Мезомерный эффект, или как рисовать резонансные (граничные) структуры
На этой страничке подробно познакомимся с такими важными понятиям, как мезомерия, резонанс, сопряжение, делокализация, мезомерный эффект, мезомерные доноры и акцепторы, граничные структуры.
Это новая редакция страницы про мезомерию, намного более подробная и длинная. На всякий случай вот ссылка на старую страницу, возможно, её кто-то читал как раз тогда, когда мне пришла блажь заменить ее на новую. Могу себе представить злость и раздражение человека, у которого из под носа вырывают инструкцию по использованию мясорубки и подкладывают вместо нее инструкцию по пользованию кухонным комбайном другой фирмы, типа, мясорубка там тоже есть, ну, может не настоящая мясорубка, но котлеты сделать можно. Извините. Вот старая инструкция.
Если с индуктивным эффектом обычно проблем не бывает, то второй тип электронных эффектов гораздо труднее поддается освоению. Это очень плохо. Теория резонанса (мезомерия) была и остается одним из важнейших инструментов обсуждения структуры и реакционной способности органических соединений и заменить ее нечем. А как же квантовая наука?! Да, правда, в нашем веке стали легкодоступными квантово-химические расчеты, и теперь каждый исследователь или даже студент, потратив весьма немного времени и сил, может бесплатно раскочегарить на своем компьютере расчеты, уровню которых еще 20 лет назад позавидовали бы все нобелевские лауреаты. Увы, результаты расчетов не так просто использовать – они плохо поддаются качественному анализу и зрительно не очень понятны. Сидеть и смотреть на бесконечные столбики цифр и рассматривать запутанные и перегруженные картинки орбиталей и электронной плотности можно долго, но пользу из этого извлекают немногие. Старая добрая теория резонанса в этом смысле гораздо эффективнее – она быстро и довольно надежно дает именно качественный результат, позволяет видеть, как распределена электронная плотность в молекуле, найти реакционные центры, оценить устойчивость важных частиц, участвующих в реакциях. Поэтому без умения нарисовать резонансные структуры, оценить их вклад, и понять, на что влияет делокализация, никакой разговор об органической химии невозможен.
Мезомерия, резонанс, сопряжение – вещи настолько важные, что имеет смысл разобраться в них получше, потратить на это время один раз, потом вернётся сторицей, потому что это работает всегда и везде, и недопонимание с самого начала потом многократно приведёт к фрустрации и вопросам, на которые непросто будет найти ответы. Учебная литература на удивление небрежно относится к этой проблеме, видимо, считая, что здесь всё очевидно, и любой может разобраться. Проблема ещё в том, что отношение органиков к мезомерии и резонансу менялось в разные времена и эпохи, и каждая такая эпоха оставила нам учебники со всей этой смутной историей вопроса. Вы можете сами это заметить, просто сравнив, как этот эффект трактуют разные книги, если они вообще хоть как-то его трактуют. Были и конкурирующие школы, предлагавшие свои интерпретации и системы обозначений, а долгий спор о том, как обозначать донора – знаком плюс или минус – до изумления напоминает спор тупоконечников и остроконечников, если конечно, не обратить внимания, что в этом случае в каждой партии состояли мощнейшие учёные, аргументы которых были вовсе не вздорны, а глубоки и неоспоримы. Но время прошло и как-то договорились не спорить, всё более-менее устоялось. Само собой получилось так, что донор стали обозначать знаком +, а акцептор –, хотя это и противоречит тому, как химик мыслит два соответствующих заряда – плюс – это пустота, или, как говорят физики, дырка (hole) – хорош донор, у которого ничего нет, даже камня не положит в протянутую руку, камня у него тоже нет. А минус – это много электронов, лишняя пара, её бы как раз отдать. Именно такие соображения заставляли когда-то партию обратных знаков истово отстаивать свою правоту. Мы ведь с ними согласны, дело говорят (говорили). Но, как ни странно, в обозначениях взяло верх как раз такое обыденное понимание знаков, не имеющее никакого отношения к зарядам и электронам: плюс означает увеличение, доброту и щедрость, минус – уменьшение, злобу, алчность. Донор – даёт, акцептор хапает и спасибо не говорит. Попробуйте так, просто сказать “злой акцептор” – и поянений не понадобится, всем понятно, что акцептор такой сильный, отнимает всё у всех, злой, да. А теперь попробуйте сказать “злой донор”. Фу, глупость какая, как же злой, если даёт, злые не дают. Язык диктует, не электроны. И так далее. Тоже неплохо. Поэтому этот спор двух партий разных знаков – это спор умных с хорошими. Хорошие победили, и выяснилось, что они не глупее умных.
Что такое вообще теория резонанса? В трудах каких классиков она описана? Это такое пафосное название для удобной и, главное, незаменимой системы рисования структур молекул (любых, устойчивых, неустойчивых, заряженных, нейтральных), которые не могут быть адекватно изображены обычной одной структурной формулой. Почему не могут? Скоро поймём. Пока просто примем это прискорбное обстоятельство – в органической химии (на самом деле, и в неорганической тоже) полно молекул, катионов, анионов и прочих частиц, которые невозможно изобразить одной структурной формулой так, чтобы не забыть что-то важное. Структурная формула, по идее, должна достаточно описывать любую молекулу или ион. Но – часто не описывает. И тогда нам требуется какая-то надстройка над классическими структурами, чтобы как-то удобно и убедительно описывать такие случаи. Вот эту роль и выполняет резонанс или мезомерия. Никакой особой теории за этим нет, как бы странно это не казалось. Когда-то теорию пытались под это подсунуть, в виде так называемой теории валентных связей. Проблема в том, что под этим пафосным называнием скрывается один из самых больших провалов в теоретической химии. Это не настоящая теория, а ее проект, очень сырой и приблизительный, из которого так ничего путного не получилось, кроме того, что мы называем граничными структурами. В этом смысле это удивительная конструкция – представьте себе. что вы решили создать какую-то теорию, нарисовали презентацию красивую, картинки, анимации, денег под это дело выбили, потом долго пыхтели и сопели, усердно копошились, рассказывали всем вокруг, как близки вы к успеху, и как скоро человечество узнает что-то невероятно важное – но, время прошло, а теорию так и не создали, не получилось, бывает. По-моему, узнаваемая ситуация. Но это тот редчайший случай, когда претензий нет, потому что презентация оказалась полезной сама по себе и зажила своей жизнью. Всё потому что презентацию эту нарисовал действительно талантливый человек, Лайнус Полинг, обладавший огромным даром доносить сложные вещи до нормальных людей. И никто не в претензии, что собственно теория не получилась, он и не собирался её создавать, он надеялся, что по его презентации это сделают другие, гораздо более серьёзные физики и математики. Но последние не поддались на эту милую провокацию, а вместо этого создали и понемногу довели до ума другую теорию – теорию молекулярных орбиталей, в которой все орбитали и так уже делокализованы, и никакой нужды в резонансе и мезомерии нет. Правда, при этом теория МО удивительно беспомощна в смысле методологии – ей чрезвычайно сложно пользоваться в повседневной работе химика – осмыслении свойств и реакций. Не невозможно – при должном усердии и склонности к разглядыванию цифр и графиков из результатов, получаемых с помощью теории МО и ее современной версии, теории функционала плотности (маститых теоретиков, если им случайно попадется на глаза это крамольное высказывание, я покорно попрошу не возбуждаться – я знаю, что эта теория не имеет никакого отношения к МО, но то, как обычные органики реально видят результаты DFT-расчетов и то, как они ими пользуются, ничем не отличается от результатов расчетов с помощью обычной теории МО – те же орбитали, те же диаграммы, те же характеристики электронной структуры) – это вполне возможно. Но – неудобно, громоздко, требует доступа к расчетам и умения ими пользоваться. А теория резонанса и мезомерия ничего подобного не требуют, и вам вполне будет достаточно почти любого способа рисования, хоть мелом на доске, хоть пальцем на песке, но только не вилами на воде.
А есть ли разница между понятиями мезомерия, резонанс, сопряжение? Да есть, и это важно понимать. Между мезомерией и резонансом разница была когда-то, но уже давно не имеет значения – сейчас это интересно только историкам химии. Будем считать, что эти понятия взаимозаменимы, можно использовать какое-то одно или оба в любых пропорциях. Один нюанс есть – когда говорят не о делокализации в общем, а об электронном эффекте заместителя, предпочитают термин мезомерный эффект (и обозначают соответственно буквой M). Кроме того, еще используют и слово “сопряжение” (точнее, π-сопряжение). Но самое важное слово в этом ряду – сопряжение (по-английски conjugation, откуда иногда и в русском всплывает слово конъюгация, но смысла в этом слове нет, и я не рекомендую его использовать; впрочем оно очень часто используется в сотаве термина гиперконъюгация, но об этом отдельно и ниже). Понять, что такое сопряжение, можно очень просто. Посмотрите на любую структурную формулу почти любого органического соединения. Вот этан, пропан, этилен, ацетилен, ацетон, этанол: 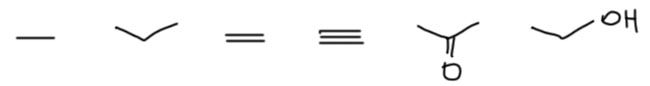 Мы рссматриваем такие структуры просто – вот атомы, вот связи между ними. Связи мы рисуем прямыми черточками со времен двух Александров, русского и шотландца, Бутлерова и Крама-Брауна, с 1860-х, с тех бурных времён, когда где-то отменили рабство, а где-то крепостное право, образовалась единая Италия, но пока без Рима, началось объединение Германии, а в Японии прогнали последнего сёгуна и приступили к обширным реформам по западному образцу – в общем, удивительное время, когда за 10 лет и мир и органическая химия изменились совершенно драматически и начали хотя бы отдалённо напоминать то, что у нас есть сегодня. И мы с тех пор структурные формулы рисуем точно так же, как тогда и придумали, ну только немного красивее научились рисовать сложные структуры, не выписывая очевидные вещи. Такие структурные формулы с полным правом можно назвать классическими структурными формулами. Их главная отличительная особенность – все связи соединяют атомы попарно, иными словами, все связи двухцентровые. Связи изображаются сплошными чёрточками, и некоторые связи подразумеваются по очевидным законам валетности и вообще не изображаютс, но в любой момент могут быть раскрыты и тоже обязательно будут двухцентровыми. И мы вполне довольны, это совершенно адекватная картина строения органических молекул, и вообще, как можно быть недовольным чем-то классическим. Каждая связь рассматривается по отдельности. Мы в реакциях разрываем отдельные связи, образуем отдельные связи. Очень удобно. И при этом, если мы знаем, как устроена самая популярная квантовохимическая теория электронной структуры, теория молекулярных орбиталей, то знаем и то, что в ней практически не бывает отдельных орбиталей связей, там почти все орбитали делокализованы, даже в таком простом соединении как метан. И как соединить привычные нам структурные формулы и молекулярные орбитали? Это не очень просто, но вполне возможно. Рассмотрим это когда-нибудь отдельно. Сейчас нам это не мешает, потому что мы пока не очень часто прибегаем к методам теории МО. Мы, как правило, оперируем структурными формулами, состоящими из отдельных связей, и вполне нормально разбираемся во всем, включая механизмы.
Мы рссматриваем такие структуры просто – вот атомы, вот связи между ними. Связи мы рисуем прямыми черточками со времен двух Александров, русского и шотландца, Бутлерова и Крама-Брауна, с 1860-х, с тех бурных времён, когда где-то отменили рабство, а где-то крепостное право, образовалась единая Италия, но пока без Рима, началось объединение Германии, а в Японии прогнали последнего сёгуна и приступили к обширным реформам по западному образцу – в общем, удивительное время, когда за 10 лет и мир и органическая химия изменились совершенно драматически и начали хотя бы отдалённо напоминать то, что у нас есть сегодня. И мы с тех пор структурные формулы рисуем точно так же, как тогда и придумали, ну только немного красивее научились рисовать сложные структуры, не выписывая очевидные вещи. Такие структурные формулы с полным правом можно назвать классическими структурными формулами. Их главная отличительная особенность – все связи соединяют атомы попарно, иными словами, все связи двухцентровые. Связи изображаются сплошными чёрточками, и некоторые связи подразумеваются по очевидным законам валетности и вообще не изображаютс, но в любой момент могут быть раскрыты и тоже обязательно будут двухцентровыми. И мы вполне довольны, это совершенно адекватная картина строения органических молекул, и вообще, как можно быть недовольным чем-то классическим. Каждая связь рассматривается по отдельности. Мы в реакциях разрываем отдельные связи, образуем отдельные связи. Очень удобно. И при этом, если мы знаем, как устроена самая популярная квантовохимическая теория электронной структуры, теория молекулярных орбиталей, то знаем и то, что в ней практически не бывает отдельных орбиталей связей, там почти все орбитали делокализованы, даже в таком простом соединении как метан. И как соединить привычные нам структурные формулы и молекулярные орбитали? Это не очень просто, но вполне возможно. Рассмотрим это когда-нибудь отдельно. Сейчас нам это не мешает, потому что мы пока не очень часто прибегаем к методам теории МО. Мы, как правило, оперируем структурными формулами, состоящими из отдельных связей, и вполне нормально разбираемся во всем, включая механизмы.
Вот молекулы посложнее: 
И эти молекулы состоят из отдельных связей. В некоторых мы узнаем важные заместители или группы, но и в них вполне конкретные связи. В органической химии с ее бесконечным разнообразием структур это просто важнейший инструмент познания свойств и реакций – мысленно разобрать молекулу на отдельные связи и группы, и разобраться с каждой по отдельности. Единственное дополнение к этому мы уже умеем применять – индуктивный эффект заместителей не изменяет структуры, но несколько модифицирует свойства и реакционную способность, немного смещая электронную плотность в пределах валентных возможностей каждого атома. Это чисто количественный эффект. Например, двойная связь остается двойной, но если к ней присоединен донорный заместитель, слабое смещение электронной плотности в сторону от заместителя создает небольшую асимметрию и наводит слабый, но заметный заряд. И мы что-то увидим в реакциях.
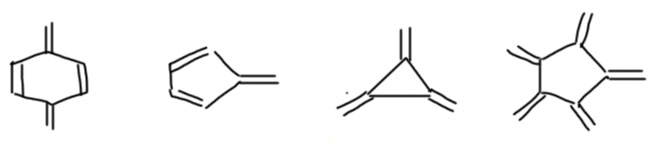
Но вот еще один набор молекул: 
У всех этих молекул есть одна особенность – они (их свойства и реакции) больше невозможно разобрать по связям и группам. Отличия от такого представления будут не только количественные, но более значительные, качественные. Взаимное влияние некоторых групп и связей в этих молекулах так велико, что нам приходится с этим считаться, определять такие молекулы в отдельные группы и мы будем постоянно с этим сталкиваться в курсе (например, мы будем выделять особые классы, такие как сопряженных диены, ароматические соединения, еноны и енали, акцепторы Михаэля, донорные нуклеофильные олефины, и т.п.). Общей особенностью таких молекул является то, что нарисованная одна структура, классическая структурная формула, больше не является адекватным изображением реальной структуры – в каждом таком случае нужно что-то придумывать еще, чтобы как-то из этого положения выкрутиться.
Когда-то это стало большой проблемой. Ведь когда в органической химии возникли структурные формулы, счастью не было пределов, и казалось, что стоит нарисовать структуру, как все про молекулу станет понятно. Первой молекулой, для которой стало очевидно, что это может быть не так и одной структуры может не хватить, был бензол. Споры о том, что нужно делать, и как рисовать молекулу бензола, затянулись на полвека. Ничего не получалось, пока не появилась квантовая механика и специальная ее отрасль для химии. Только тогда появились представления об орбиталях, о связях как воплощении перекрывания орбиталей. Стало ясно, что свойства молекулы, а возможно и ее реакции можно получить, если записать для молекулы и решить уравнение Шредингера. И немедленно стало очевидно, что с этим решением все очень непросто – не получалось его решить ни для чего кроме атома водорода, ну и с некоторыми приближениями для иона диводорода H2+. Это стало невероятным разочарованием для химиков 1920-30-х годов – инструмент есть, ясно, что он невероятно мощен, да вот воспользоваться им невозможно. С практическим решением уравнения Шредингера и вообще решения квантово-химической задачи расчета структуры и свойств молекул пришлось подождать очень долго, то поколение химиков до этого фактически не дожило. Но те химики не хотели просто ждать, и создали довольно много временных инструментов, которые позволяли на качественном уровне прикинуть, как устроена молекула, и как это может отразиться на ее свойствах и реакциях.Среди таких инструментов и гибридизация, и представления об эффектах заместителей, и сопряжение, и симметрия орбиталей, и теория возмущений, и кристаллическое поле для комплексных соединений, и много чего еще. И самое забавное, что инструменты эти оказались настолько эффективны и удобны, что когда наконец пришла эпоха мощных компьютеров и квантовые расчеты стали доступнее прогноза погоды, и показалось, что все те инструменты нужно срочно выкинуть на свалку истории, этого никто не стал делать и их по прежнему с удовольствием и толком применяют и будут применять. Очень они, в отличие от результатов квантовохимических расчетов, удобны – для того, чтобы их использовать не нужно ничего, кроме правил использования и листа бумаги.
В молекулах последней группы есть один общий признак – в них есть π-связи. Такие связи образованы чистыми p-орбиталями. Чистыми – значит негибридными. Большинство простых связей в органических соединениях – это σ-связи, образованные гибридными орбиталями. Отличие σ- от π-связей очень просто понять, если немного представлять, как устроены связевые орбитали. Связь в этом смысле – это область перекрывания орбиталей, понимаемая чисто символически – как место в пространстве, где встречаются доли орбиталей одной фазы (одного знака, одного цвета). Обратите внимание, что в этом месте не нужно думать про молекулярные орбитали, если вы успели разобраться в основах этого метода и научились отличать связывающие орбитали от разрыхляющих. В этом месте мы не используем молекулярные орбитали, а просто символически показываем возможность такого перекрывания, исходя из набора гибридных и чистых орбиталей у атома.
Вспомним про гибридизацию, если забыли
Этот набор прежде всего зависит от гибридизации. У атомов второго периода – углерода, азота, кислорода (а также бора) есть три основных типа гибридизации – sp, sp2, sp3.
- sp-гибридизация: две гибридные орбитали, и две чистые p-орбитали,
- sp2-гибридизация: три гибридные орбитали и одна чистая p-орбиталь
- sp3-гибридизация: четыре гибридные орбитали.
Как узнать, какую гибридизацию имеет тот или иной атом? В принципе, если вы хорошо владеете правилами Джиллеспи, то сами во всем отлично разберетесь. Если нет, попробуем разобрать подробнее, что бывает.
Первое, что нужно сделать – посчитать количество атомов, с которыми связан интересующий вас атом. Когда-то это называли валентностью (точнее, нужно считать количество формально одновалентных атомов, то есть аккуратно складывать все ковалентные связи, исходящие из интересующего нас атома), но увы, с тех пор это слово сильно исказило смысл, и что это такое сейчас мало кто знает. Просто считаем. Я думаю, вы разберетесь. Вот, на простом примере – атом углерода в формальдегиде имеет три соседних связанных атома (два водорода и кислород, но это не важно). Второе, что нужно сделать – посмотреть, в каком виде оставшаяся и оставшиеся электроны. Не забывайте, что нормальное состояние атома p-элемента – октет, то есть восемь электронов в валентной оболочке, или четыре пары. На каждый связанный атом уходит минимум пара. Не забывайте, что каждый связанный атом связан одной σ-связью и возможно чем-то еще, но одной σ-связью точно. В мире интересующих нас элементов 2-го периода не бывает ситуаций, когда взаимодействие между соседними связанными атомами обходится без σ-связи.
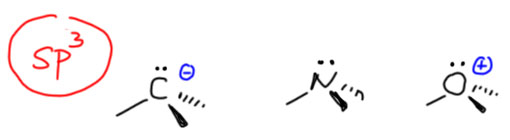
- если соседа 4, то гибридизация может быть только sp3. Все связи – σ-связи. Октет расписан целиком на 4 пары. Атомы углерода в насыщенных соединениях, атом азота в ионе аммония, атом бора в анионе тетрафторбората – все sp3.
