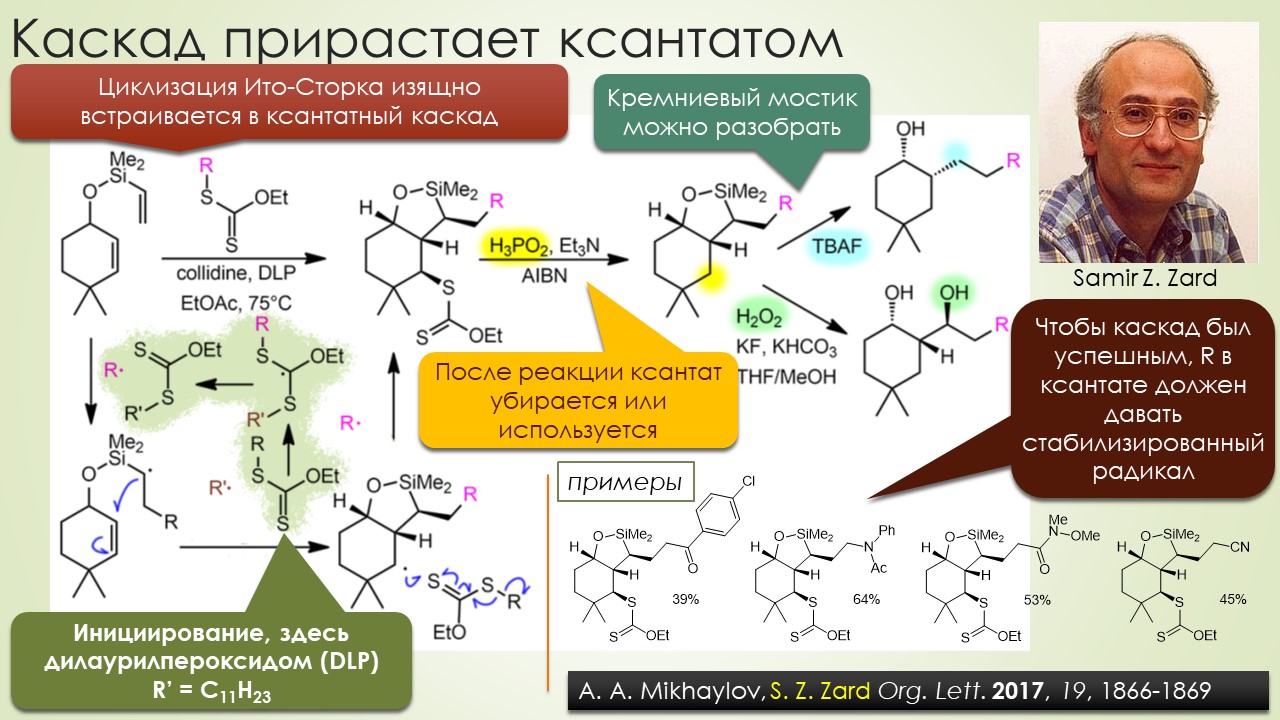
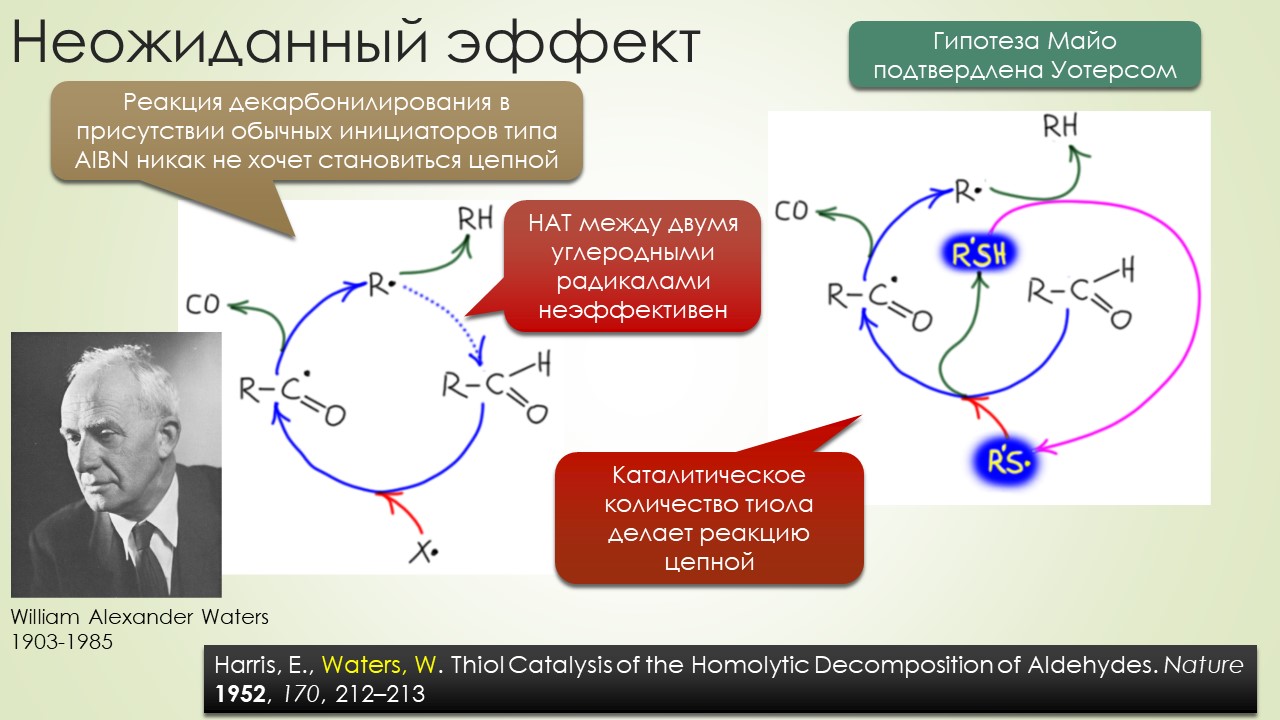
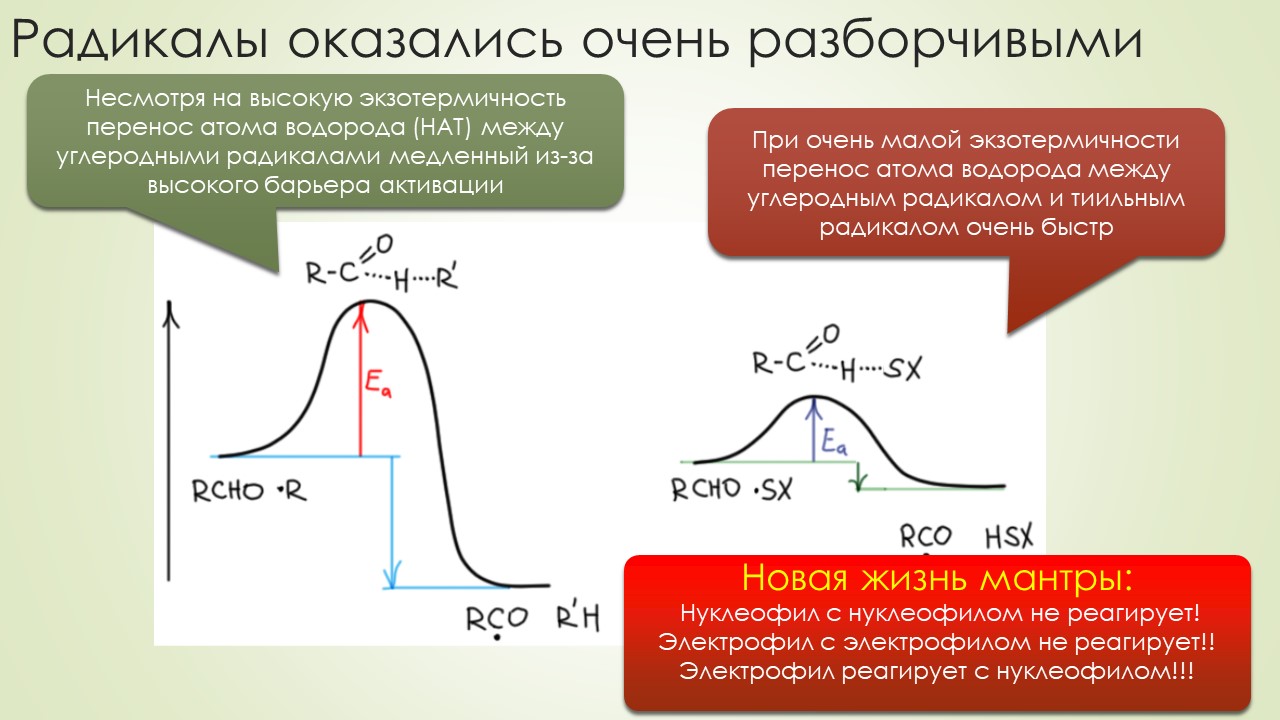
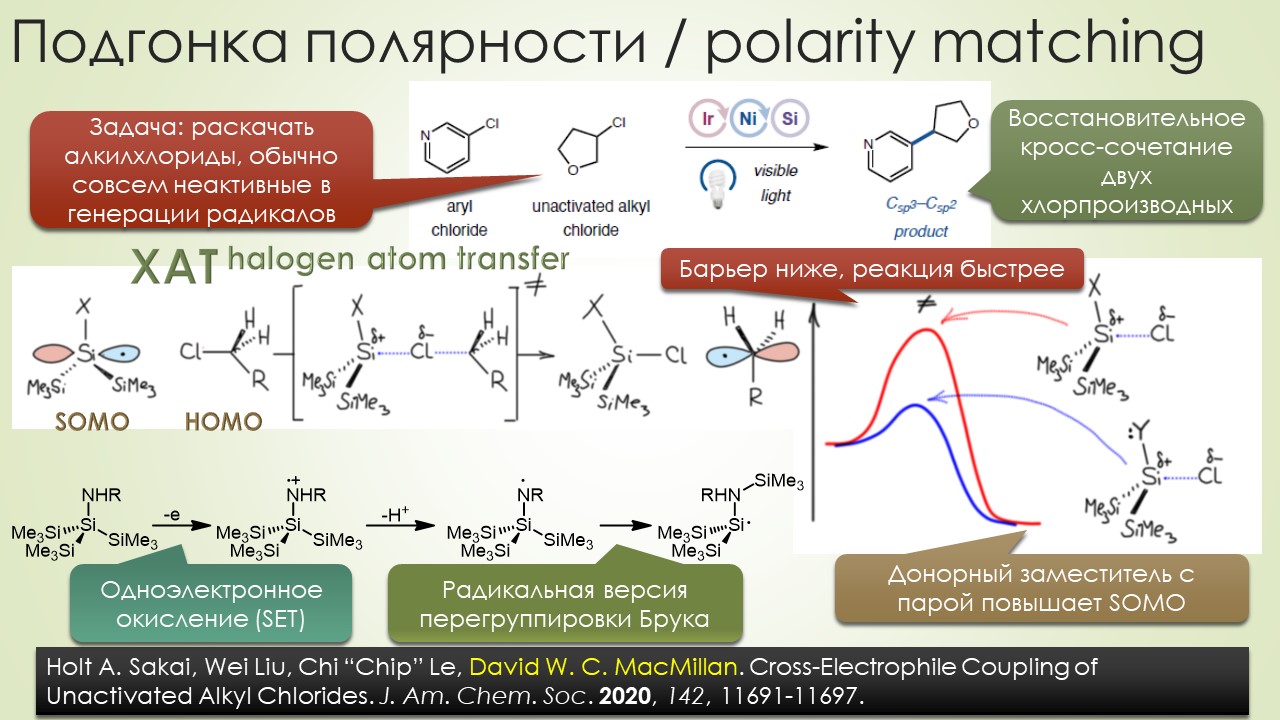
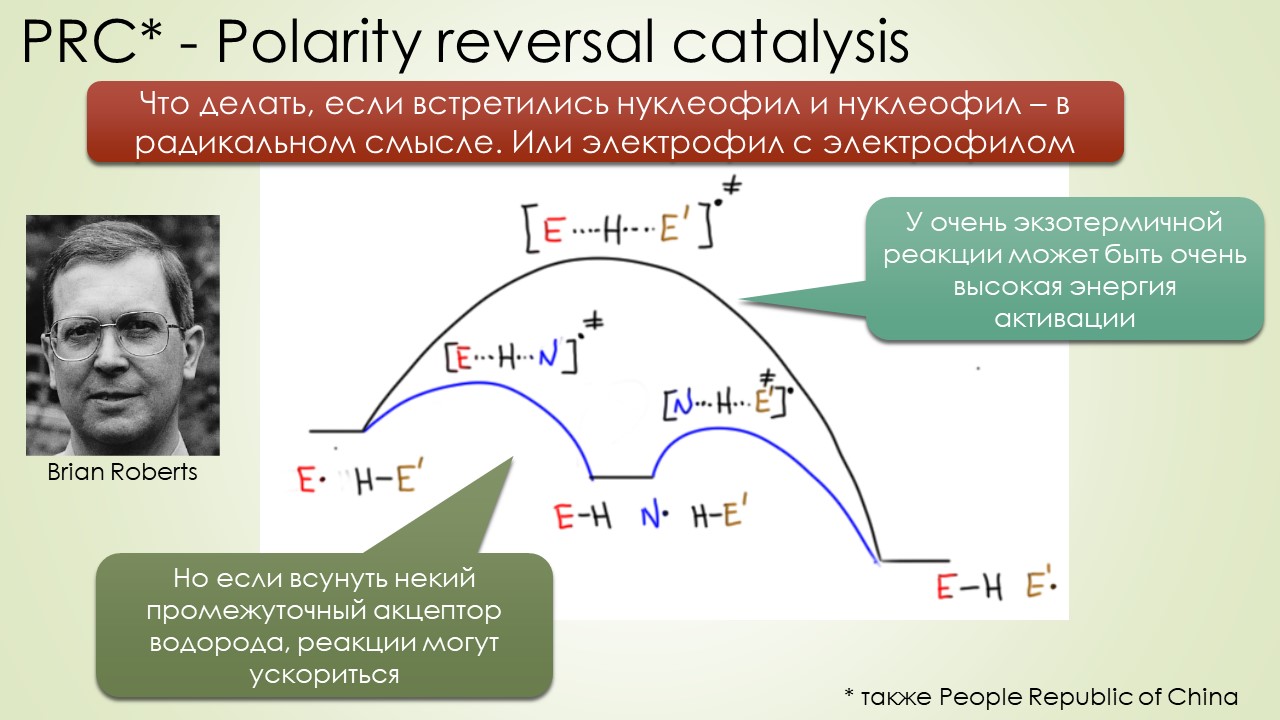
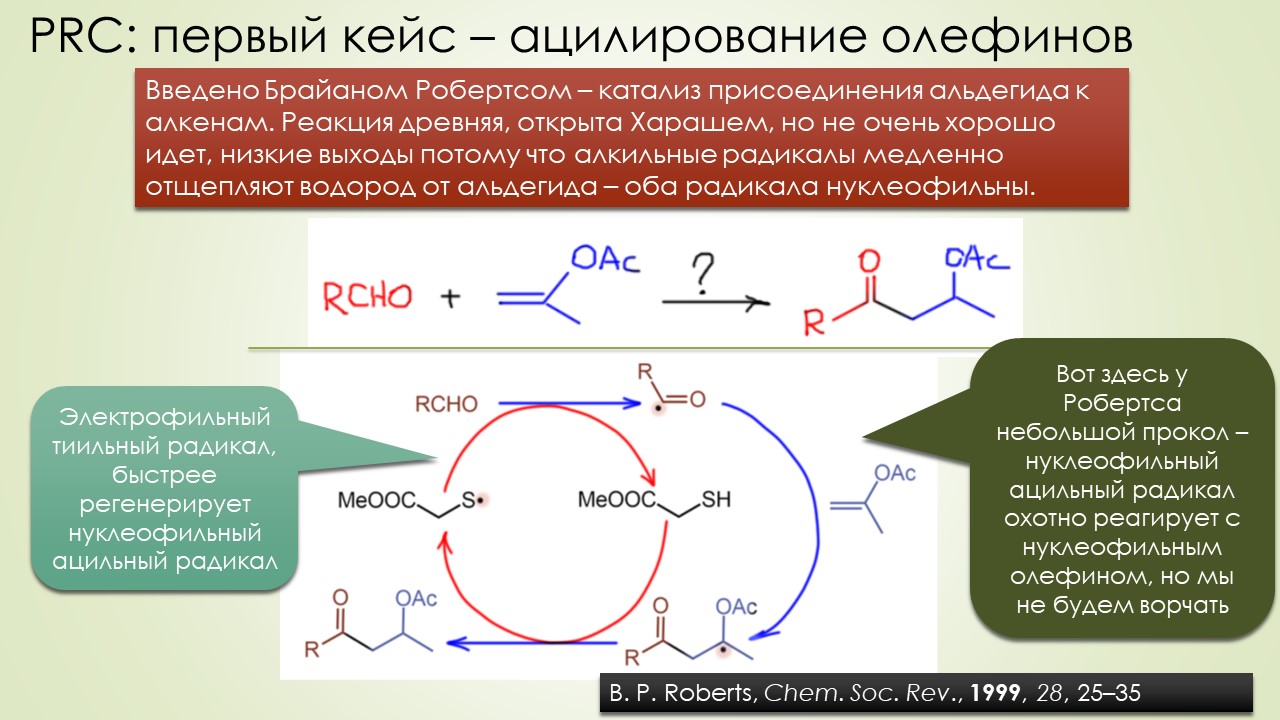
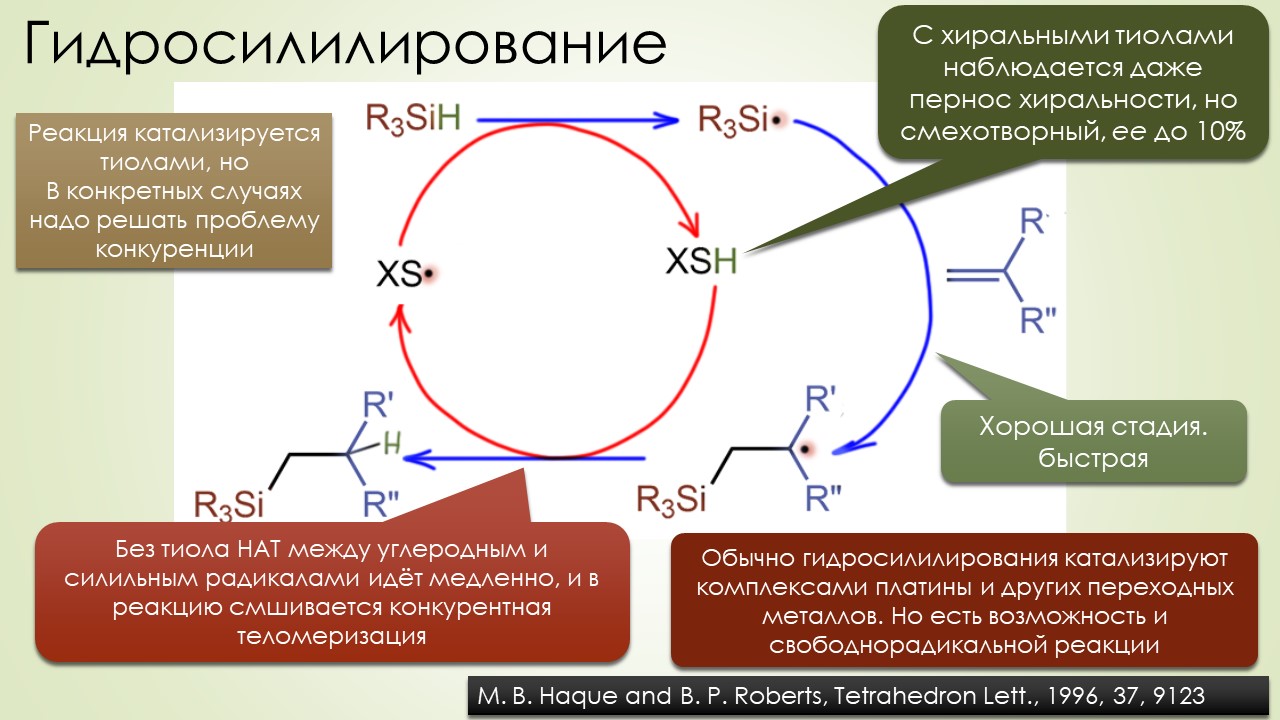
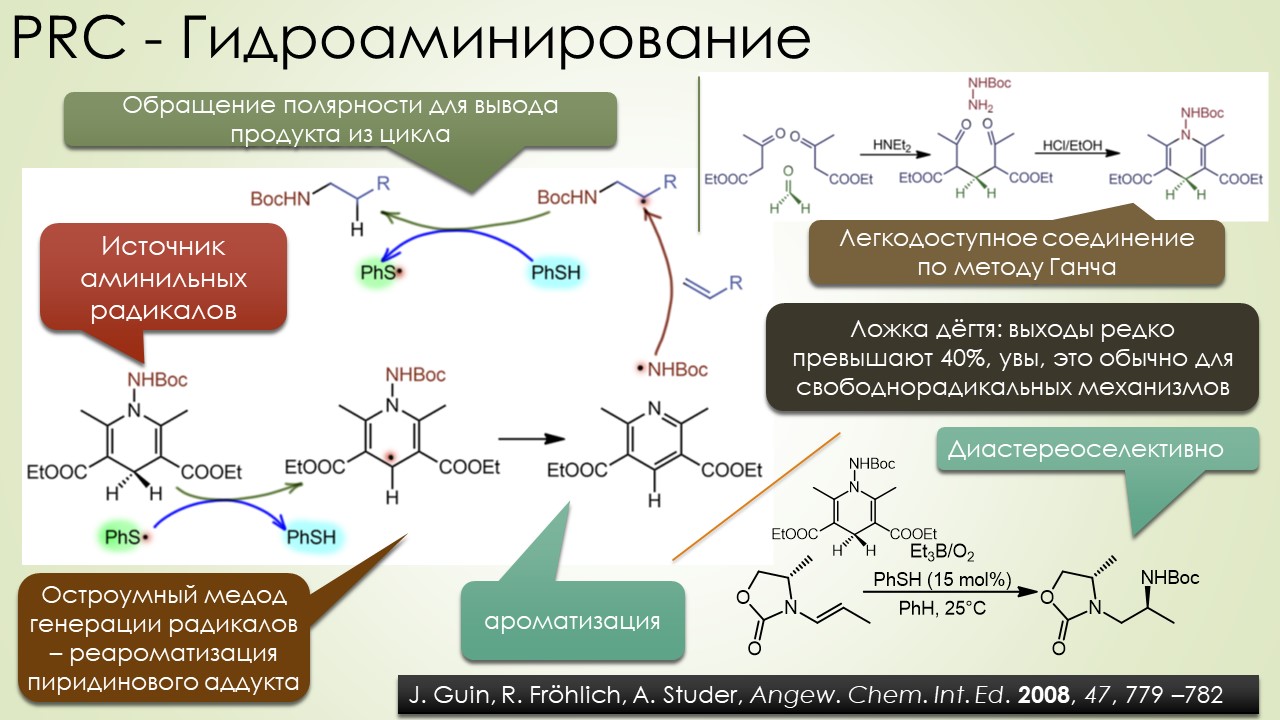
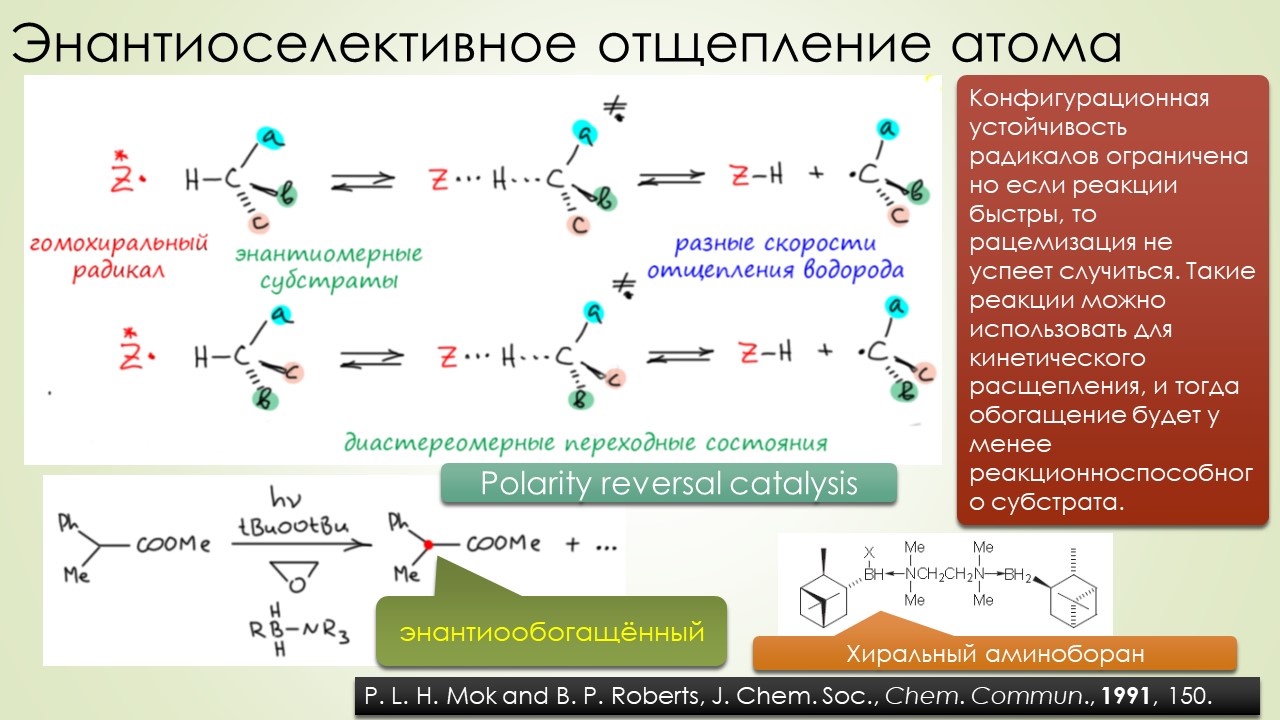
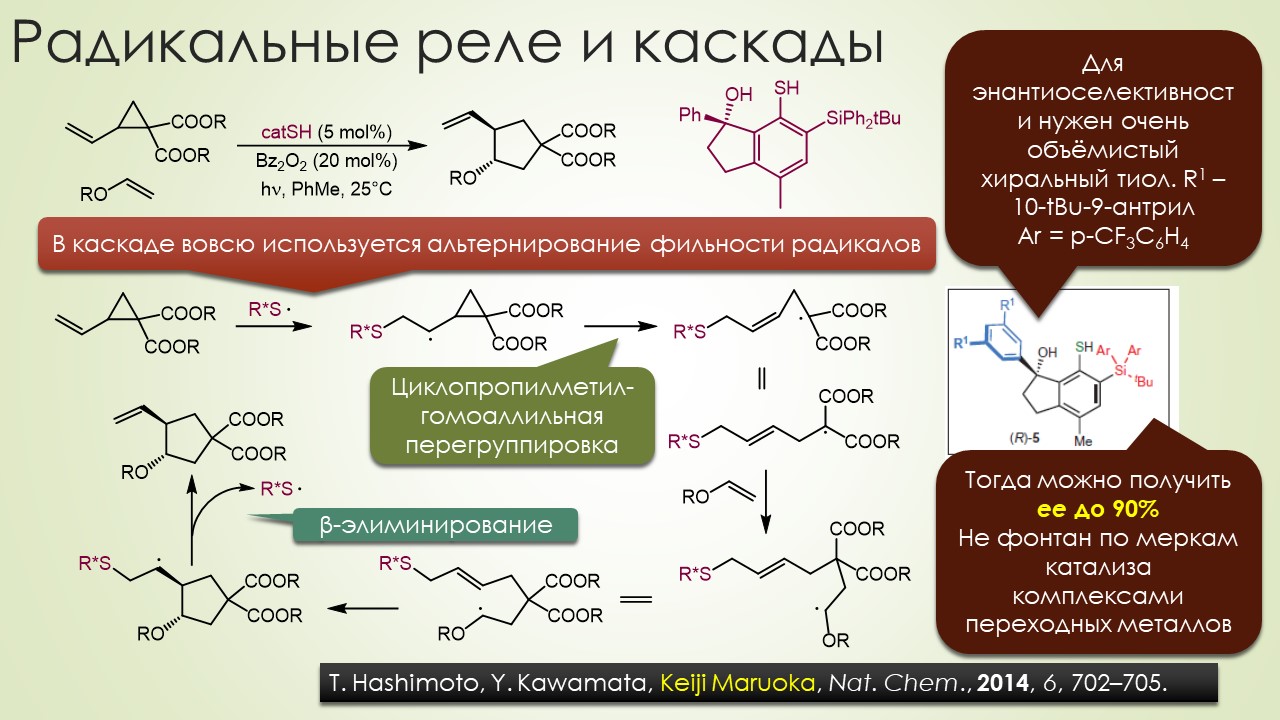
