Аминокислоты
Аминокислоты – карбоновые кислоты, содержащие амино-группу где-нибудь в молекуле. Аминокислот разных очень много, но нас здесь будут интересовать только такие, из которых состоят белки, хотя в Природе много и других аминокислот. И даже еще точнее – те, которые попадают в молекулы белков исключительно за счет всем известного процесса синтеза белков на рибосомах в результате трансляции – перевода генетического кода нуклеиновых кислот в аминокислотный код белков. Таких аминокислот – их еще называют кодируемыми, потому что каждой соотвествует трехбуквенный генетический код, или протеиногенными, потому что из них образуются белки (они же протеины – это полные синонимы слова белок) в процессе биосинтеза – ровно 20. Обратите внимание, что в состав реальных белков входит большее количество аминокислот, потому что многие белки после собственно образования полипептидной цепи в процессе трансляции на рибосоме претерпевают еще разные дополнительные превращения, их называют пост-трансляционными. Из-за этого особенно в более старых справочниках и книжках аминокислот было больше 20. Но уже довольно давно с этим окончательно разобрались, и число протеиногенных белков больше не изменяется. Набор этот, то ли выработанный эволюцией, то ли спущенный в готовом виде из каких-то неземных мест, очень остроумен. Структура входящих у него аминокислот обеспечивает максимальное разнообразие взаимодействий, как межмолекулярных, так и химических, и по этой причине белки, различаясь последовательностями аминокислот, обладают фантастическим разнообразием форм и свойств.
Что совершенно несущественно для курса органической химии? – Понятие о заменимых и незаменимых аминокислотах. Эти понятия совершенно случайно попали в программу нашего курса, так как это не про химию, а про еду. Не говоря уже о том, что сами эти термины на редкость странно выглядят. Что значит “заменимая” аминокислота? Заменимая чем? Для построения белков нужны все, и нет ни одной лишней. На самом деле, это неудачный перевод с английского (essential) или немецкого (essentielle), что буквально значит, что эти аминокислоты необходимы в пище, организм человека не может их ни откуда взять, если не получит готовыми, и не сможет развиваться, и даже просто нормально существовать. Неудачность этого перевода еще и в том, что произошел невольный перенос отрицания: их essential это наше незаменимый, а их non-essential – это наше заменимый.
Разберемся теперь с разными сторонами аминокислот и пептидов.
Раздел пополняется. Время от времени обновляйте.
Стереохимия аминокислот
Опять речь пойдет только про кодируемые аминокислоты. Одна из них, глицин, нехиральна. Все остальные хиральны. Подавляющее большинство содержат только один стереогенный центр, и именно тот, на котором висят амино- и карбоксильная группа. Эти аминокислоты в Природе являются чистыми энантиомерами. Две, изолейцин и треонин, содержат и еще один стереогенный центр. Обе являются в Природе чистыми диастереомерами.
В стереохимии кодируемых аминокислот нет ничего сложного. Как только мы разобрались со стереохимией углеводов и поняли, что такое относительная конфигурация, определяемая по отношению к эталонной молекуле (там – это D-глицериновый альдегид), то здесь нам уже ничего делать не нужно, так как эталонная молекула в ряду кодируемых аминокислот та же. Эталонная молекула – это единственное, что нужно помнить в этой схеме определения стереохимии. Все кодируемые аминокислоты кроме глицина соотносятся с L-глицериновым альдегидом а следовательно принадлежат к L-ряду. Способ такого соотнесения прост и вполне понятен – располагаем обе молекулы, изображенные проекциями Фишера так, чтобы сверху были старшие функциональные группы, альдегид или кислота. Расположение гидроксила или амино-группы подскажет нам конфигурацию. Внизу располагается все остальное. Слева – эталонная молекула, которую нужно запомнить, D-глицериновый альдегид. В середине его энантиомер – L-глицериновый альдегид. Справа, L-аминокислота, конфигурация которой соответствует L-глицериновому альдегиду. Если от относительной конфигурации перейти к абсолютной, выражаемой с помощью R/S-номенклатуры, получается тоже довольно удобно, так как в аминокислотах (подавляющему их большинству) старшинство групп на стереогенном центре убывает в ряду NH2 > COOH > R (можно себе представить группу R с большим старшинством чем у карбоксила, например, CF3, но среди кодируемых аминокислот таких представителей нет), и тогда относительная L-конфигурация соответствует абсолютной S-конфигурации.
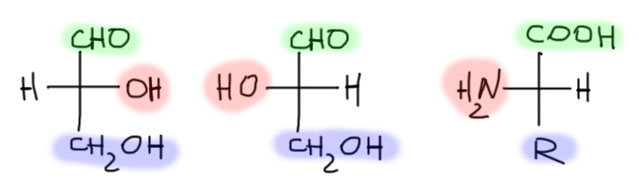
Еще раз подчеркну, что путать абсолютную конфигурацию и относительную нельзя. Простой пример это очень хорошо показывает. Простейшая хиральная кодируемая аминокислота с R = Me называется аланин. L-аланин имеет абсолютную конфигурацию S – это (S)-2-аминопропановая кислота. Если представить себе ее очень простое галогенированное производное, например, трифтор или даже 2-хлораланин, то они тоже будут принадлежать к L-ряду, но абсолютная конфигурация из-за изменения порядка старшинства станет R.
Проекции Фишера очень неудобны для изображения молекул в реакциях. Поэтому всячески рекомендуется использовать естественные проекции. Это очень просто. Расположить молекулу аминокислоты в пространстве можно разными способами, но особенно популярен один, потому что именно так располагаются остатки аминокислот в пептидах. Постоянный остов любой аминокислоты изображают в плоскости либо вверх, либо вниз, и собственно конфигурацию обозначают, показывая направление связи с переменным заместителем. Аминогруппу обычно рисуют слева. Почему – узнаем, когда дойдем до синтеза пептидов.
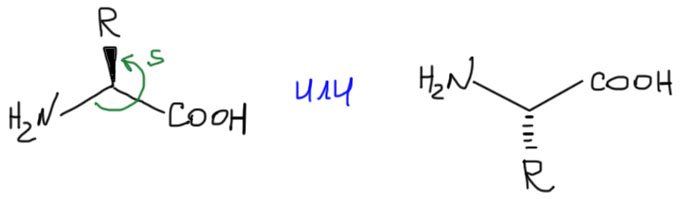
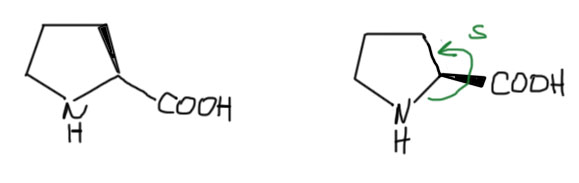
Есть одна очень необычная аминокислота – пролин. В ней нет свободной аминогруппы, а вместо нее гетероцикл пирролидин. Изобразить ее так же, как обычные аминокислоты неудобно, потому что цикл проще изображать формально плоским. Тогда конфигурацию обозначают карбоксилом. Обратим внимание, что это тоже L-аминокислота (иногда ее зачем-то обзывают иминокислотой, что совершенно неверно, вторичные амины никогда не называют иминами), и это очевидно, так как роль обычного заместителя R в пролине играет фрагмент кольца, а этот заместитель тоже имеет самое слабое старшинство после амина и карбоксила.
Стереохимия двух аминокислот с двумя стереогенными центрами, в принципе, знать не обязательно. Но с одной из них, треонином, легко и поучительно разобраться. Само название этой аминокислоты содержит всю информацию о ее стереохимии, а точнее, относительной конфигурации. Она относится к L-ряду, а это задает конфигурацию первого стереоцентра (относительно эталонной молекулы), и корень -трео-, хорошо нам знакомый по паре эритро/трео, а это не что иное, как обозначение относительной конфигурации двух соседних стереоцентров (относительно друг друга). Все, рисуем Фишера. И если очень хочется, то и естественную проекцию, пригодную для использования в цепочках пептидов.
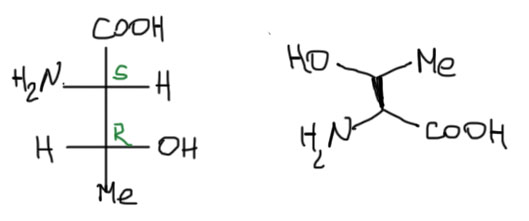 Что касается изолейцина, то конфигурация второго стереоцентра практически никого не волнует. Она известна (S), но большого значения не имеет, так как относится к довольно банальному втор-бутильному заместителю, асимметрия которого мало на что влияет. Диастереомер изолейцина, который называется L-алло-изолейцин хорошо известен и встречается в Природе, так как образуется при случайной эпимеризации настоящего изолейцина. Он настолько похож на правильный изолейцин, что их путает даже транспортная РНК изолейцина: не то, чтобы ей совсем было все равно, нет – это все-таки удивительно тонкий и точный инструмент, но где-то один раз из 20, если концентрации одинаковые, путает. Поскольку такого в клетке никогда не случается, чтобы концентрация настоящего изолейцина и аллоизолейцина почему-то сравнялись, то проблем серьезных с этим не бывает.
Что касается изолейцина, то конфигурация второго стереоцентра практически никого не волнует. Она известна (S), но большого значения не имеет, так как относится к довольно банальному втор-бутильному заместителю, асимметрия которого мало на что влияет. Диастереомер изолейцина, который называется L-алло-изолейцин хорошо известен и встречается в Природе, так как образуется при случайной эпимеризации настоящего изолейцина. Он настолько похож на правильный изолейцин, что их путает даже транспортная РНК изолейцина: не то, чтобы ей совсем было все равно, нет – это все-таки удивительно тонкий и точный инструмент, но где-то один раз из 20, если концентрации одинаковые, путает. Поскольку такого в клетке никогда не случается, чтобы концентрация настоящего изолейцина и аллоизолейцина почему-то сравнялись, то проблем серьезных с этим не бывает.
Синтез рацемических аминокислот
Или, скорее, не совсем синтез, а методы введения амино-группы рядом с карбоксилом.
Строго говоря, никаких особенных реакций для этого не нужно. Большинство таких реакций – уже вполне знакомые нам методы получения аминов с некоторой спецификой. Обратите на это внимание – вводят именно амин при уже готовой карбоксильной группе или ее производному, но не наоборот – в амины не вводят карбоксильную группу.
Еще одна важная вещь – мы не умеем сразу получать оптически чистые вещества. Именно мы не умеем, а не никто не умеет. Методы получения сразу готовых энантиомеров называются асимметрическим или энантиоселективным синтезом, и в современной органической химии таких методов очень много. Но мы их не проходим, боимся сложностей. Вместо этого мы получаем рацемические смеси, надеясь, что существуют методы их разделения на энантиомеры. Посмотрим на некоторые важные методы синтеза рацемических α-аминокислот.