- если соседа 3, то гибридизация может быть и sp2 и sp3. Поскольку соседа три, то есть три σ-связи. Итого 6 электронов, три пары. До октета остается еще пара.
- Если это действительно пара, не задействованная в связывании, то гибридизация sp3: азот в аммиаке и насыщенных аминах; углерод в карбанионе; кислород в ионах оксония.
 Если забыли, откуда на атомах заряды, напоминаю: нужно посчитать “свои” электроны и сравнить с числом электронов, положенных элементу номером группы; свои электроны считаются так, что каждая ковалентная связь дает один электрон, а несвязывающая пара – два. Вот аммиак – три + два = 5, равно номеру группы, значит азот при своих, заряда нет. Остальные две структуры – также пять электронов, и для углерода один лишний – заряд -1; для кислорода одного не хватает – заряд +1.
Если забыли, откуда на атомах заряды, напоминаю: нужно посчитать “свои” электроны и сравнить с числом электронов, положенных элементу номером группы; свои электроны считаются так, что каждая ковалентная связь дает один электрон, а несвязывающая пара – два. Вот аммиак – три + два = 5, равно номеру группы, значит азот при своих, заряда нет. Остальные две структуры – также пять электронов, и для углерода один лишний – заряд -1; для кислорода одного не хватает – заряд +1. - Если пара задействована в связывании, например, на атом заходит не одинарная, а двойная связь. Или – пара используется в сопряжении (об этом дальше, пока не расшифровываем). Тогда гибридизация sp2. Алкены, карбонильный углерод, азот в солях иминия.
- Если пара просто отсутствует, на ее месте дыра, пустое место, вакантная орбиталь, то гибридизация sp2. Бораны, карбокатионы.
- Если это действительно пара, не задействованная в связывании, то гибридизация sp3: азот в аммиаке и насыщенных аминах; углерод в карбанионе; кислород в ионах оксония.
- Если соседа два, то гибридизация может быть sp, sp2, sp3. Два соседа – две пары на σ-связях, до октета еще две пары.
- если это две неподеленные пары, то гибридизация sp3. Кислород в воде, спиртах, простых эфирах.
- если одна неподеленная пара, а вторая использована в двойной связи (или сопряжении), то гибридизация sp2. Азот в иминах и всем, что на них похоже; кислород в енолах, эфирах енолов.
- если обе пары задействованы в связях (это две двойные или одна тройная), то гибридизация sp. Углерод в алкинах, алленах, диоксиде углерода
- если одной пары из двух вообще нет, на ее месте дыра, пустое место, вакантная орбиталь, то гибридизация sp2. Вообще-то это не очень простой случай, тут бывают фокусы, но начинать всегда нужно с sp2. Это карбены.
- Если нет обеих пар, то это тяжелый случай, потому что 4 валентных электрона это очень невыгодно. Это какая-то ужасная экзотика. Не будем брать вообще.
- Если сосед один, то строго говоря, нам должно быть до лампочки, потому что в этом случае судить о гибридизации не очень полезно. Гибридизация нужна прежде всего, чтобы понимать форму молекулы, а форма в значительной степени определяется углами между связями (109, 120 или 180 градусов) и расположением связей в пространстве (тетраэдр, треугольник, линия). Если связей (соседей) хотя бы два, это имеет смысл. Но если связь всего одна, то какая разница? Забудем. И тем более, что если у нас есть π-сопряжение, то на таком атоме всегда есть доступные для сопряжения p-орбитали.
И про сигма- и пи-связи тоже вспомним
Так вот, как только мы разобрались с гибридизацией, мы увидим, что двойные и тройные связи не могут образовываться sp3-гибридными орбиталями, но только sp2-гибридными и/или sp-гибридными, то есть такими, у которых точно есть в наличии чистая p-орбиталь, одна или две.
Принципиальное отличие π-связи от σ-связи очень просто: мы должны провести линию, соединяющую два атома, и посмотреть на область перекрывания орбиталей, образующих связь. Если эта линия протыкает эту область хоть где-то, то это σ-связь. Если не протыкает совершенно нигде, а это возможно только если эта линия бежит по узловой плоскости – это π-связь. Поэтому σ-связи могут образовываться самыми разными орбитальками, и s-, и гибридными, и даже чистыми p-орбиталями. Орбитали могут встретиться даже под некоторым углом – мы встретимся с такой ситуацией в циклопропанах – но это не меняет природу связи, хотя и довольно сильно отражается на ее свойствах. А вот настоящая π-связь образуется только чистыми p-орбиталями. В принципе так могли бы расположиться и гибридные орбитали, но реальной структуры, в которой это могло бы реализоваться придумать сложно. С другой стороны, ниже мы увидим, что существует такое явление как σ-π–сопряжение, оно же гиперконъюгация, в котором как раз и встречаются соседние гибридная и p-орбиталь.
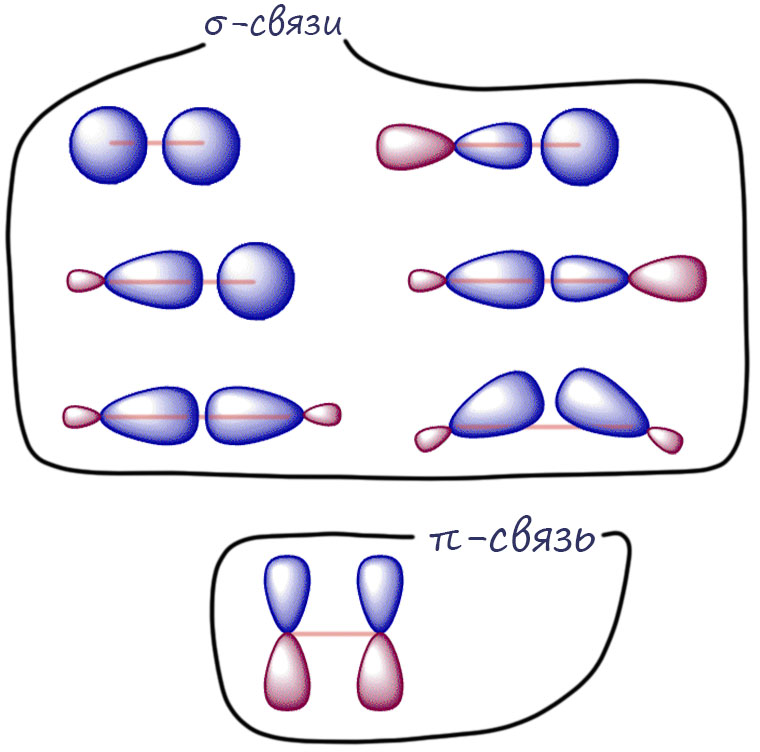
Орбитальки везде для простоты нарисуем одного размера. Размеры орбиталек – это не шутка, а вполне объективная вещь. Орбитальки, конечно, это неограниченные функции, но любые такие трёхмерные фугкции вседа представляют поверхностями, на которых функция принимает какое-то заранее согласованное значение, всегда одинаковое, которое подбирают так, чтобы картинки имели красивые размеры в выбранном масштабе. И для разных элементов размеры одних и тех же орбиталей разные – в периоде происходит сжатие, в следующем периоде мы будем брать следующий уровень и орбитальки станут больше и вновь будут сжиматься. В квантовых расчетах размеры орбиталек элементов учитывают в базисах. Но нам сейчас это не важно, важно понять разницу между σ- и π-связью, а для этого размер орбиталек до лампочки.
Если мы это поймём и усвоим, нас ждёт просто невероятный бонус – никаких других типов связей в молекулах непереходных элементов не бывает. Связь или σ- или π-, а третьего не дано.
Но даже если нас занесёт в химию переходных металлов, то связи переходных металлов с непереходными элементами тоже могут быть или σ- или π-, а третьего не дано.
И только связи переходных металлов между собой иногда могут иметь третий тип – дельта. δ-связи иногда могут быть и тогда, когда переходный металл связан не с одним атомом непереходных элементов, а сразу с целой группой атомов в так называемых гаптных комплексах. Но даже там это не очень часто встречается, и про это можно так и совсем не узнать, даже пристально занимаясь химией переходных металлов.
Итак, в органической химии даже с участием металлов, мы ничего кроме σ- или π-связей никогда не встретим. Не знаю, испытали ли вы облегчение, но вроде должны были, а если не испытали, то еще испытаете не раз. Ведь это так важно знать, что в науке в какой-то области число объектов ограничено и заранее известно. Если бы это было не так, мы могли бы напороться на молекулу, анализ связей в которой дал бы нам загадку – вот какая-то связь, не σ- или π-, а какая? И стали бы мы лихорадочно рыться в толстых книгах и вопрошать советы мудрецов, но поскольку ни толстые книги, ни мудрецы никогда не говорят просто и понятно, то ответа бы не нашли и сошли бы с ума, стали бы всем рассказывать, что открыли новый тип связи, писать статьи про это, а люди и журналы стали бы вас избегать. Печальная судьба. Так бывает. Это обычный механизм рождения такого общественного явления как “непризнанный гений”. Несчастный, скорее всего, просто не разобрался в чём-то важном. И ему стали мерещиться открытия там, где их нет уже двести лет. Но теперь нам это не грозит. Мы просто протрём глаза, аккуратнее нарисуем орбитальки, и придем к выводу, что нам показалось, и связь вполне обычна – или σ- или π. Просто потому, что других не бывает.
Про гибридизацию вспомнили, про типы связей тоже, теперь возьмёмся за сопряжение. Сопряжение – это орбитальное взаимодействие, не описываемое классическими связями. Классические связи всегда соединяют два соседних атома. Скоро, уже в теме алканы мы увидим, что бывают и неклассические, трёхцентровые (а бывают и многоцентровые) связи. Ещё сложнее бывают взаимодействия. Это сильно усложняет картину, но надо понимать, что всё, что мы обсуждаем здесь – мезомерия, резонанс, сопряжение, делокализация – это понятия химии почти столетней давности (через 10 лет еще это станет буквально верно и можно будет убрать слово “почти”), когда связи были только классические, двухцентровые. И понятие сопряжения возникло тогда, поэтому оно и отталкивается только от классических связей. А как в эту картину вписать неклассические связи? Да чёрт его знает. Видимо, никак, но нам такая проблема не встретится. Надо также хорошо понимать, что разговоры на уровне классических связей и структур, сопряжения, электронных эффектов – это очень удобная и хорошо работающая, но сильно упрощённая картина. Несложно понять, что упрощённая картина плохо подходит для описания каких-то очень необычных объектов, явно выходящих за рамки обычного. Мы ведь будем иметь дело именно с обычным – и этого обычного в органической химии горы и горные системы. Нам хватит точно. А если столкнёмся с необычным, просто поймём, что разговор должен пойти на другом уровне, и тогда точно придётся прибегнуть к настоящей квантовой химии, хорошим расчётам и всему такому, чего мы успешно избегаем в области обычного, где вместо расчётов у нас структурные формулы, электронные эффекты и сопряжение.
Вот ещё раз тот ряд структур, про которые мы уже сказали, что в них всё сложнее, чем представляется просто структурными формулами. Берём любую, например, первую. В ней две двойные связи и одна простая (про C-H связи пока забудем). Вопрос – они взаимодействуют? Да. Сильно? Не очень, но вполне серьёзно. А можем мы про это забыть? Хотелось бы, но как только мы лезем в химию и свойства бутадиена, всё время натыкаемся на особенности, которые не описываются просто классической структурой, и эти особенности определяются взаимодействием двойных связей. И это взаимодействие – не просто смещение плотности из-за разницы электроотрицательностей, а нечто более серьёзное. Это именно взаимодействие орбиталей там, где классическая структура этого не подразумевает, например, между вторым и третьим атомами углерода. По классике там простая связь, а в реальности связь заметно прочнее. А как мы это узнаем? В первую очередь, по структуре, но не нарисованной, а реальной – прочность связи определяется (или определяет) расстоянием (или расстояние) между атомами. Для простой связи между двумя sp2-гибридными атомами расстояние одно, а в бутадиене оно заметно меньше. Что значит заметно? В этом месте я начинаю заметно нервничать, и лихорадочно придумывать молекулу, в которой у двух соседних sp2-гибридных атомов углерода нет двойных связей или чего-нибудь еще более серьёзного, чтобы сравнить с ней бутадиен. И что-то не придумывается, вернее, придумывается, но нечто настолько непригодное для нормальной дискуссии, что-то такое скрученное-перекрученное, такое, что лучше не связываться. Давайте поэтому пока просто поверим в то, что есть какие-то очень серьёзные аргументы в пользу существования существенного взаимодействия между двойными связями в бутадиене и что-то подобное мы увидим для других молекул ряда (с бензолом как раз все понятно, но это отдельная проблема – не просто сопряжение, а ещё и ароматичность). Органическая химия вся такая – как только мы начинаем вести себя неприлично и подобно Пиноккио (aka Буратино) лезть туда, куда не просят, и задавать нескромные вопросы – сразу начинают появляться странные дыры и нестыковки. Ничего страшного, это просто значит, что модели и теории приблизительны и грубоваты. Но полезны.
Если бы мы могли позволить себе добавить к нарисованным структурам еще и квантовохимические расчёты, мы бы могли воспользоваться таким понятием как порядок связи. Для классических структур это очень просто – у простой связи порядок единица, у двойной два, у тройной три. И это прямо целые числа – один, два, три. С точки зрения электронов это просто число пар, обслуживающих взаимодействие между атомами – одна пара, две пары, три пары. В квантовых расчётах порядки связей посчитать можно, хотя и с некоторыми заморочками. Так вот, если бы мы посчитали порядки связей в молекулах такого типа, то увидели бы, что порядки связей существенно отличаются от классических один-два-три, причём то, что на структурах выглядит как простая связь, имело бы порядки, большие единицы, иногда немного, иногда даже близко к полутора. А то, что выглядит как двойная или тройная связь, имело бы меньшие порядки связей, иногда не сильно, а иногда и значительно. Вот это уже интересная особенность. Эту особенность – когда двойные или тройные связи как будто размазываются по соседним простым, – называют делокализацией. Кроме двойных или тройных связей делокализовываться могут неподелённые пары или вакантные орбитали. Во всех случаях мы будем видеть в расчётах изменение порядков соседних связей: неподелённые пары будут увеличивать порядки соседних связей, а вакантные орбитали – уменьшать.
Сопряжение бывает разным: нам сегодня нужно только одно
Итак, сопряжение это взаимодействие орбиталей, никак не отражаемое в классических структурных формулах.
Вопрос – какие орбитали взаимодействуют.
Ответ – любые, если они пространственно сближены, то есть находятся на соседних атомах, но иногда возможно сближение и сильно удаленных, в смысле расстояния по связям в структуре, атомов, если форма молекулы их как-то принудительно подтаскивает друг к другу. На второй возможности, не такой уж редкой, но всё же достаточно специальной, мы пока останавливаться не будем. Кроме пространственной сближенности необходима возможность перекрывания с общим ненулевым результатом. Результат перекрывания предсказывается просто. У орбиталей есть части (доли) с разным знаком (или фазой). Только у s-орбитали доля одна, у всех остальных две или более. Более – это у d-орбиталей и гибридных орбиталей с участием d-орбиталей, в нашем курсе это не понадобится. Еще раз повторю – не понадобится! Это есть только у переходных металлов, а ими мы серьёзно заниматься не будем. У элементов s- и p-блоков, то есть, в частности, у всех неметаллов и металлоидов, никаких d-орбиталей в валентной оболочке нет, и искать их не нужно. Следовательно, нам понадобятся только p-орбитали и s/p-гибридные орбитали.
Лучше всего взаимодействуют чистые p-орбитали, потому что они часто бывают параллельны, что обеспечивает максимальное перекрывание обеих долей, причём перекрываются доли одного знака (фазы). Такое перекрывание является причиной π-сопряжения. Буква π в этом случае означает, что результат перекрывания приводик к дополнительному связыванию, сильно похожему на связь π-типа – область перекрывания избегает линии, соединяющей атомы. Точнее это даже стоило бы назвать не просто π-сопряжением, а π-π-сопряжением. Так или иначе, мы будем заниматься в основном именно этим типом сопряжения, и именно этот тип сопряжения часто называют просто сопряжением, из чего можно сделать вывод, что это единственный тип сопряжения. Это не так, но с устоявшейся системой названий спорить бесполезно.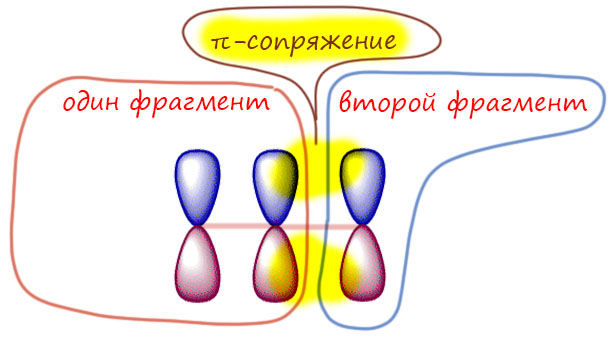
Взаимодействие чистых p-орбиталей и гибридных орбиталей тоже возможно – доли у них одинакового типа. Но, у гибридных орбиталей доли разные по размеру, а во-вторых, они всегда находятся под углом к p-орбиталям соседних атомов. Почему? Геометрия молекул так устроена, попробуйте сами покрутить, попробовав все возможности (sp-, sp2-, sp3-) и учитывая все варианты соседства атомов разной гибридизации. Если учесть, что гибридные орбитали обслуживают σ-связи, а чистые p-орбитали обслуживают π-связи, такие взаимодействия осуществляются между π-связями (двойными или тройными) и σ-связями. 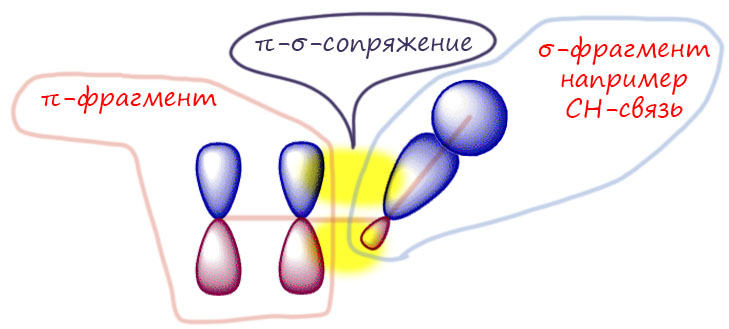 Кроме π-связей в таком взаимодействии могут участвовать одноатомные фрагменты с чистыми p-орбиталями (секстетные атомы или атомы с неподелённой парой). В любом случае такое взаимодействие, а это тоже сопряжение, потому что никак не отражается на классической структурной формуле, принято называть π-σ-сопряжением. А почему не просто σ-сопряжением по аналогии с π-сопряжением? Вообще, иногда термин σ-сопряжение найти можно, но редко. Хотя бы потому что это взаимодействие не является аналогом σ-связи, оно вообще больше похоже на π-связь. По-хорошему, это не так уж сильно отличается от обычного π-сопряжения, но обнаружено оно было немного (совсем немного, первая работа вышла в 1935, а про обычное сопряжение Лайнус Полинг начал писать с начала 1930-х) позже, чем обычное π-сопряжение, и в литературе закрепилось под названием гиперконъюгации или даже, по именам исследователей, которые впервые опубликовали признаки этого эффекта, эффектом Бейкера-Натана. Я рано или поздно сделаю отдельную страничку про эффекты, подпадающие под понятие гиперконъюгации. И поскольку с самого начала эффект считался более тонким и спорным, чем обычное сопряжение, его принудительно вывели в отдельную категорию, снабдив своими названиями, чтобы не путались.
