Обновления
Опубликована 8.04.2023
Алифатические нитросоединения
Алифатические нитросоединения – очень интересный класс органических веществ. Они считаются близкими аналогами карбонильных соединений из-за значительной CH-кислотности α-протонов (если они есть, конечно), из-за чего возникает аналогия с кето-енольной таутомерией, реакциями енолятов и так далее. И это очень поучительная история, потому что если в ней немного разобраться, оказывается, что аналогия, во-первых, не совсем полная, – в нитро-соединениях вместо карбонила нитро-группа, и с электрофильной стороной жизни возникаем много непонятных вещей – мы не очень хорошо понимаем, что делать с электрофильностью азота, если она вообще как-то проявляется. Соответственно, возникает асимметрия аналогии – основный катализ очевидно есть и енолятная аналогия хорошо работает (хоть и тоже с некоторой спецификой), а что делать с кислотным катализом, есть ли у нитросоединений аналоги енолов и тем более их производных, довольно трудно понять. Соответственно, мы не можем просто механически переносить всё, что мы узнали в химии карбонильных соединний на химию нитросоединений. И если мы в этом хотя бы немного разберёмся, это может научить нас к более культурному отношению к аналогиям, которое можно отлично описать любимой фразой сорокового президента США Рональда Рейгана, произносившейся приблизительно по-русски: “Даверрьяй но праверрьяй”. Каждую аналогию надо воспринимать серьёзно (доверять тому, что аналогия может быть полезна и даже очень), но обязательно проверить а) здравым смыслом; б) экспериментальными данными; в) пониманием структуры; г) знанием основ химии участвующих элементов (здесь, например, надо хорошо понимать, что азот это не углерод, и почему); г) любыми другими соображениями.
На этой страничке пока разобрано далеко не всё. Надеюсь, я потихоньку её доделаю, но и сейчас тут немало интересного. Страничка эта довольно подробна и поэтому вряд ли может быть рекомендована для 3-го курса. В общем, почти всё, что пригодится на 3-м курсе по химии алифатических нитросоединений, уже есть на страничке про методы. Тем не менее, наиболее любопытным эта страничка может помочь, потому что химия алифатических нитросоединений в большинстве учебников изложена очень приблизительно и оставляет множество вопросов, на которые не так легко найти ответ.
Краткое содержание
Многие свойства алифатических нитросоединений связаны с CH-кислотностью и таутомерией, и в этом надо разобраться в первую очередь. Там же мы узнаем, как можно пронитровать циклопентадиенильный анион, и что общего между аци-формами нитроалканов и Чаушеску – на вкладке . Дальше мы чуть-чуть коснёмся того, существуют ли реакции по атому азота и является ли он электрофильным центром по аналогии с карбонильным углеродом – на вкладке . Следующие вкладки посвящены получению алифатических нитросоединений, в первую очередь реакциями , первую реакцию Виктора Майера, и во вторую – пресловутой которая уже почти 150 лет продолжает скрывать от нас главную тайну – как же в ней образуются алкильные радикалы – и мы никак не сможем разрешить эту проблему, хотя и попытаемся. Действительно ли эта реакция используется.
На вкладке выясним, есть ли в химии нитросоединений аналог альдольной или сложноэфирной самоконденсации карбонильных соединений, и выясним, что такая аналогия не только есть, но что это не совсем аналогия, а в результате такой самобытной самоконденсации возникает нечто чрезвычайно полезное, и столь же опасное.
И наконец посмотрим на две важные и интересные реакции, происходящие при действии водных растворов кислот на аци-формы нитро-соединений – и . Особенное внимание уделим загадке, как в реакции Нефа может образовываться веселящий газ, но это не единственное её достоинство – реакция весьма полезна в синтезе, образуя связующее звено между химией нитросоединений и карбонильных соединений.
Таутомерия нитро-соединений
Таутомеры нитро-соединений называются или нитроновыми кислотами или аци-формой. Сама таутомерия обычно называется аци-нитро-таутомерия. Оба термина применяются одновременно в самой разной литературе, включая учебники, и не совсем ясно, зачем такая избыточность. А это просто след давнего спора двух мощных немецких химиков рубежа 19-20-го веков, Ойгена Бамбергера и Артура Ганча.
После того, как стало ясно, что у алифатических (как тогда называли – жирных) нитросоединений есть ещё одна форма, которую стали сначала называть изо-формой, Ойген Бамбергер обратил внимание на большое сходство между предполагаемой структурой этих соединений и карбоновыми кислотами. Тогда ведь еще спокойно писали в пятивалентном азоте, типа того, что в азотной кислоте, две чёрточки (потому что тогда это и были валентности). Получалось вот что:
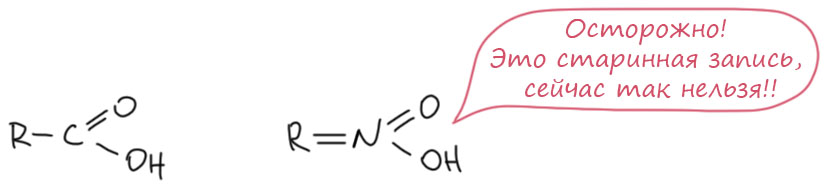
Похоже, не правда ли? Единственное отличие – на органический остаток идут две чёрточки, а не одна, но азот пятивалентен, а углерод четырёхвалентен. В общем, аналогия казалась полной. Но и Бамбергера смутила одна вещь – если это аналог карбоновых кислот, то должны быть и производные, сложные эфиры хотя бы, нитронаты. А у нитроновых кислот, как назло, не очень хорошо известны были такие производные. Бамбергер пытался их найти, но это и сейчас не очень просто – они очень неустойчивы. Спор он до конца не довёл, потому что ровно в это время, в 1905 году у него началось тяжёлое заболевание, которое фактически до конца жизни вывело его из науки – очень похоже, что он тоже чем-то отравился во время своих исследований. Но, как мы увидим дальше, его идея прижилась всё же лучше.
Название “нитроновые кислоты” страшно не понравилось другому классику, одному из первооткрывателей таутомерии Артуру Ганчу. Ему прежде всего показалось неудачной идеей придумывать особое название для того, что уже тогда стало восприниматься как общее явление – таутомерной форме. Ганч в 1905 году привел множество примеров уже тогда известных таутомеров, в том числе енольные формы альдегидов, кетонов и т.п. Фактически, он интуитивно догадался до общего свойства таутомерных систем самого разного типа – тот таутомер, который не является основным в равновесной смеси, проявляет более сильную кислотность по сравнению с основным таутомером. Ведь в большинстве случаев таутомерии в химии сначала появляется некоторое соединение, например, ацетон или нитрометан. А после выясняется, что у этих соединений есть и ещё какая-то менее стабильная форма, как говорили в 19 веке первые исследователи этого феномена – изо-форма, и нужно как-то её называть и исследовать. Название “изоформа” ушло сразу, как слишком общее, и начались споры, как это всё точнее назвать.
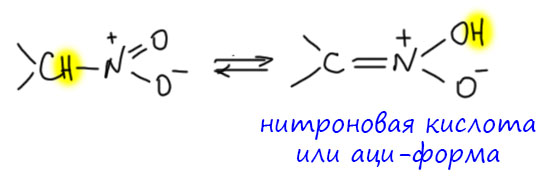
И придумал великий Ганч (который кроме своих великих работ знаменит ещё и тем, что в его фамилии Hantzsch на единственную гласную приходится аж семь согласных) , что можно просто во всех таких случаях использовать приставку аци- – то есть кислая форма. Например, енольные формы кетонов называть аци-кетонами, и так далее, ну и таутомерные формы нитро-соединений называть аци-нитросоединениями. Нитрометан – аци-нитрометан, и т.п. Мы теперь точно знаем, что предложение Ганча не нашло отклика в органической химии – нет такой общей приставки для обозначения более кислых таутомеров, но именно для нитро-соединений эта идея вполне закрепилась, и многие до сих пор предпочитают название “аци-форма” Ганча нитроновым кислотам Бамбергера. Впрочем, аци-формы проще встретить в общей литературе, например, в учебниках, а как раз специалисты по химии нитро-соединений предпочитают нитроновые кислоты, хотя бы потому что в более современной химии известно уже множество производных этого ряда, которых как раз недоставало Бамбергеру, чтобы заткнуть Ганча.
Название нитроновая кислота немного сбивает с толку, потому что создает аналогии с другими “оновыми” кислотами – карбоновыми, фосфоновыми, сульфоновыми. С одной стороны, это отчасти соответствует общей характеристике таких кислот – атом элемента связан с одним органическим остатком. С другой стороны – не совсем, потому что в “настоящих” оновых кислотах органический остаток одновалентный, и это делает такие кислоты непосредственными производными кислородных кислот в высшей степени окисления элемента. В нитроновых кислотах азот имеет степень окисления +1 (если забыли, как считать степени окисления атомов в органических соединениях, посмотрите внизу странички про валентность есть разбор простых правил). Ещё один способ посмотреть на таутомеры нитросоединений – рассмотреть их как производные неустойчивой и известной только производными азиновой кислоты H3NO2, а это просто N-оксид гидроксиламина, и в этом случае нитроновые кислоты это алкилиденазиновые кислоты.
Названия нитроновых кислот получаются добавлением к исходному названию суффикса -нитроновая кислота (используется не название радикала, а название соединения, не этил- и не этилиден-, а этаннитроновая кислота). Следовательно – это характеристическая группа высокого старшинства. Удивительная несправедливость – исходная нитро-группа – всего-навсего заместитель, всегда идущий в префиксе. При необходимости указать нитроновую кислоту в префиксе можно использовать обозначение Ганча – аци-нитроэтан. Так в более современной литературе два непримиримых классика пригодились, хотя в целом Бамбергер всё же победил.
Аналоги енолятов – нитронаты, они же соли аци-форм. В присутствии оснований таутомерное превращение происходит через анионы нитронатов, и поскольку CH-кислотность нитро-соединений весьма значительна, то оснòвными катализаторами этого превращения могут служить достаточно слабые основания типа бикарбонат-иона или аминов. В остальном закономерности таутомерного превращения почти не отличаются от закономерностей кето-енольной таутомерии в присутствии оснований. 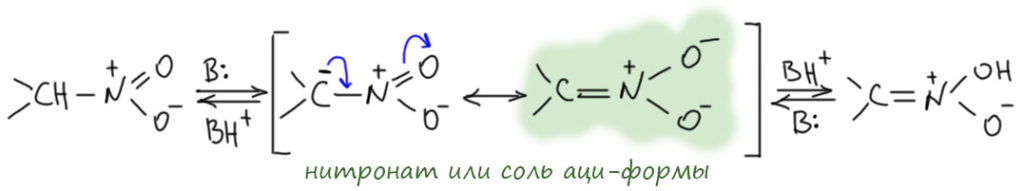
Для этой таутомерии может работать и кислотный катализ. Это далеко не так очевидно, и есть мнение, что кислотный катализ совсем не годится. Некоторые более стабильные нитроновые кислоты, что само по себе большая редкость (см. ниже), даже стабилизируются в кислых растворах, в то время как основания быстро их перегруппировывают в обычную нитро-форму. Но в этом случае мы, скорее всего, имеем дело с кинетикой – кислотно-катализируемая реакция намного медленнее основно-катализируемой, но совсем ею пренебречь нельзя, так как есть реакции нитросоединений в кислой среде, и мы тут их ниже рассмотрим. Но точно можно сказать, что кислотный катализ малоэффективен для таутомерного превращения нитроновой кислоты в нитро-форму. Причина этого довольно легко выясняется, если мы попробуем раcписать механизм действия кислотного катализа. Если мы это запишем, то увидим, что это даже рисуется не так гладко, как основный, в отличие, например, от таутомеризации кето-енольнгого типа, гда оба катализа совершенно органичны и эффективны фактически в равной степени. Ведь здесь нам придётся, во-первых, запротонировать нитро-группу. На первый взгляд, ничего страшного в этом нет, там же даже кислород отрицательный, и если на него повесить протон, получится проcто такой аммоний. Слово “аммоний” вряд ли может напугать даже работника жилкомхоза, но аммоний аммонию рознь. Здесь сложилось такое нагромождение электроотрицательных атомов, что становится не по себе. А если мы ешё вспомним, что нитро-группа когда-то давно выиграла чемпионат по акцепторности, причём сразу в обоих категориях – индуктивной и мезомерной, – и никто с тех пор эти титулы не отобрал. А теперь представим, что это ещё и запротонировали. Должен получиться просто невероятный акцептор. Протон на соседнем углероде от соседства с таким акцептором станет настолько кисл, что его отщепит каждый, у кого завалялась хоть какая-то неподеленная пара – вода та же, или анион кислоты (бисульфат-ион, например, в серной) – не удивляйтесь поэтому обозначению B: в кислотном катализе, это любое очень слабое основание. Ну и всё – нитроновая кислота образуется сразу же после отщепления протона, и граничная структура карбанионного типа рисуется только формально, в реальной структуре она значимого вклада не даёт. Из этого мы легко поймём, что кислотный катализ работает намного менее эффективно, и только в режиме настоящего протонирования (специфический кислотный катализ), то есть в присутствии настоящих сильных кислот. И точно так же мы поймём, что в обратную сторону – для превращения нитроновой кислоты в нитро-форму – это работает ещё хуже, просто потому что нужно запротонировать атом углерода, связанный с потрясающей акцепторной группой. Именно поэтому, в тех редких случаях, когда нитроновые кислоты могут быть получены в относительно стабильном виде, небольшое подкисление дополнительно замедляет их превращение в нитро-форму. А подкислять сильно нельзя, потому что начинают идти реакции типа реакции Нефа или Майера.
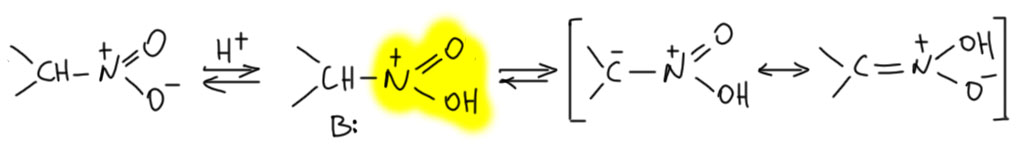
Нитроновая кислота – всегда менее стабильный таутомер, чем нормальное нитро-соединение. Это свойство очень сильно отличает эту таутомерную пару от кето-енольной. Для кето-енольной таутомерии мы знаем множество случаев большей устойчивости енола по сравнению с кето-формой просто потому, что CH-кислотность можно изменять с помощью заместителей в очень широких пределах элементарно сделать ее больше, чем OH-кислотность енола. А вот в паре аци-нитро, аци-форма (нитроновая кислота) всегда кислее как OH-кислота, чем нитро-форма как CH-кислота. Это не из чего не следует – это просто экспериментальный факт. Но обосновать его можно тем, что CH-кислотность нитро-соединений даже в самых простых нитроалканах уже настолько высока, что варьировать ее получается не так сильно (где-то 8 единиц pK весь диапазон, если не включать в него несколько совсем оcобых случаев типа тринитрометана).
