Ароматические соединения
Ароматичность – одно из ключевых понятий органической химии, а ароматические соединения совершенно вездесущи. Мы добрались до обсуждения этого явления только что, а фенильных колец в разных местах видели уже немало, и привыкли считать их просто таким заместителем, который влияет или не влияет на реакционную способность других реакционных центров. Но у ароматических соединений есть своя и очень интересная жизнь, и иногда мы обращаем внимание именно на ароматические кольца, и тогда уже все остальное становится заместителями.
В этом разделе мы попробуем разобраться в том, что такое ароматичность. К этому можно подойти легко и без претензий, и тогда больших проблем не будет: в первом приближении ароматичность – это очень просто и понятно. Но если попробовать углубиться в это понятие, то вдруг могут начать появляться вопросы и сомнения, и ароматичность перестанет казаться такой уж очевидной.
Ниже краткая программа курса по этой теме.
Программа раздела
1. Ароматичность. Ароматические углеводороды
Строение бензола. Формула Кекуле. Молекулярные орбитали бензола. Аннулены. Аннулены ароматические и неароматические. Концепция ароматичности. Правило Хюккеля. Ароматические катионы и анионы. Конденсированные ароматические углеводороды: нафталин, фенантрен, антрацен, азулен и др.. Гетероциклические пяти- и шестичленные ароматические соединения ( пиррол, фуран, тиофен, индол, азолы, пиридин, хинолин).
Антиароматичность на примере циклобутадиена, циклопропенил-аниона, катиона циклопентадиенилия. Критерии ароматичности: квантовохимический, энергетический (теплоты гидрирования) и магнитный.
Получение ароматических углеводородов в промышленности – каталитический риформинг нефти, переработка коксового газа и каменноугольной смолы.
Лабораторные методы синтеза: реакция Вюрца-Фиттига и другие реакции кросс-сочетания, алкилирование бензола и аренов по Фриделю-Крафтсу, восстановление жирноароматических кетонов.
Каталитическое гидрирование аренов, восстановление аренов по Берчу. Реакции замещения водорода в боковой цепи алкилбензолов на галоген. Окисление алкилбензолов и конденсированных ароматических углеводородов до карбоновых кислот.
2. Реакции электрофильного замещения в ароматическом ряду
Классификация реакций ароматического электрофильного замещения. Общие представления о механизме реакций, кинетический изотопный эффект в реакциях электрофильного замещения водорода в бензольном кольце. Представление о p – и s -комплексах. Структура переходного состояния. Изотопный обмен водорода как простейшая реакция электрофильного замещения. Аренониевые ионы в реакциях электрофильного замещения. Влияние заместителя на скорость и направление электрофильного замещения. Согласованная и несогласованная ориентация.
Нитрование. Нитрующие агенты. Механизм реакции нитрования. Нитрование бензола и его замещенных. Нитрование нафталина, бифенила и других аренов. Галогенирование. Галогенирующие агенты. Механизм реакции галогенирования аренов и их производных.
Сульфирование. Сульфирующие агенты. Механизм реакции. Кинетический и термодинамический контроль в реакции сульфирования на примере фенола и нафталина. Превращения сульфогруппы.
Алкилирование аренов по Фриделю-Крафтсу. Алкилирующие агенты. Механизм реакции. Полиалкилирование. Побочные процессы – изомеризация алкилирующего агента и конечных продуктов. Синтез диарил- и триарилметанов. Триарилметилкатионы, анионы и радикалы. Методы их генерирования и стабильность.
Ацилирование аренов по Фриделю-Крафтсу. Ацилирующие агенты. Механизм реакции. Региоселективность ацилирования. Формилирование по Гаттерману-Коху и другие родственные реакции.
3. Нуклеофильное ароматическое замещение
Общие представления о механизме нуклеофильного замещения.
1. Механизм отщепления-присоединения на примере превращения галогенбензолов в фенолы и ароматические амины. Методы генерирования и фиксации дегидробензола. Строение дегидробензола.
2. Механизм присоединения-отщепления SN Ar, примеры реакций и активирующее влияние электроноакцепторных заместителей. Анионные s -комплексы Мейзенгеймера и их строение.
