Некоторые полезные сведения даны на карточках. В этом разделе вообще нет стереохимии, а для синтеза с усложнением скелета фактически используется единственный метод, основанный на ацилировании по Фриделю-Крафтсу.
Обновления:
19 ноября 2020: В раздел про С-С связи добавил коментарий к одной задаче контрольной работы, вызвавший много полезных ассоциаций.
Новые C-C связи
- Выбираем...
- Ацилирование
- Алкилирование через ацилирование
- 1,3,5-триалкилбензолы
- Реакция Дильса-Альдера с участием аринов
- Комментарий к одной задаче
Самое главное здесь ацилирование, всё остальное имеет вспомогательный характер и встречается достаточно редко
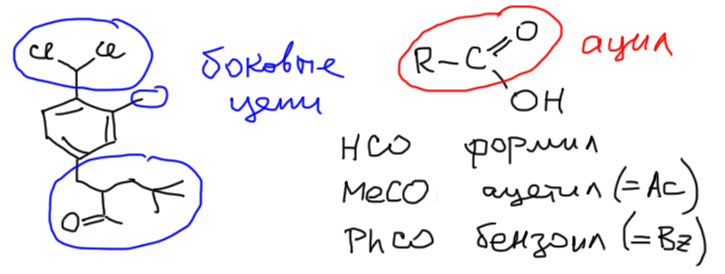
Для ацилирования используют комплекс хлорангидрида кислоты (хлористого ацила) с кислотой Льюиса, обычно безводным хлоридом алюминия. Хлорид алюминия – жесткая кислота Льюиса, которая выбирает более жесткий атом кислорода для координации (слово “жесткий” из теории ЖМКО, вспомните неорганическую химию). В продукте реакции поэтому кислота Льюиса остается координированной по атому кислорода, и реакция не является каталитической – на каждый моль хлорангидрида требуется моль хлорида алюминия (обычно даже небольшой избыток). Продукт высвобождается только при кислотном гидролизе после реакции. Если делали эту реакцию в практикуме, вспомните, как лили соляную кислоту в реакционную смесь после реакции до полного растворения выпавших основных солей алюминия.
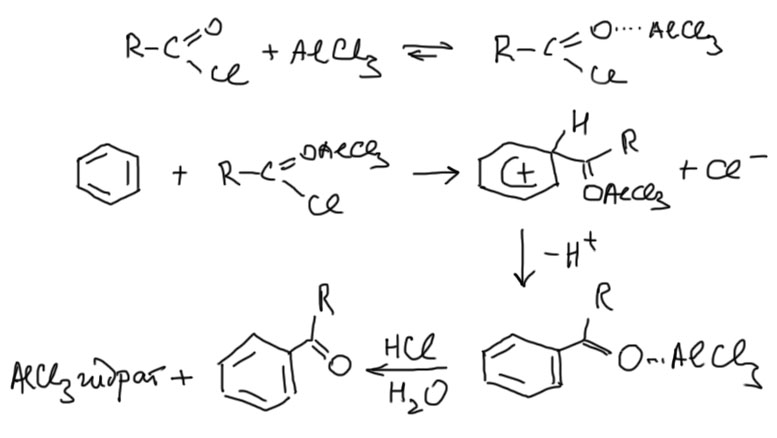
Про механизм и подробности поговорим отдельно, а пока практические вещи. Ацилировать можно любые производные бензола, содержащие орто/пара-ориентанты. Если пара-положение к самому мощному ориентанту свободно, то образуется только пара-изомер, если занято – орто-изомер. Это очень удобно, потому что не нужно делить никаких смесей, что и делает реакцию ацилирования такой популярной и надежной.
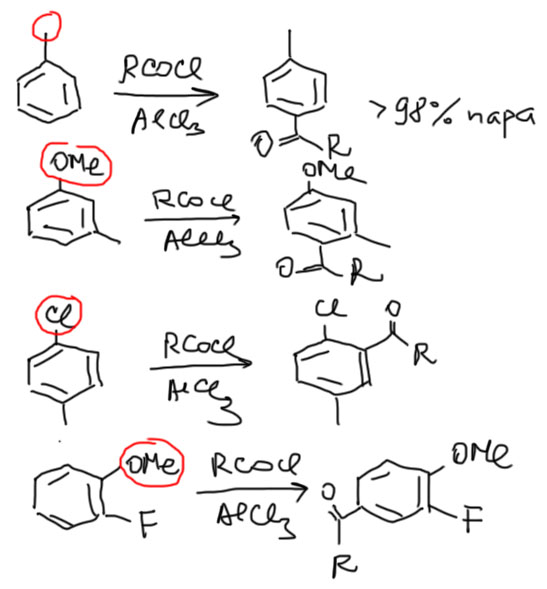
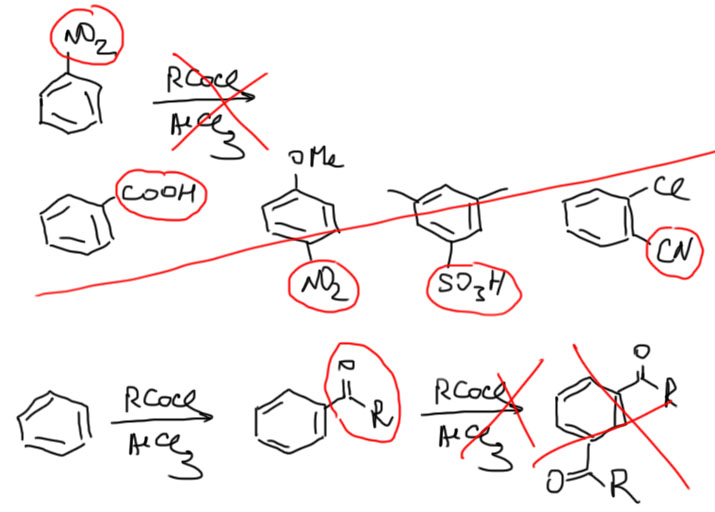
Но есть проблема – если в молекуле найдутся заместители второго рода – дезактивирующие мета-ориентанты, то реакция не идет, даже если есть еще и хорошие заместители. По этой причине не бывает двойного или тройного ацилирования – ведь ацил также мета-ориентант (нитрование, галогенирование, сульфирование, алкилирование – все эти реакции можно использовать для введения нескольких групп, а ацилирование не дает такого результата).
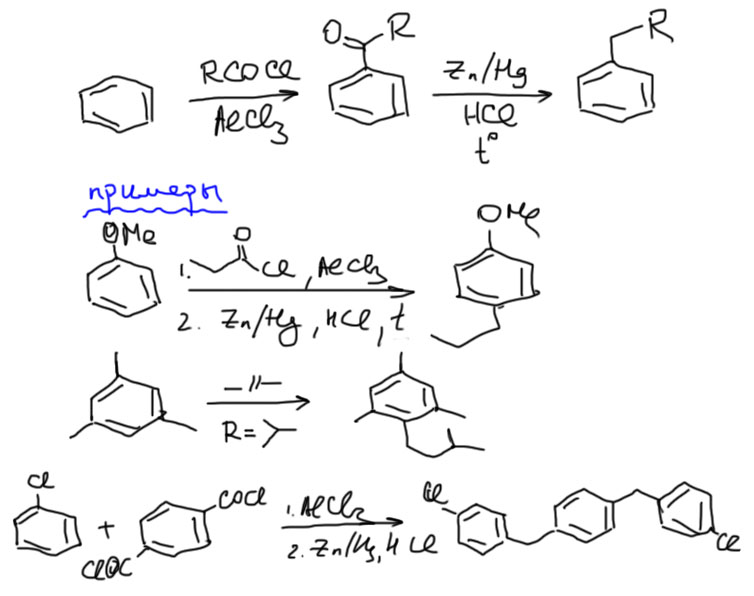
Алкилирование по Фриделю-Крафтсу – почти бесполезная реакция, но есть несколько частных случаев, когда ее можно и даже нужно применять. Один из таких случаев – быстрый одностадийный синтез простых 1,3,5-триалкилбензолов, в которых три алкильные группы находятся в мета-положениях друг относительно друга. Почему такие странные продукты получаются в алкилировании лучше почитать в теоретическом разделе.
Для получения 1,3,5-изомеров триалкилбензолов используют условия алкилирования по Фриделю-Крафтсу, то есть добавляют к бензолу или простым алкилбензолам избыток хлористого алкила и очень немного безводного хлористого алюминия. Смесь оставляют при комнатной температуре на сутки или более (можно на неделю). Потом, как обычно, тушат водой, экстрагируют и т.п. И перегоняют. Температуры кипения увеличиваются с увеличением количества алкилов в молекуле, а фракция, соответствующая трем алкилам, в основном состоит из 1,3,5-изомера. Ее обычно перегоняют повторно, более аккуратно и медленно. В схеме реакции пишем 1,3,5-триалкилбензол как основной продукт, всем остальным пренебрегаем (это правильно – в схемах реакций всегда пишут выделяемый продукт, даже если реакция неселективна, но есть заведомо известный способ выделения нужного продукта). Этим методом вводят только простые алкилы, не осложняющие жизнь перегруппировками.
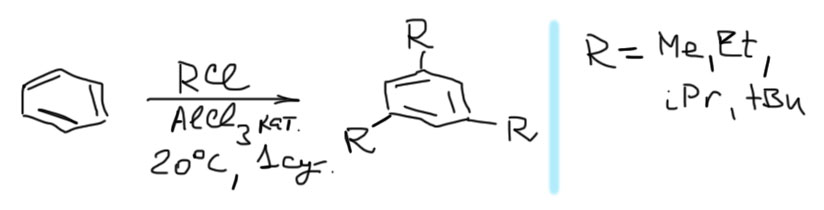
Реакцию еще можно применять к получению 3,5-дизамещенных, особенно трет-бутильных, если в исходной молекуле уже есть один простой алкил.
Дегидробензолы (арины), которые мы подробно обсуждаем в ароматическом нуклеофильном замещении, являются прекрасными диенофилами в реакции Дильса-Альдера. Проблема только в том, что это не обычные реагенты, которые можно не спеша отвесить на весах и положить в колбу. Время жизни типичного арина – миллионные доли секунды, и даже самый ловкий человек за это время не успеет ничего сделать. Этот дешевый троллинг, тем не менее, – настойчивое напоминание о том, что, когда мы замышляем реакцию с короткоживущими молекулами, мы должны тщательно продумать, как ее выполнить, иначе не миновать нам позорного провала.
Например, нельзя сначала генерировать арин, и затем добавить диен. Диен должен быть в реакционной смеси уже тогда, когда арин появляется на свет. Поскольку большинство известных методов генерирования (генерирование – это специальное слово, используемое вместо обычного “получение”, именно потому, что мы не можем получить – выделить, потрогать, понюхать, осмотреть – короткоживущие молекулы, а другого слова для этого процесса русский язык не имеет) аринов использует сильные и очень сильные основания, нужно думать о совместимости таких реагентов с диенами. Увы, обычные диены терпеть не могут сильных оснований, большинство просто банально полимеризуется, а некоторые, типа одного из любимейших диенов человечества, циклопентадиена, вообще дают ароматические анионы, которые не проявляют совершенно никаких признаков диенового характера.
Это скверная история: иметь такие мощные диенофилы и быть настолько ограниченным в выборе диенов! Химия – чертовки мерзкая гадость, в ней никак не получается свободный полет творческой фантазии: это нельзя, то нельзя, все остальное тем более нельзя, и еле-еле удается промылиться между всеми этими препятствиями, чтобы хоть что-то стало можно. Строго говоря, с аринами есть отличный выход, потому что существуют неплохие методы их генерации, не использующие сильных оснований. Да вот беда – мы их не проходим.
Итак, генерируем арины действием сильных оснований на галогенпроизводные. Основание подбираем так, чтобы оно не давало нуклеофила для конкурирующего замещения, то есть, например, бутиллитий, а не амид натрия. И достаточно 1 эквивалента (для замещения, как мы знаем, нужно два). При этом, не забываем, что во многих случаях такое отщепление дает не один арин, а два. Если хотим этого избежать и генерировать точно один арин, следует использовать чуть видоизмененный метод, когда берут орто-дигалогенбензол – в этом случае арин получается (в смысле, генерируется) один – с “тройной связью” между атомами, на которых были галогены, – и действуют на него бутиллитием (или метиллитием) – в этом случае вместо отщепления протона идет так называемый обмен галогенид-металл, а за ним, как в обычном методе, отщепление второго галогенида.
Диены придется поискать. Самые популярные диены для этого синтеза довольно нетривиальны: оба являются ароматическими соединениями, сочетающими ароматичность с ярко-выраженным диеновым характером – фуран и антрацен. Продукты этих циклоприсоединений – довольно занятные вещества, особенно аддукты в антраценом, называемые триптиценом и представляющие собой такую трехконечную звезду на бициклическом каркасе.
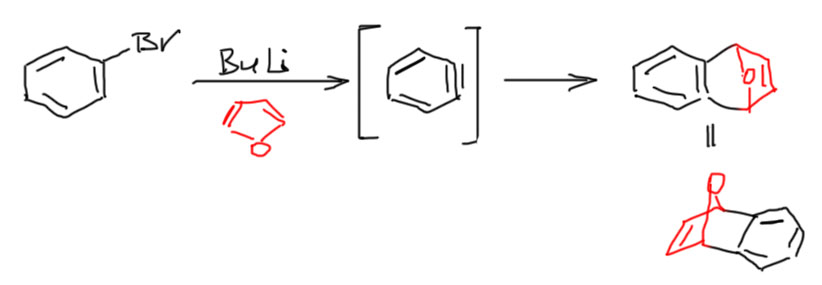
Диеновый характер антрацена обусловлен двойными связями в среднем цикле, – эта реакция выгодна, потому что взамен утраченной ароматичности всей молекулы антрацена (14 электронов во внешнем контуре) возникает два полноценных ароматических бензольных кольца. Продукт реакции, триптицен – такая трехконечная звезда на бициклическом каркасе, очень забавен тем, что в нем все три бензольных кольца совершенно одинаковы, и уже нельзя сказать (если не использовать заместителей или изотопных меток), что откуда прибыло, что из бензина, а что из антрацена.
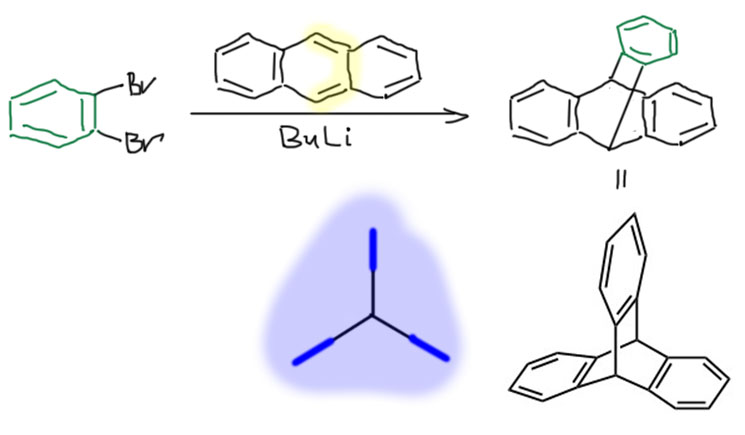
Кто-то, возможно, залезет в Википедию, и тут же обнаружит там пример циклоприсоединения арина именно с циклопентадиеном, вроде бы опровергающий то, что было сказано выше. Если почитать статью (Org.Lett. 2009, 11(1), 201), на которую ссылаются в Вики, можно понять в чем дело – там использован огромный избыток циклопентадиена, и авторы при этом жалуются на очень низкий (а very low yield) выход аддукта (зачем жалуются непонятно, у них выход приведен в 77%, а это отличный выход, отчего на самом деле cоздается впечатление, что авторы малость заврались, но это бывает). Проблема ведь в том, что кислотность циклопентадиена (pK 19) очень велика и BuLi количественно и практически моментально превращает его в анион – то есть у нас больше нет BuLi для генерации арина. Приходится признать, что раз реакция идет, циклопентадиенильное проиводное лития справляется с задачей генерации арина вместо BuLi именно как основание.