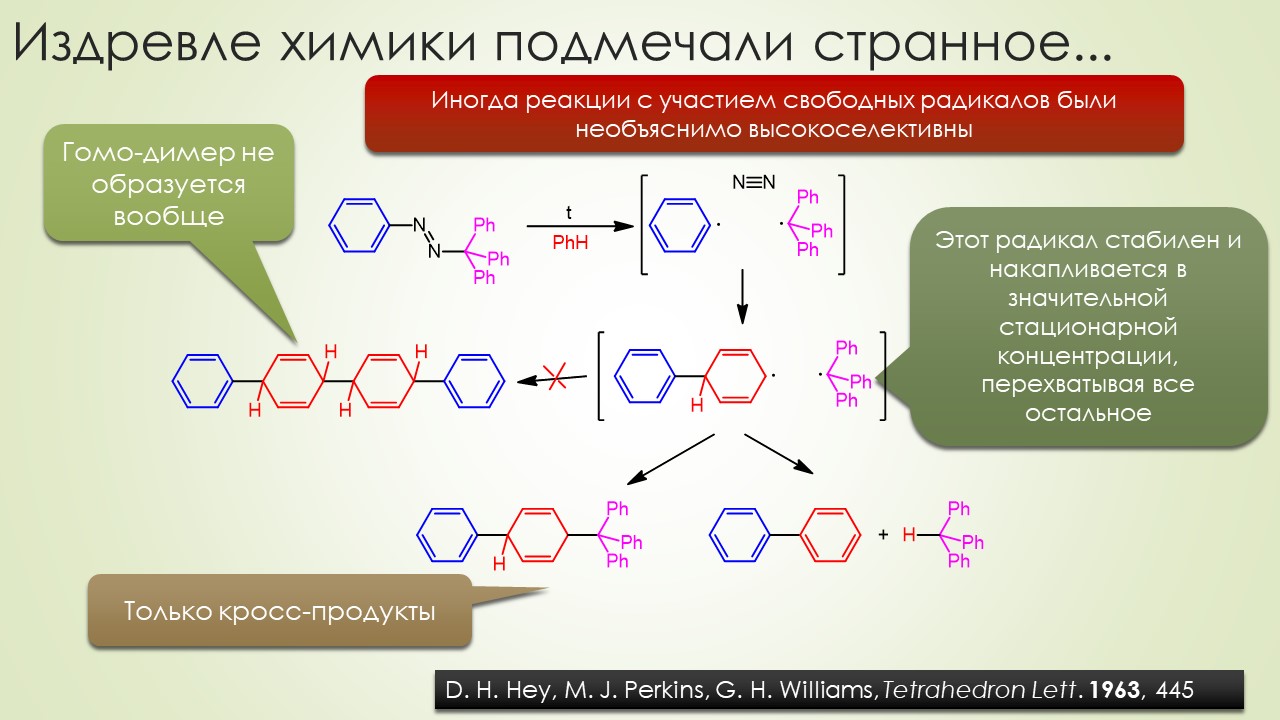
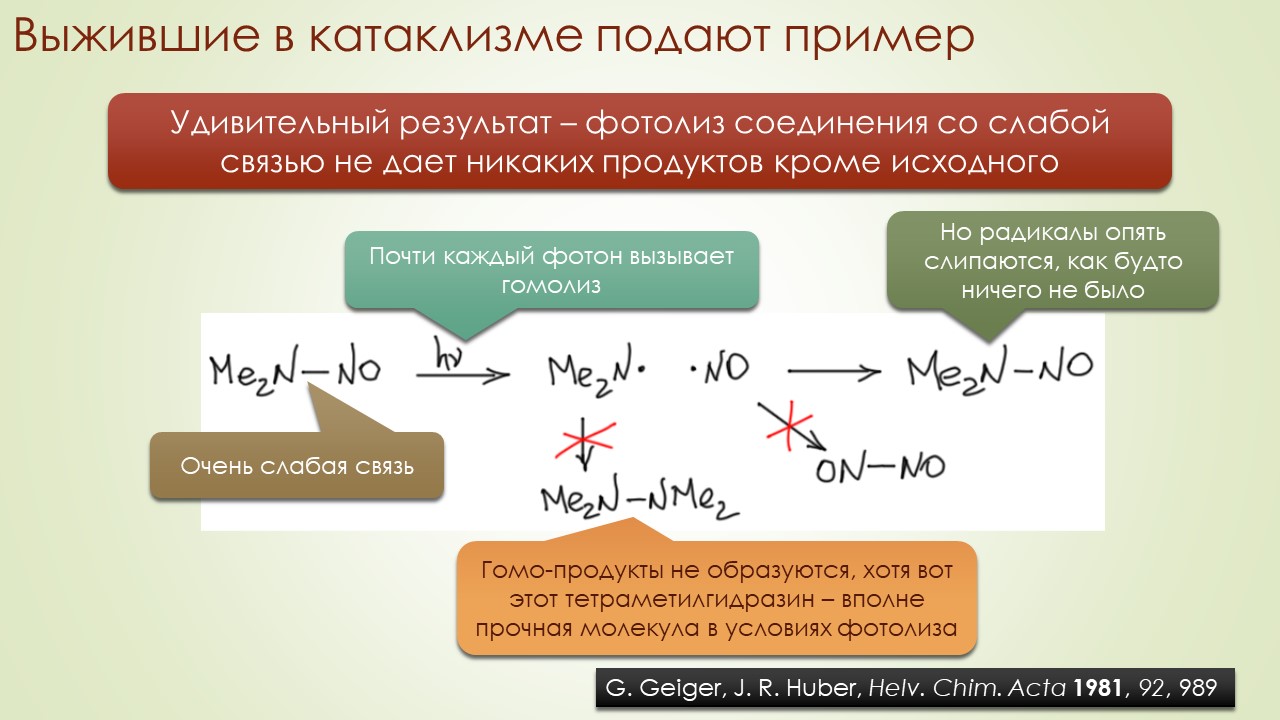
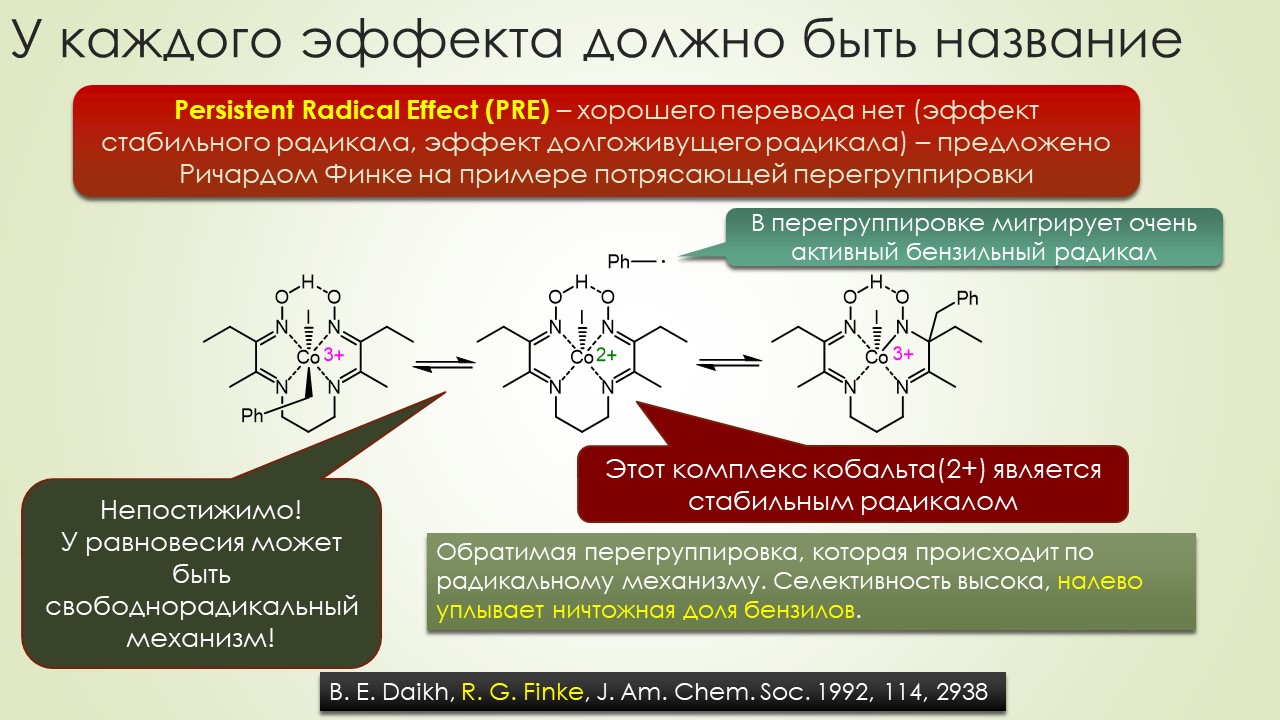
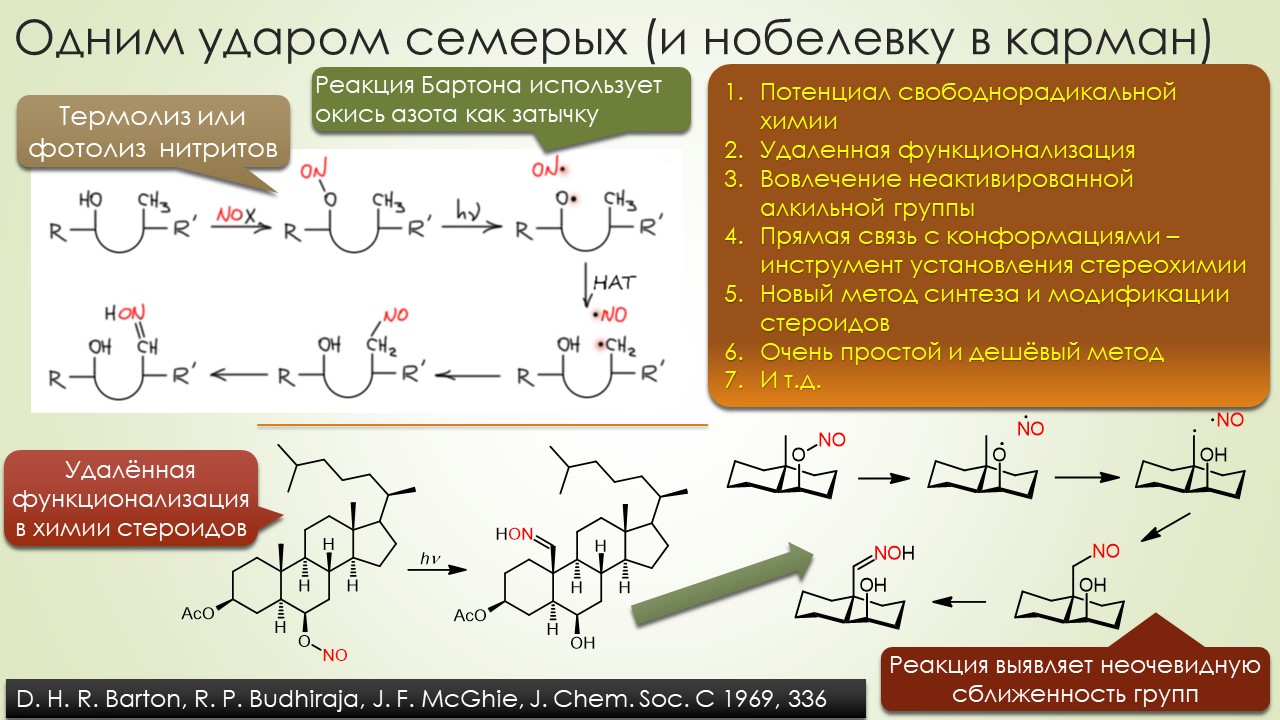
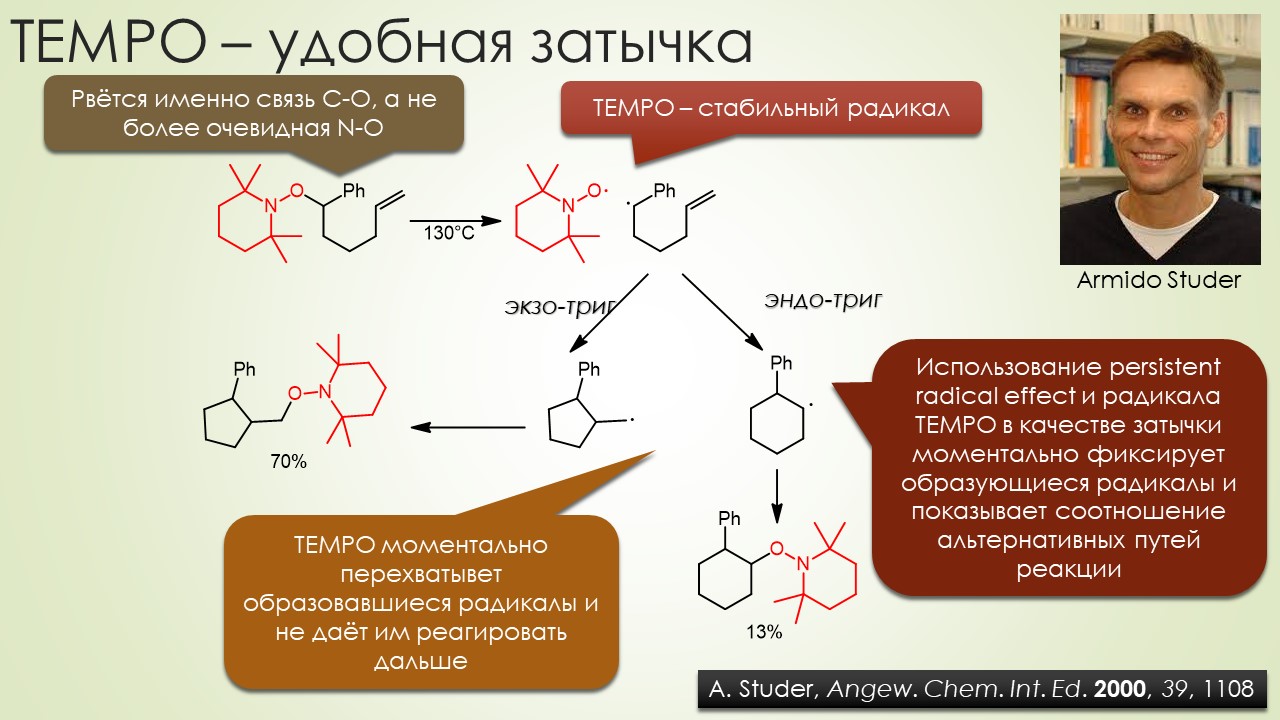
Запрягали весь 20-й век и наконец поехали, да как!
Среди ключевых реакционноспособных интермедиатов в органической химии радикалы должны были бы занимать первое место – ведь это совсем очевидный способ заставлять реагировать соединения с ковалентными связями: просто порвать одну из таких связей и пустить фрагменты искать себе судьбу во взаимодействии с другими молекулами. Уж куда проще, чем как-то делать катионы или анионы, возиться со всеми проблемами ионной химии, в том числе эффектами растворителей, противоионов и так далее. В газовой фазе вообще никаких других путей и нет – ионы там образуются только в виде исключения и только окольными путями: гетеролиз связей без содействия жидкой средой почти невозможен по энергетическим соображениям. Ну а в жидкой фазе радикалы тоже должны себя чувствовать неплохо, получая обычную помощь среды в равновесном перераспределении энергии и не испытывая никаких других сложностей. В общем, в органике радикалы должны играть очень важную роль. Должны были бы!