3. SN1-Механизм ароматического нуклеофильного замещения в реакциях гидролиза катиона арендиазония.
Как обычно, понемногу будем разбираться с отдельными проблемами
Восстановление по Бёрчу
Бензол и некоторые замещенные бензолы восстанавливаются растворами металлических натрия или лития в жидком аммиаке в присутствии двух эквивалентов этанола в 1,4-циклогексадиены.
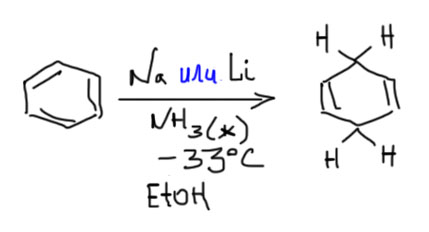
Это единственная реакция в нашем маленьком арсенальчике, в которой ароматическое кольцо теряет ароматичность, и это кажется какой-то аномалией, ведь нам настойчиво внушают, что ароматическое кольцо это нечто настолько стабильное и самодовольное, что уничтожить его можно только в печи огненной, да даже и там возможно выживет, как трое приятелей пророка Даниила. Это не так. Ароматическое кольцо теряет ароматичность в многих реакциях, и такие реакции очень часто дают весьма непростые и интересные продукты, к которым по другому и подступиться-то непонятно как. Именно поэтому такие реакции, прозванные деароматизацией, стали очень популярны в современном органическом синтезе. Восстановление по Бёрчу – один из главных прототипов всех таких реакций. Она показала чрезвычайную привлекательность получения неароматических соединений из ароматических.
Как мы уже убедились, сохранение ароматического кольца в самых разных реакциях – один из самых надежных признаков ароматичности. Но реакции, нарушающие ароматичность все-таки бывают. Для этого всегда приходится прилагать немало усилий, использовать серьезные реагенты, а иногда и просто “ловить” промежуточные продукты разных реакций, фиксируя их в неароматическом состоянии. Мы же знаем, что во многих реакциях ароматических соединений, например, в реакциях замещения, по дороге от одного ароматического соединения к другому ароматичность временно теряется (например, образуются всякие σ-комплексы, делокализованные катионы, анионы или радикалы), и мы могли бы попробовать как-то перехватить такое временное состояние, не позволить ему вернуться в ароматичность. Но – это непросто, и всегда требует достаточно точного понимания того, что происходит в реакции. Попробуем в этом разобраться достаточно детально.
По сравнению с тем, что мы обычно изучаем, это довольно современная реакция, Артур Бёрч описал это реакцию в 1944 году. Оцените, сколько всего интересного в химии произошло в годы Второй мировой войны, закрадывается даже нехорошее подозрение, что известное изречение – “Кому война, а кому мать родна” – описывает не циничных торговцев оружием и кровожадных политиков, а учёных, но это несправедливое подозрение. Причина такой бурной научной деятельности во время войны, скорее всего, намного более проста: наука для военных нужд нужна особенно, всякие исследования интенсифицируется, а параллельно с полезными для войны вещами открывается много нового. И ещё работа во время войны становится особенно отчаянной, всякие соображения про опасность уходят на второй план, а на первый выходит: мечи в колбу всё, что есть в лаборатории, враг у ворот, нечего церемониться. Если и взорвётся, никто особенно не обратит внимания. Щелочные металлы и жидкий аммиак – самое оно и есть.
