Предупреждение!
Дорогой Читатель! Если Вы случайно являетесь школьником или абитуриентом, предупреждаю Вас, что материал данной страницы может противоречить тому, чему Вас учат Ваши уважаемые Учителя. В этом случае применение материала этой страницы может принести Вам вред, затруднить прохождение экзаменов и испортить перспективы поступления в лучшее учебное заведение. Эта проблема не стоит таких жертв. Вернётесь к ней, если захотите, когда таких проблем не будет.
Предупреждение!
Дорогой Читатель! Если Вы случайно являетесь школьником или абитуриентом, предупреждаю Вас, что материал данной страницы может противоречить тому, чему Вас учат Ваши уважаемые Учителя. В этом случае применение материала этой страницы может принести Вам вред, затруднить прохождение экзаменов и испортить перспективы поступления в лучшее учебное заведение. Эта проблема не стоит таких жертв. Вернётесь к ней, если захотите, когда таких проблем не будет.
Положите валентность на место!
И больше никогда не трогайте! Что за вопль вопиющего? Не пустыня же, чай, вокруг, люди кругом, услышат, санитаров вызовут.
Какова валентность азота в азотной кислоте? Что за вопрос, со времён Берцелиуса и Франкланда пять, у Менделеева тоже пять, да и почти любой химик, выучившийся хотя бы 20 лет назад или раньше, не будет спорить, а наоборот осведомится, что за проблема с химической таблицей умножения. Так, а азота в аммонии? Три, конечно, это просто протонированный аммиак, с каких это пор протонирование изменяет валентность!? А углерода в оксиде углерода? Да два, два, эй, санитары! А кислорода в той же молекуле? Звук сирены…
Не будет никаких санитаров. Как я не давно с крайним удивлением узнал, уже очень многие считают совершенно не так, и на те же вопросы скажут – в азотной кислоте и аммонии азот четырехвалентен, это же очевидно – посчитайте связи, черточки вон вокруг буквы N не дураки же нарисовали. Очки запотели? Протрите и посчитайте, и не отвлекайте серьёзных людей от неустанных забот на ниве совершенствования методических указаний преподавания химии. А в оксиде углерода, угарном газе углерод трехвалентен, так и только так, и это очень важно, все должны это знать. И не надо тут угорать. Все современные (особое ударение на слове современные, от такого ударения слабонервные уползают под стол и больше не вякают) химики знают, как устроена молекула CO, и это очень-очень очень-очень-очень важно, это должен знать каждый школьник, а если кто-то отвечает два, то вот два ему/ей и есть самая заслуженная оценка. Закройте дверь с обратной стороны! На Луну вы не полетите.
В этом месте правда иногда возникает одна заминка. Адепты “новой валентности”, назовём их для краткости нововалентнистами, на трехвалентном углероде в СО настаивают особенно, это для них просто пароль на вход в сообщество, увы, не тайное. Те же люди обычно как-то стесняются сказать, что и у кислорода валентность три, ведь очень трудно это отрицать, если у углерода три, то и у кислорода три, молекула двухатомная, деваться некуда. Но что-то, видимо, внутренний голос говорит им, что если вслух сказать “кислород трёхвалентен, на том стою!” то некто очень знакомый, косматый и бородатый, с недоделанным чемоданом и штофом сорокаградусной в руках будет являться им в кошмарах, и призывать, как бронзовый Командор дона Хуана на дружеский ужин в очень неуютное место, где бесогенное потепление давно вышло из-под контроля. Впрочем, я уже успел повстречать и настоящих кремней, которые не моргнув глазом говорят, а какая-де проблема в трёхвалентном кислороде? Современная химия не видит проблем, она вообще ни в чём проблем не видит, современная же, а вашего Менделеева оставьте в музее, это такой патриотический истукан, годный только для дежурных славословий, но мы же понимаем… А есть такие современные хитрецы, кто отвечает что-то типа “ни два, ни три, что-то посредине”. В общем, недалек тот день, когда валентность будут каждый год назначать заново в методических указаниях по ЕГЭ. “Какая у нас нынче валентность на дворе” – станут выяснять родители выпускников, подбирая знатоков последних новинок в валентности в качестве репетиторов для своих чад.
Попробую-ка я всё же заступиться за Менделеева и напомнить, что определение валентности ни на малую толику не изменилось с 1869 года, а нововалентнисты просто зачем-то пытаются возродить споры столетней давности, да еще и с изрядной долей банальной путаницы. А попытки её переопределить очень похожи на такое известное упражнение в исторической науке как “новая хронология” – вроде предлагается революционный пересмотр сложившейся науки, а на деле получается смесь откровенного шарлатанства и извлечения на свет давно забытых концепций столетней давности. Учение “новой валентности”, к большому сожалению проникшее в школу, ужасно на это похоже и столь же бесплодно. Будем надеяться всё же, что оно сгинет бесследно в обозримом будущем.
И заодно намекну, что валентность в современной химии можно совсем не использовать, как делает, например, англо-американская система изучения химии, как несложно заметить не самая отсталая. Но уж если используете, извольте придерживаться никуда не девшейся и очень удобной традиции и не ломайте хорошую вещь, сослужившую добрую службу многим поколениям химиков. И вообще, я думаю, что она ещё послужит – это чертовски классная штука, потрясающе удобная и понятная. Не трогайте валентность, положите на место!
О чём эта страница
О валентности как одном из начальных понятий химии. Цель обсуждения – напомнить о том, что такое валентность, и почему её не нужно переопределять, как число ковалентных связей или иным способом, как, увы, стали часто делать. Ярким символом этого подхода является утвержедение о четырёхвалентности азота в азотной кислоте и аммонии. Но это самодеятельность, не подтвержадемая ни одним авторитетным источником. На этой странице подробно обсудим, почему этого делать нельзя, и почему валентность в традиционном понимании до сих пор является весьма важной характеристикой элементов в соединениях, впрочем не обязательной. Не хотите обычную валентность – просто не используйте, но не ломайте.
Кроме этого здесь быдут рассмотрены всякие полезные вещи типа , различия разных типов химической связи, обозначений в структурных формулах и т.п. Страница никак не связана с программой 3 курса, и адресована просто всем желающим.
Кратко о валентности и связанных вещах
Сначала перечислим кратко основные тезисы. Если этого окажется недостаточно, многие рассмотрены полнее на раскрывающихся вкладках внизу. Там очень много букв, всё это не обязательно читать, особенно всё целиком. Я просто ощутил потребность разобрать это так подробно, чтобы и самому ещё раз убедиться в том, насколько беспочвенной и бессмысленной является эта странная попытка грубо влезть в самые основы понятийного аппарата химии. Я задавал себе вопрос – может за этим что-то есть, может это основано на чём-то серьёзном, обсуждается в серьёзных книгах и статьях, а не только в странных пособиях для поступающих и методичках по сдаче егэ. Но нет, всё нормально, основы химии на месте, никто на них не посягает кроме нововалентнистов, которые практически исключительно являются не действующими учеными, а методистами. Это довольно странный выход за границы профессии. Работа методиста, вполне полезная и нужная, состоит в том, чтобы помогать изучать науку. Создавать и изменять понятия науки методисты не могут, это не их работа, они просто не умеют этого делать. Это все равно как если бы тренер по плаванию решил бы, что плавать теперь надо не в горизонтальных направлениях, а в вертикальных. Буль-буль, карасики… как говорили у нас в очень далёком советском детстве.
- Валентность – самая первая количественная характеристика способности атомов элементов соединяться друг с другом, образуя молекулы. Валентность не имеет никакого отношения к электронной структуре, химическим связям, типам связей, орбиталям и всему остальному, так как была введена в 19 веке до открытия электрона и структуры атома. Валентность как количественная характеристика с тех пор не пересматривалась и не будет пересматриваться, так как в этом нет никакого смысла – для описания электронной структуры и всего, что с этим связано, существуют другие понятия.
- Главная проблема с валентностью в том, что мы встречаемся с ней в ранней школе и больше никогда не возвращаемся, чтобы осмыслить. Поэтому у нас невольно возникает интуитивное понимание этого понятия, например, как числа ближайших соседей атома (что неверно, потому что это координационное число, а не валентность), или как числа связей, часто дополнительно добавляем – ковалентных связей. Но и это неверно, так определялась не валентность, а ковалентность, причём это последнее слово в этом смысле давно вышло из употребления. Ковалентные связи больше никто сегодня не считает, так как это не имеет смысла и порождает путаницу между обычными соединениями и координационными.
- В 20 веке у слова валентность появился более общий смысл, но качественный. Так получилось потому что слово валентность, первоначально возникшее как невинное сокращение слова эквивалентность, оказалось носителем более глубоких смыслов – со словами такое случается, цивилизация древнее химии, и корни многих слов успели впитать множество смыслов, мимо которых очень трудно пройти, если хоть немного об этом подумать.
Желательно не путать классическую валентность, имеющую количественный смысл (ту, которая используется в фразах типа “азот в азотной кислоте пятивалентен”, “какова валентность иода в периодате?” и т.п.), и валентность в общем смысле. Слово одно и то же, к сожалению, и понимать, какой смысл подразумевается в конкретных случаях (статьях, книгах, лекциях) приходится из контекста. Справиться можно без труда, потому что одно понятие совершенно конкретно и выражается числом, а второе представляет собой скорее нечто общефилософское. Валентность в последние 100 лет понимают чаще именно как наиболее общее понятие, выражающее способность атомов соединяться в молекулы, но это не значит, что старое понятие валентности выброшено или отдано желающим для произвольного переопределения.
Когда вспоминают про количественную сторону валентности в этом смысле, а так делают и нередко, имеют в виду то, что современная химия предлагает множество количественных характеристик, описывающих связывание. Чаще всего в этой роли выступает степень окисления, но могут быть и более сложные понятия из квантовой химии. В общем, любой исследователь или методолог может предлагать свои характеристики валентности и расхваливать их достоинства, только не имея при этом права называть это просто валентностью. Валентность в количественном смысле уже определена и это именно классическая валентность. - Классическая валентность определяется только соотношением элементов в соединении. Химия как наука возникла тогда, когда появилось понятие элементов, были определены атомные веса, и путем обобщения данных по элементному анализу быстро увеличивавшегося множества новых соединений было выяснено, что соотношение элементов не произвольно, а подчиняется простым рациональным соотношениям – отношениям эквивалентных масс или просто эквивалентов. Эти соотношения эквивалентов и получили названия валентностей. У каждого элемента одна или несколько разных валентностей.
- Классическая валентность определялась и определяется непосредственно только для нейтральных бинарных соединений элементов в том случае, если валентность одного из элементов известна, а в соединении нет связей атомов одного элемента друг с другом. Есть несколько элементов, у которых валентность всегда одинакова. Два самых важных – водород (одноваленый) и кислород (двухвалентный). Годятся также галогены, особенно фтор (всегда одновалентный). Другие галогены тоже годятся, только надо иметь в виду, что они одновалентны не всегда, но почти всегда, кроме узкого набора исключений – соединений друг с другом (интергалогенов) и оксидов. ИЮПАК предлагает в качестве стандартных элементов для определения валентности водород и галогены, хотя в традиции химии предпочитают водород и кислород. Можно спокойно использовать все шесть элементов.
- После определения валентности элементов в бинарных соединениях, можно перейти к более сложным соединениям. Это делается очень просто, если можно установить химические взаимоотношения соединений – узнать, как более сложное соединение образуется из более простого, бинарного. И далее – как следующее по сложности образуется из предыдущего. То есть узнать цепи превращений. Некоторые говорят – установить генетические зависимости соединений. Не люблю это излишне пафосное слово, но если нравится, пожалуйста. Здесь очень важна теснейшая связь валентности с реальной химией. Именно поэтому валентность так полезна при изучении химии.
- Самый основной и наиболее привычный всем путь состоит в том, что у каждого элемента сначала выписывают существующие соединения с водородом (гидриды) и оксиды. От этих соединений производят ряды производных. От гидридов путем замещения атомов водорода на другие элементы или группы производят ряды замещенных. Валентность элемента в таких рядах не изменяется. От оксидов производят гидраты оксидов (основные, кислотные, амфотерные), и далее соли. И в этих рядах валентность сохраняется. У этого правила есть мощное обобщение, основанное на особой роли в химии обобщенных теорий кислот и оснований – Бренстеда-Лоури и Льюиса. В кислотно-основных реакциях и того и другого типа валентность не изменяется. Совершенно ясно, что эти правила покрывают колоссальное количество реальных соединений, вплоть до комплексных (комплексные соединения чаще всего образуются за счет взаимодействия кислот и оснований Льюиса), поэтому определить валентности элементов можно без больших проблем для очень большого количества реальных соединений, и за бортом остаются только небольшое количество соединений с необычной структурой.
Коротко этот тезис можно сформулировать так – хотите узнать валентность элемента в каком-то соединении, разберитесь, как оно получилось. Полезнее упражнения, особенно при изучении химии, представить трудно. - Но валентность не всеобща и не всесильна. Всё же это довольно старинная штука. Валентность особенно полезна для p-элементов, в первую очередь для всех неметаллов и металлоидов. Можно даже сказать, что ничего лучше валентности для этого типа элементов химия не придумала. Валентность отлично, просто и ясно упорядочивает практически все необозримое множество соединений этих элементов друг с другом и с s-элементами. От валентности у этих соединений очень легко перейти к электронной структуре. Отсюда же следует то, что валентность очень полезна для органических соединений, можно даже сказать – особенно полезна. Углерод почти всегда четырехвалентен, всё многообразие органических соединений производится от соединений углерода и водорода, углеводородов реакциями замещения – эти два тезиса фактически организуют весь материал органической химии. Органическая химия от этого никогда не отказывалась. Другие меры валентности (в обобщенном смысле), та же степень окисления, в органике непопулярны.
- Наиболее серьёзным испытанием для классической валентности являются соединения с донорно-акцепторной связью, комплексные соединения, координационные соединения. Вопрос вроде простой – сопровождается ли образование таких соединений изменением (увеличением) валентности атома-донора. На первый взгляд, проблема кажется простой – а как иначе, ведь этот атом использует для этого валентные возможности сверх того, что были задействованы в исходном соединении. Но тут же возникает контраргумент – если это так, то и атом акцептор должен изменять валентность, ведь валентность всегда взаимна, в этом ее смысл – атомы соединяются за счет валентностей, и если один расчехлил еще одну валентность, то и другой должен ответить тем же. Но нет, так не происходит и это тоже очевидно: протонирование аммиака, например, никак не изменяет валентность атома водорода – как был одновалентным, так и остался. Можно и другие примеры попробовать, чтобы в этом убедиться. Поэтому на рубеже 19 и 20 веков после работ Вернера сложилась конвенция считать соединения, образованнные этим способом, в отдельный класс, и принимать, что классические валентности атома-донора и атома-акцептора при этом не изменяются. В терминах теорий кислот и оснований Бренстеда-Лоури и Льюиса, возникших немного позднее, это равнозначно утверждению, что взаимодействия кислотно-основного типа валентность не изменяют.
- В химии переходных металлов валентность – очень плохой помощник, ее применять можно, она даже бывает довольно полезна, но делать это надо с огромной острожностью и полным пониманием особенностей структуры соединений переходных металлов, у которых все соединения без исключения, даже с виду самые незатейливые и как две капли воды похожие на аналоги из непереходной химии, являются координационными соединениями, для корректного анализа которых нужны подходы координационной химии. Хромовый ангидрид, например, как две капли воды похож на серный, но если последний – нормальное соединение 6-валентной серы, структуру которого можно элементарно описать с помощью обычных понятий (ковалентные связи, донорно-акцепторные связи и т.п.), то первый – это триоксокомплекс хрома(6+) и для описания его структуры извольте доставать все инструменты координационной химии, кислород в нём лиганд, весьма типичный. А гидриды в химии переходных металлов вообще совсем не годятся как опорные для определения валентности. В то же время в простых соединениях переходных металлов можно аккуратно вести ряды от оксидов (оксид-гидроксид-соли), сохраняя в них валентность в кислотно-основных превращениях точно так же как в соединениях элементов главных подгрупп..
- Валентность является основой короткопериодной формы Таблицы Менделеева. В этой Таблице восемь групп, и номер группы является максимальной валентностью элемента. У каждого элемента существует оксид, соответствующий максимальной валентности. У этой Закономерности есть два важнейших исключения – фтор и кислород, но эти исключения понятны и скорее подчеркивают Закономерность, чем опровергают её. Только в восьмой группе эта закономерность работает со сбоями, но это не так важно, потому что в этой группе состоят переходные металлы, для которых валентность не очень хороший инструмент (не очень хороший – значит работает, но не всегда, см. выше) и инертные газы, у которых химия весьма скудна и очень своеобразна (впрочем у ксенона есть оксид восьмиваленого элемента, и это подтверждает, что даже инертные газы стараются подчиняться общим законам и иногда у них получается). Получается очень красивая система, которую легко запоминать и изучать. Для начального изучения химии эта система великолепна, и ломать ее не стоит. Как ломать – например, утверждать, что в азотной кислоте азот четырехвалентен, таким образом отказывая важнейшему элементу в следовании этой общей закономерности.
- Структурные формулы для определения валентности использовать можно, но очень осторожно. Дело в том, что в ходу у химиков до сих пор два типа структурных формул, между которыми различия очень трудно увидеть на глаз, не имея хорошего опыта в структурной химии. Увы, надо признать, что даже в 21 веке химия остается весьма неряшливой наукой в этом отношении – смысл символов в структурных формулах определен нечётко. Это ужасно, это никто не собирается исправлять, так как все привыкли. Все привыкли, но неопытные химики отлично попадают в ловушки, расставленные плохо определённой системой. Структурные формулы могут отображать и валентности и химические связи.
- Валентности используются в классических структурных формулах. Черта в этих формулах – не химическая связь, а единица валентности. По какой-то странной причине мы не отдаем себе отчета в том, что до сих пор предпочитаем классические структурные формулы перед структурными формулами, изображающими химические связи на основании электронной структуры. Для элементов 1-4 групп разницы между двумя типами структурных формул нет – черта одинаково хорошо описывает и единицу валентности и связь. А вот для 5-7 групп (и для редких соединений инертных газов) разница есть и большая. И мы предпочитаем именно старые формулы с валентностями, делая исключение только для одного элемента, азота. Этот парадокс в химии (а это химия важнейших элементов – фосфора и всех пниктогенов, серы и всех халькогенов, галогенов в степенях окисления выше двух, а также криптона и ксенона) удивительнейшим образом не находит ясного разрешения и постоянно становится источником дурацких недоразумений и даже ошибок. С этим же связана, хотя и косвенным образом, проблема так называемой гипервалентности, которая будет разобрана на отдельной странице.
- Валентность в рамках электронной структры имеет простое толкование – единица валентности соответствует одному валентному электрону (электрону валентной оболочки), но надежно это работает только для элементов главных подгрупп. Поэтому максимальная валентность всегда соответствует числу валентных электронов, или номеру группы в короткопериодной Таблице. Отсюда происходит такой популярный термин как валентные возможности элемента. Валентность, меньшая максимальной, соответствует неполному использованию валентных возможностей. В зависимости от числа неспаренных электронов и электронных пар, которые невозможно распарить, возникает два ряда валентностей – основная и гипервалентность (в главных подгруппах 5-8 групп). В химию переходных металлов с этим лучше не лезть, хотя и там это иногда позволяет быстро понять, например, такое хитрое явление как связи металл-металл. Впрочем и для элементов главных подгрупп это правило скорее позволяет лучше понять, почему элемент проявляет ту или другую валентность, но для непосредственного определения значения валентности этим надо пользоваться с осторожностью – определение валентности через ряды соединений намного надежнее.
- В изучении современной химии можно совсем обойтись без классической валентности, и именно так написаны все большие университетские учебники по неорганической химии и химии элементов (Коттон-Уилкинсон, Шрайбер-Эткинс, Гринвуд-Ёрншоу и др. – во всех этих отличнейших, знаменитейших и авторитетнейших книгах вы вообще не найдёте валентности ни в каком смысле кроме общефилософского). В современных университетских курсах мерой валентности (в общем смысле) обычно считают не валентность (в количественном смысле), а степень окисления, и материал упорядочивают на основании строения атома (и типичных электронных конфигураций). Периодическая таблица для этих целей используется в длиннопериодной форме, где нет главных и побочных подрупп. Химию так безусловно изучать можно, а в Университете и нужно, но на начальном этапе, в школе, это сильно усложняет задачу упорядочивания материала химии. В нашей школе к тому же это еще и категорически не умеют делать, – у нас предпочитают короткопериодную таблицу, а она очень плохо соотносится с электронной структурой, потому что отражает именно классическую валентность.