Не знаю зачем в Вики использовали такой странный и двусмысленный пример, но даже он отлично говорит о том, что проблем с выбором диена очень много. Не будем использовать циклопентадиен в этой реакции. —- В ноябре 2019 я перечитал этот раздел, и аж за голову схватился! Вот настоящее лыко в строку, говорит же Великий Вождь, что википедию надо срочно выбросить на свалку истории и срочно заменить ее большой советской энциклопедией или чем-то ещё того же типа, вероятно, откопав её на этой свалке. Вот вам ярчайший пример правоты Великого Вождя – действительно очень плохой пример привела википедия, непроверенный, ненадёжный, сбивающий с толку. Мораль – будьте осторожнее с википедией для поиска информации по органической химии. Но и большую какую-то-там энциклопедию тоже не советую – это еще хуже.
В последней контрольной работе одна вещь вызвала у меня глубокое изумление. Прошло уже несколько часов, а изумление не проходит. Чтобы как-то справиться с этим навязчивым состоянием, я решил написать вот это дополнение.
Дело в том, что в одной реакции над стрелкой была обозначена метафосфорная кислота. Вот так было написано (HPO3)n. 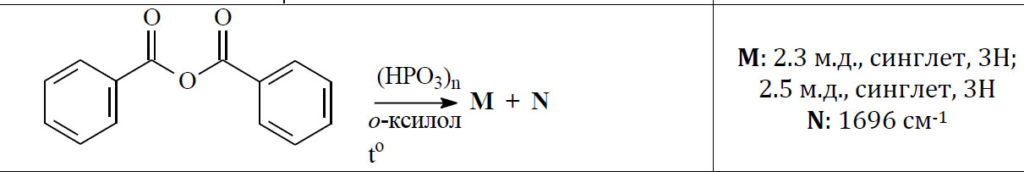 Это общепринятое обозначение полимерной метафосфорной кислоты. Что это за реагент, зачем он был взят, почему именно это и что имелось в виду? Естественно, когда так пишут, вместо обычных реагентов для, по всей видимости, обычной реакции ацилирования по Фриделю-Крафтсу, возникает масса вопросов и гипотез, уж не имел ли в виду составитель что-то гораздо более интересное. Уж больно реагент необычен. Успокоиться удалось не сразу, в голову лезли самые экзотические гипотезы типа образования ароматического цикла, состоящего из атомов фосфора и кислорода. Ну вот как-то так:
Это общепринятое обозначение полимерной метафосфорной кислоты. Что это за реагент, зачем он был взят, почему именно это и что имелось в виду? Естественно, когда так пишут, вместо обычных реагентов для, по всей видимости, обычной реакции ацилирования по Фриделю-Крафтсу, возникает масса вопросов и гипотез, уж не имел ли в виду составитель что-то гораздо более интересное. Уж больно реагент необычен. Успокоиться удалось не сразу, в голову лезли самые экзотические гипотезы типа образования ароматического цикла, состоящего из атомов фосфора и кислорода. Ну вот как-то так: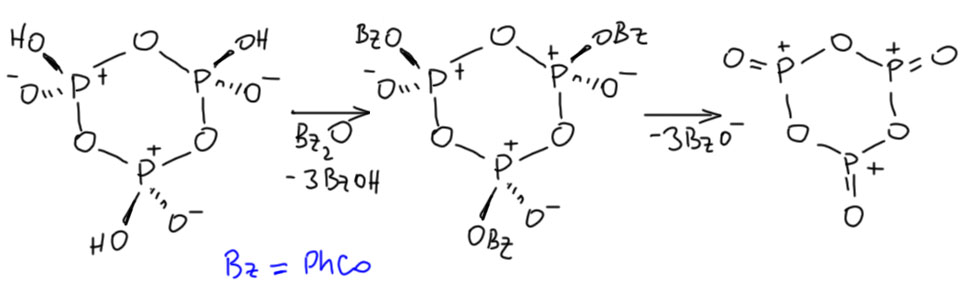
Эта циклическая хрень в конце, некий тримерный оксид фосфора, и правда может быть ароматической, потому что у кислорода есть пара, а у такого фосфора вакантная p-орбиталь, хотя бы по аналогии с серным ангидридом. Но гипотеза кажется всё равно диковатой. Такое соединение фосфора вряд ли вообще существует, хотя кое-что похожее с азотом вместо кислорода есть, и в любом случае это неорганика, а у нас вроде бы органика. Хотя, чёрт знает.
В общем, мучения становились невыносимыми, но тут я заметил, что под стрелкой написан о-ксилол. Довольно типичный выкококипящий растворитель. А вдруг это реагент? Не пишут реагенты под стрелкой, если только не хотят устроить разводку и прикол. Вроде бы контрольная не место для таких развлечений, но мы живём в непростое время, и все немного уже свихнулись, не будем строги к другим, не раскаявшись в собственных грехах. Тут я увидел и два сильнопольных синглета в условии, понял, что это ещё один прикол – забыть привести ароматические мультиплеты. В общем, в этом месте наступило просветление, обозначенное вздохами, переходящими в истерический хохот. Всё просто, оказывается, это обычное ацилирование по Фриделю-Крафтсу, в котором субстрат написан под стрелкой, реагент занимает больше места, чем иной субстрат, ЯМР-спектр подвергся цензуре, а реагент вызывает странные ассоциации. В придачу, опущено разложение реакционной смеси водой, без чего в этой смеси будет много самых неожиданных веществ. Вот что, видимо, имелось в виду.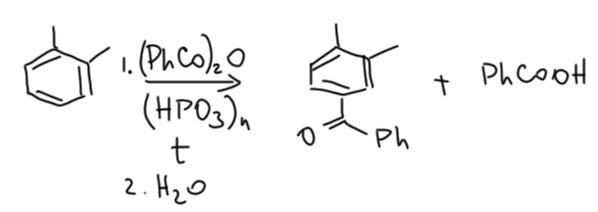
Но что это за странный катализатор полимерная метафосфорная кислота. Нельзя сказать, что это популярный катализатор. Весьма полная Энциклопедия реагентов для органического синтеза, например, никак не упоминает это реагент вообще. Точнее, упоминает, в одном месте, в стиле “отвяжитесь”. Но это не выдумка. Такой реагент есть. Более того, если за него потянуть, можно вытянуть немало интересного. Давайте разберёмся, это как раз довольно интересно, и в придачу, мы имеем шанс наконец понять, как устроены всякие загадочные полифосфорные кислоты. Это то же самое или нет? ПФК, которую мы уже часто применяли, и полимерная метафосфорная кислота это одно и то же? Самое удивительное, что оба ответа – и да, и нет можно считать одинаково верными или неверными. Есть смысл покопаться. Приступим.
Фосфорных кислот на свете довольно много. Фосфор в этом окислительном состоянии находится в sp3-гибридном состоянии, это тетраэдр, он координационно насыщен, все валентные возможности использованы. Самая обычная фосфорная кислота – орто-фосфорная H3PO4 Поэтому фосфорная кислота – просто протонная кислота, в водных растворах довольно слабая. Мы ее иногда применяем в качестве кислотного катализатора, например, для дегидратации несложных спиртов в олефины, всегда при нагревании и используя смещение равновесия, отгоняя летучий продукт. Но для ацилирования ароматики её кислотности не хватает. Для ацилирования применяют галогенангидриды и ангидриды. Мы знаем, что активация происходит за счет кординации кислотного катализатора по ацильному кислороду. Кислотный катализатор может быть или протонной кислотой или кислотой Льюиса. Электрофилом может быть или сам комплекс ацилирующего агента с кислотой Льюиса, или протонированная форма, или ацильный катион, который получается при диссоциации этих предшественников. В разных случаях бывает и то, и то. Нам сейчас это не важно. Мы просто понимаем, что, во-первых, протонные кислоты используют для ацилирования реже Льисовых просто потому, что они вызывают побочные реакции. Во-вторых, кислотность орто-фосфорной кислоты еще и маловата для этих целей.
А что мы так прицепились к фосфорной кислоте? Дёшево, доступно, невредно (каждый человек с кока-колой за жизнь выпивает десятки литров фосфорной кислоты), да и реагент почти не вызывает побочных реакций, так как не является окислителем, как серная. В общем, мечта, только не такая, которую можно ухватить руками без некоторой подготовительной работы! К счастью, орто-фосфорная кислота не одна в этом ряду. Нет ли там чего-нибудь поактивнее. А вот мета-фосфорная кислота бывает? Что это вообще? О, это совершенно удивительная штука! Это кислота Льюиса. Фосфор в этой молекуле имеет sp2-гибридное состояние, она плоская, а на фосфоре сидит огромная пустая p-орбиталь. Более того, анион этой кислоты, мета-фосфат (а она и протонная кислота тоже) тоже плоский, и поэтому изоэлектронен и изоструктурен не много не мало – с самим серным ангидридом! Вот они все три, для каждого показана только одна граничная структура, сверху они на плоскости, снизу – бочком с p-орбиталью. 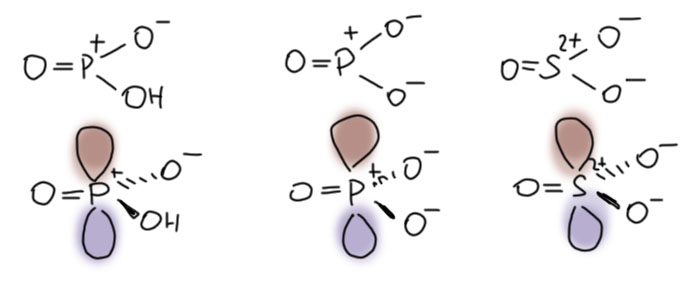
Мы знаем, что серный ангидрид – электрофил чудовищной силы, и кислота Льюиса могучая. Настолько, что применять его во всяких реакциях просто трудно, он совершенно неукротим и дров может наломать немало. Перебор. Метафосфат, напротив, электрофил и кислота Льюиса слишком деликатная, всё же в нём минусов больше, чем плюсов. А вот метафосфорная кислота, похоже, то, что нужно. Это весьма сильный электрофил и кислота Льюиса. В этом несложно убедиться, если понять, что эта молекула изоэлектронна и изоструктурна протонированному серному ангидриду. Протонированного серного ангидрида вообще-то не бывает, потому что сам серный ангидрид уже электрофил колоссальной силы и протонировать его совершенное безумие, мы бы точно переполнили чашу долготерпения богов. Но когда мы от серы переходим к фосфору и видим там изоэлектронные и изоструктурные соединения, мы понимаем, что этот переход сопровождается очень сильным снижением кислотности и электрофильности для изоэлектронных молекул. Могучий электрофил SO3 соответствует просто хорошему электрофилу метафосфат-иону PO3–, а реально несуществующий, но по определению невероятно могучнейший электрофил HSO3+ соответствует просто сильному электрофилу мономерной метафосфорной кислоте HPO3
Нарисуем реакцию с ангидридом карбоновой кислоты. Электрофильный фосфор с удовольствием цепляет кислород, фосфор очень любит кислород, дальше обычная активация , фосфорный остаток забирает карбоксилат, остаётся ацильный катион, готовый ацилировать ароматику (желающие могут нарисовать то же самое, только без образования ацильного катиона, как мы это делаем, например, а ацилировании хлорангидридом в присутствии хлористого алюминия, будет тоже самое, только более громоздко). Вот это нечто, образовавшееся, когда фосфорный остаток заберёт карбоксилат – это смешанный ангидрид фосфорной и карбоновой кислот, в виде сопряжённого основания (кто любит биохимию, знает, что там обожают рисовать фосфорные остатки именно в таком виде). И еще, напомню, что я везде рисую структуры соединений фосфора и серы, сохраняющие октет, так, что на атоме не может быть больше 4 валентных чёрточек, как, в принципе, и надо делать. 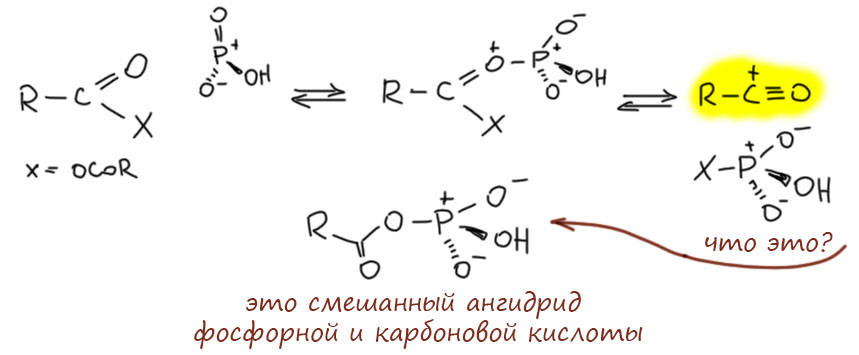
Итак, мы видим, что метафосфорная кислота вполне способна работать как кислота Льюиса и активировать ацилирующие агенты в ароматическом ацилировании по Фриделю-Крафтсу.
Но! Проблема! На свете нет свободной метафосфорной кислоты. Если ее попробовать получить, очень электрофильный фосфор спросит, а нет ли тут немного водички, и если есть, тут же присоединит её, образовав обычную орто-фосфорную кислоту. А если водички нет, то в другой молекуле метафосфорной кислоты найдётся нуклеофильный атом кислорода, и тоже пойдёт в дело – произойдёт димеризация, и на этом дело не остановится. 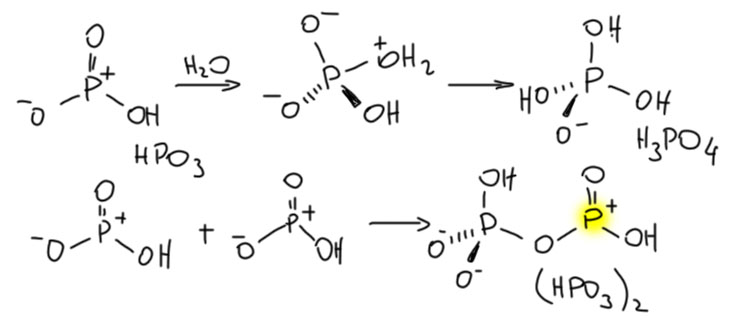
Смотрим внимательно на димер метафосфорной кислоты и видим, что один из фосфоров сохранил sp2-гибридную конфигурацию, а значит, остался сильным электрофилом и кислотой Льюиса, пригодным для активации в реакции Фрилеля-Крафтса. Но он тоже захочет нуклеофила, и если будет рядом молекула метафосфорной кислоты, присоединит её. И так и будет, пока есть свободная HPO3 – поэтому вместо мономера реально всегда быввает полимер. Вот такая метафосфорная кислота и существует реально. И не забывайте на фосфорах заряд ставить, не будьте как я. 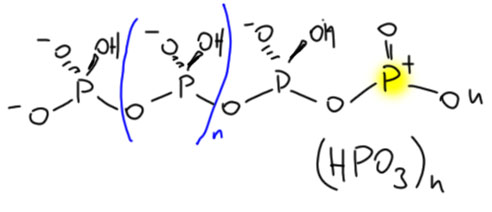
И когда такая полимерная кислота встречает, например, ангидрид, она просто сдаёт ему концевую электрофильную группу, и укорачивается на одно звено. Поскольку полимер этот не имеет определённой длины, считайте, что он этого и не заметит, а укороченная молекула будет снова иметь на конце такое звено и сможет активировать следующую молекулу. Здесь заряды не забыл. Активированный ангидрид отправится в реакцию ацилирования или сам, или диссоциировав на ацильный катион. 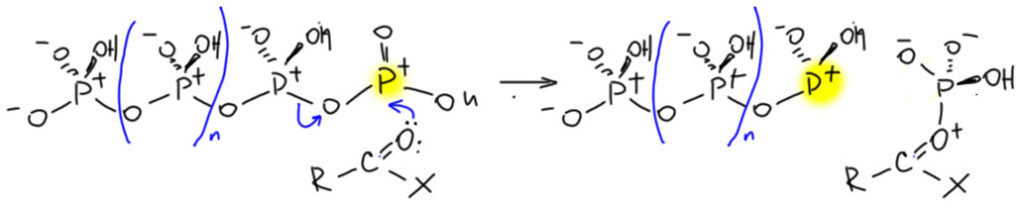
Вот так это и должно работать, если мы используем полимер метафосфорной кислоты для активации ацилирующего агента в реакции Фриделя-Крафтса.
Но это только полдела. Тут всё ещё немного запутаннее, и если хотите прочитать продолжение, в котором, например, выплывет полифосфорная кислота и фосфорный ангидрид, и даже АТФ, его можно найти здесь.