Нитроновые кислоты – действительно кислоты, чуть более сильные, чем карбоновые кислоты, что неудивительно, если учесть что азот всё же более электроотрицательный атом. А почему тогда не намного более сильные – вот ведь в паре угольная кислота – азотная кислота разница в кислотности колоссальна. А, скорее всего, именно из-за двойной связи с органической частью молекулы, обеспечивающей весьма сильное смещение электронной плоности к атому азота и далее к атомам кислорода. Граничная структура с плюсом на углероде и парой на азоте, безусовно, не даёт большого вклада в структуру нитроновой кислоты, но даже небольшой вклад сильно снижает кислотность нитроновой кислоты, смещая константы в область слабых кислот.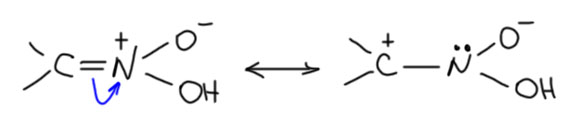
Просто сравните структуру нитроновой кислоты со структурой азотной. Изоэлектронные соединения (с точки зрения азота), не правда ли? Но нитроновая кислота слабая, а азотная сильная, и здесь мы сразу видим разницу в силе акцепторного индуктивного эффекта группы азот-двойная связь – кислород или алкилиден.

У большинства нитроновых кислот кислотность очень близка и находится в диапазоне pK 3-4, очень мало зависит от заместителей в органической части, что тоже понятно.
Забавно, что существует и с виду очень похожая таутомерия между нитрозо-соединениями и оксимами – нитрозо-оксимная таутомерия. И в этом случае равновесие почти всегда смещено в сторону оксима. А оксим – это аналог именно нитроновой кислоты. Интересно, что я не вмог найти никаких адекватных данных ни по кислотности оксимов, ни по CH-кислотности нитрозосоединений. Тем не менее, судя по результату можно предположить, что оксимы – более намного более слабые OH-кислоты, что вполне неудивительно, если учесть явную дестабилизацию соответствующего аниона за счёт отталкивания электронных пар на азоте и кислороде (α-эффекта), чего в помине нет в нитро-соединениях и нитроновых кислотах. А CH-кислотность нитрозо-соединений, видимо, весьма прилична, потому что нитрозо-группа это вполне солидный акцептор, уж точно не хуже карбонила.
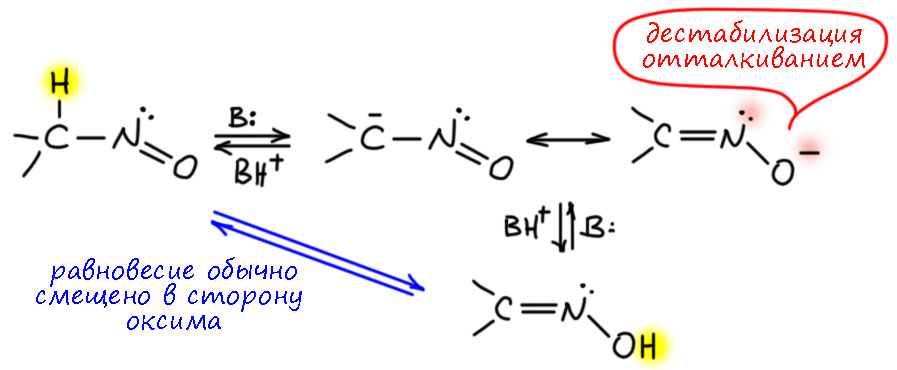
И в нитрозо-оксимной таутомерии отлично работает кислотный катализ, в отличие от аци-нитро-таутомерии. И тоже понтяно почему – здесь не работают те соображения, которые мы высказали против влияния кислот на нитро-соединения. Нитрозо-соединения обычно весьма устойчивы сами по себе, и могут храниться в чистом виде очень долго, но быстро перегруппировываются в оксимы в присутствии кислот. Но у нитрозо-соединений химия несколько сложнее, потому что есть ещё две равновесные формы – димер и нитроннная форма (продукт перемещения протона с кислорода на азот). Дальше пока не будем про эту интересную и запутанную химию.
Есть в некоторых кругах миф о том, что нитроновые кислоты устойчивы, и что скорость превращения в нормальную нитро-форму невелика. Это не так. И даже понятно, что это не может быть так. Нитроновые кислоты обладают вполне значительной кислотностью (еще раз – они кислее уксусной кислоты). Но в некоторых случаях этот процесс тормозится и тогда нитроновую кислоту можно даже выделить. Впервые сделал это Михаил Коновалов в Москве в 1896, продемонстрировав просто фантастическую по тем временам технику эксперимента (M. I. Konowalow, Chem. Ber., 1896, 29, 2193). Он взял нитронат из дифенилнитрометана и из водного раствора быстро подкислил холодной серной кислотой. Фокус в том, что такое большое соединение с двумя фенильными кольцами нерастворимо в воде и немедленно выпадает в осадок. Коновалов умудрился не только аккуратно выделить этот осадок, но даже перекристаллизовать и определить температуру плавления, единственный инструмент установления аутентичности в те времена, показав, что это изомер исходного нитросоединения с отличными от него свойствами. Например, оно, как типичная кислота с немаленькой кислотностью, быстро и легко растворяется в водном растворе карбоната натрия. Нормальные нитросоединения тоже растворяются в растворе соды, но далеко не быстро, а такое большое как исходный дифенилнитрометан – так и вообще крайне медленно.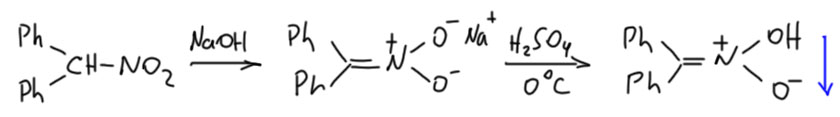
Тем не менее, и такая нитроновая кислота и в твёрдом состоянии, и еще быстрее в растворах возвращается в нормальную нитро-форму. Поэтому и удивительно, как сноровистый химик Коновалов успел проделать с этим веществом все необходимые манипуляции и вполне убедительно доказать, что получил он именно то, что хотел. А ведь никакой спектроскопии в подспорье у него не было и он ориентировался только на реальные свойства веществ, верно оценивая тенденции в те времена, когда теория химии еще даже не была в колыбели – её, теорию, ещё только предстояло родить и туда, в колыбель, положить, хорошенько спеленав, чтобы поменьше брыкалась во младенчестве.
Еще более стабильную форму получили тридцать лет спустя Колер и Стоун (J. Am. Chem. Soc. 1930, 52, 761), действуя очень похоже на эксперимент Коновалова, но получив нитронат немного иначе – 1,4-присоединением гриньяра к непредельному нитропроизводному. Заодно видим, что и такая аналогия с химией карбонильных соединений есть в химии нитросоединений. Дальше полученный нитронат магния осторожно и на холоду подкисляют и получают нитроновую кислоту, которая еще более устойчива и хранится несколько недель. Впрочем, и она медленно разлагается и при хранении и в растворе, а в присутствии оснований быстро таутомеризуется в нитро-форму.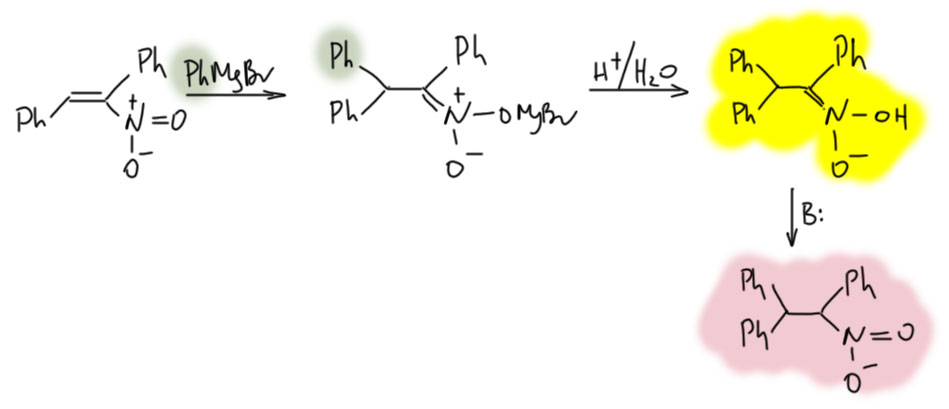
Оба этих исследования показывают довольно простую вещь – таутомерной превращение требует протонирования по углероду, и это может быть медленным, если на углероде висят объёмные заместители, а протонированию подвергается сама нейтральная нитроновая кислота, у которой этот углерод скорее электрофильный, и протон не очень жалует. В этом случае нитроновая кислота становится довольно устойчивой и ее можно выделить и некоторое время в неё играться. Протонирование становится быстрым, если протонируется не кислота, а отрицательный нитронат, и здесь уже стерика не очень помогает.
Есть и очень необычные случаи. Например, нитронат циклопентадиенильного ряда. Этот нитронат и сам по себе интересен, и получается необычно. Это сделал ещё Иоханнес Тиле в 1900 году – он тогда выдвинул совершенно гениальную идею о сходстве циклопентадиена с обычными метиленовыми компонентами типа кетонов (Thiele, J. Chem.Ber., 1900, 33, 666), и так и ароматичность небензоидную предвосхитил и до фульвенов добрался. И сделал много реакций, подтверждающих эту аналогию (мы еще обсудим эту работу подробнее в другом месте), в том числе то, что мы никогда не делали – реакцию с эфиром азотной кислоты. И получил не совсе то, что хотел (хотел получить нитроциклопентадиен), но почти то – он получил нитронат. Это выдающееся исследование настолько опередило своё время, что сам Тиле не мого точно разобраться в том, что получил, но настолько хорошо всё описал, что в другие времена всё это с удовольствием воспроизвели и доразобрались.
Итак, в реакции циклопентадиенилнатрия с этилнитратом он получил нитронат. А нитронат ли это? Посмотрим. На схеме я рисую локализованную форму в том же смысле, в котором в реакциях бензола рисуют формулы Кекуле. Циклопентадиенил-анион, безусловно делокализовани и симметричен, но в тот момент, когда молекулы вступают во взаимодействие локализация фактически происходит. И что мы видим? Мы видим электрофильное ароматическое замещение, вот ровно такое, какое мы видели в классических реакциях нитрования производных бензола. Здесь у нас, конечно, не нитроний-катион, для такого сильнодонорного ароматического соединения как циклопентадиенил-анион брать такой электрофил – безумие, поэтому мы и берём мягкий электрофильный реагент с ковалентно-связанной нитро-группой. Ну мы и в химии бензола для сильнодонорных молекул иногда брали ковалентный нитрующий агент, ацетилнитрат.Здесь молекула ещё более донорная, и агент еще более мягкий. Но дальше мы видим самый обычный σ-комплекс (ароматический контур разорван, углерод, на котором происходит замещение, меняет гибридизацию на тетраэдр). Кто-то обязательно скажет – там σ-комплекс это делокализованный карбокатион, а здесь? А здесь такой частный случай, когда исходное было карбанионом и карбокатион с ним сложился в нейтральную (в цикле) частицу. Согласимся, что это отличие, но сходства всё же больше. Дальше убираем протон и получаем циклопентадиенил-анион (ароматический) и висящую на нём полноценную нитро-группу. Это не что иное как продукт электрофильного нитрования ароматического соединения. Если мы считаем, что циклопентадиенил-анион – не просто какая-то отдельная ароматическая молекула, а основа ряда большого количества производных точно так же, как бензол – основатель ряда производных бензола, то полученное нитро-производное точно так же относится к исходной молекуле ряда, как нитробензол к бензолу. Оказывается, для циклопентадиенил-аниона вполне работает то, что мы называем химическим критерием ароматичности – участие в реакциях замещения, даже электрофильных, даже таких прямо самых классических – нитровании. И понятно, что рисовать это как нитронат – а это просто вторая граничная структура, – лучше не надо, так как в этой структуре теряется ароматичность, и мы отлично понимаем, что это невыгодно. Но оцените фокус – нитронат может оказаться нитро-соединением. Вот что ароматичность делает! 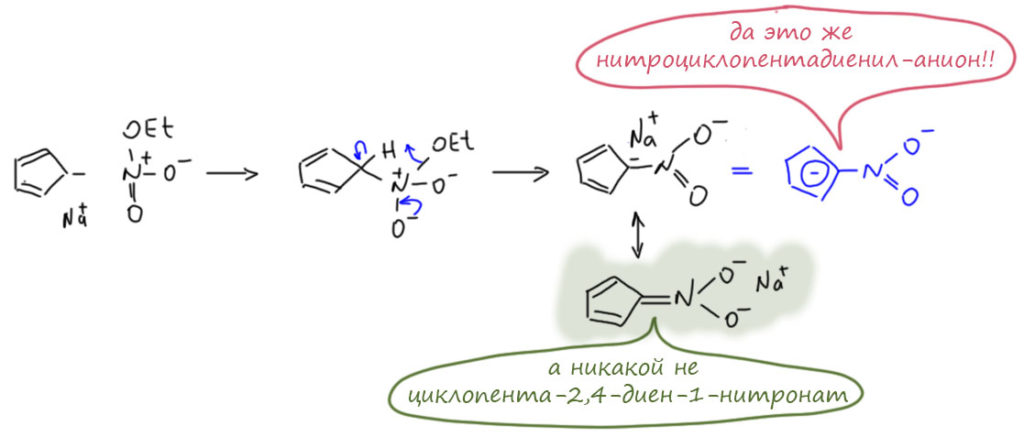
Когда этот результат Тиле воспроизвели в эпоху ЯМР, результаты получились еще более интересны (R. C. Kerber and M. J. Chick, J . Org. Chem., 1967, 32, 1329). У этого не-нитроната, то есть нитроциклопентадиенил-аниона есть ещё одно удивительное свойство – эта соль легко растворима в воде, этот раствор весьма стабилен, и вообще не изменяет нейтральной реакции воды, то есть не показывает ни малейших признаков гидролиза. Это говорит о том, что соответствующая ему кислота, а это нитроциклопентадиен, имеет просто ошеломляюще высокую кислотность – это просто сильная кислота, авторы дают оценку pK не выше ноля. В принципе, ничего удивительного в этом нет – если у обычных нитроалканов pK в районе 10, то что ждать от молекулы, где анион стабилизирован и ароматичностью и сопряжением (ох, внимательный читатель может здесь усмотреть лукавство – ведь если карбанион сопряжен с внешней нитро-группой, один из электронов секстета как будто уходит, и как тогда собрать великолепную шестёрку? – но не забудьте, что в ароматичности нитробензола никто никогда не сомневался).