- При обсуждении валентности иногда всплывает ещё один термин – ковалентность. Под ковалентностью в первой половине прошлого века понимали число ковалентных связей атома. Вот именно ковалентность в этом смысле у азотов аммония и азотной кислоты равнялась четырем, а у углерода и кислорода в оксиде углерода – трём. Поскольку число ковалентных связей у p-элементов должно подчиняться правилу октета, такое понимание ковалентности приводило к тому, что у всех элементов 5-8 групп, главных подрупп, ковалентность не могла быть больше четырёх, да собственно в большинстве соединений и равнялась четырём (углерод везде, азот в аммонии, диоксиде азота, азотной кислоте, нитросоединениях, нитратах, сера в серной кислоте, хлор в хлорной кислоте и перхлоратах, фосфор в фосфорной и фосфоновых кислотах и так далее), большого смысла в этой системе найти не удалось, и она как-то сама без посторонней помощи тихо слилась. У этой системы есть и ещё один недостаток – она невольно вызывает яростные споры по поводу того, что и когда можно считать ковалентной связью. Споры эти довольно бессмысленны, но это не мешает им вспыхивать с новой силой.
В современной химии под ковалентностью понимают совершенно иное – это антоним к слову ионность, используемый в рассуждениях о том, чем отличается полярная ковалентная связь и ее разновидности от чисто ионной (электростатической). У чисто ионной связи ковалентность равна нулю. Но валентность натрия в хлориде натрия равна единице. И хлора тоже. Одно это ставит крест на отождествлению ковалентности с валентностью в осовремененном виде. Такого отождествления никогда не было и нет.
Каков смысл термина "валентность" и как он изменился за 200 лет химии
Слово “валентность” чрезвычайно важно в химии, потому что именно в этом слове скрывается сама химия. Нет валентности нет химии. В самом глубоком смысле этого слова оно сейчас столь же актуально, как и тогда, когда впервые возникло в самом начале 2-й половины 19 века. Но смысл его стал намного глубже.
Валентность в первоначальном понимании – следствие того, что элементы в химических соединениях входят в состав в дискретных пропорциях, кратных атомным массам элементов. После установления этих соотношений кратности все время расширявшееся множество химических соединений было упорядочено, все 1860-е разные исследователи пытались найти секрет этого упорядочивания, шаг за шагом приближаясь к окончательной формулировке Д.И.Менделеевым Периодического закона и установления формы Периодической таблицы в конце того десятилетия. Это произвошло очень быстро как только в конце 1850-х появилось и утвердилось само понятие валентности, было признано, что эта величина у каждого элемента может принимать одно или несколько значений, и в этих величинах угадывается некая закономерность. Это очень важно: химия в научном виде стала формироваться уже в конце 18 века, но 50 лет с лишним потребовалось на то, чтобы выработать понятие валентности и определиться с ним. И как только это было сделано и за несколько лет овладело химками, равитие химии, и органической и неорганической буквально полетело – будет время и желание попробуйте посмотреть несколько статей даже начала 1860-х и несколько статей 10 лет спустя – просто наугад, откройте любые научные журналы тех времен, – увидите, как из просто рассказов об опытах вдург появляется уже вполне привычный нам способ изложения, с схемами реакций, структурными формулами, рассуждениями о тенденциях и закономерностях. Ключ к этому взрыву – валентность. До открытия электронной структуры, а это произойдёт еще через полвека с лишним, только валентность и тащила на себе всю химию, и классно тащила – тогда были открыты все недостающие элементы, получены тысячи соединений, открыты все классические реакции, появилась термодинамика и кинетика, катализ и ошеломляющее количество информации.
Всё это время и во многих краях и сильно после, у слова валентность только один, классический смысл – закономерность соотношения элементов в соединениях, кратность эквивалентных весов.
Но вот возникает представление об атоме и электронах, и отчетливое осознание того, что именно электроны делают всю работу химии. Тогда и стало ясно, что такое собственно химия, о чем она. Физика разобрала всю материю на маленькие частицы, разобралась в том. как они взаимодействуют, собрала из этих частиц ядра, установила, что каждый тип ядра соответсвует элементу, а если в поле этих ядер запустить электроны, получаются атомы. И атомы соответствуют элементам. И уже самая ранняя квантовая физика вполне разобралась с тем, как устроены атомы, и нашла причину того, почему атомы можно расположить в упорядоченной таблице, только таблица эта выглядела не как Таблица Менделеева, она же короткопериодная, а как таблица строения атома, упорядоченная по значениям квантовых числе или по заполнению оболочек – длиннопериодная таблица. Но пока это отдельные атомы, это физика, а не химия. Атомные спектры, например, хорошо объясняет.
И тут, чисто из этой физики выяснилась совершенно сногсшибательная вещь – если два атома окажутся достаточно близко друг к другу, часть электронов вдруг вместо того, чтобы оставаться в поле своего ядра, вдруг “привязываются” к обоим атомам, их состояние и движение, в квантовом смысле, больше нельзя описать в рамках “своего”ядра, но приходится решать более хитрую задачу – электрон в поле двух ядер (и всех остальных электронов). И такое сближение атомов оказалось может дать устойчивую двух-атомную систему, и в этой системе химики, разинувши рты, узнали свои молекулы. Атомов может быть не два, а больше, и тоже часть электронов всех атомов перепривязывается к нескольким атомам, и это более сложные молекулы, причем это происходит только если атомы находятся в конкретных положениях друг относительно друга.
Из этого следуют две вещи, одна хорошая, другая плохая, но никто точно не знает какая. Во-первых, следует, что химия содержится в физике вот прямо вся целиком, и нет ничего такого, что было бы нужно еще добавить. Эта мысль свела с ума многих. Во-вторых, когда разобрались чуть получше, то выяснили, что содержится таким хитрым образом, что достать одну из другой категорически невозможно – химия в физике и правда содержится, но не просто так, а в зашифрованном виде, и шифр этот имеет сложность, несовместимую с имеющимися у человечества возможностями расшифровки. Довольно типичная проблема, однако. Вот сейф, у него замок с шифром, внутри сейфа нечто невероятно ценное – задача открыть сейф за какое-то разумное время. Строго говоря, время не ограничено, число попыток любое – сиди и открывай. Сто лет прошло с тех пор, пока не открыли, хотя пока одни ломают голову над шифром, целая толпа более нетерпеливых со всех сторон крушит сейф динамитом, сверлит, пилит, режет, гнёт фомой дверь – через щели и дырки многое уже видно, но далеко не всё.
И вот мы сидим в самом начале этого пути. Мы только что выяснили, что атомы могут взаимодействовать, образуя молекулы. Но мы это и так уже давно знаем – мы знаем что атомы соединяются и в определенных соотношениях, называемых валентностью. Но тут мы узнали, что причина этого – электроны. Электроны из атомов придают им способность соединяться. В этом момент возникает очень богатая идея. В наиболее ясном виде ее изложил в 1927 году совершенно гениальный английский даже не химик, а скорее мыслитель Невил Винсент Сиджвик. Сиджвик написал книгу под невероятно амбициозным названием Электронная теория валентности. Не на пустом месте, напротив, сразу после появления модели атома сразу несколько нехилых химиков – Льюис, Лангмюр, Коссель – принялись думать, а как через эту модель добраться до химии. Льюис уже в 1923 написал книжку с названием Валентность, но он еще не очень далеко зашёл, и этот текст выглядит немного архаично. Именно Сиджвик всего несколько лет спустя первый всё это собрал, сам как следует передумал, и изложил одной книгой, и в общем-то именно с этой книги и начинается вся современная химия.
В этой книге Сиджвик делает мощное обобщение – он понял, что знакомое каждому химику понятие валентности (в смысле количественного соотношения эквивалентов) выражает фундаментальное свойство атомов соединяться в молекулы, заложенное в структуре атома, и связанное с электронами. Поэтому валентность теперь имеет куда более фундаментальный философский смысл – Valency is a general term used to describe the power which atoms possess of combining with one another to form molecules. Кокетливые люди однако все большие философы – он пишет used, используемая, но никто до тех пор так валентность не понимал, валентность понимали только в количественном смысле соотношения эквивалентов, это именно Сиджвик захотел, как у нас любили говорить про всяких вождей, – возвыситься до обобщения и задать новый смысл привычному понятию. Удивительно, но Сиджвик немного похож на Ленина, особенно молодого, времён третьего съезда РСДРП. Но внешность обманчива, к счастью, и из двух внешне похожих людей один был озабочен тем, как облагодетельствовать абстрактных людей в будущем за счёт массового убийства и ограбления современников, а другой усердно закладывал основы одной из важнейших наук, которая точно поможет сделать много важного и полезного и изменить мир к лучшему (да, химия много пачкает, но по мере развития и убирает за собой и многое изменяет к лучшему). И возвысился. Именно поэтому так называется его книга – он в ней собрался не разбирать, отчего один элемент одновалентен, другой двухвалентен и так далее, а куда выше – вообще понять, где заложена эта способность атомов. Атом – физическое понятие. Молекула – химическое. Валентность – это то, что делает химию из физики. Не было бы валентности (в общем смысле Сиджвика), не было бы химии. Так и была бы Вселенная, звёзды были бы, а вот планет уже нет – не сделаешь даже самую простую планету из отдельных атомов, даже газовые планеты состоят не из атомов, а из простых молекул типа воды, метана, азота, аммиака.
От Сиджвика далее валентность приобрела этот общий смысл. За 20 век было написано несколько фундаментальных книг, раскрывавших валентность именно в этом смысле, и почти повторявших называние книги Сиджвика. Теория валентности – это фактически синоним квантовой химии или теории химической связи. Многие крупные ученые стеснялись старого понимания валентности, и видели в этом слове только этот, обобщенный смысл. Это касается прежде всего университетской химии. Учебник Коттона и Уилкинсона (Advanced Inorganic Chemistry, есть несколько русских переводов с разных изданий), а это не просто учебник, а фактически целая революция в химии элементов, вообще брезгует старым понятием валентности, не считает нужным даже упоминать это. Многие другие знаменитые и большие учебники (Гринвуд-Ёрншоу, Виберг-Виберг-Холлеман, Шрайвер-Эткинс) тоже относятся к валетности как к не очень нужной архаике. Я немного еще коснусь этой темы ниже на вкладке сопоставления валентности истепени окисления. Но так или иначе, это задает отношение в среде профессиональных химиков с серьёзным университетским бэкгрундом – валентность это или чрезвычайно общая вещь, абстрактная основа химии, скорее философская абстракция, которую каждый мыслитель волен наполнять новым смыслом; либо то, что знает любой с первых шагов в химии, и что не требуется как-то повторять или заново рассматривать, ежу понятно, что кислород двухвалентен и так далее.
Проблемы с этим словом у профессиональных химиков, постоянно связанных с научной работой и читающих много статей и монографий, тоже есть и они намного сложнее. Проблема в том, и я это уже не раз писал и говорил, что в серьёзных науках нет общепризнанных авторитетов на всю науку целиком и особенно на ее основы. В конкретных областях и направлениях авторитеты есть, правда и там они не навсегда, и постоянно меняются и конкурируют. А в науке в целом нет даже и этого. Ох, как это не нравится в остающих странах, склонных к авторитаризму в политике – так в этих странах кажется странным и невозможным, что в такой важной вещи как наука (а то, что наука важна понимают уже кажется все, не поручусь за талибов и Мадуро, но, например, покойный архетип великого диктатора Фидель Кастро понимал отлично) нет главного ученого, с которым могли бы говорить вожди и непосредственно давать задачи на опрежающее развитие местной науки. Вождям это позарез нужно. Но вот нет, ни фига из этого не выходит. Даже Самых Главных Ученых успешно назначают для общения с вождями. А дальше все как-то не проходит. Не тех, видимо, находят. Но нет, не в том дело. Кого ни найди будет всё одно и то же. Наука или свободна и быстро развивается, или выстроена в отраслевую вертикаль и столь же быстро превращается в инструкцию по использованию купленных в других странах прибамбасов (гаджетов, самолётов, разнообразных модных таратаек, станков, лекарств, приборов, заводов и т.п.).
Поэтому как только в науке возникает вопрос, а что такое нечто важное, то ну никак не появляется руководящее указание, действительное во всём вменяемом мире, однозначно определяющее эту важную вещь или понятие. Попробуйте, например, однозначно понять, что такое энергия – уж кажется важнее понятия в науках нет – и не найдёте одного толкования. И так почти про все. И чем фундаментальнее понятие, тем больше мнений, тем сложнее найти равнодействующую. Как же быть?
В общем довольно просто, хотя и не так просто, чтобы просто посмотреть в самую главную энциклопедию (такой нет и ни в коем случае не считайте таковой википедию!!). Надо попробовать понять, какие мнения существуют, то есть попробовать разобраться в том, каким смыслом наполняют понятие разные серьёзные люди (профессионалы в данной науке, желательно действующие, действующим профессионалам всегда доверие больше, чем пенсионерам, профессиональным преподам, никогда не работавшим в исследовательской науке, журналистам, просто любителям), есть ли у этих мнений общий вектор, есть ли разные школы, какими смыслами оперирует каждая.
Что такое классическая валентность
Валентность не имеет никакого отношения к числу связей, типу связей, и всему, что связано с электронной теорией структуры. В момент появления валентности ничего этого не было, а когда это появилось валентность пытались переопределить с помощью терминов электронной структуры и квантовой химии, но не трогая саму валентность, а вводя новые характеристики – электровалентность, ковалентность, степень окисления, порядок связи, координационное число и т.п. Обо всем этом ниже. Поскольку у слова валентность появился качественный, обобщающий смысл для количественной характеристики валентности в этом смысле стали вводить меры валентности. Наиболее общеупотребительной мерой валентности стала степень окисления, которая в современной химии определена с высокой степенью детальности. Но осталась и старая классическая валентность, которую теперь точно так же понимают как еще одну меру валентности в общем смысле.
Валентность в количественном смысле определяется из стехиометрии соединений. Только для простых бинарных соединений это делается прямым расчетом, исходя из элементного состава. Этому упражнению учат в школе, и все его знают. Смысл его в том, что в состав бинарных соединий элементы входят в соотношении эквивалентов – у каждого элемента с атомной массой А есть один или несколько эквивалентов. Эквиваленты определяются делением атомной массы на некоторое небольшое целое число от 1 до 8. У каждого элемента есть свой набор этих чисел. Эти числа были найдены в опытах – исследователи открывали новые соединения, определяли их состав, и находили эти все время повторяющиеся (в пределах точности) величины. Это произошло очень рано в химии – в первой половине 19 века произошло это первичное накопление химического капитала. Никого не убили и не ограбили, между прочим. И вот эти числа связаны с эвивалентами. И атомную массу элемента можно представить как валентность * эквивалент. Элемент так и называли сначала: скажем, кислород, валентность два, эквивалент равен 8.0, атомный вес (назовём по-старому, сейчас называем атомной массой) тогда равен двум эквивалентам, и так говорили сначала – кислород двухэквивалентен. Это быстро немного сократили для удобоговорения – двухвалентен. Кислород двухвалентен. А, например, азот бывает двух, трех, четырех, пятивалентен. Это значит, например, что у азота есть оксиды, состав которых состоит из соответствующих эквивалентов.

Таким способом было для всех тогда известных элементов найдены валентности. В этом процессе выяснилось, что есть некоторые элементы у которых только одна валентность. В первую очередь это водород – он всегда одновалентен. Поэтому в соединениях элементов с водородом число атомов водорода равно валентности элемента. Во вторую очередь кислород – он всегда двухвалентен. Ну и еще много других элементов тоже, но водород и кислород хороши тем, что у почти всех элементов есть соединения и с тем, и с другим. Поэтому эти два элемента стали опорными для определения валентностей других элементов.
The maximum number of univalent atoms (originally hydrogen or chlorine atoms) that may combine with an atom of the element under consideration, or with a fragment, or for which an atom of this element can be substituted. – Валентность это максимальное число одновалентных атомов (в оригинале водорода или хлора) которое может соединяться с атомом элемента, или с фрагментом, или на которое можно заместить атом элемента.
Это ужасное определение, крайне двусмысленное и неудобное, но оно важно тем, что мы видим, что ИЮПАК не планирует переосмыслять валентность на основе подсчета связей, электронов или чего-то подобного. Это по-прежнему соотношение эквивалентов, упрощенное за счет использованием одновалентного атома в качестве опоры расчета. ИЮПАК использует слово “combine” специально для того, чтобы не вдаваться в подробности, КАК атомы соединяются – здесь вообще нет даже слова “связь”, “связывается” – и это правильно, потому что в понятие валентности это не входит.
Внесем в это ясность на основе сложившейся традиции. Нам ведь нужно оперировать валентностью не только в бинарных соединениях. Система общеизвестна, просто напомню. Валентность элемента понимается как способность соединяться с другими элементами, причем способность имеет количественное выражение – у каждого элемента есть одна или несколько единиц валентностей, в сумме равных валентности. У одновалентного элемента одна валентность, у двухвалентного две валентности, у трехвалентного три валентности, и т.д. И элементы соединяются именно единицами валентности, часто просто говорят – валентностями. Для образования устойчивого соединения все валентности элементов должны быть использованы, не может остаться ни одной неиспользованной валентности. Этот подход использовался с 1860-х для рисования структур, и, как ни странно, используется до сих пор практически без изменений. Черточки в этих структурах – это валентности, а не связи. Подробнее об этом на отдельной вкладке.
Безусловно тут не все просто, и химии пришлось решать много проблем, в частности устанавливать, как связаны атомы в соединениях, в каком порядке. Собственно, в этом и состоит очень важная часть химии – теория строения. Установили, например, что в кислородных кислотах и гидроксидах, элемент связан с кислородом и водороды связаны с кислородом, за редкими исключениями. Тогда стало просто рисовать структуры из валентностей и определять валентности элементов, причем удобно начинать именно с кислорода. Например, серная кислота: четыре кислорода имеют восемь валентностей, две из них уходят на водороды, остается шесть – атом серы обязан выставить шесть своих валентностей. Этот алгоритм проходят в школе, не будем пережевывать сто раз жеваное. Хотя тут всё такое, но пришло же кому-то в голову опять вытаскивать из пыльных сундуков старую добрую валентность. У азотной кислоты три кислорода, шесть валентностей, одна – водороду, на азот осталось пять. Как можно найти в азотной кислоте четырехвалентный азот? Да, чёрт знает!
Такие же соображения применяют, когда вдруг находят что-то нестандарное. Например, у азота есть еще один оксид, закись азота, вроде азот в нем одновалентен. Но еще в 19 веке выяснили, что это необычный оксид, в нем кислород связан только с одним азотом. А вообще азот чаще всего трехвалентен. Ну и попробовали собрать молекулу из двухвалентного кислорода, трехвалентного азота, и еще одного азота – какого? Задачка на комбинирование палочек. Ну вот так как-то можно попробовать. Пятивалентный азот в середине получился. Так и рисовали. В 20-м веке выясниилось, что там есть одна проблема, которую чаще всего принято называть гипервалентностью, я подробно разберу это отдельно. Но на валентность это никакого влияния не оказало. Она по-прежнему пять, что в азотной кислоте, что в закиси азота.
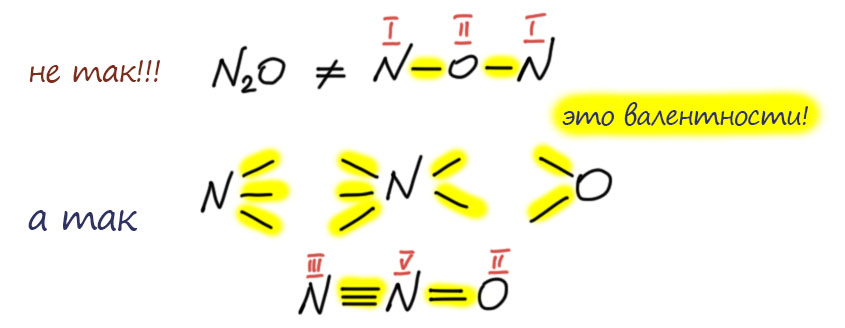
Следующий шаг в валентности очень важен. Этот шаг говорит, что для того чтобы знать валентности атомов в соединениях, даже весьма сложных, нужно установить связи этого соединения с более простыми соединениями, используя общеизвестные соображения о взаимопревращениях соединений. Такие связи устанавливаются в хорошо известных рядах превращений. Валентность в таких рядах сохраняется. Валентность издревле обозначают римской цифрой в скобках или надстрочнике. К сожалению в литературе последних 70 лет эту конвенцию соблюдают не всегда – об этом на вкладке про степень окисления. Мы будем ее соблюдать.