Синтез Штрекера
Это единственный (у нас) метод, в котором одновременно возникают обе функциональные группы. Удивительно древняя реакция, одна из древнейших вообще в органике – Адольф Штрекер открыл ее в 1850 году, когда еще не было даже структурных формул, и статьи писались просто словами с редкими вкраплениями брутто-формул. Часто говорят, что эта реакция, типа, просто аналог хорошо нам известного синтеза циангидринов из карбонильных соединений и HCN.
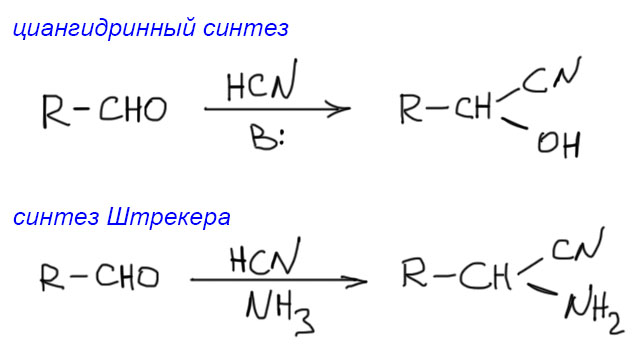 Строго говоря, это не совсем так – синтез Штрекера открыт намного раньше циангидринного синтеза (строго говоря, это не совсем так, потому что циангидринный синтез известен в Природе, он стоит за так называемыми цианогенными гликозидами, самым известным из которых является амигдалин из горького миндаля, а маслом горького миндаля и его превращениями много занимались самые ранние химики – это интересная история, я когда-нибудь расскажу ее или в разделе про циангидрины, или про бензоиновую конденсацию), и тогда уже именно последний является аналогом первого. И это не удивительно – синтез Штрекера проще и легче идет, чем образование циангидринов. В отличие от циангидринов эта реакция фактически необратима. Легко понять почему. В синтезе Штрекера цианид-ион присоединяется не к альдегиду, а к имину, и промежуточный анион на азоте имеет гораздо большую основность и немедленно выхватывает протон из растворителя (Штрекера обычно делают в водных растворителях), смещая равновесие в сторону аддукта.
Строго говоря, это не совсем так – синтез Штрекера открыт намного раньше циангидринного синтеза (строго говоря, это не совсем так, потому что циангидринный синтез известен в Природе, он стоит за так называемыми цианогенными гликозидами, самым известным из которых является амигдалин из горького миндаля, а маслом горького миндаля и его превращениями много занимались самые ранние химики – это интересная история, я когда-нибудь расскажу ее или в разделе про циангидрины, или про бензоиновую конденсацию), и тогда уже именно последний является аналогом первого. И это не удивительно – синтез Штрекера проще и легче идет, чем образование циангидринов. В отличие от циангидринов эта реакция фактически необратима. Легко понять почему. В синтезе Штрекера цианид-ион присоединяется не к альдегиду, а к имину, и промежуточный анион на азоте имеет гораздо большую основность и немедленно выхватывает протон из растворителя (Штрекера обычно делают в водных растворителях), смещая равновесие в сторону аддукта.
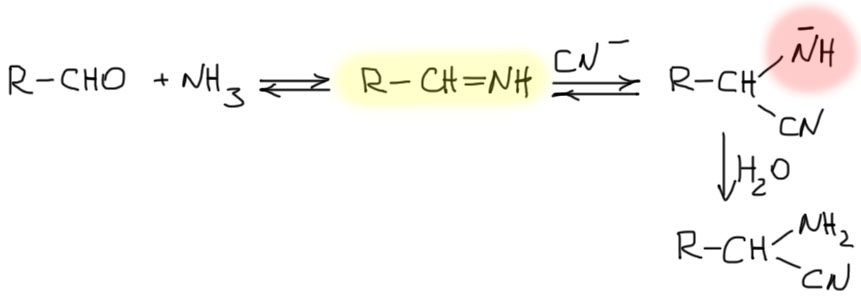
Более того, как правило в синтезе Штрекера не останавливаются на нитрилах, а немедленно гидролизуют аддукт разбавленной соляной кислотой. Гидролиз идет очень легко и дает рацемические аминокислоты, естественно, в виде солей. Обычно всю реакцию делают так:
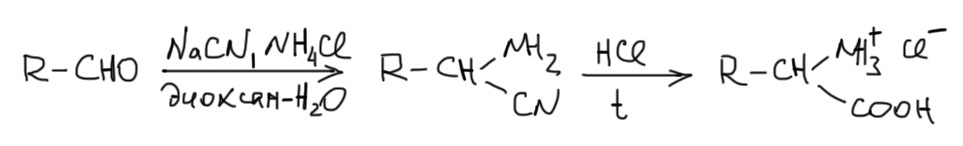
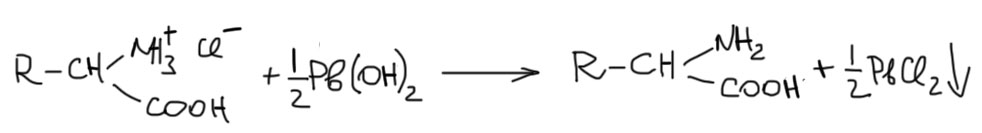
При желании получить свободную аминокислоту, а не ее соль, можно легко наломать дров. Вроде бы, что может быть проще – добавить эквивалент щелочи, и дело в шляпе. Конфуз будет в шляпе, а не дело! Проблема в том, что в аминокислотах есть и кислотная, и основная группы, причем их pK различны. У каждой аминокислоты есть свое соотношение кислотности и основности (обсудим эту проблему подробнее в другом месте), но здесь просто отметим, что для каждой аминокислоты пришлось бы подбирать свое pH, чтобы получить суммарно электронейтральную молекулу аминокислоты, а не преобладание катионной или анионной формы. Но издревле эту проблему решают остроумнее – используют для вытеснения свободной аминокислоты из солянокислой соли такое странное основание, как гидроокись свинца. Ничего странного – это, во-первых, слабое основание, которое само по себе не может оторвать протон от карбоксила, а только забирает противоион из-за низкой растворимости хлорида свинца, ну и с противоионом эквивалент протонов. Только нужно это делать аккуратно и действовать расчётным количеством гидроксида свинца, иначе можно молучить свинцовый хелат аминокислоты, который придётся разлагать обратно и оказаться в той же точке, с которой начали.
По какой-то загадочной для меня причине стал часто всплывать один специфический вариант реакции Штрекера, который называют методом Тимана. Этот метод опубликовал в 1880 Фердинанд Тиман (Tiemann, F., Chem.Ber., 1880, 13, 381.), которого мы знаем по реакции Реймера-Тимана в химии фенолов. Ничего особенного в этом варианте нет, это именно минимальная модификация, а не принципиальное расширение. Вообще, этот Тиман – довольно скользкий персонаж, любитель примазываться к чужим работам. И к реакции Реймера он приписался задним числом, и здесь просто подсуетился. Суть метода Тимана проста – берется сразу именно готовый циангидрин и обрабатывается метанольным раствором аммиака. 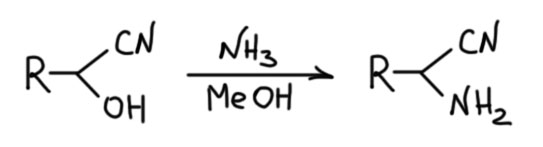 Что здесь происходит совершенно очевидно – в присутствии основания образование циангидрина обратимо, ну и равновесный альдегид вступает уже в реакцию Штрекера по обычному механизму через имин. Равновесие сдвинуто именно в сторону аминонитрила, но это мы уже и так поняли. Услуги кислотно-основного катализа и перетаскивания протонов с места на место обеспечивает пара аммиак-аммоний.
Что здесь происходит совершенно очевидно – в присутствии основания образование циангидрина обратимо, ну и равновесный альдегид вступает уже в реакцию Штрекера по обычному механизму через имин. Равновесие сдвинуто именно в сторону аминонитрила, но это мы уже и так поняли. Услуги кислотно-основного катализа и перетаскивания протонов с места на место обеспечивает пара аммиак-аммоний.
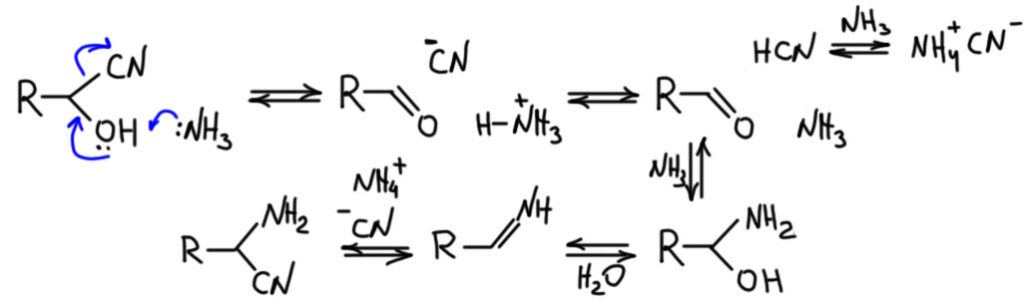 Вообще, справедливовсти ради заметим, что это сейчас мы такие умные, чуть что сечём механизм и в три пинка всё объясняем, а в 19 веке всё это было только в проекте и каждую реакцию открывали с чистого листа. Поэтому Тиман все же вполне хороший химик, и оставил своё имя вечности незадаром. Большим достоинством этого метода является крайняя простота и выполнения (просто крутим при комнатной температуре или очень мягком нагревании) и выделения продукта – всё летучее кроме ожидаемого аминонитрила, поэтому просто упариваем все на роторе и продукт очищаем обычными способами.
Вообще, справедливовсти ради заметим, что это сейчас мы такие умные, чуть что сечём механизм и в три пинка всё объясняем, а в 19 веке всё это было только в проекте и каждую реакцию открывали с чистого листа. Поэтому Тиман все же вполне хороший химик, и оставил своё имя вечности незадаром. Большим достоинством этого метода является крайняя простота и выполнения (просто крутим при комнатной температуре или очень мягком нагревании) и выделения продукта – всё летучее кроме ожидаемого аминонитрила, поэтому просто упариваем все на роторе и продукт очищаем обычными способами.