Изменения реакционных центров
Электрофильное замещение
В электрофильном замещении очень важно не забывать про реакционную способность и ориентацию замещения. Ароматические соединения очень сильно различаются по реакционной способности в реакциях электрофильного замещения, и ваша задача – правильно подбирать реагент, не использовать одно и то же для метоксибензола и нитробензола. От вас никто не требует запоминать десятки вариантов реагентов, которые можно найти в литературе буквально для каждого из субстратов, но важно хотя бы запомнить и применять два-три варианта реагентов – для активированных производных (одна или больше донорных групп), для дезактивированных производных (одна или больше акцепторных групп, включая галогены), и для серединки (сам бензол и производные одновременно с донорными и акцепторными группами). Также вашей заботой должна быть и селективность замещения – избегайте получать смеси изомеров, которые вы не знаете, как разделять. Те случаи, когда выделение конкретных изомеров возможно и относительно легко, разобраны ниже в секции про Вспомогательные методы. Во всех остальных случаях предполагается, что разделение невозможно, и нужно выбирать другой подход к синтезу.
Сюда входит также сульфирование, но мы так редко применяем эту реакцию, что совсем забыть о ней – не велика потеря, всё равно вряд ли пригодится. Кроме бромирования есть еще хлорировавние и иодирование, но я специально выделил только бромирование, потому что бром – самый удобный галоген в ароматической химии, и если нет особой необходимости в других галогенах бром стоит предпочесть безусловно.
Ароматическое нитрование – чрезвычайно важная реакция хотя бы потому, что нитро-группа легко восстанавливается в амино-группу, а из ароматической амино-группы можно сделать вообще все на свете, в чем мы убедимся во 2 семестре. Пока разберемся с нитрованием.
Нитровать можно и активированные, и дезактивированные производные бензола. Нитрование ароматических аминов и фенолов – особая задача, которую мы разберем отдельно в соотвествующих разделах.
Для нитрования бензола и его активированных производных используют простые нитрующие реагенты. Основной нитрующий реагент – обычная нитрующая смесь, смесь концентрированной азотной кислоты (у такой кислоты приблизительно 67%-ная концентрация и плотность 1.41, это прозрачная бесцветная подвижная жидкость, почти не дымящая на воздухе) и концентрированной серной кислоты (с концентрацией 96-98% и плотностью 1.86). Мононитрование бензола и его активированных производных (алкил, алкокси-бензолов) делают такой смесью при температурах не выше комнатной, или при охлаждении. Чем активнее субстрат, тем тщательнее нужно держать температуру, иначе неизбежно получите динитро, а иногда и тринитросоединение – это не только уменьшает выход, но и опасно, при перегонке продуктов возможны неприятности. Всегда образуется смесь изомеров согласно правилам ориентации
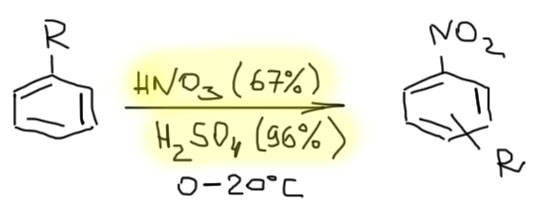
Если производное бензола совсем активно, например, содержит несколько донорных групп, нитровать можно чистой азотной кислотой без серной кислоты, во всяких растворителях, совместимых с азотной кислотой – уксусной кислоте, хлорпроизводных (растворители из спиртов, эфиров, кетонов, производных кислот с азотной кислотой несовместимы, а их применение может привести к плохим последствиям – воспламенению или даже взрывам. Вообще будьте осторожны – химия нитрования чревата неприятностями и расслабленного отношения к подбору условий реакции не прощает – если будете делать нитрование в практикуме, особенно тщательно относитесь к соблюдению условий реакции и выделения продукта, указанные в методике. Если вы упустите температуру в других реакциях, наказанием будет осмоление, низкий выход, плохое качество продукта. Если вы упустите температуру в нитровании, наказанием может быть нечто более серьёзное.
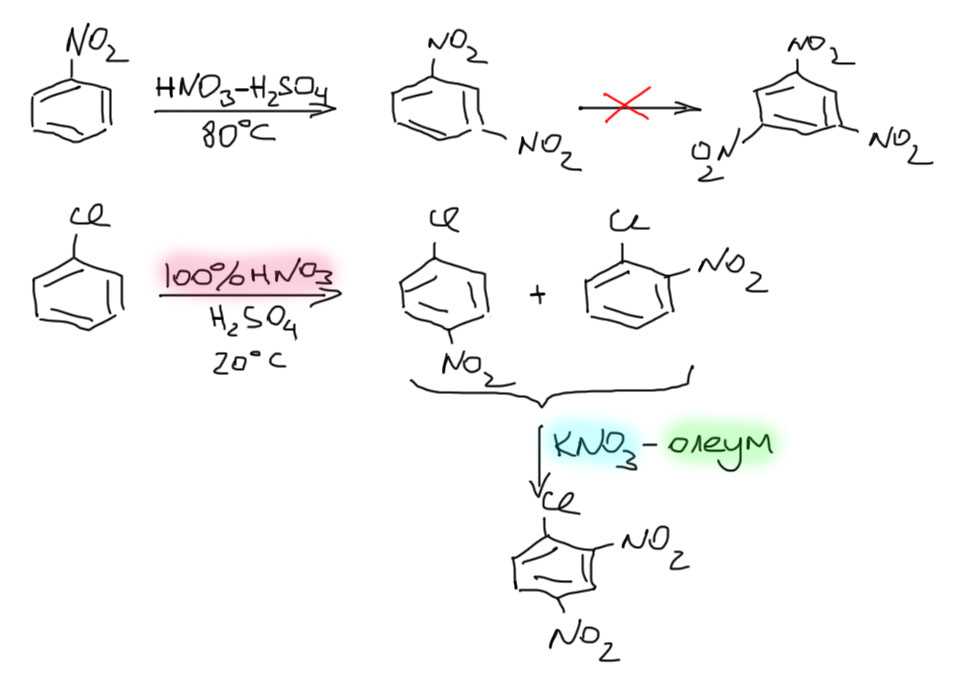
Дезактивированные производные бензола нитруют более активными нитрующими агентами. Их много, но принцип их составления прост – меньше воды и больше кислотность. Поэтому концентрированную азотную кислоту заменяют на безводную, 100%-ную азотную кислоту. Такую кислоту часто еще называют дымящей – она почти всегда, если только не свежеперегнана с использованием веществ, убирающих низшие степени окисления азота, имеет бурый цвет и активно дымит. Плотность такой кислоты 1.51. Это – очень неприятное вещество, весьма опасное в обращении, активно окисляющее почти любую органику, например, резиновые шланги. Работать с ней нужно или очень аккуратно и вдумчиво, или не работать вовсе. Вместо такой кислоты в нитрующих смесях высокой реакционной способности часто используют твердые нитраты щелочных металлов, селитры, это гораздо удобнее и безопаснее, но имеет свои сложности в виде затвердевания реакционных смесей, затрудняющих перемешивание. Концентрированную серную кислоту заменяют 100%-ной или олеумами. Про эти вещи подробнее поговорим в сульфировании. Чем ниже реакционная способность, тем более жесткий нитрующий агент подбирают, одновременно немного увеличивая температуру. Температурой при этом злоупотреблять нельзя – выше 70-80ºС начинаются почти всегда крайне мерзкие и опасные побочные процессы в том числе частичной деструкции кольца, и образуются полинитрованные соединения, вплоть до тетранитрометана C(NO2)4 – нитрование, вышедшее из-под контроля нередко становится билетом в рай в один конец. Именно это, и ничто иное ограничивает наши возможности ужесточения условий нитрования, и, например, не дает нормально вводить три нитрогруппы в бензол и менее реакционноспособные соединения при том, что третье нитрование реально идет, но с низкой скоростью, а еще повысить температуру, чтобы эту скорость подстегнуть и получить приличный выход, мы не можем, потому что жить хочется. Поэтому тринитробензол, тринитрохлорбензол и похожие соединения получают окольными путями. Зачем? Это очень важные субстраты в нуклеофильном замещении, см. соответствующую вкладку. Вот несколько типичных примеров нитрования дезактивированных производных бензола.
Бромирование – самая полезная из реакций ароматического галогенирования. Если вам нужен галоген в ароматическом кольце, и не важно, какой конкретно (например, вы хотите иметь магний или литий-органику, или нуклеофильное замещение), выбирайте бром. Реакции бромирования – самые селективные из всех реакций галогенирования, а реагенты бромирования имеют самый большой диапазон – бромировать доступными бромирующими реагентами можно и очень активные и сильно дезактивированные производные бензола. Бром очень легко вводить по одному атому столько, сколько нужно – дибром-, трибром-, тетрабром-, вплоть до полностью бромированных по всем доступным положениям соединений.
Как всегда в этой теме убираем из рассмотрения ароматические амины и фенолы.
Самый простой бромирующий агент – сам бром, желательно в протонных растворителях типа уксусной кислоты, которые помогают разделить молекулу брома на электрофильную часть и уходящий бромид. Реакционной способности брома в уксусной кислоте вполне достаточно для активированных производных бензола с алкильными и алкоксильными заместителями, хотя моноалкилбензолы реагируют довольно медленно. Как всегда получается смеси изомеров, определяемые правилами ориентации. По сравнению с нитрованием в бромировании всегда получается больше пара-изомера, но не настолько, чтобы считать реакцию селективной. В лаборатории такие смеси обычно разделить невозможно (ничего невозможного нет, но приходится думать, сколько это будет стоить, и ответ будет неутешителен). Поэтому выбирайте для бромирования такие соединения, которые дают один основной изомер.
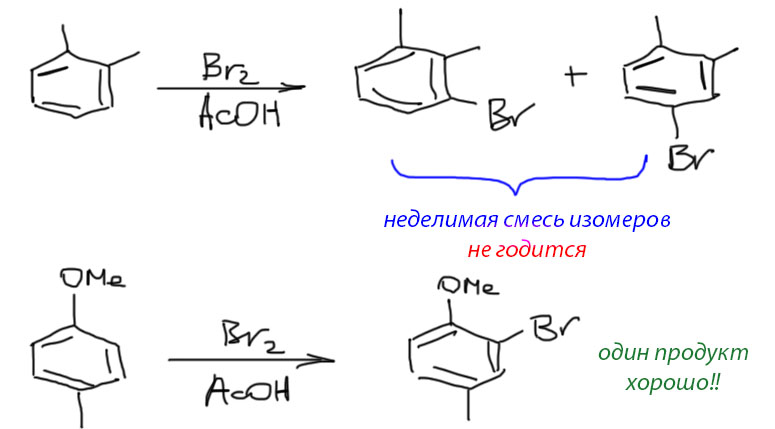
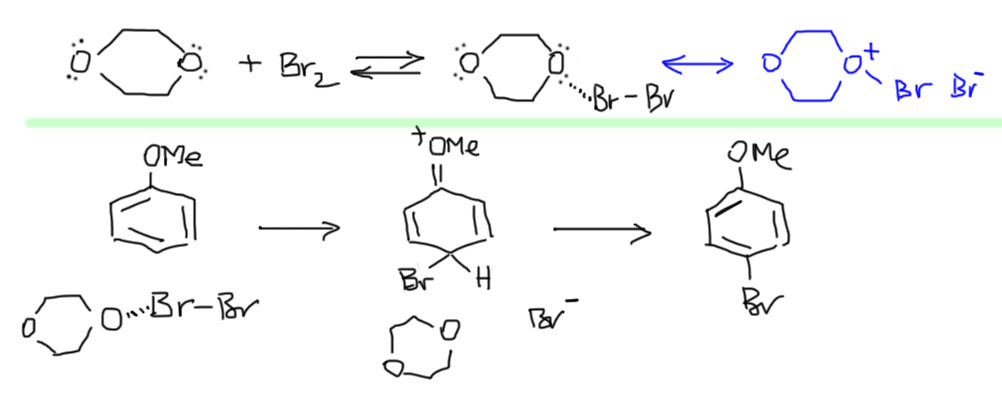
Для селективного бромирования в пара-положение к главному ориентанту есть довольно много специальных реагентов. Все они устроены одинаково – к электрофильному брому лепится что-нибудь крупное, и тогда направление реакции будет в основном определяться стерическими препятствиями – в орто-положения такой реагент просто не лезет. Самый простой из таких реагентов – комплекс брома с 1,4-диоксаном, дешевым растворителем эфирного типа. Это очень слабый, так называемый молекулярный комплекс, в котором молекула брома слабо взаимодействует с молекулой диоксана через неподеленную пару кислорода в основном за счет слабой поляризации молекулы брома, что обусловливает диполь-дипольное взаимодействие. Структуру комплекса можно было бы представить иначе, если предположить, что связь между кислородом и бромом – настоящая донорно-акцепторная, что привело бы к разрыву связи Br-Br и образованию оксониевого иона с высокополярной связью O-Br и весьма электрофильным атомом брома (в этой структуре формальный заряд рисуется на атоме кислорода в соответствии с правилами образования донорно-акцепторной связи, но реальный положительный, электрофильный центр будет на броме из-за разности электроотрицательностей). Такая структура отлично объясняла бы, почему такой комплекс электрофилен. Увы, реальная структура гораздо ближе к слабому молекулярному комплексу с очень слабой наведенной поляризацией. Мы имеем право нарисовать обе структуры и считать их граничными, но не можем забывать, что вклад синей структуры очень мал.
Тем не менее этот комплекс, несмотря на его слабость, очень легко получается просто при смешивании брома и диоксана в виде оранжевого порошка. Нет ли здесь противоречия – ведь слабость подразумевает равновесие, смещенное в сторону исходных? Нет, противоречия нет, в растворе равновесие действительно так и устроено, но комплекс выпадает в виде твердого осадка и равновесие смещается. Твердый комплекс довольно устойчив и может даже недолго храниться при отрицательной температуре, хотя его стараются использовать сразу после приготовления и короткой сушки. Если будете его получать, не оставляйте при комнатной температуре даже “до следующего практикума” – улетит, и записочки на прощание не оставит. И не забывайте – и комплекс диоксана, и другие такие реагенты работают только для активированных ароматических соединений. Строго говоря, даже для моноалкилбензолов (толуола и др.) этот реагент слабоват, и реакция бромирования очень медленная.
Для менее реакционноспособных производных бензола, начиная от моноалкилбензолов в сторону дезактивированных производных (например, в кольце и донорные, и акцепторные заместители, или только один акцепторный, там же и сам бензол) нужно повышать электрофильность. Делают это, добавляя к молекулярному брому кислоты Льюиса. В принципе, годятся многие, но поскольку задача довольно простая, редко берут что-то кроме бромного железа или алюминия. Бромное железо даже не нужно где-то брать отдельно как готовый реактив – из-за огромной гигроскопичности безводный бромид жедеза(III) умудряется нахватать воды и превратиться в мерзкую коричневую жижу даже в с виду плотно закрытых банках. Но достаточно в реакционную смесь просто добавить металлическое железо в виде порошка или опилок, и оно там само прореагирует с бромом и даст нужный катализатор бромирования. Именно катализатор – кислоты Льюиса в электрофильном галогенировании не расходуются, и их можно использовать в небольших количествах относительно количеств основных реагентов. С бромистым алюминием проблема похожа, так он тоже чудовищно гигроскопичен, но здесь не получится обойти это препятствие таким же способом – реакция алюминия с бромом настолько экзотермична, что контролировать ее невозможно. Поэтому в большинстве случаев выбирают именно смесь железо-бром, а к безводному бромистому алюминию прибегают только в особо сложных случаях, так как он дает намного более активную бромирующую смесь. Пара типичных примеров:
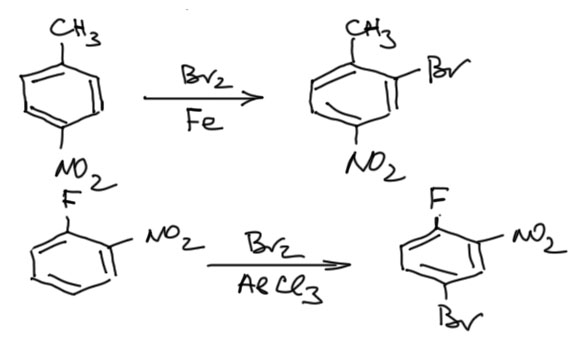
И наконец, хотя мы и не будем часто встречаться с такими случаями, если нужно вогнать бром в совсем дезактивированное соединение уже с двумя-тремя акцепторами, придется использовать совсем тяжелую артиллерию. На кислоты Льюиса в этих случаях надежды мало, и приходится пытаться получить бром в степени окисления 1+ прямо в реакционной смеси. Если бы кислоты Льюиса были в состоянии растащить молекулу брома на две части – бромид и Br+ – то это было бы именно, то, что нужно. Увы, молекулу брома (и других галогенов) невозможно полностью растащить на ионы – на этот процесс есть фундаментальный запрет во всех известных религиях, включая верования в Великого Макаронного Монстра, и он, процесс диссоциации, никогда не происходит. Более того, мы не можем растащить на ионы и так называемые интергалогены, например, BrF, из-за чего эти самые интергалогены, которые сначала кажутся прямо идеально предназначенными для таких целей (мы же знаем, насколько сильно различаются по электроотрицательности, например, бром и фтор, и кажется – такие молекулы должны прямо сами разваливаться на анион и катион, но нет – не разваливаются, ни сами, ни с помощью кислот Льюиса) – вообще ни на что серьезное неспособны и из-за этого почти совсем не применяются в химии ароматического галогенирования.