А теперь посмотрим на нитроновую кислоту в этом ряду. Что будет, если подкислить этот нитро-Cp (Cp – это общепринятое сокращение циклопентадиенила)? Будет нитроновая кислота или нитро-Cp? Ещё Тиле нашел, что при подкислении “нитроната” ничего хорошего не получается. В 1967 с ЯМР получилось веселее – Кербер и Чик нашли что Тиле был прав, но при некоторой сноровке кроме кучи какой-то дряни удалось получить в раствор немного настоящего нитроциклопентадиена и определить его структуру. 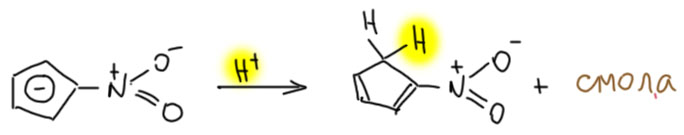
По структуре получалось, что нитро-Cp протонируется по соседнему атому углерода. И это вполне подтверждается результатами по дейтерообмену, который очень медленно происходит в растворе тяжёлой воды именно в это положение. Забегая вперёд в химию гетероциклических соединений, отметим, что это удивительным образом согласуется с местом протонирования пятичленных гетероциклов – фурана, пиррола и тиофена. А почему удивительным – углерод с присоединённой нитро-группой, которая эффективно увеличивает электроотрицательность углерода, можно отлично представить аналогом гетероатома, а во всём остальном системы изоэлектронны. А если системы изоэлектронны, то можно ожидать и аналогий в поведении и свойствах (на этом пути только никогда не надо заходить слишком далеко – любые успешные аналогии в химии опъяняют и тянут продожить банкет, – но важно вовремя остановиться и сказать себе: “Аналогии – инструмент хороший, но поверхностный, а Козьма Прутков учил “зрить в корень”, то есть копать глубже поверхностных аналогий”).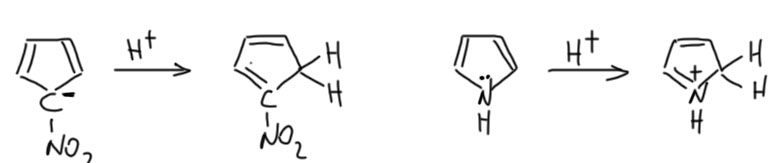
А почему же получается в основном смола? Видимо, потому, что в основном протонирование даёт нитроновую кислоту, а не нитро-форму. Но это же противоречит тому, как устроена соль! Нисколько, если предположить, что в этом случае аномалия еще и в том, что кислотность нитро-формы выше кислотности нитроновой кислоты (это не удивительно, так как депротонирование нитро-формы идёт из Cp-кольца, а в нитронате протон висит на обычной нитроновой группе), а следовательно таутомерное превращение смещено в сторону нитроновой кислоты, и это фатальная ошибка Того, Кто Создал Все Молекулы и Всё Остальное (это я невольно вспомнил своё советское школьное детство, когда мы, бывало, на уроках литературы рассказывали, в чём ошибался Толстой или Достоевский, так что привычка ниспровергать авторитеты должна была однажды докатиться и до Творца). Оказывается, законы таутомерных превращений и вообще кислотности по Бренстеду-Лоури могут классно противоречить закономерностям ароматичности. Обратите на это внимание – это интересный и поучительный случай, говорящий нам о том, что стабильность – вещь относительная. То, что стабильнее в одном смысле (в кислотно-основном равновесии) может оказаться нестабильнее в ином смысле (например, как здесь, в отношении антиароматической дестабилизации). Ведь если мы нарисуем нитроновую кислоту, и обычную граничную структуру нитроновых кислот, мы поймём, что эта структура антиароматична, а это всегда ведёт к пониженной стабильности и большой склонности вступать в реакции, например, олигомеризоваться через реакцию Дильса-Альдера. Вот ровно это, видимо, и происходит, образую ту самую кучу дряни. 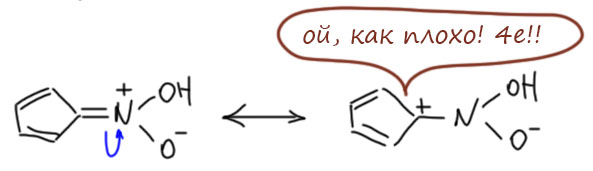
Не менее поучительна история с дибензо-производным циклопентадиена, флуореном. Это соединение должно себя вести довольно двусмысленным образом – соответствующий карбанион это, с одной стороны, вроде бы производное циклопентадиенил-аниона, следовательно не чужое ароматической стабилизации. С другой стороны, тот же карбанион просто и банально стабилизирован сопряжением с двумя бензольными кольцами – это так называемый бензгидрильный карбанион, да еще и дополнительно стабилизированный тем, что бензольные кольца смонтированы на жёстком основании и идеально сопряжены. И поди пойми, что важнее. В химии так очень часто бывает, когда два или даже больше факторов играют в одну сторону. Но химия нитропроизводного даёт неожиданный ключ к этой загадке. Нитронат из флуорена впервые получил не менее знаменитый немецкий химик Вильгельм Вислиценус-сын (Wislicenus, W., Waldmüller, M. Chem.Ber. 1908, 41, 3334; Вислиценус-сын, конечно, менее велик чем Вислиценус-отец, подаривший нам, в частности химию малонового эфира, но тоже ничего), практически точно так же, как Тиле получил производное циклопентадиена. 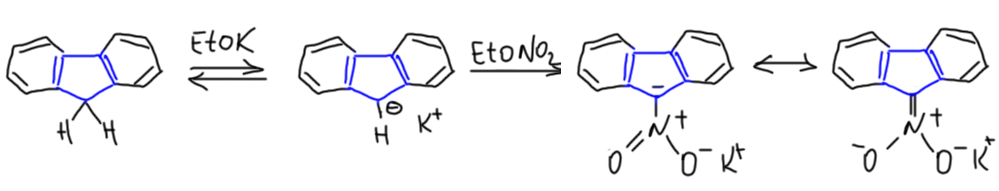
Сам Вислиценус сообщил, что из этой соли аци-формы, свойства которой, кстати, совершенно не отличаются от такого же производного циклопентадиена, можно легко получить соответствующую нитроновую кислоту. Соль отлично растворима в воде, и не изменяет pH раствора, то есть, видимо, также является солью сильной кислоты. При подкислении выпадает осадок, который был определён как нитроновая кислота. Вислиценус также сообщил, что при стоянии раствора нитроновой кислоты медленно происходит таутомерное превращение в 9-нитрофлуорен. И здесь ничего не противоречит тому, что мы знаем про нитроновые кислоты и аци-нитро-таутомерию. 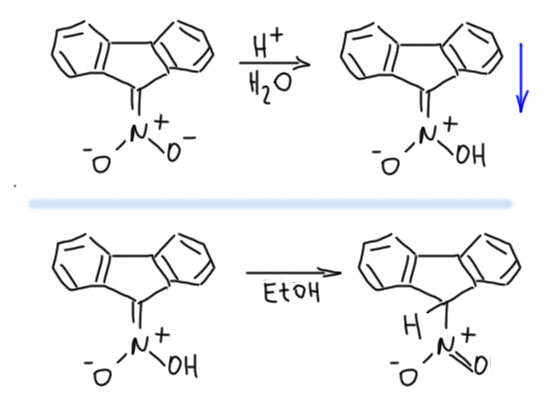
Но в 1930-х румынский химик Костин Неницеску вдруг сообщил, что флуорен-9-нитроновая кислота, полученная по Вислиценусу, совершенно стабильна, что в твёрдом виде, что в растворах. Эта точка зрения стала доминирующей, причём утвердилась точка зрения, что эта нитроновая кислота представляет собой исключение из общего правила – она стабильнее соответствующей нитро-формы. Почему эта нитроновая кислота действительно может быть такой? Потому что карбокатионная граничная структура в этом случае никакая не антиароматическая, а наоборот, очень стабильная, потому что это карбокатион бензгидрильного типа, отлично делокализованный по двум бензольным кольцам. Мы хорошо знакомы с таким влиянием конденсированных колец на антиароматические системы – их влияние перебивает эффект антиароматической дестабилизации. 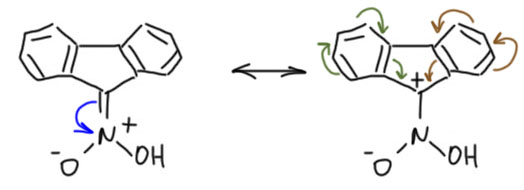 Если это так, то 9-нитрофлуорен является очень интересным примером нитропроизводного с более стабильной аци-формой. Но в этом есть большие сомнения. Во-первых, ранний результат Вислиценуса по изомеризации вряд ли является недостоверным. Во-вторых, сам Неницеску уже в социалистическое время в 1958 вдруг опубликовал статью, причем на русском языке в советском журнале Известия АН СССР в которой рассказал, что таутомерное превращение зависит от растворителя, причем в неполярных растворителях (в бензоле), в равновесии преобладает нитро-форма, а в полярных (в качестве которых фигурируют абсолютный этанол и диоксан) преобладает аци-форма. К сожалению, методы исследования в статье вполне допотопные (титрование бромом аци-формы), но сам вывод необчайно странен. Мы хорошо знаем, что соотношение таутомерных форм в равновесии определяется очень просто отношением констант кислотности. Константы кислотности, конечно, зависят от растворителя, но представить себе настолько широкое изменение, что их отношение меняется на обратное, не просто сложно, а почти невозможно. Видимо, в это время Неницеску уже настолько стал начальником марсксистко-ленинского типа, что всякие мелочи стали ему неинтересны, а подчинённые подсунули ему туфту. Так эта история и зависла. Насколько я знаю, в ней так никто до конца и не разобрался, и мы имеем вроде бы такой аномальный кейс стабильной нитроновой кислоты, идущий от откровенно топорной работы.
Если это так, то 9-нитрофлуорен является очень интересным примером нитропроизводного с более стабильной аци-формой. Но в этом есть большие сомнения. Во-первых, ранний результат Вислиценуса по изомеризации вряд ли является недостоверным. Во-вторых, сам Неницеску уже в социалистическое время в 1958 вдруг опубликовал статью, причем на русском языке в советском журнале Известия АН СССР в которой рассказал, что таутомерное превращение зависит от растворителя, причем в неполярных растворителях (в бензоле), в равновесии преобладает нитро-форма, а в полярных (в качестве которых фигурируют абсолютный этанол и диоксан) преобладает аци-форма. К сожалению, методы исследования в статье вполне допотопные (титрование бромом аци-формы), но сам вывод необчайно странен. Мы хорошо знаем, что соотношение таутомерных форм в равновесии определяется очень просто отношением констант кислотности. Константы кислотности, конечно, зависят от растворителя, но представить себе настолько широкое изменение, что их отношение меняется на обратное, не просто сложно, а почти невозможно. Видимо, в это время Неницеску уже настолько стал начальником марсксистко-ленинского типа, что всякие мелочи стали ему неинтересны, а подчинённые подсунули ему туфту. Так эта история и зависла. Насколько я знаю, в ней так никто до конца и не разобрался, и мы имеем вроде бы такой аномальный кейс стабильной нитроновой кислоты, идущий от откровенно топорной работы.
Нитронаты – безусловные нуклеофилы, хотя и довольно слабые. А вот нитроновые кислоты – соединения во многом парадоксальные, потому что они скорее являются аналогами карбонильных соединений, такими специальными иминами с кислородными заместителями на азоте. Это делает таутомеры нитроалканов прелюбопытными соединениями, так как они могут быть и нуклеофилами и элестрофилами на одном центре, причём переключение между этими типами – а это, как мы помним, в органике любят называть немецким словом Umpolung – происходит просто при протонировании/депротонировании, то есть в условиях кислотно-основного катализа. Современные любители красивых словечек любят называть такие реагенты (реагенты, способные проявлять любой из основных типов реакционной способности в зависимости от условий)) хамелеонами. Сами они хамелеоны, мы не будем бросаться такими словами, а лучше разберёмся, как это происходит и что из этого получается.
Итак, нитронат – скорее нуклеофил, аналог енолята, и это проявляется в многочисленных реакциях, в которых он ведёт себя как слабый углеродный нуклеофил, подобный сильно стабилизированным енолятам.
Нитроновая кислота, особенно в присутствии кислот или электрофильных активаторов, ведёт себя скорее как электрофил, и это не имеет прямых аналогий в химии карбонильных соединений, в которой сделать α-углерод электрофилом можно только с помощью серьёзного изменения структуры, например, введением галогена. Здесь скорее аналогия другого типа – нитроновая кислота и ее производные ведут себя как аналоги самих карбонильных соединений, как активированные имины. Электрофильность нитроновых кислот проявляется, например, в реакции Нефа и других интересных превращениях..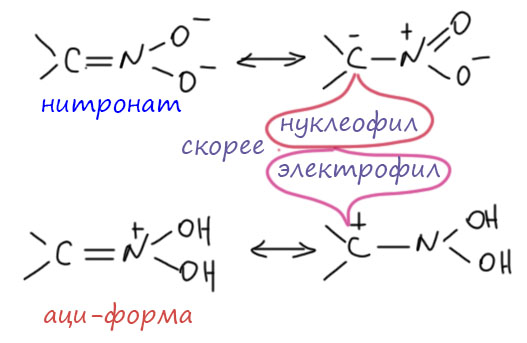
Реакции по электрофильному азоту
Аналогия с карбонильными соединениями какая-то однобокая. Карбонильные соединения очень важны как электрофилы – это врождённое свойство карбонильного углерода. В у нитро-соединений есть такие реакции, азот нитро-группы электрофилен?
Да, конечно, и как минимум, это проявляется в реакциях восстановления нитро-группы. Большинство таких реакций начинаются с переноса электрона на нитро-группу, но есть и такие, которые включают перенос гидрида на атом азота, например, восстановление боргидридами. Здесь мы про это не будем, надо будет однажды разобрать подробнее всё это восстановление.
А вот с гриньярами нитро-соединения реагируют? Судя по всему да, потому что мы всегда напоминаем, что нельзя получить гриньяры с нитро-группой в молекуле, по крайней мере при температуре выше -50 градусов, а лучше -100. А этот запрет обычно связан с тем, что гриньяр просто реагирует с запретной группой. Эту реакцию пытались провести почти с самого рождения гриньяров, то есть с начала прошлого века, но результаты получались противоречивые. Более-менее разобрались с этой реакцией сильно позже (G. D. Buckley J. Chem. Soc., 1947, 1492), причем оказалось, довольно неожиданно, что гриньяр не действует как основание, отрывая такой доступный протон с образованием нитроната магния, хотя разница в кислотностях здесь огромна. Еще раз убеждаемся, что отрыв протона определяется не только термодинамикой, но и кинетикой, а кинетика отрыва протона может быть вовсе не так быстра, особенно если собственно основный центр (здесь – карбанионный углерод) закрыт металлом. Вместо продукта депротонирования образуется продукт присоединения к азоту, полностью аналогичный аддукту гриньяра с карбонильной группой. Можно даже и здесь назвать его тетраэдрическим, потому что азот в нём тоже sp3-гибридный, тетраэдрический. Проблема этого аддукта в том, что ему не соответствует никакого устойчивого соединения – если вы это подкислите, то получите странную молекулу, которая представляет собой N-оксид дизамещенного гидроксиламина – ту самую азиновую килоту. Поэтому на этом не остановились, а добавили еще гриньяра, и произошло достаточно лёгкое восстановление этого N-оксида в просто гидроксиламин (механизм этой реакции возможно очень похож на реакции диметиосульфоксида – нуклеофильное замещение на кислороде, но это никто кажестя не исследовал). Впрочем, реакцию нитроалканов с гриньярами после еще несколько раз исследовали с весьма противоречивыми результатами, и применения она поэтому не нашла, хотя, может быть, в совсем свежей химии ей кто-нибудь опять балуется.