Восстановительное аминирование
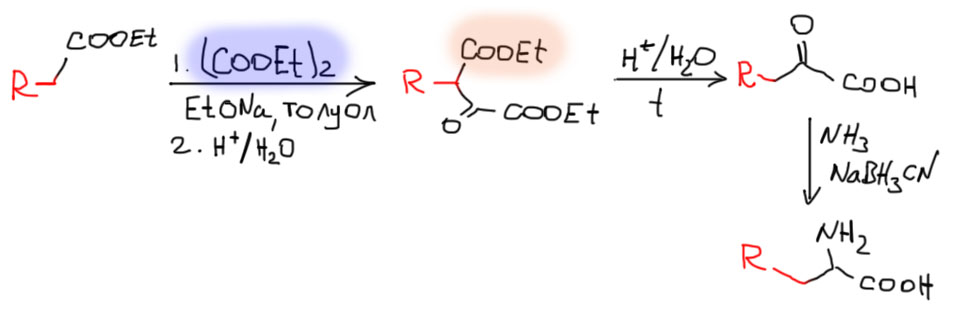
Если у нас есть α-кетокислота, то можно применить восстановительное аминирование. Этот простой путь очень важен, хотя бы потому что именно так многие аминокислоты образуются в Природе – это едва ли не основной путь биосинтеза. Но и в колбе все должно быть неплохо, если у нас есть α-кетокислота – все дело только в ней. Умеем ли мы делать α-кетокислоты? Честно говоря, не очень. Другие кетокислоты мы делали в огромных количествах: β-кетокислоты из сложноэфирной конденсации, γ-кетокислоты и еще более далекие кетокислоты – из продуктов сложноэфирной конденсации. А вот с α-кетокислотами все не так весело. Конечно, их можно получать окислением α-гидроксикислот, но беда в том, что их мы делаем гидролизом циангидринов, а зачем нам это нужно, если мы владеем гораздо более прямым синтезом Штрекера.
Один метод все-таки есть, и мы его знаем, хотя и основательно забыли. Сложноэфирная конденсация с эфиром щавелевой кислоты (диэтилоксалатом) дает двойной кето-эфир, который обычно подвергают нагреванию с элиминированием CO. Но можно этого и не делать, а просто провести гидролиз с декарбоксилированием (не забывайте главное правило, по которому определяют, какие карбоксилы отлетают при нагревании – посмотрите на страничке про сложноэфирную конденсацию). Дальше проводим восстановительное аминирование.
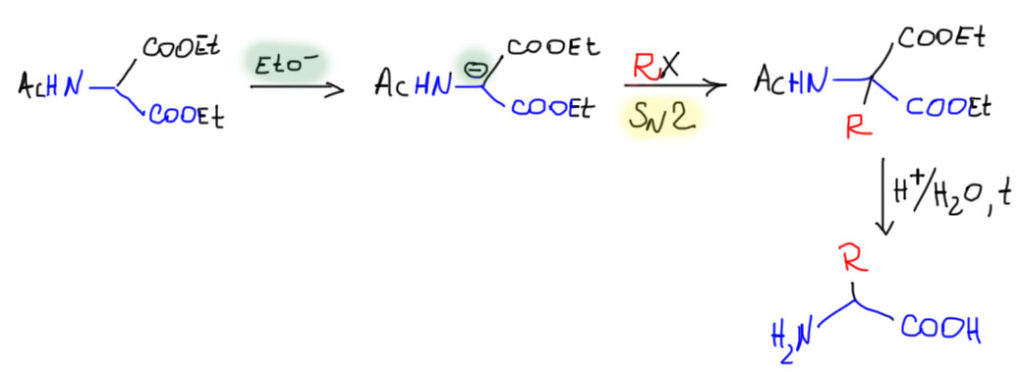
Разновидность малонового синтеза
Синтезировать кислоты через малоновый эфир мы умеем очень хорошо. Точно так же можно синтезировать и рацемические аминокислоты, если взять вместо малонового эфира его легкодоступное производное с защищенной аминогруппой – ацетамидомалоновый эфир. Эта штука исключительно легко получается из малонового эфира очень простыми реакциями – нитрозированием, и восстановлением нитрозо-производного цинком в присутствии уксусного ангидрида (запрещен в Российской Федерации).
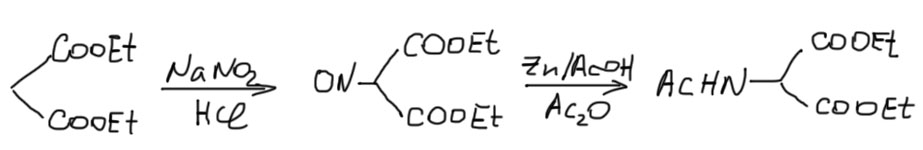
Ацетамидомалонат точно так же как и обычный малоновый эфир обладает повышенной CH-кислотностью, легко депротонируется самым незатейливым основанием, и реагирует как нуклеофил в SN2-замещении. В этом месте хорошую службу служит ацетильная защита, которая одновременно ослабляет донорный эффект неподеленной пары азота, которая без этого сильно ослабляла бы CH-кислотность, и одновременно мешала бы в замещении. Как обычно в таких случаях напомню, что реагент для замещения нужно проверять на SN2. Впрочем, кроме SN2 здесь можно использовать и присоединение по Михаэлю. Дальше следует обычный кислотный гидролиз с декарбоксилированием, и одновременно и амин высвобождается из под ацетильной защиты. Это очень удобный метод синтеза разнообразных α-аминокислот, известный как метод Сёренсена.
У этой реакции есть весьма популярная разновидность, использующая фталимидное производное малонового эфира – фталоиламиномалоновый эфир, который легко получают из малонового эфира свобонорадикальным бромированием с последующей реакцией Габриэля.
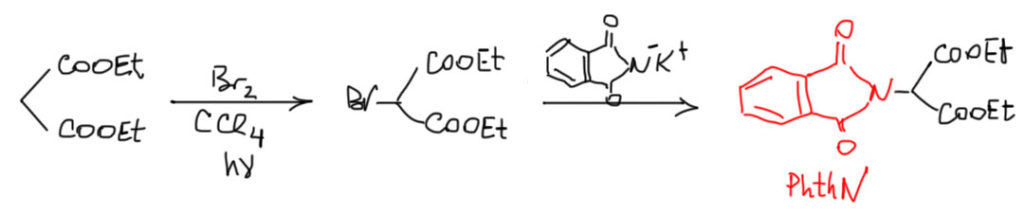
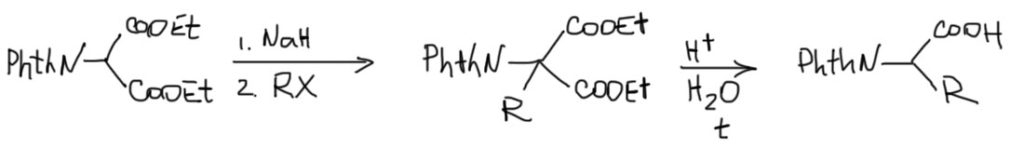
Эта группа сокращается PhthN и довольно часто используется, как защитная для амино-группы. В этом прелесть этого варианта синтеза – вы получаете защищенную аминокислоту, которую можете использовать в синтезах, например, пептидов, и не снимать эту защиту сразу.
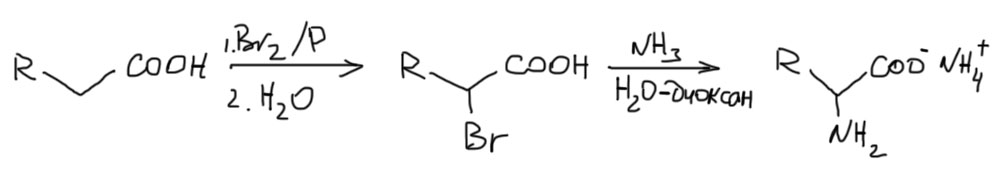
Нуклеофильное замещение
Это самый простой метод просто введения аминогруппы в уже готовую структуру карбоновой кислоты. Кислоты легко бромируются по методу Гелля-Фольгарда-Зелинского. Получающиеся α-бромкислоты, как и все подобные галогенпроизводные, очень хороши для SN2-замещения, настолько хороши, что они отлично реагируют даже с обычным аммиаком (напомню, что в синтезе других аминов этот путь не рекомендован из-за множнства сложностей). Единственная проблема – образуется не сама аминокислота, а ее аммонийная соль, и здесь уже придется подбирать pH для каждой аминокислоты для получения электронейтральной формы желаемой кислоты.
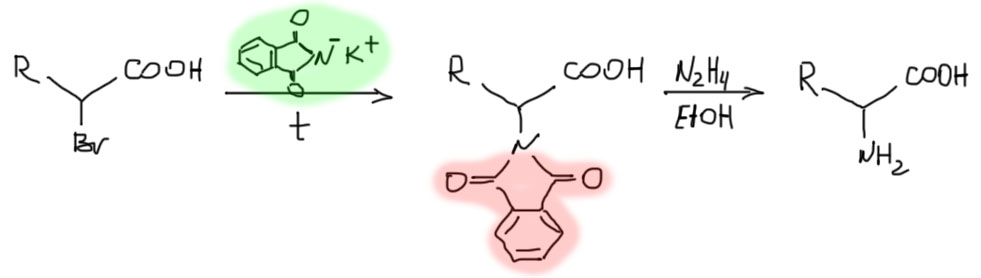
Разновидностью этого метода является использование фталимида в методе Габриэля. Фталимид – очень слабый нуклеофил (фталимид калия – легкодоступный реактив), но для такого замечательного по реакционной способности бромпроизводного он вполне годится. Достоинством метода является то, что фталоильная группа является отличной и очень стабильной защитной группой, и этот метод выбирают тогда, когда образующееся производное планируют дальше использовать в каких-то синтезах, для которых свободная аминогруппа может быть помехой. Тогда фталоил снимают (обычно действием гидразин-гибрата) только после всех превращений.
Оптически активные аминокислоты
Как получить рацемическую аминокислоту, ясно. А что делать, если нужна оптически чистая аминокислота, такая, как в Природе? Один путь понятен – у Природы же ее и позаимствовать. Выделить из гидролизата какого-нибудь легкодоступного белка (этот путь условно можно назвать – из рогов и копыт, так как это как раз и есть легкодоступные белковые материалы). Но этот путь подразумевает необходимость разделения смесей аминокислот. Для некоторых аминокислот с особыми свойствами из-за наличия в молекуле той или иной функциональной группы, это вполне возможно и даже довольно просто. Но тут еще одна засада – в обычных белках, тем более таких незатейливых, как просто строительные материалы для тех же рогов и копыт, ассортимент аминокислот довольно беден, и основу их составляют самые простые аминокислоты. Более богатый выбор можно получить, если обратиться к биотехнологии – заставить, например, какой-нибудь микроорганизм производить конкретные аминокислоты. И т.п.