Приходится прибегать к другому приему – пытаться получать бром в степени окисления +1 (но не сам катион Br+, а что-то молекулярное, условно говоря, производное гипобромистой кислоты HOBr) окислительно-восстановительной реакцией, используя бромид и сильный окислитель в таком соотношении, которое дает как раз эту степень окисления (да, придется вспомнить, как такие реакции уравниваются, иначе не попадете). И в качестве растворителя брать то, что дополнительно повышает электрофильность, например, серную кислоту или даже олеум (сульфирования не боимся – сильно дезактивированные соединения практически не сульфируются). Одна из популярных смесей такого типа бромид-бромат.
Нуклеофильное замещение
- Выбираем...
- Активированное нуклеофильное замещение
- Ариновый механизм
- Ариновое замещение
- Кине-замещение: орто в мета
- Внутримолекулярное ариновое замещение
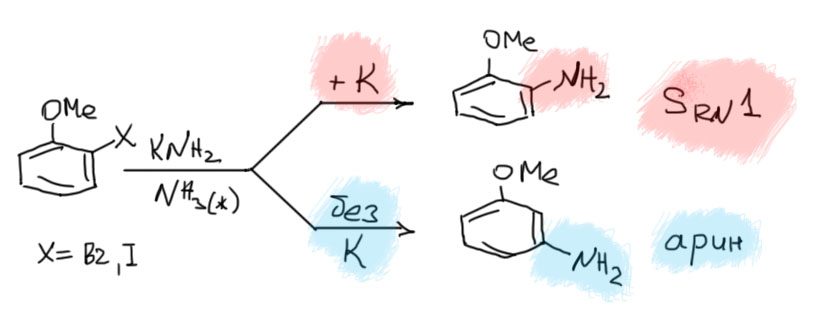
- SRN1 - восстановительное нуклеофильное замещение
Обязательно разберитесь с нуклеофильным замещением, с тем, как мы направляем реакцию по тому или иному механизму, и что от этого зависит. Немного более подробно тема разработана на отдельной странице
Активированное нуклеофильное замещение – очень частная реакция, имеющая, тем не менее большое значение. Механизм этой реакции сильно напоминает механизм реакции электрофильного замещения, являясь во многих смыслах его антиподом (это особенно ярко проявится, когда мы доберемся до химии гетероциклических соединений). Для реакции необходимо два условия – наличие уходящей группы, как правило, атома галогена, и в орто- или пара-положению к ней акцепторных заместителей. В качестве акцепторных заместителей почти всегда используют нитро-группы. Особенно хорошо, когда акцепторов (нитро-групп) в таких положениях две, а особенно три. Когда нитро-групп три, реакция становится настолько легкой, что позволяет использовать и другие уходящие группы, и очень широкий ассортимент нуклеофилов. Механизм реакции включает обратимое образование σ-комплекса (очень похожего на σ-комплекс ароматического электрофильного замещения, только там делокализуется катион, а здесь анион), с последующим, как правило необратимым уходом уходящей группы. Медленной стадией всегда является стадия образования σ-комплекса, а необратимость второй стадии имеет очень важные последствия, например, не свойственную другим реакциям нуклеофильного замещения возможность замещения фторида. Запишем пример механизма с пара-замещенным нирогалогенбензолом. В σ-комплексе сразу обозначим делокализацию минуса из кольца в нитро-группу.
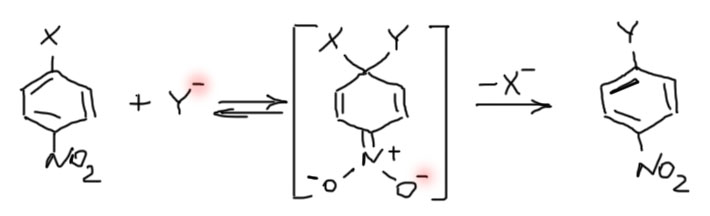
Еще раз повторю, что яркой особенностью этого механизма является очень легкое замещение фтора – и это очень явно выраженный эффект. Реакционная способность фторпроизводных на два-три порядка превосходит реакционную способность хлорпроизводных (с одинаковыми заместителями, естественно), а это колоссальная разница, позволяющая, например, селективно замещать фтор, если в молекуле есть атомы фтора и других галогенов (но не забываем, что замещаются только галогены в активированных положениях). А вот между остальными галогенами разница небольшая и непостоянная, и ею проще пренебрегать, чем пытаться что-то из этого вытащить. Ряд реакционной способности получается такой:
F >>> Cl ≈ Br ≈ I
Именно поэтому эту реакцию в самом общеупотребительном варианте обычно очень легко опознать, а опознав, очень легко использовать. Для ее осуществления не нужно ничего, кроме подходящего субстрата и нуклеофила. Нуклеофилы используют точно те же самые, что и для SN2-замещения, но при этом совершенно не нужно беспокоиться о конкуренции с элиминированием – поэтому годятся и нуклеофилы, являющиеся сильными основаниями. К сожалению, не годятся карбанионы в виде литий- и магнийорганических соединений, потому что они реагируют с нитро-группой (опять таки, когда мы доберемся до химии гетероцикличесих соединений, мы увидим, что там полно субстратов для активированного нуклеофильного замещения, и при этом, там не нужны нитро-группы, и там как раз можно использовать карбанионы). Еще одна странность – цианид-ион, который при уже привыкли считать очень ценным и понятным нуклеофилом в алифатическом нуклеофильном замещении, очень странно и необычно ведет себя в ароматическом. Просто не будем его использовать, а интересующиеся могут поискать в литературе необычные реакции цианида с субстратами нуклеофильного ароматического замещения.
Если мы имеем дело с неактивированным производным бензола, в котором есть атом галогена и либо вообще нет заместителей, или есть донорные или слабо-акцепторные заместители, то в обычных условиях такие соединения не вступают в реакцию нуклеофильного замещения, остаются инертными к действию нуклеофилов в полном соответствии с тем, что мы обсуждали в теме алифатическое нуклеофильное замещение – говорили же тогда, что если галоген висит на sp2-гибридном углероде, то с места его не свернешь, гиблое дело.
Свернуть однако все же можно, и даже не одним способом, но всякий раз для этого потребуются ухищрения. Самый полезный метод использует, как это часто называют, грубую силу (brute force). Мы отлично знаем, что можно сделать, когда замещение не идет – тогда может идти элиминирование. Элиминирование в целом более универсально чем замещение, и если взять основание посильнее, то почти всегда есть шанс оторвать сначала протон из соседнего положения, если он там есть, конечно. Но что будет, если это сделать в ряду бензола? Должна получиться тройная связь. Но это невозможно – в циклах размером меньше 8 атомов настоящей тройной связи быть не может! Напомню, что такое настоящая тройная связь. Это связь между атомами в состоянии sp-гибридизации, что означает, что эти атомы и непосредственно связанные с ними лежат на прямой линии, и ничего с этим сделать нельзя. Если по какой-то причине эти 4 атома отклоняются от прямой, то в середине уже не настоящая тройная связь, а какое-то недоразумение и посмешище, готовое от стыда и позора провалиться под землю, – ну или быстренько с чем-нибудь прореагировать, если уж провалиться не получится. Вот поэтому настоящей тройной связи в небольших циклах (до 7-членных) быть не может – вы просто не сможете соединить прямой четырехуглеродный фрагмент в цикл, распялив между 1 и 4 атомами линейного фрагмента оставшиеся вам атомы – это такая геометрия нехитрая, обойти которую нельзя.
Тем не менее, если невозможна настоящая тройная связь, может быть возможна какая-нибудь другая, послабее. Посмотрим. От хлор- или бромбензола действительно элиминировать можно, и молекула, которая при этом получается не является ацетиленом, хотя ее рисуют с тройной связью, и называют бензином. Да, именно так, и всем плевать на страдания носителей русского языка, у которых этим словом обозначается одна из самых священных жидкостей – в других языках такой коллизии нет. Будем поэтому мучиться и вздрагивать – этот бензин на бензоколонке не продают, потому что он очень неустойчив и живет малые доли секунды. Обладая совершенно гигантской реакционной способностью, бензин (или в общем виде арин), всегда найдет во что превратиться, и за это особенно ценим химиками.
Итак, во-первых, бензин – это не ацетилен, не алкин, в нем нет настоящей тройной связи между углеродами в состоянии sp-гибридизации. Чтобы понять, что это достаточно просто подумать, как он получается. У галогенбензола с двух соседних атомов уходит протон (а электронная пара связи остается на месте), и галогенид, уносящий пару с собой. При этом, все это происходит в плоскости кольца, то есть на месте остаются гибридные орбитали углерода, ранее отвечавшие за эти связи. Сравним это с тем, что происходит, если мы элиминируем от галогеналкена – нечто похожее, но на месте бывших связей C-H и C-X появились p-орбитали (так как весь фрагмент распрямился, связи выстроились в линию), перекрывание которых дает полноценную π-связь – образуется настоящая тройная связь. В бензоле уже такое быть не может – потому что связи кольца имеют углы 120º и не собираются от этого отказываться, и в линию вытянуться не могут, а без этого нет sp-гибридизации. Гибридизация углерода остается sp2, и вместо p-орбиталей мы имеем гибридные орбитали под углом, что не дает им эффективно перекрываться и образовывать полноценную связь. Поэтому бензин – это не алкин, хоть именно так его и положено изображать, просто потому что это удобно. Но реально это устроено скорее как нечто среднее между двумя граничными структурами, в которых эти гибридные орбитали попеременно несут пару и дырку (минус и плюс, карбанион и карбокатион). Одна из этих граничных структур просто отражает, как образовался бензин – от соседних атомов ушли две уходящие группы, оставив на месте как раз пару и дырку. Поскольку в бензине два атома углерода одинаковы, законы квантовой науки не дают нам возможности сказать, на каком из углеродов что конкретно находится – после ухода протона и галогенида бензин “забывает”, что и где у него было до ограбления. Это “забывает” мы и изображаем в виде граничных структур. Или, чтобы не мучиться, в виде такого фейкового алкина с тройной связью. Главное, чтобы у нас память оказалась не так коротка, как у бензина, и мы помнили и что случилось, и что это значит. Итак, принимаем, что в бензине есть связь, обслуживаемая перекрыванием гибридных орбиталей в сумме несущих пару электронов (мы уже много раз убеждались, что это важный признак химической связи – именно пара электронов в общем пользовании двух, а иногда и более атомов), но эта связь слаба, что делает такую молекулу короткоживущей и невероятно реакционноспособной.
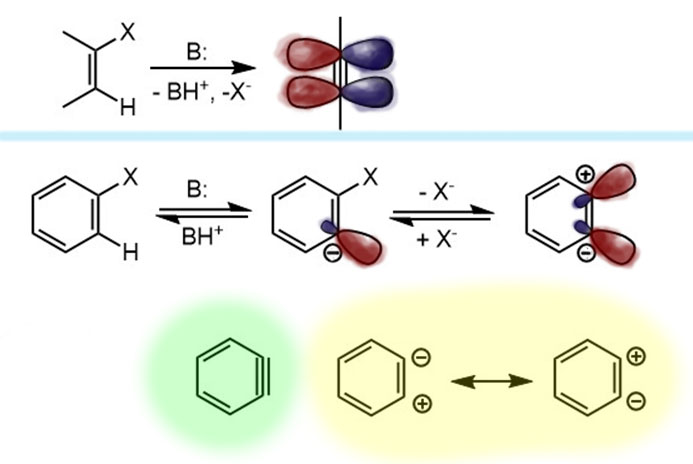
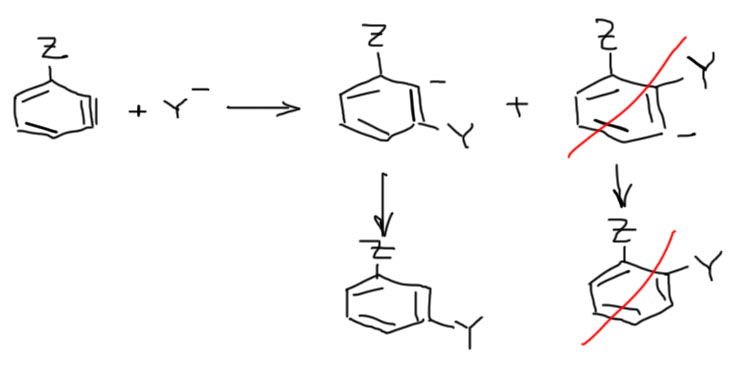
У самого бензина два углерода, образующие “тройную связь” совершенно одинаковы, и это означает, что вклад двух граничных структур совершенно одинаков, и на атомах реально нет зарядов. Но все может быть иначе, если в молекуле бензина есть заместитель рядом с этой “тройной связью”. В этом случае углероды становятся разными, различимыми, и законы квантовой науки более не заставляют нас ничего забывать. А так как у заместителей бывают донорные или акцепторные эффекты, граничные структуры становятся неравноценными. Например, если заместитель акцепторный (речь идет об индуктивном эффекте, так как мезомерный в такой конфигурации не действует), то граничная структура с плюсом рядом с заместителем дестабилизируется и понижает свой вклад за счет второй – это означает, что ближний углерод становится немного карбанионным, а дальний карбокатионным. Эта поляризация “тройной связи” имеет интересные последствия, которые мы обсудим чуть дальше.
Теперь можно нарисовать полный механизм нуклеофильного замещения с участием бензина (арина). Сначала для галогенбензола без других заместителей.
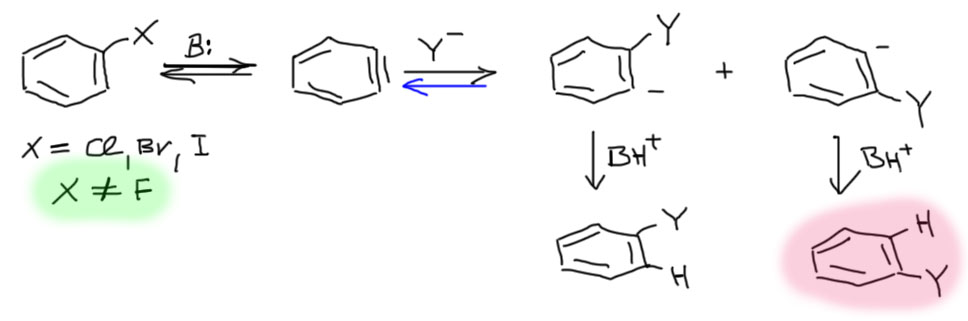
При отщеплении протона от галогенбензола (фтор не годится(примеры таких реакций в литературе найти можно, но они редкие и довольно спорные)) и быстрого ухода галогенида (фторид просто не уходит и поэтому фтор не годится) образуется промежуточный бензин, который немедленно присоединяет к своей “тройной связи” нуклеофил из реакционной смеси. Поскольку углерода “тройной связи” неразличимы, то нуклеофил с равной вероятностью присоединяется либо к одному, либо к другому. При этом образуется карбанион. Стадия присоединения нуклеофила обычно необратима, но в тех случаях, когда она бывает обратима, случаются интересные реакции (например, бешеная пляска галогенов, посмотрите одну из задач внизу и разберитесь сами, как это может быть). Карбанионы эти обладают огромной основностью и немеделенно находят протон, хотя бы в сопряженной кислоте основания, которое отрывало протон от исходного галогенбензола. Из-за равновероятного присоединения получается два продукта, которые можно различить, использовав изотопные метки в исходном галогенбензоле. В одном из продуктов нуклеофил садится туда, где была уходящая группа – это нормальное замещение. В другом – нуклеофил садится не туда, где была уходящая группа. Этот сенсационный результат невозможно объяснить по другому, и именно он стал ключевым доказательством этого механизма, который без этого доказательства так и остался бы безумной гипотезой. Путь, ведущий к этому продукту получил особое название кине-замещения.