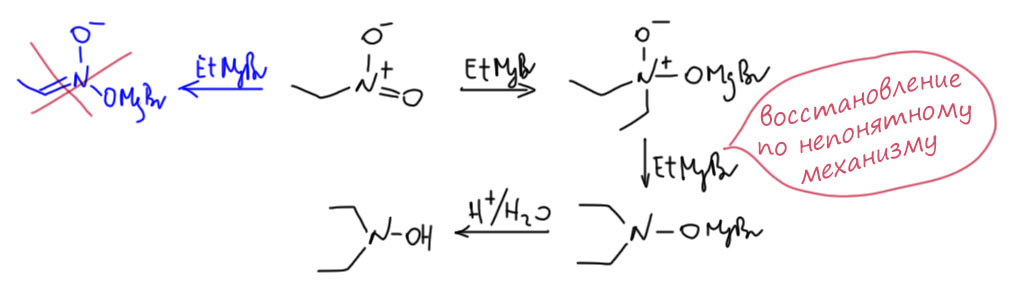
Может быть и другой путь – из аддукта может произойти элиминирование. При этом получится весьма занятное соединение, нитрон. Но он реализуется не всегда, а только тогда, когда элиминирование даёт двойную связь, сопряжённую с другой двойной связью или с ароматическим кольцом. Элиминирование происходит при послереакционном подкислении аддукта как раз из этого неустойчивого N-оксида гидроксиламина.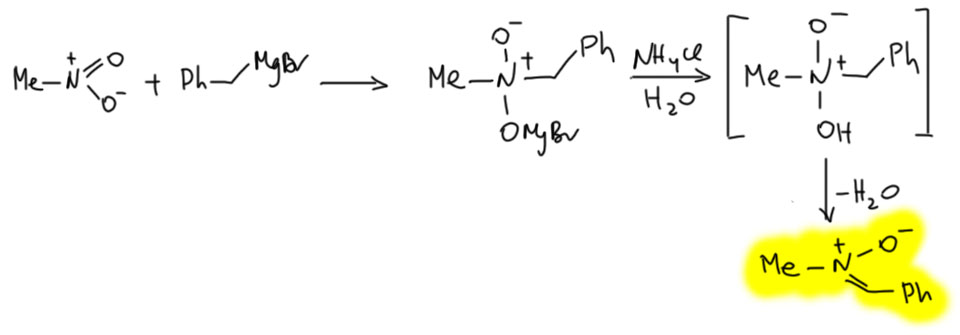
Получающееся соединение относят к классу нитронов. Очевидна попытка намекнуть на аналогию с кетонами, точно такая же как с нитроновыми кислотами. Аналогия вполне прозрачная, особенно очевидная, если взять старинный способ написания соединений пятивалентного азота. Так рисовать сейчас не нужно, но из химии невозможно выбросить ее историю, хотя бы потому что она объясняет многие устоявшиеся названия. 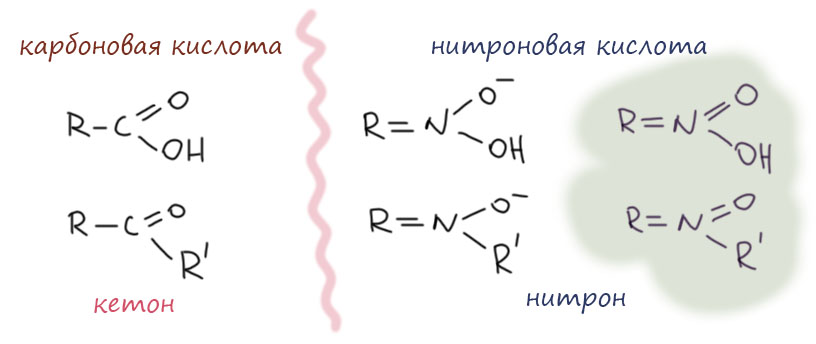
Нитроны – очень интересные соединения. Но мы ими займёмся как-нибудь отдельно, если выйдет какая-нибудь занятная оказия.
Получение нитросоединений реакцией замещения
Нитрит-ион это амбидентный нуклеофил, который может дывать в нуклеофильном замещении как нитросоединения, так и эфиры азотистой кислоты, нитриты. Это одна из реакций, которая стала причиной появления мифа о ЖМКО как принципа, определяющего селективность. В таких случаях, когда альтернативные пути дают разные классы соединений (как здесь – нитросоединение и сложный эфир), такая селективность называется хемоселективностью. Вся эта история разобрана на отдельной страничке, и должна рассматриваться только как курьёз. Факт состоит в том, что нуклеофильное замещение с нитритами даёт именно нитро-соединения как основные, а как правило, и единственные продукты. А эфиры азотистой кислоты, очень полезные соединения сами по себе, которые даже однажды сделали важнейший вклад в одну нобелевскую премию, получаются совершенно элементарно нирозированием спиртов производными азотистой кислоты.
Впервые нитроалкан получил Виктор Майер в 1872 году. Это такой показательный пример того, как зарождалась органическая химия. Начало 1870 это как раз то время, когда химики только что научились рисовать структуры и понимать, как одни соединения превращаются в другие. Но многое еще было очень формально. Майер поставил задачу так: теоретически, есть два изомера азотистой кислоты, и у каждого изомера должны быть эфиры. Обычные эфиры азотистой кислоты уже были известны, они совершенно элементарно получаются действием солей азотистой кислоты на спирты в присутствии серной кислоты, и Майер решил получить то, что он считал эфирами изомерной формы азотистой кислоты. И открыл новую реакцию, известную под его именем – реакцию галогенпроизводных с нитритом серебра. Первым нитроалканом был нитропентан. 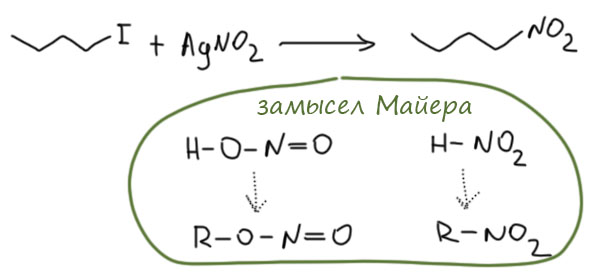
Эта реакция в практически неизменном виде сохранилась до сих пор, и носит она называние реакции Майера (есть и вторая реакция Майера, и о ней мы ещё вспомним). Прежде чем мы ее немного обсудим, пара слов о самом Викторе Майере.
Итак, реакция галогенпроизводных на насыщенном углероде с нитритом серебра – это чистое SN2-замещение, несмотря на то, что иногда пишут, особенно в более старых учебниках, что серебро оттаскивает галогенид и реакция идет через карбокатион – не идёт!! Не исключено, что ион серебра координируется по галогену в галогенпроизводном, и помогает его уходу как уходящей группы, но сама реакция идет только как SN2-замещение. Вообще тут не всё просто, потому что есть большая уверенность в том, что нитрит серебра совсем не растворим в тех растворителях, которые применяют для этой реакции, а значит, реакция идёт на поверхности частичек нитрита серебра за счёт сорбции галогенпроизводного, а это взаимодействие точно включает координационное взаимодействие Ag(+) на поверхности с атомами галогена. Теория таких реакций на поверхности до сих пор почти не разработана, потому что это чрезвычайно сложно моделировать. Оставим поэтому как есть. Подробнее приключения нитрита серебра и других солей серебра с амбидентными анионами можно почитать на страничке про амбидентные нуклеофилы.
Поэтому выбор субстрата диктуется правилами SN2-замещения: первичные, бензильные, аллильные, альфа-замещенные карбонильные соединения и т.п. Вторичные тоже реагируют, но медленно и выходы ниже. Реакции обычно проводят в суспензии в простых растворителях типа бензола или эфира, обычно без нагревания; в конце реакции просто отфильтровывают осадок галогенида серебра, и выделяют продукт. Если будете делать сами, пожалуйста, помните, что все нитросоединения потенциально опасны, особенно те, что поменьше. Надо быть очень осторожным, если потребуется нагревать, например, при перегонке. Вакуум должен быть хорош, и все предосторожности (экран, очки, перчатки) на месте.
А почему не использовать обычные соли азотистой кислоты? Потому что и нитрит натрия, и нитрит калия очень плохо растворимы в органике. Когда субстрат маленький, а ещё лучше водорастворимый, реакция отлично идет в воде или водном спирте. Но для большинства органических галогенпроизводных такой растворитель не годится. Проблему решили в 1950-х, когда появились полярные растворители, сначала ДМФА, который впервые использовал Корнблам, тот самый, который запустил в химию всю эту лабуду про амбидентные нуклеофилы, а он как раз занимался в первую очередь алифатическими нитросоединениями. По этой причине реакцию галогенпроизводных с нитритами щелочных металлов называют реакцией Корнблама. Немного позднее стали использовать и ДМСО. В ДМСО реакции с бром и иодпроизводными идут просто отлично (опять надо не забывать про правила SN2). ДМФА растворяет нитрит намного хуже, и требуются всякие дополнительные ухищрения, например, добавляют мочевину, которая тянет нитрит-ион в раствор водородными связями, и заодно подчищает побочно образующиеся эфиры азотистой кислоты, хотя в реакциях щелочных нитритов это почти никогда не происходит. Реакция Корнблама хороша ещё тем, что в ней можно использовать тозилаты вместо галоенпроизводных, а вот с нитритом серебра тозилаты бесполезны.
Так можно вполне хорошо в большинстве случаев обойтись без нитрита серебра, хотя в тонких синтезах, когда вещество ценнее реагента, до сих пор часто используют соль серебра.
Реакция Коновалова
Реакцию Коновалова часто считают скучной данью необходимости упоминать достижения отечественных учёных. Это совершенно неверно сразу в двух смыслах – это действительно достижение великолепного химика, который, наряду с Виктором Майером, Нефом, Ганчем, Бамбергером создавал химию органических производных окисленного азота (есть куча функциональных групп, где азот связан хотя бы с одним атомом кислорода). И это очень интересная реакция, которая требует вдумчивого погружения в химию свободнорадикальных реакций и никак не позволяет отбрехаться банальностями, как это часто бывает с реакциями второго ряда важности – нарисовали что-то отдалённо правдоподобное и, типа, жрите, другой химии нет.
Реакцию Коновалова мы уже обсуждали в самом начале курсе в теме алканы. Реакция не очень полезная в лабораторном синтезе (не совсем бесполезная тем не менее, есть вполне свежие примеры нитрования даже довольно сложных алканов в условиях Коновалова), но очень интересная, отчасти даже загадочная. К тому же, это, наверное, первая именная реакция, открытая в МГУ. Приват-доцент Московского университета, ученик Марковникова, Михаил Коновалов обнаружил эту реакцию в конце 1880-х и опубликовал результате в ряде очень подробных и тщательно выполненных статей на главном языке химии в главном химическом журнале тех времён, Berichte. Коновалов, как хорошо видно из его работ, был весьма искусным и опытным экспериментатором, его исследования хорошо обоснованы и проработаны, и это очень важно, потому что в те времена не было никаких способов понимать, как идут реакции, и исследования строились на интуиции и оптыте исследователя. В этом смысле работы Коновалова безупречны и соответствуют уровню, достигнутому к этому времени самыми передовыми учёными. А это, в свою очередь, весьма помогает разобраться в том, что было сделано тогда уже следующим поколениям на новых уровнях.
С этой реакцией связано немалое количество мифов. Некоторые из них я старательно воспроизвел в тексте на странице Алканы. Не буду исправлять, пусть остаётся в назидание. Ничего особо преступного там нет, просто несколько заблуждений.
Во-первых, эта реакция не парофазная, а самая обыкновенная жидкофазная. И ничего опасного в ней нет совсем. Коновалов использовал разбавленную азотную кислоту, обычно где-то 12%-ную (плотность 1.075), но иногда пробовал и более крепкую, до 35%. B температура там не очень большая – обычно чуть больше 100ºС. Коновалов не объяснил, почему использовал запаянные трубки вместо обычной колбы для самой обычной реакции с небольшим нагреванием, но, кажется, никакой особой идеи за этим нет – скорее всего, это связано просто с тем, что в те времена ассортимент лабораторного стекла для органических реакций был весьма скудным, никаких трёхгорлых колб с мешалками и прочей знакомой нам сбруей ещё просто не было. И даже если в распоряжении Коновалова были круглодонные колбы с обратным холодильником, соединялось всё на пробках, скорее всего, корковых, а все это совсем плохо держит горячие пары азотной кислоты. Поэтому проще запаять в толстостенную трубку и осторожно нагревать. Такой способ проведения реакций был весьма популярен в старой химии. У оригинальной методики Коновалова есть один большой изъян – она гетерогенная, разбавленная азотная кислота не смешивается с алканами (никакая не смешивается). Возможно, это даже хорошо, потому что иначе продукты реакции могли не сохраниться до ее конца, а так они частично изолированы от самой азотной кислоты в углеводородной фазе, ведь алифатические нитросоединения реагируют с сильными кислотами, правда только тогда, когда у них есть альфа-водороды. Выходы в реакции тоже обычно не очень большие, что тоже не просто так – если бы большая часть углеводорода превратилась в нитросоединения, природа органической фазы изменилась бы очень сильно, сильно-полярные нитроалканы растворяют и азотныю кислоты тоже, правда с водой не смешивается доже нитрометан, и это тоже привело бы к разложению продуктов.