Вообще-то это просто еще один пример восстановления непредельных соединений щелочными металлами в жидком аммиаке. С одной такой реакцией мы хорошо знакомы – именно так восстанавливают ацетилены в транс-олефины. В таких восстановлениях используют в качестве основного реагента ни более ни менее, а просто электрон, самый настоящий электрон, по две штуки на каждую молекулу восстанавливаемого соединения. То есть чтобы восстановить по Бёрчу один моль бензола, надо взять два моля электронов (сколько это, кстати, весит, на тот случай, если в практикуме придется требование выписывать, перемножьте на досуге массу электрона на число Авогадро.). Понятно, что это не так просто, электроны в кулёк не насыпешь, электроны, как и протоны, бывают сами по себе, но только не в колбе. Электроны еще бывают внутри металлов, они там даже почти свободно перемещаются большими толпами, образуя электрический ток. А вот в жидкой фазе, в растворах свободных электронов как правило не бывает, потому что они тут же “прилипают” к молекулам растворителя, восстанавливая их. Способность молекул связывать электрон называется сродством к электрону, и это почти всегда положительная величина – почти все молекулы с большим или меньшим удовольствием присоединяют электрон, отправляя его на самую нижнюю свободную, обычно разрыхляющую орбиталь. Или ноль – есть молекулы, к которым электрон не клеится. Одна из таких молекул как раз аммиак. К молекуле аммиака электрон не пристаёт, и никак ее не разрушает, не восстанавливает – слишком высоко находятся разрыхляющие орбитали N-H связей. Но когда молекул амиака много в жидкости, электроны удерживаются в таком растворе, сольватируются молекулами аммиака, как какой-нибудь обычный ион, хотя бы за счёт электростатики заряд-диполь. Не последним в процессе взаимодействия металла и аммиака является и то, что катиону металла очень хорошо в жидком аммиаке, и даже щелочной металл находит возможность в виде катиона образовывать донорно-акцепторные связи с молекулами аммиака. Имеем ещё один пример очень приятной ситуации, когда что-то, связанное с возникновением двух противоположных зарядов (катиона и аниона, или катиона и электрона) происходит потому, что среда очень сильно благоприятсвует в основном одному, оставляя второго почти без присмотра для всяких разнузданных бесчинств. Не так ли устроен в нуклеофильном замещении и элиминировании межфазный катализ? действие ДМСО и других спецрастворителей? Катион пристроен, анион проявляет необузданную нуклеофильность или основность. Вот и в растворении щелочных металлов в жидком аммиаке видим мы еще один пример этой истории. Электрон выходит на авансцену химии практически свободным, еле-еле прикрытым молекулами аммиака, а их можно стряхнуть с себя при первой возможности приклеится к чему-нибудь поинтереснее.
Итак, электрон приклеивается к первой свободной орбитали. Фокус в том, что такие орбитали почти всегда являются разрыхляющими. Орбитали вообще всегда возникают парами, и большинство орбиталей обслуживает какие-нибудь связи. Каждая пара обслуживает какую-нибудь связь, и в нормальном состоянии молекулы нижняя орбиталь занята парой – это именно то, что мы называем химической связью с парой электронов. Но и парная к этой свободная орбиталь тоже обслуживает ту же связь. Вернее не обслуживает, а наоборот отвечает за отталкивание связанных атомов. Пока она пуста, она не работает, связь связывает. Стоит на нее закинуть электрон, возникнет отталкивание, связь ослабнет. Если закинуть пару, то совсем лопнет, но это чрезвычайно трудно сделать, можно даже считать, что невозможно.
Но одного электрона достаточно, чтобы связь ослабла, и молекула начинает искать пути выхода из трудной ситуации. А ведь там еще и заряд возник, электрон принёсс собой один минус.
Посмотрим, что получится, если к молекуле бензола приклеить электрон. Молекула бензола электрон примет, потому что у этой молекулы, как и у всех непредельных молекул, сродство к электрону положительное. Когда к молекуле прилипает электрон, образуется анион-радикал, то есть частица, обладающая и зарядом (лишний электрон придает ей отрицательный заряд) и неспаренным электроном, или, как говорят, спином (у молекулы все электроны были спаренные, почти все устойчивые молекулы обладают четным числом электронов, значит, добавление еще одного электрона делает общее число электронов нечетным). Как устроен анион-радикал бензола? Очень просто. Возьмем для начала одну двойную связь. Присоединение электрона к двойной связи отправит этот лишний электрон на разрыхляющую орбиталь π-связи, которая немедленно разрыхлится, и поскольку мы не умеем рисовать связи, которые разрыхлились, мы просто делаем вид, что связи больше нет, и каждый из атомов углерода получил наследство от этой связи, то есть один электрон, плюс оба получили еще один электрон. Вдвоем они таким образом получили три электрона. Нельзя по справедливости разделить три электрона на двоих, электроны неделимы. Придется одному дать один, а второму два. А кто из них первый, и кто второй? В общем случае мы сказать не можем, поэтому придется нарисовать две граничные структуры, и говорить, что в анион радикале есть неспаренный электрон и свободная пара, и оба этих объекта делокализованы по двум углеродам бывшей двойной связи. И все это ужасное мучение только потому, что мы не умеем рисовать “неправильные” связи, с большим или меньшим чем два числом электронов. А почему до сих пор не научились? – спросите вы наверное, сделав строгое лицо. Вас ждём, займитесь на досуге. А пока опять за старое.