Кроме π-связей в таком взаимодействии могут участвовать одноатомные фрагменты с чистыми p-орбиталями (секстетные атомы или атомы с неподелённой парой). В любом случае такое взаимодействие, а это тоже сопряжение, потому что никак не отражается на классической структурной формуле, принято называть π-σ-сопряжением. А почему не просто σ-сопряжением по аналогии с π-сопряжением? Вообще, иногда термин σ-сопряжение найти можно, но редко. Хотя бы потому что это взаимодействие не является аналогом σ-связи, оно вообще больше похоже на π-связь. По-хорошему, это не так уж сильно отличается от обычного π-сопряжения, но обнаружено оно было немного (совсем немного, первая работа вышла в 1935, а про обычное сопряжение Лайнус Полинг начал писать с начала 1930-х) позже, чем обычное π-сопряжение, и в литературе закрепилось под названием гиперконъюгации или даже, по именам исследователей, которые впервые опубликовали признаки этого эффекта, эффектом Бейкера-Натана. Я рано или поздно сделаю отдельную страничку про эффекты, подпадающие под понятие гиперконъюгации. И поскольку с самого начала эффект считался более тонким и спорным, чем обычное сопряжение, его принудительно вывели в отдельную категорию, снабдив своими названиями, чтобы не путались.
Если идти по этому пути дальше, можно и две гибридных орбитали таким образом заставить взаимодействовать. Никакой принципиальной разницы нет, и в тех же терминах это будет σ-σ-сопряжение (точнее, σ-σ*-сопряжение, но такие детали отложим для отдельной странички). Это очень тонкий эффект, мы с ним точно никогда не встретимся, хотя он, например, сильно влияет на барьеры вращения и относительную устойчивость конформаций. В современной химии таким эффектам стали уделять внимание – химиков стало так много, что в дело пошли и самые дохлые эффекты и явления, иначе новым ордам химиков нечего будет исследовать, а там, сзади, уже и следующие напирают.
Поищем сопряженные фрагменты в реальных молекулах. Все просто – находим кратные связи, атомы с парами и секстетные атомы, находящиеся рядом друг с другом в любых (пока) комбинациях. Важно, что наблюдатель, идущий по цепи сопряжения, не должен наступать на атомы, не принадлежащие к этим трем типам. Как только встречаем такой атом, сопряжение заканчивается.
Разновидности сопряжённых систем: цепь сопряжения и кросс-сопряжение
Напоминаю, что мы говорим о π-сопряжении. В этом разделе и почти везде на этой странице под сопряжением имеется в виду только π-сопряжение.
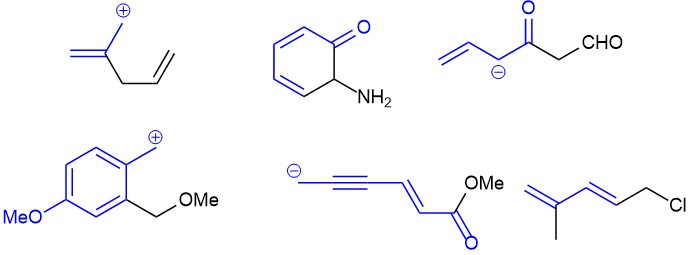
Сопряжение возникает тогда, когда рядом находятся атомы с p-орбиталями. Рядом – значит подряд, два, три, четыре, сто, тысяча, сколько угодно, но подряд. Стоит хоть одному не дать p-орбиталь как сопряжение нарушается. Не дать – значит у этого атома ее или совсем нет, или, по каким-то причинам она расположена перпендикулярно соседним. Такой случай есть, например, в 1,2-диенах (алленах). В этих диенах нет π-сопряжения. π-Связи в них изолированы. Значит ли это, что они вообще не взаимодействуют? Нет, не значит, но это взаимодействие в любом случае более сложное и слабое, и скорее всего, даже отрицательно влияющее на устойчивость таких соединений, дестабилизирующее. Когда дойдём до алленов, попробуем в этом разобраться, а пока просто отложим в сторону, потому что сейчас мы занимаемся π-сопряжением, а это почти всегда эффект, увеличивающий устойчивость, стабилизирующий, выгодный. 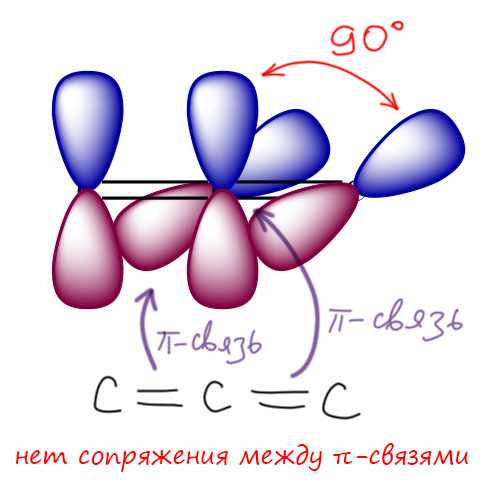
Следующая важная вещь – подряд, это понятно, но может быть два случая: цепь и разветвление. Например, возьмём четыре атома с p-орбиталями, пока просто абстрактно, не думая о конкретных соединениях, но возможные примеры молекул с такими ситуациями приведем.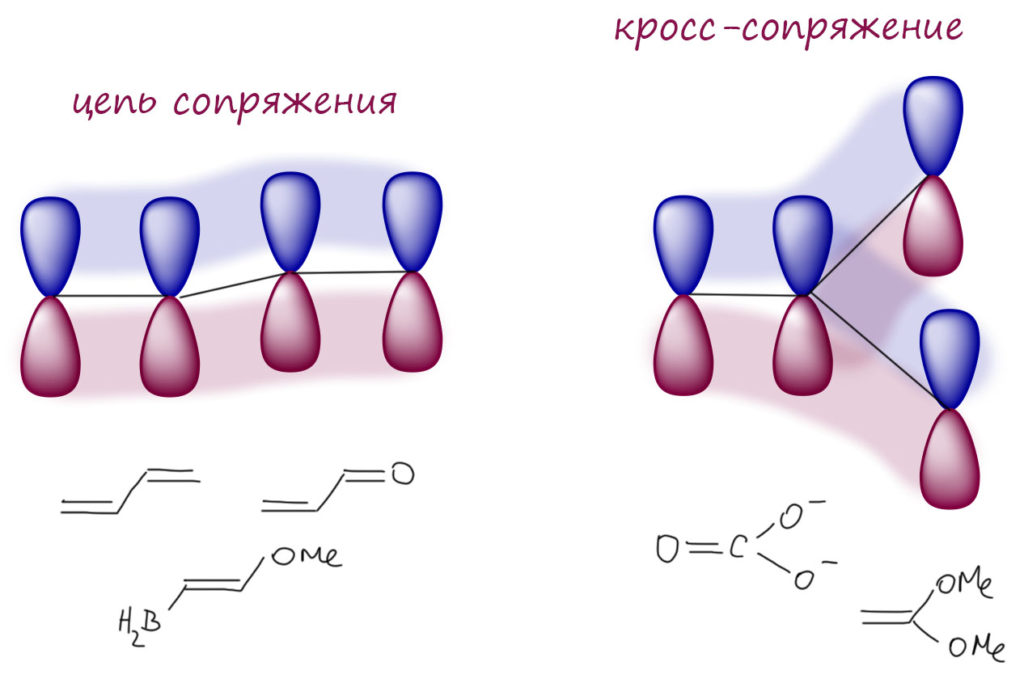
Видим, что в обоих случаях перекрывание p-орбиталей обеспечено их сближенностью. Кажется даже, что второй случай должен быть в этом смысле даже еще лучше – p-орбитали так красиво сплотились вокруг одной p-орбитали, прямо как мы любим. Но не все так просто. И это совершенно принципиальный момент. Поэтому остановимся, вздохнём поглубже, переведём дух – сейчас нам предстоит сделать важное заявление, которое потом будет множество раз аукаться во всем курсе органической химии. Чем лучше мы это поймём, тем меньше удивлений будет потом. Итак, сначала пара определений.
Сопряжение по цепи атомов – цепь сопряжения, всегда линейная конструкция, один за другим, у каждого атома не более двух соседей.
Сопряжение с разветвлением – кросс-сопряжение. У некоторых атомов в сопряженной системе может быть три соседа.
Итак, сопряженные системы бывают двух типов – цепи сопряжения и кросс-сопряженные системы.
Так вот, только цепь сопряжения является безусловно выгодным, стабилизирующим типом взаимодействия. Поэтому мы везде будем стараться не пропустить наличие цепей сопряжения, и должны будем уметь их анализировать с помощью мезомерии и рисования граничных структур.
Кросс-сопряжение – случай, намного более сложный. В принципе, кросс-сопряжение можно представить как взаимодействие нескольких цепей сопряжения, разбить полную структуру на такие цепи и анализировать сопряжение внутри таких цепей. Понятно, что общая система будет наложением (суперпозицией) сопряжения в цепях. Граничных структур поэтому будет много, но не это главное. Проблема в том, что системы с кросс-сопряжением, несмотря на большое количество формально рисуемых граничных структур, часто дестабилизированы. Предвкушаю, что в этом месте найдётся тот, чей взгляд скользнёт на схему выше, на которой в качестве примера системы с кросс-сопржением нарисован карбонат-ион. “Это карбонат-ион-то дестабилизирован, а как же из карбонатов сложены целые горные массивы, в частности тот несчастный шихан Куштау, который так настойчиво пытаются спасти жители Стерлитамака!!” – непременно вскричит такой читатель – “Караул! Крамола!! Ересь!!! Полиция, хватайте еретика!!!!” Не торопитесь, дорогой читатель. В химии не бывает абсолютной стабилизации и дестабилизации. Всякая частица сравнивается не с абсолютным мерилом стабильности, которого нет, а с близкими по строению и составу, причем такими, в которые можно превратиться или из которых можно образоваться. Дестабилизировнность карбонат-иона вполне отчётливо проявляется, например, в таком его свойстве, как весьма сильная основность – протонирование по первой ступени ослабляет кросс-сопряжение и поэтому весьма выгодно. Мы еще обсудим это, когда дойдём до карбоновых кислот.
А с ароматическими кольцами что делать?
Ароматические (и антиароматические) кольца – это, на самом деле, третий тип сопряжённых систем. Такое сопряжение называют циклическим. С виду это очень простая вещь – цепь сопряжения скручиваем в бараний рог и даже сильнее – в полное кольцо. При этом происходит замечательная вещь – в цепи сопряжения были концевые атомы с единственным соседом, в циклическом сопряжении больше нет концевых атомов.
У циклического сопряжения есть одна невероятно важная особенность – такая сопряженная система может стать ароматической или антиароматической. К нашей сегодняшней теме это никакого отношения не имеет. Ароматичность (и антиароматичность) не имеет отношения к мезомерии и граничным структурам, и рисовать их, например, для бензола, бесполезно, они ничего не говорят.
Но у любого ароматического кольца есть две стороны – ароматичность, которая присуща кольцу целиком (точнее, циклической сопряжённой системе) и то, что любая часть кольца представляет собой цепь сопряжённых атомов, поэтому любая часть кольца может участвовать в цепях сопряжения с приделанными к кольцу атомами и группами.
Но стоит нам приделать к ароматическому (давайте для простоты всегда иметь в виду бензольное, а другими ароматическими системами разберёмся в своём месте) кольцу что-нибудь, имеющее p-орбиталь, один атом или цепь сопряжения, как происходит удивительная вещь – сопряжение этого фрагмента и кольца с образованием более длинной цепи сопряжения. И нам может понадобиться проанализировать этот случай с помощью мезомерии и граничных структур. Зачем? Для того чтобы понять, насколько присоединение такого фрагмента меняет свойства, выгодно это или нет. Например (пока без граничных структур): 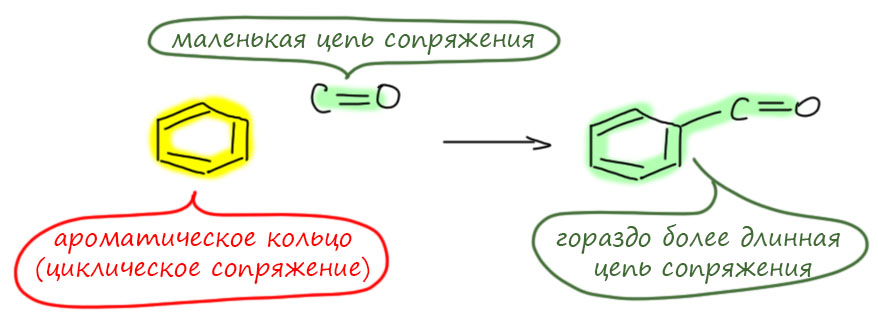
Очень важно понимать, что ароматичность (циклическое сопряжение внутри цикла) никуда не делось. Но к нему, как бы сверху, добавилась новая цепь сопряжения. Ничего необычного в этом нет – не забывайте, что хотя мы и рисуем такие понятные структурки, но речь ведём фактически о квантовых вещах, квантовых состояниях. А в квантовой науке нет ничего необычного в том, что в одной вещи мы можем углядеть и одно и другое одновременно. Когда-то это красиво называли квантовой суперпозицией (наложением) квантовых состояний. Химики пугаются таких слов, и услышав их с утра, могут так разволноваться, что прольют очередной реактив мимо горлышка колбы. Поэтому нас и не пугают, а просто показывают как правильно рисовать граничные структуры – нам тогда кажется, что мы не лезем ни в какую квантовую лабуду, а привычно двигаем структурки прямо как отец наш А.М.Бутлеров. Но ведь, сами того не зная, лезем. Сталина на нас нет.
Еще интереснее получается, когда к бензольному кольцу присоединяют два фрагмента с p-орбиталями (две цепи сопряжения). Тогда, если присоединение произошло так, что образовалась орто- или пара-замещенная система, в ней возникает общая длинная цепь сопряжения. Даже две таких цепи. Что с этим делать разберемся, когда дойдем до рисования граничных структур. Пока просто заметим, что придется нарисовать граничные структуры, соответствующие каждой из цепей, но если некоторые из них получатся одинаковыми, такие можно опустить. 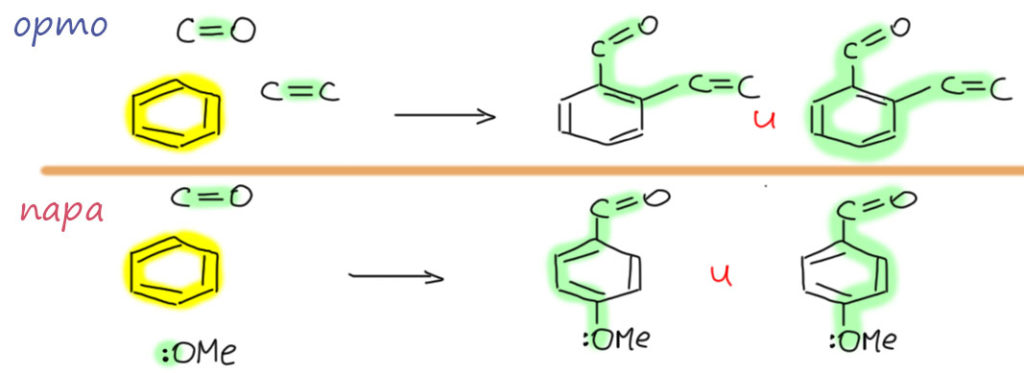
А вот если получается мета-замещенная система, то общей цепи сопряжения не возникает. Возникают две отдельные, с каждым из фрагментов по отдельности, и взаимодействуют по принципам кросс-сопряжения, что, как мы уже поняли, морока, головная боль, и вообще, отстаньте. Мета-замещение действительно почти всегда намного менее выгодно, чем орто или пара, за исключением случаев, когда по каким-то причинам общая цепь споряжения как раз не нужна. Всякое бывает. Но редко.
А если фрагментов (цепей) три или больше. Тогда, как ни замещай, тоже не получится единая цепь сопряжения. Придётся рисовать много разных, это опять будет кросс-сопряжение. В некоторых случаях какие-то два фрагмента могут образовать такую прекрасную цепь, что остальным можно будет пренебречь. А как отличить прекрасные цепи от тех ужасных, типа тех, которые никак не получается потерять пролетариату, мы скоро разберёмся.
Из чего состоят сопряженные системы
Сопряженные системы состоят из атомов с p-орбиталями, это понятно. Но лучше построить более точную систему.
Когда возникает сопряженная система, почти всегда это приводит к смещению электронной плотности от одних частей к другим. И в этом смысле все сопряженные системы удобно делить на части, имеющие конкретные функции:
- фрагменты или группы, которые являются донорами электронной плотности – электронная плотность всегда смещается от таких фрагментов или групп. Это – всегда доноры. В тех случаях, когда мы их рассматриваем как заместители, их называют мезомерными донорами и обозначают символом +M.
- фрагменты или группы, которые являются акцепторами электронной плотности – электронная плотность всегда смещается к таким фрагментам или группам. Это – всегда акцепторы. В тех случаях, когда мы их рассматриваем как заместители, их называют мезомерными акцепторами и обозначают символом -M.
- фрагменты или группы, которые могут быть как донорами, так и акцепторами в зависимости от ситуации, и эффект таких заместителей, донорный или акцепторный, также зависит от ситуации
- фрагменты или группы, которые выполняют роль соединяющих элементов в сопряженных системах – они соединяют доноры и акцепторы в единое целое, сопряженную систему, цепь сопряжения или несколько взаимодействующих цепей сопряжения. Такие фрагменты не считаются заместителями и им не приписываются эфффекты заместителей.
Теперь разберёмся с каждой из групп.
Всегда доноры
Всегда донорами являются атомы с неподеленными парами. Такие атомы могут нести отрицательный заряд, а могут быть нейтральными.
Во втором периоде, а мы в основном будем сидеть в нём, бор не бывает донором, углерод может быть мезомерным донором только в виде карбаниона, кислород может быть анионом и нейтральным донором, азот, хотя и может быть анионным донором, но это большая редкость, фтор – только нейтральный донор (фтор – донор?! да, конечно, есть неподеленная пара и вопрос закрыт, донор).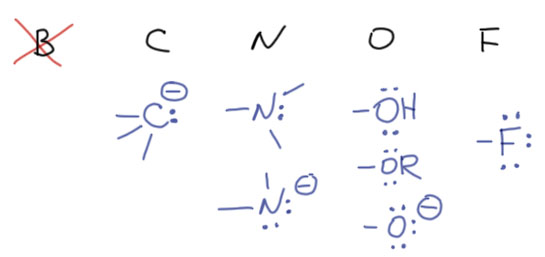
Существуют качественные тенденции, которые позволяют на глазок сравнивать силу доноров. Учтите, что здесь речь идёт не о тонких эффектах, а именно об очень сильной зависимости – можно считать, что доноры занимают ступени лестницы, и донор с верхней ступеньки настолько сильнее донора с нижней, что в случае прямой конкуренции даже не заметит его присутствия.
Для одного элемента атом с отрицательным зарядом всегда более сильный донор, чем нейтральная форма. Во втором периоде это прежеде всего касается кислорода – анион на следующей ступени относительно гидроксида и алкоксида. С анионным азотом у нас мало шансов встретиться.
Для похожих заместителей донорность связана с электроотрицательностью элемента – чем она больше, тем донорность слабее. Но сравнивать можно только похожие заместители, например, только нейтральные. Фтор поэтому очень слабый донор, и очень непростой. Это атом с максимальной электроотрицательностью и сильнейший индуктивный акцептор. Он с очень большой неохотой отдает пару даже сильнейшим акцепторам, а одновременно действующий индуктивный эффект одновременно сильно дестабилизирует одну из граничных структур. Поэтому хотя фтор безусловно является мезомерным донором и доказательсвт этому в химии немало, но затейливое соечетание донорного и акцепторного эффекта приводит кл всяким всяким аномалиям поведения фторпроизводных. Мы не будем туда лезть, хотя это и очень интересно, в нашем курсе такие молекулы не встретятся.
Всегда акцепторы
Всегда акцепторы бывают двух основных типов:
Электронодефицитные атомы. У атомов второго периода электронодефицитность означает одно – шесть электронов в валентной оболочке вместо положенного октета. Не устаю повторять – все, что с нечетным числом электронов, радикальные частицы мы откладываем в сторону, там всё особенное. Поэтому после полной оболочки, октета, сразу следует 6. Четыре электрона в валентной оболочке при обычных условиях (не в газовой фазе, не в плазме) не бывает, слишком мало, два – тем более, ищите такие частицы в межзвёздном газе, и если правда пойдёте искать, можете не возвращаться.