Высокая селективность реакции была замечена самим Коноваловым – он заметил, что если в углеводороде нет третичных атомов водорода, реакция идёт туго, требует более высокой температуры, или вовсе не идёт. Но если есть, получается смесь продуктов нитрования по третично и вторичному положению, которую легко разделить, потому что нитроалканы, способные образовать нитронат-ион растворяются в щелочах, а те, которые не могут, не растворяются. Например, при нитровании диизоамила (2,7-диметилоктана) получаются третичные и вторичные нитро-соединения, а также вполне определённый динитро-продукт. Вторичное нитро-производное в схеме нарисовано одно из двух возможных, просто потому, что Коновалов в то время не смог точно установить его структуру, поэтому скорее всего, это смесь двух возможных вторичных нитро-соединений. 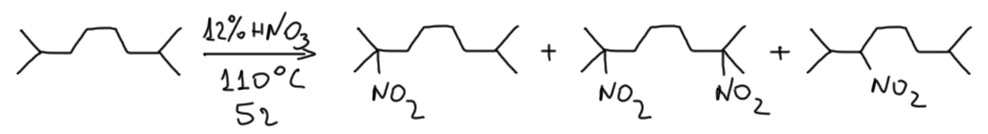
На этой схеме отсутствуют выходы. Коновалов их не определял. Тогда так было можно. Впоследствии было показано, что они довольно невелики, в сумме всех нитро-продуктов не превышают 30-40%. И в методе Коновалова с этим сделать ничего нельзя – если просто нагревать подольше, просто происходит окисление и выходы даже снижаются.
В некоторых случаях, когда в молекуле немного положений, в результате нитрования можно получить вполне конкретные вещества. Например, при нитровании 2,2-диметилбутана получается одно нитро-соединение. 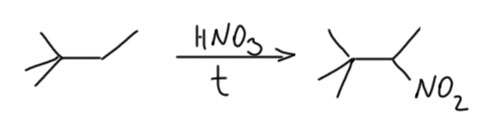
В некоторых случаях получаются довольно необычные результаты – образуются вполне конкретные полинитро-производные, в тех случаях, когда они оказываются твёрдыми кристаллическими веществами и выкристаллизовываются из реакционной смеси. Например, однажды Владимир Ипатьев, уже в США, так доказал структуру 2-метилпентана, получившегося у него в новой реакции алкилирования олефинов алканами (Grosse, A. V., Ipatieff, V. N., J. Org. Chem., 1943, 8, 438). В этой работе Ипатьеву помогал персонаж, не намного менее интересный, чем сам великий отец промышленного катализа – Аристид фон Гроссе, ядерный химик родом из Риги, сын российского дипломата, работавшего в Китае и Японии, подданный Российской империи, никогда в России-СССР не бывавший, но много скитавшийся по миру. Именно он, по его воспоминаниям, и был тем человеком, который уговорил Ипатьева уехать из СССР в 1930-м году и осесть в США. Гроссе занимался в основном ядерной химией, первым получил металлический протактиний, был близок к кругам в Германии, где появились идеи о делении ядер, перебрался в Штаты, и некоторое время по знакомству подрабатывал у Ипатьева в лаборатории с обычной химией. Вот они исследовали продукты алкилирования алкенов алканами, а поскольку никаких спектроскопий тогда не было, Ипатьев придумал нитровать продукты почти по Коновалову только 37%-ной азоткой, и выделять третичные нитроалканы, а иногда и продукты более глубокого нитрования. Например, из метилпентана получилось кристаллическое тринитропроизводное, которое они смогли идентифицировать. Этот результата совершенно не говорит ничего про то, что в этой реакции больше ничего не получилось, но всё остальное осталось в реакционной смеси и не было выделено.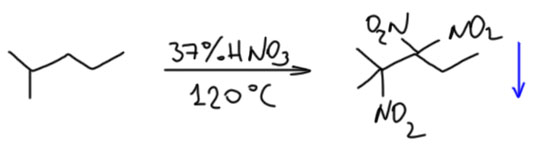
Реакция Коновалова еще интересна тем, что в ней можно нитровать и алкилбензолы. Как и следовало бы ожидать, реакция идёт в боковую цепь и весьма селективно – только через наиболее стабильный бензильный радикал. И в ароматическое ядро разбавленная азотная кислота такие субстраты не нитрует, хотя когда Коновалов взял триметилбензол, три метильные группы уже активируют ядро достаточно для конкуренции нитрования в боковую цепь и ядро, и получилась сложная смесь. Но для моноалкилбензолов реакция вполне селективна, например: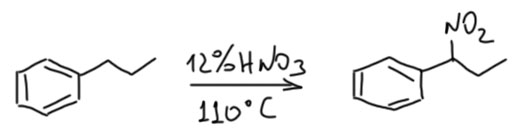
Впоследствии реакцию Коновалова много раз пытались исследовать и усовершенствовать. Предложили, например, пробулькивать пары концентрированной азотной кислоты через нагретый алкан. Этот метод работает несколько лучше исходного метода Коновалова, в частности с его помощью можно пронитровать нормальные алканы (получается смесь всех возможных вторичных нитро-производных практически в статистическом соотношении, но первичный продукт практически не получается (не более 2%), что ещё раз говорит о довольно высокой селективности метода.
А некоторые, видимо, совсем отмороженные господа придумали нитровать алканы пентоксидом азота (L. B. Haines, H. Adkins, J. Am. Chem. Soc, 1925, 47, 1419), и это получается сделать без нагревания (и очень хорошо, потому что иначе разбирать продукты реакции пришлось бы следователям и криминалистам на огромной воронке, оставшейся от лаборатории). Но я думаю, что в этом случае реакция вообще не была радикальной, она была, видимо, электрофильной). Так или иначе, эксперимент был описан настолько безобразно – типа, реагирует количественно, но что получилось, мы не разобрали – что можно забыть об этом. В 1930-х реакцию Коновалова много исследовали американские химики с целью сделать её пригодной для промышленного использования. Именно тогда реакцию стали делать в паровой фазе, пропуская смесь алкана и азотной кислоты или диоксида азота через нагретые трубки, причем температура реакции увеличивалась до более 200 градусов. В этих работах уже много от инженерии, исследователей интересует уже не только выходы, но и безопасность – изучаются пределы воспламенения таких смесей, и безопасные режимы работы. Именно эти исследования привели к промышленному процессу нитрования лёгких алканов, который используется до сих пор (об этом ниже).
Возвращаясь к механизму реакции, признаем еще раз, что как было непонятно, что ее инициирует, так и осталось непонятно, точнее, осталось на уровне гипотез – определенного ответа не было получено. Напомню, что мы категорически отмели диоксид азота, страшно ленивый стабильный радикал. Я совершенно недоумеваю, почему это продолжают писать даже в довольно современных публикациях, хотя далеко не все Термохимия диоксида азота известна в деталях. Можно просто посмотреть в сторону такой замечательной науки, как химия ракетных топлив, где диоксид азота очень часто используют как окислитель, хотя чаще вместе с диметилгидразином, где химия совсем другая; но и с углеводородами типа специального ракетного керосина тоже. Условия реакций там совсем другие – высочайшая температура, в таких условиях вообще нет проблем с инициированием.
И не согласились с выбором гидроксильного радикала на эту роль (посмотрите еще раз на страничке про алканы аргументы против). Напомню, что этот радикал должен быть совершенно неселективен из-за чрезвычайно высокой энергии связи H-O в воде – стадия отщепления атома водорода будет чрезвычайно экзотермична.
Про диоксид азота все не так однозначно, потому что в тех же 1930-х было найдено, что алканы можно нитровать чистым диоксидом азота в газовой фазе при темературе выше 200ºС. Последующие исследования показали, что распределение продуктов нитрования при этом получается точно такое же как при нитровании азотной кислотой по Коновалову. Иными словами, это одна и та же реакция с точки зрения инициирования – почти наверняка в них работает на отщепление водорода от алкана один и тот же радикал, и это довольно селективный радикал. И с диоксидом азота вроде вообще нет других реагентов, и кажется что просто больше нечему эту реакцию инициировать кроме самого диоксида азота. В серии работ в 1950-х (Bachman, G. B.; Hewett, J. V.; Millikan, A. G., J. Org. Chem., 1952, 17, 935 и т.д.) тем не менее было показано, что эта реакция ускоряется, если в смеси присутствует или кислород, или галогены. Вот это, видимо, и есть разгадка инициирования. Да даже если там правда нет кислорода как примеси, диоксид азота разлагается на оксид азота и кислород при температурах выше 300ºС с заметной скоростью, а это значит, что при 200ºС эта реакция может идти в небольшой степени, но много ли нужно для инициирования. А кислород – намного более селективный радикал, чем гидроксильный, и мог бы показывать приблизительно такую же селективность. Получается, что у реакции Коновалова вот такой незамысловатый механизм:
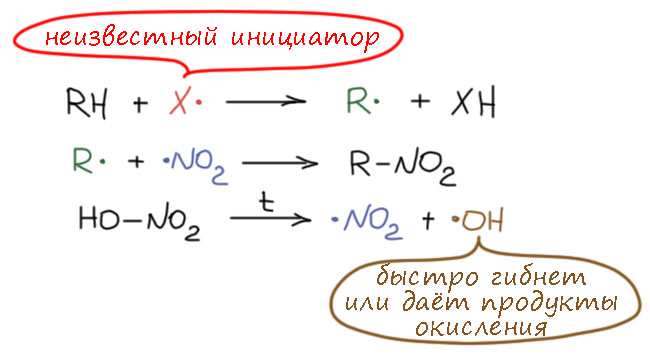
Алкильные радикалы образуются при отщеплении атома водорода неопределенным инициатором (не диоксидом азота и не гидроксильным радикалом, возможные кандидаты – кислород, гидропероксильный радикал, атом брома, или что-то ещё), и тут же рекомбинирует с диоксидом азота. Поэтому это не цепной механизм, и радикалы нужно генерировать внось и вновь для каждой молекулы продукта. Диоксид азота или используется сразу, или получается при термическом гомолизе азотной кислоты.
Если лабораторное значение реакции Коновалова невелико, она имеет важное промышленное применение. Условия промышленной реакции сильно отличаются от условий оригинальной реакции, но это несомненно она и есть. В промышленности применяют газофазное нитрование пропана, при этом получают смесь четырёх нитроалканов. Реакцию ведут смешивая пропан с парами концентрированной (не разбавленной, как у Коновалова) азотной кислоты при высокой температуре, и быстро охлаждая продукты. Состав реакционной смеси подбирают так, чтобы в ней не могло возникнуть пламя. Вряд ли можно забыть, что предельные углеводороды (керосин) с азотной кислотой – реактивное топливо. Но очень важно, чтобы в составе паровой смеси были пары воды – она забирает энергию и не дает реакциям в такой системе разгоняться до температуры самоподдерживающейся плазмы (пламени), а то и до чего ещё похуже. Уже в концентрированной азотной кислоте 60%-ной концентрации воды для этого вполне достаточно, поэтому реакция идёт спокойно и стационарно, и ничего никуда не взрывается, хотя для разработки безопасного процесса потребовались годы исследований и проектирования реактора. Использовать же оригинальные условия реакции Коновалова просто неэкономично – с такой разбавленной кислотой будет слишком низкая концерсия пропана. В промышленной химии за счет тщательной работы над оптимизацией всех параметров процесса всегда достигают максимизации производительности при сохранении безопасного режима работы.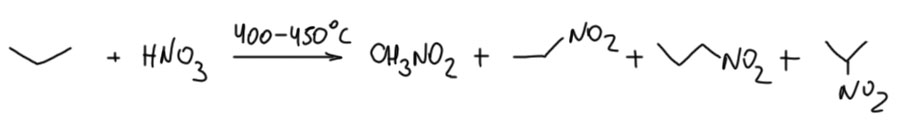
Очевидный вопрос – а почему не метан или этан? Ну, в таких случаях ответ может быть не только химический, но экономический или инженерный. Экономический – потому что на рынке есть спрос на все четыре нитроалкана, они используются как специальные растворители и полупродукты синтеза. Экономичнее делать их все четыре в одном процессе. Возможно также, что условия нитрования метана или этана хуже или опаснее – это знают только инженеры, работавшие над задачей. В принципе, это понятно, реакция Коновалова в любом виде терпеть не может первичные положения, и скорее всего вообще их не берёт. И уже тем более сам метан. Занятно, что на примере этой реакции мы видим, что иногда бывают случаи, когда низкая селективность даже приветствуется. Но это единичные случаи и почти всегда промышленные. Они никак не посягают на общее правило химии: Селективность Превыше Всего.
Почему получается нитрометан и нитроэтан? Считается, что реакция алкильных радикалов с NO2 может давать не только нитро-соединение, но и нитрит за счет не рекомбинации радикалов, а радикального присоединения к атому кислорода. Это вполне возможно, хотя и требует обоснования. Пока поверим. И если так, то образующиеся нитриты имеют слабую связь N-O и поэтому термически неустойчивы, и в тех условиях немедленно разлагаются (есди бы у молекул были мозги, стали бы они образовываться, чтобы тут же распасться, но у молекул нет мозгов, а, с другой стороны, редко ли существа, имеющие мозги, поступают так же – делают что-то важное в условиях, когда успеха нет и быть не может). Алкоксильные радикалы при высокой температуре тоже неустойчивы, разлагаясь на карбонильное соединение типа формальдегида и алкильные радикалы меньшего размера. В продуктах реакции нитрования пропана действительно есть немаленькое количество спиртов и формальдегида, которые отделяют и утилизуют. 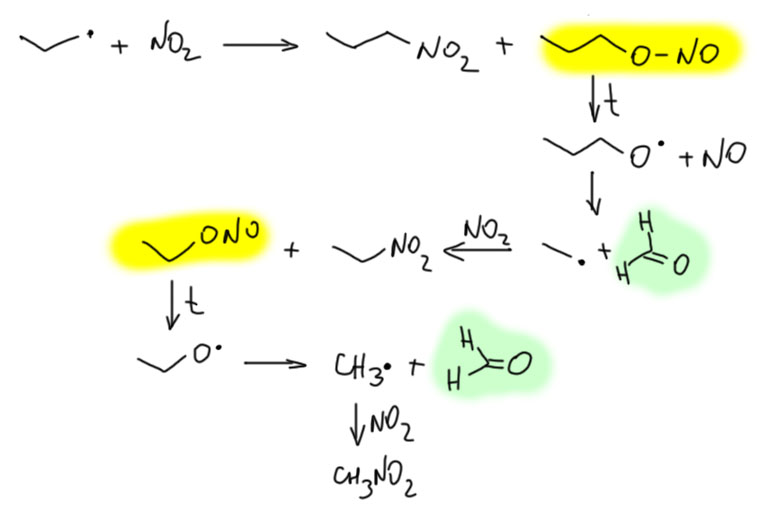
В завершение замечу, что химия не стоит на месте, и уже в новом столетии японские исследователи во главе с Ясутакой Исий всё-так нашли разумный лабораторный протокол свободнорадикального нитрования и именно за счет поиска хорошего инициатора (Sakaguchi, S.; Nishiwaki, Y.; Kitamura, T.; Ishii, Y., Angew. Chem. Int. Ed., 2001, 40, 222). В качестве такового они использовали весьма модное соединение – N-гидроксифталимид (NHPI), который довольно легко даёт при отщеплении атома водорода нитроксильный радикал. Радикал этот чрезвычайно моден в современной химии, где его гоняют через каталитические циклы фотокатализа с большим успехом. Поэтому, как и полагается модной молекуле, ему выдали игривую аббревиатуру PINO (очевидный намёк на весьма популярные в виноделии сорта винограда, а значит и на сами вина), весьма корявый акроним, но что не сделаешь ради понтов. И это весьма селективный радикал – энергия соответствующей связи O-H около 88 ккал/моль, а это как раз и есть уровень энергии, позволяющий легко отличать более слабые C-H связи от более сильных. Реакции ведут с азотной кислотой или лучще диоксидом азота в присутствии воздуха и инициатора при 70°С очень долго (что говорит о неэффективности самого инициирования), причем получаются нитроалканы или продукты нирования алкилбензолов в боковой цепи с селективностью, типичной для Коновалова, наряду с очень значительными количествами продуктов окисления (спиртов, альдегидов, кетонов). И мы опять видим, что этот инициатор, конечно, отщепляет водород от алкана, но кто инициирует сам инициатор, осталось неизвестным – я предположу, что именно кислород воздуха.