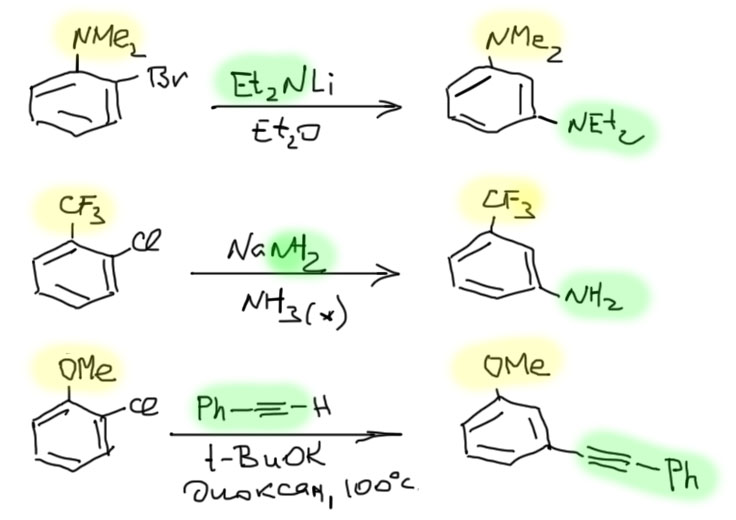
С ариновым механизмом замещения разобрались, теперь посмотрим на то, как это делают. Уже ясно, что нужно сильное основание и нуклеофил. Как мы знаем из алифатического нуклеофильного замещения, нуклеофилы иногда бывают сильными основаниями, и там это было почти проклятием, потому что не давало провести замещение, но здесь это стало благословением, потому что нам ровно это и нужно. Основания требуются довольно сильные: амиды щелочных металлов, простые литийорганические соединения, трет-бутилат в ДМСО. Амиды щелочных металлов чаще всего берут в растворе жидкого аммиака, в основном потому что эти ионные соединения с очень прочной кристаллической решеткой малорастворимы в обычных растворителях. Самый простой вариант – берем два моля основания-нуклеофила на моль галогенбензола. Пара примеров:
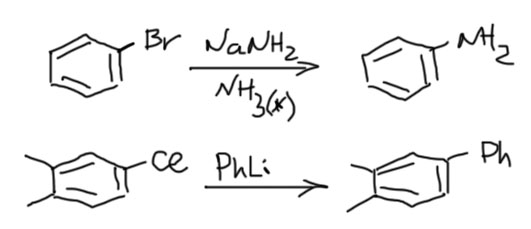
Так можно получать самые разные амины, вначале получив из амина его соль с щелочным металлом (такие соли называют амидами, и нужно быть очень внимательным, чтобы не путать эти амиды с амидами карбоновых кислот).
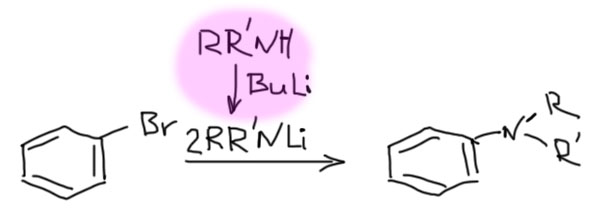
Иногда можно разделить основание и нуклеофил, первый использовать для генерации арина, второй для дела. Таких реакций довольно много, но планировать их трудно, и нам стоит избегать этот вариант, просто потому что у нас не хватит знаний корректно предсказать результат таких реакций. Посто для примера приведу образование сульфида. В этом случае все более-менее просто, потому что основность сульфида недостаточна для депротонирования, а нуклеофильность у него ого-го какая! А у трет-бутилата все наоборот. Из этой реакции мы можем сделать весьма важный вывод: несмотря на очень высокую реакционную способность аринов, они неплохо разбираются в нуклеофилах и выбирают те, что получше. В этом смысле они сильно и в лучшую сторону отличаются от совсем неразборчивых карбокатионов в SN1-замещении.

Все это совершенно замечательно, и кажется, что цены нет такому потрясающему методу. В современном синтезе эту реакцию не очень любят из-за низкой экономичности – один моль основания-нуклеофила теряется впустую (да, это серьезный аргумент). Нам это не очень важно. Но стоит нам ввести заместители в бензольное кольцо, как начинаются проблемы с селективностью, а это уже проблема тяжелая и, мы поклялись ею не пренебрегать. Как мы видели в механизме реакции, арин присоединяет нуклеофил к двум разным углеродам, и получается или нормальный продукт, или продукт кине-замещения, а чаще всего их смесь. Как мы видели в структуре арина на углеродах “тройной связи” может быть больше карбокатионного, или больше карбанионного характера, в зависимости от заместителей. В реальности, если заместитель находится в пара- или мета-положении, его эффект на поляризацию “тройной связи” не очень велик, и почти всегда образуются смеси, и нам точно нет смысла разбираться в том, чего там может быть больше и когда, да и не только нам, а и в большом органическом синтезе вся эта суета очень не нравится – не любят синтетики эту реакцию, грязная она. Вот, например, что получается из мета-изомера в типичной реакции с ариновым механизмом.
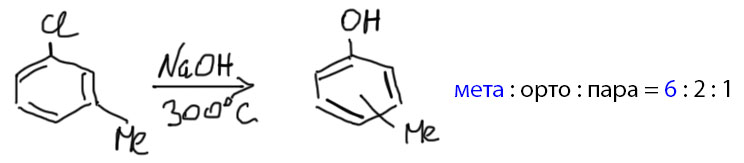
Высокая температура нужна, чтобы заставить щелочь – недостаточно сильное основание – реагировать по этому механизму. А что, температура так сильно влияет на основность??? Да, но только в том случае, когда отщепление протона необратимо, и в дело вступают законы кинетики (помните заклинание: При повышении температуры на десять градусов скорость химической реакции увеличивается в 2-4 раза – повторять натощак каждый день десять раз, выходные и праздники можно пропустить). На равновесия температура влияет, но далеко не так сильно. В случае аринового механизма отщепление протона необратимо, потому что за ним немедленно следует отщепление галогенида и реакция арина. И это делает возможным раскочегарить примитивную щелочь на ариновый механизм. Этим активно пользовались в допотопной промышленности для синтеза фенолов. После потопа изобрели более изящные методы, о которых мы поговорим в своем месте.
А вот пример ариновой реакции с пара-замещенным. А продукт основной – мета, увы, далеко не чистый, и пара-изомера тоже немало.
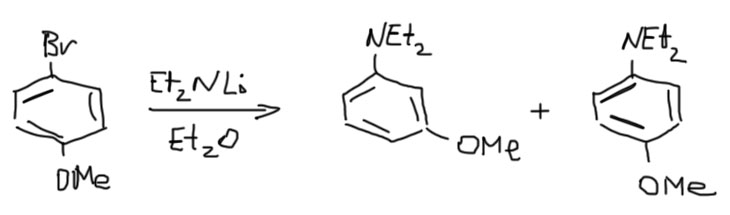
Ну и зачем нам такая химия? К счастью в некоторых частных случаях все получается гораздо чище и определеннее. Обсудим их подробнее на отдельной вкладке: Кине-замещение.
Ариновое замещение может быть полезным в тех случаях, когда присоединение нуклеофила к “тройной связи” чем-то управляется, так, чтобы сделать преобладающим только одно направление – либо нормальное замещение, либо кине-замещение. При этом нормальное замещение не так ценно как кине-замещение, потому что существует, особенно в современной химии, немало способов гарантированно добиться этого результата. А вот кине-замещение – характернейший признак только аринового механизма, и если есть случаи, когда оно идет чисто, то такие случаи обязательно навлекут на себя любовь и признательность синтетиков. И да, такие случаи есть. Самый известный и надежный, обнаруженный задолго до открытия аринового механизма – образование мета-изомеров из орто-галогензамещенных с группами, имеющими акцепторный индуктивный эффект, а это прежде всего алкокси-группы и амино-группы.
Но это же и мезомерные доноры, и сильнейшие! Это не важно, в ариновом механизме мезомерный эффект не работает, так как все события замещения происходят в плоскости ароматического кольца, в так называемой узловой плоскости π-орбиталей, а это обусловливает полное отсутствие сопряжения.
То, что важен именно индуктивный эффект экспериментально проверяется, например, и на трифторметильных производных – этот заместитель в принципе не обладает мезомерным эффектом, а результат замещения получается тот же. Отдельный вопрос – если заместитель фтор. Здесь, к сожалению, картина довольно запутанная, так как реальных данных немного, и иногда наблюдается замещение, а иногда – образование уже другого арина за счет ухода двух галогенов, и все очень сильно зависит от условий реакции. Нам такие подробности точно ни к чему, поэтому цсловно признаем фтор заместителем того же типа, и будем ожидать чистого кине-замещения.
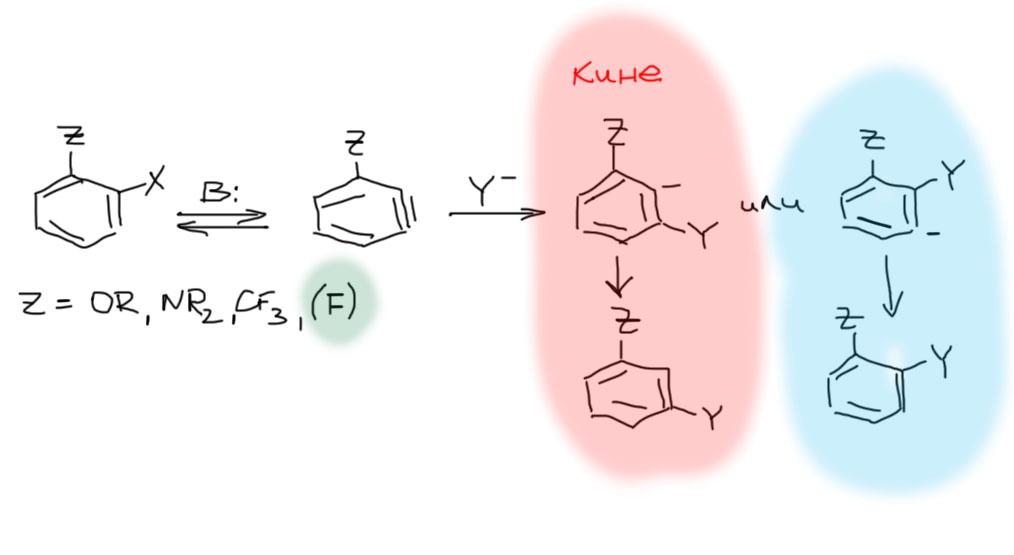
Почему это происходит. Есть два простых объяснения. Первое более популярно в научной литературе и восходит к ранним трудам классиков, оно основано на поляризации “тройной связи”, которое легко представить, как мы уже делали, двумя граничными структурами в представлении арина в виде такого карбанионо-карбокатиона. Мы помним, что в незамещенном арине вклад обеих структур был одинаков, но если рядом индуктивный акцептор, то структура с карбанионом рядом стабилизируется, а с карбокатионом рядом дестабилизируется. В результате арин действительно начинает напоминать настоящий карбанионо-карбокатион, и естественно приглашает нуклеофил по карбокатионному центру.
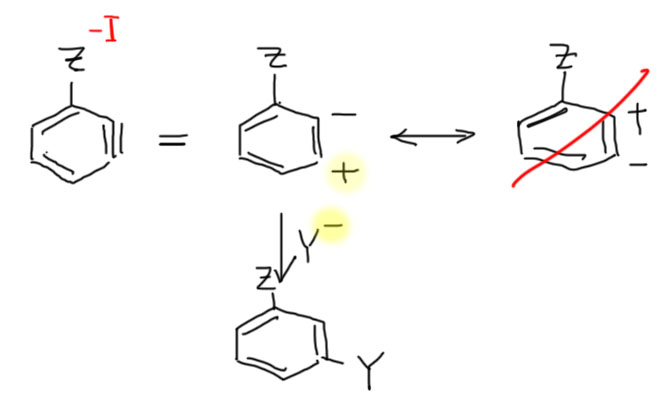
Второе объяснение более популярно в учебной литературе. Оно предполагает, что нуклеофил уже присоединился к “тройной связи”, при этом сначала образуется карбанион. Альтернативные пути присоединения дали бы два разных карбаниона. Тогда мы сравнили бы эти карбанионы по устойчивости и выбрали бы тот, который более устойчив, а это тот, у которого рядом индуктивный акцептор. И результат получился бы точно таким же. Это объяснение очень похоже на все, что мы делали раньше, например, с присоединением к двойной связи, где мы тоже выбираем более устойчивый карбокатион (если присоединение электрофильное) или карбанион (если нуклеофильное).
Так какое же объяснение верно! – обязательно воскликнет любитель ясности. Увы, после того как нас безвременно покинули товарищи Ленин и Сталин, которые в любом деле всегда знали единственно верное объяснение на основании диалектического и исторического материализма, никакой ясности не стало, и непонятно, откуда ее можно взять. Серьезная наука всегда работает с гипотезами, проверяя их на экспериментальном материале, и любая гипотеза всегда имеет вероятностный и приблизительный характер – может так, а может и не так. Та гипотеза, которая больше объясняет известных фактов и выбирается как рабочая. Но мир так сложен, что обязательно найдутся факты, которые плохо согласуются с рабочей гипотезой. Придется выдумывать новую, помощнее. В ариновом замещении все то же самое. Простые факты неплохо согласуются с этими двумя гипотезами, с первой получше, со второй немного похуже хотя бы потому что в ней напрочь игнорируется эффект входящей группы. Но уже много накопилось данных, которые плохо объясняются что одной, что другой. И да – уже есть и другие объяснения, но они сложны и требуют гораздо более глубокого знания того, как устроены молекулы. Пока оставим это, но не думайте, что на старой телеге можно далеко уехать. Химия довольно быстро развивается, и лет через 10-20 вы посмеетесь над всем этим архаичным наивом.
В конце приведу несколько примеров реакций с ариновым механизмом и кине-замещением ((1) J. Am. Chem. Soc. 1946, 68, 143; (2) J. Am. Chem. Soc. 1949, 71, 3838; (3) Org. Lett., 2011, 13, 4172).
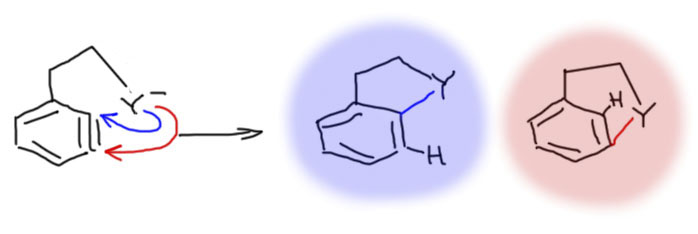
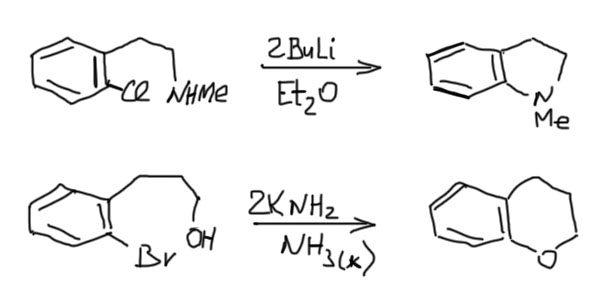
Ариновый механизм очень неплохо работает, когда нуклеофил находится в той же молекуле (по английски это совершенно официально назвали бы tethered nucleophile, а по нашему мы это игриво и почти буквально переведем – нуклеофил на веревочке, и опять предупреждаю – у нас это не термин, не тащите его на люди, вас никто не поймет – вместо этого вам придется серьезно говорить про нуклеофил, связанный с бензольным кольцом тем-то и тем-то, ну или переходить на английский), и в результате получается новый пяти или шестичленный цикл.
Новый цикл должен быть соединен со старым одной общей связью (такое соединение циклов часто называют “конденсированным”) – это важно, потому что можно два цикла склеить и двумя общими связями, но такие структуры в бензольном ряду получаются плохо, а точнее совсем не получаются, потому что бензольное кольцо невозможно деформировать, и атом водорода на среднем атоме углерода оказался бы внутри нового цикла – а это невозможно, потому что у небольших циклов нет никакого “внутри”, они непустые – там электронная плотность окружающих атомов и связей, там даже для водорода места нет. Эта демагогия имеет прямое отношение к делу. Посмотрим, как мог бы образоваться цикл из ариновой “тройной связи”. Если мы сделаем тройную связь прямо рядом с нуклеофилом на веревочке, впритык к веревочке, то цикл мог бы замкнуться и на соседний углерод, и на следующий – в обоих случаях получаются 5 или 6-членные циклы. Но второй вариант невозможен!