А если без помощи Природы. Тогда нужно разделять рацемические смеси на отдельные энантиомеры – заняться расщеплением. Основная проблема здесь в том, что индивидуальные энантиомеры обладают совершенно одинаковыми свойствами по отношению ко всему нехиральному. Поэтому их просто так нельзя разделить обычными физическими методами – перегонкой, хроматографией, перекристаллизацией. С перекристаллизацией есть некоторая зацепка, потому что иногда смесь энантиомеров кристаллизуются не в виде рацемата, а отдельными кристаллами (это называется спонтанным расщеплением), – получается смесь кристаллов, которую можно попробовать разделить визуально, потому что у кристаллов и форма тоже бывает асимметрическая и можно найти кристаллики, которые являются зеркальным отражением друг друга. Именно так впервые и разделил энантиомеры винной кислоты Луи Пастер аж в 1849 году. Это красиво, но невероятно утомительно, хотя бы потому что когда кристаллы выпадают из раствора, только немногие имеют правильную красивую форму, а остальные выглядят далеко не так выставочно и поди пойми, где там какой энантиомер. У Пастера получилось сделать еще красивее: найдя по несколько хороших кристаллов каждого вида, он использовал их как затравку и получил отдельно кристаллизацию каждого из энантиомеров. Это выглядит уже намного производительнее, но требует просто филигранной экпериментальной работы – не забудем, что растворимость у каждого из энантиомеров одинакова, и раствор, насыщенный по одному, будет насыщенным и по другому, а кристаллизация вызывается не только затравкой, но и массой других случайных причин, а после начала кристаллизации одного из энантиомеров, раствор по нему сразу станет ненасыщенным, а по второму продолжит таковым оставаться. В общем, тот, кто сможет повторить трюк Пастера в хоть сколько-нибудь приличном масштабе, точно заслужит титул гроссмейстера перекристаллизации. То, что эту процедуру проделывают (точнее, проделывали) в промышленных масштабах при производстве L-глутаминовой кислоты, натриевая соль которой обильно применяется как вкусовая добавка, не стоит расссматривать, как признак того, что это очень просто – в промышленных условиях любой процесс сначала годами отлаживают в лабораториях и пилотных установках пока не будет достигнута полная воспроизводимость, а сам процесс тщательно контролируют.
Кристаллизация диастереомерных производных
Более удобный и универсальный метод состоит в разделении не энантиомеров, а диастереомеров или веществ, похожих на диастереомеры. В очередной раз повторим, что в отличие от энантиомеров, диастереомеры обладают разными, иногда очень сильно различающимися свойствами, совершенно всеми свойствами, включая растворимость. Поэтому если мы сможем превратить энантиомеры в диастереомеры, то точно сможем их разделить, подобрав условия кристаллизации. Можно получить какие-то производные или по амино-группе (амид с оптически активной кислотой) или карбоксильной (сложный эфир с оптически активным спиртом), раскристаллизовать их на два диастереомера, и провести гидролил, чтобы достать уже оптически активную кислоту. Просто как пример посмотрим на эфиры с легкодоступным природным энантиомерно чистым спиртом, ментолом (это та самая штука, которая отвечает за мятный вкус и странное ощущение так называемой “свежести” везде и всюду, от конфет и жвачки до лекарств, шампуней и зубной пасты). Получают эфиры с помощью тех же методов сшивки, как и в синтезе пептидов, предварительно закрыв амино-группу, чтоб не мешалась, ацилированием. У ментола есть еще две группы с точным расположением в пространстве (это RSR-стереоизомер), но это не важно, потому что мы расщепляем не ментол, а аминокислоту, поэтому обратим внимание только на расположение гидроксила.
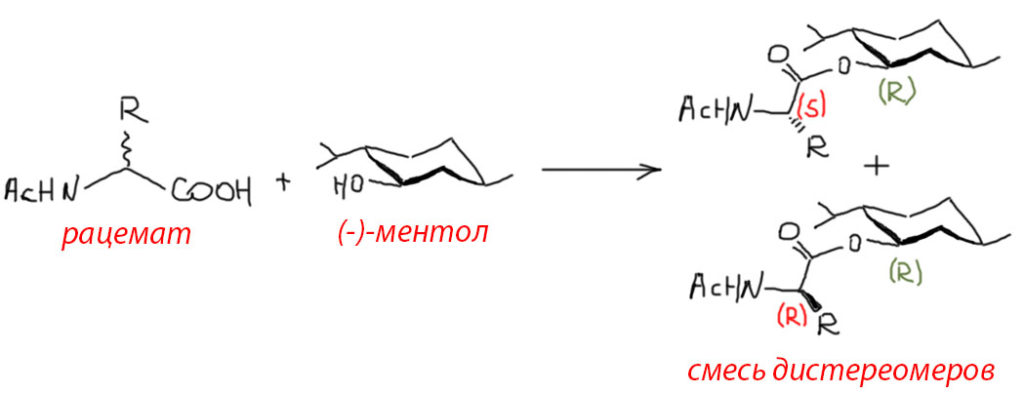
Получаем смесь диастереомерных эфиров, которую можно раскристаллизовать, потому что диастереомеры имеют разную растворимость. Растворитель и условия подбирают экспериментально.
Обратим внимание на то, что задача изначально непроста, потому что обычно перекристаллизацией отделяют вещество, которое преобладает в смеси, а все остальное является примесями. В этом же случае есть два вещества (диастереомеры), содержание которых в начальной смеси заведомо одинаково, а разница в растворимости вряд ли очень велика, так как вещества опять таки заведомо очень похожи. Поэтому кристаллизацию нельзя вести так, как мы это обычно делаем в практикуме – растворяем в кипящем растворителе (и хорошо если в минимально возможном количестве, а то, как мы любим, так и вообще “на глазок”) и просто охлаждаем вплоть до льда, чтобы побольше выпало. Это даже в простых случаях техника плохая, но она оправдана, когда нужно просто убрать несколько процентов примесей в основном веществе. Но в случае раскристаллизации диастереомеров это заведомо обречено на провал. В этом случае важно даже не минимальное количество растворителя, а медленное и осторожное охлаждение, лучше при перемешивании, что создает максимально близкие к равновесным условия – даем молекулам в растворе возможность образовать однородные по составу зародыши, на которых будут расти кристаллы. И отделяем кристаллическую фазу без форсированного охлаждения, лучше отобрать несколько фракций, образующихся при постепенном снижении температуры.
Точно так же экспериментально определяют, какой диастереомер выкристаллизовывается, и какая на нем висит аминокислота. В растворе остается смесь, сильно обогащенная другим диастереомером. После уже дополнительными перекристаллизациями достигают желаемой оптической чистоты каждого из диастереомеров. Повторные перекристаллизации позволяют дочистить до высокой чистоты как тот диастереомер, который выпал в первой перекристаллизации, так и второй, который остался в растворе в смеси с остатками того, который выпал. Это очевидно, потому что повторные перекристаллизации уже проходят в обычном режиме, когда в смеси преобладает одно вещество, а второе является примесью. Опытный и упорный экспериментатор может достичь почти полного разделения смеси, но это требует высокого мастерства и тонкого понимания процесса кристаллизации.
Перекристаллизация (как и любой другой физический или химический процес разделения) никогда не может дать чистых диастереомеров (энантиомеров) со 100%-ным содержанием. Довольно легко и быстро достигается 85-90% разделение, а дальше уже требуется то самое высокое мастерство, чтобы получить 97-98%. Что это за проценты? Это не выход чистого диастереоизомера, а оптическая чистота или энантиомерный избыток, сокращается ee. Это очень простая и очевидная характеристика, измеряемая в процентах, как и обычный выход. Даже если мы забыли, что это такое, моментально вспомним, просто имея в виду очевидную вещь – она равна 0% для рацемической смеси и 100%, если энантиомер совершенно чист. Такую величину очевидно можно получить, просто определив разность между содержанием того энантиомера, которого больше, и того, которого меньше. А экспериментально это элементарно определяется измерением угла вращения смеси. У рацемата она равна 0, у чистого энантиомера – удельному углу вращения этого энантиомера. В промежутке имеем простую линейную зависимость как раз от энантиомерного избытка.
Кристаллизация солей с оптически активными кислотами или основаниями
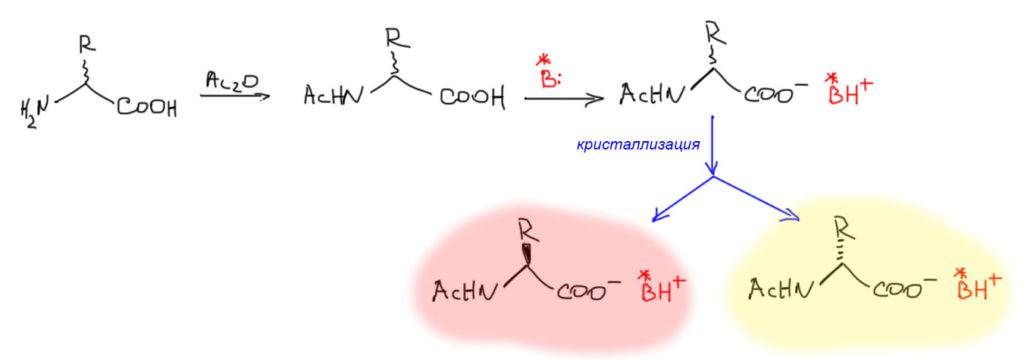
Раскристаллизация диастереомерных производных – метод хороший, но очень трудоемкий. Производные нужно сначала получить, потом раскристаллизовать, потом обратно превратить в аминокислоту. Есть способ проще, хотя и еще более чувствительный к качеству работы экспериментатора. Это кристаллизация солей с оптически активными кислотами или основаниями. Если то, что вам нужно разделить – кислота, то выбираем основание, Если основание – то кислоту. А если, как в случае аминокислот, и то, и то? То и выбрать можно и то, и то, но гораздо удобнее сначала сделать аминокислоту только кислотой (подавив основность амино-группы, например, ацилированием), или только основанием (убрав свободный карбоксил в сложный эфир). То есть, все равно какую-то химическую работу сделать нужно, но это очень простая работа. Вот как-тот так: берем рацемическую аминокислоту, обрабатываем ее уксусным ангидридом, убираем основную амино-группу, добавляем оптически чистое основание в количестве, необходимом для образования соли из обеих энантиомеров рацемата, и создаем условия кристаллизации (концентрируем раствор до начала кристаллизации, охлаждаем, трём стенки стеклянной палочкой, и т.п.) и в случае успеха получаем кристаллы и маточник. Для обоих определяем удельный угол вращения (или, если мы в 21 веке и у нас есть хроматограф с хиральной колонкой, определяем стереоизомерный состав хроматографически), и пытаемся понять, во-первых, получается ли вообще разделение, или падает смесь, не обогащенная ни ожним из энантиомеров, и, во-вторых, если получается, то где какой энантиомер. Предсказать это невозможно, если только вы просто не воспроизводите уже известную методику. Если все получается, продолжаем кристаллизацию, собираем фракции, кристаллизуем их еще раз или несколько раз, так, как это описано выше для кристаллизации диастереомерных производных. В конце обрабатываем соль щелочью для отделения хирального основания (которое собирают, чистят и используют для следующих разделений), гидролизуем защиту аминогруппы и перекристаллизовываем уже чистый энантиомер аминокислоты.