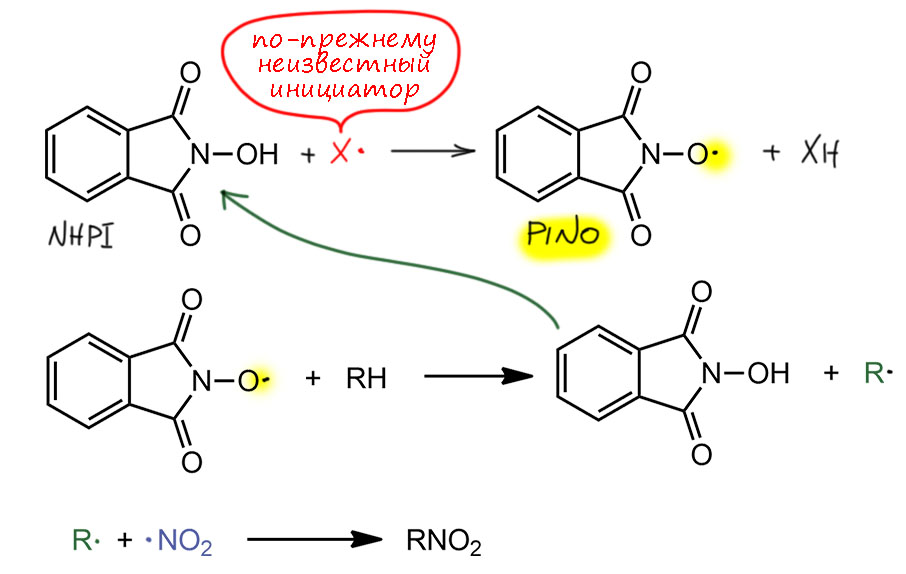
Увы, статьи написаны совершенно безобразно, и вопросов в оставляют очень много. Хотя работа подана с пафосом и опубликована в Ангевандте, убедительного впечатления действительно серьёзного движения вперёд в исследовании свободнорадикального нитрования она не оставляет.
Вот так мы и живём с реакцией Коновалова – недалеко ушли от работ нашего соотечественника и даже фактически коллеги по факультету (правда, химфака тогда ещё не было). Боюсь, что ему стыдно бы за нас стало, если бы узнал, что мы так толком не разобрались в том, как идёт открытая им реакция. Но сама история весьма поучительна и лишний раз нам напоминает, что в химии немало мест, где как было много вопросов, так и осталось.
Самоконденсация нитроалканов
Ещё один набег в аналогию между карбонильными соединениями и нитроалканами приводит нас к вопросу – что у них с лицом самоконденсацией? О, это очень интересный след, надо по нему пойти. И мы в очередной раз увидим, что аналогия оборачивается драматическим отличием. Нам для самоконденсации нужен и нуклеофил, и электрофил. Нуклеофил это нитронат. А электрофил – сама нитроновая кислота. И, что самое крутое – электрофил и нуклеофил находятся на одном атоме углерода.
Наиболее известна такая реакция для самого нитрометана. Ученик Виктора Майера итальянец Марко Лекко так впервые (Chem.Ber. 1876, 9, 705) описал продукт самоконденсации нитрометана под действием спиртовой щёлочи. В те времена способные следать работу и написать статью молодые химики часто печатали работы под своими именами, не включая руководителя, и прямо в тексте сообщая о полученных советах и указаниях. Так и здесь – автор в работе один, сам Лекко. Последующими исследованиями была установлена структура этого соединения, и найдены более удобные методы его получения. Больший выход получается при конденсации нитрометана под действие водно-спиртового раствора аммиака, или, еще лучше, при действии недостатка соляной кислоты на раствор щелочной соли нитрометана. Схему образования можно легко записать, ведь в этих случаях очевидно, что в смеси одновременно присутствуют и нитроновая кислота и нитронат. Один электрофил, другой нуклеофил. То, что нитроновая кислота очень быстро таутомеризуется в нитрометан, мы знаем, но знаем и то, что небольшая равновесная концентрация вполне достаточна для реакции, и если реакция сама необратима (или обратима, но обратная реакция достаточно медленная), то можно ожидать образования продукта за счет смещения равновесия. 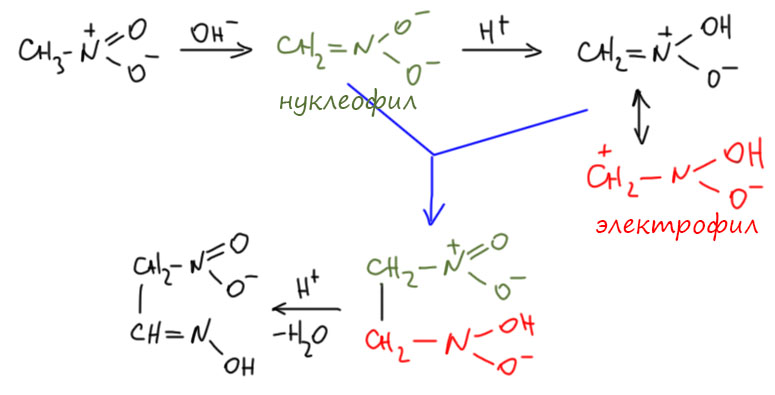
Лекко назвал продукт метазоновой кислотой чисто временно, он не мог понять, как устроена эта молекула, но видел, что она даёт соли, и произведена от нитрометана (мет- -азо-…). Но название осталось даже тогда, когда структуру установили, а это произошло только тогда, когда появился ЯМР (S.Brownstein J. Org. Chem. 1963, 28, 2919–2921). По структуре это оксим нитроацетальдегида. В этой молекуле мы видим очень интересное сочетание нитро- и нитрозо-групп, причем для нитрозо-группы устойчивее аци-форма (оксим), а для нитро-группы исходная нитро-форма. Всё ровно так, как мы и разобрали на вкладке про таутомерию.
В литературе этой реакции приписывают механизм, нарисованный впервые ещё Ганчем – димеризация нитроната. Этот механизм исследовали даже кинетически, причём схема механизма рисовалась так (J. Am. Chem. Soc. 1955, 77, 2622):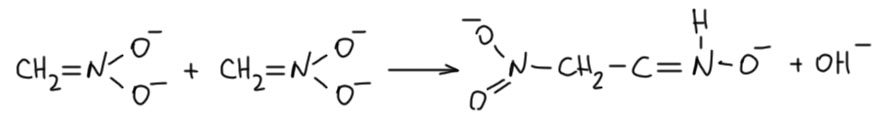
Если у вас глаза вылезли на лоб, это не я виноват, я это очень добросовестно перерисовал. Но это же джакс! Ну, в 1955 джакс ещё не был тем джаксом, которым он стал ближе к концу 20 века. В те годы это был просто химический журнал, не лучше и не хуже других. Померили люди кинетику, расписали кинетические уравнения, а как от них перейти к механизму, понять трудно. Кинетика вообще не очень хороший инструмент исследования механизма, хотя из многих учебников может сложиться обратное впечатление – кажется, что чуть не основной. Но даже если это перерисовать по-человечески, всё равно трудно понять, почему нуклеофил вдруг стал реагировать с нуклеофилом, к тому же, и откуда там может образоваться оксим. А меж тем, в условиях этой реакции можно спокойно предположить образование небольшой равновесной концентрации нитроновой кислоты, и нормальный механизм, подобный тому, что я нарисовал выше.
В более современной химии метазоновая кислота пришлась очень кстати – это такое легкодоступное бифункциональное соединение, которое так и хочется засунуть в какую-нибудь циклизацию, получив какой-нибудь гетероцикл. И таких работ можно найти немало.
Но метазоновая кислота известна не только этим. Соли этой кислоты чрезвычайно взрывчаты, и очень чувствительны, поэтому очень опасны в обращении. И с этим возможно связана опасность самого нитрометана. Нитрометан сам по себе очень сильное взрывчатое вещество, пишут, что не уступающее тротилу, но при этом настолько нечувствительное, что грохнуть его можно только хорошей бомбой. Впрочем, его применяют как добавку к другим композициям. В США в прошлом веке один ультраправый дегенерат типа тех, которых так любит в наше время собирать под свои знамёна Трамп, устроил кошмарный теракт, подорвав целый грузовик аммиачной селитры, пропитанной нитрометаном – он хотел наказать каких-то ненавистных ему федеральных чиновников, но первым на пути взрывной волны оказался детский сад. В Штатах легко достать нитрометан в больших количествах – там находится один из немногих заводов, где как раз делают парофазное нитрование пропана (см. на вкладке про реакцию Коновалова). И именно в Штатах произошёл в далёком 1958 году чудовищный взрыв цистерны с нитрометаном, к счастью, на далекой безлюдной сортировочной станции в Иллинойсе, так что погибла только локомотивная бригада. До этого взрыва никто не считал нитрометан опасной жидкостью, и перевозили его цистернами вообще без предосторожностей. Так и тогда, цистерны обычно сортировали на горке – это когда вагоны своим ходом скатываются, и стрелками их распределяют по составам. Очередная цистерна поехала, и когда докатилась до сцепки и резко тормознула, произошёл чудовищной силы взрыв – это как несколько тонн тротила бабахнуло. С инцидентом долго разбирались, пытаясь понять, почему нитрометан стал чувствителен к удару (когда жидкость в сосуде резко встряхивает, возникает гидравлический удар и кавитация – в микропузырьках созникает высокая температура и давление). Одна из основных гипотез – в нитрометане из-за примесей идет самоконденсация и образуются соли метазоновой кислоты – они то и инициировали взрывной распад самого нитрометана. После этого нитрометан стали, во-первых, стабилизировать против самоконденсации. И во-вторых, перевозить по особым протоколам безопасности. Вроде больше инцидентов в мире не было. Но в лаборатории, где мы с удовольствием используем нитрометан как уникальный высокополярный экстрагент (он не смешивается с водой и экстрагирует из воды даже многие соли), нужно очень внимательно относиться к этой жидкости. Контакт с основаниями особенно может сделать ее опасной при перегонке. Я вообще не советую перегонять нитрометан ни при каких обстоятельствах, ни в коем случае не связываться со старым реактивом, требующим очистки, не регенерировать его после работы. Ну а если необходимо, обзаведитесь всеми средствами защиты, не перегоняйте досуха, не перегревайте.
Umpolung наоборот: реакция Нефа
У нас в программе нет реакции Нефа и напрасно – это очень интересная реакция, которая очень много говорит о том, как устроены нитроалканы. К тому же, про существование этой реакции знают многие, в том числе преподы, и они иногда забывают, что ее нет в программе и норовят как-то использовать в задачах и синтезах. Разберёмся в этой реакции. Это тоже факультативный материал, но, скажем так, условно факультативный. Когда вас на экзамене спрашивают что-то и вы с ужасом видите, что не проходили и не задавали, не всегда получается убедительно изобразить оскорблённую добродетель, страдающую от вопиющей несправедливости и немотивированного насилия. Может быть, проще знать немного побольше.
Реакцию Нефа тоже открыл Михаил Коновалов, чисто случайно, в 1893 году. Он пытался восстановить соль 2-нитрогексана цинком в уксусной кислоте. Если вы поморщитесь и скажете что-то типа, как это можно, соль в кислоте? Ну, во-первых, в 19 веке еще не было представления об относительных кислотностях и основностях (их и сейчас у многих нет, но живут же…), а во-вторых, если бы были, то тем более в этой реакции нет ничего криминального потому что кислотности аци-формы и уксусной кислоты сравнимы, и аци-форма имеет даже несколько большую кислотность, то есть уксусная кислота далеко не нацело протонирует соль аци-формы. И получил Коновалов кроме аминогексана некоторое количество циклогексанона, и добросовестно и точно это описал в статье в главном химическом журнале того времени, в Берихьте, но дальше исследовать не стал – он был занят другой реакцией, получившей потом его имя. Это хорошее назидание нам, действующим и будущим исследователям. В исследованиях всегда важно сделать правильный выбор, а то не видать вам реакции имени себя. Спустя год реакцию решил исследовать Неф, обративший внимание на результаты Коновалова.
Этот Неф – весьма любопытный персонаж. Даже не сам по себе, а как замечательный пример того, что всё одинаково. И что когда-то США были в том же положении, что и многие другие страны позже – надо было начинать превращаться из традиционной сельскохозяйственной страны в высокоразвитую. Мы вот все время упоминаем того или иного крупного ученого, и говорим, что вот он перехал в США, как правило, из России, но попадаются из Германии, Франции, Бельгии, и т.д. Так вот Неф – из Швейцарии, из немецкого протестантского кантона, такого эталона зажиточности и спокойствия двести лет назад. В США переехал его отец, Иоганн Ульрих Неф, но не от бедности, а в поисках перспектив, он был по современному выражаясь менеджером на фабрике текстиля, а в США в то время открылись возможности развития этой отрасли, хлопка там было много, сами знаете откуда, если забыли, перечитайте или пересмотрите Унесённых ветром. Вот так, идут века, в США все открываются и открываются возможности, и едут и едут туда и предприимчивые, и талантливые. Сам юный Неф, на американский манер переименованный в Джона Алрика Нефа, прошел все то, что проходили и другие желающие чего-то добиться в жизни. Так он (сын небогатого иммигранта из глухой провинции, где даже школы путной не было) и поступил в Гарвард, чтобы стать медиком, и там познакомился с наукой химией. Мы тут уже где-то писали про то, что США в 19 веке – не то место, где можно было изучить химию, а уж про органическую химию в США не знал практически никто. Точнее, не никто, а один единственный человек, Чарльз Лоринг Джексон представлял собой всю органическую химию США в конце 19 века. И на него и нарвался молодой Джон Неф. И сразу после поступления в Гарвард так хорошо прявил себя, что ему дали стипендию для образования заграницей. Неф решил изучать химию, и – куда нужно ехать в 19 веке, чтобы изучать настоящую химию из первых рук у великих учёных – ежу понятно, в Германию. Так Джон Неф (в Германии, наверняка, вспомнил про свои корни и опять стал Иоганном Ульрихом, как отец) оказался в Баварии, в Мюнхене, у великого из великих, у Адольфа фон Байера, и так тому понравился, что стало ясно, чем будет заниматься Неф дальше. И вот он выучился и вернулся в Америку, где был нанят профессором в один из новых частных университетов в чистом поле – в Университет Пердью в Лафайетте в Индиане. И выделили ему неплохое жалование и новую лабораторию (старых там не было, да и химией там никто не занимался). Это сейчас этот университет среди очень престижных, а тогда это была обычная провинциальная дыра. Неф создал там химическую лабораторию, но не остался, а переехал в другой университет, затем еще в другой, в совсем новый университет в Чикаго. Так, с самого начала в Штатах сложилась та научная система, которая и сделала эту страну передовой – там все всегда в движении, люди получают финансирование под новые проекты, перемещаются, ищут возможности. Почти никто не сидит на одном месте всю жизнь. Это дало потрясающий результат – менее чем за полвека США превратились в ведущую научную державу, сначала сравнялись с Германией, а после спокойно обогнали, и оставили далеко позади. Тем более что Германия, выбравшая вместо свободы и развития путь деспотизма и войны, сама вышибла себя из этой гонки. Это вообще очень надёжный способ. Если какой-то стране в какой-то период вдруг кажется, что она слишком быстро развивается, и это может оставить без работы и заработка множество милых традиционных людей, не желающих учиться, а также кучу агрессивных шарлатанов и бездельников, присосавшихся ко всяким бесполезным государственным кормушкам, то можно устроить марш в обратную сторону, к благостной старине, а для верности ещё и развязать войнушку – дело в шляпе, о развитии можно будет забыть на десятилетия, бездельники и шарлатаны получат новые подряды и должности, а милые традиционные люди – вещмешок, винтовку, и осиновый крест на чужбине. Работает надёжно. Проверено многократно.