- Ряды: элемент – оксид – гидроксид или кислородная кислота – соли
Например: азот – азотный ангидрид – азотная кислота – нитраты (5-тивалентый азот)
хром – Cr2O3 – Cr(OH)3 – соли хрома(III) - Ряды: элемент – гидрид – продукты замещения атомов водорода на другие элементы или остатки
Это очень большие ряды, связывающие огромное количество соединений. Достаточно только сказать, что ряд от гидридов к продуктам замещения создает практически все органические соединения: углерод четырехвалентен. Эта идея в начале второй половины 19 века произвела революцию в органической химии, фактически создав ее в том виде, которым мы и сейчас пользуемся.
Кроме этих рядов, производящих огромное количество соединений, есть еще одна важнейшая конвенция, возникшая из споров о том, как устроены комплексные или координационные соединения. Суть ее в том, что кислотно-основные взаимодействия не изменяют валентность атома-донора и атома-акцептора. Это относится и к теории Бренстеда-Лоури, и к теории Льюиса. Это дает нам, например, ряд азот – аммиак – аммоний, бесконечно продолжающийся через замещение на амины и замещенные аммонии, гидразин и его призводные, гидроксиламин и его производные и т.д. И, с другой стороны, ряд железо – оксид железа(II) – гидроксид железа(II) – цианид железа(II) – жёлтая кровяная соль и другие комплексные соединения железа(II) вплоть до ферроцена, который получается кислотно-основным взаимодействием Льюиса иона железа(II) и циклопентадиенильного аниона. Как видим, эта простая конценция позволяет легко обозначать валентность даже в очень нетривиальных соединениях. Напомню в очередной раз – валентность это про стехиометрию соединений, а не про связи.
Почему валентность так важна в химии?
Это очень просто – валентность организует материал химии. Мы изучаем эту науку как химию элементов, и у каждого элемента смотрим на множество соединений. Способ их упорядочивать изобрёл, как известно, Менделеев. Этот способ называется Периодической таблицей элементов, а сейчас нам уже нужно уточнять – короткопериодной формой Таблицы. Вот она, например, такая знакомая. В России мы именно такую используем со школы.
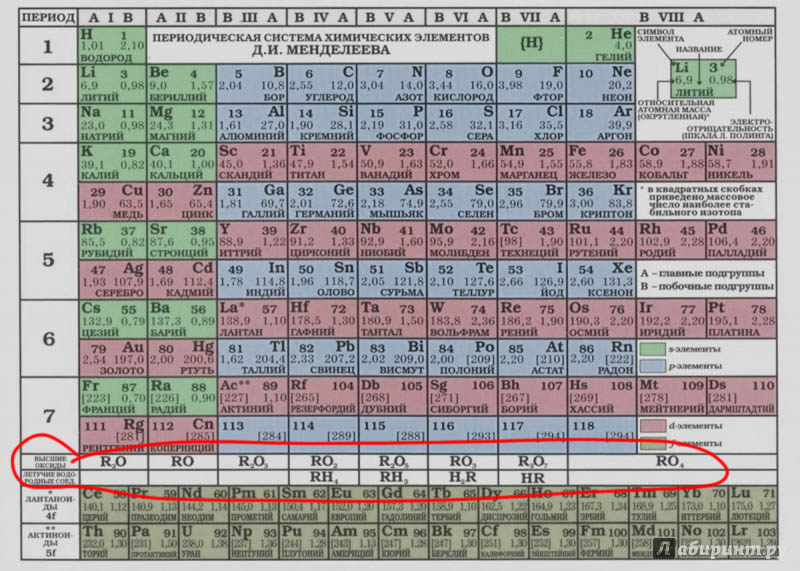
И хотя это совершенно очевидно, но приходится напоминать, что эта классическая Таблица построена именно на валентности. Хотя к этому подбирались и предшественники Менделеева, в первую очередь Лотар Мейер, именно Менделеев довел систематику элементов до окончательного вида, причем он был настолько уверен в этом решении, что оставил пустыми места, в которых должны будут расположиться еще неоткрытые тогда редкие элементы. Такую уверенность дал именно принцип расположения элементов по валентности в типичных соединениях. Никаких других характеристик атомов тогда не было. Атомная масса природной формы элемента – только подспорье в первоначальной сортировке, причем мы знаем, что валентность заставляет переставлять некоторые элементы, нарушая порядок сортировки по массам. Хорошо бы эту потрясающую вещь осознать: мы так сейчас привыкли к тому, что о химии надо рассуждать с помощью электронов, орбиталей и тому подобных вещей, что можем немало изумиться тому, что самый главный инструмент химии, Периодическая таблица, возникла тогда, когда ничего этого не было даже в проекте, а все делалось только на основе анализа закономерностей в стехиометрии соединений, то есть классической валентности. И ничего, Таблица сразу получилась правильная, и нам с тех пор только и осталось, что наполнять её новыми смыслами, да пытаться как-то по-новому нарисовать, в точности сохраняя главное. Ни один (!) элемент с тех пор не поменял места в Таблице, и все добавленные элементы легли в свои клетки, как влитые. Вот что валентность животворящая делает!
Напомню опять некоторые общеизвестные вещи, на которых Таблица и основана. Все эти вещи – про валентность.
Во-первых, это потрясающая закономерность – у многих важных элементов, неметаллов и металлоидов, сумма валентностей элемента в гидриде и высшем оксиде равна восьми (метан и углекислота, аммиак и азотный ангидрид, сероводород и серный ангидрид, хлористый водород и хлорный ангидрид). Это наблюдение фактически предвосхищает открытие электронной структуры атомов и правила октета Льюиса-Лангмюра. Таблица поэтому основана на особой важности числа 8 – в ней восемь групп, и номер группы не формален – это возможность максимальной валентности. Элемент из группы N может иметь соединения с максимальной валентностью N. Сейчас мы отлично знаем, что не все элементы пользуются этой возможностью, но во времена Менделеева это еще никто не знал, сведения об элементах были отрывочны – что кто случайно получил, а после Менделеева поиск новых соединений и еще не открытых элементов стал систематическим. И этот поиск показал, что система работает с какой-то ужасающей надежностью, в том смысле, что отклонений от нее – нарушения максимальной валентности практически найдено не было. Собственно, есть только одна группа, в которой есть такая проблема – восьмая. Но мы давно знаем, что восьмая группа не такая как все остальные. Это такая помойка, в которую свалили все, что не поместилось в нормальные группы, причем во времена Менделеева в этой группе еще не было главной подгруппы. Потом она появилась, и сейчас мы вполне знаем, что и тут все сошлось – есть валентность восемь у ксенона, и у некоторых металлов тоже.
Число восемь проявляется еще в одной важнейшей закономерности. Валентность в гидридах, как известно, растет сначала до 4 группы и затем так же уменьшается. Период поэтому ведет себя как такая валентная горка: 1, 2, 3, 4, 3, 2, 1. Эти числа (добавим справа ещё ноль для восьмой группы) играют еще одну важнейшую роль – это самая распространенная валентность в группе. Выпишем их по группам вместе с примерами типичных соединений. И под ними выпишем максимальную валентность в группе, тоже вместе с примерами типичных соединений. И тогда мы увидим интересную вещь – для первых четырех групп нормальная валентность совпадает с максимальной. А для последних четырех эти величины расходятся: нормальная валентность уменьшается, а максимальная растет. Вот эта странность последних 4 групп была давно замечена, но объяснение получила только во второй половине 20 века, когда возникло такое понятие как гипервалентность. Из этой картинки мы видим почему это так называется – это как будто некая избыточная (гипер) валентность по сравнению с нормальной. То есть в последних 4 группах обычная валентность встречается в большинстве соединений, но есть возможность потрудится и разбудить дополнительные валентные возможности. Это настолько интересный феномен, являющийся причиной совершенно дикой путаницы даже в весьма серьезных источниках, настолько важен, что я вынес его обсуждение на отдельную страницу.

Надо только заметить, что когда появилась электронная теория и понимание сути валентных оболочек, стало ясно, что у d- и f-элементов есть вроде бы большие резервы для образования связей. Но фокус в том, что эти резервы практически не используются для образования обычных соединений, которые входят в тем самые ряды по валентности. Пока же просто отметим, что только металлы подгруппы меди, находящиеся в Таблице Менделеева в 1-й группе, нарушают правило максимальной валентности, проявляя и в простых соединениях (оксидах, галидах, простых солях) валентности 2 и 3. И на этом всё.
Это удивительно и надо понять, что это ни из чего более-менее простого не следует. Чтобы разобраться в возможностях образования разных электронных конфигураций потребовалась самая современная квантовая наука, и ее выводы невозможно нормальным языком довести до нормального человека, даже профессионального химика. Эта современная наука просто говорит нам – спокойно, мы всё проверили, Менделеев оказался прав, так бывает, некоторые люди по каким-то неведомым причинам умудряются советоваться с богами и получать от них гарантии. Химия оказалась заперта внутри Таблицы Менделеева, она может выйти за ее пределы только вверх, надстраивая систему простых соединений, которая задана Таблицей, только сверху – координационными соединениями, молекулярными комплексами, супрамолекулярными структурами, макромолекулами, и переходя на уровень организации вещества от отдельных молекул. Получается колоссальная и всё время растущая наука, но стоит она на малюсенькой и компактной площадке, которая совершенно незыблема, не расширяется, не расползается. Эта площадка организована на основе валентности.
Теперь давайте все это сломаем. Азот у адептов “новой валентности” не бывает более чем четырехвалентным. Ниже я покажу, что проблема не только в азоте, но даже если бы только азот проявил такую странную строптивость, это бы одно ломало всю систему Периодической таблицы. Мы знаем, что у трех следующих элементов – кислорода, фтора, неона – бывает только нормальная валентность (у кислорода есть одно соединение-исключение), и это вписывается в схему. Но валентность четыре не вписывается ни в одну схему: это все равно больше, чем нормальная валентность для 5 группы, три. Но не равно номеру группы. И что мы достигаем такой “реформой”?
Валентность и структурная формула: можно ли определить валентность по структурной формуле?
Можно, но очень осторожно. Соответствие между структурной формулой и валентностью атомов весьма запутано, потому что с момента появления электронных представлений о химической связи, единых правил рисования структур так и не появилось, и в химической литературе весьма причудливым образом болтаются остатки сразу нескольких систем, бывших в употреблении, и мало кто обращает на это внимание. Может и не нужно? А это ваше дело, как к этому относиться, это зависит от того, что собственно вы хотите от структуры.
Важно понимать, что валентность старше структуры. Старше и в том смысле, что появилась раньше, и в том, что валентность можно и нужно устанавливать до структуры по химическим взаимоотношениям соединений в рядах, восходящих к простым бинарным соединениям. Азот в азотной кисоте пятивалентен потому что азотная кислота происходит от азотного ангидрида, валентность в котором устанавливается непосредственно по соотношению эквивалентов азота и кислорода. Я постоянно повторяю этот пример, потому что на меня произвело огромное впечатление стремление современных методистов доказать, что азот в азотной кислоте четырёхвалентен, поставив структуру, весьма поверхностно понятую, впереди валентности. И сломав тем самым очевидный ряд. О других, не менее поразительных примерах поговорим дальше.
Мы сейчас понимаем структуру однозначно: как картинку атомов, соединённых химическими связями. Дальше мы начинаем думать, что такое химические связи и какие они бывают, очень часто на этой дороге возникают вопросы, мы сначала честно их пытаемся разрешить, не всегда получается, и в конце концов смиряемся с тем, что всё сложно, а жить надо, и принимаем какую-нибудь упрощённо-компромиссную картину связывания, не всегда корректную.
А вот до появления этих представлений структуры рисовали вполне определенным образом, соединяли элементы черточками, количество черточек соответствовало валентностям. Каждая черточка – единица валентности. Ещё раз – не связь! – а единица валентности. А почему не связь? Некоторые ученые 19 века использовали и такое слово, но по этому поводу были большие споры, а можно ли вообще использовать такое понятие как связь. Не первый раз пишу, что ученые 19 века были строгими позитивистами в философском смысле, за что их совершенно страстно ненавидел Ленин и даже книжку по этому поводу написал, весьма злобный памфлет под названием Материализм и эмпириокритицизм. Ученые настаивали на том, что единственный источник заний – опыт, эксперимент, и его интерпретация, вновь проверяемая опытом. Если что-то нельзя установить опытом, это не предмет науки, вненаучная фантазия. Ленин же настаивал на том, что существует некоторая истина, материя, и существует она сама по себе. Знания о материи истинны. Ленин не озабочивался вопросом, а откуда эти знания берутся, и что делать, если их не получается установить. В этом месте может показаться, что противоречие между позитивистами-учеными и шарлатаном Лениным чисто кажущееся, просто де Ленин намекает на то, что ученые придумают новые эксперименты и разберутся, наука всё время развивается, какие проблемы. Типа, как говорят любители Ленина – смотрел в будущее, “в черепе сотней губерний ворочал…взвешивал мир в течение ночи”. Фокус в том, что великий вождь Ленин и его поклонники, малые вождьки, были совершенно уверены в том, что они имеют инструмент отделения верных знаний от неверных, что они, конечно, не знают каких-то деталей, но имеют некую целостную картину материи, и могут на этом основании отделять правильную науку от неправильной. Ленин на этом пути установил планку и весьма высоко, в памфлете он страстно клеймит великих физиков Больцмана и Клаузиуса, фактически обвиняя их в непонимании физики. Он, типа, лучше понимает Второе начало термодинамики, чем ученые, которые это начало и открыли и разработали. Это совершенно дикая самоуверенность, почти на грани душевной болезни. Не понимая ни одной формулы физики, на основании своих вульгарно-материалистических построений, он крушит всё, что ему не нравится, и что подвергает сомнению его собственную мечту кроить мир в соответствии со своими завиральными идеями. Мы отлично знаем, что было дальше, с каким удовольствием этим же стали пользоваться дальше уже совсем убогие вождьки, и какой страшный урон это нанесло российской науке, вполне передовой в начале 20 века.
Так вот настоящие учёные в 19 веке были позитивистами. И очень осторожно относились к понятию “связь” – в то время трудно было себе представить эксперимент, который можно было бы использовать для обоснования этого понятия. В 19 веке вполне поняли масштаб размеров атомов и молекул, Иоганн Лошмидт в 1865 году оценил диаметр молекулы кислорода в число порядка одного нанометра, одной миллионной части миллиметра. Оптику в те времена знали уже очень хорошо, и без лишних споров поняли, что визуально молекулы рассмотреть не получится возможно никогда. И как ещё их можно рассмотреть тоже понятия не было никакого. Для каких-либо экспериментов на уровне молекул и для их корректной интерпретации нужна квантовая физика, и это всё тогда было сильно впереди и за горизонтом. Поэтому химики того времени старались придерживаться того, что было для них ясно из их экспериментов. Было ясно, что атомы элементов могут каким-то образом связываться с атомами других элементов, но ничего кроме электричества, зарядов, притяжения и отталкивания зарядов для того, чтобы попробовать объяснить как атомы связываются друг с другом, не было. Электричество и электростатика возникли намного раньше электронов. Ученые того времени естественно ходили вокруг электричества, тем более что представление о ионах тоже уже тогда было. Но электричество не может объяснить одну важную вещь – почему атом притягивает (связывает) всегда одно и то же число других атомов. Можно это выразить так – электростатическое взаимодействие не является насыщаемым – пока есть заряд и место вокруг него, он будет притягивать другие заряды. А вот взаимодействие атомов как раз является насыщаемым – как только атом притянет (свяжет) положенное количество других атомов, он больше не хочет, он насытился. И наличие места ни при чём -для некоторых атомов для насыщения достаточно одного другого атома, для других – двух, или трех, или четырех. Все это слишком мало, чтобы занять все место. Явно не в месте дело.
В общем, пришлось признать, что разобраться, как связаны атомы, есть ли между ними какая-то связь, и какова природа этой связи, в 19 веке было невозможно, это было за пределами опыта и разумной интерпретации, и настоящие позитивисты, а это, повторю еще раз, было что-то типа правил приличия для ученого 19 века, признали, что единственное, что они знают и установили из опытов – это соотношения элементов в соединениях, то есть валентность. Валентность – это свойство атомов элемента, определенное экспериментально. Вполне в рамках позитивизма, основной философии научного познания 19 века. Мы не знаем почему это так, но знаем что это именно так из надежных и многократно повторенных опытов. Кто-то считает, что у водорода может быть валентность два – отлично, предъявляйте ваш эксперимент (представление о водородной связи, особенно сильной как в дигидрофторид-ионе появилось уже в 20-м веке, а это можно попробовать интерпретировать как наличие у водорода более одной валентности и потребуются дополнительные соображения чтобы доказать, что это не так). У кислорода три? – данные на стол! – нет? – вон отсюда!!
Если есть валентность, получается весьма стройная теория структуры молекул, не выходящая за рамки опыта и его разумной интерпретации. У каждого атома есть своя валентность, то есть способность соединяться с определенным числом других атомов, в соответствии с их валентностями. Атомы соединяются валентностями. Валентность изображается прямой чертой. Получается то, что мы узнаем как структурные формулы, только у нас в это понятие вложено гораздо больше смысла – мы видим молекулу как конкретный геометрический объект, знаем какие там связи, какие у них длины, какие углы, двугранные углы, знаем кое-что о внутреннем вращении и так далее, и всё это можем увидеть в экперименте с помощью разных дифракционных методов, и с успехом объясняем с помощью какой-нибудь квантовой теории, более простой, типа правил Джиллеспи-Нюхольма, или более сложной типа теории МО или квантовых расчетов разной степени сложности. А в те времена под структурой понимали просто соединение атомов, соответствующее валентностям. И никакой геометрии за исключением одного важного случая – теории Вант-Гоффа и Ле Беля об асимметрическом атоме углерода, это 1870-е, изрядная старина, несколько лет всего после Таблицы Менделеева, тем более это невероятно гениальная теория. Гипотеза Вант Гоффа – Ле Беля потребовалась, чтобы объяснить экспериментальный факт – наличие оптических изомеров. Раз такие изомеры – а это экспериментальный факт – есть, нужно это объяснить. У нас нет ничего кроме валентности (углерод 4-валентен) и из этого не следует ничего, кроме того, что рядом с углеродом 4 других атома. Всё, больше ничего нет. Объяснение, пришедшее в голову одновременно голландцу и французу – образец лаконичного и прямого мышления. Вокруг насыщенного атома углерода четыре валентности и атомы на них образуют тетраэдр.
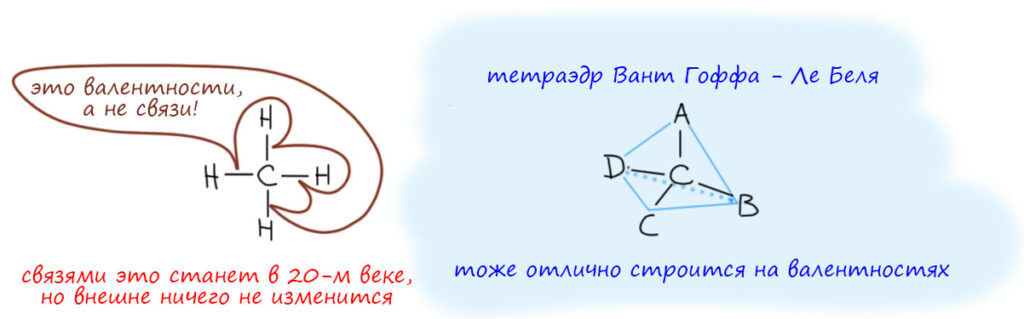
Для этого, обратите внимание, не потребовалось петрить в гибридизации и орбиталях. Вант Гофф и Ле Бель и не петрили, и отлично себя чувствовали, Вант Гофф (точнее ван’т Хофф) даже в конце получил нобелевку, правда не только за это, а по совокупности заслуг перед химией, которых хватило бы на дюжину крупных учёных. Вообще для этого не нужно ничего, кроме догадки о том, что тетраэдр – самое естественное расположение четырех атомов (шариков) вокруг одного атома (шарика): тетраэдр – это просто самая симметричная фигура с четырьмя вершинами, делающая эти четыре вершины наиболее равноправными в пространстве. Самая позитивистская гипотеза из всех возможных – если мы не знаем ничего, кроме того, что вершин четыре, самая простая фигура в пространстве – это тетраэдр. И немного повертев тетраэдр в руках, выясняется, что при четырех разных вершинах получается два разных тетраэдра. Любая другая будет сложнее, потому что придется объяснять, отчего какие-то атомы получили какое-то особое расположение, из чего это следует, откуда данные, из какого эксперимента? Ах нет эксперимента, кроме существования оптических изомеров при четырех разных вершинах, тогда берите самое простое – тетраэдр, и не морочьте нам голову! Всё гениальное просто, не в том смысле что это может понять любой самозваный шарлатан, помусолив на ночь популярную книжицу, а в том смысле, что гениальное всегда проще негениального. Гениальное можно понять, затратив усилия, а негениальное нельзя – в негениальное можно только верить.