А почему не играют. Точнее, не играли ещё недавно. Вспомним, много ли мы узнали и много ли пользовались свободнорадикальными реакциями и механизмами в основном курсе органической химии. Ну, там была химия алканов, которую мы пролетели как мимолетное и не очень приятное впечатление пассажира скоростного поезда, который видит в окно вдали какую-то серую дымящую конструкцию неясного назначения, и гораздо раньше чем в мозгу возникнут гипотезы о том, что это было, оно рассеивается как мираж. Протрите глаза, вам не показалось? Нет, вроде было что-то, дымило, но зачем и почему и куда это – нет ответов. И даже там нам говорят, что первый класс органических соединений, от которого всё остальное и производится, алканы они бедные такие, парафинами назвали их древние – parum affinis – убогие в сродстве: сродством, напомню, древние отцы химии называли реакционную способность. Парафины могут расcчитывать в химии только на свободнорадикальные реакции и поэтому они такие убогие в сродстве, малореакционноспособные. Так говорила химия полтора века своего развития. И когда полвека назад Джордж Ола развил идеи Владимира Ипатьева о том, что алканы можно еще раскачать электрофильно, это вызвало такой неподдельный интерес химиков всего мира – наконец этот несчастный класс органических соединений выпустили из свободнорадикального узилища – что герою немедленно всучили нобелевку, сопроводив это дифирамбами, приличествующими больше правителям и победоносным военачальникам, чем учёному.
А была ли нобелевка за радикалы? Была конечно, нобелевка Николая Семёнова и Сирила Хиншельвуда 1956 года так прямо и заявленная как “за исследования механизмов реакций”, до этого года единственная нобелевка по химии российскому учёному. И вроде бы она прямо за это – за исследование цепных свободнорадикальных реакций, разве хлорирование метана это не то? Именно то, но есть проблема, не для выдающихся лауреатов, конечно, а для нас, обычных органиков, – их исследования про кинетику и другие физико-химические особености таких реакций с совершенно очевидным уклоном даже не в химию, а в физику цепных процессов, в первую очередь, того, что происходит в процессах горения и всем, что с этим связано. Обычные органики из работ лауреатов узнают немного, кроме того, что некоторые из них делают вещества, используемые как топливо для реактивных двигателей и тому подобного, а дальше уже должны прийти химические физики и на основании работа Семенова и Хиншельвуда рассчитать процессы, происходящие при горении топлива, чтобы достичь максимального импульса и избежать проблем неустойчивости горения и перехода горения в взрыв. Но органикам-то обычным какое до этого дело – мы ведь даже от газовых горелок почти полностью в лабораториях отказались, разве что капилляр оттянуть, да и то необязательно. Была еще одна нобелевка, которую можно было бы счесть свободнорадикальной – Дереку Бартону, но там формулировка чисто стереохимическая, и то, что самых важных результатов в своих работах Бартон достиг с помощью невероятно изящных свободнорадикальных реакций, знают далеко не все. Во всяком случае именно Бартон оставил неслабую школу, которая стала серьёзно развивать возможности свободнорадикальных реакций и понесла это в новый век.
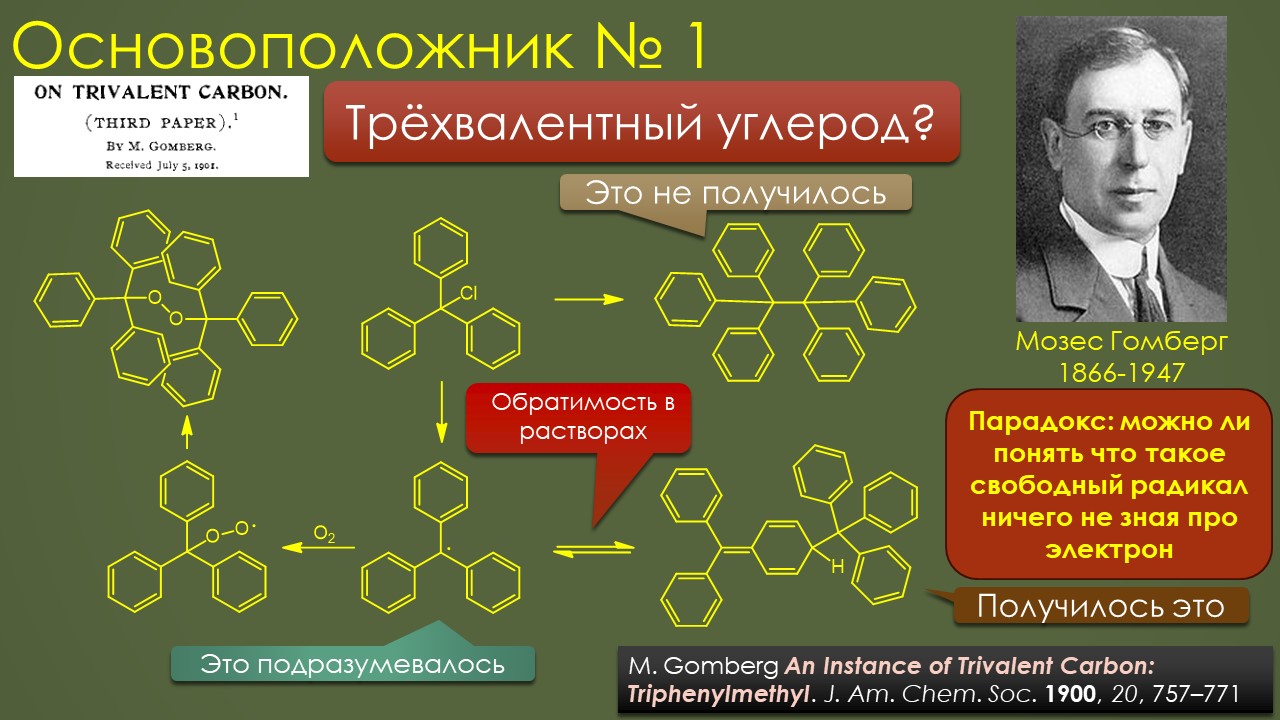
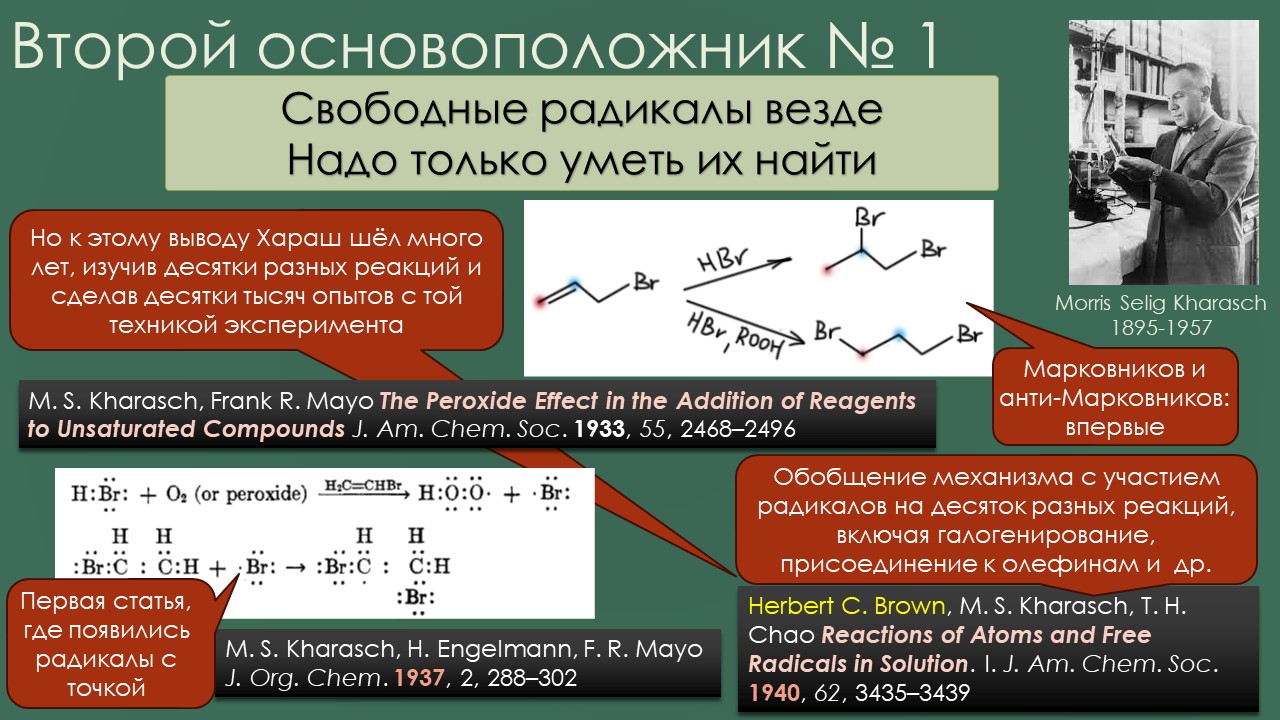
Так, ну и где ещё мы встречаемся со свободнорадикальной химией на 3 курсе? Конечно это один из гвоздей программы – присоединение HBr к олефинам против Марковникова, так говорят, хотя, конечно, никто не против Марковникова, просто хотят сказать, что присоединяется не так, как диктует правило Марковникова. Это действительно то, что называется по-английски seminal work – работа, ставшая семенем, из которого произросло древо. Совершенно бесспорно то, что химия свобоных радикалов в органике вышла из работ Хараша, а началось это именно с обнаружения “перекисного эффекта” в присоединении к олефинам, и дальнейшие исследования именно Хараша и его коллег и вытащили оттуда фактически всё, что и стало разделом про свободнорадикальную химию в учебниках. Сначала Хараш обнаружил эффект, а после в результате почти десятилетних усилий доказать коллегам органикам, что это не ошибка эксперимента, ему пришлось не только сделать совершенно беспрецедентное количество экспериментов, но и раскопать причину – и это и было первое явление радикалов в органической химии.
Сам же Хараш невольно и стал виновником того, что эта наука почти никому не понравилась. Он попробовал огромное количество вариантов, пытаясь присоединять к двойным связям множество других соединений, наоткрывал кучу новых реакций – и показал всем, насколько они капризны. Парадоксальной проблемой стало то… что Хараш и его коллеги были совершенно непревзойденными экспериментаторами, и просто мало кто мог воспроизвести их результаты, потому что делать все настолько аккуратно и тщательно могут немногие. Совершенно комические многократные попытки опровержения Хараша – а у нас всё по другому, Хараш ошибался – публиковались многократно в основных журналах середины прошлого века – и всякий раз оказывалось, что авторы просто что-то плохо очистили, или вообще не знают, как нормально работать в лаборатории. Эффект Хараша это такой удивительный тест на состоятельность химика как экспериментатора – и слишком многие его не проходили. Обидно!
Ещё мы из того же времени знаем и пользуемся реакцией аллильного (и бензильного) бромирования Воля-Циглера – той самой, где NBS, и с одной стороны, её нахваливаем, а с другой – парадоксальным образом никто не знает, что эта реакция даёт в хоть чуть-чуть более сложных случаях с несимметричными олефинами. И такое складывается впечатление, что и дела никому до этого нет, как нет и понятных закономерностей, а наши простые соображения типа мезомерии аллильных радикалов почему-то в этой реакции почти не работают. Мезомерная делокализация есть, а соотношение продуктов бромирования почему-то ей не следует. Только одного этого достаточно, чтобы работающие химики прониклись глубоким недоверием к свободнорадикальным реакциям.