По той же причине в принципе невозможно замкнуть цикл на “тройную связь”, смещенную относительно веревочки. Это очень хорошо и резко сужает возможные варианты. Циклы можно замкнуть только если галоген в исходной молекуле находится в орто или мета положении относительно веревочки, причем продукт получается один и тот же. Веревочка может быть из двух атомов (не считая нуклеофила) – тогда получится пятичленный цикл, или из трех – шестичленный. Всё. В литературе при большом желании можно найти единичные примеры 7-членных циклов, но это исключение, очень редкое, и выходы обычно жалкие. Мы не будем. Обратим еще внимание, что нуклеофил обычно берут в протонной форме, который прямо на месте депротонируют еще одним эквивалентом основания.
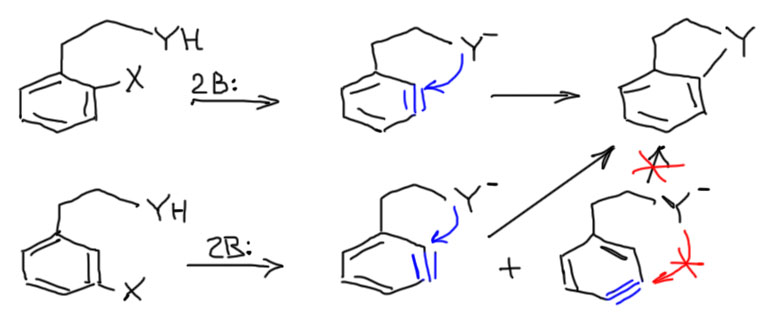
Естественно, в случае мета-галогенпроизводного аринов при отщеплении будет два (молекулы не знают, что от них хотят люди, и продолжают жить своей жизнью), только один из них пойдет в циклизацию, а второй превратится в какие-то побочные продукты. Поэтому предпочтительно использовать орто-галогенпроизводные – выходы будут лучше.
Несколько примеров:
Реакции нуклеофильного замещения в присутствии источников электронов, обычно называемые SRN1-замещением имеют весьма ограниченное применение в реальном органическом синтезе, но в учебных целях их ценят, как пару к реакциям, идущим через арины, так как при очень похожих условиях выполнения, у этих реакций драматически отличаются результаты: ариновое замещение часто сопровождается кине-замещением, а в восстановительном замещении этого нет и нуклеофил вступает строго на место уходящей группы. На словах это красиво, но всё это наблюдается только в одной реакции – замещении галогена (брома или иода) на амино-группу:
Механизм восстановительного нуклеофильного замещения будет разобран на отдельной страничке.
Иногда попадается еще одно применение этого механизма, действительно довольно интересное. Эти м способом можно присоединять фенилы или замещенные фенилы или некоторые другие ароматические остатки к α-углероду кетонов или других похожих соединений (сложных эфиров, амидов и т.п.). В самое ближайшее время мы серьезно возьмёмся за химию карбонильных соединений и узнаем, среди прочего, что такое енолизуемые соединения, и как правильно отрывать от них протон. Пока не будем сильно заморачиваться этими проблемами, тем более, что и в том, что касается химии SRN1 никто тоже этого не делал. Чтобы не связываться с конкуренцией отрыва протона от двух разных групп при карбониле, здесь использовали или только метилкетоны с второй группой без таких протонов, или с симметричными кетонами типа ацетона или циклогексанона. Это также позволяет очень сильное, но стерически не затруднённое основание амид калия в жидком аммиаке, тем более что это именно то, что мы уже использовали. Реакцию делают просто. Сначала в жикий аммиак кидают калий, и немного соли железа, которая катализирует образование амида калия вместо обычного раствора электронов. Когда раствор обесцветится (признак образования амида калия – это важно потому что электроны из синего раствора просто восстанавливают кетон). Тогда добавляют кетон, который очень быстро депротонируется амидом. Пока мы не разобрались в тонкостях карбонильной химии просто нарисуем депротонированный кетон в виде карбаниона, стабилизированного мезомерным акцепторным эфектом карбонильной группы.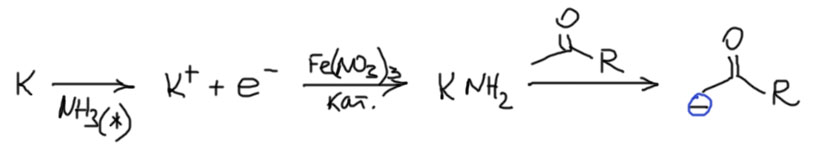
После добавляют галогенпроизводное и еще калий. В этих условиях идёт в основном восстановительное нуклеофильное замещение и образуется замещённый кетон с арильной группой.
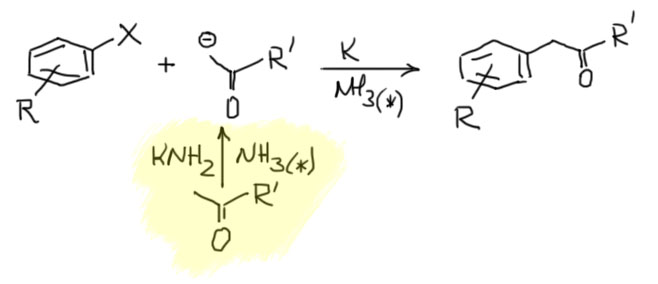
Как и другие примеры восстановительного нуклеофильного замещения эта реакция также не очень чувствительна к стерике, и это позволяет получать производные с орто-замещёнными арилами, например:
Как и всё остальное, связанное с SRN1, и в этом случае в реальной жизни всё проходит не так гладко – выходы оставляют желать лучшего, образующийся кетон частично восстанавливается в спирт, часть галогенпроизводного тоже восстанавливается. В задачах, тем не менее, эту реакцию любят, считая одним из вершинных достижений органической химии.
Реакции в боковой цепи
Реакциями в боковой цепи называют любые реакции в присоединенных к ароматическому кольцу неароматических заместителях, даже если они не очень похожи на цепи, с которыми никак не может расстаться пролетариат. Например, в фенилциклогексане циклогексильный остаток вполне квалифицируется как боковая цепь. Да и такая маленькая фитюлька как метил – тоже вполне себе боковая цепь.
Бензильное бромирование очень похоже на аллильное, которое мы изучали в алкенах. И там и здесь, это свободнорадикальная реакция, использующая радикал, стабилизированный сопряжением с двойной связью там, или ароматическим кольцом здесь. Поэтому и здесь можно использовать бромсукцинимид (NBS) в растворе четыреххлористого углерода при кипячении с радикальным инициатором типа перекиси бензоила или AIBN. Но, в отличие от аллильного бромирования, где по другому делать нельзя, в химии ароматических соединений можно обойтись без бромсукцинимида, а взять просто бром и инициировать реакцию мощной лампой (годятся, например, галогенные светильники ватт на 150, а лучше побольше). Если будете делать это в лаборатории, будьте осторожны, они очень сильно греются и могут устроить хороший пожар. Бромсукцинимид поэтому удобнее и безопаснее, и это очень дешевое вещество, так что не экономьте на мелочах. Как писали раньше на железнодорожных платформах: “Сэкономишь минуту, потеряешь жизнь!” В данном случае не минуту, а рубль.
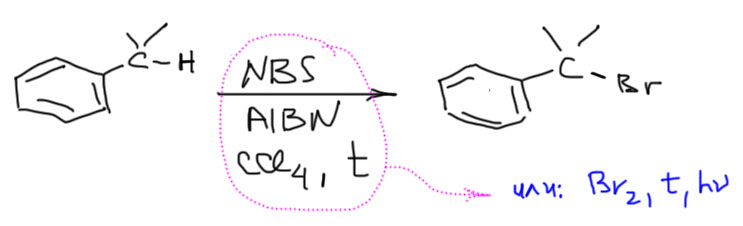
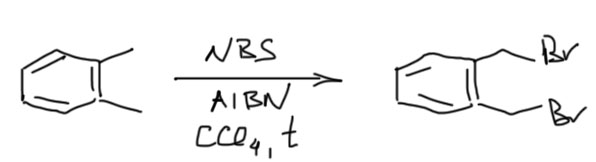
Реакция затрагивает только атомы водорода на первом к кольцу углероде (их называют бензильными). Трет-бутильная группа поэтому в этой реакции совершенно мертва. Если у нас на первом углероде два атома водорода, то взяв второй эквивалент брома и увеличив время реакции можно получить дибромпроизводное. Второй бром входит тяжелее первого почти наверняка по стерическим причинам, так как на стабильность радикала замещение бромом почти не влияет. Метильную группу можно превратить в трибромпроизводное, но это потребует еще больших усилий. То, что второй бром тяжело входит на тот же бензильный углерод позволяет делать очень полезную вещь – превращать ксилолы довольно чисто в бис-бромметильные производные, очень полезные исходные вещества во множестве занятных синтезов.
Что окисляется?
Любая боковая цепь, кроме третичного алкила, окисляется до карбоновой кислоты сильными окислителями. В боковой цепи может быть всё, что угодно, но одна вещь совершенно обязательна – на первом углероде должен быть хотя бы один атом водорода. У третичного алкила, например, трет-бутила такого водорода нет, и он не окисляется. Это обстоятельство заставляет предположить, что механизм окисления является свободнорадикальным, и начинается с отщепления этого атома водорода какой-нибудь радикальной частицей. И действительно, среди окислителей, использующихся для этой реакции, больше всего всяких свободнорадикальных реагентов – смесей перекиси водорода с солями металлов, разбавленной азотной кислоты, даже кислорода с разными катализаторами. Мы не будем в этом разбираться, это противная и мутная химия в основном промышленного характера. А вот окислители, не замеченные в свободнорадикальных фокусах, например, всё, что касается шестивалентного хрома (хромовый ангидрид, хромат, бихромат, хромовая кислота и т.п.), для этой реакции не используют.
Итак, что окисляется в карбоновую кислоту, а что не окисляется или окисляется, но не туда (это нам просто не интересно, поэтому игнорируем такие случаи).
- окисляется любой заместитель, у которого на первом атоме, присоединенном к ароматическому кольцу есть хотя бы один водород;
- но не окисляется CH2 группа, зажатая между фенилами;
- окисляется любой ацил у которого на следующем атоме углерода после карбонильной группы есть хотя бы один водород
- но не окисляется ацил, у которого за карбонилом следует неокисляемый остаток (фенил, трифторметил и тому подобное)
- не окисляется третичный алкил или любой другой заместитель, присоединенный углеродом без атомов водорода
Другие заместители в молекуле останутся. Вот несколько примеров молекул с боковыми цепями, которые можно окислить. Окислено в карбоксил будет то, что выделено красным, и любой такой заместитель, сколько бы в нём ни было атомов углерода, превратится в одну карбоксильную группу. Если заместитель занимает не одно место, как например, неароматический цикл, будет два карбоксила. Всё остальное останется. 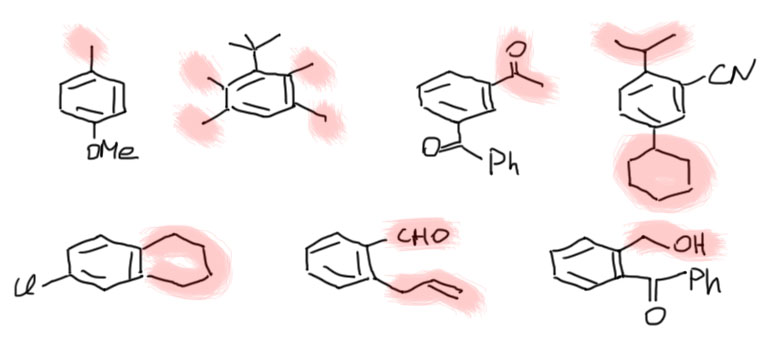
Чем окисляется?
Окислителей для этой реакции предложено очень много, в основном потому что эта реакция имеет довольно серьезное промышленное значение, но мы не будем во всём этом разбираться, и ограничимся перманганатом калия. И мы не будем разбираться в механизме его действия. Может ли он быть свободнорадикальным? Может, потому что у марганца полно степеней окисления, и нарезаны они через один – семь, шесть, пять, четыре, три, два. В таких случаях окислительно-восстановительная реакция вполне может быть одноэлектронной (сопровождаться изменением степени окисления на 1), а это то же самое, что сказать, что в ней участвуют свободные радикалы. Точнее это никто пока не исследовал.
Когда говорят про перманганат калия, практикующие химики обычно недовольно морщатся. Не буду скрывать, это очень неудобный окислитель. Чрезвычайно популярный в элементарной химии, но в химии посложнее капризный и неудобный. Он дорогой, его трудно достать в приличных количествах, он числится в каких-то противных списках, так что если вы его сдуру закажете у алибабы, то вместо перманганата к вам придут люди в чёрных шапочках с болгаркой. Кроме того, у него очень плохой окислительный эквивалент (в органике он обычно восстанавливается до Mn(IV) и то не количественно), так что его нужно довольно много. Окисление самой маленькой боковой цепи, метила в карбоксил, например, это 6 электронов, то есть вам потребуется не менее 2 молей марганцовки на моль окисляемого соединения. При молярной массе сильно за сто, это больше 250 грамм на моль. Если вы когда-нибудь уже имели дело с марганцовкой, идея взять 250 грамм этого вещества должна вас сильно напугать. Если справитесь со страхом и сделаете реакцию, вся посуда будет загажена мелким коричневым осадком, стенки колб и вы сами будете покрыты коричневым налётом (легко смывается щавелевой кислотой, если что). А если боковая цепь длиннее, то перманганата понадобится вообще целая гора.
Но всё это – не главное. Главное в том, что перманганат ни в чём кроме воды не растворяется, и в ней-то растворяется через пень колоду, как типичная соль калия с большим анионом. Есть, правда, перманганат натрия, который на порядок лучше растворим, но это изрядная экзотика. А органика, как мы уже сто раз повторяли, обычно не растворяется в воде. Вот и сидят они в разных фазах: перманганат может окислить, но не хочет, а ароматика с боковой цепью хочет окислиться, но не может. Если ароматика у вас небольшая, типа толуола или чего-то не намного более сложного и к тому же жидкая, можно окислять перманганатом в щелочной среде при хорошем перемешивании и нагревании – реакция пойдёт на поверхности капелек, это не очень быстро, но так как щелочь будут удалять продукт окисления в воду, капельки понемногу растворятся. Не переборщите с щелочной средой – если взять просто щёлочь, перманганат в сильнощелочной среде неустойчив и будет разлагаться, а так как основная реакция гетерогенная и знать не знает ни про какие проблемы перманганата, значительная часть окислителя будет израсходована впустую. Поэтому берут просто соду, карбонат натрия – этого достаточно. После окончания реакции фильтруем осадок двуокиси марганца, раствор подкисляем и получаем продукт – бензойные кислоты обычно плохо растворимы в воде. На схеме R – это боковая цепь, но не третичный алкил, X – любой заместитель, выдерживающий окисление перманганатом – свободная гидрокси или амино-группа (под свободной амино-группой имеется в виду любой амин, замещенный или незамещенный, первичный, вторичный или третичный). 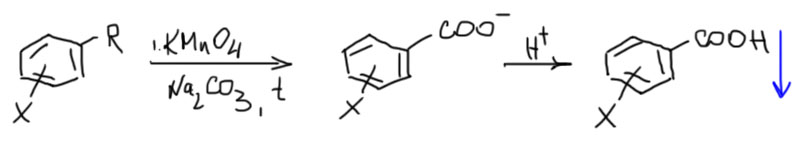
Но если амино-группа ацилирована, то окисление становится возможно, но только в нейтральной среде (J.Org.Chem., 1989, 54, 3744), так как в щелочной происходит гидролиз амида. Выходы поганенькие, но методика удобна и заслуживает внимания. 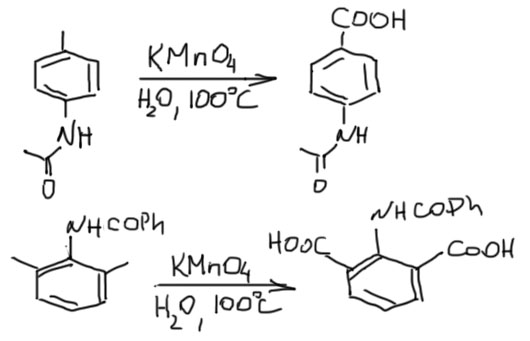
Зачем окисляется?