Итак, первые структурные формулы отражают именно валентности, а вовсе не химические связи, о которых до появления квантовой теории никто ничего не знал. И в рамках позитивистского мышления не имел права выдвигать гипотезы, не проверяемые опытом. Забудем поэтому пока про связи и убедимся, что с помощью валентностей получается весьма стройная картина структуры на том уровне знаний. В этом месте у многих возникнет вопрос, причём очень горячо сформулированный – а зачем мы копаемся в этом старье, 19 век, позитивизм, ленины всякие, валентности – зачем нам это в 21 веке, мы же в зиллион зиллионов раз умнее тех, сколько знаем всего. Бозон Хиггса нынче в тренде (уже успел выйти, но ещё пахнет), а не доисторические валентности. Не торопитесь. Химия – очень консервативная наука в своих основах, она прочно держится за понятия как раз 19 века, довольно часто даже почти их не изменяя. Мы часто даже не догадываемся, насколько недалеко ушли от тех основоположников и их идей, что говорит не только о нашей косности, но и о том, что идеи были хороши и построены на очень прочном основании непосредственной связи с опытом.
Вот, например, еще раз вспомним, как рисовалась структура азотной кислоты, и увидим понятную картинку. Азот в азотной кислоте 5-валентен. Это очевидно, потому что азотная кислота происходит от азотного ангидрида N2O5, а в нём азот очевидно пятивалентен, потому что на два азота приходится пять кислородов, кислород двухвалентен по определению, на десять валентностей кислорода приходится два азота. Поэтому структура азотной кислоты рисовалась с азотом, у которого 5 чёрточек-валентностей. Мы знаем, что сейчас категорически запрещается так рисовать азотную кислоту, потому что теперь мы рисуем связи, и соблюдаем правило октета, чёрточек-связей поэтому не может быть больше 4-х, получается известная всем формула с разделением зарядов. Отлично! И мы понимаем, почему азот в азотной кислоте пятивалентен, как её ни рисуй. Потому что не может валентность зависеть от способа изображения структуры. Если способ правилен, соответствует представлениям, в рамках которых он используется. Здесь оба способа правильны! Это очень важно понимать. Структура с пятью черточками правильна, если мы оговариваем, что изображаем валентности. Эта структура была в ходу очень долго, и в России (СССР) так азотную кислоту споскойно рисовали еще тридцать-сорок лет назад почти все, а когда кто-то шибко умный рисовал формулу со связями и соблюдением октета, иной раз мог и на проблемы нарваться. Конечно, с тех пор всё же все решили, что с октетом по жизни шагать лучше, чем без октета и теперь проблемы будут у того, кто нарисует с пятью черточками. И правильно будут, потому что в 21 веке уже наверняка никто не помнит, что раньше в структурных формулах изображали валентности.
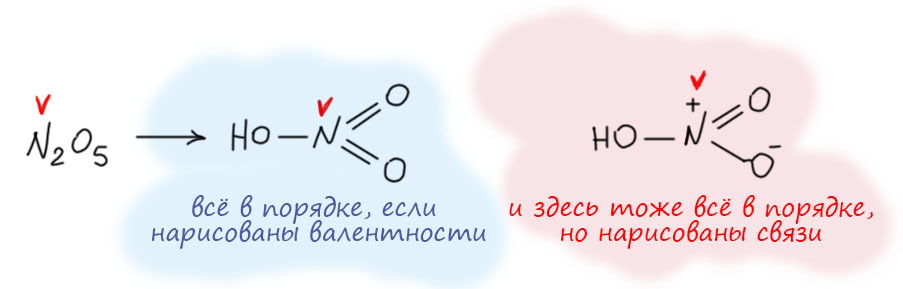
И всё было бы совершенно здорово, если бы не одно странное обстоятельство. А что если нам нужно изобразить структуру не азотной, а фосфорной кислоты. В 21 веке! Сейчас. К сожалению, придется рисовать не мета- а орто-фосфорную кислоту, потому что мета-фосфорная кислота HPO3, прямой аналог азотной, по каким-то причинам страшно неустойчива, а точнее, не неустойчива, а чудовищно реакционноспособна, и воду вырывает даже у песков пустыни Сахара, образуя орто-фосфорную. Ну или полимеризуется, если воды нет вовсе. Фосфор в фосфорной кислоте пятивалентен, с этим точно никто не спорит, хотел бы я посмотреть на того, кто скажет, что в фосфорной кислоте фосфор четырёхвалентен. Я хочу видеть этого человека! Нет такого человека. В азотной кислоте азот нынче черырехвалентен, а в фосфорной фосфор пятивалентен. Вот как интересно стало в российском образовании, Периодический закон что дышло, вертим как хотим, мы не ждём милостей от химии, мы ею помыкаем. И всё же, вот как нарисуют структуру орто-фосфорной кислоты почти все:
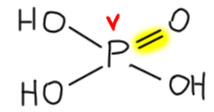
Согласимся, что нам привычна такая формула, мы и сами ее нарисуем, и школьника похвалим, если нарисует. И в литературе мы почти всегда видим такие формулы фосфорной кислоты, фосфатов, полифосфатов. В химической и биохимической литературе, в последней вообще фосфаты, фосфорильные остатки встречаются чаще воды, и рисуют их только так, в нуклеотидах, нуклеиновых кислотах, АТФ, мириадах фосфорилированных биомолекул, и т.п. Отлично.
Только погодите, а что означают эти черточки между фосфором и кислородом? Это связи или валентности? Какие могут быть валентности, что за старьё, в самых современных книжках и журналах только так рисуют, видели последний номер Nature? Любой, ведь какая-нибудь статья про какие-нибудь биомолеклы всегда найдётся, значит будут фосфорильные остатки, и если их разрисуют структурой, у фосфора будет один из кислородов на двойной связи. Не может же в таком современнейшем научном издании быть – валентность!? Значит это связи, ковалентные связи. А почему их пять? Почему у азота нельзя рисовать пять, а у фосфора можно и нужно? Фосфор же прямой аналог азота, прямо под ним стоит хоть в длинной, хоть в короткой Таблицах. В этом месте обычно начинается вкрадчиво-сбивчивый лепет про d-орбитали, типа они, конечно, высоко, но если нужно, а нужно ведь не кому-то там, а фосфорной кислоте и фосфатам, жизнь на Земле на кону, вы что, не понимаете, а вот d-орбитали всё понимают, поэтому так чуть-чуть присели, электрончик с дважды-занятой p-орбитали взяли и тут же использовали на связь с кислородом. А что, это тогда будет sp3d-гибридизация, это же наверное не тетраэдр, а антипризма какая-нибудь, а как вообще выглядит ион фосфата, это же вроде всё же тетраэдр? Да и, ой, а какие же у фосфора d-орбитали?? 2d – так таких не бывает. 3d – так эти только через пять элементов спустятся с небес и начнут принимать электроны, а пока они высоко-высоко, никакой роли играть не могут, что и показано за последние 70 лет всеми возможными способами. Да, был грех, была такая гипотеза, но она давно скисла и растворилась без остатка в анналах химии. Нет никаких сомнений в том, что валентная оболочка фосфора соответствует его статусу p-элемента и состоит, точно так же как у азота из s- и p-электронов, только не из второй, а из третьей оболочки, но это никак не меняет дела – фосфор есть p-элемент и обязан подчиняться правилу октета точно так же как азот. И если рисовать именно связи, нужно соблюдать правило октета, и вот что получится:
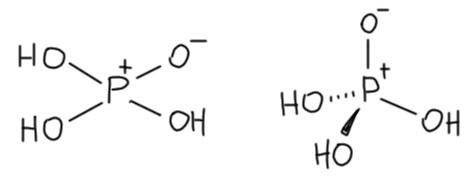
Заодно нарисовали эту молекулу в стереохимической проекции, чтобы увидеть, что форма этой молекулы – тетраэдр. Да, это правильная структурная формула. И здесь черточки изображают не валентности, а настоящие связи, ковалентные. И это не фантазия, а да, так и надо, это полностью соответствует всей совокупности известных данных про эту структуру. Да, рисовать надо именно так, и с этим согласны все, кто хоть немного серьёзно интересовался этой проблемой. А почему так не рисуют? Рисуют иногда, но чаще рисуют по старому, не отдавая себе отчета, что вместо связей рисуют валентности, как это и пошло с 19-го века. Никакого криминала здесь нет, такова традиция, кому надо тот рисует корректную структуру и использует именно ее в работе. Но традиция занятна – вот почему у азота обязательно надо рисовать, соблюдая октет и связями, а у фосфора не обязательно или вообще в голову не берем, поэтому рисуем как в 19 веке валентностями. Такие вещи очень трудно изменить. Особенно это касаеся биохимии, в которой всем до лампочки какие там связи и валентности, им важны фосфорильные остатки для своих нужд, и для них это чисто вспомогательная информация, недаром они часто вообще этот остаток сокращают как P в кружочке.
Ну и никто пока, видимо, не догадывается, что если так нарисовать, то выскочат продвинутые методисты и заявят, что фосфор отныне везде четырёхвалентен. Не буди лихо, хотя, к сожалению, оно уже давно не тихо, но хотя бы в эту сторону не смотрит.
И теперь мы увидим очень простую вещь – октет надо соблюдать для всех p-элементов. Для всех элементов пятой, шестой, седьмой, даже восьмой групп, главных подгрупп. Для фосфора, мышьяка, серы, селена, теллура, даже полония, хлора, брома, иода, даже астата,
сурьмы, висмута. Везде октет, везде максимум четыре ковалентные связи, а если есть пары, то меньше. Попробуем составить табличку из типичных, важных и часто встречающихся соединений самых важных элементов из этого набора, нарисуем структуру из валентностей, и правильную структуру, союлюдающую октет. Слева, на розовом фоне будут формулы, использующие валентности, как в 19 веке постановили их рисовать, в эпоху до открытия природы химической связи. Справа, на голубом, нормальные структурные формулы, соблюдающие октет и дополнительно показывающие стереохимию. Выборка отчасти случайная, можно было бы еще множество соединений добавить и других элементов. Но понятно, что фосфор, сера, хлор важнее всего. Ну и для полноты картины знаменитый тетраоксид ксенона. Везде я пометил валентности красными римскими цифрами (случайно в сернистой кислоте зачеркнуто, это просто перо сорвалось). Видим, что валентность не зависит от способа написания формулы.
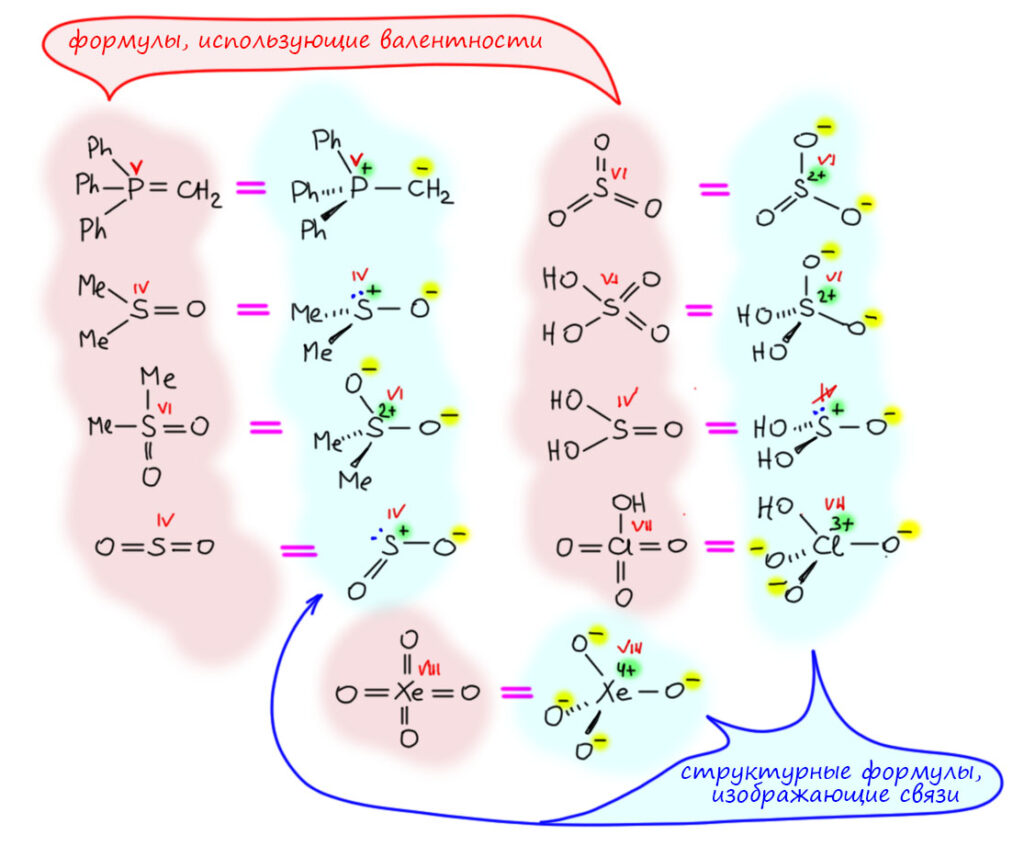
Посмотрим на эту табличку и покопаемся в памяти – к каким формулам мы привыкли. Думаю, почти все скажут, почти всегда к старым, с валентностями, а что они неправильные? Ещё раз – они правильные, но там валентности, а не связи. Если для каких-то целей желательно иметь более точное представление о структуре, тогда нужны связи, а не валентности, и это структуры на голубом фоне, соблюдающие правило октета. Видим презанятнейшую вещь – минимум для двух соединений из этой таблицы, для илидов фосфора и для сульфоксидов структуры сейчас чаще рисуют со связями и соблюдением октета. Вот как все повернулось – выбрали по каким-то причинам два соединения (два типа соединений) и настаивают, чтобы вместо валентностей рисовали связи. Для всех остальных как будто и не настаивают. Так получилось чисто случайно, просто в истории и илидов и сульфоксидов были настойчивые и влиятельные ученые, которые всех приучили рисовать корректные структуры со связями вместо валентностей. Во всех других случаях этого не произошло, пока не произошло. Особенно забавно, например, что для сульфоксидов теперь настаивают на формуле с октетом, а для сульфонов – вообще об этом не вспоминают. Так получилось потому что несмотря на близость сульфонов и сульфоксидов, химия и применение у них драматически различны, и исследователи в обоих областях разные.
Одной из причин массового нежелания использовать октетные формулы явно является необходимость писать заряды на атомах, для шестой-восьмой групп заряды получаются неуютно большими, не всякая рука хладнокровно выведет +2 (на сере, селене и т.д..),+3 (на хлоре, броме, иоде) и уж совсем караул +4 на ксеноне! Фффигня какая-то!! Да нет, все нормально. Это ведь формальные заряды, а не настоящие. Формальные заряды, напомню, рисуются из предположения, что ковалентная связь симметрична, пара электронов поровну делится между атомами. Мы просто считаем свои электроны и сравниваем с числом своих электронов в исходном атоме, разница и будет формальным зарядом. У ксенона, например, четыре ковалентные связи, восемь электронов всего, половина из них ксеноновы, половина от восьми это 4, а в исходном атоме ксенона своих электронов все восемь. Восемь минус четыре равно четырём, это и есть формальный заряд. У кислорода наоборот одна ковалентная связь и три пары свободных (потому что тоже полный октет) – значит “своих” электронов 6+1=7, а у свободного атома своих электронов (6-я группа) шесть. Один электрон сверху получился, формальный заряд -1. Но если мы заинтересуемся не формальными (на бумаге), а фактическими зарядами, которые будут проявлять себя в разных свойствах молекулы, например, в том же дипольном моменте, придется задуматься о распределении электронной плотности. Ведь по-настоящему связь несимметрична, плотность смещена в сторону электронодефицитного атома, во всех этих случаях плотность от кислорода с отрицательным зарядом смещается к центральному атому. Это вполне обычный индуктивный эффект, как мы его понимаем в органике. Этот эффект будет частично гасить положительный заряд на центральном атоме. Фактический заряд будет меньше. Насколько – это зависит от того, как мы выполним деление пространства между атомами (partitioning), это не самая простая задача, ведь между атомами нет границ и нет таможни, которая пропускает или не пропускает чужую плотность в область своего атома, и не берет взяток, не берет потому что ее нет. Была бы, брала бы. В квантовой науке используется несколько разных теоретических схем деления пространства молекулы на атомы – Малликена, Лёвдина, Бейдера. Не будем здесь об этом, мы здесь про самое простое в химии, про валентность, а это к ней не имеет никакого отношения. Но важно не бояться формальных зарядов, если они расставлены правильно. Не позволяйте развести себя на ехидные вопросы типа, а, хлор с тремя плюсами! разве так может быть (десять знаков ? и в голосе слышится: А вы, товарищ, не иностранный ли агент, не враг ли народа, вредитель, гвозди в масло трудящимся не ты положил, сволочь!). Спокойно объясните про то, что такое формальный заряд в химии, не поддавайтесь на провокации.
Увидим и еще одну прикольнейшую вещь. Если бы о таких структурах узнали методисты, продвигающие понимание валентности как подсчет ковалентных связей, вышла бы презабавнейшая вещь, даже две – у всех неметаллов не может быть “валентности” выше 4-х; валентность 4 в этой логике будет признана присущей подавляющему большинству соединений элементов от бора до ксенона, и поди тогда пойми зачем все эти периодические таблицы, коль везде четыре. Вот так и возникают верования. Образовалась бы партия Методистов Свидетелей Четвероицы. Вывод – от греха подальше не рассказывайте ЕГЭшным методистам про то, как надо правильно рисовать структуры соединений неметалов в высоких степенях окисления. А мы еще раз обсудим эту проблему на страничке про гипервалентность, которая скоро тоже будет.
И ещё одна вещь, довольно странная. Есть такая международная общественная организация, ИЮПАК, которая занимается в том числе стандартизацией номенклатуры и вообще всех формальных вещей, которые связаны с химией. Довольно часто получается неплохо, например, та же номенклатура органических соединений, которой мы пользуемся, хотя и видим в ней кучу недостатков. ИЮПАК это не большой-пребольшой дом в одной из пафосных столиц мира, в котором снуют уполномоченные миром специально обученные люди, которые за огромное жалованье неустанно сочиняют все эти правила и рекомендации. Это просто весьма свободное сообщество ученых, которые в свободное время соглашаются делать что-то полезное, бесплатно (иногда им компенсируют расходы на участие в ассамблеях) и так как получится. Получится хорошо, хорошо. Не очень хорошо, тоже хорошо. Совсем нехорошо, всё равно спасибо, ведь старались люди. Большое количество таких документов-рекомендаций просто публикуются в журнале ИЮПАК и остаются незамеченными. Никто не может заставить ни одного ученого, даже самого скромного (напоминаю, что скромность ученого не украшает) пользоваться рекомендациями ИЮПАК или хотя бы знать об их существовании. Номенклатурой – да, заставляют, на этом просто настаивают редакции журналов. Но не очень настойчиво. Один только пример: ИЮПАК требует называть фосфины фосфанами. Ну и часто вы это слово видели в статьях? Только одно издательство, Wiley, в своих журналах требует фосфанов, все остальные даже не замечают, если мы продолжает называть эти важнейшие соединения и лиганды фосфинами. А знаете, как те же правила рекомендуют называть воду? Ни в жисть не догадаетесь и бьюсь об заклад (надеюсь, это мягкая штука), что вы никогда не видели этого слова – оксиданом. Хотели назвать оксаном, но доперли, что ИЮПАК раньше это слово уже успел заиграть за одним гетероциклом.
Так вот это я к чему? К тому, что есть пафосная Рекомендация ИЮПАК-2008, составленная полутора десятками видных ученых из множества стран, и необычайно мелочно описывающая, как нам рисовать химические структуры и формулы (Brecher, J. Graphical representation standards for chemical structure diagrams (IUPAC Recommendations 2008). Pure Appl. Chem. 2008, 80, 277–410). Невероятно мелочно, вплоть до требований соблюдать определенные расстояния между чертами двойной связи и так далее. Большинство этих рекомендаций – просто дань здравому смыслу и обычному вкусу, мы и без них всю дорогу так рисуем. Но есть и крайне странные вещи. Например, рекомендации не рекомендуют использовать заряды рядом с атомами там, где этого можно избежать. И совершенно конкретно – не рисовать вот те самые структуры, соблюдающие октет, если для этого требуются заряды. В этих правилах написано – используйте в таких случаях двойные связи. Причем авторы рекомендаций понимают, что они таким образом рекомендуют нарушать октет – так и пишут, рисуйте двойные связи даже если это нарушает октет. Но не у соединений азота, здесь они крепко держатся за структуры с четырьмя связями. Более того, даже в таких соединениях как сульфоксиды, где уже давно все привыкли рисовать структуру с зарядами, тоже ИЮПАК это не рекомендует. Не просто не рекомендует, а пишет – Not acceptable, то есть неприемлемо. И делает это исключительно исходя из соображений эстетики, как её понимают авторы этих рекомендаций. На мой взгляд, здесь есть одна проблема – вот это слово “неприемлемо” по отношению к вещи, которая обусловлена законами химии. Я точно имею право нарисовать фосфат, сульфат, и так далее как структуру с соблюдением октета. Просто потому что это правильно, хотя и немного громоздко. Но могу согласиться этого не делать по эстетическим причинам, если ИЮПАК мне гарантирует, что всем понятно, что таким образом рисуются не связи, а валентности. Это было бы нормально, но увы, ИЮПАК ни мне, ни кому бы то ни было ещё ничего не гарантирует. Но так консервирует эти формулы с валентностями вместо связей. И продолжают у многих химиков поддерживать неверное представление об абсолютной уникальности азота, для которого нарушать октет – ни-ни, низззя!! – а для всего остального не просто можно, а нельзя по другому.