Атомы с 6 валентными электронами в валентной оболочке называются секстетными (секстет – шесть). Во втором периоде секстетные атомы это обычный бор с тремя связями (бораны) и углерод в виде или карбокатиона, или карбена. У азота тоже есть такие состояния (нитрен и нитрений), но они встречаются достаточно редко и в системы сопряжения мы их включать не будем, хотя это и возможно. Секстетное состояние кислорода (оксений) имеет очень высокую энергию и встречается только в виде гипотетических частиц в механизмах перегруппировок, а в цепи сопряжения такой кислород вставить можно только в рамках праздного любопытства. Про секстетный фтор лучше не задумываться – это страшные мысли, от которых закипают мозги, вернуться к нормальной жизни уже не получится. На схеме отсутствующая пара электронов обозначена красным квадратиком. Увы, официального символа для вакансии так и нет.
Хотя это и очевидно, но лишний раз напоминаю, что обычные ониевые ионы – аммоний и оксоний – имеют октетные атомы и никакого отношения к электронодефицитности не имеют. 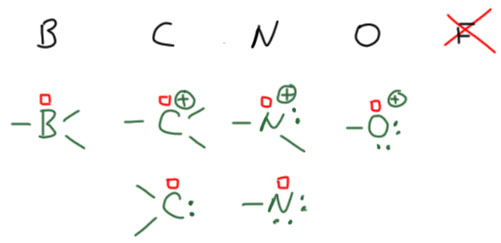
Кратные связи с гетероатомами. Любые группы, в которых двойными или тройными связями связаны два гетероатома или один гетероатом и углерод, являются обязательными акцепторами. Поскольку мы говорим о втором периоде, то выбор довольно ограничен. У бора нет кратных связей, у фтора тоже нет. Остается кислород и азот. Самая главная группа этого типа – карбонил. Но таких групп намного больше, большинство с участием азота.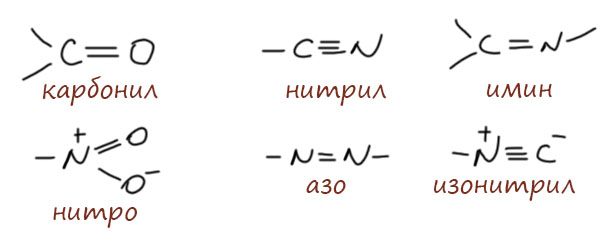
Чего изволите: можем донором быть, а можем и акцептором
Несколько фрагментов могут быть и тем, и тем. Это двойная и тройная связь углерод-углерод, а также фенил. А как же тогда узнать, какую роль выполяют эти фрагменты в конкретных молекулах? Да очень просто. Мы же уже выяснили, что должны везде искать цепи сопряжения. И вот – нашли мы такую цепь, на одном конце у неё обязательный акцептор -M, а на другом нет обязательного донора, но есть вот такая группа. Должно же там что-то быть. В этом случае такая группа возьмёт на себя роль донора. И наоборот, если на одном конце цепи обязательный донор +M, а на другом нет акцептора, то такая группа станет акцептором.
Очень удобно. Символически это означает, что такие группы могут быть и +M и -M. Возьмём пока очень короткие цепи сопряжения.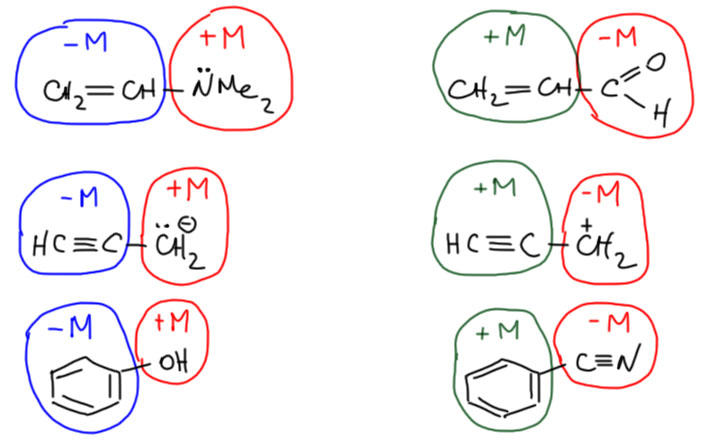
Конечно, остаются еще вопросы. Например, а что будет, если такие группы на обоих концах цепи сопряжения? Тогда они будут перетягивать друг у друга эти звания. Пока отложим, но обязательно разберёмся.
Соединительные элементы
Доноры и акцепторы соединяются в цепь сопряжения или прямо, или с помощью дополнительных вставок. Роль вставок могут играть двойные связи С=С. Так из любой пары донор-акцептор мы можем получить много аналогов. Есть даже специальное слово, обозначающее такую аналогию – винилогия (от слова винил, остаток этилена, что, кстати, не совсем корректно, потому что двухвалентный остаток этилена называется не винил, а винилен, но так сложилось очень давно). Это явление обнаружил 75-летний классик Людвиг Кляйзен в 1926 году, как и положено старому немецкому классику ничего не зная про электронное строение, мезомерию и всё это, а просто по аналогии химических свойств, оставив объяснение потомкам; слово впрочем придумал не он.
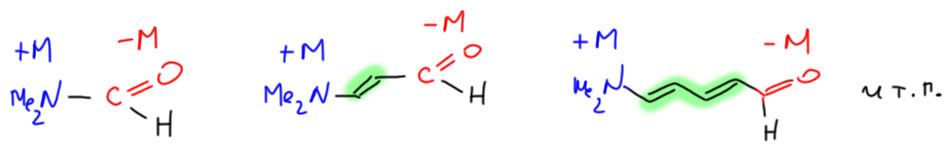
Не менее эффективной соединялкой является остаток ацетилена этиндиил. Красивого слова для этого не придумалось, но сам по себе ацетиленовый удлинитель даже еще более эффективен, чем двойная связь, потому что совершенно не связан никаким требованиями к нахождению сопряженных групп в одной плоскости – линейная ацетиленовая группа всегда находится в плоскости.

Третим типом соединяющих элементов является бензольное кольцо, но только в том случае, если донор и акцептор присоединяются или орто- или пара-способом. Некоторая сложность бензольного кольца состоит в том, что в нем как будто две дорожки для сопряжения, одинаковые в случае пара-замещения, или сильно разный в случае орто. В реальности, естественно, сопряжённая система едина, но нет никаких проблем в том, чтобы немного упростить себе жизнь, и смотреть на такие дорожки просто потому, что с двойными связями более-менее понятно, что делать, а с целостной ароматической системой обращаться непросто. 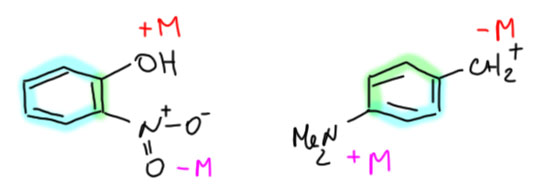
Все эти соединялки можно комбинировать самыми разными способами, и это дает возможность получать огромное количество разнообразных сопряжённых систем с самыми разными свойствами.
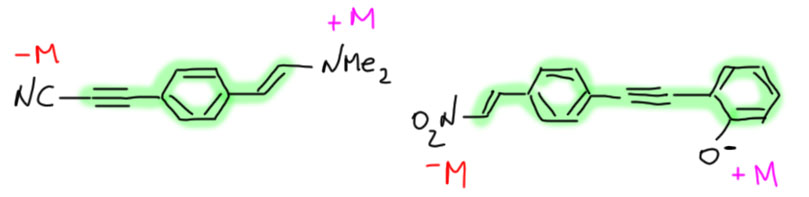
Конечно, в реальности дела немного сложнее. Не стоит забывать, что для возможности сопряжения фрагменты должны находиться или в одной плоскости, или хотя бы достаточно близко к ней. Но в очень больших системах этому может активно мешать, во-первых, стерика, и, во-вторых, энтропия. Энтропия всегда играет против сопряжения (эффект сопряжения – стабилизация – чисто энергетический) просто потому, что максимальное сопряжение будет в максимально упорядоченной, плоской молекуле, но это как раз невыгодно с энтропийной точки зрения особенно, если в частях молекулы есть гибкость. А гибкость есть, например, в соединении двойных связей. Поэтому очень длинные цепочки сопряженных двойных связей не очень-то и устойчивы – энтропия стремится разупорядочить их, хаотично разворачивая отдельные двойные связи друг относительно друга, при этом молекула ищет оптимум, жертвуя частью энергии сопряжения, но возвращая себе энтропию. В отличие от двойной связи, тройная лучше работает, потому что, как её ни крути, сопряжение остается. Но там тоже есть проблема в граничных структурах алленового типа, которые далеко не так хороши из-за взаимной дестабилизации двойных связей. Тем не менее, тройные связи очень популярны в современной химии сопряженных систем, именно как эффективные соединители.
Доноры, акцепторы - немного подробнее
Хотя мы имеем полное право не различать разные виды доноров и акцепторов, используя всего два символа для обозначения эффекта +М и -М, есть довольно значительная разница, которая собственно и объясняет, отчего некоторые являются обязательными, а некоторые приспосабливаются к обстоятельствам. И еще есть разница в том, как осуществляется π-сопряжение.
Попробуем немного прикрутить сюда орбитали. Не полноценные молекулярные орбитали, а их заготовки, такие воображаемые орбитали, из которых можно было бы построить настоящие. Квантовая химия и метод МО отлично позволяют это делать, потому что в основе этого метода как раз и лежит построение орбиталей более сложной системы из орбиталей фрагментов, причём это не обязательно атомные орбитали, но могут быть и молкулярные орбитали фрагмента. Так, мы можем построить диен не только из четырёх атомных p-орбиталей, но и из двух (наборов) молекулярных орбиталей двойных связей.
Итак, что такое акцептор? Это фрагмент, который может принять пару на вакантную орбиталь. Вакантные орбитали акцептора могут быть двух основных типов:
- атомная p-орбиталь – и это случай секстетных атомов типа карбокатиона
- разрыхляющая π*-орбиталь кратной (двойной, тройной) связи
А что такое донор? Это фрагмент, который может отдать пару. Такой фрагмент тоже может быть двух типов:
- атомная p-орбиталь с неподелённой парой
- связывающая π-орбиталь кратной (двойной, тройной) связи.
Поэтому то, что мы называем π-сопряжением можно для точности разделить на несколько более узких типов. Для определённости всегда будем донор писать перед акцептором.
- p-p-сопряжение (секстетный атом в сопряжении с атомом с неподелённой парой)
- π-p-сопряжение (кратная связь в сопряжении с секстетным атомом)
- π-π*-сопряжение (сопряжение кратных связей)
Зачем это нужно? Без этого отлично можно жить и дальнейшее изложение случаев сопряжения это не учитывает. Но если вы самостоятельно попробуете различать эти случаи, то классифицировать сопряжённые системы и рисовать граничные структуры или мезомерные стрелочки станет ещё легче. Вы точно поймёте, в чём разница разных случаев.
И ещё – чем отличаются обязательные доноры и акцепторы от тех, что по вызову. Очень просто – доступностью орбиталей для сопряжения. У тех фрагментов, где участвуют атомные p-орбитали, вопросов нет – это или есть, или нет. Но фрагменты с кратными связями очень даже могут быть разными. У обязательных акцепторов доступны только разрыхляющие π*-орбитали (связывающие π-орбитали у них лежат слишком низко и поэтому отдать ничего не могут).
У необязательных доноров и акцепторов (кратных связей между атомами углерода или ароматическими кольцами) доступны и связывающая π-орбиталь, и разрыхляющая π*-орбиталь. Поэтому и та, и та могут участвовать, если рядом находится подходящий фрагмент для сопряжения.
Так, чисто для корректности замечу, что у ароматических колец дело немного сложнее, но не принципиально. В них нужно говорить не о π-орбиталях связей, а о π-орбиталях ароматической системы. Пока оставим это, но когда-нибудь разберёмся.
И еще – какова здесь роль соединительных фрагментов? Они предоставляют свои π-орбитали для построения общих орбиталей цепей сопряжения. Это несколько усложняет дело, но тоже непринципиально, и на классификацию типов сопряжения не влияет.
Сопряжение можно изобразить разными способами, самый распространённый и надёжный из которых – граничные структуры и резонансные стрелочки. Работает это довольно просто – если мы не можем адекватно изобразить молекулу одной классической структурной формулой, попробуем изобразить ее несколькими, каждая из которых отображает что-то важное. И дальше мы можем нарисовать между этими структурами такие специальные стрелочки, резонансные стрелочки, которые будт нам говорить – не просто говорить, а орать прямо в ухо – это не настоящие структуры, а выдуманные, настоящая где-то между ними, настоящая может быть похожа на одну из этих структур, а может и быть где-то на полпути от одной к другой. Граничными структурами можно попробовать изображать вообще любую структуру, даже такую, в которой нет никакого сопряжения. Например, молекулу водорода. Вот две граничные структуры. Атомы водорода стоят на месте, а электроны изменили свое расположение, например, сместились к одному из атомов. Что мы хотим этим сказать? То, что мы пока не знаем, какая связь в молекуле водорода, ковалентная или ионная. Вы можете от возмущения просто лопнуть – как это мы не знаем, откуда в водороде взялась ионная связь, что за бред???!!! Погодите, в химии бывает много интересного. Прямо сразу лучше не возмущаться. Молекула водорода, возможно, находится в каких-то необычных условиях, может на поверхность какую угнездилась или ещё куда-нибудь попала, что-то с ней происходит, и нам просто позарез понадобилось об этом подумать. И вот мы и используем такой инструмент, чтобы подумать о том, не может ли связь в молекуле водорода стать не ковалентной неполярной, а ковалентной с долей ионности. И изображаем это именно так – две граничные структуры и мы можем попробовать оценить вклад каждой в настоящую структуру: если преобладает вклад первой, то водород остается тем водородом, к которому мы привыкли со школы, если второй – то что-то заставляет молекулу поляризоваться (например, поверхность какая-нибудь).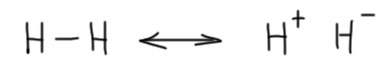
Использование резонанса и граничных структур позволяет нам рассуждать о том, как устроены молекулы (сами по себе или в каких-то особых обстоятельствах), если для этого не хватает одной классической структурной формулы. Естественный вопрос – а почему бы не придумать какие-нибудь символы или ещё какие-нибудь финтифлюшки, которые прямо на одной структуре помогут понять, что в ней что-то есть еще кроме двухцентровых связей, зарядов и неподелённых пар. Символов и финитифлюшек придумали немало, это и пунктирные связи, означающие делокализованные связи с нецелым порядком связи, и мезомерные стрелочки. Но эти обозначения страдают или произвольностью и низкой степенью информативности, или некоторой произвольностью. Мезомерными стрелочками мы будем иногда пользоваться, ими можно пользоваться, хотя лучше делать это уже как следует разобравшись в граничных структурах. Пунктиры я бы порекомендовал полностью исключить. Там дальше мы поподробнее разберёмся почему. Из всех способов граничные структуры – самые честные и самые понятные, они рисуются по весьма строгим правилам и не допускают произвола. В общем, это отличный, пожалуй даже гениальный инструмент анализа структуры, в котором заложен еще и огромный потенциал – эти структуры можно успешно приспособить не только для сопряжения.
Итак, что важно:
- граничные структуры всегда классические – в каждой все связи двухцентровые, валентности соблюдены, заряды расставлены
- в каждой граничной структуре строго соблюдается правило октета – ни один из атомов p-элементов не может иметь больше 8 валентных электронов (меньше может)
- каждая следующая граничная структура соотвествует минимально возможному, но полному смещению пары электронов
- пара на атоме – связь на том же атоме
- связь – соседняя связь на том же атоме
- строго соблюдается сохранение суммарного заряда (анион остается анионом, катион катионом, нейтральная структура может перейти в цвиттер-ион и обратно), заряды на отдельных могут возникать или исчезать, но суммарный заряд (арифметическая сумма плюсов и минусов) у всех граничных структур должен быть одинаков
А правда, что чем больше граничных структур, тем лучше
Есть весьма распространённое мнение, что чем больше граничных структур можно нарисовать тем лучше, тем стабильнее соответствующая молекула или ион. Из той же серии высказывание: делокализация всегда выгодна. Нередко эти высказывания возводят в абсолют и придают ему значение чуть ли не закона, или, по крайней мере, очень важного правила. Так говорят: “Нарисуйте граничные структуры, и там, где их получится больше, будет более устойчивое соединение или ион.” Слышали? Я сам не только слышал, но и много раз говорил такие вещи, и мне не стыдно. Я всегда имел в виду конкретные ситуации, в которых сравнивались похожие системы с одинаковыми способами делокализации и приблизительно одинаковыми вкладами граничных структур. Тогда это неплохо работает. Сформулируем, когда это должно работать.
Если вы сравниваете две аналогичные структуры, в каждой из которых есть очевидное π-сопряжение, а природа частей каждой рассматриваемой системы (доноры, акцепторы, удлинители сопряжения) одинакова или хотя бы очень похожа, но количество граничных структур приблизительно одинакового веса (вклада) в одной структуре больше, чем в другой, так структура, у которой больше граничных структур, скорее всего, более стабильна, чем та, у которой их меньше.
В этом высказывании есть вполне произвольное определения – “аналогичные”, “похожие”. Это как? Это никак нельзя формализовать, поэтому смотрите на это, как на призыв задействовать здравый смысл. Смысл здравого смысла в этом случае вполне здрав – чем более структуры кажутся вам похожими, аналогичными, тем лучше, тем надёжнее. И, наоборот, чем больше у вас сомнений в том, что структуры похожи, аналогичны, тем больше шансов, что это не сработает и нужно искать другие способы анализа проблемы. Да и не забывайте, что химия, да благословят уполномоченные боги создателей этой науки, наука не умозрительная, а экспериментальная, и вам никогда не придётся гадать нечто абстрактное, но у вас будут какие-то реальные данные – энергии, реакции, свойства, и т.д., и вам нужно будет как-то разумно объяснять то, что реально существует. Поэтому даже если вы ошибётесь в своём объяснении, ничего страшного не случится, экспериментальные данные не вывернутся наизнанку, а просто будут ждать более адекватного анализа.
Но стоит это начать обобщать, как может получиться или даже обязательно получится нелепица на нелепице. Но вообще это высказывание так привычно, что долго это повторяешь, поневоле обобщая и не задумываясь, а откуда это собственно взялось, и почему это должно быть верно.
Ответ обескуражит. Это ниоткуда не взялось, и является, строго говоря, просто произвольным обобщением нескольких частных случаев. А в общем случае это не только не верно, но и часто дает совершенно неверную оценку устойчивости. Вот мы сейчас разберёмся в разных ситуациях, возникающих в сопряжении и поймём, что лучше всего сопряжение работает в цепях сопряжения с донором и акцептором на концах цепи. И в этих случаях почти всегда доминирует одна граничная структура, остальные можно даже не рисовать. А молекулы и ионы, которые имеют такую особенность, почти всегда весьма стабильны, и отличаются в этом смысле в лучшую сторону от цепей, в которых нет такого расположения донора и акцептора, и где нужно рассматривать две или более полноценных граничных структур.
Само высказывание в этом смысле очень похоже на другую популярную в химии мантру: более реакционноспособный реагент менее селективен, чем менее реакционноспособный (обратная зависимость реакционной способности и селективности). Современная химия давно вынесла приговор этому принципу – он просто неверен и полностью уводит анализ в сторону от истинных источников реакционной способности и селективности. Все такие высказывания, часто высокопарно называемые принципами, появились в химии 1950-70-х, когда из химии ударными темпами пытались сделать чуть ли не точную, но, по крайней мере, количественную науку, с законами, уравнениями, числами, числами, числами. Крупные ученые выступали со смелыми обобщениями, каждый хотел открыть какой-нибудь закон химии. Это хорошо и правильно, хороший ученый и должен искать обобщений. Ученый, как и политик, должен быть дерзок и нагл, искать чести и славы. Скромность украшает кого угодно, но не ученых и не политиков. Тут только одно важное замечание – это правильно работает только в высококонкурентной среде, когда вокруг все такие же дерзкие и наглые, жаждущие чести и славы. Тогда получается общее движение вперед, а иначе получится или узурпация, или то самое страшное, что бывает в науке – доминирование авторитетов. Тогда пиши пропало – будете учить гениальные мысли авторитетов и не высовываться. Результатом будет застой и отставание. Авторитеты, кстати, вовсе не обязательно являются блатными шарлатанами (такое бывает), но могут быть и действительно великими учёными, даже реально величайшими. Именно это в первой половине прошлого века почти убило великую немецкую науку, Гитлер только немного помог ее добить. Это удивительная ирония истории – великую немецкую науку загубили ее величайшие представители с помощью одного гнусного мерзавца. И не только с немецкой наукой приключилась такая история, просто это самый яркий пример того, как работает монополизация мысли и воли даже в самой передовой науке в мире.