Это очень удобный способ быстро создать одну или несколько карбоксильных групп, если вам по какой-то причине доступны производные бензола с боковыми цепями. Обычно это метил, – это экономнее всего. Но вполне может оказаться, что откуда-то прямо в руки падает какое-то более сложное производное, и его можно окислить. Два карбоксила рядом (это называется фталевыми кислотами), например, можно получить не только из орто-ксилола, но и из тетрагидронафталина, чрезвычайно легко получаемого из нафталина, да и кучей других способов.
Есть и еще одна важная причина – управление ориентацией замещения. Алкилы это орто/пара-ориентанты и активирующие заместители, а карбоксилы – мета-ориентанты и дезактивирующие заместители. Поэтому мы можем вводить разные заместители, подбирая время окисления, и получать разные продукты.
Разные вспомогательные методы
- Выбираем...
- Получение чистых орто-изомеров через маскировку пара-положения
- Пара-изомеры
- Восстановление по Бёрчу
- Ароматизация
Сюда мы как всегда сваливаем всё, что не удаётся легко классифицировать, всякие подсобные инструменты. Но они очень важны для решения реальных задач, поэтому не пренебрегайте.
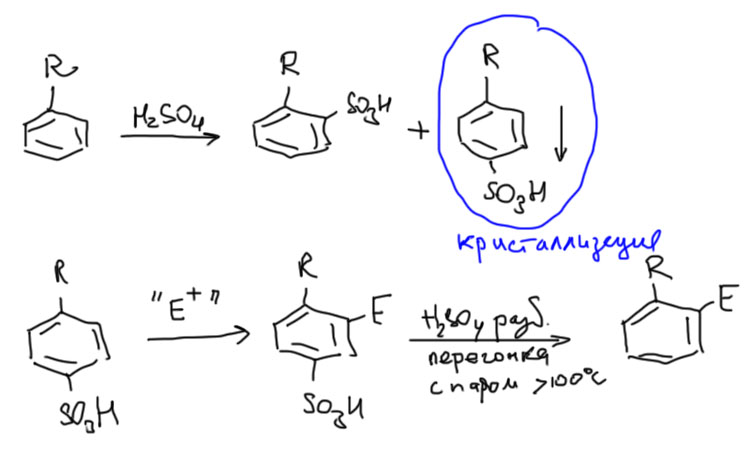
Для этого существует группа методов, в которых пара-положение временно закрывают заместителем, который не влияет на ориентацию замещения (предполагается, что основной заместитель – более сильный ориентант). Тогда реакция замещения направляется в свободные орто-положения. Можно даже сделать две последовательные реакции, или одну реакцию с двумя эквивалентами реагента. Далее, блокирующий пара-заместитель удаляют. Таких временных заместителей известно несколько. Пока что для нас годятся два – сульфо-группа и трет-бутил.
Сульфо-группа получается сульфированием, причем получается, как положено, смесь орто- и пара-сульфокислот. Более симметричные легко выкристаллизовываются из этой смеси, из-за чего часто возникает неверное представление о высокой пара-селективности сульфирования. Но нам это до лампочки – нам важно, что пара-замещенные сульфокислоты легко получаются. Далее делают нужные реакции, продукты вчерне выделяют и удаляют сульфо-группу. Удаление сульфо-группы объясняется тем, что реакция сульфирования обратима, и чтобы пустить ее вспять, нужно воспользоваться принципом Ле Шателье. При прямом сульфировании используют концентрированную серную кислоту (или даже олеум), и в реакции выделяется вода. Поэтому для обратного сульфирования добавляют избыток воды – используют разбавленную серную кислоту, а продукт удаляют прямо из реакционной смеси отгонкой с перегретым водяным паром (дуют в колбу водяной пар, который еще и подогревают горелкой перед вводом в колбу).
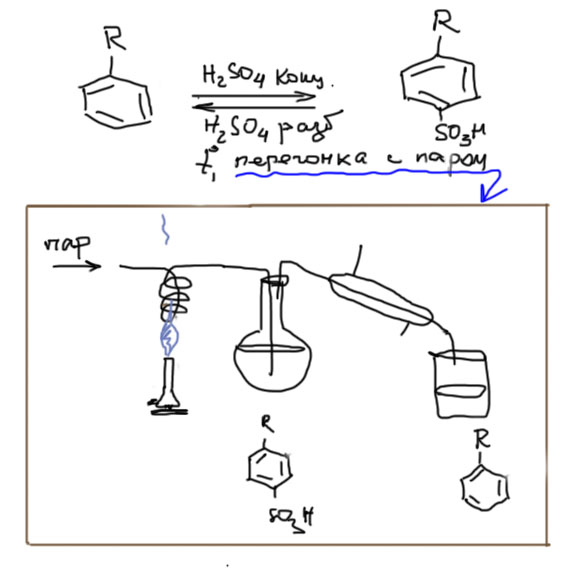
Метод в целом так и работает – сульфируем, замещаем, снимаем сульфо-группу. Часть исходного выбрасываем вместе с орто-изомером сульфокислоты. Метод не годится для алкилирования и ацилирования, так как эти реакции несовместимы с сульфо-группой.
Второй метод использует трет-бутильную группу. Трет-бутил вводится алкилированием. Мы знаем, что алкилирование по Фриделю-Крафтсу – очень ненадежная, неселективная реакция. Но трет-бутил как раз вводится довольно чисто. Это исключение связано с тем, что трет-бутильный катион довольно стабилен и реакция идет в более мягких условиях, чем алкилирование другими алкилгалогенидами. Для повышения селективности еще и применяют специальный растворитель – очень полярный нитрометан, дополнительно стабилизирующий трет-бутильный катион. Большой размер трет-бутила имеет и еще одно положительное следствие – в орто-положение к трет-бутилу никто не лезет, и сам он не лезет в орто-положение к другим группам, поэтому довольно чисто получается пара-изомер. Его вводят в электрофильное замещение, которое в этом случае идет в орто-положение к первому заместителю. В конце трет-бутил снимают. Это – очень непростая проблема. Снятие трет-бутила предполагает протонирование под действием сильной кислоты (используют хлорид алюминия и капельку воды – в результате гидролиза образуется HCl, которая в присутствии хлорида алюминия становится очень сильной кислотой), но трет-бутильный катион необходимо перехватывать, иначе он усядется обратно. Для перехвата требуется ароматическое соединение, заведомо более реакционноспособное, чем то, что образуется в результате снятия трет-бутила. Поэтому здесь нельзя указать один 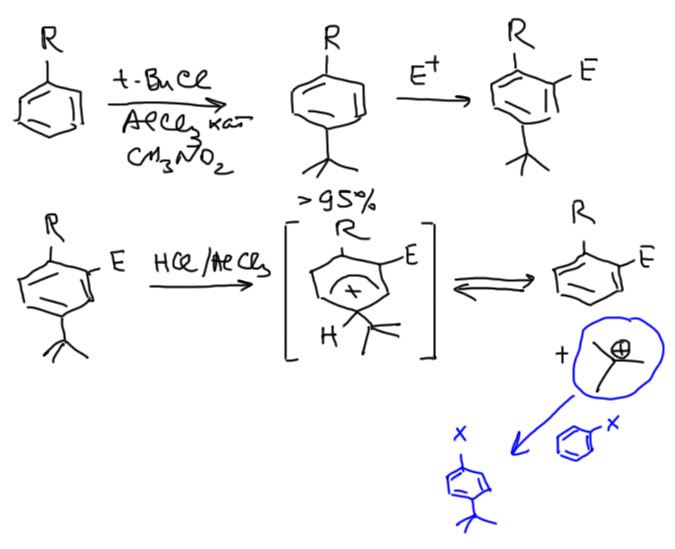 реагент на все случаи жизни. Это очень неудобно. В реальной синтетической химии этот прием используют очень редко. В качестве примера посмотрим галогенирование толуола. Ожидаемый продукт – о-хлортолуол. Трет-бутил тогда можно пересадить на метоксибензол, потому что это соединение гораздо более реакционноспособно чем хлортолуол. В других случаях может понадобится еще более реакционноспособное соединения для перехвата трет-бутила.
реагент на все случаи жизни. Это очень неудобно. В реальной синтетической химии этот прием используют очень редко. В качестве примера посмотрим галогенирование толуола. Ожидаемый продукт – о-хлортолуол. Трет-бутил тогда можно пересадить на метоксибензол, потому что это соединение гораздо более реакционноспособно чем хлортолуол. В других случаях может понадобится еще более реакционноспособное соединения для перехвата трет-бутила.
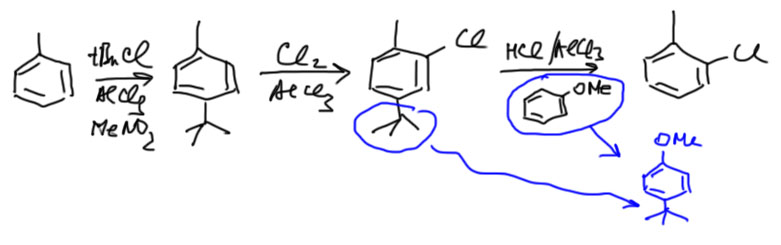
В конце не могу не заметить, что в реальной химии оба метода применяют очень редко. Реальных примеров описанных синтезов очень мало. Есть другие способы синтеза орто-изомеров, более надежные и эффективные. Но как пример метода эти вполне годятся и мы можем ими пользоваться в решении задач очень широко.
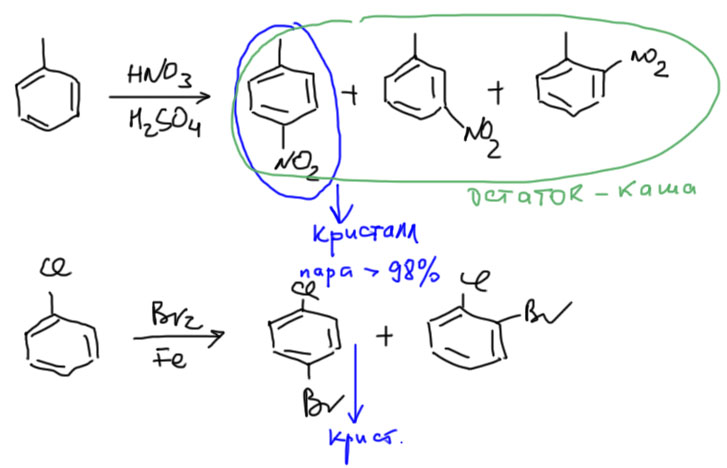
Вывод такой – реакции замещения можно делать даже если ожидается смесь, но вам требуется чистый пара-изомер. Просто обозначьте выделение пара-изомера кристаллизацией. И не обольщайтесь – в остатке после кристаллизации пара-изомера нет чистого орто-изомера. Кристаллизация никогда не бывает количественной. Поэтому в остатке будет каша орто плюс пара, а иногда еще и мета. Место такой каше – в банке для сливов. Ничего сделать с ней в лаборатории нельзя. Хроматографией это делить никто не будет. Температуры кипения у изомеров редко отличаются больше чем на 2-3, максимум 5 градусов, и лабораторной перегонкой разделить это невозможно.
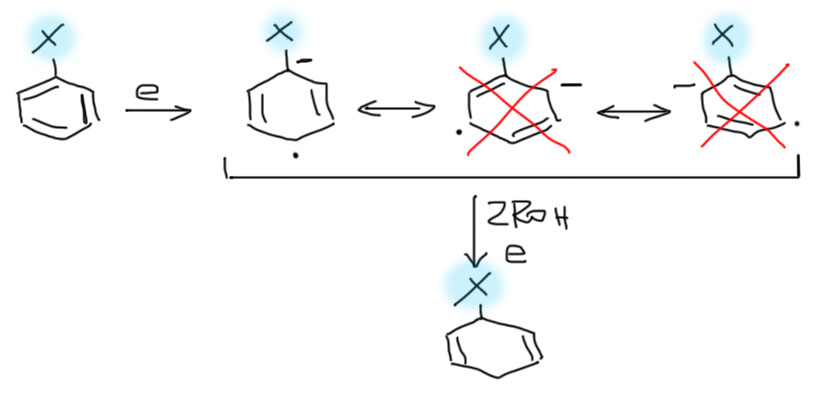
Бензол и некоторые замещенные бензолы восстанавливаются растворами металлических натрия или лития в жидком аммиаке в присутствии двух эквивалентов этанола (или трет-бутанола) в 1,4-циклогексадиены.
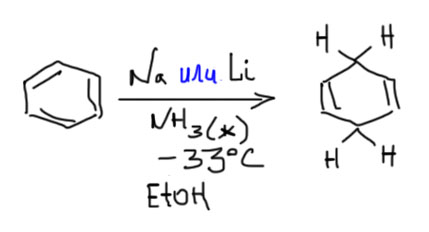
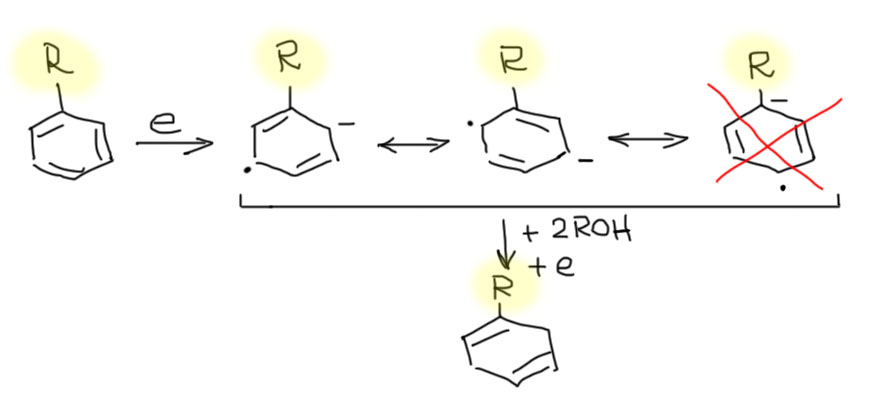
Жидкий аммиак как растворитель играет в этой реакции ключевую роль. Дело в том, что реальным восстановителем является вовсе не натрий или литий, а просто электрон. Поскольку восстановление по определению является реакцией, к которой молекулы приобретают электроны, понятно, что трудно представить себе более чистый образ восстановителя, чем сам электрон. Но химикам немного трудно думать про электрон как про обычный реагент. Мы предпочитаем говорить об источниках электронов, восстановителях, а электрон у нас неявно переносится в реакции, точнее, в полуреакции, когда мы разделяем окислительно-восстановительную реакцию на две половинки: окисления воссановителя и восстановления окислителя. Еще очевиднее это делают в электрохимии, когда каждую полуреакцию выполняют изолированно на своём электроде, а вместе всё это собирают в обычную окислительно-восстановительную реакцию с помощью куска провода с нагрузкой и электролитного мостика.
Но в реакции Бёрча происходит нечто весьма необычное. Восстановителем здесь является, безусловно, металлический натрий, но он передаёт электроны не прямо окислителю, а окислителем является ароматическое соединение, но выдаёт их в раствор, где они достаточно свободно существуют, пока им не предложат какое-то соединение, к которому они смогли бы приклеиться. Электроны – исключительно сильные восстановители и клеиться могут к большинству органических соединений. Не хотят принимать электронов только простые связи с другими элементами второго периода и водородом: C-H, C-C, C-O, C-N, а почти всё остальное электрон принимает с удовольствием. Подробнее как идёт эта реакция разберёмся на отдельной вкладке на основной странице, а здесь ограничимся прктическими вещами.
Кроме бензола можно восстанавливать и другие ароматические соединения, но, во-первых, сразу поймем, что весьма жесткие условия восстановления фактически свободными электронами большинcтво заместителей просто не выдержит, поэтому мы ограничены только небольшим набором донорных заместителей – алкилов и алкокси-групп. И еще более узким набором акцепторных заместителей, в первую очередь карбоксилов (большинство других акцепторных групп типа альдегидов и кетонов, сложных эфиров, нитрилов, нитро-групп сами восстанавливаются свободными электронами, и предсказать продукты таких реакций очень непросто – мы не будем их даже пробовать). И тогда все получается совершенно элементарно: из шести равноценных граничных структур, которые мы рисовали для изображения делокализации анион-радикала бензола, остается гораздо меньше. Когда заместители донорные, минус будет избегать попадания под такой заместитель, и останутся только те граничные структуры, в которых этого нет. Конечный продукт будет соответствовать такой картине, и донор будет висеть в продукте на углеродах с двойными связями. И наоборот, если заместитель акцептор, минус будет стараться попасть под него, и в продукте акцептор окажется на насыщенных углеродах. Это очень неплохо работает и дает возможности получать весьма интересные и полезные замещенные 1,4-циклогексадиены. Вот как это будет для донорных заместителей R = алкил или алкокси.