Вывод из этого прост как кирпич: валентность нас всех переживёт, даже если мы об этом и не догадываемся. И – не читайте рекомендаций ИЮПАК ни до, ни после чего бы то ни было.
Валентность и степень окисления
Претензии к валентности, к тому что это какое-то замшелое старьё, не имеющее простого определения, которое как-то неприятно использовать в век искуственного интеллекта и бороздящего просторы вселенной Илона Маска, выражаются в поисках другой универсальной характеристики атома в молекуле. И эти поиски довольно естественно приводят к степени окисления. Вот вроде бы отличная характеристика, во-первых, заведомо помоложе валентности, во-вторых, в отличие от валентности, определение которой в реальных соединениях требует анализа химических взаимоотношений соединений в рядах, степень окисления определяется из структуры конкретного соединения и никаких дополнительных действий не требует. Если кто-то не знает, как определять степень окисления атомов в молекулах, рецепт один – узнайте. Это несложно.
Напомню, зачем нам валентность. Чтобы упорядочивать множество простых соединений элементов, группировать их в ряды. Например, “соединения трехвалентного азота”, “соединения четырехвалентного азота”, “соединения пятивалентного азота”. В каждом таком ряду группируются соединения, связанные родством, близостью структуры, взаимопревращениями. Трехвалентный азот: аммиак, галогениды азота, азотистая кислота, нитриты, нитрозо-соединения и т.п. Соединения ряда связаны друг с другом реакциями замещения, иногда реальными, иногда формальными. За каждым стоят длинные ряды производных – от аммиака идут амины и так далее, от азотистого ангидрида азотистая кислота и нитриты; галогениды азота это такая промежуточная ступень между аммиаком и оксидом азота(III) (в этом месте есть одна проблема, потому что реальная структура этого оксида сложнее, в ней есть связь азот-азот, а следовательно простые правила перестают работать). Такие ряды есть у каждого элемента, у каждой валентности. Мы привыкли к этому, проходим эти ряды на уровне подсознания, на этих рядах строится наше понимание химии, а в учебниках, даже самых огромных и современных так расположен материал.
Теперь заменяем валентность степенью окисления. Ряд тут же разваливается. Аммиак – азот(-3), галогениды азота – азот(-3) и азот(+3) в зависимости от галогена; азотистый ангидрид – азот(+3)(а на самом деле даже +2 и +4). Соединения, принадлежащие ряду трехвалентного азота, такие как гидроксиламин и гидразин имеют азот(-1) и азот(+2). И так далее. Ничего общего, как это собрать вместе? И так будет буквально везде.
Сульфоновые кислоты совершенно естественно считаются аналогами серной кислоты, и то, и то относится к ряду шестивалентной серы. Но степень окисления у серной кислоты +6, у сульфоновых кислот +4, и что нам – сближать сульфоновые кислоты с сернистым ангидридом и сульфитами. Нет, нам это не нравится, мы знаем о том, что сульфоновые кислоты намного ближе к серной по свойствам (это сильные кислоты) и структуре (тетраэдрическая сера, нет свободной пары, не нуклеофилы).
Ещё одна проблема возникает с молекулами и ионами, которые нельзя предствить одной структурой, те, для которых нужна мезомерия. Оказывается у атомов в граничных структурах разные степени окисления, и это уже не может не внести большую сумятицу в использовании такой характеристики. Попробуйте, например, граничные структуры закиси азота, диазометана, аллильного аниона и т.п., убедитесь, что степени окисления атомов разные. Внизу на вкладке про то, как считать степень окисления, я расскажу, что с этим делать. Это совсем скверно. А вот валентность не изменяется – в граничных структурах она постоянна.
Степень окисления неплохо заменяет валентность в производных металлов, для большинства таких степень окисления это просто валентность без знака. Мы часто этим пользуемся, обозначая металл в каком-то состоянии то так, то так, например, Fe(3+) и Fe(III), на это никто вообще внимания не обращает, считая оба обозначения фактически тождественными. Но и у металлов, стоит нам копнуть соединения похитрее, начинаются фокусы, особенно в чрезвычайно модных в последние полвека соединениях со связями металл-металл (кластерах). Например, хорошо известный ацетат молибдена представляет собой димер с четверной связью металл-металл, степень окисления там Mo(2+), а валентность молибдена – шесть, вполне приличествующая элементу 6-й группы. Удивительно, но именно старинная валентность помогает понять модные кластеры лучше, чем степень окисления.
Удивительно, но большой вклад в путаницу валентности и степени окисления внесли два крупнейших ученых 20 века, Коттон и Уилкинсон, которые в своем великом учебнике неорганической химии буквально прямо пишут, что валентность – это только качественное абстрактное понятие, а в реальной химии мы должны использовать степени окисления, причём плевать, как мы ее обозначаем – хотите римскими цифрами, хотите обычными, положительный знак можно не указывать и так далее. Этим основоположникам просто неинтересно было копаться в таких азах, ведь они писали не просто учебник, а книгу, которая перевернет все понимание химии. До них неорганическая химия считалась такой наукой про простые производные элементов, что-то типа развернутого описания Периодической таблицы, и, как бы это ни было неприятно неорганическим химикам, главной химией считалась органическая. Так сложилась история химии, что именно достижения в органической химии создали множество фундаментальных вещей в химии вообще, от структуры до механизмов реакций. Да и количество органических соединений далеко опережало (сейчас это уже не совсем так) количество неорганических. Один элемент против всех остальных побеждает с разгромным счетом. Точнее, побеждал. Все стало изменяться во второй половине 20 века, когда Уилкинсон поучаствовал в открытии принципиально нового структурного принципа – гапто-связывания в ферроцене и других аценах, и стала с бешеной скоростью развиваться новая координационная химия, основная идея которой в том, что существуют металлы, и вся химия это химия металлов, все остальные молекулы для которых являются лигандами. И если вы нашли молекулу, которая пока не входила ни в один комплекс, то вы “просто не умеете ее готовить”, поищите получше металл для этой молекулы, или поработайте над техникой эксперимента, возможно, комплексы этой молекулы имеют необычные свойства и трудноуловимы. Коттон и Уилкинсон переосмыслили нерганическую химию, включив в нее координационные соединения, металлоорганические соединения и все, что над ними. И только эпизодический выплеск совершенно несвойственной этим двум крупнейшим ученым скромности помешал им сделать заявление, что органической химии больше нет, а это просто такое приложение к неорганической – глава про химию лигандов, надо же их самих как-то готовить.
Не могу не заметить, что книга Коттона и Уилкинсона (в последнем издании, вышедшем уже без Коттона и Уилкинсона добавлены еще два соавтора, но они только добавили новый материал, не трогая ни буквы в основном священном тексте) у всех в основном стоит на полке как Священное писание, мало кто изучает по ней химию, просто неудобен такой радикальный разрыв с традиционной методологией. Другие же крупные учебники (Химия элементов или Неорганическая химия Эткинса-Шрайвера) рассматривают все как мы привыкли, но уходят от валентности в пользу степени окисления с помощью очень простого приёма – в них сразу подробно разбирают электронную структуру, используют не короткопериодную, а длиннопериодную таблицу, построенную именно на структуре атома. И дальше группируют соединения элементов по типам: галогениды, оксиды, другие бинарные соединения, гидроксиды, кислородные кислоты и т.д. В каждом разделе соединения рассматривают в порядке увеличения степени окисления. Это разрушает горизонтальные связи (ряды по валентностям), но создает новые отношения – в рядах, где соединения связаны окислительно-восстановительными реакциями. Так тоже можно, но, на мой взгляд, это годится для больших университетских курсов химии там, где химия – основная профессия. Но для школы или облегченных курсов это тяжеловато, и не создает правильного образа периодичности свойств.
Вывод, на мой взгляд, простой: не годится степень окисления на замену валентности, разрушает она систему. Причина этого проста: степень окисления – величина совершенно формальная, зависящая от упорядочивания множества элементов по электроотрицательности, для близких в Периодической таблице элементов одной подгруппы электроотрицательность изменяется очень широко и это приводит к разведению аналогичных производных элементов по разным величинам степени окисления. Ну и совсем уж конфуз с тем, что шкала электроотрицательностей не одна. А вот валентность сведет их в одно множество. Валентность просто заставляет нас видеть аналогии, а это очень мощный инструмент мышления в химии.
Есть и еще одна причина, довольно издевательская. Если очень хорошо вдуматься, то для того, чтобы узнать степень окисления, надо знать валентность, которая помогает нарисовать структурную формулу. Не наоборот. Опять таки, любой химик делает это на автомате, не вдаваясь в суть процесса, и в разницу между валентностью и степенью окисления, как мсье Журдэн не знал разницы между прозой и поэзией.
Валентность и вопросы языкознания
Валентность – совершенно искусственное слово, возникшее именно в химии, за пределами химии его или вообще нет, или оно заимствовано прямо из химии (в филологии, например, есть такой термин). Повторю, что это просто сокращение слова эквивалентность, сначала такой типичный профессиональный жаргон химиков. Профессионалы всегда придумывают свой собственный язык, чтобы отличаться от простых смертных, проблем с этим нет, так как в любой профессии всегда полно специальных терминов. Но профессионалы и более общие слова иногда специфически коверкают, переносят ударение (осужденные, компас и т.п.), сокращают. Вот валентность – слово этого ряда. Этимология его идет к слову эквивалентность. Но фокус в том, что слово эквивалентность сложное, имеет два корня, экви – равный, и -вал- или -валент-, а это очень важный корень в европейских языках, происходящий из латыни, и далее вглубь тысячелетий куда-то к самым древним языкам. Этот корень несет важнейший, первобытный, основной смысл – смысл силы, действия, способности к действию, активной стороны жизни. Оттуда все производные смыслы в современных языках и производных словах – мужества, доблести, законной силы, права на действие и так далее. Покопайтесь сами, слов с таким корнем во всех языках десятки.
Именно так стали переосмысливать слово валентность в 20 веке после появления электронных и квантовых теорий связывания. Это слово стало обозначать в самом общем виде способность атомов соедняться друг с другом. Сказать, что атомы или элементы обладают валентностью всё равно, что сказать: Атомы и элементы способны образовывать соединения, давайте разбираться как они это делают. Чем дальше, тем таких способов больше. Мы сегодня очень хорошо понимаем, почему атомы образуют конкретное число связей, какие соединения устойчивы, а какие нет, и всё остальное. Структурных тайн почти не осталось. Поэтому и слово валентность перестало так часто звучать, и больше никто не называет так книги (последней была книга Коулсона, изданная в 1960-х), все стало намного конкретнее и детальнее.
Это очень глубоко сидит в подсознании. На мой взгляд, именно поэтому слово так быстро прижилось и стало одним из основных понятий. Химики имеют дело с атомами, способными взаимодействовать и образовывать молекулы. Химик очень хочет выразить это свойство атомов – способность к взаимодействию и соединению с другими атомами, такая химическая сила, и даже, не побоюсь этого слова, доблесть. Фтор или кислород вот, например, такие удалые, что и криптон и ксенон могут нагнуть. Вот какова у них валентность! Слово валентность просто попало в точку – и стало ровно так и пониматься, как способность атомов к самому главному действию в химии – соединению с другими атомами в молекулы. И совершенно шикарно, что это качество по определению количественно и дискретно – атом может уметь соединяться с одним, двумя, тремя, и т.д. другими атомами прежде всего в бинарных соединениях. И это очень удобно выражается с помощью приставок – двухвалентный, значит способный соединяться с двумя другими атомами, трехвалентный – с тремя. Здесь важна способность этого термина выражать именно потенциальную способность. Поэтому на вопрос, какова валентность водорода, мы говорим один, даже не спрашивая, о каком соединении идет речь. Кислорода? – два. То же самое. Он может, они могут. Дайте им другие атомы и они смогут. Мы знаем, мы в них верим, они не подведут, у них есть валентность. В химии эту способность иногда выражают понятием валентные возможности, не подозревая, что это тавтология, избыток смысла, потому что уже в слове валентность содержится смысл возможности действия. Атом имеет валентность – может соединиться с другим атомом. Необходимость в таком дополнении понятно чем обусловлена – тем, что валентность приобрела количественный смысл, это одновременно как будто одна единица валентности. У атома кислорода две валентности, он двухвалентен. И так далее.
Итак, в химической литературе слово валентность имеет три основных смысла:
- самый основной и первоначальный смысл: способность атома элемента образовывать с атомами других элементов бинарные соединения, в состав которых каждый элемент входит эквивалентным весом (массой); отношение атомной массы к эквивалентной это и есть валентность в количественном смысле; от бинарных соединений к более сложным мы переходим считая валентности как единицы связывания.
- валентность = сокращение термина “единица валентности”. Каждый элемент входит в состав соединений, соединяясь с другими соединениям за счет имеющихся единиц валентности. В устойчивых соединениях все единицы валентности использованы.
- последний смысл, чисто качественный и скорее философский – сама способность атомов образовывать связи с другими атомами. В этом употреблении валентность не имеет количественного измерения, не может сочетаться со словами, обозначающими степень владения этой способностью: нельзя поэтому говорить про большую, малую, сильную, слабую, большую, меньшую валентность в этом смысле (в первом, количественном смысле валентности, конечно, можно говорить, например, про большую валентность углерода в углекислоте по сравнению с угарным газом). Быть восьмивалентным не означает быть в в восемь (или сколько угодно) раз лучше, чем быть одновалентным. Фтор всегда одновалентен, но кто устоит?
Валентность и электроны
Когда в химии появились электроны, а это случилось сто лет назад, всего сто лет назад – появилось естественно и потребность переосмыслить валентность в терминах химических связей и электронной теории. Но поскольку за дело тогда взялись не егэ-методисты, а крупнейшие ученые, им не пришло в голову самое простое решение – теперь мы знаем, что такое связь, так давайте просто считать связи в молекулах, сколько связей, такова и валентность. Не пришло, потому что они не посчитали возможным подкладывать под уже устоявшееся и чрезвычайно важное понятие валентности принципиально другое содержание. Очевидно, что при этом получилось бы нечто иное, и с какой стати называть это уже использованным и небесхозным словом.
Посмотрим на проблему поближе. Первое, что бросается в глаза, это соответствие между максимальной валентностью и количеством электронов на внешней оболочке. Точно соответствие наблюдается для s- и p-элементов. Отсюда появляется понятие валентной оболочки. Сколько электронов, такова и максимальная валентность. Для d- и f-электронов от этого простого соответствия есть отклонения, но мы и так уже признали, что для этих элементов старая добрая валентность не вполне адекватна обилию вариантов связывания.
Вернемся к s- и p-элементам. Соответствие между количеством электронов и максимальной валентностью очевидно, не может быть случайным. Но – как это связать друг с другом? Как электроны связаны с валентностью? Попробуем разобраться, и для этого просто пройдемся по типичному периоду, возьмем третий, потому что во втором у нас в конце нет максимальных валентностей.
Начнем с s-элементов. Очевидно, что для этих элементов свойственны ионные связи, то есть валентные электроны полностью передаются акцептору. Образующиеся соединения ионные. Но никто никогда не оспаривал, что валентность в таких соединениях натрия – один, магния – два. Это очевидно из стехиометрии связывания в бинарных соединениях: оксидах, гидридах, галогенидах и т.п. Получается, что ионная связь не противоречит валентности. Хотя мы не рисуем черточку в соединениях с ионной связью, вместо этого рисуем заряды. Классики начального периода электронной теории, Лангмюр, Сиджвик назвали это электровалентностью. В электровалентности и ионной связи есть одна серьёзная проблема, даже две, и я ещё к ним вернусь. Но пока зафиксируем – для s-элементов валентность равна числу валентных электронов. Слово максимальная можно не использовать, потому что у них другой и нет. Поскольку при образовании соединений валентные электроны полностью переходят к акцептору, нам не важно, спарены они или нет на валентных оболочках исходных атомов.
Следующий элемент, алюминий образует и ионные и ковалентные связи. Ещё следующий, кремний, в основном только ковалентные.
Ковалентная связь – нечто новое, этот термин появился тоже в самом начале электронной теории в 1918-1919. Слово придумал Лангмюр, но сама суть дела была выявлена чуть раньше Льюисом. Суть дела – связь образуется за счет совместного владения двумя атомами парой электронов. Смысл слова очевиден: ко-валентность (так писали сначала) – совместное владение валентностями. У одного есть единица валентности (в старом смысле) и у другого единица валентности – атомы скидываются этими единицами и делают связь между собой. Валентные электроны вкладывают в это смысл: единица валентности – это валентный электрон. У одного атома есть электрон, у другого электрон – возникает ковалентная связь, то есть пара электронов в совместном владении пары атомов.
Для атомов 3-й и 4-й группы мы обычно предполагаем, что есть возможность распаривания s-электронов на p-уровень, так что атом 3-й группы может образовать три ковалентные связи и этим объясняется валентность 3 (каждая связь может считаться электронным объяснением единицы валентности), а атом 4-й группы – четыре, чем объясняется четырехвалентность. Хоть все это со школы затвердили, но всё равно нарисую.
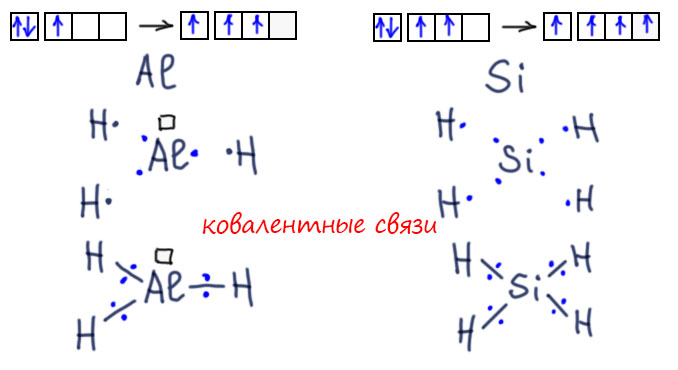
В этом месте есть одна тонкость. Предполагается, что в молекуле атомы сохраняются прямо целиком – в том числе сохраняют все свои электронные оболочки в неизменности, в том числе и валентную. Поэтому никаких новых ячеек не образуется, и электроны в молекуле обязаны довольствоваться имеющимися. Из этого следует, что для образования ковалентной связи соответствующие валентные электроны должны быть неспаренными. Мы со школы помним трюк, который применяют для того, чтобы образовать максимально возможное количество ковалентных связей – электроны из ячеек со спаренными электронами распределяют по свободным, если такие есть. Для атомов третьей и четвертой группы есть.
Это работает, но заслуживает маленького комментария. Вообще-то эта гипотеза (о полном сохранении всех оболочек атома в молекуле) неверна, и это следует из совершенно любой квантовой теории структуры, даже самой простой, теории молекулярных орбиталей в любом виде. Во всех этих теориях внутренние оболочки атомов сохраняются, но валентные полностью растворяются в новых квантовых состояниях, уже привязанных не к атомам, а к молекуле в целом или ее фрагментам. И нам, казалось бы, и дела никакого не должно быть до валентных состояний атомов, атомных орбиталей, так называемых ячеек и прочей школьной милоты. Стройте молекулу, решайте, находите молекулярные орбитали или даже что-то над ними, электронные состояния, и уж в этом копайтесь в своё удовольствие. И это так и есть, но удивительным образом, это почти никогда не опровергает простые соображения, построенные именно на атомах и их валентных оболочках. То же правило октета, например, (или 18-электронное правило химии переходных металлов) основано на атомных орбиталях – именно их ёмкость мы якобы не можем преодолеть. Но если в молекулах их больше нет, какое нам дело до этого, давайте искать ответов на молекулярном уровне. Хорошая идея, но сложная, а правило октета – вот оно, проще не придумаешь, работает, стоит как скала, чёрта с два сдвинешь. В устойчивых молекулах валентные состояния всех атомов p-элементов соблюдают правило октета, так и хочется сказать, добровольно. Вот это дисциплина! Не было ли у них там, внутри молекул, товарища Сталина, что так все по струночке ходят, а может он и сейчас там живет, в электронных облаках молекул, как в Океане Соляриса у Станислава Лема. Но нет, не в этом дело, Сталина и на них нет, всё гораздо проще. Секрет прост, и особенно хорошо виден с помощью молекулярных орбиталей. Подробнее как-нибудь отдельно, но главное в том, что связывающие МО, с помощью которых собственно и устроены молекулы, строятся из атомных орбиталей валентной оболочки; каждый атом отдает свои валентные орбитали, и количество связывающих МО равно количеству АО пополам. Ну и поэтому связывающие МО могут вместить ровно столько же электронов, сколько их могли вместить валентные оболочки исходных атомов. Все лишние пойдут на несвязывающие орбитали (в этом состоит фокус гипервалентности, об этом отдельно) или на разрыхляющие, а это недопустимо в устойчивых молекулах. Получается, что правило октета верно, потому что всесильно. А всесильно оно потому, что прочно опирается на квантовые законы строения атомно-молекулярных структур, получая каждый раз новую интерпретацию, как только мы забираемся на новый уровень теории.