Так вот, в те годы и возникли эти обобщения. История принципа реакционная способность – селективность в этом смысле особенно характерна, потому что известно, кто выдвинул этот принцип, и как его обосновывал, любой неленивый желающий может просто пройти тем же путем и понять, чем обоснован этот принцип. Займёмся этим когда-нибудь в другом месте. В данном случае это не имело плачевных последствий, потому что возникло как раз в конкурентной науке, и хотя автором этой идеи был один из самых крупных авторитетов (нобелевский лауреат Герберт Браун), навязать ее не удалось, и она так и осталась просто одним из обобщений – хотите пользуйтесь, не хотите – игнорируйте или критикуйте.
- научиться рисовать граничные струкры
- оценивать их вклад в реальную структуру и отбрасывать неважные
Итак, перед нами некоторая молекула, в которой очевидна возможность π-сопряжения. Это первое. Нет π-сопряжения, рисовать нечего. С тем, что такое π-сопряжение мы разобрались. Если нет, повторяем.
Второе. Везде ищем цепи сопряжения. Для цепей сопряжения граничные структуры рисуются легко и действительно помогают понять настоящую структуру. А вот если встречаем кросс-сопряжение, относимся к этом у с большой настороженностью. Граничные структуры для кросс-сопряжения рисуются не сложнее, чем для цепей сопряжения, но они обычно мало что выражают. К счастью, мы крайне редко будем встречать примеры кросс-сопряжения. Если до этого момента не разобрались, чем отличаются цепи сопряжения от кросс-сопряжения, повторяем и разбираемся. Это очень важно, и в будущем избавит вас от бессмысленной фрустрации.
Цепи сопряжения тоже бывают разные. Посмотрите еще раз, что такое обязательные доноры и акцепторы, доноры и акцепторы по обстоятельствам, и соединительные элементы цепей сопряжения на вкладке Из чего состоят сопряжённые системы.
- самая лучшая цепь сопряжения та, у которой на концах обязательный донор и акцептор.
- может иногда быть так, что после обязательного донора или акцептора, или обоих, цепь сопряжения продолжается.
- с одной стороны обязательный донор или акцептор, с другой необязательный.
- обязательный донор или акцептор внутри цепи сопряжения
- цепь сопряжения не содержит ни одного обязательного донора или акцептора
Разберемся с этими случаями. Прежде чем перейти непосредственно к разбору типичных цепей сопряжения, заметим еще одну важную вещь. Сопряжение можно изображать граничными структурами, а можно кривыми резонансными стрелочками, изображающими смещение электронной плотности в цепи. Оба способа законны и широко применяются. Тем не менее я весьма категорично рекомендую считать основным способом только граничные структуры, а резонансные стрелочки рисовать только тогда, когда вы имеете дело с хорошо понятной цепью сопряжения, для которой вы уже рисовали граничные структуры и хорошо их прочувствовали. Проблем кривых стрелочек в том, что они только обозначают смещение, но не говорят, как оно происходит и насколько оно обосновано. Обратите внимание на то, что граничные структуры всегда должны соответствовать законам химии, например, они не имеют права нарушать правило октета (во втором периоде, с остальными мы пока решили не связываться). А когда вы рисуете мезомерные стрелочки нарушить правило октета можно очень легко, потому что вы никак графически не обозначаете смещения электронной плотности.
Обязательные донор и акцептор на концах цепи сопряжения
В таких цепях граничных структур всегда две – исходная и та, которая изображает полное смещение пары от донора к акцептору. Одна из них почти всегда преобладает, и может считаться адекватным изображением реальной структуры. В это есть сразу два скрытых парадокса:
- суть мезомерии, как следует из названия (мезо – середина, ни то, ни сё) состоит в том, что реальная структура не выражается одной из граничных структур, но где-то посредине. Но в случае цепи донор-акцептор это часто не так – в этих случае реальная структра очень близка к одной граничной структуре, а влияние второй можно определить только по косвенным признакам.
- стабилизация в случае таких цепей всегда очень явно выражена несмотря на минимальное количество граничных структур
Следующий важный вопрос – какая из двух граничных структур является преобладающей. Обычно та, в которой смещение от донора к акцептору произошло полностью. Особенно это касается структур, в которых акцептором является секстетный атом – в этих случаях полное смещение электронной плотности приводит к заполнению валентной оболочки, и вторая структура уже не будет иметь секстетных атомов – это очень выгодно.
Начнём с цепей, в которых обязательные донор и акцептор соединены непосредственно. Пример 1 – донор (+М) кислород, акцептор (-М) сектетный углерод, карбокатион. Преобладает вторая структура, в которой нет секстетных атомов (посчитайте электроны сами, если не верите). Когда рисуем мезомерные стрелочку, показываем откуда смещается электронная плотность (от неподелённой пары кислорода) и куда смещается (на π-связь между кислородом и углеродом). Неверно смещать ее от атома на атом, так как в этом случае кислород стал бы секстетным. 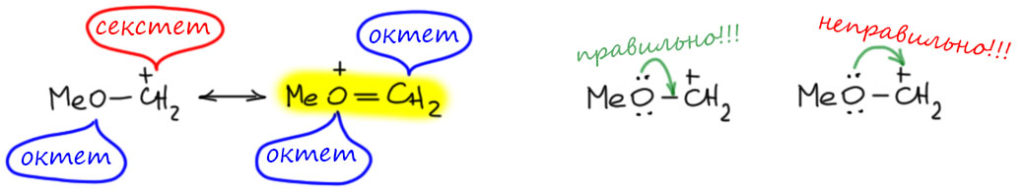
Пример 2 – донор (+М) карбанион, акцептор (-М) карбонил. В этом случае секстетных атомов нет, но преобладает всё равно вторая структура, и очень понятно почему – минус перемещается на кислород, элемент с намного большей электроотрицательностью. При использовании мезомерных стрелок, их потребуется две штуки. 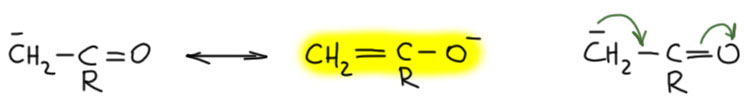
Третий пример очень похож, но донором является кислород с минусом. Узнаём карбоксилат-анион. Всё то же самое, кроме одной важной вещи – обе граничные структуры одинаковы, а значит ни одна из них не преобладает, реальная структура ровно посредине, минус в равных долях делокализован по двум атомам кислорода. 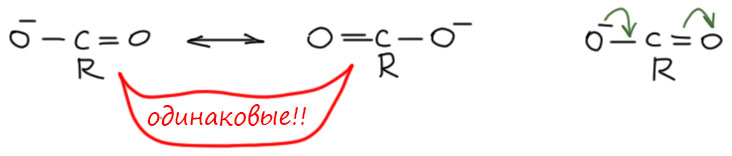
Четвёртый пример бьёт в ту же систему. Донором возьмём азот, акцептор оставим тот же. В этом случае смещение тоже происходит в сторону более электроотрицателнього атома, но разница уже не так велика, как между кислородом и углеродом в предыдущем примере. Мы не можем сказать, что вторая структура преобладает, но и одинаковыми структуры уже не являются. Они вносят разный вклад, причёс структура с отрицательным кислородом не дает большего вклада. Это легко понять, если обратить внимание на то, что во второй структуре произошло разделение зарядов, и кроме минуса на кислороде возник плюс на азоте. Общая тенденция (не правило! нет такого правила, в каждом случае надо смотреть отдельно, но именно тенденция, то есть наблюдение, что в среднем чаще бывает так, а не наоборот) состоит в том, что граничные структуры с разделёнными зарядами дестабилизированы относительно тех, у которых явного разделения зарядов нет. Поэтому мы можем сказать, что обе структуры вносят большой, но неравный вклад, и несколько преобладает исходная. Вообще это невероятно важная штука, потому что это амидный фрагмент, ключевой структурный мотив белков (что такое “структурный мотив” попробуйте догадаться сами, и вообще привыкайте, в современной науке обожают такие игривые словечки, ну и вам никто не мешает ввернуть это как-нибудь на коллоквиуме). 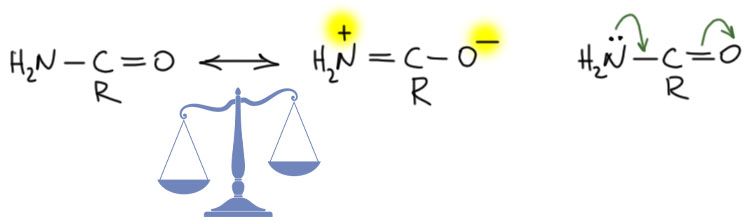
Донор и акцептор разделены удлинителем. Самый простой удлинитель – двойная C=C связь, одна или несколько, чередующихся с простыми. Посмотрим, что будет, если мы уже в разобранные примеры вставим такой удлинитель. Карбокатион на одном конце, мезомерный донор MeO на другом. Формально получаем еще одну граничную структуру, соответствующую смещению плотности в самом удлинителе. Но вес этой структуры невелик, и основной структурой будет точно так же, как в ситуации без удлинителя, последняя структура с полностью смещенной плотностью и отсуствием секстетных атомов. Обратите внимание, что мезомерные струлочки всегда рисуются сразу все и наличие промежуточной граничной структуры в явном виде не показывает. В этом месте у нас может возникнуть вопрос – а откуда мы знаем, какая структура дает какой вклад. Это фантазии, или все же реальность. Фантазии, конечно, мы же сразу сказали, что мезомерия это методический приём, позволяющий легко прикинуть то, как устроена частица (молекула) без применения более надёжных методов, а именно квантовохимических расчетов высокого уровня. Если бы мы сделали такие расчеты, мы увидели бы реальную структуру, прежде всего длины связей, которые нам сказали бы, какие из них ближе к одинарным, а какие ближе к двойным. Мы бы могли также посмотреть на распределение заряда в ионе. И так далее. Кроме этого, мы могли бы посмотреть на реакции, в которых можно предположить образование такого иона, но это данные косвенные, и их не так просто правильно интерпретировать. Поэтому согласимся пока, что рисование граничных структур и оценка их относительного вклада (веса) прото дает нам инструмент, и когда мы реально встретимся с частицами такого типа (встретимся-встретимся), мы смело начнем анализировать их структуру, свойства и реакции, и речи наши будут наполнены смыслом и разумными доводами. 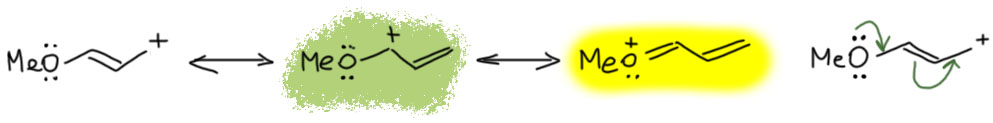
Второй пример – карбанион через удлинитель сопряженный с акцептором, карбонильной группой. Стрелочки уже не буду рисовать отдельно, нарисую на первой структуре. Вообще, эти два способа 9стрелочки и граничные структуры) смешивать не рекомендуется, но мы этого и не делаем, просто вместо отдельной картинки совместим с первой структурой. Струлочки опять нарисованы все. А граничная структура и здесь появилась промежуточная, мы имеем право ее нарисовать и можем поискать признаки ее вклада в реальных свойствах такого аниона. Но основной структурой будет тоже последняя.  Третий пример, который мы получим вставкой удлинителя в карбоксилат-ион, очень нас удивит. Я даже думаю, что некоторые особо чувствительные могут испытать головокружение и другие признаки эйфорического возбуждения. Химия умеет кружить голову неожиданностью сопоставлений, там, где этого никак не ждёшь. Вот ведь что мы получим:
Третий пример, который мы получим вставкой удлинителя в карбоксилат-ион, очень нас удивит. Я даже думаю, что некоторые особо чувствительные могут испытать головокружение и другие признаки эйфорического возбуждения. Химия умеет кружить голову неожиданностью сопоставлений, там, где этого никак не ждёшь. Вот ведь что мы получим: 
Да, это анион соединения с двумя карбонильными группами на одном атоме углерода. Такие соединения в органике знамениты, мы будем много ими заниматься, это, например, ацетилацетон, ацетоуксусный эфир и т.п. На схеме нарисован такой прототип всех этих соединений. Обычно, когда обсуждают такие соединения, говорят про то, что они обладают повышенной CH-кислотностью, от них легко оторвать протон, птому что получается анион, сопряжённй с двумя карбонильными группами. Но сейчас, зайдя с другой стороны, мы получаем даже более точную картину свойств анионов таких соединений. Оказывается, это отличная цепь сопряжения с донором и акцептором на концах. Две крайние граничные структуры преобладают в реальной струкруре таких анионов, и поскольку они очень похожи, то и вклад вносят, если не одинаковый, то сопоставимый, а вот как раз средняя форма с анионом на углероде, имеет очень малый вклад, потому что углероду сложно конкурировать аж с двумя, очень прожорливыми на электронную плотность, атомами кислорода. Углероду взять хотя бы немного минуса себе все равно, что маленькой собачке пытаться отнять кусок мяса у двух голодных тигров. Как бы саму не сожрали.
С двойными связями пока хватит, попробуем удлинитель в виде тройной связи. Будут немного непривычные структуры, но мы не должны их пугаться. Впрочем, в нашем курсе у нас мало шансов встретиться с такими, поэтому скорее для полноты картины и для общего образования, ведь химия 3 курсом не ограничивается, а в заголовке сайта написано …and beyond. Вставим тройную связь между карбанионом и карбонилом. 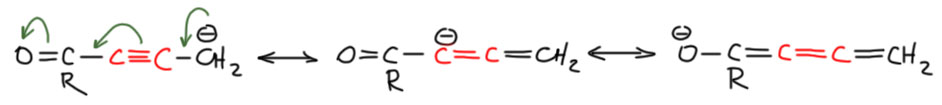 Как видим, при полном переносе пары от донора к акцептору получается довольно странная штуковина с тремя двойными связями подряд. Ничего страшного в этом нет. Кто-то вспомнит, что в алленах двойные связи не сопряжены. Точно так, поэтому черные двойные связи не сопряжены с красной, но между собой они сопряжены, а цепь сопряжения именно так и образуется. В общем, тройная связь – вполне зачетный удлинитель сопряжения.
Как видим, при полном переносе пары от донора к акцептору получается довольно странная штуковина с тремя двойными связями подряд. Ничего страшного в этом нет. Кто-то вспомнит, что в алленах двойные связи не сопряжены. Точно так, поэтому черные двойные связи не сопряжены с красной, но между собой они сопряжены, а цепь сопряжения именно так и образуется. В общем, тройная связь – вполне зачетный удлинитель сопряжения.
Вариант с бензольным кольцом в качестве удлинителя сопряжения рассмотрим на отдельной вкладке, чтобы не захламлять эту, где и так уже тесно.
Продолжение - донор, акцептор, а между ними бензольное кольцо
Мы уже разобрались с тем, что бензольные кольца тоже работают как удлинители цепей сопряжения, причем только при орто- или пара-замещении. Попробуем рисовать резонансные структуры. Опять возьмем уже разобранный случай – карбокатион и мезомерный донор, метокси-группа. Посмотрим на орто-замещение. Разрисуем граничные структуры по двум путям, короткому и длинному. Видим, что как ни рисуй, конечная структура с полным переносом пары с донора на акцептор одна и та же. Все остальные структуры реально не играют и их можно не рисовать. 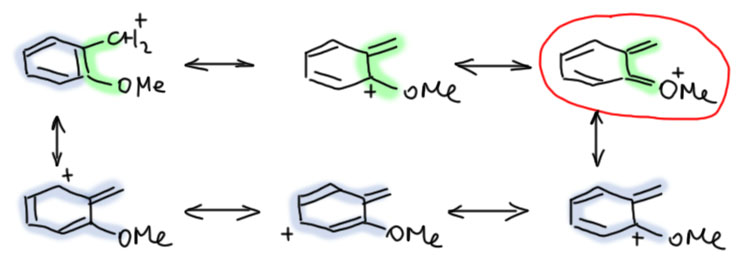
В отличие от более простых удлинителей, бензольное кольцо нельзя назвать индифферентным удлинителем, котору главное – служить сопряжению, а остальное не важно. Обратим внимание на то, что в конечной структуре колько принимает так называемую хиноидную форму. Точнее, орто-хиноидную. Хиноидных форм есть две – орто и пара-хиноидная. Вот как они выглядят – будем узнавать их везде, они часто встречаются. Когда видите хиноидную форму, можно не вспоминать правила вежливости – на хиноидные формы можно и даже нужно показывать пальцем. И громко восклицать: “О, орто-хиноидная форма!” – или – “А, пара-хиноидная форма!!” И все окружающие будут кивать головами и одобрять сделанное вами открытие, и не будут вас порицать за то, что вы отвлекли их от тягостных раздумий о судьбах Родины. Ведь хиноидные формы в химии ароматических соединений играют множество интересных ролей. 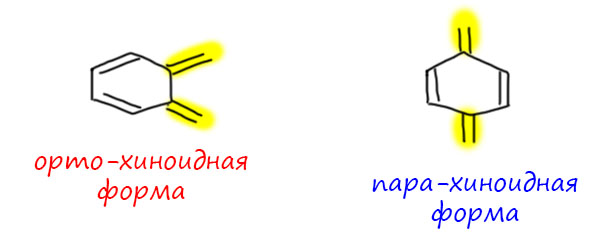
Признаком хиноидных форм являются две двойные связи, торчащие наружу, при сохранении ненасыщенности и сопряжения в остальной части кольца. Главный фокус хиноидных форм состоит в их принципиальной неароматичности, отсутствии ароматической стабилизации (подробнее мы это обсудим где-то в октябре, когда будет ароматичность). Иными словами, они невыгодны, а граничные структуры с такой формой дестабилизированы. Это приводит к тому, что в отличие от форм с более простыми удлинителями, где форма с полным переносом пары доминирует без вопросов, в этом случае это не так, и доминирует исходная форма с секстетным углеродом, а хиноидная форма только дает некоторый вклад в сопряжение и делокализацию.
Еще раз, чтобы убрать вопросы: в изображенном случае и похожих на него, в резонансе есть две основные структуры – исходная и конечная. У каждой есть одно преимущество и один недостаток. У исходной преимущество – ароматическое кольцо, недостаток – секстетный атом. У конечной преимущество – отсутствие секстетных атомов, недостаток – хиноидная форма. Результат – баланс этих преимуществ и недостатков, но ароматичность – обычно самый сильный вклад: где она лежит, там и самая лучшая граничная структура.
Ну, теперь нарисуем пара-замещение, и уже понимаем, что получится. Поскольку оба пути сопряжения одинаковые, можно не рисовать лишних граничных структур. Возьмём для разнообразия другой донор – амин, более сильный мезомерный донор, чем кислород. 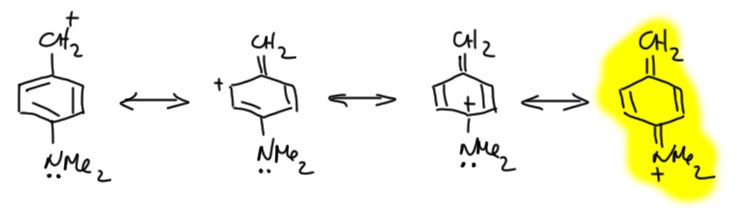
Видим, что конечная структура имеет пара-хиноидную форму. И всё уже сказанное относится и к этому случаю, но намного более сильный донор не мог не повлиять на соотношение вкладов граничных структур – в этом случае вклад даже хиноидной формы намного больше, хотя тоже не может быть доминирующим.