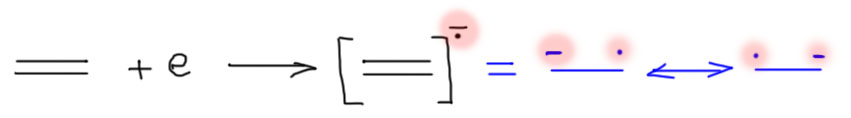
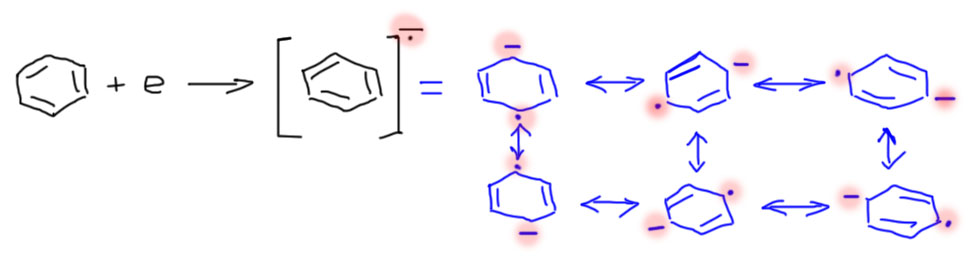
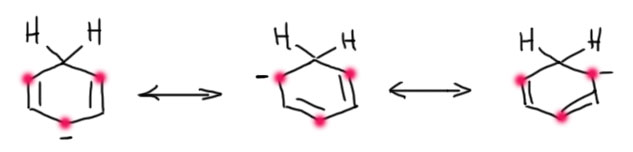
Если это понятно, то мы сможем приклеить электрон и к бензолу. У бензола не одна двойная связь, а целая ароматическая система, и электрон приклеится именно к ней. В этом месте нас ждет просто убийственный удар по тому, что мы так усердно изучили и успели поверить. Бензол ароматичен, значит особенно стабилен, именно потому, что в ароматической π-системе 6 (шесть) электронов. Когда электрон приклеился – их стало семь! А значит – прощай ароматичность! – значит, что мы потеряли ароматическую стабилизацию, что, по всем нашим теориям очень невыгодно, и должно быть немедленно вознаграждено какой-нибудь достойной наградой в виде новой химической связи, потому что энергия полноценной химической связи всегда больше чем любая ароматическая стабилизация. Хорошо, новую связь найти просто, так как в анион-радикале есть анион на углероде, а это – карбанион, а это – очень сильное основание, и его очень легко заткнуть кислотой Бренстеда, например, спиртом. Тогда мы получим на одном из атомов углерода кольца новую связь C-H (это очень хорошая, выгодная вещь) на насыщенном атому углерода. Осталось еще решить, как делокализованы минус и неспаренный электрон в шестичленном кольце. Очень просто – по всему кольцу. Каждый из них – по всему кольцу, но они не независимы друг от друга по очень простой причине – фундаментальным свойством электронов является священная ненависть этих частиц друг к другу, которая называется разными красивыми и умными словами, но для нас в этом главное – они (минус и радикал) стараются держаться друг от друга максимально возможно далеко. В шестичленном кольце нет ничего дальше, чем пара-положения. Поэтому нам пришлось бы нарисовать 6 одинаковых резонансных структур, в которых минус обегает цикл по кругу, а радикал делает то же самое на максимальном расстоянии от минуса. В бензоле все эти 6 структур одинаковы, поэтому можно их все не рисовать (но при этом знать, что они все есть, и мы не можем сказать, на каком из углеродов минус – это легко проверить, если приклеить электрон к бензолу с одним изотопным углеродом – в продуктах будет равномерная смесь всех возможных структур).
Анион-радикал получили – и тут же затыкаем минус протонированием слабой кислотой типа спирта. Остается радикал. И куча неизрасходованных электронов вокруг. Углеродные радикалы, причем любые, очень легко принимают еще один электрон и становятся карбанионами. Как мы уже поняли, можно это другими словами сказать так, что углеродные радикалы всегда имеют положительное сродство к электрону. Карбанион тут же затыкается еще одной молекулой слабой кислоты – получается продукт восстановления. Структура анион-радикала, требующая максимального удаления между анионным и радикальным центрами, диктует и структуру продукта, который всегда представляет собой 1,4-циклогексадиен. В этом месте остановитесь и оцените необычность результата – всегда получается несопряженный диен, в то время как мы знаем, что сопряженные диены выгоднее.