Теперь возьмём элемент 5-й группы, например, фосфор. У фосфора 5 валентных электронов, и, как известно из химии фосфора – максимальная валентность пять. А может это случайное совпадение? Ведь валентные электроны в случае фосфора в одной ячейке спарены, и распарить их невозможно. Очень долго в химии держалась, а в некоторых уголках и до сих пор упорно держится теория о том, что каким-то непостижимым образом на помощь приходят d-оболочка, видимо, 3d, но этого быть не может ни при каких обстоятельствах, потому что эта оболочка в этом месте Периодической таблицы ещё так высока по энергии, что до нее не допрыгнешь никак, даже накушавшись мельдония и с шестом. Здесь надо четко ответить на простой вопрос – а у фосфора вообще-то есть 3d-оболочка? Есть, конечно, она и у водорода есть. Это ведь так и называется – водородоподобный атом, такая физико-математическая абстракция, сферический конь в вакууме (как хорошо – атомы всегда рассматриваются в вакууме, а то, что атомы сферические сомнению не подвергнет никто – так вот что такое, оказывается, пресловутый сферический конь в вакууме – это просто водородоподобный атом). Итак, 3d-оболочка, и даже 5f-, 8s-, 22d- и так далее, есть у любого атома, в решении атомного уравнения Шредингера. Только они пустые, виртуальные (то есть существующие на бумаге), хотя и играют роль в так называемых ридберговых состояниях, но это не про химию, а про физику. Они лежат высоко по энергии, возбудить электрон на самые низкие из них можно, но это точно не то, что происходит при обычных химических реакциях. Фосфор горит очень ярко, образуя как раз пятивалентный оксид, энергии выделяется пропасть, но, во-первых, она выделяется, а не поглощается, и во-вторых, по сравнению с энергией возбуждения электрона на 3d-уровень это очень мало, пустяк. Этого мало даже для разрушения связей P-P в решетке красного фосфора, ярко и жарко горит только белый. А энергия химических связей по сравнению с энергией возбуждения электронов тоже пустяк. В общем, есть 3d-уровень у сферического коня в вакууме номер 15, сокращённо фосфора, да не допрыгнешь. И никакой роли в валентности фосфора этот уровень не играет и играть не может, это не только очевидные общие соображения, но и факт, установленный на всех возможных уровнях теории (не только для фосфора, но и для всех других p-элементов).
Ну и как тогда с валентностями фосфора. Три неспаренных электрона дают три ковалентные связи, и это основная валентность в соединениях фосфора (и других элементов 5 группы, начиная с азота). Остается пара электронов. Это должно давать еще две единицы валентности – две валентности, и суммарно с обычной – максимальную валентность, равную 5. Но как? Просто так пару невозможно использовать для образования двух ковалентных связей или двойной ковалентной связи (двойная связь это все равно две ковалентные связи, сигма и пи), пять ковалентных связей это пять пар, десять электронов и прощай, октет. Октет на это вам заметит: Сами прощайте, точнее, проваливайте, нет октета кроме Октета, и Льюис пророк его, да пребудет навеки, а все враги его да расточатся. Дан вам октет и извольте соблюдать, а если не умеете, то это ваши проблемы. Давайте, поэтому соблюдать. Это несложно, хотя почему-то многие об этом до сих пор не догадываются. Пятивалетный фосфор (как и азот) бывают далеко не так часто как трехвалентные, и вот эти дополнительные валентности могут разбудить только немногие элементы. Проще всего и чаще всего это делает вице-король неметаллов, кислород. А почему не сам король, фтор? Фтор тоже, но это сложнее, и мы разберем это отдельно и даже на другой странице, про гипервалентность.С кислородом проще. Атом кислорода можно представить в двух состояниях: обычном, с двумя неспаренными электронами (такой атом еще называют триплетным) и состоянии с немного большей энергией со всеми спаренными электронами и одной вакантной атомной орбиталью (такой атом называют синглетным или оксеном).
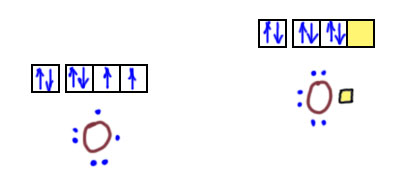
Для образования связей с участием неподеленных пар другого элемента нужен как раз такой синглетный атом – тогда пара и вакансия подойдут друг к другу как… как сами хотите что. Как двухвалентный донор и двухвалентный акцептор. А почему двухвалентные? А потому что используют два валентных электрона, а это, как мы уже договорились, эквивалентно двум единицам валентности. Вы спросите, а что, это такая реакция бывает? Во-первых, действительно бывает, например, в том самом адском пламени горения белого фосфора фосфор превращается в фосфорный ангидрид в частности с помощью таких реакций, при высокой температуре молекула кислорода распадается на атомы, и атомы эти частично имеют синглетное состояние (не путать с молекулярным синглетным кислородом!!!). Во-вторых, речь идет не о реакции, а о способе образовать связь или связи, точно так же, как для образования ковалентной связи мы формально рисуем встречу двух атомов с неспаренными электронами.
Вот так можно получить пятивалентный фосфор из трехвалентного, использовав пару вместо двух неспаренных электронов. Образуется ковалентная связь (одна!), и всего 4 пары, то есть полный октет, ни больше не меньше. И разделение зарядов на связаннных атомах, обеспечивающее дополнительную связь. Поскольку такое расширение валентных возможностей происходит сверх обычной валентности (напомню, что она равна 8 минус номер группы), обеспеченной неспаренными электронами, поэтому такие валентности часто объединяют термином “гипервалентность”. Я подробно разберу это структурное явление отдельно.
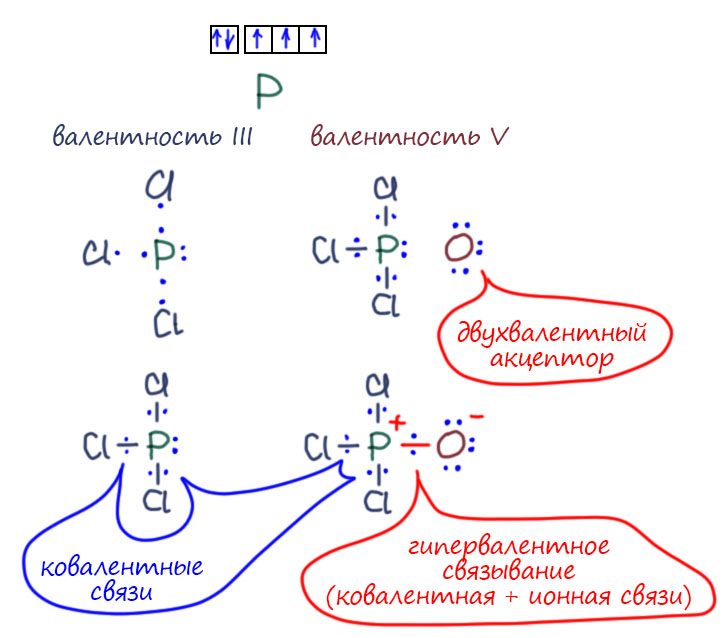
Замечу, что такую связь, которая получается при взаимодействии двухвалентного акцептора с пустой орбиталью и донора с парой, в химии после Лангмюра и Сиджвика называли семиполярной. Это довольно странный и неудачный термин, даже странно, что такие великие основоположники нашли такой неудачный теримн. Семи- значит полу-. Имелось в виду, что связь полуковалентная-полуионная, и то, и то нехорошо, но мы, я думаю, поняли, что имелось в виду. Они тогда считали, что это такая разновидность донорно-акцепторной или дативной связи (тогда предпочитали термин “дативная связь”, в последние десятилетия этот термин понемногу вышел из употребления, потому что он ничего не добавляет к более распространенному термину “донорно-акцепторный”, но до сих пор иногда встречается). А вот термин семиполярная связь, и активно использовавшаяся для ее обозначения стрелка, нынче совсем вышли из употребления, вплоть до явной рекомендации ИЮПАК не применять этот символ в структурах. К сожалению, у нас стрелка до сих пор в ходу и в школе, и не только. Это создает ложное впечатление, что это какая-то совершенно особенная связь, особенный тип связи. Но это не так, связь самая обычная, ковалентная, но с дополнительным ионным, электростатическим связыванием, которое легко обозначается просто зарядами на атомах. Особым является только способ образования такой связи, впрочем, это широко распространенный способ, которым образуются все валентности от V до VIII у p-элементов 5-8 групп. О том, как это устроено, кратко можно прочитать на вкладке про валентность и структурные формулы, и в деталях на отдельной странице про гипервалентность.
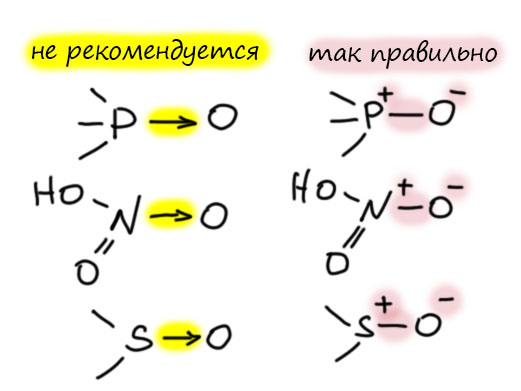
В общем, резюмируя: в химии s- и p-элементов, или по короткопериодной Таблице Менделеева, элементов главных подгрупп есть совершенно кристально ясное взаимоотношение между валентностью и числом валентных электронов. Оно очень простое – каждый электрон валентной оболочки это и есть та самая единица валентности, одна валентность (еще раз напомню, что у слова валентность есть два значения – общее понятие и единица валентности, о том, какое значение применяется почти всегда можно понять по контексту). Для переходных металлов система немного посложнее, мы ее здесь рассматривать не будем, желающих отсылаю к своему второму сайту. Поскольку валентных электронов у элементов главных подрупп может быть не более восьми, соответственно и валентность может быть максимально восемь. Такая валентность в 8 группе есть только у самого тяжелого и наименее электроотрицательного инертного газа, ксенона, и более ни у кого. Это понятно, потому что в конце периода оболочки настолько сильно сжимаются, что забрать электроны становится невероятно трудно. Инертный газ и так находится в состоянии с заполненной валентной оболочкой, о чем только мечтают все остальные неметаллы, включая кислород и фтор. Поэтому электроны у них можно только отобрать, а для этого нужен второй атом с огромной электроотрицательностью. Понятно, что для тяжелых инертных газов это фтор и кислород, а для легких – просто нет такого элемента на свете, Творец забыл создать, думал видимо, что фтора и кислорода хватит на всех, немного отвлекся и не заметил, что на гелий, неон и аргон нужно что-то еще круче, но тут кончились Дни Творения. И, как мы знаем, опочил Творец от трудов своих навеки, увидев, что то, что уже получилось, и так хорошо весьма. И чёрт с ними, с лёгкими инертными газами – обращайтесь в преисподнюю, если приспичит заняться химией неона (у аргона, кстати, кое-какая химия всё же есть, но в основном координационная). Мы полностью согласны с этим мнением и думаем, что большой проблемы в том, что нет такого элемента, который мог бы отодрать электроны от легких инертных газов, нет, и к врагу рода человеческого обращаться не станем.
Валентность и ковалентность
Когда возникли электронные представления в химии (по иронии истории это произошло почти одновременно с октябрьским переворотом в России и последующим ускоренным уничтожением великой и быстро развивавшейся, хотя и несколько заблудившейся страны), основоположники новых воззрений, Льюис, Лангмюр, Сиджвик, сильно озадачились тем, как перейти от такого понятия как валентность, на котором успешно построили всю доэлектронную химию, к новым понятиям химии, химической связи и ее разновидностям. ТО, что химическая связь – штука непростая, и есть не один ее тип, стало ясно немедленно. И основоположники сразу отбросили идею, что они быстренько пересадят валентность в новую эпоху и наполнят ее новыми смыслами. Пусть валентность остается на месте, а мы вместо этого лучше введем несколько новых понятий.
Проблема возникла сразу, как только увидели, что одновалентным, например, является и водород или хлор, и литий или натрий. А двухвалентным и кислород или сера, и магний или кальций. Но связи в молекулах,например, HCl и LiCl разные. В одной мы видим полное перемещение валентного электрона от одного атома к другому, и взаимодействие ионов чисто электростатическое. Валентные электроны не делятся атомами, они полностью переходят от одного атома к другому. В таких случаях связь не принято обозначать чертой. И соотвествующую валентность основоположники решили называть электровалентностью. На отдельной вкладке разберемся, что там не так, и в чем там проблема. Но в том, что валентность остается на месте проблемы нет.
В другой молекуле мы видим, что электроны в виде пары находятся в распоряжении обоих атомов. Сейчас мы будем рассуждать об электронной плотности, тогда говорили о паре электронов, но так или иначе смысл един – пара (или плотность) разделяется атомами, причем возможно смещение пары (или плотности) к одному из атомов, более электроотрициательному, но никогда не полное. Диссоциация молекулы на ионы к этому не имет никакого отношения, потому что это более сложный процесс, для которого обязательно требуется взаимодействие ионов с растворителем. Поэтому полярная ковалентная молекула и ионная молекула (ионная пара) это принципиально разные вещи.
Связь назвали ковалентной. А валентность – ковалентностью. И в отличие от валентности, ковалентность можно и нужно устанавливать, подсчитывая ковалентные связи. И даже прилагательные появились. Азот в азотной кислоте пятивалентный, но четырехковалентный. Углерод в угарном газе двухвалентный, но трехковалентный. Именно так. Такие термины отлично можно найти в статьях, книгах, учебниках в 1920-х по приблизительно 1960-е, бывает и позже залетает. Но ковалентность в этом смысле в химической литературе не прижилась, и сейчас это слово в этом смысле можно увидеть только у каких-то немного потерявшихся во времени авторах, зацепившихся духовными скрепками за милую архаику более полувековой давности. Ковалентность в этом смысле не прижилась, потому что этот термин довольно бесполезен для понимания структуры. Это связано с тем, что для p-элементов есть проблема с пониманием правила октета и гипервалентности. К сожалению в литературе, особенно учебной распространена путаница, которую я подробно разберу на страничке про гипервалентность. Но право на жизнь имеет только одна точка зрения – правило октета нужно соблюдать для всех p-элементов, хоть это и дико неудобно. Но если это так, то максимальное число ковалентных связей у p-элемента – четыре. А значит и ковалентность должна изменяться в диапазоне от нуля до 4, причем значение 4 будет у огромного количества совершенно разных соединений. И даже еще хуже – теория гипервалентности предполагает широкое распространение трехцентровых и многоцентровых связей, например, в таких соединениях как PF5, SF6 и т.п. С валентностью в таких соединениях все понятно, а с ковалентностью? Сколько там ковалентных связей? Опять четыре? Ну и зачем тогда нужна характеристика, которая будет равна 4 для половины соединений, и не будет нормально определена для большого количества весьма важных соединений?
Для иллюстрации просто пометим валентность (красным) и ковалентность (зеленым) для нескольких обычных молекули ионов. В основном пометим центральный элемент молекулы. Видим, что в ионных соединениях с валентностью все привычно, а ковалентность равна нулю. Видим, что расхождения между валентностью и ковалентностью встречаются очень часто, что ковалентность часто равна 4, и что для многих гипервалентных соединений мы вообще не понимаем, как определять ковалентность, а валентность определяем без проблем.
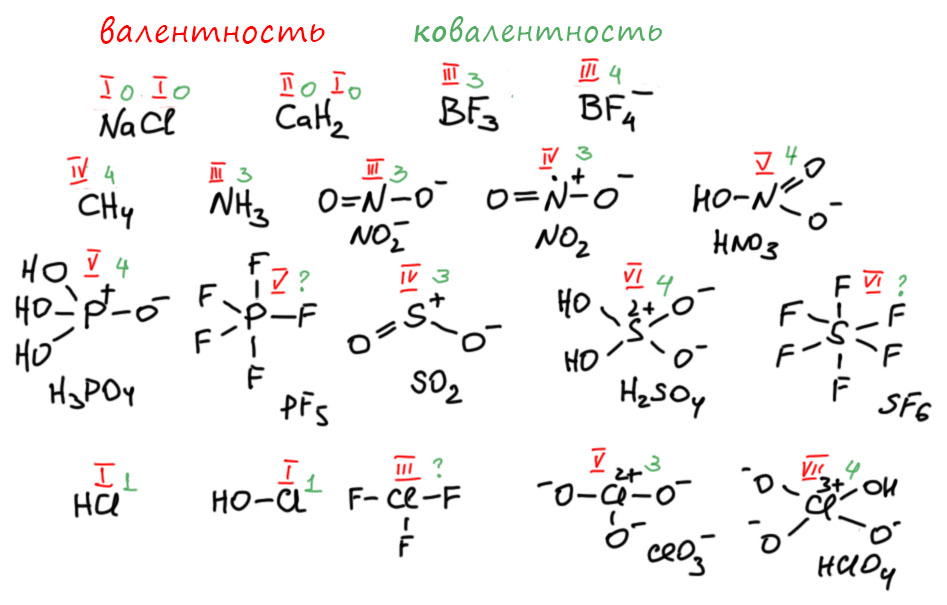 В современной литературе термин ковалентность есть, нечасто, но есть, но смысл у него совершенно иной. Под ковалентностью понимают очень непростую вещь – есть ли у какой-то нековалентной связи хоть какая-то степень ковалентности, то есть совместного владения электронной плотностью. Нековалентные связи это, например, ионные, или шире, электростатические, водородные и другие слабые межатомные взаимодействия; агостические; и т.п. Взаимодействий в химии полно, особенно тех, которые над обычной валентностью. И в современной химии все чаще возникают вопросы, а как та или иная связь устроена. Это не так просто. Та же водородная связь, которую мы считаем чем-то соврешенно обыденным, – но как она устроена, почему молекулы цепляются друг за друга атомами кислых водородов. И тогда производят теоретические расчеты очень сложными методами, применяют не менее изощренные теории, позволяющие установить, как распределяется электронная плотность, и пытаются найти, нет ли хотя бы небольшой доли плотности, которая находится в “совместном пользовании”. Если находят, говорят, вот, типа, раньше думали, типа, что это просто электростатика, а тут, наше вам с кисточкой, полпроцента электронной плотности обнаружились в непонятном месте, значит, связь имеет некоторую долю ковалентности. В общем, это чистая теория, но любопытная, для тех, кому интересны детали строения молекул и веществ.
В современной литературе термин ковалентность есть, нечасто, но есть, но смысл у него совершенно иной. Под ковалентностью понимают очень непростую вещь – есть ли у какой-то нековалентной связи хоть какая-то степень ковалентности, то есть совместного владения электронной плотностью. Нековалентные связи это, например, ионные, или шире, электростатические, водородные и другие слабые межатомные взаимодействия; агостические; и т.п. Взаимодействий в химии полно, особенно тех, которые над обычной валентностью. И в современной химии все чаще возникают вопросы, а как та или иная связь устроена. Это не так просто. Та же водородная связь, которую мы считаем чем-то соврешенно обыденным, – но как она устроена, почему молекулы цепляются друг за друга атомами кислых водородов. И тогда производят теоретические расчеты очень сложными методами, применяют не менее изощренные теории, позволяющие установить, как распределяется электронная плотность, и пытаются найти, нет ли хотя бы небольшой доли плотности, которая находится в “совместном пользовании”. Если находят, говорят, вот, типа, раньше думали, типа, что это просто электростатика, а тут, наше вам с кисточкой, полпроцента электронной плотности обнаружились в непонятном месте, значит, связь имеет некоторую долю ковалентности. В общем, это чистая теория, но любопытная, для тех, кому интересны детали строения молекул и веществ.