Попробуем донорный кислород и акцепторный карбонил в орто-расположении. Заодно еще применим мезомерные стрелочки. И когда нам захочется рисовать эти стрелочки, мы поймём, зачем мы парились рисовать промежуточные граничные структуры – мезомерные стрелочки всегда точно следуют именно всем промежуточным структурам – невозможно нарисовать сразу конечный результат смещения, использовав стрелочки. Поэтому мы ещё раз убеждаемся в то, что граничные структуры – более удобный и адекватный способ изображения сопряжения, чем стрелочки. В незнакомом случае всегда сначала рисуйте граничные структуры, и только разобравшись в очередном шаблоне, можете экономить время стрелочками. 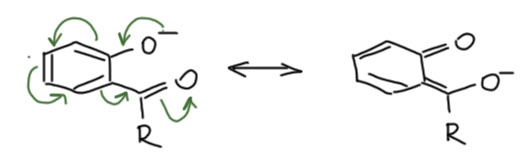
Пара-замещение, и возьмём карбанион (+М) и нитро-группу (-М). Всё то же самое, но можно запутаться в плюсах и минусах. Не забывайте убедиться, что суммарный заряд рассматривамой структуры не изменяется. В исходном два минуса и плюс, суммарно один минус. По дороге плюс с минусом могли състь друг друга, но остался бы минус. Здесь – не съели, и в конце те же два минуса и плюс. Соображения про хиноидную структуру повторять не будем. 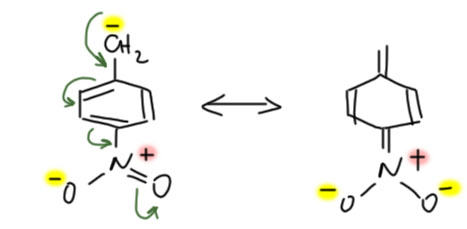
Обязательный донор или акцептор в цепи один. Совсем один.
Очень часто в цепи сопряжения на одном конце обязательный донор или акцептор, а в остальной цепи ничего нет – только кратные связи и/или ароматические кольца. К этому типу относится несколько важнейших сопряженных систем, например, аллильные анион и катион, бензильные анион и катион, енолят, нукелеофильные олефины и ацетилены и т.п. Органическая химия ломится от таких систем, они в ней везде, на каждом шагу, в каждом втором механизме, в сокровенных мыслях и чаяниях органического химика. С раннего утра и до поздней ночи органик думает о таких сопряжённых системах, что бы ещё с ними сделать или как бы от них избавиться. На следующий день всё повторяется, и так всю жизнь.
Ситуация с такими системами очень проста, но имеет один нюанс, слегка отличающий ее от того, что происходит в цепях сопряжения с настоящими донором и акцептором на концах. Этот нюанс мы легко поймём, когда разберёмся с несколькими важнейшими системами.
Итак – самое главное: если с одной стороны цепи нет обязательного донора или акцептора, то эту роль на себя берёт двойная или тройная связь или фенил, таким образом, чтобы в результате получилась нормальная цепь донор-акцептор.
Сначала разберёмся с теми случаями, когда в цепи больше ничего нет. Пример 1 – – акцептор, секстетный углерод (карбокатион), донор – двойная связь. Получается одна из священных коров органической химии – аллильный катион. Двойная связь берёт на себя функцию мезомерного донора (+М). И что мы видим – две совершенно одинаковые граничные структуры, можно даже сказать идентичные, если мы не предпримем какие-нибудь усилия по различению атомов, например, пометим один из конечных атомов углерода изотопом. Если мы так сделаем, то сможем различить (на бумаге!!! граничные структуры “существуют” только на бумаге!!!) две структуры, но в реальном аллильном катионе плюс будет поровну делокализован между двумя углеродами. Двумя! Не тремя, как явно следует из очень популярного способа изображения делокализации пунктирной скобкой. Я в очередной раз призываю не пользоваться этим ленивым, но некорректным способом. В лени ничего плохого нет, но в откровенной некорректности такого способа есть много плохого. В советской литературе, в том числе учебной, этот способ особенно распространён, можете сами в этом легко убедиться. Это такое странное следствие борьбы марксизма-ленинизма против квантовой науки – делокализация в аллильном катионе и подобных структурах была очевидна всем, но изображать ее граничными структурами или мезомерными стрелочками, которые явно говорили “о свободе воли электрона и прочей чертовщине”, было категорически невозможно, но вот кинуть стыдливый пунктирчик с плюсиком можно было попробовать, поди пойми, что это значит, может и не противоречит всесильному учению. Нам это, слава богам, больше не нужно, и давайте похороним эти пунктирчики на свалке истории. 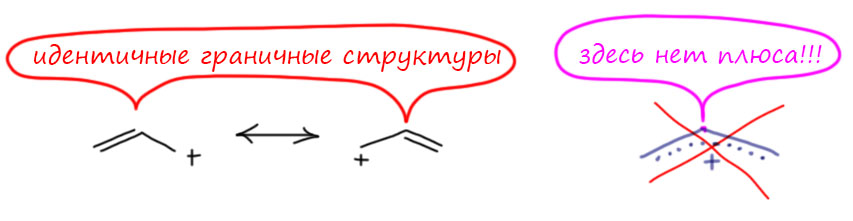
До сих пор мы рассматривали только цепи сопряжения и не касались того, как на цепь сопряжения может повлиять какой-нибудь заместитель. Речь не идёт про заместители, способные к сопряжению – такие заместители изменяют цепь сопряжения, и мы будем такие случаи анализировать, рассматривая новую цепь сопряжения. Но что будет, если заметитель попроще, индуктивный донор или акцептор? Ничего особенного, цепь сопряжения не изменится, граничные структуры останутся теми же, но их относительный вклад уже не будет равным. Вот, например, добавим пару метилов на один конец аллильного катиона. Дальше мы фактически смотрим на граничные структуры так, как будто это реальный ион или молекула, и применяем наши обычные представления о стабилизации и электронных эффектах. Тогда левая структура станет выгоднее. Что это значит? Не то, что какое-то реальное равновесие между двумя разными изомерными формами сместилось в сторону левой формы (в этой гипотезе нет ничего криминального, имено так когда-то и думали, но за последние 70 лет эта гипотеза была многократно опровергнута), а то, что реально существующий единственный делокализованный диметилаллильный катион немного ближе по структуре к левой граничной структуре, чем к правой. В конкретных реакциях с участием такого катиона это может стать очень важным фактором. До этого мы доберемся в будущем, но пока учтём, что хотя граничные структуры являются чисто “бумажными”, их относительную энергию можно неплохо прогнозировать с помощью обычных представлений о стабильности катионов или анионов. 
Попробуем аллильный анион. Здесь донор – карбанион, а двойная связь становится акцептором. Только что была донором, а тут стала акцептором. Да, так это устроено. В этом заключена очень с важная характеристика углерод-углеродных π-связей – лёгкость смещения электронной плотности в любую сторону. Это делает из двойных и тройных связей удивительно мощный фрагмент в органической химии, способный к самым разным реакциям с разными типами реагентов.
Итак, рисуем две граничные структуры, и тоже видим, что они идентичны, а значит реальная структура строго посредине, минус делокализован в равных долях. И так же не стоит рисовать пунктирчик, по той же причине – в середине нет минуса, а пунктирчик как бы говорит нам, что минус “размазан” по всей тройке атомов.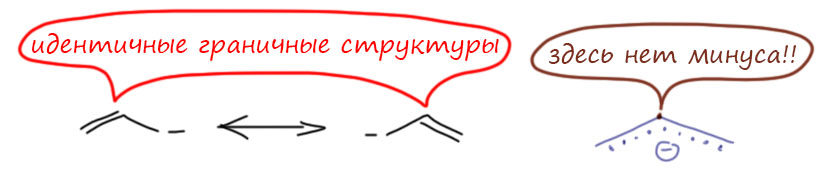
И точно так же мы можем посмотреть, что будет если в сопряженной системе есть заместители, тоже индуктивные, для простоты и сравнения возьмём те же. Результат будет противоположным – в реальной структуре будет преобладать вклад аниона с минусом на менее замещённом атоме, потому что во второй структуре индуктивные доноры дестабилизируют минус. 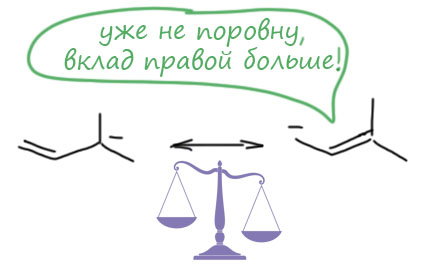 Все остальное сказанное выше про катион, справедливо и здесь, повторяться не будем. Ну, чуть-чуть – это тоже не равновесие, а именно резонанс, структуры не настоящие, а бумажные, воображаемые, анион один. Почти как в одной уважаемой религии повторяем: делокализованный аллильный анион – анион един. И делокализованный аллильный катион – катион един. Надеюсь, мы никого не оскорбили. Точно не собирались. Нам просто до чёртиков надо крепко усвоить, что мезомерия и резонанс это не равновесие, и что граничные структуры это не настоящие изомерные молекулы или ионы.
Все остальное сказанное выше про катион, справедливо и здесь, повторяться не будем. Ну, чуть-чуть – это тоже не равновесие, а именно резонанс, структуры не настоящие, а бумажные, воображаемые, анион один. Почти как в одной уважаемой религии повторяем: делокализованный аллильный анион – анион един. И делокализованный аллильный катион – катион един. Надеюсь, мы никого не оскорбили. Точно не собирались. Нам просто до чёртиков надо крепко усвоить, что мезомерия и резонанс это не равновесие, и что граничные структуры это не настоящие изомерные молекулы или ионы.
Попробуем еще одну цепь – донорный кислород с минусом сопряжён с двойной связью, которая берёт на себя роль акцептора. Рисуем вторую граничную структуру и… видим, что мы это уже рисовали, только в обратном направлении. Вторая граничная структура изображает цепь сопряжения – донор-карбанион и акцептор-карбонил. Ничего страшного в этом нет, так часто будет, ведь для изображения цепи сопряжения мы можем использовать любую из граничных структур, а поскольку в крайних структурах пара полностью перешла с донора на акцептор, то донор и акцептор закономерно меняются местами. При этом, как ни нарисуй цепь сопряжения, мы понимаем, что мы обсуждаем некую реальную молекулу или ион, и с помощью мезомерии пытаемся понять, как он устроен – ближе к одной из граничных структур или где-то посредине. В этом случае мы уже договорились, что преобладает стуктура с отрицательным кислородом, и никак этот вывод не меняется от того, что мы нарисовали граничные структуры в обратном направлении. 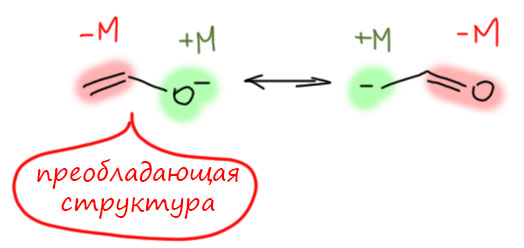
Попробуем тройную связь в качестве конечного фрагмента цепи сопряжения. Нарисуем тоже катион, такой катион называется пропаргильным и тоже часто встречается в реакциях. Нарисовав, видим, что отличие от аллильного катиона весьма велико – у аллильного катиона две идентичные граничные структуры, вклады которых в настоящую структуру одинаковы. У пропаргильного граничные структуры принципиально разные, вторая имеет структуру аллена, и плюс (секстетный атом) находится на атоме с sp-гибридизацией. Эти два фактора сильно дестабилизируют вторую граничную форму и делают ее вклад существенно меньше в настоящую структуру. Именно поэтому этот катион называют пропаргильным, а не алленильным и не пропаргильно-алленильным. 
Воодушевлённые пропаргильным катионом, попробуем посмотреть на такой же анион. Формально это похоже, но фактически разница велика – одно дело делокализовать катион, секстетный углерод. Другое – анион, мезомерный донор. Тройная связь берёт на себя роль акцептора с большей охотой, чем роль донора. B здесь тоже две граничные структуры различаются, и алленильная тоже дестабилизирована алленовым типом соседства двух двойных связей, но при этом анион гораздо комфортнее себя чувствует на sp-гибридном углероде. Поэтому в данном случае у каждой граничной струкры есть и достоинстваи недостатки, они более-менее компенсируют друг друга, и поэтому структура такого аниона не близка одной из граничных форм, а находится где-то посредине. Поэтому в литературе такие анионы часто называют двойным эпитетом пропаргильно-алленильные. Мы скоро встретимся с этими анионами в пропаргильно-алленильной перегруппировке Фаворского. 
Ну и последний важный фрагмент, приспосабливающийся к потребностям цепи сопряжения – фенил. Поскольку это очень важгая вещь, вынесем ее на отдельную вкладку.
Обязательный донор или акцептор в цепи один. Но цепь удлиннённая.
Разобрались с очень распространённой ситуацией, когда единственный обязательный донор или акцептор сидит на одном конце цепи, в которой присутствует только ещё одна двойная связь (винильная группа) или тройная связь (этинильная группа) или ароматическое кольцо (фенил). Теперь уже обычным образом вставим в серединку удлинитель. Ограничимся двойными связями. С тройными и промежуточными бензольными кольцами справитесь сами, но нам такие молекулы встретятся вряд ли. Возьмём аллильный катион и вставим одну двойную связь. Конечная двойная связь примет обязанности донора, а двойная связь – удлинитель свяжет эти два фрагмента в цепь сопряжения. Всё будет так же, как в случае, когда такой удлинитель связывает обязательные донор и акцептор, но с одним отличием – в такой цепи придётся рисовать и рассматривать и промежуточные граничные структуры, и они могут быть вполне хороши и давать значительный вклад в настоящую структуру. Вот пентадиенильный катион имеет три граничные структуры, две из них одинаковы, но и средняя тоже выглядит неплохо. Вывод простой – в таком катионе плюс будет делокализован на трёх углеродах, первом, третьем, пятом. То же самое будет с анионом. 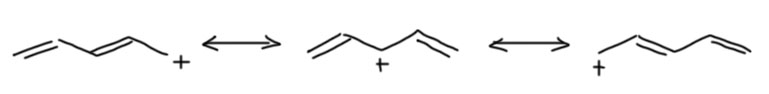
То же самое будет и с другими донорами и акцепторами. Вот случай с донором – кислородом с минусом. Опять три структуры, из них одна, первая, исходная имеет минус на кислороде, две других – на углеродах. Более короткую цепь сопряжения этого типа мы уже рассмотрели, и договорились, что минусу комфортнее на кислороде, и что именно эта структура преобладает. Но две другие дают небольшой, но весьма заметный вклад, причём мы можем осторожно предположить, что средняя может давать больший вклад, чем крайняя. Почему? Мы уже несколько раз подмечали, что хотя граничные структуры это не настоящие соединения, и к ним нельзя применять понятие стабильности в прямом смысле этого слова, но никто не мешает оценивать их относительную (относительно других граничных структур) выгодность, а эта энергия (выгодность всегда оценивается в терминах энергии, но только в качественном смысле, никаких цифр!!) дает нам возможность оценивать относительный вклад данной граничной структуры – чем структура выгоднее, тем вклад больше. Извините, что я это уже в сотый раз повторяю, но это важно. Мы договорились, что важные вещи будем часто повторять. Так вот, средняя граничная структура должна иметь более весомый вклад, чем последняя, потому что минус дополнительно стабилизируется акцепторным индуктивным эффектом карбонила. Мы имеем право рассматривать дополнительное влияние индуктивного эффекта на относительную выгодность граничных структур, потому что это эффект другой природы, не связанной с сопряжением. А вот мезомерию, резонанс, сопряжение мы не имеем право дополнительно использовать (типа аргумента, что в средней минус сопряжен с двумя двойными связями – это плохой аргумент) потому что мы и так именно это и анализируем, и если попробуем сюда же затащить еще сопряжения, получим гнилой логический тупик под названием рекурсия – это когда нечто пытаются аргументировать им же самим (типа: некий вождь велик, потому что вождь не может не быть велик, и не может быть невелик, и кто велик, если не вождь, а вождь только этот, потому что не этот – не вождь, и так далее) 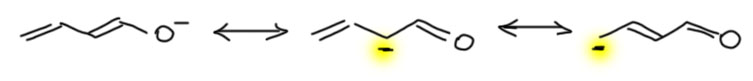
А это как-то проявляется в реальных свойствах таких ионов? Да, безусловно, и хотя мы не будем в нашем курсе явно упоминать такие анионы (это еноляты, полученные из непредельных карбонильных соединений), но в литературе вы такие вещи можете встретить, и увидеть, как в реальных реакциях проявляют себя такие удлиннённые еноляты – они действительно дают продукты, соотвествующие такой картине делокализации отрицательного заряда.
Ещё один пример – обязательный акцептор в виде карбонила, двойная связь на конце цепи и двойная связь – удлинитель. Видим нечто очень похожее на уже рассмотренный удлиннённый аллильный катион. Ну и знаем по уже рассмотренному карбонилу с одной сопряжённой двойной связью, что в настоящей структуре преобладает вклад первой граничной структуры, но две другие, сравнимые по вкладам дают очень важный вклад в свойства и реакции таких соединений. 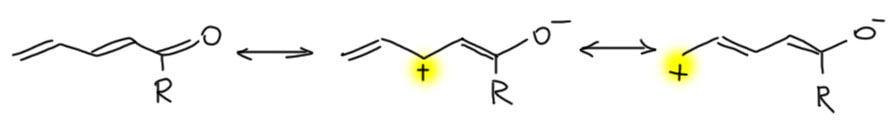
Дальше всё будет в том же духе. Можете сами поиграться, только имейте в виду, что чем длиннее цепь сопряжения, тем меньше шансов с ней встретиться, потому что длинные цепи сопряжения имеют проблемы, в частности, с энтропией, и их нужно дополнительно стабилизировать, например, включая в жесткие структуры, состоящие из сочленённых циклов.
Обязательный донор или акцептор, один, на бензольном кольце
Фенил точно так же берёт на себя роль донора или акцептора, если настоящего донора или акцептора в сети нет, а имеющийся один. Посмотим на примеры таких систем, тем более что среди них много очень важных. Специфика фенила в качестве такого конечного элемента цепи сопряжения в том, что он даёт не одну, а три практически равноценных граничных структуры.
Начнём с катиона, сопряжённого с фенилом – бензильного катиона. Фенил становится донором, и мы рисуем три граничные структуры, в которых электронная плотность из фенила затыкает дыру на снешнем секстетном атоме углерода. Секстетность от этого никуда не девается, она частично распределяется между тремя атомами углерода внутри кольца, находящихся в орто и пара-положениях относительно боковой цепи. 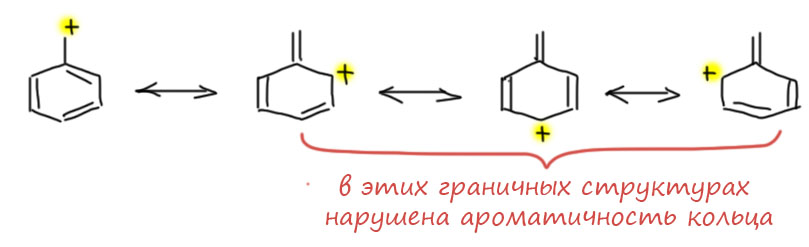
Итого мы видим четыре граничные структуры. Можем сравнить это с аллильным катионом, в котором структуры только две, но одинаковые. В бензильном катионе граничные структуры далеко не одинаковые. Те три, в которых плюс внутри кольца, на первый взгляд очень хороши, но на второй – мы замечаем, что кольцо в них не является ароматическим, оно теряет нужную для ароматичности циклическую делокализацию с правильным числом электронов. Пока серьёзнее не будем в этом разбираться, достаточно того, что мы и так, невооружённым глазом видим, что эти структуры что-то не очень похожи на ароматические. Напомню в очередной раз, что граничные структуры это не настоящие соединения, и мы не оцениваем их настоящую стабильность – граничные структуры это воображаемые объекты, но мы пришли к выводу, что мы можем применять к ним обычные способы качественной оценки стабильности молекул и ионов, и это даёт нам представление об относительном вкладе, вносимый этими структурами в настоящую структуру. Итак, мы приходим к выводу, что в бензильном катионе преобладающей является все же исходная структура с внешним плюсом и нетронутым ароматическим кольцом, а три дополнительные структуры участвуют в общей стабилизации и делокализации, но достаточно деликатно. Об этом говорят и расчетная структура бензильного катиона (основная доля положительного заряда остается на внешнем углероде), и реакции, в которых принимает участие такой катион – вы почти никогда не встретите реакций, в которых какой-то реагент цепляется за заряд в кольце. Конечно, это не так просто, и там работают и другие факторы, но всё же. Нам это точно стоит понимать – если аллильный катион в реакциях проявляет делокализацию, в частности, через образование двух продуктов (такие реакции называют аллильными перегруппировками), то с реакциями с участием бензильного катиона ничего подобного не наблюдается – в реакциях с участием бензильного катиона продукты практически всегда будут образовываться с участием бензильного положения, и никаких перегруппировок мы не увидим. У бензильного катиона есть другая интересная возможность – превращение в изомерный и полностью ароматический, а значит точно более стабильный катион циклогептатриенилия (тропилия), но это отдельная тема.