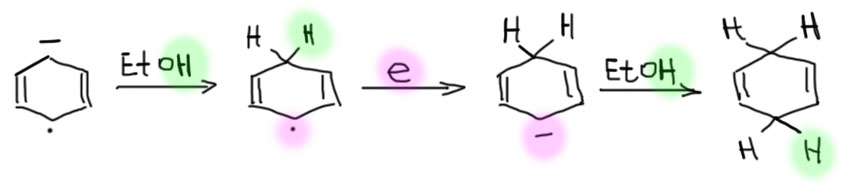
Если мы не любим задавать лишних вопросов, то все вроде понятно – в анион-радикале было так, и продукт поэтому получает два протона в пара-расположении. И хорошо. Но обязательно найдутся проницательные читатели, которые увидят во всей этой схеме довольно мерзкий подвох. В прекрасном и далеком прошлом на попытку протащить такое объяснение последовал бы удар канделябром по голове объясняющего и возмущенный возглас: “Вы шулер и подлец, вы нам дадите сатисфакцию – к барьеру!” Не годится такое торопливое объяснение, которое совершенно не учитывает того, что после затыкания протоном первого аниона образуется новый делокализованный радикал или анион, – нужно же было нарисовать граничные структуры, и увидеть, что второй анион делокализован по трем углеродам и уже “забыл”, как были расположены минус и точка в анион-радикале. И тогда по всем законам химии должен был бы получиться не 1,4-диен, а более устойчивый сопряженный 1,3-диен. А реально получается 1,4-диен. Что делать, и как изменить механизм, чтобы уйти от этого неприятного момента? Давайте разберёмся, потому что оставлять за спиной мутные места нехорошо, недостойно людей по настоящему взыскующих истины.
Согласимся с тем, что делокализованный анион быть на схеме механизма должен. А как он устроен и как будет реагировать? Запишем делокализацию граничными структурами:
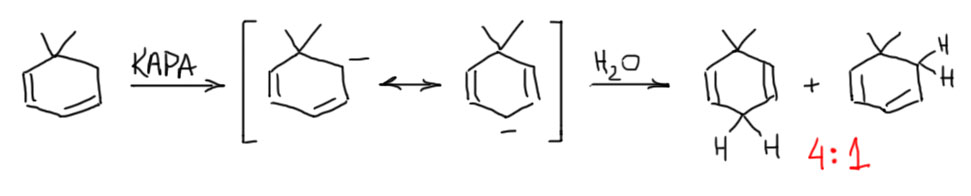
Протонирование такого аниона может дать два продукта: сопряженный диен, который более стабилен, и несопряжённый, который менее стабилен. А какой получится? Если бы мы не знали экспериментального ответа, то скорее согласились бы с первым, потому что двойные связи всегда стараются оказаться в сопряжении – это нехилый выигрыш в энергии. Ну или боязливо предложили бы смесь. Но в этом месте нужно остановиться, и подумать еще об одной важной вещи: протонирование этого аниона обратимо или нет. Потому что аргумент о большей стабильности продукта играет роль только в ситуации термодинамического контроля, когда реакция может побывать в обоих продуктах, узнать, кто из них лучше (стабильнее) и выбрать именно его просто с помощью константы равновесия. Это работатет только так. Обратите внимание, что стрелки равновесия я рисую из всего аниона (условно из центра), а не из конкретных граничных структур – не устаю напоминать, что граничные струткруы это не реальные формы молекулы, а просто условное обозначение делокализации, за неимением более компактного способа изображения.  Если же реакция необратима, то смотрины продуктов отменяются, и исходное (в данном случае, делокализованный анион) ничего не знает о том, что получится, когда его уже не будет на свете. Реакция пойдёт в ту сторону, куда быстрее, где больше константа скорости. Это – кинетический контроль. А что мы про это знаем? Увы, ничего конкретного, поэтому придётся ограничиться оценками. С одной стороны, перенос протона всегда принципиально обратим. С другой, если разница в pK много порядков, то фактически необратим. В этом случае взаимодействие сильного основания с кислотой, которой соотвествует намного менее сильное сопряжённое основание можно считать необратимым. Это как аммиак и серная кислота. Такой перенос протона настолько быстр, что по английски для описания такого используют метафорическое слово quench – тушение, так и говорят – тушить основание (люди, склонные к кулинарному творчеству могут это превратно понять, поэтому еще раз – это про окунание тлеющей спички в ведро воды). Итак, основание это делокализованный карбанион, кислота – это этанол – какая между ними разница в кислотности/основности? Точно мы этого не знаем, но можем оценить pK циклогексадиена как величину, близкую к pK аллильной системы, из-за дополнительного сопряжения скинем 2-3 единицы: получим что-то около 40. С этанолом всё