Неф прожил не очень долгую жизнь, но очень много успел в химии. Публиковался он кстати в основном по-немецки в немецких журналах, самых лучших в его время, а не в только недавно возникших и пока еще очень дохлых американских. Это очень верный выбор. Лучшие исследования должны публиковаться в лучших журналах, и пресловутый патриотизм тут ни причём, а точнее даже очень при чём – это и есть он самый, патриотизм – умудрился сделать что-то хорошее, позаботься, чтобы об этом знал весь мир, а не только соседи по бараку. А будет процветать наука, появятся и отличные журналы, не наоборот.
Итак, что открыл Неф в Чикаго. Он не собирался открывать никакой реакции Нефа. Он решал куда более интересную задачу – пытался понять, какова структура солей нитроалканов. В то время считали, что если какое-то органическое соединение может давать соли за счёт атома водорода на атоме углерода (такие соединения тогда называли псевдокислотами, совершенно напрасно, как мы знаем, это самые настоящие кислоты, CH-кислоты), то металл в соли находится на атоме углерода. Сейчас бы мы назвали такие соединения металлоорганическими. Но Неф решил опровергнуть эту гипотезу, он уже давно к этому присматривался, ещё со времени работы с Байером. Он считал, что некоторые псевдокислоты, в старой терминологии, могут дывать другой тип солей, в которых металл сидит на атоме кислорода. Сейчас мы бы сказали не так – мы знаем, что ион щелочного металла штука подвижная, особенно в растворах, и где он там сидит знает только он сам, но вот что точно – отрицательный заряд сидит не на атоме углерода, а на атоме кислорода, ну а металл это просто противоион. Иным словами, именно Неф предложил знакомую нам формулу нитроната (аниона аци-формы), и доказал это несколькими реакциями.
Например, он решил приготовить ртутную соль нитрометана, и неожиданно вместо ожидаемое соли нитрометана получил уже хорошо тогда известное соединение, фульминат ртути, гремучую ртуть. Нитронат ртути просто самопроизвольно теряет воду. Поскольку в структуре фульмината тогда тогда рисовали связь ртуть-кислород, он посчитал это доводом в пользу своей гипотезы. Эх, а ведь он был неправ, и как раз фульминат ртути образуется по атому углерода – мы бы сейчас этому не удивились и стали бы с упоением рассуждать про жесткие и мягкие кислоты и основания, и про то, что мягкая ртуть соединяется с мягким атомом углерода амбидентного аниона изоцианата. Но в конце 19 века идея о том, что соли одной кислоты с разными металлами могут иметь разные структуры, казалась несущественной. А правильная формула фульмината ртути вообще потрясающа – получается, что фульминат-ион – это N-окись цианид-иона. Неудивительно, что это так хорошо бабахает. И N-окись цианид иона изоэлектронна закиси азота: слышите, какая ирония структур – ион фульмината изоэлектронен молекуле веселящего газа! А до него мы скоро тоже доберёмся.

Но Неф привел и другой довод, к которому уже даже мы не подкопаемся. Производит вообще мощное впечатление этот Неф – копает глубоко, мыслит нешаблонно, излагает ясно, гораздо яснее большинства куда более знаменитых современников, даже делит длинный текст на разделы, экперимент описывает подробно и аккуратно. Пожалуй, он вполне достоин того, чтобы быть одним из отцов-основателей американской химии. Итак, Неф показал, что при действии разбавленной серной или соляной кислоты на соли нитроалканов образуются альдегиды или кетоны, в зависимости от органического остатка, первичного или вторичного. Из соли нитрометана получается формальдегид. Реакция идёт мягко, фактически это просто гидролиз (ещё раз напомню, что структуры я пишу в современном виде, а не так как у древних). 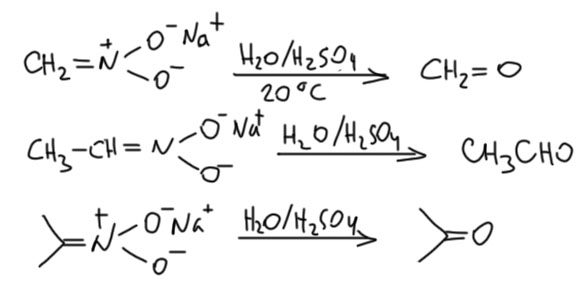
Так, хорошо, а азот-то куда девался? Вот это еще одно свидетельство совершенно потрясающей способности Нефа разбираться в результатах эксперимента. Неф не наплевал на этот азот – ведь ему интересна была органическая часть, и какая-то неорганическая часть, которая просто теряется – да другой бы даже не вспомнил про это. Но Неф не наплевал, он хотел написать именно уравнение новой реакции со стехиометрическими коеффициентами, а не просто приблизительную схему. И он определил, что азот уходит в виде … закиси азота, молекулы с двумя азотами. Тогда получилось написать и уравнение реакции. 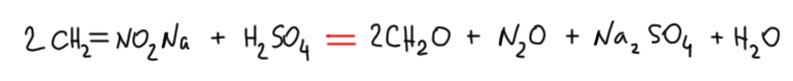
Во времена Нефа такое уравнение не вызывало никаких вопросов – получается, и получается, почему нет. Но когда возникла наука о механизмах, и потребовалось написать механизм, вопросы возникли. В виде чего уходит азот? Какова уходящая группа, ведь это замещение. Получается, что в реакции участвуют две молекулы нитроната, чтобы как-то обеспечить образование молекулы с двумя азотами. Или это после происходит, а уходит что-то другое. Надо разобраться в механизме.
За это взялись только в 1950-е. Юджин Ван Тамелен, весьма знаменитый химик, ученик Сторка, точно подметил сходство между реакцией Нефа и реакциями гидролиза производных карбонильных соединений (Van Tamelen, E. E.; Thiede, R. J. J. Am. Chem. Soc. 1952, 74, 2615). И правильно решил, что в реакцию вступает не нитронат, а нитроновая кислота. Поскольку мы знаем, что нитроновая кислота очень быстро перегруппировывается в обычную форму нитроалкана, это значит, что реакция Нефа, ее скорость-определяющая стадия очень быстра, чтобы перехватывать нитроновую кислоту раньше. В этом уже есть очень вадное отличие от химии енолизуемых карбонильных соединений, где ничего не нужно перехватывать, там просто реагирует равновесная форма енола, которая образуется и в присутствии оснований, и в присутствии кислот. В случае нитроалканов кислотный катализ таутомерного превращения неэффективен, и сами нитро-формы в реакцию Нефа не вступают (они вступают в реакию Виктора Майера, но об этом на отдельной вкладке). Аналогия действительно довольно прозрачная. Запишем это, чуть-чуть осовременив, и, как мы умеем, перекидывая протоны в места с наибольшей основностью, не выписывая промежуточные формы. 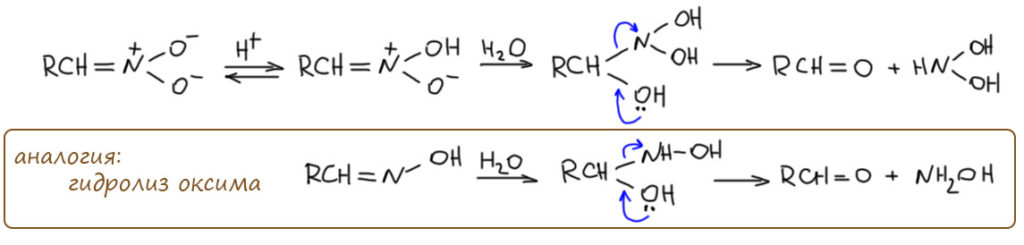
Нитроновая кислота выступает как производное карбонильного соединения, электрофил на атоме углерода. Как мы отлично знаем, исходный нитронат это нуклеофил на том же атоме углерода. Используя современную терминологию, видим, что это самый настоящий Umpolung – изменение типа реакционной способности на каком-то атоме. Нитроалканы исключительно интересны тем, что для такого изменения не требуется никакая серьёзная реакция, получение производного или еще что-то, сопровождающееся изменением структуры, превращением в другое соединение – нет, здесь это просто следствие кислотно-основного равновесия: сопряженная кислота и основание имеют разные реакционные способности на одном и том же углероде.
В качестве дополнительного доказательства ван Тамелен привёл относительные выходы реакций разных нитронатов, увидев неплохую качественную корреляцию со скоростями гидролиза соответствующих оксимов. В некоторых случаях эффект заметителя в реакции Нефа даже более силён, чем в гидролизе оксимов. Так, оксим бензальдегида реагирут очень медленно, потому что сопряжение с бензольным кольцом сильно снижает электрофильность углерода. А вот нитронат фенилнитрометана вообще ничего не даёт в реакции Нефа. В структуре этой нитроновой кислоты точно так же должна работать делокализация, снижающая эффективный заряд и электрофильность.  Немного домыслив за ван Тамелена, предположим, что в этом месте как раз проявляется очень важная особенность реакции Нефа – нитроновая кислота или присоединяет нуклеофил (воду), или перегруппировывается (в условиях реакции необратимо) в нитро-форму. Я думаю, что если присоединение нуклеофила идёт медленно, он просто не успевает за конкуренцией с перегруппировкой.
Немного домыслив за ван Тамелена, предположим, что в этом месте как раз проявляется очень важная особенность реакции Нефа – нитроновая кислота или присоединяет нуклеофил (воду), или перегруппировывается (в условиях реакции необратимо) в нитро-форму. Я думаю, что если присоединение нуклеофила идёт медленно, он просто не успевает за конкуренцией с перегруппировкой.
И то же самое происходит в других случаях, когда есть сопряжение нитронатного фрагмента с ароматическим кольцом. Реакция Нефа в оригинальном варианте в этих случаях просто не работает (а что работает рассмотри ниже).
Механизм реакции Нефа окончательно уточнил большой специалист в химии нитроалканов Нэйтан Корнблам (Kornblum N., Brown, R. A. J. Am. Chem. Soc. 1965, 87, 1742). Строго говоря, он почти ничего не добавил, но уточнил детали и нашёл дополнительные доказательства. Самое время, наконец, разобраться с тем, откуда там закись азота. Корнблюм ничего нового про это не сказал, но, подтвердив механизм целиком, показал, что искать каких-то экзотических путей не стоит. Всё должно быть довольно просто, надо только точнее понять химию некоторых простых соединений азота. Азот вообще-то штука удивительная, вроде бы такой простой элемент, между углеродом и кислородом, но насколько богата у него химия. Я, естественно, не хочу сказать что у его соседей химия скучная, я хочу сказать, что она совсем другая. Формальная причина этого довольно проста – столько степеней окисления нет больше ни у одного из элементов второго периода, а у элементов следующих периодов они, конечно, есть, но азот фактически третий или четвёртый элемент по электроотрицательности. Сильно электроотрицательный неметалл с богатейшими валентными возможностями – это он, азот, и он один такой. Азот в своих многочисленных реакциях свободно скачет между своими многочисленными степенями окисления, и просто голова идёт кругом от этой кутерьмы. Я смотрю на это и с ужасом понимаю, что если химию углерода я как-то научился ближе к закату немного понимать, то химия азота остаётся во многом выше моего понимания, и уже нет времени в ней разбираться.
Перепишем ещё раз механизм, но уже аккуратнее будем обращаться с азотной частью. И тогда увидим, что азот с двумя гидроксильными группами это как будто гидрат нитрозо-соединения, очень похоже на гидрат карбонильного соединения. Скорее всего, эта штука легко теряет воду, превращаясь в нитрозо-соединение. Нитрозо-соединения имеют зеленовато-голубой оттенок и некоторые даже уверяют, что во время реакции Нефа такое окрашивание действительно возникает. И дальше мы видим обычную карбонильную химию, предполагая, что нитрозо-группа может стать уходящей, и тогда мы увидим обычный распад тетраэдрического аддукта с образованием карбонильной группы и какого-то непонятного соединения NOH. 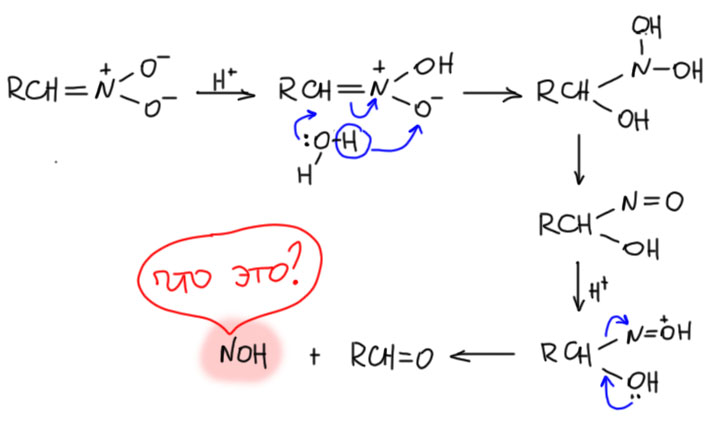
А где же закись азота? Пока что у нас только один атом азота в каком-то непонятном огрызке, который больше похож не на соединение, а на какую-то аббревиатуру, типа National Organisation of Harrassment или что-то типа того. А нам это надо? Мы же органики, что нам париться про соединение азота? Хороший органик всегда должен хорошо понимать, как идёт реакция, и что в ней уходит, и реальное ли это соединение, а может вздор какой-нибудь. Это нужно знать и чтобы понимать, как улучшать реакцию, и чтобы не зевнуть что-то опасное, и вообще так приятнее.