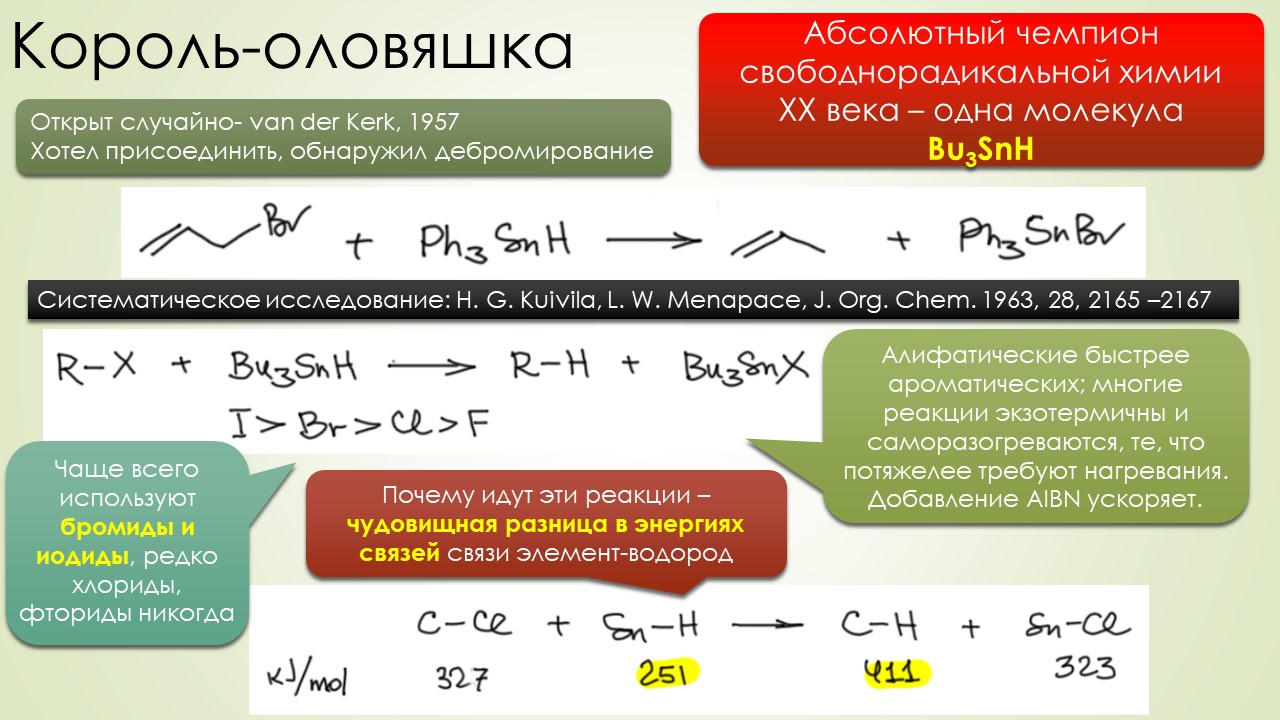
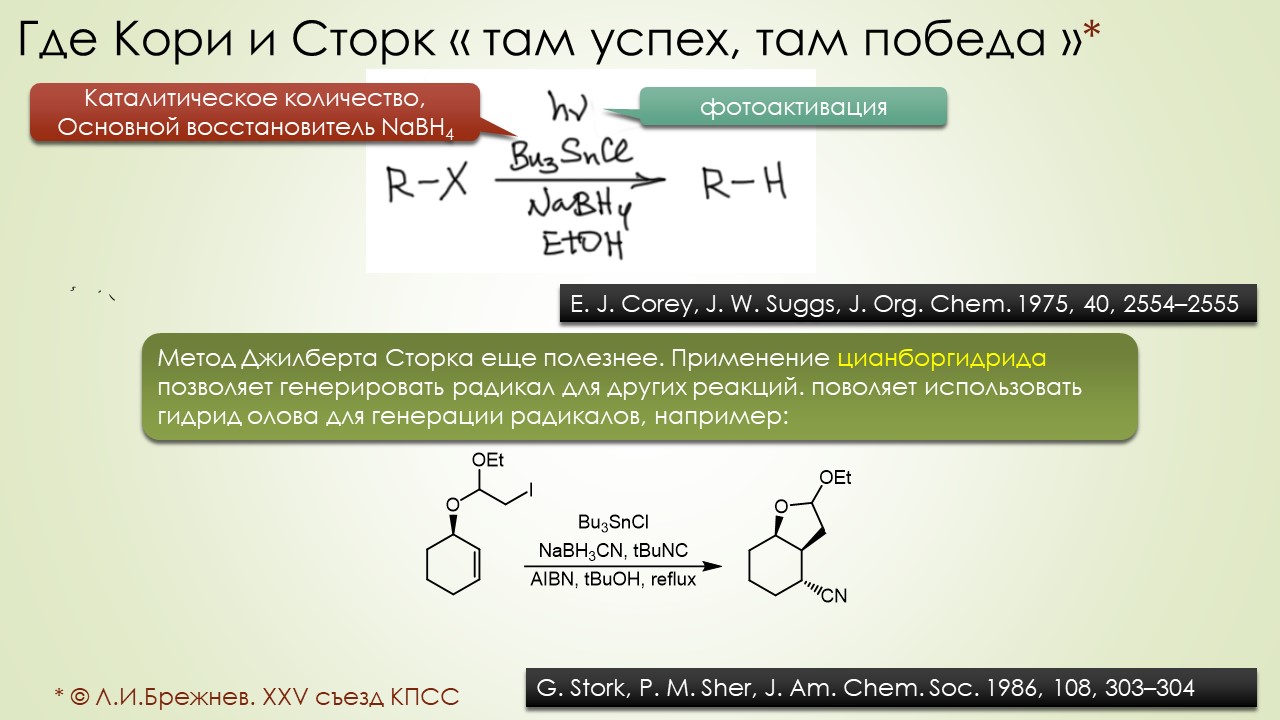
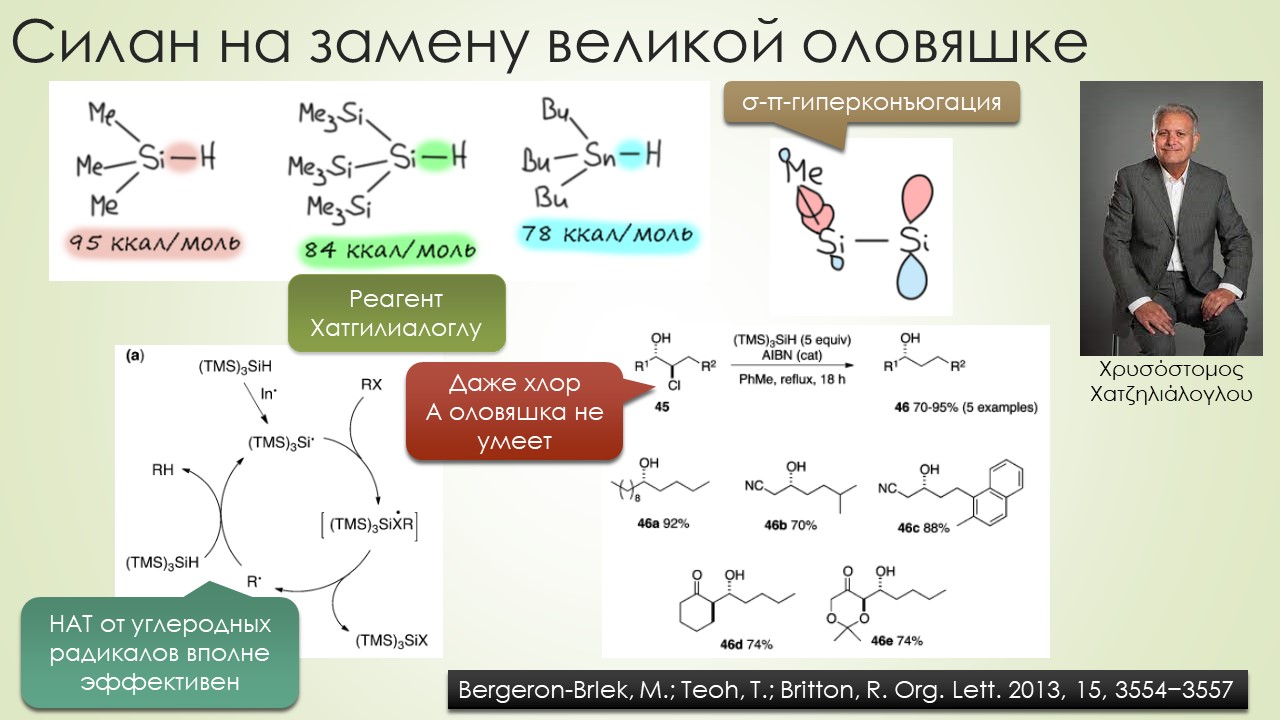
Вторые полвека 20-го столетия не прошли для свободнорадикальных реакций незамеченными. Был открыт самый главный реагент этой химии, трибутилоловогидрид, и вот с ним стало всё получаться настолько хорошо, что свободными радикалами наконец серьёзно заинтересовались самые крутые синтетики этого золотого века (полувека, если быть точным) органического синтеза. С свободнорадикальными реакциями в арсенале стали делать красивые синтезы, но – сравните количество таких реакций на одной стороне, и реакций с соеиненями, сохраняющими в реакциях заполненные электронные оболочки (а это все нуклеофильно-электрофильные и согласованные реакции типа перициклически) в синтезах – счёт будет даже не неприличным, а неправдоподобным, что-то типа сто к одному. В курс органической химии больше никаких серьёзных вторжений не было, хотя какие-то мелочи типа окисления солью Фреми, или, или, или (натужно вспоминаю) ну прямо чёрт знает что ещё.