Что такое оптически активные основания для этой работы? Это могут быть и очень простые вещи типа 1-фенилэтиламина, энантиомеры которого можно недорого купить (смотрим в каталог реактивов, вот (S)-энатиомер 99% оптической чистоты около тысячи долларов за полкило, (R)-энантиомер вообще почти даром, грамм за доллар). А могут быть и вещи посложнее, природные вещества, в растениях полно всяких энантиомерно чистых азотистых оснований, называемых алкалоидами. Особенно популярен алкалоид бруцин, который вместе с всем известным стрихнином выделяют из плодов тропического дерева рвотный орех. Это очень сложное и красивое энантиомерно чистое соединение, и, кстати, совсем недорогое, просто потому что его выделяют вместе со стрихнином из этих орехов в очень больших количествах. Во, какое чудище.
Я насчитал 6 хиральных атомов углерода и один хиральный азот, верхний, то самый, который и протонируется и отвечает за основность бруцина (нижний, который индольный, он плоский, нехиральный). Но сколько там стереогенных центров не важно, важно то, что в целом эта молекула очень сильно асимметрична, и когда, в протонированном виде, образует соль с асимметричным анионом, то разные энантиомеры по разному взаимодействуют с такой сложной формой. Любопытства ради посмотрим, как выглядит эта молекула в пространстве (по данным рентгеноструктурного анализа, структура 197896 Кебриджской базы данных), и чтобы было яснее, покрутим ее. Видим, что она сильно неплоская, и напоминает такой затейливый кривой черпачок, или даже такую полуоткрытую горсть руки, и это хорошее сравнение, потому что хиральность всегда ассоциируется с асимметрией руки. 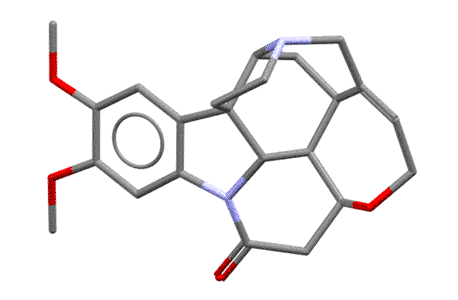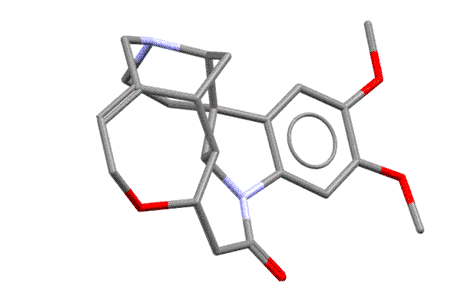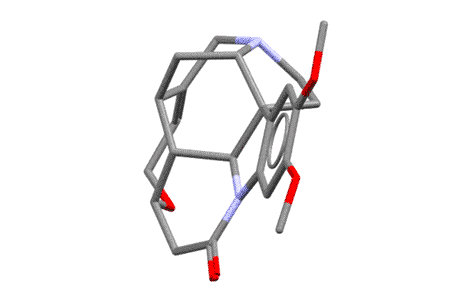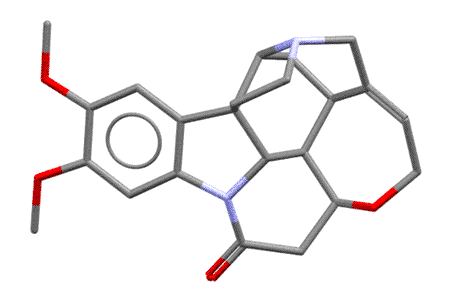 Представьте теперь две другие несимметричные штуковины, совершенно одинаковые по форме, но представляющие собой отражения в зеркале – пойдут, например, руки другого человека, левая и правая (если найдете что-то другое того же типа, пойдет и это). Попробуйте вложить в раскрытую горсть по очереди два таких объекта, левый и правый – сразу увидите, насколько по разному они вписываются в асимметричную горсть. Вот это ровно то, что нужно – это показывает, что взаимодействие одного асимметричного объекта с парой взаимно зеркальных других асимметричных объектов, сильно отличается. А когда что-то отличается, то всегда одно будет лучше другого (в одной паре взаимодействия будут сильнее чем в другой паре). Так приблизительно и работает различение (дискриминация) двух энантиомеров одной молекулы другой хиральной молекулой. Молекула бруцина в этом смысле очень хороша, она большая, имеет очень сложную форму и при этом ее соли хорошо кристаллизуются, поэтому бруцин очень часто применяют для расщепления на энантиомеры рацемических смесей всяких органических кислот. Единственный ее недостаток – очень большой молекулярный вес, нужно много (в граммах) бруцина, чтобы расщепить несколько граммов органической кислоты. Более простые оптически активные основания типа уже упомянутого 1-фенилэтиламина тоже используют, и принцип их действия тот же – взаимодействие асимметричного катиона фенилэтиламина с энантиомерами анионов какой-то хиральной кислоты попарно различается, одна пара будет лучше другой, энергия взаимодествия будет больше, кристалл получится прочнее, а более прочный кристалл (кристалл с большей энергией кристаллической решетки) менее растворим и легче выкристаллизовывается. Но когда асимметрия не так сильно выражена, как у того же бруцина, разница будет меньше, разница растворимостей меньше, и раскристаллизовать труднее.
Представьте теперь две другие несимметричные штуковины, совершенно одинаковые по форме, но представляющие собой отражения в зеркале – пойдут, например, руки другого человека, левая и правая (если найдете что-то другое того же типа, пойдет и это). Попробуйте вложить в раскрытую горсть по очереди два таких объекта, левый и правый – сразу увидите, насколько по разному они вписываются в асимметричную горсть. Вот это ровно то, что нужно – это показывает, что взаимодействие одного асимметричного объекта с парой взаимно зеркальных других асимметричных объектов, сильно отличается. А когда что-то отличается, то всегда одно будет лучше другого (в одной паре взаимодействия будут сильнее чем в другой паре). Так приблизительно и работает различение (дискриминация) двух энантиомеров одной молекулы другой хиральной молекулой. Молекула бруцина в этом смысле очень хороша, она большая, имеет очень сложную форму и при этом ее соли хорошо кристаллизуются, поэтому бруцин очень часто применяют для расщепления на энантиомеры рацемических смесей всяких органических кислот. Единственный ее недостаток – очень большой молекулярный вес, нужно много (в граммах) бруцина, чтобы расщепить несколько граммов органической кислоты. Более простые оптически активные основания типа уже упомянутого 1-фенилэтиламина тоже используют, и принцип их действия тот же – взаимодействие асимметричного катиона фенилэтиламина с энантиомерами анионов какой-то хиральной кислоты попарно различается, одна пара будет лучше другой, энергия взаимодествия будет больше, кристалл получится прочнее, а более прочный кристалл (кристалл с большей энергией кристаллической решетки) менее растворим и легче выкристаллизовывается. Но когда асимметрия не так сильно выражена, как у того же бруцина, разница будет меньше, разница растворимостей меньше, и раскристаллизовать труднее.
И если уж мы сюда заехали, то разберемся еще в одной вещи, почему-то всегда вызывающей вопросы. Соли, состоящие из энантиомерно чистого основания и пары энантиомерных кислот не являются диастереомерами в строгом смысле этого слова, по крайней мере, в растворе, где ионы достаточно свободны или даже совсем свободны. Растворите такую пару солей, и в растворе ионы тут же перемешаются. Разделение происходит только при кристаллизации. А в кристалле это можно назвать диастереомером? Нет. Если вы когда-нибудь видели, как устроены кристаллы органических соединений, то понимаете, что молекулы там расположены в большом порядке, каждая на своем месте, и если это ионы, то и ионы будут занимать определенные положения, и каждый будет на своем месте, – но сказать, какой катион принадлежит какому аниону невозможно. Хотя мы тут развели всякие метафоры про черпачки и горсти, но реальность сильно сложнее – энергия кристаллической фазы определяется не только парными взаимодействиями анион-катион, но всей суммой взаимодействий находящихся рядом в кристалле молекул и ионов. Кристалл – это чертовски сложная вещь, и анализ взаимодействий в ней – невероятно сложная задача. Именно поэтому кислоты или основания для кристаллизации подбирают только экспериментально, и сказать заранее, какой из раделяемых энантиомеров пойдет в кристалл, а какой останется в растворе, невозможно.
Синтез пептидов - общие идеи
Пептиды – это аминокислоты, соединенные в линейную цепь одним и только одним способом, пептидной связью. Пептидная связь – это просто амид – аминогруппа от одной аминокислоты, и карбоксил от другой. Вот, например, пептид, состоящий из трех аминокислот – трипептид. Пептиды обычно рисуют одним способом, слева направо, слева располагают свободную аминогруппу (это называется N-конец – N-terminus), справа – свободную карбоксильную группу (это называется C-конец, C-terminus – почему C, а не O, ведь на конце именно O? – потому что реакционным центром при образовании амидной связи является атом углерода, электрофильный карбонильный углерод). Обратим внимание на стереохимическую конфигурацию заместителей, которую очень легко изображать, когда мы уже договорились о том, как изображать L-аминокислоты.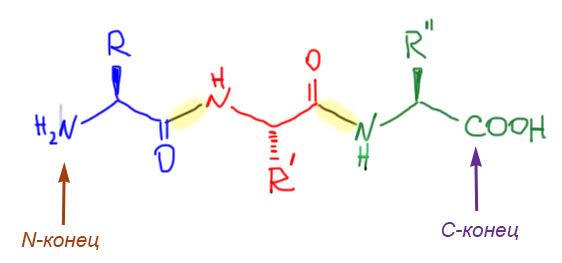
Такое расположение цепи всегда используется и в названии пептидов – остатки перечисляют от N-конца. Каждый остаток называют от аминокислоты, обычно заменяя -ин на -ил:
глицин – глицил, лейцин – лейцил, валин – валил (не валинил, хотя это название и можно иногда встретить), и т.п.;
но, цистеин – цистеинил, а не цистеил;
триптофан – триптофанил;
и еще проблема с глутаминовой кислотой и глутамином – остаток от первой называется глутамил, а от второго глутаминил;
аналогично, аспарагинованя кислота – аспарагил, аспарагин – аспарагинил.
В Природе, в клетке любых организмов пептиды синтезируются в результате процесса трансляции на рибосомах. Хотя это изумительно сложный процесс, в его основе лежит вполне определенная химическая реакция, которая нам очень хорошо знакома. Когда мы хотим сделать амид из кислоты и амина, мы всегда делаем одно и то же – берем кислоту и получаем из нее более реакционноспособное производное, обычно хлорангидрид, и вводим его в реакцию с свободным амином. Этот процесс – получение хлорангидрида или другого производного из карбоксила – называют активацией карбоксила. Вот и в Природе происходит ровно то же самое: аминокислота сначала подхватывается транспортной РНК именно за карбоксильную группу, и это огромное сооружение, аминоацил-тРНК, и есть активированная форма по карбоксилу, такой природный аналог хлорангидрида.