Что-же такое NOH. Видимо, это результат протонирования аниона NO– – нитрозильного аниона. А это что за зверь? Используем изоэлектронные аналогии – это молекула, изоэлектронная молекуле кислорода, только не обычного, триплетного, а синглетного, того, который рисуется с двойной связью. Если нарисовать и нитрозил-анион и NOH, мы увидим, что для них напрашивается мезомерия, и вторая граничная структура, особенно хорошо выглядящая как раз для NOH, имеет секстетный атом азота – атом с шестью электронами. Если бы это был углерод, мы бы назвали такую структуру карбеновой, а для азота аналогия будет очень ясной – это нитреновая структура. 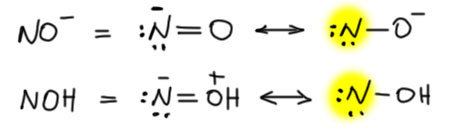
Вот оно – кажется, становится понятно, откуда берётся закись азота. Ведь карбены димеризуются в алкены – образуется двойная связь. Вот и нитрены должны вести себя так же. Димеризация тогда легко пишется и даёт уже вполне хорошо известное соединение, гипоазотистую кислоту. А она совсем неустойчива и тут же теряет воду. Вот нам и закись азота, чрезвычайно странное, между прочим, соединение – N-окись молекулярного азота. Одна мысль о том, что у азота может быть N-окись, как у какого-нибудь пиридина или триэтиламина, просто необычайно смешная. Просто подумайте об этом, и точно начнёте ржать, как ненормальные. Веселящий газ во всех смыслах однако. Всё, все секреты реакции Нефа открыты. 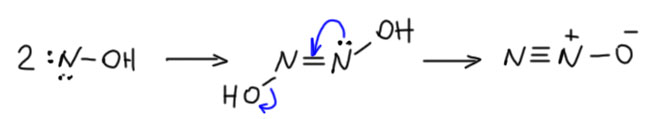
А если без веселящего газа
Оригинальная реакция Нефа весьма полезна, потому что позволяет создавать карбонильную группу на месте нитро-группы. Делают её просто – генерируют нитронат, действием основания на нитросоединение, и выливают раствор нитроната в избыток разбавленной сильной кислоты типа соляной или серной, обычно при охлаждении (в растворе плавают кусочки льда). После непродолжительного перемешивания экстрагируют продукт как обычно.
Но у оригинальной версии реакции Нефа есть очень существенные изъяны. Во-первых, в ней выделяется веселящий газ, и экспериментатор может им надышаться и вести себя плохо, приставать к коллегам, насмехаться над начальством, подвергать сомнению миролюбивую внутреннюю и внешнюю политику. Во-вторых, условия этой реакции – действие избытка сильной кислоты, хотя и сильно разбавленной – нельзя назвать мягкими и в сложных молекулах с другими чувствительными местами это может быть причиной побочных реакций. Одна из таких реакций – это как раз реакция Виктора Майера, разобранная на отдельной вкладке. В-третьих, реакция Нефа у условиях кислотного гидролиза нитроновой кислоы далеко не всегда дает высокие выходы, и мы уже видели, что в случае арилнитроалканов она вообще ничего не даёт потому что жестко зависит от конкуренции присоединения нуклеофила и таутомерного превращения, и последнее очень часто выигрывает, тогда мы просто получаем обратно исходное нитропроизводное.
Чтобы это избежать было придумано много других способов превращения нитроната в карбонильное соединение. Даже очень много. Кстати, сам по себе такой интерес очень ясно говорит о большой полезности этой реакции. Основных путей реформы реакции Нефа два – окислительный и восстановительный. И в том, и в другом избегают не только промежуточной нитроновой кислоты, которая может ускользнуть из гидролиза, но еще и избегают этой странной уходящей группы NOH. В окислительном пути просто находят окислитель, который разрывает двойную связь C=N. Это довольно простая реакция, и ее способны осуществить многие окислители, но самым популярным является банальный перманганат. Этому очень сильно способствует то, что нитронаты очень хорошо растворимы в воде, а это, как мы знаем, лучший растворитель для перманганата, который больше ни в чём и не растворяется. Чтобы избежать щелочной среды, в которой перманганат ведёт себя непредсказуемо, добавляют сульфат магния, работающий как ограничитель щёлочности – избыток гидроксид-ионов связывается ионами магния в слабое основание.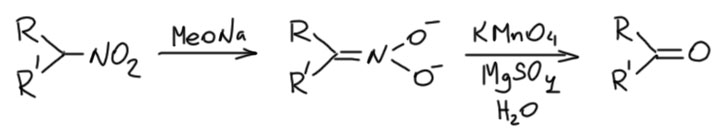
Еще более популярны восстановительные завершения реакции Нефа. Это ещё проще – нитронат просто восстанавливается до какого-то более простого производного карбонильного соединения, например, оксима, который спокойно и без приключений гидролизуется. Но может быть даже ещё проще – нитросоединение само может првращаться в карбонил без предварительного обрзования нитроната. Так действует самый популярный реагент, раствор трихлорида титана. Всё, что связано с восстановленными формами титана, открыл Мак Мерри (или Мак Мурри), и это не исключение. Реакция эксплуатирует нездоровую склонность титана цепляться на кислород везде, где можно. И забирать его себе (это называется дезоксигенированием, и соединения титана очень часто делает это). Остаётся нитрозо-группа, которая быстро таутомеризуется в оксимную, а та гидролизуется. Ацетат аммония добавляют для того, чтобы ограничивать кислотность и делать условия реакции более мягкими. 
Вот и всё. Но это не всё. Всяких фокусов с реакцией Нефа очень много. Это отличная, жирная на развитие реакция, приносящая много радости новым поколениям исследователей, которые пишут все новые статьи и защищают все новые диссертации и не устают славить блистательных отцов-основателей химии алифатических нитросоединений Виктора Майера, Михаила Коновалова и Джона Нефа, немца, русского и американца, прям как в многочисленных скабрезных анекдотах.
Реакция Нефа легко сочетается с другими
Очень простые условия реакции Нефа делают возможным легко соединять её с другими реакциями нитроалканов – Неф завершает процесс, выходя на карбонильное соединение. Это делает реакцию Нефа таким переходником между двумя химиями, и создаёт возможность использовать возможности реакций нитроалканов для синтеза карбонильных соединений, а там ничего лишнего не бывает.
Отлично сочетается реакция Нефа, например, с присоединением по Михаэлю. Если брать нитронаты в качестве нуклеофилов, то иногда можно даже использовать то основание, которое берут для отщепления протона от нитроалкана, или сначала ведут присоединение, а затем просто экстрагируют нитронат в разбавленную водную щелочь и выливают растор на холоду в избыток разбавленной кислоты – условия настолько мягки, что ничего лишнего не гидролизуется. Это сочетание забавно ещё тем, что Неф и Михаэль – первые американские органики (мы знаем Михаэля по немецкому произвношению его фамилии, но на самом деле он был Артуром Майклом, и работал он в основном в США, хотя статьи печатал в лучших немецких журналах), реализовавшие свои научные амбиции в лоне немецкой химии,
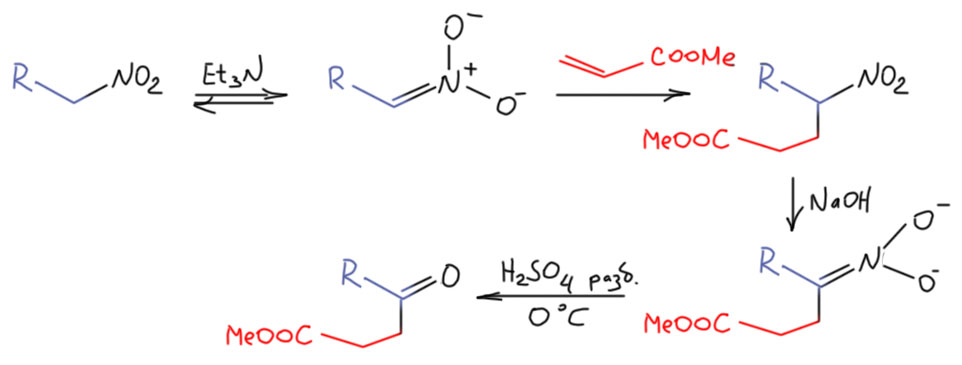
Более современная версия этой комбинации предложена японскими исследователями (M. Miyashita, T. Yanami, A. Yoshikoshi J. Am. Chem. Soc. 1976, 98, 4679–4681), причем здесь выбор реагентов обратный – акцептор Михаэля это нитроолефин, а нуклеофил – это силиловый эфир енола. Для таких реакций, как мы знаем по альдольной конденсации Мукайямы, нужен кислотный катализ кислотами Льюиса, которые активируют электрофильный реагент, в данном случае это нитроолефин. Лучшей кислотой Льюиса для этой реакции оказалось четырёххлористое олово. После собственно реакции Михаэля реакционную смесь просто разбавляют водой; продукты гидролиза хлорпроизводных кремния и олова дают кислотность, нужную для реакции Нефа с образовавшимся нитронатом олова. Образуются 1,4-дикетоны с очень хорошими выходами.
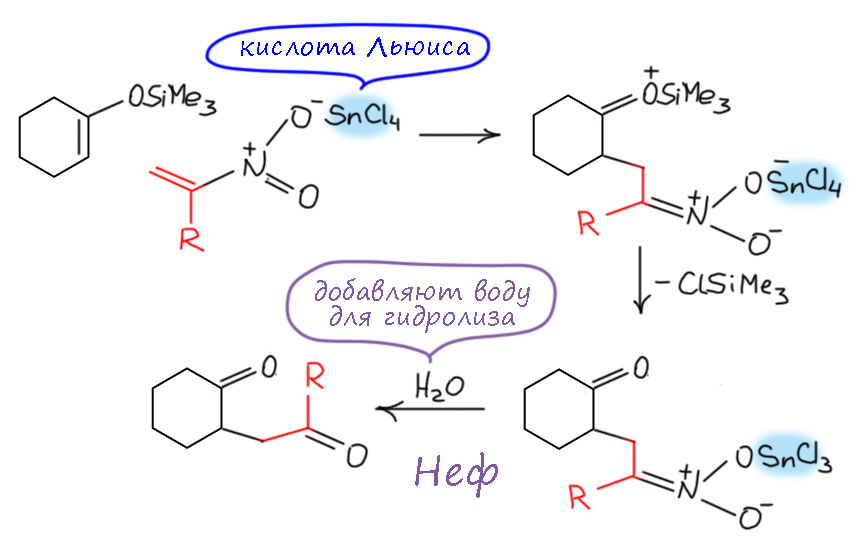
Как видим, именно лёгкость реакции Нефа, для которой нужна только небольшая кислотность и больше ничего, делает эту реакцию таким удобным завершением более сложных процессов. В литературе можно найти еще немало таких трюков.
Вторая реакция Майера
В отличие от реакции Нефа, которая имеет в синтезе хоть и ограниченное, но довольно важное применение, превращение первичных нитроалканов в карбоновые кислоты малоизвестно, а в синтезе практически вовсе не используется. Я бы никогда не стал обсуждать здесь эту реакцию, если бы она неожиданно не всплыла однажды во вполне учебной цепочке, вызвав мое искреннее изумление и даже негодование. А ведь такая реакция есть, и она даже иногда (про нее вспоминают в среднем один рав в четверть века) упоминается как именная – реакция Виктора Майера -именно так, видимо, чтобы не путать с другим Майером, Лотаром Майером, которого в некоторых странах вполне справедливо считают вторым создателем Периодической таблицы элементов. Реакцией Виктора Майера чаще называют другую, гораздо более известную реакцию – получение нитросоединений действием нитрита селебра на галогенпроизводные.
Виктор Майер весьма рано обнаружил реакцию нитроэтана с крепкой серной кислотой, собственно потому что он же этот нитроэтан и получил годом раньше реакцией иодэтана с нитритом серебра (V. Meyer and C. Wurster, Chem. Ber. 1873, 6, 1168). Получается, что эта реакция на 20 лет старше реакции Нефа. Но после открытия эта реакция почти не привлекала внимания по причине невысокой полезности – карбоновые кислоты гораздо доступнее, чем нитроалканы, да и сама реакция не была толком исследована. В современной химии её снова стали применять, в основном потому что научились не доводить её до конца, а перехватывать более интересные промежуточные продукты типа гидроксамовых кислот и их производных, или N-оксидов нитрилов, которые иногда даже бывают стабильны, или их тоже можно как-то перехватить.
Как видим, реакция идёт через протонирование нитроновой кислоты (об этом подробнее в обсуждении реакции Нефа), гидратации по альфа-углероду, нитрозо-оксимной таутомерии, приводящей к гидроксамовой кислоте или продукту ее дегидратации, нитрилоксиду. Дальше обычно идет полный гидролиз в карбоновую кислоту. Фактически это окисление альфа-углерода нитросоединения, а окислителем служит сама нитро-группа, азот заканчивает реакцию в виде гидроксиламина (его соли).
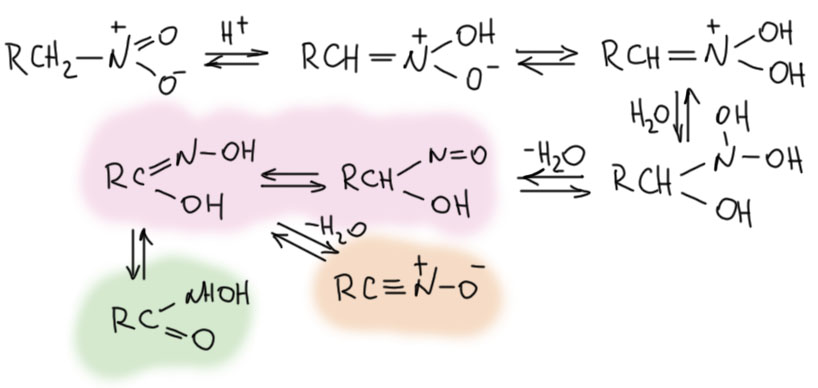
Как видим, большая часть механизма этой реакции практически идентична механизму реакции Нефа. Разница только в самом конце – вместо расщепления изо-формы гидроксамовой кислоты происходит дегидратация с последующим гидролизом. Реакция Майера поэтому естественный конкурент реакции Нефа, но для того, чтобы эта реакция стала главной, требуется более высокая кислотность и более жёсткие условия. Поэтому для реакции Нефа всегда выбирают более мягкие условия, разбавленные кислоты, охлаждение, и тогда проблем с Нефом не бывает. Раакцию Майера в её оригинальном виде с выходом на карбоновую кислоту в современной химии, кажется, не применяют совсем, но есть разные ухищрения, которые позволяют не доводить её до конца и останавливаться на более ценных промежуточных продуктах.