похуже, потому что мы ничего не знаем про жидкий аммиак, а такой растворитель должен сильно влиять на pK этилата. Но в любом случае это не будет больше чем pK в ДМСО, хотя бы потому что аммиак является донором водородной связи, хотя и очень слабой, но сольватировать анион этилата будет толее сильно чем ДМСО. Верхняя оценка поэтому около 30, скорее всего немного меньше. Итого разница в 10 единиц pK, а скорее не меньше 12-14. Это очень большая разница, и можно считать, что этанол протонирует карбанион с огромной скоростью в режиме quench – тушения. Я надеюсь, что на вас произвел удручающее впечатление этот жалкий лепет про оценку pK не очень сложных кислот и оснований с точностью до третьего-четвёртого знака перед запятой. Ну что вы хотите, сейчас всего-навсего начало 21 века, органической химии еще и 200 лет не исполнилось, приходите ещё лет через 200, померим поточнее. А хотите быстрее, так возьмитесь сами, только трезво оцените, насколько человечество в этом нуждается. А пока и так сойдёт. Нам ведь цифры не нужны, нужно только ситуацию оценить, а это вполне получается и с такой точностью. Следовательно, мы можем обоснованно считать, что равновесия не будет, а контроль будет кинетическим. Осталось понять, какой продукт получается быстрее – сопряжённый или несопряжённый. Чтобы это понять, можно заняться пресказаниями, а можно посмотреть, нет ли какого эксперимента. Для предсказания неплохо как-то узнать, как распределён отрицательный заряд в таком карбанионе, например, с помощью квантово-химических расчётов: такие расчёты сделать несложно, и они показывают что в пара-положении заряд больше, чем в орто (цифры упоминать на будем, потому что между расчётными цифрами и реальными выходами количественной связи нет никакой, роль играет только волшебное слово “больше”). Надёжнее эксперимент, и такой есть (J.Org.Chem., 1988, 53, 5972), только с диметил-производным, но это не должно приводить к сильному изменению делокализации аниона. Взяли сопряжённый диен, депротонировали очень сильным основанием, той самой KAPA, которой мы сдвигаем тройную связь. И анион погасили водой, что гарантирует кинетический контроль протонирования. В результате получили в основном несопряжённый диен.
Если же реакция необратима, то смотрины продуктов отменяются, и исходное (в данном случае, делокализованный анион) ничего не знает о том, что получится, когда его уже не будет на свете. Реакция пойдёт в ту сторону, куда быстрее, где больше константа скорости. Это – кинетический контроль. А что мы про это знаем? Увы, ничего конкретного, поэтому придётся ограничиться оценками. С одной стороны, перенос протона всегда принципиально обратим. С другой, если разница в pK много порядков, то фактически необратим. В этом случае взаимодействие сильного основания с кислотой, которой соотвествует намного менее сильное сопряжённое основание можно считать необратимым. Это как аммиак и серная кислота. Такой перенос протона настолько быстр, что по английски для описания такого используют метафорическое слово quench – тушение, так и говорят – тушить основание (люди, склонные к кулинарному творчеству могут это превратно понять, поэтому еще раз – это про окунание тлеющей спички в ведро воды). Итак, основание это делокализованный карбанион, кислота – это этанол – какая между ними разница в кислотности/основности? Точно мы этого не знаем, но можем оценить pK циклогексадиена как величину, близкую к pK аллильной системы, из-за дополнительного сопряжения скинем 2-3 единицы: получим что-то около 40. С этанолом всё похуже, потому что мы ничего не знаем про жидкий аммиак, а такой растворитель должен сильно влиять на pK этилата. Но в любом случае это не будет больше чем pK в ДМСО, хотя бы потому что аммиак является донором водородной связи, хотя и очень слабой, но сольватировать анион этилата будет