Поэтому я не соверую сейчас использовать термин ковалентность в смысле числа ковалентных связей. Хотя это хотя бы имеет историческое оправдание – во-первых, так и правда делали; во-вторых, никто и никогда не командовал прекратить так делать; термин просто сам немного перекрасился, потому что старый смысл потускнел и вышел из употребления, и нашлись ученые, которые его подобрали, протёрли, и “заставили сиять заново” в совершенно другом смысле.
Валентность и ионная связь
Ионная связь – чрезвычайно коварная штука. Очень простая, проще не придумаешь. Есть ионы, у них заряды, эти заряды притягиваются по закону Кулона. Ионы подходят друг к другу на расстояние суммы ионных радиусов и пришвартовываются. Ближе – ни-ни, для этого уже нужно ковалентное взаимодействие, та самая ковалентность. Но в чистой ионной связи ковалентность равна нулю. Такие вещи устанавливаются в очень серьёзных расчетах, на очень высоком уровне теории, и время от времени, как только возникают новые вычислительные возможности, пересчитывают. Это, как ни странно, важный теоретический вопрос. Если поищите в литературе, найдете немало очень тяжелых работ, в которых, например, в очередной раз выясняют, нет ли хоть немного ковалентности, скажем, в молекуле NaCl и тому подобных галогенидов щелочных металлов. И не находят.
И тогда вопрос становится совсем интригующим. А что такое вообще молекула NaCl (или LiF, или CsI и т.п.)? Ведь чистая электростатика радикально отличается от ковалентного взаимодействия. Ковалентное взаимодействие использует конкретные орбитали, требует наличия доступных электронов на этих орбиталях. Следовательно оно конкретно, направлено в пространстве определенным образом, с чем связана геометрия молекул. Как только ковалентное взаимодействие состоялось, возможности оказались использованы, и других не будет. Это выражают термином “насыщаемо”, не в смысле насыщенности-ненасыщенности связей, а в смысле “съел и… порядок!” Больше не хочу. Хотел связь, вот она связь, мне больше не положено, орбитали уникальны и если мы их использовали, то всё. Берите готовую молекулу, разглядывайте, нахваливайте, пользуйтесь.
В отличие от этого ионная связь никаких орбиталей не использует. Последний раз речь шла об орбиталях, когда электрон перемещался от донора к акцептору (восстановителя к окислителю). После этого это уже просто два заряженных предмета. Если атомы, то шарика. Могут быть более сложные ионы, но тогда это некая форма, иногда весьма сложная, но с зарядом. И вот два иона сблизились, сначала случайно, потом почувствовали заряды друг друга в полном соответсвии с законом Кулона. Притянулись до касания ионными размерами (радиусами, если шарики) и успокоились. Если не шарики и форма сложная, заряд как-то по ней распределен, то немного поелозили друг около друга, чтобы найти взаимное расположение максимального притяжения. Это довольно иногда бывает хитрой задачей, но сейчас нам не важно. Важно то, что когда это случилось, заряды никуда не исчезли. Они не погасили друг друга, они как были так и остались. Но! На большом расстоянии эта штука уже видится как какая-то незаряженная фиговина, и еще один заряд издалека не видит смысла к ней приближаться. Фокус в том, что в реальном веществе все частицы снуют как ненормальные, и так получится, что иногда просто сближаются без всяких надежд на продолженние отношений. Просто случайно, в процессе хаотического теплового движения. И тогда, когда еще один ион случайно приблизится, то он увидит заряд – вблизи это видно именно как заряд, потому что это и есть заряд. Если посмотрите курс электростатики, увидите как ведут себя силовые линии электростатического поля, если два заряда притянулись друг к другу. Увидите, что силовые линии со стороны взаимодействующих зарядов замкнутся друг на друге, но с другой стороны они как фигачили в бесконечность, так и продолжают фигачить, как будто с другой стороны не произошла стыковка двух заряженных шариков (тел). И второй заряд притянется. Фокус в том, что и при этом заряды останутся неизменными. Издалека это будет выглядеть даже как противоположный заряд, и еще один заряд испытает отталкивание, но не такое сильное, чтобы помешать ему носится в тепловом движении и случайно таки сблизиться (у частиц есть кинетическая энергия, приобретаемая тоже случайно при столкновениях, и кто застрахован, что еще один шарик шмякнет в бок так, что возникнет вектор скорости по направлению к уже ионному тройнику. В общем, понятно, что один положительный ион может вокруг себя собрать столько отрицательных, сколько там вообще может поместиться. Это хорошо известная геометрическая задача: если шарики двух размеров, один несколько меньше другого, то вокруг одного шарика может собраться до шести других. Это и есть октаэдр. Но не совсем так просто, потому что конечно, шесть катионов натрия не притянутся к одному хлориду, потому что тогда уже на третьем-четвертом заряд ионного агрегата станет великоват и следующий катион уже никто не сможет пихнуть в эту сторону – отталкивание пересилит кинетическую энергию. Но ионы обоих зарядов есть, и точно так же ионы хлорида будут липнуть к этому агрегату со сторон, где виден положительный заряд, общий заряд при этом уменьшится, появится возможность очередному иону натрия к этому прилипнуть.
Вот так ионы случайно и попеременно будут налипать на растущий ионный агрегат. Это реально происходит с огромной скоростью – в результате образуется хорошо нам знакомый кристалл хлорида натрия. С другими ионными соединениями будет происходит нечто похожее.
Из этого следует вывод – кристалл это и есть обычная форма существования того, что мы называем, например, хлоридом натрия. А молекула такая есть? Есть, но это просто ионная пара, и ее можно наблюдать в высоком вакууме, если мы, например, попробуем испарить кристаллик NaCl. Тогда в парах у нас будут ионы, эти ионы будут слипаться в ионные пары, тройки, четверки и т.д., но если вакуум хорош, и у нас есть какой-то инструмент, который позволяет разделять эти агрегаты (это просто масс-спектрометр, но немного более хитрый, потому что нам нужно выделять из пучка нейтральные ионные пары от заряженных троек и т.д.) и как-то их измерять, то мы сможем реально зафиксировать такую штуку как молекула NaCl. И понять, что это просто ионная пара, а хороший расчет подскажет нам, что там нет никакой ковалентности. И если прибор, с помощью которого мы разбираемся в том, что происходит в парах хлорида натрия, хорош и позволяет видеть и нейтральные, и заряженные частицы, мы увидим, что там “молекула” NaCl вовсе не основной компонент, и что там много ионных троек обоих зарядов и даже агрегатов более высокого уровня.
А в растворах не может быть молекул NaCl? Нет, не может. Чтобы разрушить очень прочную кристаллическую решетку NaCl нужен растворитель, очень сильно взаимодействующий с ионами, желательно с обоими. В общем-то кроме воды на это мало кто способен. В воде ионы и натрия и хлорида будут иметь оболочку из молекул воды. Такие гидратированные ионы так слабо притягиваются, что фактически становятся свободными в растворе, по крайней мере, разбавленном. Мы это отлично знаем, Сванте Аррениус научил. Молекул в таком растворе нет, есть ионные пары, но очень слабые, намного более слабые, чем в парах, в вакууме, где нет этого барьера диполей воды вокруг каждого иона.
Делаем вывод – так называемая ионная связь это не совсем химия, это просто закон Кулона, электростатика. Ионная связь ненаправлена, потому что не связана ни с какими орбиталями, ни с каким определенными в пространстве взаимодействиями – у сферического заряда сферическое поле, оно везде и везде одинаково. Ионная связь ненасыщаема, – притяжение двух зарядов не отменяет возможности притяжения следующих со свободной стороны, поэтому заряды слипаются в оргомных количествах, образуя кристаллы, в которых у каждого заряда вокруг него все пространство занято зарядами противоположного знака, а у них, в свою очередь, то же самое.
В этом месте у нас возникают вопросы. А какова валентность иона натрия (или хлора). Я уже слышал от реальных людей, совсем не чужих химии, ответ шесть. Ведь столько у каждого иона ближайших соседей, которые с этим ионом связаны. Ведь верно? Что не так?
Нет, не верно. Этот ответ построен на основе ложной интерпретации валентности. Валентность – это прежде всего стехиометрия, и во-вторых, принадлежность молекулы к понятному ряду. В данном случае соединение бинарное, второй элемент относится к стандартным даже по рекомендации ИЮПАК. У соединения NaCl валентности обоих элементов – единица. Погодите, ведь только что сказали, что нет такой молекулы, что это просто ионная пара, да ещё и чёрт знает где – в каких-то парах, в вакууме, при высокой температуре. Опять неверно. Когда мы пишем NaCl, мы не имеем в виду молекулу, мы имем в виду реальное вещество, обычно при стандартных условиях, а это кристаллы. Состав кристаллов точно соответствует формуле NaCl, кристалл электронейтрален как целое, хотя на его поверхности могут быть избытки ионов одного знака, но это пренебрежимо малые количества по сравнению с общим количеством ионов в кристалле. Так что с классическим смыслом валентности все нормально, как было, так и осталось, и всё понятно.А вот с “новой валентностью” вообще полный облом. Считаем связи? Какие связи, сколько там связей, в чём считать связи??? Что говорят нововалентнисты? Разное говорят, но одинаково бессмысленное. Ну зачем вы ломаете работающую вещь, совершенно не представляя, что будет на ее месте! И вообще не понятно зачем эта суета. Есть вполне понятное понятие координационного числа, применимое к атомам в координационных соединениях и кристаллах, и это действительно число ближайших соседей или же лигандов. К.ч. натрия в хлориде натрия шесть, хлора (хлорида) тоже шесть. Всё, проблема закрыта.
Есть одна связанная проблема. Валентности в хлориде натрия и всех простых солях определяются элементарно. Из этого можно было бы сделать вывод, что ионная связь порождает валентность точно так же как ковалентная. Недаром основоположники ввели два понятия – электровалентность (как раз по ионной связи) и ковалентность (по ковалентной), когда искали как найти связь классической валентностью с электронной теорией химических связей. Ну и что тогда с хлоридом аммония? А это не то же самое, что хлорид натрия или калия? Почему мы не делаем вывод, что ионная связь в этой соли добавляет одну единицу валентности к валентности азота? Может она и правда четыре, как учат нововалентнисты? Тогда уж пять, потому что нововалентнисты сначала считают чёрточки у азота, чёрточек уже 4. Ну и ионная связь в плюс. Пять? В 19 веке до Вернера так ведь и рисовали структуру солей аммония. В этом месте нововалентнисты что-то не докрутили, потому что я точно знаю, что они учат валентности 4 в аммонии. Ну не будем к ним строги, они не очень точно формулируют свои воззрения, ограничиваясь банальностями, а на все вопросы обычно делают очень важный вид и говорят: “Видите ли, это сложный случай, в науке нет определенного мнения…” В 21-м веке, нет мнения по поводу аммония. Погодите до 28-го, будет.
На самом деле, всё гораздо проще. Во-первых, ионная связь автоматически валентность не порождает. Валентность возникает не от ионной связи, а от стехиометрии соединения элементов в соединении. И в NaCl валентности по единичке появились не от того, что там ионная связь, а от того, что реакция элементов идет со стехиометрией 1:1 (по эквивалентным весам). А уж как там атомы связаны, это дело другое, это не про валентность, а про другие свойства элементов – электроотрицательность и прочее. В хлориде аммония ничего подобного нет, потому что это соединение образовалось не так. Вот если бы был какой-то “аммоний”, который реагировал бы с хлором с образованием хлорида аммония, то да, возможно. Кстати, ведь долго была такая идея что есть некий свободный “аммоний”, есть даже сказка с более чем вековой историей про “амальгаму аммония”, типа раствор свободного “аммония” в ртути, получающийся при реакции амальгамы натрия с солями аммония. Но этот материал, действительно образующийся таким способом – просто какая-то непонятная и неустойчивая каша, возможно, какие-то амидные комплексы ртути, растворенные в самой ртути. Никто и никогда не пробовал разобраться с тем, что это такое, потому что в химии вообще не очень принято разбираться в невразумительных смесях непонятно чего – возни много, а результат, скорее всего, будет скучной констатацией о том, что это смесь малоинтересных соединений – но есть вполне устойчивый консенсус, что это точно не то, за что это издревле выдавали легковерные.
А всё остальное мы про хлорид аммония уже знаем – это продукт кислотно-основной реакции, которая не изменяет валентности. Не будем повторять уже многократно сказанное.
Валентность и озон
Озон – видимо, единственная простая и известная всем молекула, которая серьёзно испытывает классическую валентность на прочность. Мы же знаем, что кислород всегда двухвалентен, на этом основана валентность, хотя ИЮПАК исключил кислород из элементов, по которым определяется валентность, но мы не обязаны следовать этой дурацкой рекомендации. Галогены, которые нам ИЮПАК суёт вместо кислорода, гораздо хуже, у них, кроме фтора, гипервалентность распространена очень широко и обильно, больше чем у любых других элементов. Кислород же всегда двухвалентен. Или нет? Действительно, и на кислород случилась проруха, и у него есть одно соединение с непонятной, на первый взгляд, валентностью. Это озон. Впрочем, если сохранять спокойствие, то никакого ущерба можно не допустить. Соединение это известно с середины 19 века, и действительно многие десятилетия напрягало всех причастных химиков, пытавшихся понять, как оно устроено и как это объяснить.
Что такое озон, тем не менее, вполне легко понять. Аналогии проще всего поискать, обратившись к химии следующего по электроотрицательности элемента, азота. Мы это тут сделали отдельно, обсуждая гипервалентность. У азота есть такое занятное соединение, тоже долго напрягавшее иследователей – закись азота. Как мы выяснили (в статьях на английском в таких случаях пишут загадочные слова vide infra – что означает – поглядите где-то тут, не знаем где, если бы знали сказали бы, но точно где-то тут, или там, но точно есть…) закись азота – типичное гипервалентное соединение, полученное из молекулярного азота реакцией с двухвалентным акцептором, синглетным атомом кислорода. При этом образуется то, что раньше называли семиполярной связью и обозначали стрелочкой, но с некоторых пор так делать перестали, и рисуют это как ковалентную связь на атомах с разделенными зарядами, то есть дополнительной ионной компонентой связывания. Валентность атома-донора при этом увеличивается на 2.
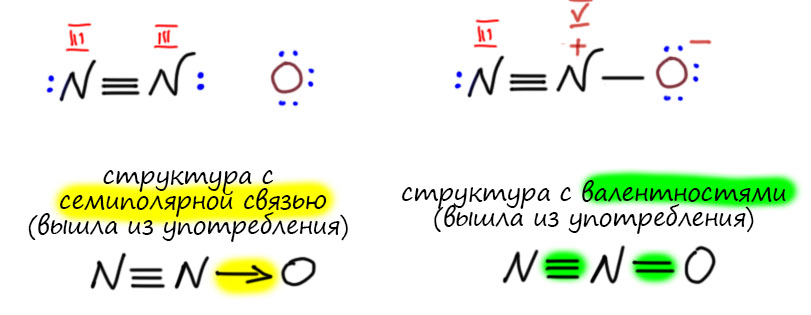
Центральный азот в закиси азота пятивалентен. Ничего себе закись! Можно сравнить только с пресловутой хлорноватой кислотой, название которой предполагает самую низкую валентность и степень окисления из возможных, но нет – это кислота пятивалентного хлора, до вершины один шаг, просто так получилось, к моменту ее открытия все названия уже расхватали и пришлось давать такое странное. Соли этой кислоты, впрочем, называют хлоратами, и это название в свою очередь поставило в неудобное положение уже соли кислоты с высшей валентностью.
Закись азота поэтому относится к обширному классу гипервалентных производных азота, часто называемых N-оксидами. Закись азота это N-оксид азота.
Теперь к озону. И мы сразу узнаем то же самое. Озон построен точно так же, как закись азота. Берется синглетный кислород (с двойной связью между атомами кислорода, единственное отличие от азота состоит в том, что там исходили из основной формы молекулы азота в основном состоянии; а здесь исходят из возбужденной формы молекулы кислорода) и соединяется по тому же принципу пара – вакансия с синглетным атомом кислорода. Ну и какова валентность среднего кислорода? Нет ни малейшего шанса уйти от ответа четыре. Четыре!!!!? Да. Было два, взаимодействие с двухвалентным акцептором увеличило на 2, итого 4. Именно так, кстати, и рисовали озон в доэлектронную эпоху, когда царили валентности. Теперь мы нарисуем структуру с одной ковалентной связью и разделением зарядов. И ещё, осознав, что молекула симметричная, применим граничные структуры, чтобы это и показать. Но на состояние центрального атома это не повлияет никак. И да, озон – гипервалентное производное кислорода. У кислорода тоже есть гипервалентность.
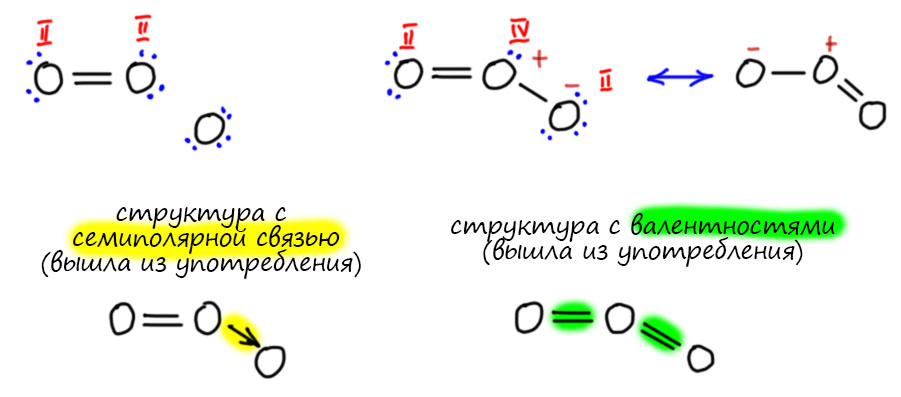
Получается, что озон это O-оксид кислорода! Кто окислил кислород? Кислород. Вы издеваетесь? Мы восхищаемся. Восхищаемся мощью и изобретательностью языка химии, в том естественно, случае, если мы его хоть немного поняли. Кстати, это даже не совсем формальное уравнение реакции. Ведь именно так образуется и распадается озон в верхних слоях атмосферы – там пониженное давление и очень много энергии солнечных лучей. Поэтому там много молекул и атомов образуется в возбужденных состояниях, могут встречаться и реагировать. Там это такое равновесие, обеспечивающее озоновый слой, озоно-кислородный цикл или цикл Чепмена. В это равновесие, кстати, вмешиваются всякие чужие молекулы, смещая его в сторону распада озона, и в этом состоит проблема устойчивости озонового слоя и озоновой дыры, которая вроде бы наконец стала зарастать.
А что, озон – единственное производное 4-валентного кислорода? Устойчивое, да, но в химии есть еще всякие реакционноспособные частицы, участвующие во всяких механизмах. Одна из таких частиц участвует в хорошо известном механизме озонолиза олефинов, механизме Криге. Подробнее разберем его в своём месте, но сейчас просто обратим внимание, что на второй стадии механизма первый озонид распадается с образованием довольно курьёзных интермедиатов – карбонил O-оксидов. Один из атомов кислорода в этих частицах точно такой же как в озоне, четырехвалентный гипервалентный. Эти молекулы очень реакционноспособны и немедленно дают второй озонид, уже устойчивый.
Ковалентная и донорно-акцепторная связь
Вопрос, который почему-то ставит многих в тупик – это разные связи или одно и то же под разными вывесками.
Одного ответа на это нет. Но не потому что здесь есть какие-то неопределенности и двусмысленности – их нет, все однозначно и довольно просто. Но есть нюанс – что имеется в виду. Как связь образуется или что из себя представляет, когда уже образовалась.
И тогда ответы разные. По способу образования это два принципиально разных способа. По одному электрону от каждого партнера, или от одного пара, от второго пустая орбиталь (так и хочется сократить в пустайку, соблазн велик, но пока не будем).
А вот когда связь уже образовалась и мы созерцаем готовую молекулу или ион, нам в принципе уже все равно, как она образовалась. Это довольно очевидная вещь. Можно взять хрестоматийный пример частицы, которая образуется за счет донорно-акцепторной связи, например, ион аммония. Никто не будет спорить, что ион аммония образуется из аммиака и протона (кислоты Бренстеда). Представить себе другой способ очень трудно, для этого потребовался бы катион-радикал аммония и атом водорода, но хоть это и не совсем банальная реакция, но не невозможная. Катион-радикал аммония существует, это просто молекула аммиака от которой забрали один электрон, от пары остался неспаренный электрон, ну и атом водорода. Ион аммония будет одинаковым, что по стандартному пути, что по этому. Получается, что связь можно образовать хоть донорно-акцепторным, хоть ковалентным способом.