Если мы хотим сделать пептид, даже самый простой из двух аминокислот, дипептид, на придется сделать то же самое – у той аминокислоты, которая даст карбоксил в пептидную связь, нам нужно этот карбоксил активировать. Но мы не сможем это сделать просто так, так как в той же молекуле есть амино-группа, которая тут же с такой активированной группой прореагирует – мы просто получим какой-то дурацкий полимер, полиамид. Это очень типичная ошибка, которую мы очень часто делаем, когда пытаемся получить соединение, содержащее группы, которые друг с другом охотно реагируют, например, пытаемся получить хлорангидрид из амино или гидроксикислоты. Не выйдет. Группу, которая в данный момент не используется, необходимо защитить. Во второй молекуле нам понадобится амин, и казалось бы, карбоксил мешать не будет. Будет, еще как будет – просто вспомните химию производных карбоновых кислот, там все время попадаются равновесия типа кислота плюс производное другой кислоты –туда-сюда– производное кислоты плюс другая кислота. Поэтому мы получим не только то, что ожидаем, но и частично перенос активированной группы на другую молекулу и образование пептида из двух одинаковых аминокислот: реакция будет неселективна. В химии пептидов этого боятся как огня, потому что с пептидами работать очень непросто, и если сразу получать не очень чистые, то представьте, какая помойка получится вместо чистого пептида, если мы захотим не дипептид, а что-нибудь подлиннее. Поэтому и вторую молекулу аминокислоты тоже придется защитить, только по карбоксилу. И только тогда мы сможем сделать реакцию и получить амид, активировав карбоксильную группу в молекуле со свободной карбоксильной группой.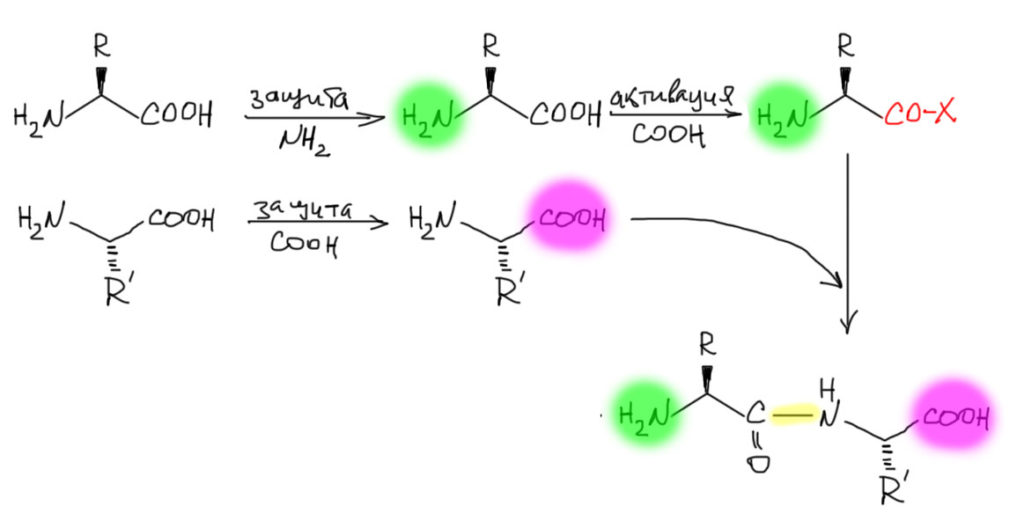
Пока у нас дипептид вопросов вообще нет. Но как только мы захотим добавить еще одну аминокислоту, они возникнут. Во-первых, у нас сейчас с обеих сторон группы закрыты защитами. И чтобы туда еще что-то присобачить, нужно сначала защиту снять. Сразу встает вопрос, откуда ее снимать, с амина или карбоксила. Можно и так, и так, и это имеет принципиальное значение, потому что определяет, в каком направлении мы собираемся строит пептид, от амина к карбоксилу (или от N- к С-концу), или наоборот (от С- к N-концу). Известны и широко применяются оба способа, но с одним важным замечанием: построение пептидной цепи реакциями в растворах, иными словами, обычными методами органической химии (взял колбу, загрузил туда реактивы и растворитель, поставил на мешалку, дождался конца реакции, обработал реакционную смесь и выделил из нее продукт, очистил его и определил качество подходящими методами) почти всегда делают от N- к С-концу, и именно поэтому так обычно и располагают пептидные цепи. Но есть и другой способ сделать эту работу, неудачно называемый твердофазным синтезом, и в нем как раз предпочитают обратный способ.
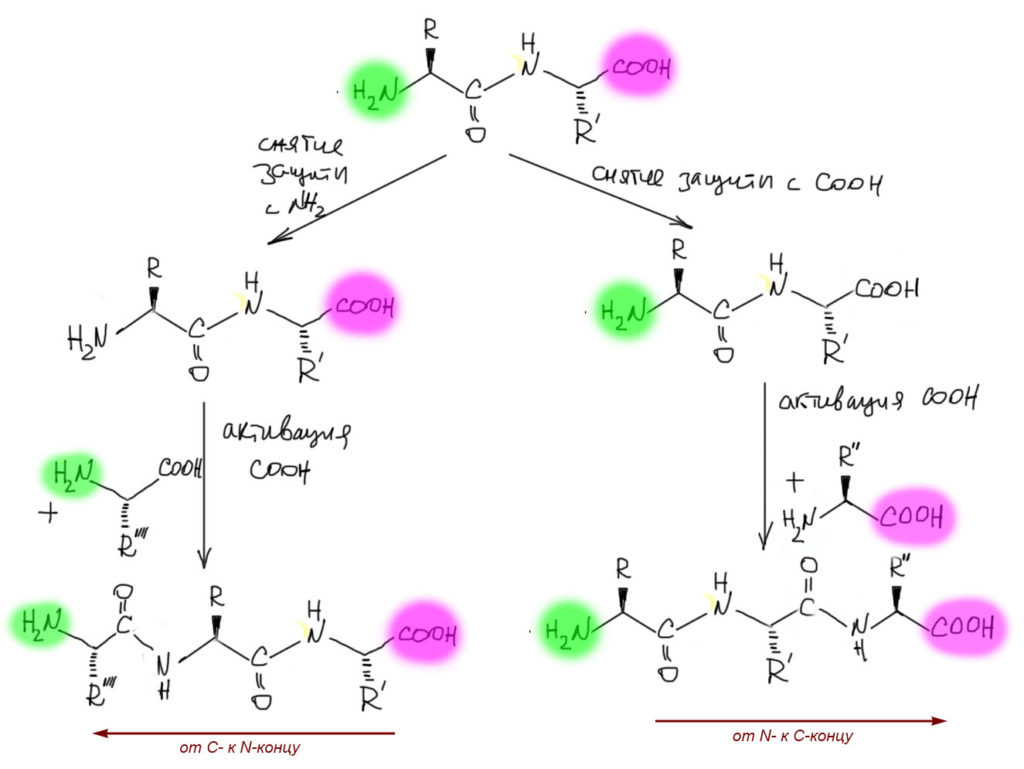
Синтез пептидов в растворах: как это делают
Теперь разберемся, как конкретно собирают пептиды. Скажем сразу, что не будем слишком сильно в это углубляться, потому что проблема эта чрезвычайно сложна, и в ней невероятное количество деталей. Одна из проблем в том, что из 20 аминокислот больше половины содержат еще какие-то реакционноспособные группы: гидроксилы (серин, тирозин, треонин), амины (лизин), карбоксилы (аспарагиновая и глутаминовая кислоты), амиды (аспарагин и глутамин), гуанидин (аргинин), живой имидазол с NH-группой (гистидин), тиол (цистеин), и живой индол с NH-группой (триптофан). И эти группы активно мешают пептидному синтезу, и их нужно прикрывать своими защитными группами. И в разных сочетаниях аминокислот в пептидной цепи происходят всякие побочные реакции, и все это нужно знать и учитывать, и есть даже такие сочетания аминокислотных остатков, что как их ни соединяй, все равно получается криво. Синтез пептидов поэтому далеко не всесилен, и реально получать этим способом можно далеко не любые пептиды, особенно если они более-менее длинные. А в некоторых случаях синтез наоборот упрощается, например, если пептидная цепь содержит остатки глицина – в этом случае синтез можно вести кусочками, и соединять их потом на глициновых фрагментах. Тот же фокус проделывают с пролином.
Не будем тратить на все это время. В реальной науке в наше время синтез пептидов вот такой классической сборкой из аминокислот делают не так часто.
Посмотрим на самую простую процедуру. Соберем пептид из аминокислот, не содержащих никаких лишних активных функций, а таких тоже немало: глицин, аланин, валин, лейцин, изолейцин, фенилаланин, метионин, да и триптофан, в принципе, тоже годится. Нам нужно разобраться в нескольких вещах.
Первое – как защищается амин
Ограничимся самой популярной защитной группой. Для любопытствующих я попозже отдельно разберу немного более широкий ассортимент. Во-первых, амины почти всегда защищают в виде производных угольной кислоты, полуамидов-полуэфиров, также называемых уретанами. Самая популярная защитная группа – трет-бутилоксикарбонил, сокращенно Boc. Эту группу обычно вешают с помощью такого своеобразного ангидрида моно-трет-бутилового эфира угольной кислоты (по-другому на это смотрят как на трет-бутиловый эфир воображаемой пироугольной кислоты). Еще нужно основание, чаще всего диизопропилэтиламин (DIPEA).
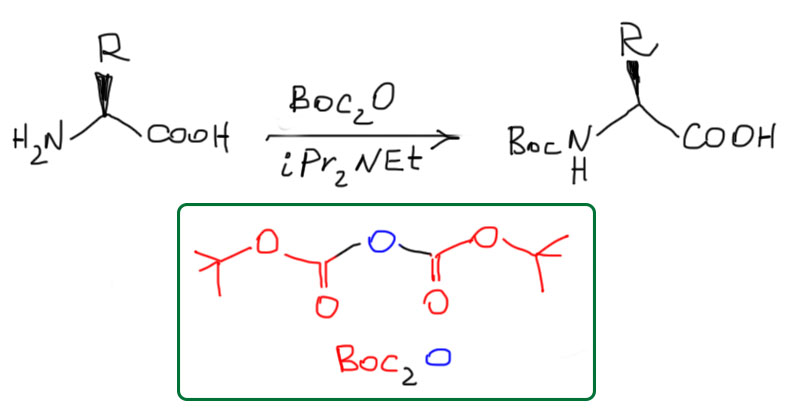
Защитная группа Boc очень хорошо сидит, и нормально выдерживает восстановители, нуклеофилы, основания, и даже мягкий кислотный гидролиз. Снимается она в условиях принудительного SN1-замещения (можно освежить в памяти SN1 и что такое принудительная SN1 реакция), например, действием трифторуксусной кислоты.