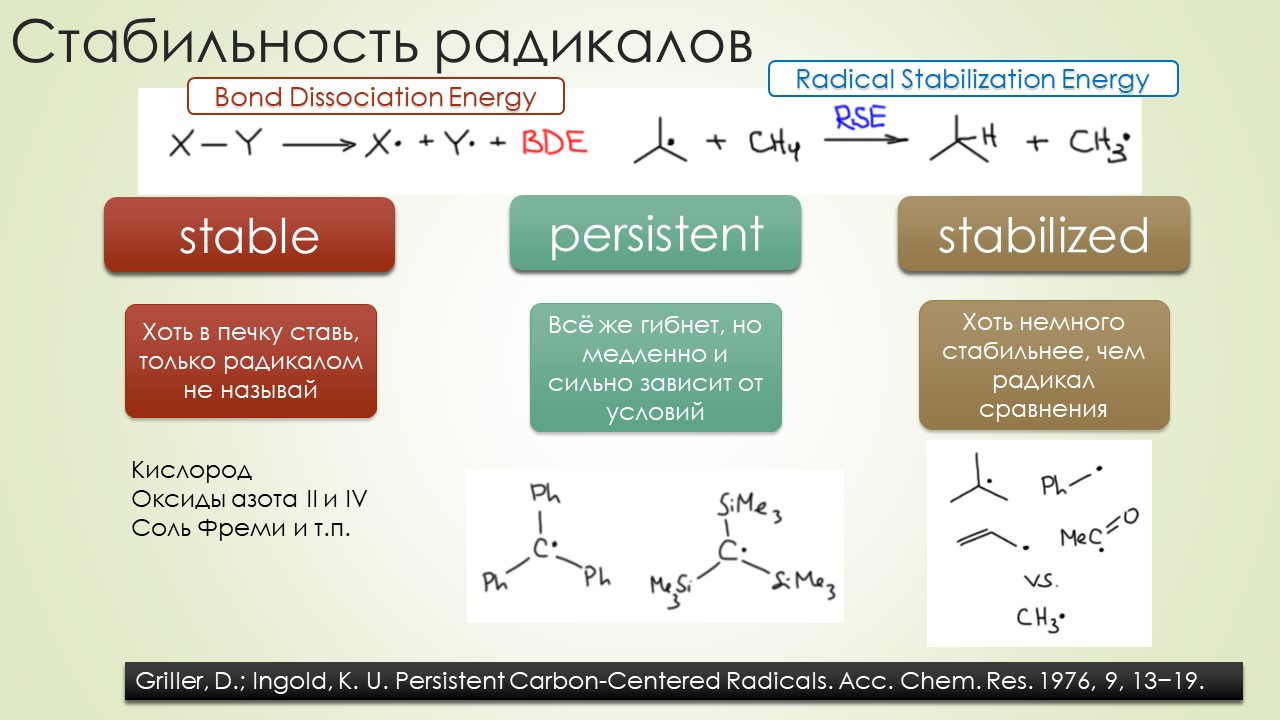
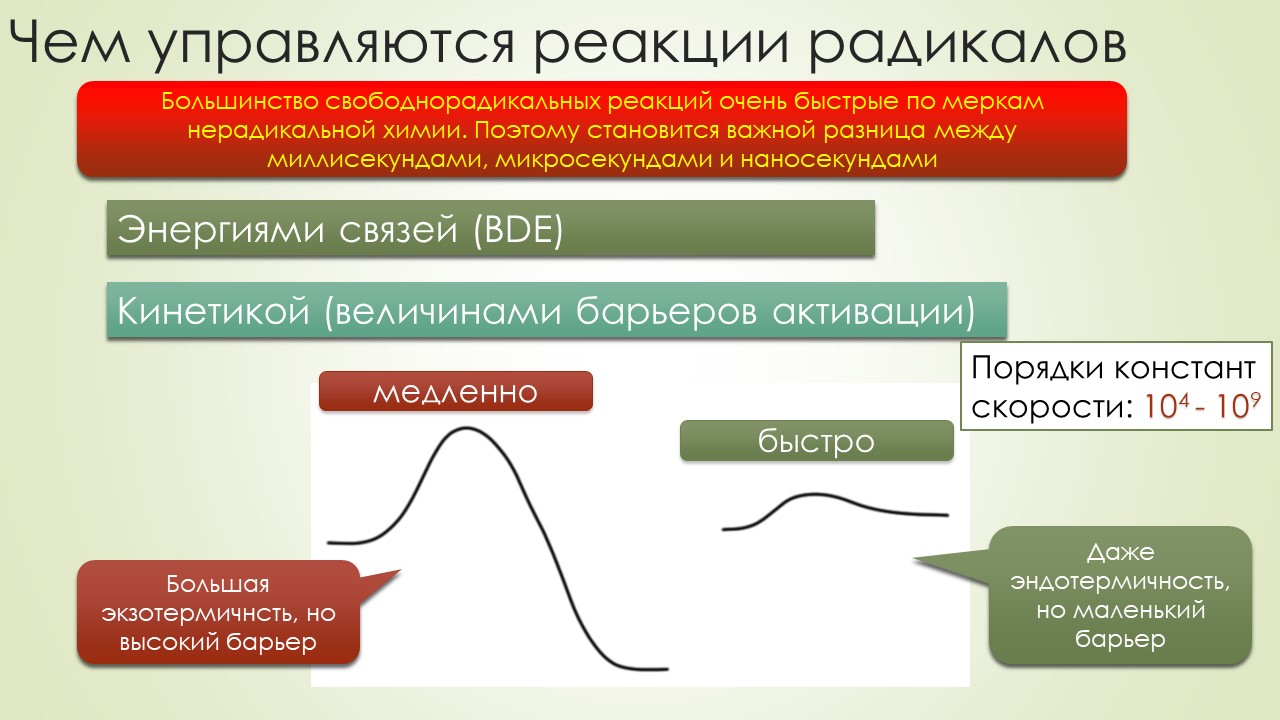
В то же время не у учебной, а в исследовательской химии работа всё же шла. Одна из немного странных побудительных причин к исследованию реакций свободных радикалов состояла в том, что возникла гипотеза о том, что многие обычные реакции типа того же нуклеофильного замещения идут не так как мы учили – пара туда, пара сюда – а через перенос одного электрона (single electron transfer, SET), а при этом неизбежно должны образовываться радикалы. И многих страшно заинтересовал вопрос времен жизни конкретных радикалов и констант скоростей типичных реакций радикалов, потому что без этого невозможно было как-то объективно разобраться в проблеме, имеем ли мы право допускать мимолётное образование радикалов в обычных реакциях. Появилось много методов таких иследований, куча всяких радикальных ловушек и радикальных часов с разными временами жизни, намерили дикое количество скоростей разных элементарных и не очень свободнорадикальных реакций. Появилась некоторая почва не только для обобщений, но и для удивления – свободнорадикальная химия оказалась не такой простой, и закономерности не всегда поддавались объяснениям. С другой стороны, накопление данных по самым разным реакциям показало, что скорости реакций варьируют в очень широких пределах, оставаясь при этом очень большими. Фокус в том, что скорость реакции с константой 10 в девятой отличается от реакции с константой десять в четвёртой (и то, и другое это очень быстро, фактически моментально, доли секунды) на пять порядков, то есть настолько, что если это мысленно перенести в обычные масштабы времени для привычных нам реакций, то одну реакцию мы назвали бы очень быстрой, а вторую очень медленной, и действовали бы соответственно. Получается, что у свободнорадикальных реакций просто другой масштаб времени, и когда мы пытаемся в голове уместить происходящее в радикальной химии, приходится признать, что тот, кто использует обычную, нерадикальную химию, подобен человеку, научившемуся управлять велосипедом, даже скорее трёхколёсным, такие там скорости. А тот, кто собирается научиться использовать и понимать свободнорадикальную химию замахивается на пилотирование сверхзвукового самолёта, надеюсь, среди них есть не только истребители, но и что-нибудь мирное, чтобы просто лететь куда-нибудь с ветерком, никого не убивая и ничего не ломая.