толее сильно чем ДМСО. Верхняя оценка поэтому около 30, скорее всего немного меньше. Итого разница в 10 единиц pK, а скорее не меньше 12-14. Это очень большая разница, и можно считать, что этанол протонирует карбанион с огромной скоростью в режиме quench – тушения. Я надеюсь, что на вас произвел удручающее впечатление этот жалкий лепет про оценку pK не очень сложных кислот и оснований с точностью до третьего-четвёртого знака перед запятой. Ну что вы хотите, сейчас всего-навсего начало 21 века, органической химии еще и 200 лет не исполнилось, приходите ещё лет через 200, померим поточнее. А хотите быстрее, так возьмитесь сами, только трезво оцените, насколько человечество в этом нуждается. А пока и так сойдёт. Нам ведь цифры не нужны, нужно только ситуацию оценить, а это вполне получается и с такой точностью. Следовательно, мы можем обоснованно считать, что равновесия не будет, а контроль будет кинетическим. Осталось понять, какой продукт получается быстрее – сопряжённый или несопряжённый. Чтобы это понять, можно заняться пресказаниями, а можно посмотреть, нет ли какого эксперимента. Для предсказания неплохо как-то узнать, как распределён отрицательный заряд в таком карбанионе, например, с помощью квантово-химических расчётов: такие расчёты сделать несложно, и они показывают что в пара-положении заряд больше, чем в орто (цифры упоминать на будем, потому что между расчётными цифрами и реальными выходами количественной связи нет никакой, роль играет только волшебное слово “больше”). Надёжнее эксперимент, и такой есть (J.Org.Chem., 1988, 53, 5972), только с диметил-производным, но это не должно приводить к сильному изменению делокализации аниона. Взяли сопряжённый диен, депротонировали очень сильным основанием, той самой KAPA, которой мы сдвигаем тройную связь. И анион погасили водой, что гарантирует кинетический контроль протонирования. В результате получили в основном несопряжённый диен.
Восстановление по Бёрчу замещенных бензолов рассмотрено на страничке про методы.
Немного подробнее о том, как всё это устроено и почему именно такая система используется в этом восстановлении. Откуда берут электроны? Ну, это совершенно понятно – из металлов, особенно самых электроположительных, щелочных. В принципе, и так часто и делают – просто берут восстанавливаемый реагент, немного подходящего растворителя типа ТГФ и добавляют понемногу щелочной металл, натрий или литий (калий, а тем более все, что еще ниже в Таблице, тревожить не стоит, потому что есть большой шанс не донести их до колбы, в которой мы планируем реакцию – эти металлы так щедры и так любят дарить всем свои электроны, что могут по дороге отдать их молекулам кислорода в воздухе, или лично вам – и это не то, что нам нужно). Первая проблема, с которой нам придется справиться, состоит в том, что просто приклеивание электронов к молекулам, как правило, не дает ничего стабильного – получаются или анион-радикалы, или дианионы, обычно очень неустойчивые. Чтобы получить что-то стабильное, нужно на эти анионы тут же положить что-то положительное, проще всего протоны. Эти протоны мы не можем взять из более-менее сильных кислот, потому что все они сами сразу реагируют со щелочными металлами. Но можем взять очень слабые кислоты, например, спирты (почему не воду, я надеюсь, пояснять не нужно, впрочем органическая химия настолько велика, что можно найти пару примеров восстановления по Бёрчу в присутствии воды, но это нам не стоит перенимать). Но и здесь проблемы не заканчиваются – щелочные металлы ведь и со спиртами реагируют, особенно натрий. Появляется обычная проблема в органической химии – идут параллельно две реакции (восстановление нужной молекулы и реакция со спиртом с образованием водорода и алкоголята, то есть восстановление спирта) и это заставляет брать большие избытки щелочного металла.