Второе – как защищается карбоксил
И это мы отлично знаем – в виде сложного эфира. Годятся самые простые, метиловые или этиловые. Получают их весьма просто, но не совсем просто – не годится обычный наш способ, как мы это пишем, H+ и все, имея в виду, что карбоновую кислоту просто нагревают в избытке спирта в присутствии какой-нибудь сильной кислоты. В данном случае такая неряшливость не катит. Нужно делать так же, только точнее – раствор аминокислотыв в спирте насыщают сухим газообразным HCl (не соляной кислотой!). Другой вариант, более удобный, так как не требует всей этой занудной сборки промывалок для сушки газов, состоит в том, что в раствор аминокислоты в спирте прикапывают эквивалент SOCl2, который как раз и дает при взаимодейтвиии со спиртом искомый HCl. В этих процедурах получается протонированная форма эфира аминокислоты, и это нужно учитывать при проведении реакции ситеза амида – там понадобится лишний эквивалент основания.
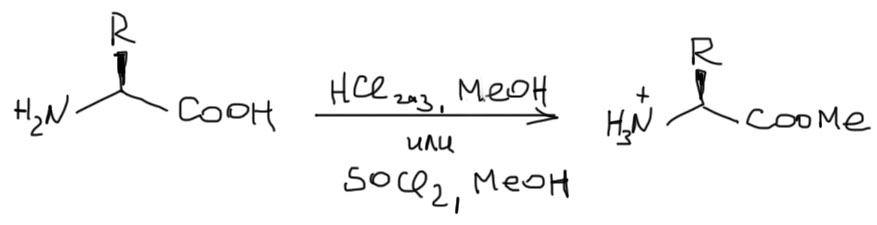
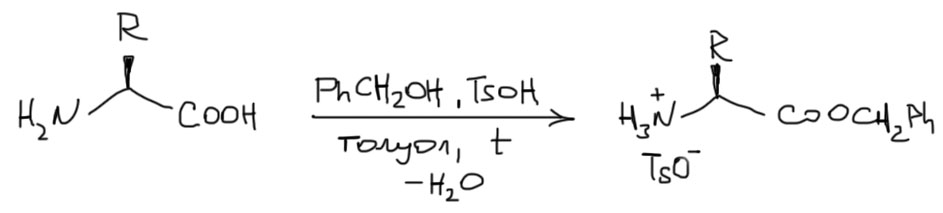
Еще более популярны бензиловые эфиры. Их получают тоже очень просто – нагревают с насадкой Дина-Старка раствор аминокислоты и толуолсульфокислоты с небольшим избытком бензилового спирта в бензоле или толуоле – вода отгоняется в боковой приемник. Бензиловый эфир тоже получается в виде соли с толуолсульфокислотой.
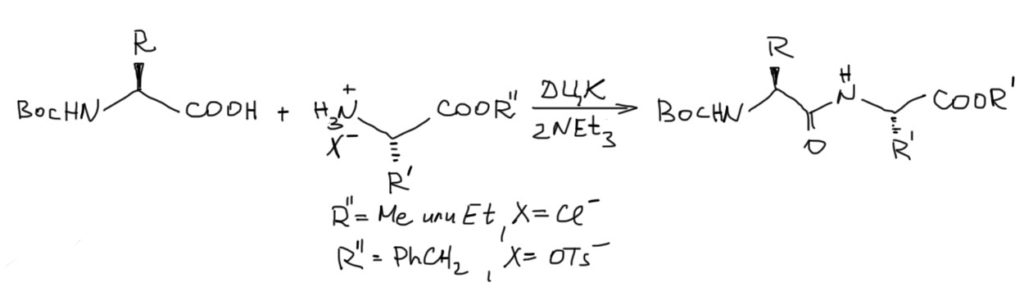
Третье – как активируется карбоксил.
Самый простой и распространенный метод нам уже известен – использование карбодиимидов. Этот прием мы разобрали в теме карбоновые кислоты. Коротко и практично: берутся аминокислота с защищенным карбоксилом и карбокислота с защищенным амином, и в растворе дихлорметана добавляют дициклогексилкарбодиимид (ДЦК) и основание, обычный триэтиламин (два эквивалента, так как нужно еще нейтрализовать кислоту из соли защищенной по карбоксилу аминокислоты). Крутим, отфильтровываем осадок дициклогексилмочевины, и в растворе получаем искомый пептид.
Что дальше?
В зависимости от того, что нам нужно – именно дипептид или мы хотим продолжить наращивание пептидной цепи. Если мы уже приехали, идем ниже в пункт про снятие защит в конечном продукте. Ели хотим продолжать, то
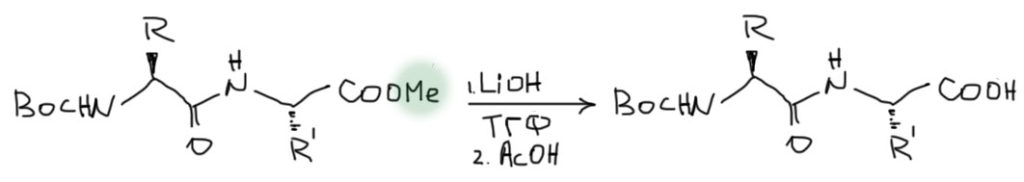
Присоединяем еще одну аминокислоту
Для этого нужно снять защиту с карбоксильной группы, но оставить защиту на амино-группе. Метиловые или этиловые эфиры гидролизуют гидроксидом лития в растворе ТГФ. Почему не обычный наш способ – гидроксидом натрия или калия в спирте? Потому что в этом случае часто происходит рацемизация – основание отнимает протон от α-углерода в аминокислотных остатках, и через енолизацию остаток теряет оптическую чистоту. Это очень плохо. Такой пептид нам не нужен. Поэтому берут гидроксид лития, – он тоже щелочь, но маленький размер катиона лития и его очень приличная льюисова кислотность держат этот катион вместе с гидроксид ионом, эффективно снижая основность, а следовательно и побочные реакции, связанные с депротонированием. А вот для нуклеофильности, а расщепление эфира требует именно ее, это все даже и неплохо, потому что катион лития тем же способом активирует карбонил эфира к атаке гидроксидом. Поскольку амино-группа пока еще защищена и основность ее подавлена, проблем с добычей карбоксильной группы из аниона нет
Бензиловые эфиры расщепляют гораздо мягче и без проблем с возможной рацемизацией – именно за это их и любят. Связь бензила с килородом расщепляется в условиях гидрирования водородом с палладиевым катализатором – но только это не гидрирование (гидрирование – это присоединение водорода к кратной связи), а гидрогенолиз (то есть, расщепление водородом). Это очень чистая реакция в результате которой получается сразу карбоксил.
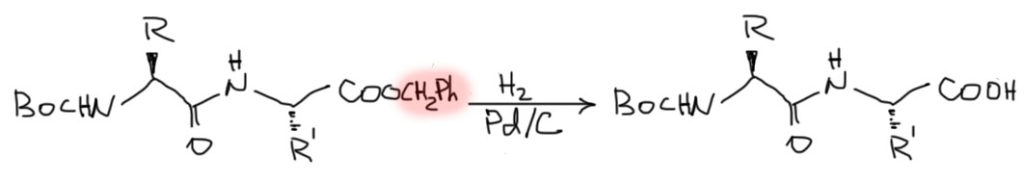
После снятия защиты с карбоксила можно насаживать следующую аминокислоту в виде метилового, этилового, или бензилового эфира. И так далее, до достижения необходимой длины.
Окончательное снятие защит
Когда необходимая длина пептида достигнута, снимают защиты с обоих концов, используя соответствующие методы. Полученный пептид подвергают очистке специфическими методами работы с такими молекулами, например, хроматографией на сшитых полимерах. Не будем про это – это не наша наука.
Защитные группы: подробнее
Синтез пептидов из аминокислот – почти идеальный повод для обсуждения защитных групп. В синтезе пептидов, особенно с включением аминокислот с дополнительными функциональными группами, используют огромное количество всяких защитных групп фактически всех возможных типов. Совсем глубоко мы в это не полезем, но несколько важных принципов рассмотрим. Один из важных вопросов, который всегда встает, когда мы смотрим на все эти странные сокращения в схемах современных синтезов, а защитные группы принято не писать структурами, а именно сокращать несколькими буквами – зачем их так много.
Ответ очень простой. В длинных синтезах сложных молекул с множеством разных функциональных групп они активно мешают друг другу. Хотите оторвать откуда-то протон, а в молекуле есть гидроксилы и амиды, и протон отрывается не там, где нужно. Хотите окислить спирт в кетон, а в молекуле есть амин, который тоже легко окисляется. И т.д., и т.п. В идеале мы должны иметь возможность защитить, то есть фактически временно убрать со сцены, любую из функциональных групп, и точно так же в любой момент вернуть ее обратно, потому что хотим наконец заняться именно ей. И точно так же мы должны иметь возможность по желанию снять защиту с любой из групп, не затрагивая защиты остальных. Такое свойство защитных групп – не мешают друг другу при введении и при снятии – красиво называется ортогональностью. Тем, кто привык к математической терминологии, это не нужно объяснять: ортогональность, она же перпендикулярность – свойство независимых измерений, в трехмерном мире таковых три, соотносимые с осями x, y, z; в многомерном независимых измерений может быть много. Это понятие вполне метафорически переносится на химию. Если превращения одной группы никак не затрагивают другую, их можно считать ортогональными. Беда этого определения в том, что его можно понять буквально: группы ортогональны, если одна никогда не мешает другой. Но это совсем не так. Химия – наука практическая, нестрогая. И ортогональность в ней понимается гораздо более либерально – не никогда не мешает, а найдется хотя бы одна реакция, в которой не мешает. Если найдется, именно эта реакция будет рекомендована, как метод снятия данной группы. Для хорошо нам уже знакомой группы Boc, например, кроме рекомендованного SN1-сольволиза трифторуксусной кислотой, ее можно снять и обычным гидролизом, как любой амид, хотя и в достаточно жестких условиях (мы это разбирали в теме Производные карбоновых кислот – амиды гидролизуются практически так же, как сложные эфиры, но намного медленнее, поэтому не составляет труда гидролизовать сложный эфир в присутствии амида). Поэтому при выборе метода снятия защитных групп это имеют в виду – подбирают условия снятия для каждой комбинации. В сложных случаях это становится головоломно-творческой задачей. Виртуозная игра на защитных группах – признак выдающегося мастерства синтетика.
Мы не будем сразу ломиться на Олимп, а попробуем разобрать несколько популярных комбинаций защитных групп только для аминокислот.
Защита амино-группы
Амино-группу чаще всего защищают в виде амидов полуэфира угольной кислоты – такие полуэфиры, полуамиды называются уретанами, а названия их получаются из обозначения группы R’ за которой следует –оксикарбонил-, поэтому в английских сокращениях обычно есть –oc. Самые популярные защитные группы этого типа – уже знакомый нам Boc. Бензильное производное обычно сокращают в Cbz, и есть еще третье, наиболее занятное, производное углеводорода флуорена – флуоренилметилоксикарбонил, сокращается соответствеено в Fmoc.