И еще один интересный вопрос можно задать, он просто напрашивается – какой катион стабильнее, аллильный или бензильный. Увы, на этот вопрос почти невозможно ответить хоть сколько-нибудь убедительно (с помощью квантовохимических расчетов, безусловно, можно посчитать энергетику модельной реакции аллильный катион плюс толуол = пропен плюс бензильный катион, и фактически на этот вопрос ответить, но как это использовать в реальной жизни решительно непонятно). В химии сравнивают аналогичные объекты, как-то связанные друг с другом – реакциями, замещением, систематическими структурными изменениями. Аллильный катион в этом смысле слишком далёк от бензильного, это производные разных рядов, структурно весьма далёкие. Более того, это и не нужно, потому что вы никогда не встретитесь с реакцией в которой такая разница в стабильности могла бы как-то проявиться. Не забывайте, что химия – наука экспериментальная, а не умозрительная, и вопросы, не имеющие отношения к наблюдаемым явлениям или реакциям, в химии не пользуются популярностью, хотя заданы, безусловно, имеют право быть.
Бензильный анион в этом смысле ничем не отличается от бензильного катиона. Можно было бы даже не рисовать. Те же четыре граничные структуры, то же соображение о том, что преобладает та, у которой минус снаружи. И всё остальное, что сказано про бензильный катион почти без изменения справедливы и для аниона. 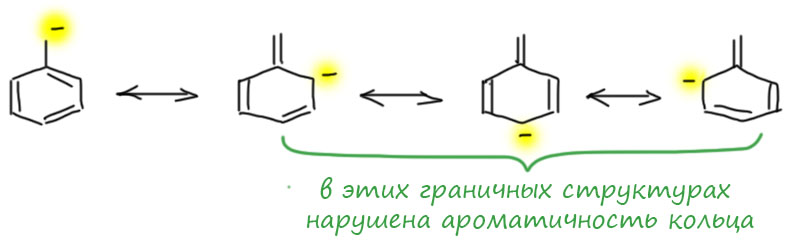
Что-то мы совсем перестали мезомерные стрелочки рисовать. Вспомним эту добродетель, нарисуем для бензильного катиона и аниона. Вот катион. Стрелочка потребуется не одна, а три. Со стрелочками ситуация сильно отличается от граничных структур в том, что когда нарисованы все необходимые стрелочки, они показывают (все сразу) полное смещение электронной плотности в цепи сопряжения – и в случае фенильного кольца оно ведет себя как цепь из 6 атомов углерода. По сути так оно и есть, но нам нужны в этом случае и во многих других и промежуточные граничные структуры, а не только исходная и последняя. Поэтому, когда мы рисуем стрелки, мы фактически рисуем три этапа смещения плотности, соответствующие граничным структурам. Вот так – я разметил последовательность стрелочек, и соответствующие граничные структуры: как видим, соотвествие полное, и так и долно быть. Именно поэтому граничные структры нужно рисовать всегда и в первую очередь, и только потом, при желании, разметить то же самое стрелочками. Потом для типичных структур и цепей стрелочки запомнятся, и можно будет рисовать только их. Но, особенно в новых случаях, никогда не начинайте со стрелочек, всегда сначала нарисуйте граничные структуры, рассмотрите их, убедитесь в том, что они хороши и разумны, ничего не нарушают.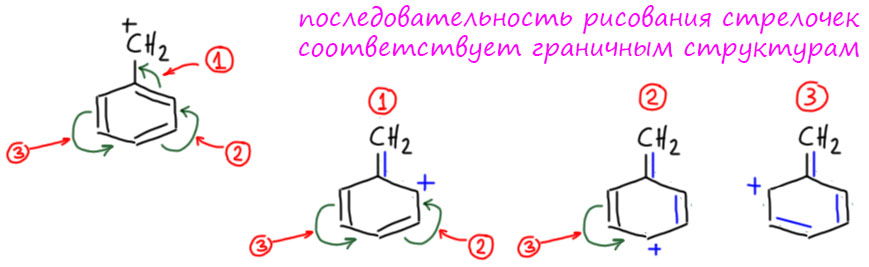
То же для аниона. Стрелочки всегда идут от донора к акцептору, поэтому направление другое. И в этом случае немного сложнее, стрелочки не прямо преобразуются в граничные структуры. Стрелочки рисуют всегда все, как показано слева зелеными стрелочками. Но в отличие от ситуации с акцептором – секстетным атомом мы не можем каждую стрелочку мысленно “заморозить” и получить все промежуточные структуры просто потому, что мешает следующая по очереди связь, создавая избыток плотности и нарушение октета. Впрочем, очень простой приём позволяет и в этом случае примирить стрелочки и граничные структуры – мы немного переставляем очередную стрелочку, заставляя ее тыкать не в следующее межатомное пространство, пока занятое, но на атом углерода, обозначая перемещение пары на атом. Кстати, самая последняя стрелочка в любом случае рисуется именно так. 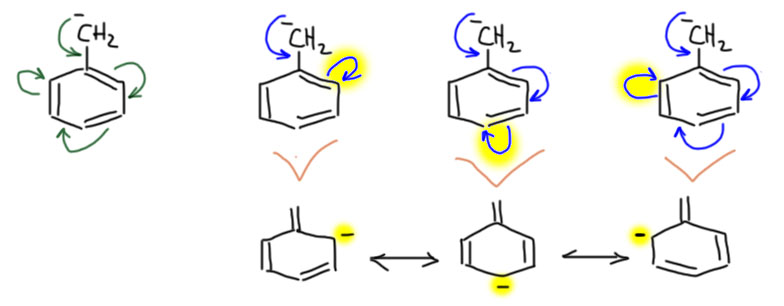
Нужно ли обязательно уметь рисовать стрелочки? Нет, не обязательно, хотя этот способ очень популярен, но далеко не у всех. Среди химиков всегда были партии “стрелочников” и “гранично-структурщиков”, когда-то эти партии даже выясняли отношения, и каждая числила в своих рядах несколько очень мощных учёных, приблизительно как феодальные армии двух королей, задумавших выяснить отношения красивым сражением, числили каждая на своей стороне несколько очень важных герцогов, маркизов и графов, причем каждый из этих спесивых сеньоров мог в следующем сражении вполне оказаться на другой стороне. В общем, так же кончились и споры про стрелочки и резонанс. Стрелочки остаются вспомогательным способом, основной – граничные структуры.
Посмотрим еще на одну важную вещь, которую нужно хорошо понимать. Ароматическое кольцо при рисовании и граничных структур и стрелочек очень похоже на обычную цепь сопряжения, у которой вместо ароматического кольца двойная связь, отделённая от обязательного донора или акцептора удлинителем в виде двух двойных связей. Вот это в линейном варианте, и я специально согну цепь колечком, чтобы совсем было похоже, хотя это не две совсем одинаковых цепи – в одной одна связь цис-, в другой все транс, но сейчас это совсем не важно. 
Видно, что формально очень похоже. Но отличие принципиально. Фенил, бензольное кольцо – целостная структура, двойные связи в ней рисуются для удобства и на уровне рисования мезомерии это отлично работает, но важно то, что мы в кольце не знаем, где конец, и где начало – кольцо симметрично, и электронная плотность в нем распределена равномерно и симметрично. Поэтому мы можем для обозначения мезомерии проходить кольцо, представляя его обычной цепью, что в одном направлении, что в друом, и эти направления заведомо идентичны, и мы рисуем одно, а не два именно потому, что так хорошо это значем, что не имеем необходимости изображать эту симметричность ещё раз. И поэтому, в разомкнутом случае все граничные струкры приблизительно равнозначны и дают сравнимые вклады. А в ароматическом случае, мы знаем, что граничные струкруры, нарушающие ароматичность, менее выгодны и вносят меньший вклад.
Обязательный донор или акцептор как-то оказался внутри цепи сопряжения
Мы договорились, что лучшие епи сопряжения те, в которых обязательный донор или акцептор находятся на концах цепи сопряжения. И что бывают цепи, в которых обязательный донор или акцептор только один, и тогда второй конец цепи берёт на себя отсутствующую функцию.
Но раз цепи сопряжения понятно из чего состоят, почему бы уже готовую цепь не взять, и не продолжить? Попробуем? Попробуем…
Начнем с цепей, в которых донор или акцептор – секстетный атом или карбанион. Добавим, например, на короткую цепь секстетный атом – акцептор / метокси – донор винильную группу, или, что то же самое, добавим на аллильный катион метокси-группу. Результат сильно напоминает простую комбинацию двух более коротких цепей сопряжения, и собственно её и является. Но интересно, что если нарисовать три граничные структуры, то одна из них, которая на схеме ниже сидит в середине, является нормальной цепью сопряжения с обязательным акцептором, двойной связью C=O (положительный заряд на кислороде никак не изменяет акцепторную природу такой связи, но даже ее усиливает) и просто двойной связью, которая, как мы уже привыкли, берёт на себя роль донора, и мы получаем самое обычное смещение пары от донора к акцептору, что собственно и выражено третьей граничной структурой. И третья граничная структура тоже представляет нормальную цепь сопряжения между донором (MeO) и акцептором (секстетным углеродом) с промежуточным удлинителем. В этом месте нас может озарить смутная догадка – а не может ли быть так, что почти любую цепь сопряжения можно представить так, чтобы донор и акцептор оказались на концах цепи? Ведь из любой граничной структуры можно получить все остальные, и поэтому любая граничная структура является полномочным представителем всего набора структур, а значит и настоящей структуры. Из любой мы нарисуем все и оценим их вклады, и это никак не изменится от того, с какой из структур мы начали. 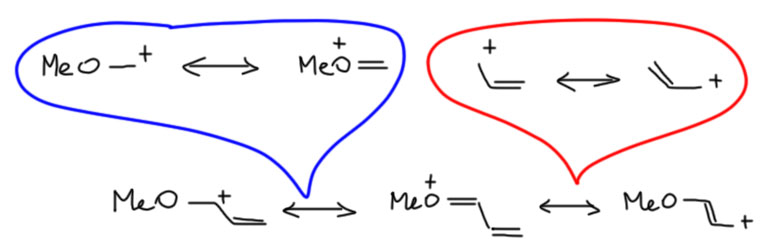
Попробуем ещё что-нибудь. Например, возьмём цепь карбанион – донор / карбонил – акцептор, и достроим его слева винильной группой. Получим карбанион, одновременно сопряжённый с обязательны акцептором и необязательным. Это отразят три резонансные структуры, попарно соответствующие двум более простым цепям. И – действительно видим, что крайние граничные структуры тоже соответствуют ситуации, когда донор и акцептор оказались на концах цепи, то есть получаем нормальную цепь сопряжения и приступаем к оценке вклада каждой из трёх граничных структур в настоящую структуру. 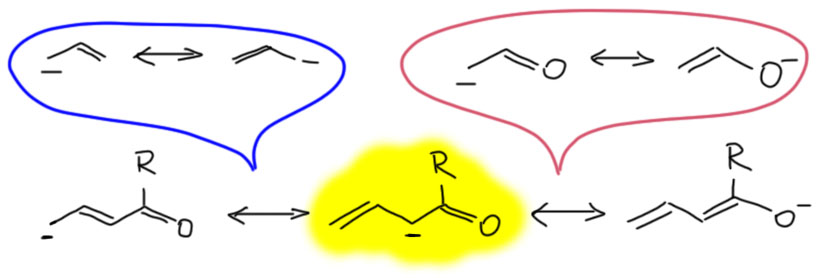
Как видим, граничные структуры обладают одним удивительным свойством – они являются чисто абстрактным, умозрительным объектом, не существующим в реальности, выдуманным для того, чтобы помогать нам качественно оценивать реальную структуру одной какой-то молекулы или иона. Но при этом они совершенно реально связывают совершенно реальные явления. Например, последний анион, один единственный, но представленный тремя граничными структурами, можно реально получить самыми разными способами из самых разных исходных. И действовать мы будем так, как будто собираемся получить одну из граничных структур, но получать каждый раз будем одно и то же – один единственный сопряжённый анион. Хотя пока мы не знаем нужной для этого химии, просто намекнем, что анионы на углероде чаще всего получают действием сильных оснований, отрывающих протон от положений с повышенной кислотностью (а такие положения чаще всего и соответствуют мезомерно-стабилизированным анионам). А анионы на кислороду можно получить, например, расщеплением сложных эфиров соответствующих спиртов. Например, так (LDA – это как раз очень сильное основание, потом разберёмся, что это). 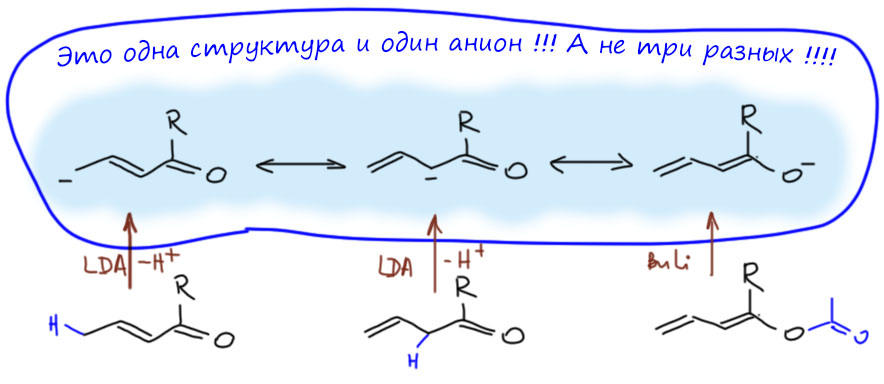
Вот это фокус! В одну реку нельзя войти дважды, а в одну структуру можно, и даже трижды, а может быть и чёрт знает сколькожды, если мы правильно поняли, как устроено сопряжение.
Цепь сопряжения не содержит ни одного обязательного донора или акцептора
Такое бывает и довольно часто. Самый простой пример – 1,3-бутадиен или вообще любой сопряжённый диен. Или стирол (фенилэтилен) или винилацетилен. Начнём с бутадиена. Итак, в молекуле нет обязательного донора или акцептора. Есть двойные связи, которые должны теперь сами решить, кем они будут. Как же им поступить? Монетку бросить, наверное, придётся. Вот, бросили, решили, что левая будет донором, а правая – акцептором. А в другой раз выпадет наоборот: левая акцептором, а правая донором. Вот и получаются граничные структуры, три штуки, исходная и две взаимно симметричные с разделением зарядов. Какая из них преобладает (даёт максимальный вклад) в реальной. Совершенно очевидно, что исходная. Прсто представьте себе, что преобладала бы левая. А что бы тогда сказала правая – ведь она точно такая же. Нет, так не может быть. ну тогда пусть обе преобладают, левая и правая. Так, и что тогда будет с зарядами, ведь они в этих граничных структурах велики, что, так бы и шарахались с одной стороны на другую. Ну это же электронная плотность, квантовая штука, от нее можно ожидать и не такого, может это такая суперпозиция, одновременно и то, и то. Чёрт его знает, что можно возразить на такой изощрённый аргумент, но в таких случаях, когда чувствуете, что еще немного и вы будуте разбиты, и что пол уже уходит из под ног, а из головы никак не хотят лезть острые иголки, как у одного знаменитого мудреца, вспоминайте, что химия – наука экспериментальная, и любое blah-blah в химии должно не противоречить экспериментальным данным. Эксперимент в таких случаях – это собственно молекулярная структура, которая для таких важных молекул известна давно и с высокой точностью. И если бы граничные структуры с разделением зарядов преобладали бы, средняя связь была бы очень похожа на двойную, была бы короче простой. Но это не так – средняя связь длиннее, и сильно, и вообще не очень сильно отличается от обычной простой. Подробнее мы это обсудим в теме диены, а сейчас просто скажем, что когда обязательного донора или акцептора нет, нет и существенного смещения электронной плотности в одну из сторон, да и не может быть, потому что молекула просто не может решить в какую. 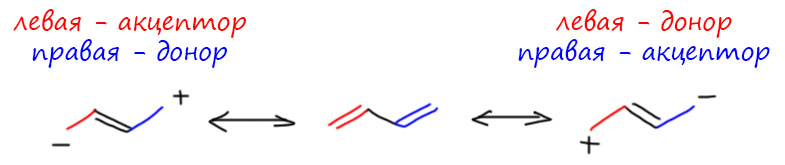
Если мы вспомним про мезомерные стрелочки, то и тут есть проблема. Когда в цепи есть обязательный донор или акцептор, направление стрелочек задано и проблем нет. А когда нет, стрелочки можно рисовать в любую сторону, и информации они несут немного.
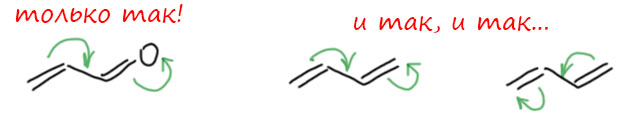
Мы могли бы ещё рассмотреть стирол, но будет такая же ситуация, но граничных структур придётся нарисовать 6 штук – три на фенил-донор, три – на фенил-акцептор. И все эти граничные структуры в общем почти никакого вклада не дают, а реальная электронная структура очень близка к исходной граничной структуре, которую ни за какие деньги не отличить от того, как мы вообще рисуем стирол, даже и не подозревая, что мы что-то забыли.
Но тогда возникает вопрос – а зачем нам вообще как-то рассматривать сопряжение в таких молекулах? Чтобы знать, что оно слабо, но оно есть и может в чём-то важном проявиться. Мы ещё вернёмся к этой интриге в теме диены.
Следующие две вкладки факультативны и только для особо любопытных и морально устойчивых.
Отдельная связь как цепь сопряжения
Химия – не математика, она не любит слишком свободного полёта фантазии и слишком широких обобщений. Всё долно быть в рамках. Свободный полёт фантазии в химии ограничивает экспериментальный характер этой науки – только куда-нибудь разлетишься, как реальность кричит вам вослед: “Алё, куда намылился, вот данные экспериментальные, структуры, реакции, они не хотят туда лететь, давай, возвращайся…” Это вам не теоретическая физика. Скучно. Но зато крыша будет всегда на месте и никуда не поедет.
Мы сказали, что π-сопряжение это взаимодействие связей или атомов, имеющих p-орбитали, из чего вроде бы ясно следует, что сами связи нельзя рассматривать как маленькие, но полноценные цепи сопряжения. И так это и должно быть, это правильно. Но есть одна лазейка. Летать нам не разрешают, это мы поняли, но тихонько проползти скрытной лазейкой запретить трудно. Попробуем.
Вот двойная связь в карбонильной группе. Попробуем переместить пару от углерода к кислороду и нарисовать граничную структуру, соответствующую этому перемещению. 
И как только мы это сделаем, сразу с удивлением обнаружим, что эта граничная структура полностью удовлетворяет тому, как мы понимаем цепи сопряжения, причём самые лучшие, с обязательными донором и акцептором на концах. Более того, мы тут же поймём, что очень похожую цепь сопряжения мы уже рассматривали в самом начале:
И мы тогда договорились, что в таких цепях, где есть акцепторный секстетный атом и есть возможность в результате смещения пары от секстета избавиться, преобладать будет граничная структура без секстетного атома. Что в применении к карбонильной группе означает, что структура карбонильной группы очень адекватно отображается… самой обычной структурой с двойной связью C=O. В применении к карбонильной группе, а не тому катиону с метокси-группой, это даже еще более верно, ведь донор здесь, анионный кислород, ещё более мощен, чем нейтральный кислород в метокси-группе, и он совсем вряд ли потерпить секстетный карбокатион рядом с собой.