Но есть один растворитель, в котором щелочной металл растворяется очень необычным, почти фантастическим способом – жидкий аммиак. Аммиак сжижается, и соответственно, кипит при -33ºС. Жидкий аммиак продается в баллонах, из которых можно просто налить (точнее, передавить – перевернув баллон, соединить его трубкой с приемником, и аккуратно открыть вентиль – оттуда прямо польется жидкость) в самую обычную колбу, жидкий аммиак. На колбу придется водрузить обратный холодильник, но охлаждать его придется не водой, а чем-нибудь, что обеспечивает конденсацию аммиака, то есть имеет температуру ниже -33ºС. В современных лабораториях с этим нет проблем – есть низкотемпературные термостаты (криостаты), которые обеспечивают циркуляцию какой-нибудь незамерзающей жидкости типа спирта с любой температурой. Тогда обратный холодильник потребуется обычный. Если такого криостата нет, то используют своеобразный обратный холодильник, рубашка которого широкая и открытая сверху. Туда просто заправляют охлаждающую смесь типа ацетона или спирта с сухим льдом или небольшим количеством жидкого азота. Все это, естественно, очень быстро сверху покрывается красивой шубой снега и живописно дымит, но в целом не доставляет особенных хлопот, и реакции ведут практически так же, как в обычном растворителе. Единcтвенная серьезная проблема – внутренность такой колбы нужно жестоко защищать от влаги воздуха, иначе вода начинает быстро намораживаться внутри и все портить. Прибор должен быть хорошо собран на надежных шлифах, все соединения защищены от выбивания пружинками или спецзажимами (не резинками – они замерзают и становятся хрупкими), а соединение с атмосферой устроено через жидкостной затвор.
Итак, жидкий аммиак у нас есть. Кидаем туда аккуратненько маленькие кусочки натрия или лития, хорошо очищенные от корки. Видим, что металл быстро растворяется, а раствор приобретает темно-синий цвет, очень быстро становится настолько темным, что видеть там уже что-трудно, остается верить, что щелочной металл растворился весь. Этот синий раствор, наверное, одна из самых удивительных вещей, которую можно наблюдать в колбе – это почти в самом прямом смысле раствор электронов в аммиаке. Натрий или литий переходят в катионы, координированные с молекулами аммиака, как положено в химии – эта координация обеспечивает выгодность перехода металлического металла в катионы в растворе. А куда девается электрон? Это самое интересное. Проблема в том, что в аммиаке нет ничего кроме аммиака (это не мерзкий троллинг, а констатация химического факта, ведь, например, в воде есть кроме воды еще и гидроксид ион, и гидроксоний, а если взглянуть поподробнее, то и много других частиц, а аммиак, хоть и является ближайшим аналогом воды в смысле периодичности свойств элементов, гораздо проще устроен прежде всего потому, что не образует серьёзных водородных связей). Электрону поэтому деваться некуда, кроме как обратиться к молекуле аммиака за приютом. Увы, не даст молекула аммиака приюта электрону. У молекулы аммиака сродство к электрону равно нулю, и электрон ей даром не нужен. Бывают такие молекулы, которые не принимают лишний электрон даже на временное хранение, у таких молекул сродство к электрону равно нулю, то есть при возможном присоединении электрона к такой молекуле никакой энергии не выделяется, а вот энтропия в таком процессе реально уменьшалась бы, а это плохо. К счастью, то, что невозможно для одной молекулы, может осуществиться, когда таких молекул много, в конденсированной фазе, в жидкости. В жидком аммиаке электроны слабо взаимодействуют с молекулами среды, иными словами, они сольватируются. Получается раствор сольватированных электронов. Так обычно говорят, хотя это и не совсем верно, потому что в том же растворе плавают сольватированные катионы металла, образуя с этими электронами, как минимум, аналог ионных пар. Эти катионы в любом случае играют важнейшую роль в стабилизации этих растворов, и превращение металла в ионы металла требует небольших затрат энергии на выход электрона (это называется потенциалом ионизации, у щелочных металлов очень низкие потенциалы ионизации), и сопровождается очень хорошей компенсацией за счет взаимодействия катиона металла с молекулами аммиака, работающими как настоящие лиганды, связанные с металлом полноценной координационной связью. Итого, со стороны металла процесс очень выгоден. Со стороны электрона выгода очень мала, и состоит в очень слабой энергии сольватации, скорее всего за счет электростатического взаимодействия заряда с полярной молекулой (есть и другие, более тонкие квантовые модели таких взаимодействий). Поэтому электроны в таких растворах почти свободны, и с огромным удовольствием присоединяются к любым молекулам, обладающим ненулевым сродством к электрону, а таких молекул подавляющее большинство, в частности, это молекулы с кратными связями. Мы будем использовать минимум три таких реакции – восстановление алкинов, восстановление ароматических соединений, восстановление сопряженных непредельных карбонильных соединений.