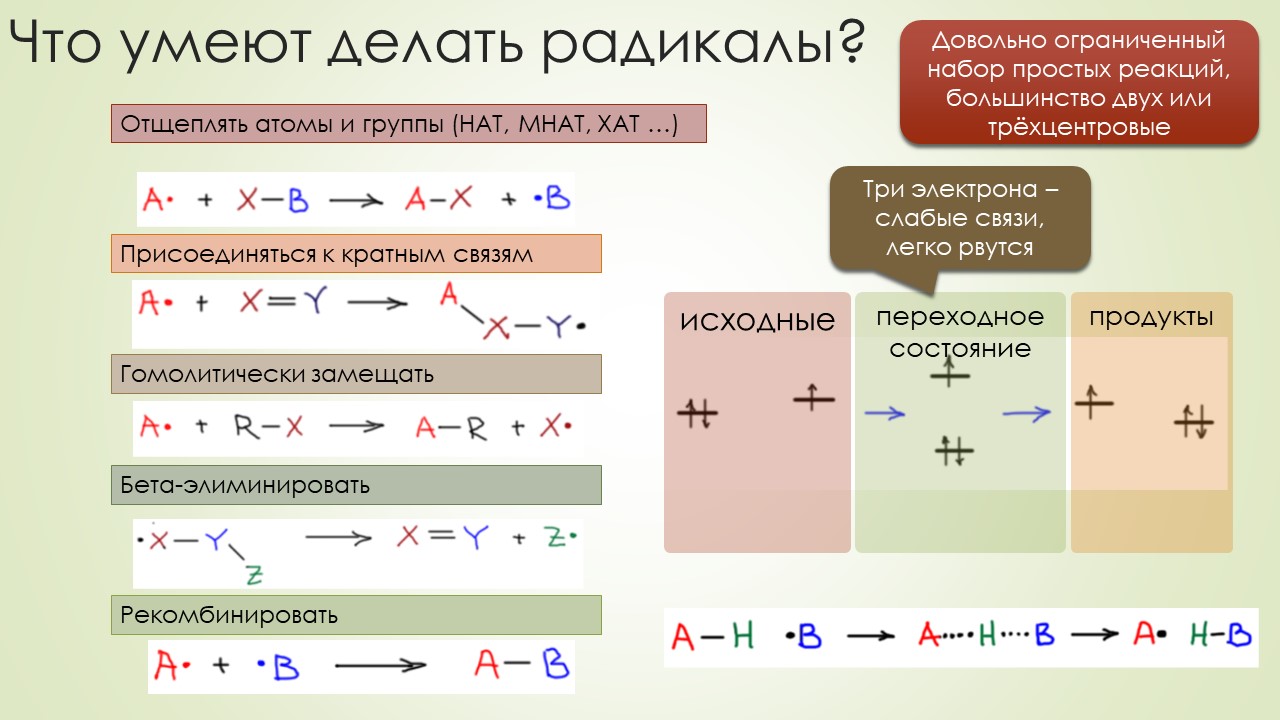
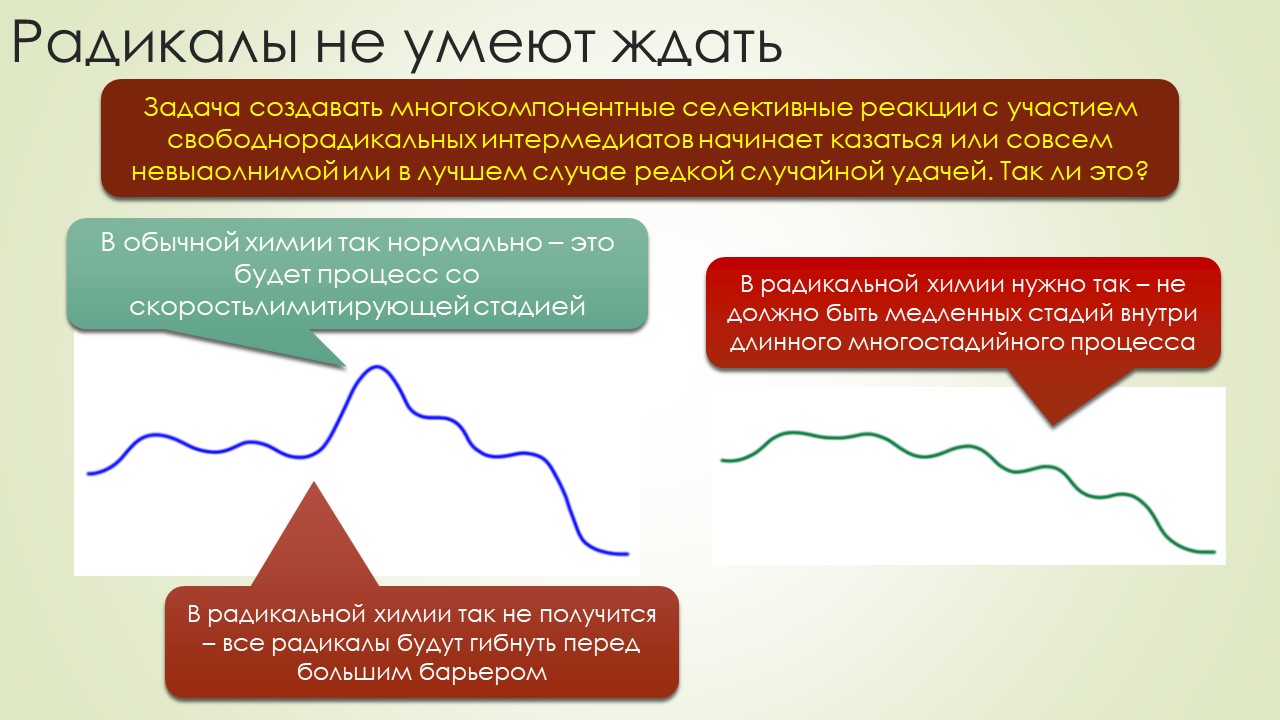
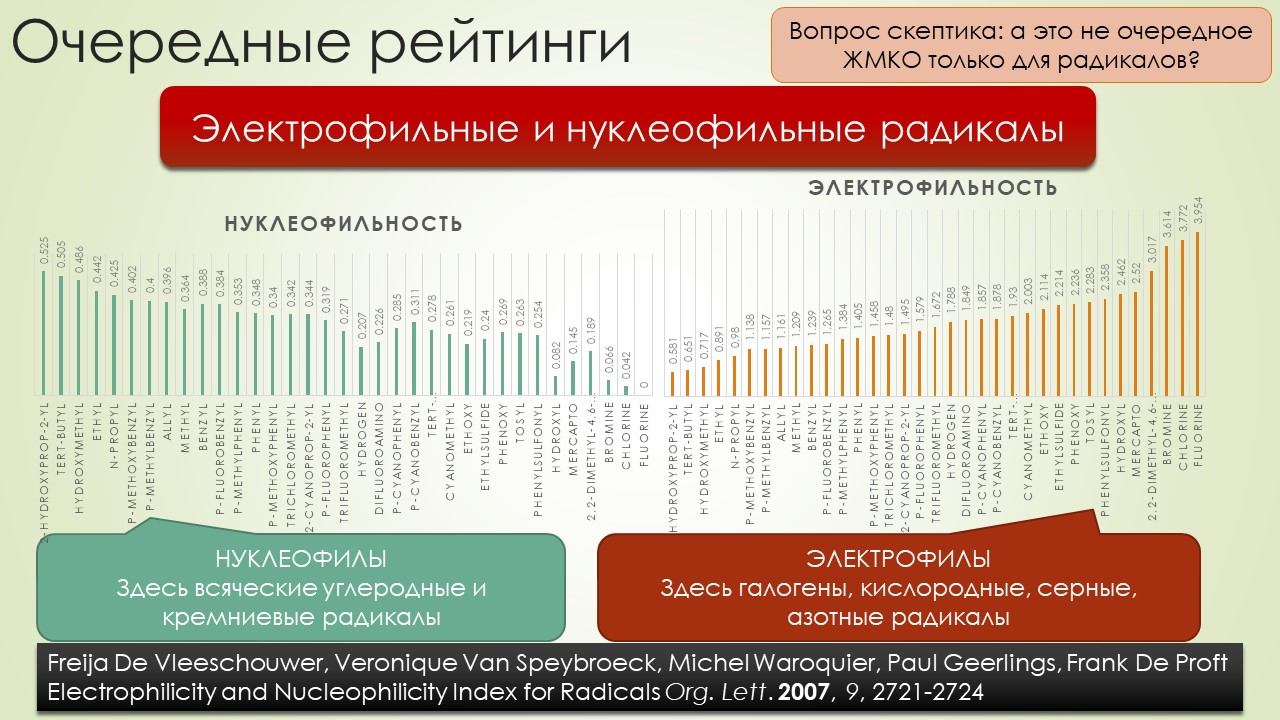
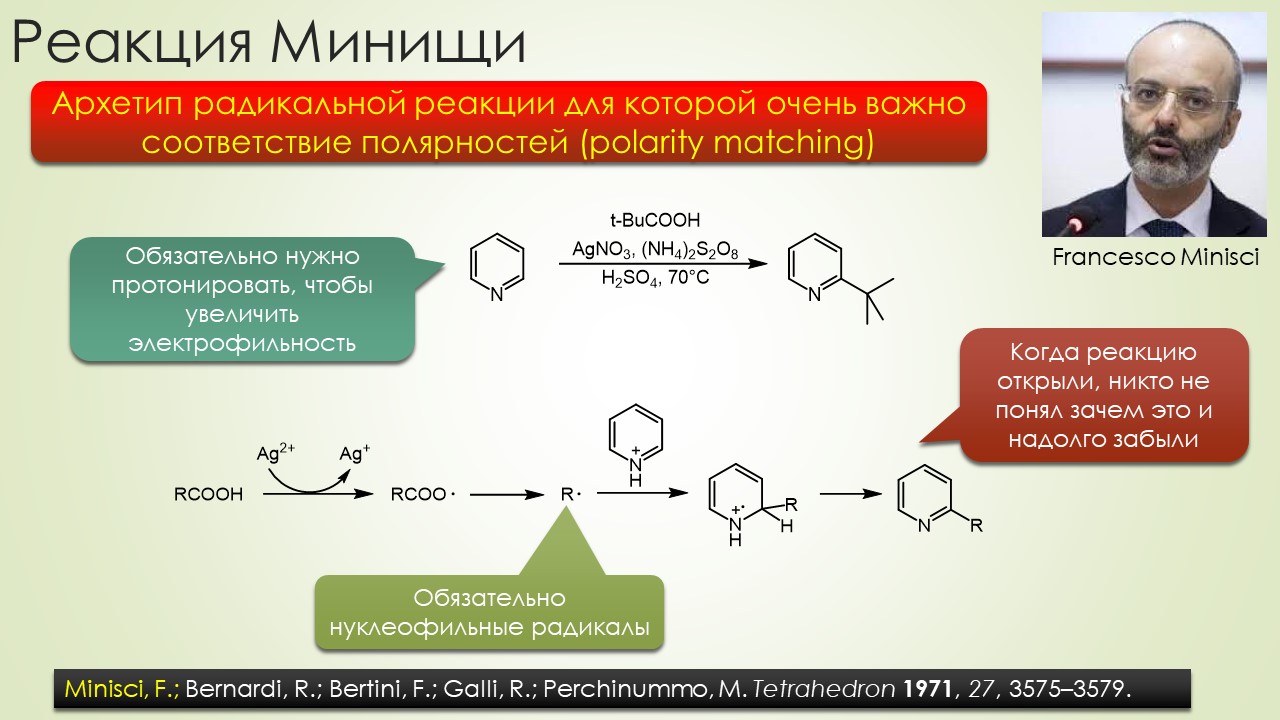
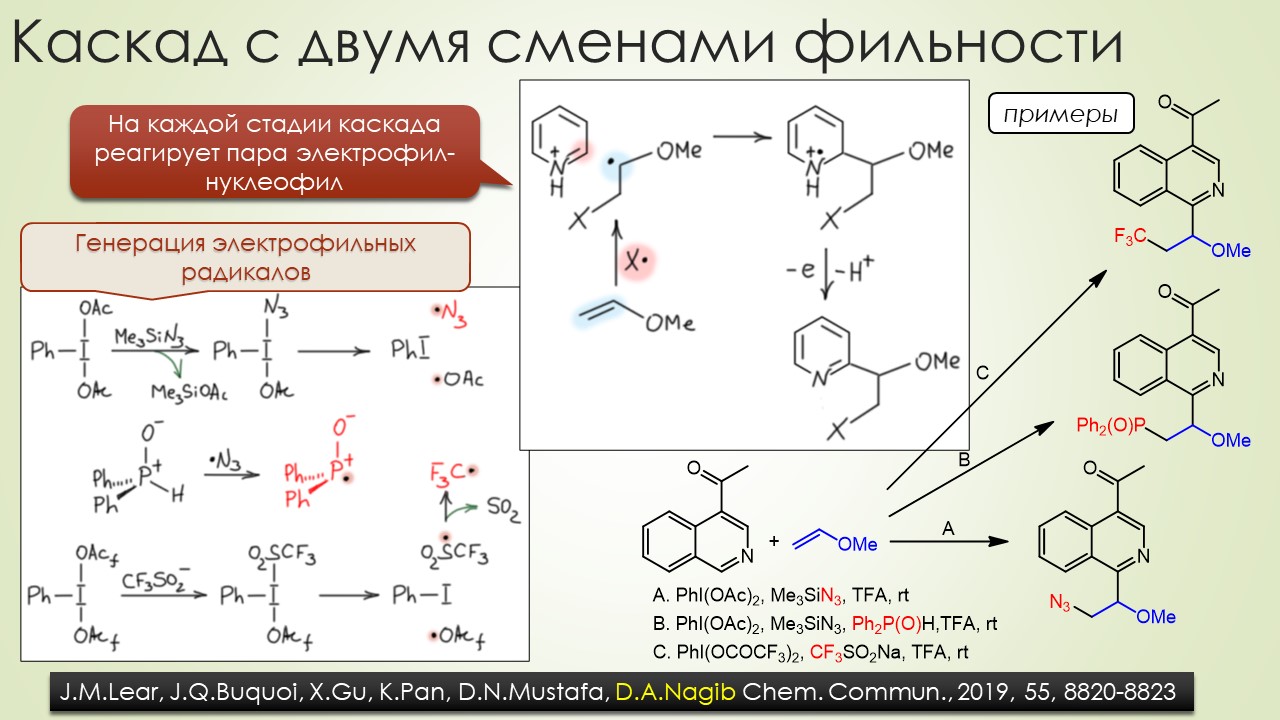
В общем, вполне достаточно, чтобы отбить всякую охоту сюда лезть. Но всё же попробуем, ведь наверное и в этой химии, как и в самолёте, есть какие-то средства управления, помогающие не разбиться в лепёшку на первой секунде полёта или даже прямо на разбеге. Попробуем немного в них разобраться, и в очередой раз убедиться, насколько милы и привычны органическому химику понятия из обычной, нерадикальной химии, раз и здесь мы ищем электрофилов и нуклеофилов, и обнаружив, не можем сдержать слёз радости – работает! Паршивенько, если честно (я не буду пока на этом останавливаться), но работает, позволяя хоть немного разобраться в разных радикалах и обнаружив, что и им свойственна некоторая избирательность при взаимодействии с обычными соединениями – они тоже ищут противоположность, хоть это и не так строго выражено, как в химии частиц с заполненными оболочками. Без этого было бы вообще худо, потому что нас ждёт, например, фокус, основанный на несоблюдении принципа кинетического контроля – в отсутствие равновесия, оказывается, не обязательно преобладает продукт, образующийся с наибольшей скоростью, а в радикальной химии торопиться медленно это и есть самая умная стратегия. И мы убедимся, что в этой химии ситуация, когда вас поджидает ловушка, совсем не такая плохая – в этой химии ловушки добрые и куда надо, туда и затягивают – одного даже в Стокгольм на нобелевскую церемонию – уметь надо ловушки выбирать.
В общем, не химия, а сплошное подтверждение любимых нынче принципов “всё не так однозначно” и “это другое”. Поэтому полезем очертя голову и не боясь трудностей и соблазнов, вдруг понравится так что потом не оторвёшь. В современной химии свободнорадикальных затей стало настолько много, что это неплохая идея, будет чем заняться ещё долго.