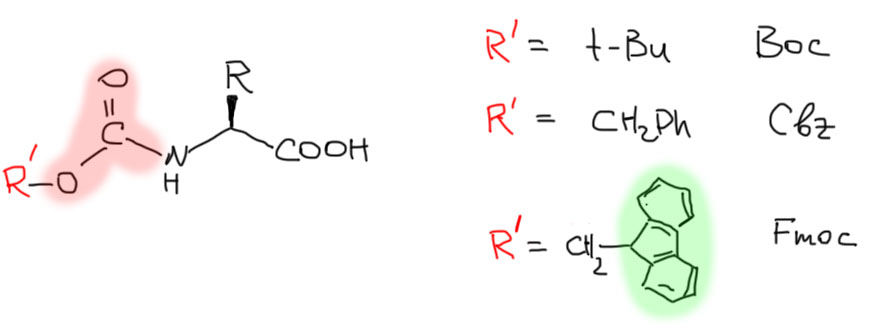
Почему берут амиды угольной кислоты, а не обычных карбоновых кислот? Потому что после отщепления R’ образуется неустойчивая карбаминовая кислота, которая тут же теряет CO2 и реакция становится необратимой (протон берется из среды или реагентов). К тому же возникает много возможностей, как отщепить этот R’. Если бы это был обычный амид, его можно было бы отщепить только с помощью гидролиза, а эта реакция обратима.
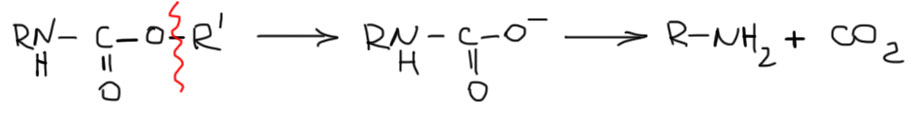
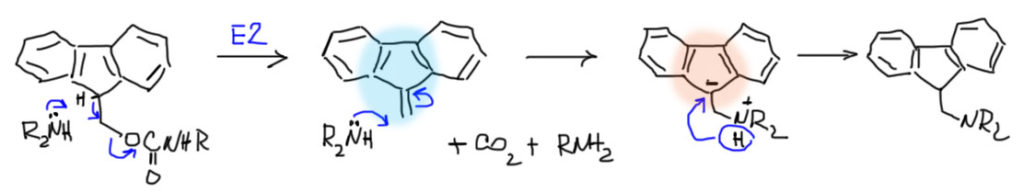
Поскольку все такие группы амиды и одновременно сложные эфиры, они плохо выдерживают кислотный и щелочной гидролиз. Boc снимается в условиях SN1-сольволиза трифторуксусной кислотой в полном соответствии с наличием в составе трет-бутильной группы. Cbz – производное бензила, а бензил сразу намекает на SN2-замещение, поэтому эту защиту снимают всегда в присутствии сильных нуклеофилов и кислотного катализа, помогающего расщепить связь Bn-O. Одна из таких снимающих смесей – раствор HBr (бромид – нуклеофил) в уксусной кислоте. Но и кроме того, как все бензильные производные, эта группа расщепляется гидрогенолизом H2/Pd/C. Очень особое место занимает Fmoc – эта группа довольно устойчива к условиям снятия двух других, и расщепляется вторичными аминами типа пирролидина через E2-элиминирование. Это очень занятная реакция, которая лишний раз показывает, как все в химии взаимосвязано. При элиминировании получается углеводород, являющийся производным фульвена, с которым мы хорошо сталкивались при обсуждении ароматических систем. Но на этом дело не кончается. Фульвен ведет себя как заправский акцептор Михаэля по очевидной причине – присоединение нуклеофила дает ароматический анион, производное циклопентадиенил-аниона. Поэтому это и идет так легко. Когда это обнаружили, Fmoc стал одной из самых популярных защит, потому что вторичные амины не снимают более никаких групп, и в этих условиях Fmoc ортогонален вообще всем остальным защитным группам. Fmoc стал основной защитой амино-группы в твердотельном синтезе пептидов.
Как вешают все эти защиты? Очень просто, через соответствующие ангириды (Boc2O) или хлорангидриды (Cbz-Cl и Fmoc-Cl), в присутствии оснований. Хлорангидриды реагируют совсем просто, как и обычные хлорангидриды с аминами, в условиях Шоттен-Баумана (водная щелочь в качестве основания).
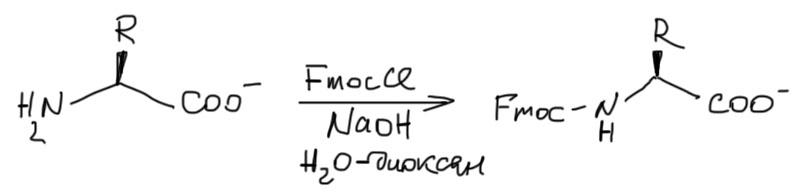
Защита карбоксила
Карбоксил мы договорились защищать сложными эфирами, и уже научились пользоваться метиловыми и бензиловыми. Добавим еще один, для пущей ортогональности, трет-бутиловый. Ой, а как его делать, он же не получается ни обычной этерификацией, ни, тем более SN1-замещением. Да, это редкостная морока, о чем лучше всего свидетельствуют цены – посмотрите на досуге и убедитесь, что трет-бутиловые эфиры аминокислот стоят сильно дороже и метиловых, и бензиловых. Но они же есть в продаже! Значит их как-то все же делают. Делают. Как такие вещи принято называть – грубой силой. Берут такую толстостенную банку с завинчивающейся крышкой, кладут туда огурцы, аминокислоту, при охлаждении нагоняют туда жидкий изобутилен в избытке (т. кипения -7º, при такой температуре кипения давление в банке при комнатной температуре будет небольшим, и если банка хорошая, то выдержит; если плохая – разнесет в мелкое крошево и поубивает всех вокруг; кстати, при отсутствии под руками специальной банки такие реакции раньше делали в бутылках из-под шампанского, закупоривая их пробками, примотанными хорошей проволокой), и немного концентрированной серной кислоты. Кислота протонирует изобутилен, образуется трет-бутильный катион, которому просто некуда деваться, кроме как сесть на единственный нуклеофильный атом (вспомните, как устроены реакции SN1 – если катион образовался, он не выбирает, куда сесть, и хватает любой нуклеофильный атом, а азот аминогруппы протонирован).
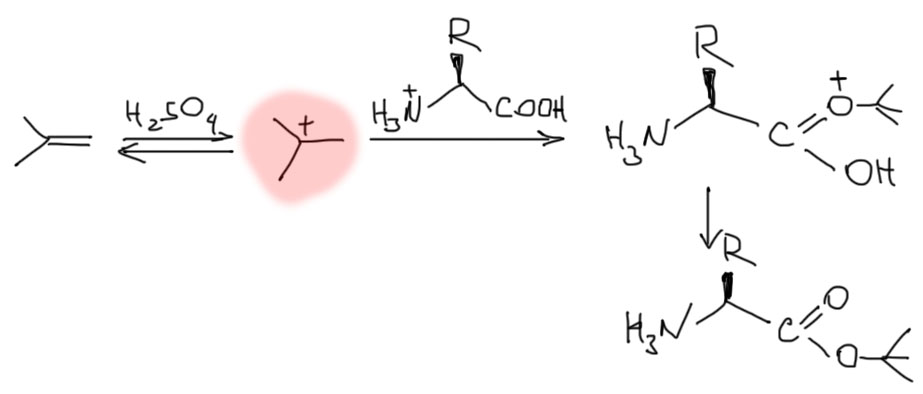
Как снимают трет-бутил должно быть понятно без лишних слов – точно так же, как Boc, в условиях SN1-сольволиза действием трифторуксусной кислоты.
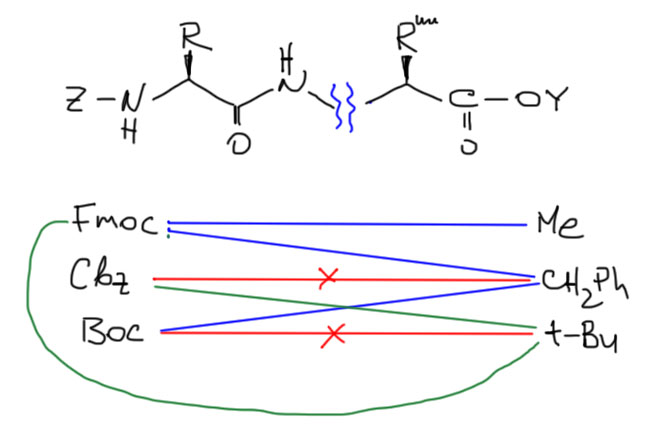
Сочетания защитных групп
Теперь у нас есть по три защитных группы для амина и для карбоксила, и мы можем подобрать пары для пептида, так чтобы можно было снимать защиту по желанию с N- или C-конца.
Основные способы снятия защит мы уже видели, соберем их в одном месте:
- SN1-сольволиз – трифторуксусной кислотой, снимаем Boc с азота, и трет-бутил с карбоксила. Эти две группы не ортогональны и использовать их вместе не имеет смысла;
- гидрогенолиз, H2/Pd/C, снимаем Cbz с азота и бензил с карбоксила. И эти две группы не ортогональны, и использовать их вместе не имеет смысла;
- щелочной гидролиз снимает метил с карбоксила, но также расщепит и Fmoc и Cbz. Эти группы не ортогональны, если нам нужно сначала убирать метил;
- действие вторичного амина снимает только Fmoc – в этих условиях эта защита ортогональна всем остальным, в том числе метилу на карбоксиле, если мы снимаем защиту на азоте первой;
- всякие прочие методы, которых еще полно, оставим в покое;
Получаем несколько вполне годных для использования комбинаций. В реальном синтезе пептидов проблема на этом не заканчивается, так как приходится еще защищать функциональные группы аминокислот, и эти хащиты должны сидеть до конца синтеза, то есть быть ортогональными к защитам двух концов. Задача сложная, про нее написаны целые книги и справочники. Но общие принципы, я надеюсь, вполне понятны.
Твердотельный синтез пептидов
Задачи на применение методов
Как обычно, название блока содержит намек на основной метод, который стоит использовать для решения
Синтез Штрекера
1. Получите из толуола и д.н.р. рацемический фенилаланин.
Малоновый синтез
1. Из малонового эфира, бромуксусной кислоты получите рацемическую аспарагиновую кислоту
2. Из малонового эфира и этилакрилата получите глутаминовую кислоту
Восстановительное аминирование
Из этилакрилата и диэтилоксалата получите рацемический метионин
Синтез пептидов
Из соответствующих аминокислот получите L-валил-L-фенилаланил-L-метионин.