Но совсем забывать про эту вторую граничную структуру не стоит. В обычной карбонильной группе она вряд ли даёт заметный вклад: часто упоминаемую идею о том, что так можно объяснить поляризацию карбонильной группы и частичные заряды, чем мы будем очень активно пользоваться, вряд ли можно считать хорошо обоснованной – та же поляризация гораздо проще объясняется полярной природой ковалентной связи, а городить два объяснения в одну сторону наука со времен преподобного Вильгельма Оккамского (1288-1347), вооружившего нас знаменитой бритвой, категорически не советует – говорит, достаточно одного и более простого. Тем не менее, эта структура ещё может всплыть в тех случаях, когда карбокатион может для чего-то понадобиться серьёзного, например, для образования ароматической системы. Вернёмся к этому однажды.
Ну и для обычной двойной связи мы тоже можем нарисовать вторую граничную структуру, представляющую собой минимальную цепь сопряжения секстетный карбокатион – карбанион. Здесь нас ждёт еще та же проблема, как в бутадиене – поскольку связь симметрична, мы не знаем, в какую сторону смещать пару, а значит должны нарисовать обе граничные структуры. И такие граничные структуры не имеют шансов как-то проявляться в обычных двойных связях, но, возможно, тоже однажды вылезет в каких-то более сложных случаях, где одна из них по каким-то причинам получит преимущество. Но точно так же, как в случае с карбонилом, этот резонанс не нужен для объяснения поляризации двойной связи под действием индуктивных доноров или акцепторов – вполне достаточно индуктивного эффекта. 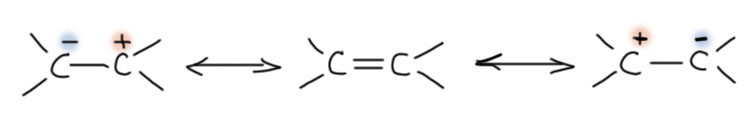
Немного про кросс-сопряжение (спойлер: это редкостная гадость, натощак не принимать)
Прежде всего замечу, что термин “кросс-сопряжение” не является общепринятым. ИЮПАК признаёт это понятие, но даёт ему такое описание и определение, из которого понять вообще ничего нельзя. Впрочем, эта замечательная международная организация и в обычном сопряжении и мезомерном эффекте умудрилась запутаться так, что единственный вопрос, который возникает к авторам соответствующих статей в так называемой Золотой книге ИЮПАК (справочнику по терминологии физической органичесской химии), это не злоупотребили ли они чем-нибудь запрещённым в Российской федерации. В любом случае, если решите прочитать этот раздел здесь (это строго факультативно и только для любопытных), не злоупотребляйте и не огорошивайте добрых людей, в том числе ваших замечательных преподавателей, речами знаменитого Вовочки из скабрезных советских анекдотов, типа: “А вы тут сидите и не знаете, что blah-blah-blah … кросс-сопряжением называется blah-blah-blah!” Добрые и очень знающие люди имеют право ничего не знать про кросс-сопряжение, или даже знать, но категорически не принимать это понятие. Это спорное понятие. В программе нашей его точно нет, и если бы его кто-то решил ввести, я бы стал брюзжать и советовать этого не делать. В нём легко запутаться, и тогда может пострадать действительно важная вещь – настоящее π-сопряжение в цепях сопряжения. Но если стало любопытно, прочитайте, и судите сами, насколько это убедительно и может быть важно. На мой взгляд, если органическая химия вам важна, и вы хотите в ней разобраться, нелишне познакомиться с этим понятием.
Кросс-сопряжение – это тоже сопряжение. Сопряжение (π-сопряжение), напоминаю, это взаимодействие π-орбиталей двойных или тройных связей и p-орбиталей отдельных атомов. В этом смысле сопряжение будет всегда, когда такие атомы или связи оказываются рядом вне зависимости от того, как они расположены. Кстати, если полезете в Википедию, там написано, что в кросс-сопряжении части не сопряжены. Это – совершенно полная фигня, и любой может в этом убедиться, просто повертев такие молекулы в руках и подумав, как они устроены. Да и спрашивается, если не сопряжены, то о чём вообще речь. Лишний раз убеждаюсь, что Википедия – очень ненадёжный источник по проблемам химии.
Но мы уже выделили самый важный вид π-сопряжения – цепь сопряжения. Цепь сопряжения не имеет разветвлений, и даже когда заходит в ароматическое кольцо, сохраняет это свойство – неразветвлённость. Как это применить к кольцу, мы тоже, надеюсь, поняли.
И еще мы поняли, что цепи сопряжения обычно имеют донор и акцептор, обязательный или ставший таковым по необходимости, на концах цепи. И что даже в случае продолжения цепи после обязательного донора или акцептора в таких случаях, ее можно представить хотя бы одной граничной структурой с донором или акцептором на концах.
И такие цепи сопряжения составляют абсолютное большинство реальных π-сопряженных систем. И образование таких сопряжённых систем почти всегда приводит к стабилизации молекулы или иона, и к делокализации электронной плотности. Почему так происходит, и почему мы так трясём этим донором-акцептором как главным условием хорошего сопряжения? Это имеет довольно простое квантово-химическое обоснование. Длаеко мы не полезем, но основу этого рассуждения поймём легко. Взаимодействие донор-акцептор в квантовых понятиях всегда можно свести к обобщению взаимодействия двух орбиталей: заполненной орбитали донора и вакантной орбитали акцептора (надеюсь, такое соответствие очевидно – донором может быть тот, у кого что-то есть, а акцептором тот, у кого есть пустой карман, в который можно положить то, что дал донор). Квантовая наука говорит нам, что при взаимодействии двух орбиталей получаются две новые, одна ниже, другая выше, так, чтобы в первом приближении суммарная энергия не изменилась. И тогда два электрона попадут на нижележащую орбиталь, и это очевидно приводит к более стабильному состоянию. 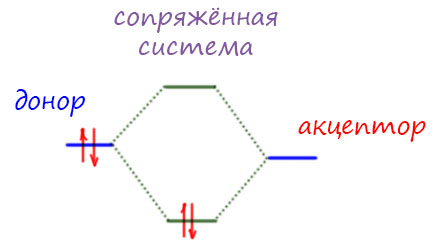
А теперь представим себе более сложную сопряжённую систему, в которой нельзя выделить одну цепь с донором и акцептором. Такую ситуацию можно увидеть довольно часто. Такой немного парадоксальный пример – угольная кислота. Молекулу угольной кислоты легко представить, как единую сопряжённую систему, но в ней невозможно выделить одну цепь сопряжения. В то же время молекулу можно мысленно наспилить на обычную цепь сопряжения – акцепторный карбонил – донорный гидроксил, и добавить к этой цепи еще одну донорную гидроксильную группу. Обратим внимание на то, что это можно сделать двумя способами, что и отображается что граничными структурами, что мезомерными стрелочками. Но – нет способа так нарисовать что граничные структуры, что стрелочки, так чтобы были одновременно задействованы все части общей сопряжённой системы! Мы всегда фактически делим общую систему на две части, хотя комбинируем их по-разному. Это такой визуальный признак того, что мы имеем дело с кросс-сопряжением – сопряжением двух (или более) более-менее самостоятельных π-систем. Это очень скользкое место. Если вы дочитали до него, и у вас возникли отрицательные эмоции – это хорошо, это значит, что вы не просто читаете, но и думаете и пытаетесь понять, не кормят ли вас туфтой. Возможно, кормят. Поэтому продумайте этот момент сами – видите ли вы действительно явное и действительно довольно драматическое отличие этой ситуации от нормальных цепей сопряжения. Если видите, то попробуйте сами поразмыслить о том, в чём это отличие состоит. 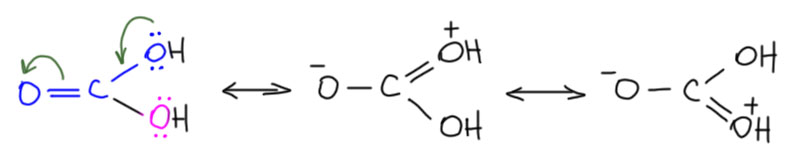
А если мы смирились с возможностью кросс-сопряжения, тогда подумаем, к чему такое сопряжение может привести. Есть одна проблема – второй донорной группе особенно некуда девать свою пару – в той цепи, к которой она присоединилась все валентные возможности уже задействованы, все атомы имеют октеты, и никому эта дополнительная плотность не нужна. Возникает ситуация взаимодействия двух фрагментов с заполненными оболочками. Это хорошо известная ситуация, упрощённо она выражается такой схемой взаимодействия верхних заполненных орбиталей. Вроде всё то же самое, что при взаимодействии донор-акцептор, но на двух новых орбиталях не два, а четыре электрона. В первом приближении мы можем решить, что энергия вообще не изменилась – одна пара пошла вниз (стабилизировалась), вторая вверх – дестабилизировалась. Но есть один вклад, который на такой простой схеме не учитывается – его можно представить просто как дополнительное отталкивание электронов, оказавшихся на одной сопряжённой системе, а значит, поневоле в одной области пространства. А можно использовать красивый термин электронная корреляция. Но смысл один – этот дополнительный вклад всегда дестабилизирующий. И такое сопряжение – дестабилизирующее. Сопряжение может стать источником дестабилизации. А зачем оно тогда? А куда от него деться – молекула образовалась, образовались нормальные химические связи, все валентные возможности каждого атома задействованы по полной. И в целом молекула такая стабильна и реально существует, ей соответствует минимум энергии на энергетической поверхности. Но она одновременно и дестабилизирована, минимум не так глубок, и если встать на дно этого минимума (квантовая наука предлагает нам для этого удобную полочку нулевого уровня колебательной энергии), то можно заглянуть через бугорок энергии активации и увидеть, что там – за бугром, а там распад на какие-то более устойчивые куски. Знаем же мы, что угольная кислота весьма легко распадается на диоксид углерода и воду. Это, конечно, не самый безупречный аргумент, ведь эти два простых оксида обладают настолько большой отрицательной свободной энергией образования, что относительно них почти любая молекула окажется дестабилизированной. С другой стороны, такая нестабильность относительно ангидрида и воды довольно необычна – все окружающие кислородные кислоты наоборот весьма стабильны относительно соответствующих ангидридов и воды (кремниевую не берём, там что ангидрид, что кислота образуют чрезвычайно устойчивые полимерные твёрдые фазы и равновесие нельзя прочитать относительно индивидуальных мономерных соединений). Когда доберёмся до карбоновых кислот, ещё раз обсудим дестабилизирующее кросс-сопряжение на примере карбонат-аниона и енолятов производных карбоновых кислот. 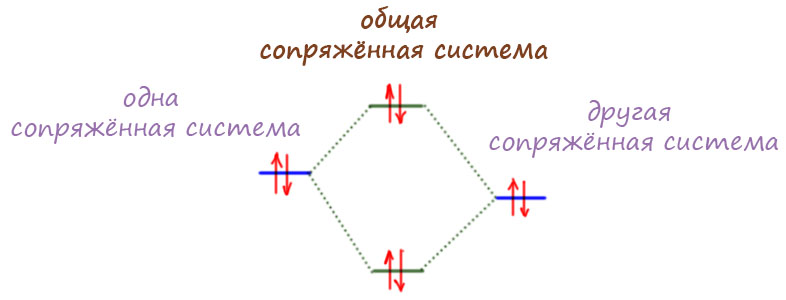
Других молекул, в которых соединяются и не могут избежать π-сопряжения цепи и фрагменты, имеющие полный комплекст электронной плотности и следовательно скорее донорные, чем акцепторные, можно найти немало. Все такие молекулы будут иметь какие-то свойства и реакции, довольно откровенно говорящие о желании избавиться хотя бы от части этой плотности. Время от времени будем на это натыкаться.
В другой стороны, встреча двух фрагментов или цепей, один из которых обладает признаками донора, а другой акцептора, но при этом не образующих единую цепь сопряжения, очень часто бывает. В таких случаях сопряжение должно приводить к стабилизации, хотя и не такой сильной, как при образовании настоящей цепи сопряжения донор-акцептор. Такая ситуация часто встречается в ароматическом ряду с мета-замещением. Как мы знаем, мета-расположение заместителей не даёт единую цепь сопряжения. Но это не мешает кросс-сопряжению. Любой бензол с +M и -M-заместителями в мета-положении иллюстрирует такую ситуацию. Обратите внимание, что вы можете нарисовать цепь сопряжения отдельно донора с бензольным кольцом и отдельно акцептора, и все эти граничные структуры правильные, но ни в одной из них не задействованы одновременно и донор и акцептор. А это и есть признак кросс-сопряжения. Мы совершенно уверены в том, все три компонента этой молекулы – амино-группа, нитро-группа и бензольное кольцо образуют единую сопряжённую систему, но не можем изобразить её ни граничными структурами, ни мезомерными стрелочками (пропробуйте сами). Это значит ровно одно – данная сопряжённая система целиком не является цепью сопряжения. Бензольное кольцо не является здесь удлинителем, вместо этого это скорее соединитель – фрагмент, который соединяет фрагменты в единую сопряжённую систему, не объединяя их. Если бы мы умели говорить языком молекулярных орбиталей, мы бы это очень хорошо увидели – вместо единого набора π-МО делокализованного по всей системе, мы бы увидели два набора π-МО, которые были бы тоже делокализованы, но один набор имел бы большие веса на одной части молекулы, а второй – на другой. Это говорит нам, что взаимодействие есть, и это взаимодействие стабилизирующее, но намного менее эффективное, чем в обычных цепях сопряжения. Это можно понять хотя бы по тому, что рассматривая два набора граничных структур, несложно заметить, что плюсы в одном оказываются на тех же атомах, что минусы в другом. Это хорошо, так как приводит не к локализации, а к ослаблению зарядов. Не забывайте, что настоящая структура это нечто усреднённое между граничными структурами. 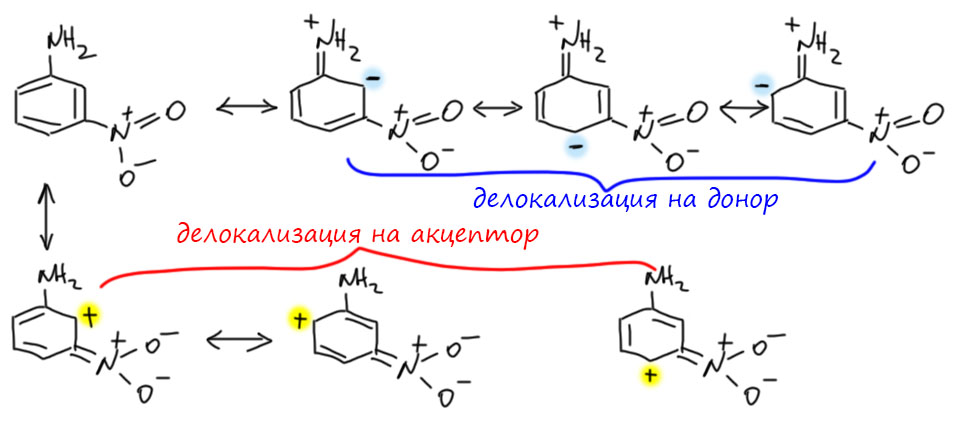
Ситуацию акцептор и акцептор представить невозможно. Вернее, возможно – это когда несколько попрошаек с пустыми карманами сошлись на дороге – и что они будут делать, делить-то нечего. И тогда они либо побазарят и несолоно хлебавши разойдутся, либо обнаружат заначку у одного из них, и примутся драться и делить её. Это значит, что встреча двух или более акцепторов либо ничем не кончится, либо в очередной раз реализуется бессмертный лозунг из Скотного двора – все акцепторы равны, но некоторые акцепторы акцепторнее, чем другие акцепторы. И тогда такие акцепторы, которые акцепторнее других акцепторов, останутся акцепторами, а остальные станут поневоле донорами, и из их вывернутых карманов выкатится завалявшаяся денежка, то есть электронная пара, которой они и не собирались делиться. Такая страшная ситуация случилась, например, в солях диазония с нитро-группой в пара- или орто-положении. И нитро-группа и диазоний – сильнейшие акцепторы, но когда они оказываются в одной молекуле в одной цепи сопряжения, нитро-группа оказывается круче и заставляет диазоний стать донором. Пока не будем с этим разбираться, оставим до 2 семестра, но граничные структуры нарисуем. Хотя бы для того, чтобы можно было убедиться, насколько такая ситуация необычна, и насколько большое унижение испытывает диазониевая группа, ставшая +M-заместителем, и вынужденная для того чтобы изыскать ту самую завалявшуюся денежку, то есть пару, ободрать дальний азот до секстетного состояния. 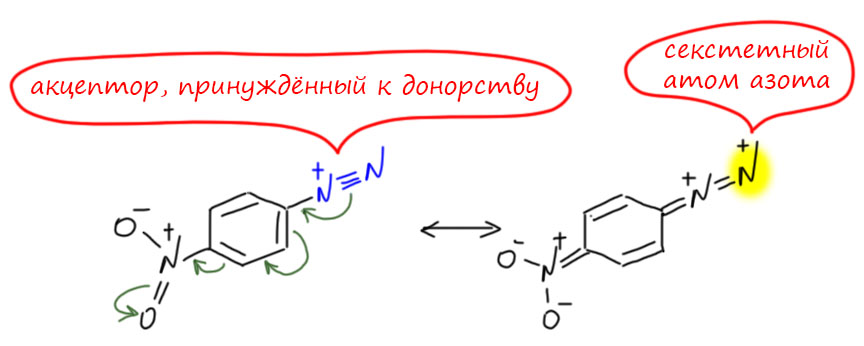
К системам с кросс-сопряжением относятся и многочисленные нелинейные нагромождения двойных связей, в которых в принципе мы не можем выделить доноров и акцепторов. Самый простой пример – дивинилэтилен. Эта молекула представляет собой как бы два бутадиена, соединённых как сиамские близнецы – что-то у них общее, что-то раздельное. Красная и зелёная двойные связи вообще кажутся несопряжёнными, и так про них часто и пишут. Это соершенно удивительное заблуждение – конечно же, они сопряжены, так как атом между ними имеет p-орбиталь, коллинеарную орбиталям обеих π-связей. То, что они сопряжены, легко видеть, если разрисовать граничные структуры, и мы сначала это сделаем, разрисовав независимо оба бутадиена, верхний и нижний. И тогда мы увидим, что граничные структуры из верхнего набора отлично и непосредственно (то есть через перемещение пары) связаны с граничными структами нижнего набора. Итого – это единый набор граничных орбиталей единой сопряжённой системы. И то, что это не обычная цепь сопряжения, а комбинация двух цепей сопряжения (кросс-сопряжение), видно из того что ни одна из граничных структур не затрагивает одновременно обе эти двойные связи. 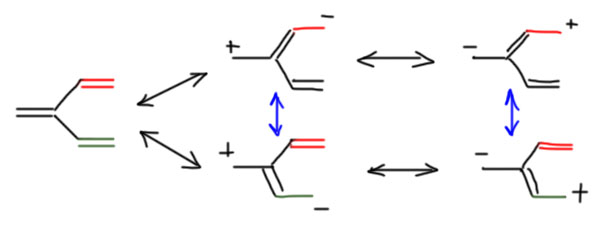
Систем таких в химии пруд пруди. Чаще всего они встречаются среди циклов. Это и хиноидные структуры (хинодиметаны), и фульвены, и радиалены и много чего ещё. Всё это очень красиво… но обычно малоустойчиво. Кросс-сопряжение в данном случае дестабилизирует. Впрочем, умеренная дестабилизация, которая не приводит к полной неустойчивости, всегда придаёт молекулам много интересного – они охотно участвуют в самых разных реакциях, в том числе весьма необычных, они проявляют множество интересных свойств и так далее. Поэтому такие соединения, в которых сидит такой червячок нестабильности и будоражит их изнутри, – любимый и лакомый объект исследований.