А вот как для акцепторных типа карбоксила. Типа карбоксила в этом контексте означает карбоксил, потому что практически все остальные группы, включая производные карбоновых кислот, нитрилы, и т.д. дают более сложные продукты. Обратим только внимание на то, что в условиях реакции карбоксил будет превращаться в соль, но карбоксилат – все равно мезомерный акцептор и нормально делокализует анион.
Если в бензольном кольце есть и донорные, и акцепторные заместители, приоритет будет у акцепторных, так как мезомерный эффект сильнее индуктивного. Но реальных примеров таких реакций не очень много, поэтому не увлекайтесь.
Ароматизацией называется превращение циклического неароматического соединения в ароматическое, например, циклогексана или циклогексена в бензол. Это очень полезная реакция, позволяющая делать разные ароматические соединения из неароматических, а это полезно потому что химия у ароматических и неароматических рядов принципиально различна. Например, мы можем использовать какие-нибудь циклизации карбонильных соединений для синтеза производных циклогексана, а затем ароматизовать протмежуточный продукт и получить производное бензола. Реакцию также часто применяют в синтезе гетероциклических ароматических соединений и всяких ароматических соединений небензольного ряда. Про реакцию ароматизации сразу нужно понять две вещи:
- Эта реакция ни разу не всесильна, и применять ее нужно с осторожностью. Общее правило при этом простое – ароматизовать нечто максимально близкое к желаемому ароматическому соединению, например, если у вас есть выбор между циклогексаном, циклогексеном, и циклогексадиеном, выбираем последнее, а если оно недоступно, то предпоследнее. Первое выбираем только если категорически не можем получить ничего другого. Если начинать издалека, то в продукте можно недосчитаться многих из заместителей, потому что все без исключения реагенты ароматизации имеют очень грубые и неселективные механизмы действия.
- это окислительная реакция, и все реагенты для этой реакции кроме одного, палладия, являются окислителями, а следовательно могут быть проблемы с другими легкоокисляемыми группами. С другой стороны, если до ароматичности остается один шаг, то есть, например, в молекуле уже есть две двойные связи, то ароматизация часто протекает настолько легко, что с ней справляются самые разные окислители, и в литературе можно найти примеры использования самых разных реагентов, азотной кислоты, хлорного железа, других солей-окислителей. Это частные примеры конкретных реакций, которые нельзя обобщать и использовать к другим исходным, потому что оценить на глазок легкость ароматизации произвольного недоароматического соединения не просто очень трудно, а почти невозможно. Используйте только стандарные реагенты!
Само существование реакции ароматизации обусловлено тем, что ароматические соединения обладают повышенной стабильностью, и их образование выгодно, оно всегда экзотермично (если быть совсем корректным, то экзотермично на последней стадии, если процесс ароматизации занимает несколько стадий, потому что сопровождается, как говорили в древности, высвобождением энергии ароматической стабилизации (напротив, чтобы убить ароматическое соединение, нужно эту энергию затратить, и поэтому, например, бензольное кольцо не гидрируется в условиях гидрирования кратных связей, и опять, оговоримся, что гидрирование бензола до циклогексана – экзотермическая реакция, эндотермической является только первая стадия, гидрирование бензола до циклогексадиена, та самая, в которой и теряется ароматическая стабилизация – как только вы смогди это сделать, дальше гидрирование идёт очень легко, поэтому бензол практически невозможно гидрировать частично до диена или олефина; и не придирайтесь, реакция Бёрча – это не гидрирование). При всей важности этой реакции для синтеза, ассортимент реагентов для этой цели потрясающе беден. Нам хватит и того, что есть, но в реальном органическом синтезе это часто бывает большой проблемой.
Дихлордицианхинон (ДДХ, DDQ) – самый универсальный реагент ароматизации
Самый популярный и наиболее универсальный реагент ароматизации – производное бензохинона с электроноакцепторными заместителями, 2.3-дихлор-5,6-дицианхинон. Естественный вопрос – почему такой странный, почему не тетрахлор или тетрациан, – имеет простой ответ; первый из них, называемый старинным именем хлоранил, легкодоступен и дешев, но слабоват; а второй просто не может быть получен в существенных количествах и очень дорог. Вот этот ДДХ или DDQ получается простой, но длинной и противной методикой, и поэтому это единственный сильноакцепторный хинон, который можно получить в товарных количествах, или купить в магазине. Стоит дорого. Но альтернативы нет – хотите что-то ароматизовать, так придется раскошелиться. Все остальное гораздо хуже.
Механизм действия DDQ простой, но он требует наличия в ароматизуемой молекуле хотя бы одной двойной связи или второго ароматического кольца (есть некоторые другие вещи, за которые он может зацепиться, но мы обойдемся, чтобы не усложнять). Тогда он забирает водород из аллильного положения с образованием аллильного катиона (чтобы не париться можно представить это как знакомый нам гидридный перенос из аллильного положения, хотя есть и другие гипотезы, но это неважно), и сам же забирает протон, фактически в E1-элиминировании. Обратите внимание, что он и сам при этом ароматизуется. И продолжает в том же духе до победного конца, как экскаватор вычерпывая лишние водороды. Наблюдали ли вы когда-нибудь, как работает экскаватор? О, это завораживающее зрелище? очень советую, не оторвётесь! Раз – опускает ковш, два – загребает грунт, три – поднимает и насыпает в самосвал, и так далее в том же духе, пока не получится котлован. Котлован – это хорошая метафора, это глубокая яма. Любое хорошее ароматическое соединение – это тоже глубокая энергетическая яма, это место, куда можно упасть, но из которого непросто выбраться. Итак, если DDQ это экскаватор, то за один раз в ковш ему помещается ровно два атома водорода, и если нужно убрать больше, то копаем дальше.
Кратко запишем это так. Обратите внимание, как часто схематически изображают реакции, в которых участуют два существенных реагента, каждый из которых превращается в что-то свое, используя такие касательные стрелки. О том, почему хинон превращается в моноанион ароматического диола (гидрохинона), поговорим, когда доберемся до хинонов.
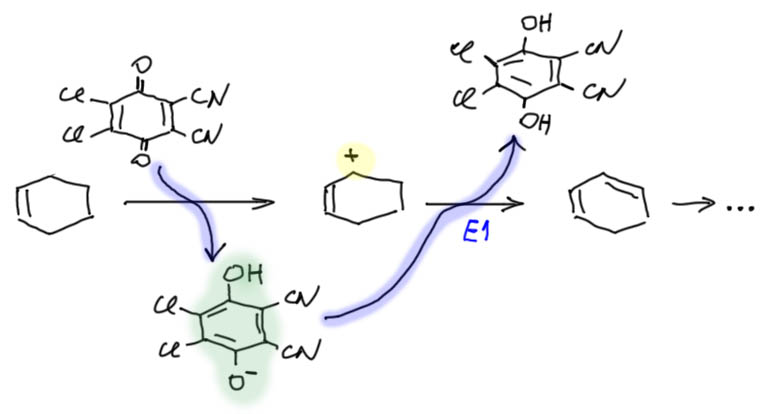
DDQ превращается в гидрохинон (это общее название для пара-дигидроксибензолов), и при желании сэкономить этот продукт можно собирать и окислять обратно в DDQ азотной кислотой, и так сделать процесс довольно привлекательным в экономической точки зрения.
То же самое возможно, если есть бензильное положение, например, в случаях, когда ароматическое кольцо конденсировано с ароматизуемым или является заместителем (сократим моноанион и конечный гидрохинон очевидным образом). Тогда вместо аллильного получается бензильный катион. Уже имеющееся бензольное кольцо или двойная связь работают как такая затравка для ароматизации гидрированного кольца.
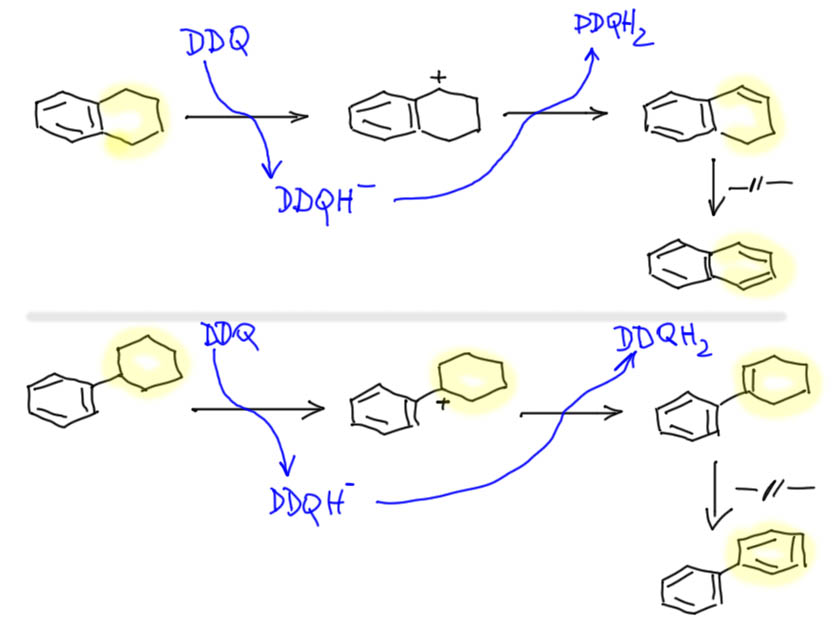
Реакции с DDQ проводят нагреванием в обычных растворителях (уксусной кислоте, толуоле, хлороформе, ТГФ – обычно растворитель выбирают просто по растворимости исходных соединений). Условия довольно мягкие, так что большинство заместителей остаются на месте, хотя на особо легкоокисляемые группы типа аминов и фенолов стоит навесить защиту. На каждую новую двойную связь берут один эквивалент DDQ плюс маленький избыток, процентов 10. Большой избыток не берут, это бесполезно и дорого.
DDQ бессилен, если в исходном соединении нет затравки (двойной связи или другого ароматического кольца), и уж совсем бессилен, если на каком-нибудь из атомов кольца есть два заместителя – DDQ умеет сносить только атомы водорода, например:
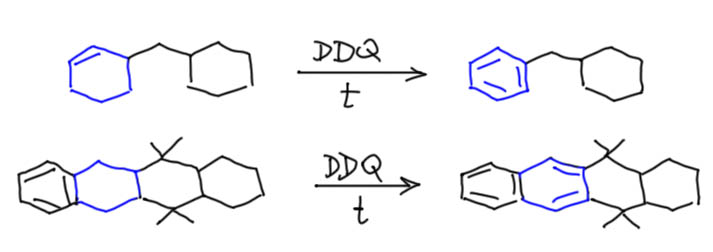
Электрофильное замещение - ориентация
- Из анизола (метоксибензола), хлористого ацетила и днр получите три разных изомера
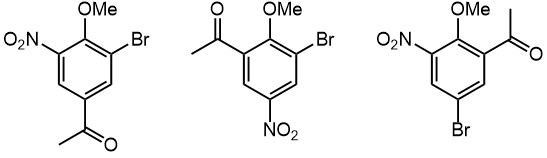
Маскировка пара-положения
1. Получите из бензола 1-бром-2-хлор-3-нитробензол.
2. Получите из анизола 2-метокси-3-бромацетофенон
Алкилирование через ацилирование
- Из бензола и хлорангидрида бутановой кислоты (бутаноил хлорида, н-бутирил хлорида) получите 1,2,4-три-н-бутилбензол
- Из бензоил хлорида и хлорангидрида 3-фенилпропановой кислоты (3-фенилпропаноил хлорида, β-фенилпропионил хлорида) получите 5-бензилиндан (нарисован внизу, хотя можно и прогуглить, что такое индан)
- Из изобутирил хлорида (догадайтесь сами, что это такое), бензоил хлорида и бензола получите 1-бензил-4-изобутилбензол.
- Из хлорангидрида терефталевой кислоты (слева) и бензола получите 1,4-бис(п-бромбензил)-2-нитробензол (справа)
Нуклеофильное ароматическое замещение
- Напишите механизм реакции и ожидаемый продукт взаимодействия 1,3-динитро-2,5-дихлорбензола с аммиаком
- Превратите фторбензол в 2-бром-4-нитроанизол (последний раз пишу: анизол – это метоксибензол).
- Не забываем про законы ориентации в электрофильном замещении. Превратите бензол в 3,4-динитрофенетол. Путь синтеза должен проходить через 3-хлорнитробензол. Фенетол – это как анизол, но с этилом вместо метила.
Арины
- Превратите фторбензол в о-фторбромбензол и далее в м-фторанилин.
- Хлорбензол, содержащий один атом дейтерия в орто-положении подвергли действию амида натрия в жидком аммиаке. Какие продукты получились, сколько их и в каком соотношении (возможным влиянием атома дейтерия на реакционную способность пренебрегаем, считайте, что это просто водород такой, типа, меченый). Напишите механизм реакции.
- Образование арина из галогенбензолов обратимо. Под действием сильных оснований, например, амида натрия в жидком аммиаке или трет-бутилата калия 1,2,4-трибромбензол кроме образования продукта замещения частично изомеризуется в 1,3,5-трибромбензол (это явление забавно называется “пляска галогенов”). Придумайте механизм, объясняющий этот процесс. Почему именно такой изомер образуется?
- Следующее превращение происходит под действием амида натрия. Предложите механизм. Сколько молей амида натрия нужно добавить на моль исходного спирта.
1,3,5-триалкилбензолы и другие реакции
2. Из толуола, трет-бутилхлорида, метилацетилена получите (E)-1-(3,5-ди-третбутилфенил)-2-бутен.
Восстановление по Бёрчу и другие реакции
Окисление боковой цепи, нуклеофильное замещение
1. Из п-бромтолуола получите 3-бром-4-этокси-5-нитробензойную кислоту. (Внимание: до 21.11.2019 на этом месте была другая, некоррекная задача, она снята.)
2. Из тетралина и изобутилена получите 4-трет-бутилфталевую кислоту
Небензоидная ароматика, ориентация замещения, ЯМР
1. Вместо того чтобы опять что-то синтезировать, потренируемся разбираться в экспериментальных данных с помощью ЯМР и общих представлениях о том, как идут реакции электрофильного и нуклеофильного замещения (данные из cтатьи Lok C.M. et al, Rec. Trav. Chim. Pays-Bas, 1973, 92, 340 doi:10.1002/recl.19730920314). Есть такой очень занятный изомер нафталина, азулен (гугл в помощь, если не встречались). Он считается ароматическим и действительно проявляет многие ароматические признаки. Например, очень легко вступает в реакции электрофильного замещения: нитрования, сульфирования, бромирования, меркурирования, дейтерообмена, ацилирования и т.п. Настолько легко, что, во-первых, приходится брать реагенты помягче и условия понежнее, а во-вторых, очень легко берет не один, а два заместителя. Вот с ним и проделали такие превращения: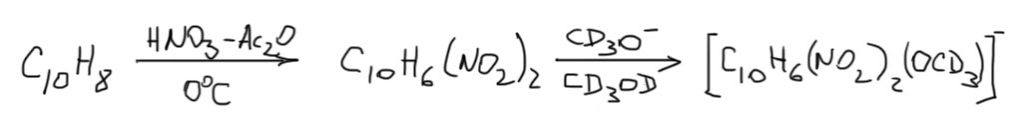
Протонный ЯМР динитропроизводного имеет синглет при 9.11 м.д., дублет при 10.0 м.д., и два более сложных мультиплета, похожих на триплеты при 8.17 и 8.25 м.д. Соотношение сигналов в порядке упоминания 1:2:2:1.
Протонный ЯМР аниона, вполне устойчивого при комнатной температуре в растворе дейтерометанола: 7.63 (синглет, 1 протон), 7.09 (дублет, 2 протона), 5.40 (немного кривой дублет, 2 протона), 3.44 (что-то похожее на триплет, 1 протон).
Попробуйте выжать как можно больше из этих данных: в идеале предложить структуры, объяснить как и почему они образовались, и почему образовались именно они.