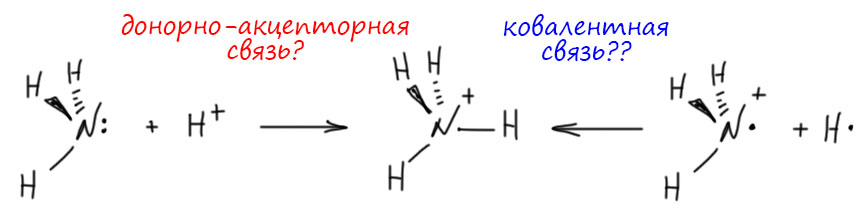
Второй довод еще проще – в иона аммония все четыре связи одинаковые, они неразличимы. Если мы возьмем всесто протона дейтерон, образуется дейтерированный ион аммония, но он будет в равновесии с обычным протоном и дейтерированный аммиак. Третий способ убедиться использует понятия изоэлектронности и изоструктурности – с ионом аммония в таком соотношении находится, например, метан. То, что в метане четыре связи одинаковые не будет оспаривать никто. Метан мы обычно образуем из метильного радикала и атома водорода, ну и как бы это нам говорит, что связь ковалентная. Но кто нам мешает образовать тот же метан из метильного карбаниона и протона – тогда связь образуется донорно-акцепторным способом, совершенно аналогично образованию аммония из аммиака и протона. Метан можно даже получить еще одним способом – из метильного катиона и гидрид-иона, способ тоже донорно-акцепторный. Метан везде будет один и тот же, все связи в нем одинаковые, и если без изотопов, то сразу после образования связи вы уже не скажете, какая из них новая, а какие были тут раньше.
Вывод отсюда только один. В готовых молекулах (и ионах) связи между атомами ковалентные. Ковалентность одначает одну простую вещь – электроны (электронная плотность) находится в общем пользовании обоих атомов.
Валентность и граничные структуры
У поклонников “новой валентности” одна из навязчивых идей – трёхвалентность углерода в оксиде углерода. В обычной химии с валентностью углерода в этом простейшем соединении проблем нет – углерод в ней двухвалентен, так как соединяется с одним атомом кислорода, который всегда двухвалентен и является одним из эталонных элементов для определения валентности. Очевидно, что трехвалентностью углерода подчеркивают то, что в этой молекуле наблюдается очень высокая степень троесвязанности за счёт высокого веса троесвязанной граничной структуры:

Эта особенность строения молекулы СО отлично известна, и на это обращали внимание ещё Лангмюр и Сиджвик, очень задолго до разработки теории резонанса. Основоположники просто обратили внимание на поразительное сходство физических свойств оксида углерода и азота, по разным простым характеристикам типа температуры кипения это как будто одно вещество. Позже, когда стали доступны различные методы изучения структуры, и спектральные и расчетные, это было многократно подтверждено. Наиболее удобный способ это выразить – мезомерия, мы рисуем две граничные структуры, исходную с двойной связью, и вторую, где за счет пары электронов кислорода и вакантной орбитали углерода образована дативная связь, и образовались формальные заряды на атомах. Мы уже разобрались в том, что термин “дативная”, или “донорно-акцепторная” относятся к способу образования связи, но не к ее природе, по природе такая связь является обычной ковалентной, то есть образованной парой электронов и двумя центрами (атомами). Следовательно, если хотите, можете использовать термин ковалентность, который как раз и определяет количество ковалентных связей. Углерод и кислород в этой молекуле трёх-ковалентны. Не трёхвалентны, а трёхковалентны. Это правильно, и так говорили. Но давно. Этот термин практически вышел из употребления уже лет 50 назад, вышел сам собой, просто оказался лишним. Если хотите возродить – пожалуйста. Но выглядеть это будет не как введение чего-то современного, а как использование архаизма. Идём по стопам А.И.Солженицына, который пытался вернуть в русский язык множество устаревших слов и снова актуализовать их через новое словообразование. У писателя почти ничего не вышло. Кстати, жаль, много интересных слов могло получиться. Но не для научной речи, а для повседневной и для беллетристики. А использовать тот же прием в науке и образовании довольно странно.
Поэтому еще раз – не путайте валентность с понятиями, построенными на теориях химической связи. Валентность хороша сама по себе, потому что она подчеркивает положение соединений в рядах родственных молекул. С оксидом углерода, в котором углерод двухвалентен, это очень полезно, потому что ставит оксид углерода в ряды производных двухвалентного углерода, карбенов. Оксид углерода – весьма типичный стабильный карбен. Это такой очень важный тип соединений, в которых вакантная орбиталь на двухвалентном углероде стабилизирована мезомерией от соседнего атома-донора. Основной граничной структурой у таких молекул как раз и является такая форма с разделением зарядов, часто называемая илидной. В этом сопоставлении выявляется много общих черт оксида углерода и таких молекул, в частности очень интеерсные сопоставления их как лигандов для переходных металлов и т.п. Назовите углерод трехвалентным и вы потеряете эти сопоставления. Поэтому в сотый раз повторю – не ломайте то, что не вами придумано и хорошо служило. Ничего нового кроме путаницы и бардака не получится.
Как определить степени окисления элементов в органических соединениях
Очень полезно уметь быстро определять степени окисления элементов, прежде всего углерода, в органических соединениях. Это помогает, например, быстро и однозначно относить реакции к окислительно-восстановительным (редокс) или не-редокс. А это не только полезно для расчета стехиометрии, но и для отлова глупейших ошибок, когда какое-то соединение с важным видом окисляют … восстановителем.
Степень окисления, как ни странно, довольно новая характеристика. В 19-м веке в классической химии ее не было. Было понятие окислительного числа, опреляемого по количеству валентностей от атома на атомы кислорода, и в отрицательную сторону – на атомы водорода. Эти величины для простых соединений (оксидов, гидроксидов, гидридов, продуктов замещения) совпадут с тем, что мы нынче называем степенью окисления, но общих правил не было. Это была эпоха валентностей, для того уровня понимания химии валентности было вполне достаточно. И не будем задирать нос, потому что из реакций, котоые мы знаем и используем, сильно больше половины было открыто именно тогда.
С появлением электронной теории связей появилась и харктеристика, которая должна была с некоторой долей условности отображать смещение электронов на ковалентных связях и при этом быть достаточно удобной и быстро определяемой. Всякие частичные заряды, то, что мы любим обозначать как δ+ и δ-, не годятся, потому что не понятно, откуда их брать. Сейчас доступны квантово-химические расчеты очень серьёзногь уровня; в этих расчетах совершенно автоматически получается распределение электронной плотности, и казалось бы, нет никакой проблемы вытащить оттуда, куда и насколько она смещена. В реальности, это оказывается не так просто, потому что в этой теории есть несколько разных способов определения границ атомов – где кончается один и начинается другой; поэтому однозначной характеристики не получается, да и кроме того нам бы сильно не понравилось, если бы для простых умозаключений нам обязали бы доставать результаты расчетов и усердно их анализировать. Положение спас великий посредник между большой наукой и нормальными людьми Лайнус Полинг. Он поступил просто, настолько просто, насколько может поступить только человек, которому не в чем ни перед кем оправдываться – не хотите не пользуйтесь, его дело предложить. Что-то типа умного голосования. Суть предложения проста – для каждой связи сделайте простую вещь: найдите более электроотрицательный атом и считайте связь полностью ионной в пользу этого атома. Это называется ионной моделью. Понятно, что эта модель не имеет никакого отношения к реальной структуре молекул, но это и не нужно. Степень окисления – величина формальная. Вся ее сила в двух вещах – простоте и стандартности. Как только мы договариваемся о схеме деления связей, все будут делать это одинаково. Ну, или скорее так, возможно и не одинаково, потому что мы сейчас увидим что и здесь есть некоторый произвол в выборе шкал электроотрицательности, но в рамках одной задачи это можно делать одинаково, и очень легко понять, в чем причина расхождений, если таковые возникнут.
Что нужно для определения степеней окисления? Структурная формула, использующая только настоящие связи, изображаемые сплошной чертой. Структура должна удовлетворять правилам валентности, и в тех случаях, когда необходимы формальные заряды, структура должан быть размечена зарядами. Интересно, что структурные формулы, использующие вместо связей валентности отлично годятся для разметки степеней окисления атомов, и ничем не хуже структурных формул, изображающих связи. Эти два типа структур дают одинаковые значения степеней окисления. В тех случаях, когда соединение невозможно изобразить одной структурой, и нужны граничные структуры, бывают некоторые сложности, но мы и с этим справимся.
Что не годится для определения степеней окисления? Структуры с неполноценными связями – пунктирами, гайками и прочей нестандартной лабудой. Я терпеть не могу весь этот мусор, и всячески рекомендую им не пользоваться. При этом, от гаек иногда почти невозможно уйти, например, в ароматических ионах, но если приспичит определять степени окисления, придется вместо гайки нарисовать граничные структуры. А вот пунктиры всех типов – однозначно гадость, использование которой не говорит ни о чём, кроме неразборчивости и да, наверное, банальной лености изображающего – типа, не хочу я думать, как тут распределяется плотность и заряды, делокализованы и славно, тыц-тыц-тыц-тыц… (рисует пунктир).
Что ещё нужно? Таблица электроотрицательностей элементов. Какая? их же много – Полинга, Малликена, Олреда-Рохова, Аллена и еще есть и до сих пор находятся желающие выкатить новую шкалу. Ответ простой – любую, какая больше нравится. Большинство шкал, хоть и основаны на совершенно разных физических принципах, линейно коррелируют друг с другом. Более того, в самой важной части – популярных в органике неметаллов (C, N, O, F, Cl, Br, I, S, P, B) практически все шкалы дают один и тот же порядок следования величин, а следовательно и сортировки элементов по электроотрицательности. И это тем более верно, что нам практически никогда не придется связываться с необычными связями, например, As-H или Sb-B. Или, тем более, связями металл-металл. Вот там разнообразие велико и есть проблемы, только дела до этих проблем нет никому кроме очень узких специалистов.
Если хотите большей определенности, то две шкалы имеют больше прав чем другие. Первая шкала – шкала Полинга – чаще всего использовалась с начала, потому что именно Полинг и заварил эту кашу, занявшись вплотную зависимостью прочности ковалентных связей от электроотрицательности элементов. И он же придумал само понятие электротрицательности, как мы уже видели, и принцип, лежащий в основе определения этих величин. Мы привыкли именно к этой шкале, когда нам нужно оценить полярность связи. Можно сказать, что мы впитали эту шкалу с самыми первыми напитками, у кого какой был – молоком матери, кефирчиком, пивом – и прямо на автомате выдаем порядок электроотрицательности именно по этой шкале. Это была первая шкала. Моя первая шкала, скажет любой химик и сладко потянется, вспомнив далекую юность.
Но на свете есть ИЮПАК. Теоретически и в идеале – чтобы помогать химикам все делать максимально правильно. В реальности часто получается не как лучше, а как всегда – ИЮПАК путается под ногами и мешает, подсовывая вместо чего-то привычного что-то странное. Надо сказать, что если к валентности отношение ИЮПАК довольно разумное – пользуйтесь привычной валентностью, хотя в официальном определении, как мы уже видели, кислород зачем-то заменен на галогены, но это не принципиально. То в отношении степени окисления ИЮПАК зачем-то страшно возбудилось и совсем недавно, и поручило заинтересованным ученым написать подробные рекомендации. Ученые справились на славу, выдав многостраничный и крайне мутный документ, ставший официальной рекомендацией ИЮПАК. Надо сказать, что большинтво того, что в нем есть мало кого волнует – там рассматриваются всякие экзотические вещи типа полиметальных кластеров и т.п. – хочется надеяться, что на свете есть боги, которым это угодно. Привычные основы этот документ практически не трогает. Но там есть одна рекомендация, мимо которой не пройти – шкала электроотрицательностей. ИЮПАК довольно строго рекомендует шкалу Аллена, довольно позднее изобретение Лиланда Аллена (1989 год), основанное на спектроскопических оценках энергий валентных электронов. ИЮПАК обосновывает это так: электроотрицательность это свойство элемента, и определять его надо из самой простой формы элемента, свободного атома. А не как Полинг – по реальным соединениям, в которых есть весьма значительные вариации, которые не так уж понятно, как учитывать. В принципе, с этим можно согласиться. Шкала Аллена хороша кроме прочего и тем, что в ней можно определить электроотрицательность совершенно любого элемента. Например, гелия, неона, аргона, радиоактивных неустойчивых элементов. И предаться сладким мечтам о том, что когда вы наконец получите гелистый неон (или неонистый гелий?) или аргонид гелия (или гелид аргона?), сможете ответить на вопрос короля Швеции в понятно какой момент, какова степень окисления этих элементов в этих соединениях. Итак, давайте будем использовать Аллена, раз так просит такая уважаемая международная организация. В том, что касается элементов главных подгрупп получается вполне разумная картина, практически неотличимая от привычной. Но постойте – что там, иод менее электроотрицателен, чем углерод?! Галоген слабее углерода?! Что ж получается, в иодистом метиле на углероде отрицательный заряд?! Фигня какая-то. Ладно оставим это пока. Если честно, это не так важно. Разница маленькая, связь малополярна. Что ещё? Ну, понятно, чемпион теперь неон. А не фтор. Чёрт с ним, все равно из неона ничего не сделаешь. Но, например, у аргона электроотрицательность получилась вполне невыдающаяся. Ну и где у нас фториды и оксиды аргона? Даже у гелия электроотрицательность, конечно, приличная, но фтору должно было бы быть по силам справиться. Фторид гелия? Не слышал. Ладно, не будем придираться, химия сложнее простых шкал. В целом шкала вполне нормальная, можно пользоваться.

Но всё же поглядим, насколько это отличается от старой доброй шкалы Полинга. Ой, почти никак. Только два отличия. Правда одно очень важное. По Полингу все-таки иод был более электроотрицателен, чем углерод, и нам это, конечно, гораздо привычнее. Галогены всё же хочется считать более серьёзными неметаллами, чем углерод, все галогены. Астат никого не волнует, хотя может вполне быть так, что электроотрицательность астата не меньше, чем у иода. Там внизу групп у очень тяжелых элементов включаются релятивистские поправки из-за того, то в сильно сжатых атомах с огромным зарядом ядра внутренние оболочки станосятся очень компактными, электроны носятся там как ненормальные, и правда становятся ненормальными, релятивистскими. Не будем про это, хотя эти эффекты весьма важны для тяжелых элементов. Второе место, где у Полинга и Аллена разные мнения, это азот и хлор. По Аллену азот круче хлора, по Полингу хлор круче азота. Соединения со связями N-Cl в химии встречаются, это хлорамины и хлорамиды, но встречаются редко. Для них степень окисления азота и хлора, почитанная с помощью Полинга и Аллена будут разными. Ну и хорошо. Как уже много раз сказано, степень окисления не является причиной ничего, это чисто формальная величина. Важно только, чтобы если вы ее применяете, везде применялась одна схема расчета. Выбирайте любую и используйте. Ошибок не будет. Ну а во всех остальных случаях разницы нет никакой, и ИЮПАК, право, напрасно все это затеяло, только иод смутили, разжаловали в более рядовые неметаллы, чем углерод. В этом весь ИЮПАК. Впрочем, большинство нормальных людей ничего не знает про эти рекомендации и не придает им никакого значения, продолжая спокойно пользоваться Полингом.
Итак, переходим к правилам подсчета степени окисления. Они просты.
- Выписываем структуру
- Каждую связь делим так, что более электроотрицательный атом получает -1 в свою степень окисления, а менее электроотрицательный +1.
- Если связи кратные, делаем это для каждой связевой черты
- Если есть заряды, добавляем их у каждого атома, на котором они есть, просто прибавляем с учетом знака
- Связи между одинаковыми элементами ничего не дают в степень окисления, в каком бы валентном состоянии ни были элементы – электроотрицательность в этом деле всегда рассматривается как неизменное свойство элемента;
Посмотрим, что получится. Берем метан. Каждая связь С-Н дает вклад -1 в степень окисления атома углерода. Раз связей 4, то и суммарная степень окисления -4. У атомов водорода степень окисления +1. У водорода почти всегда +1, кроме соединений с менее электроотрицательными элементами, напрмер, бором и кремнием. Это дает нам возможность больше не разрисовывать, как в школе все атомы водорода, а просто для каждого атома углерода, азота, кислорода, серы, на которых могут быть атомы водорода, просто добавлять число водородов с минусом к степени окисления элемента. Вот следующее соединение, метилэтиламин. С-С связи не дают ничего в степень окисления углеродов. С-N связь дает +1 углероду, -1 азоту. Суммируем для каждого атома. Видим, что степень окисления углерода изменяется довольно сильно в зависимости от заместителей. Еще одну вещь видим и это не частный случай, а общее свойство степени окисления, – сумма степеней окисления по всем(!) атомам, включая водороды, для нейтральной молекулы равна нулю. Если бы это был ион – то заряду иона. А если отделить атомы водорода, то сумма степеней окисления атомов без водородов равна числу атомов водорода с знаком минус. Вот в этом амине сумма степеней окисления (-3-1-3-2 = -9) равна числу атомов водорода (9) со знаком минус. Дальше все понятно. Не смущаемся двойной связью. Если она между углеродами, то ничего не дает. Если двойная связь с кислородом (карбонил) то дает +2 углероду, и -2 кислороду. У кислорода, впрочем всегда будет степень окисления -2, кроме соединений со связями O-F, но это мы вряд ли увидим.
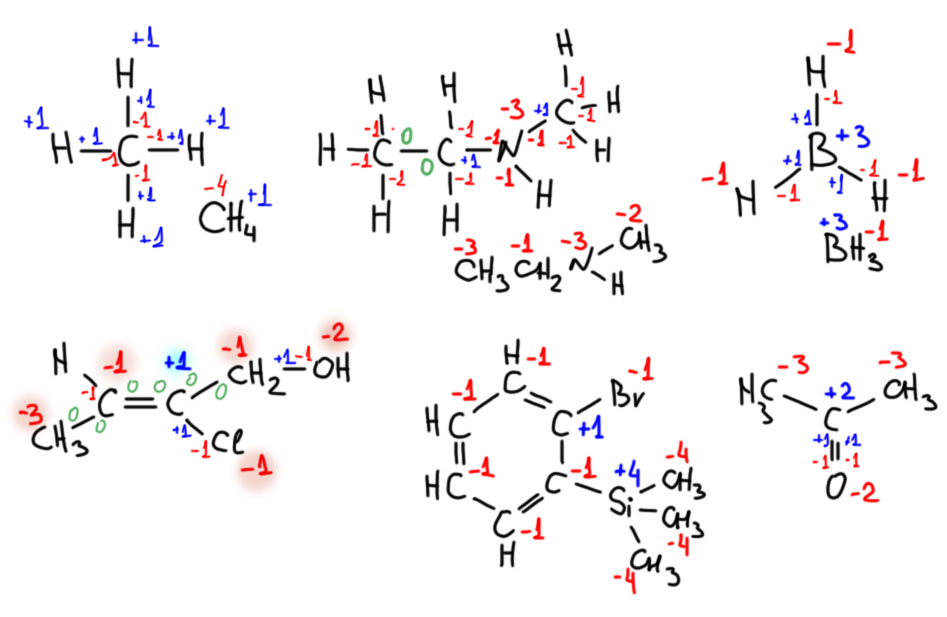
Степень окисления углерода в органических соединениях изменяется в полном диапазоне от -4 до +4. Вот небольшой список типичных примеров (углерод с данной с.о. помечен желтым). Видно, что в одну группу попадают совершенно разные типы соединений, и в этм состоит значительное неудобство степени окисления – всё же это весьма формальная величина. Но не все так плохо – тенденции в рядах есть, видно, что движение в сторону окисления (например, от углеводородов к спиртам, дальше к альдегидам, кетонам, карбоновым кислотам и производным, и наконец к углекислоте) сопровождается увеличением степени окисления.
Со степенью окисления иногда бывают неприятные фокусы, когда в молекуле или ионе наблюдается не фиксированное положение связей, а делокализация. В этих случаях мы пользуемся мезомерией и граничными структурами, или вообще имеем что-то типа ароматической делокализации. Возьмём, например. такое немудреное соединение как пиридин. Вот какая у него степень окисления углеродов рядом с азотами? С остальными все просто, там будет как у бензола -1. У нижних углеродов степень окисления получается разной (-1 от водорода, но +1 или +2 от азота в зависимости от связи, двойной или одинарной). Но мы знаем, что эти углероды одинаковы, и следовательно у них не может быть разной стпени окисления. Мы понимаем, что ароматическая делокализация делает эти углероды одинаковыми. Но как это учесть? Довольно просто, если мы вспомним главное правило – сумма степеней окисления по всем атомам равна нулю (или заряду, если это ион). Поскольку степени окисления у всех остальных атомов вполне определены, то на эти два в сумме остается +1.
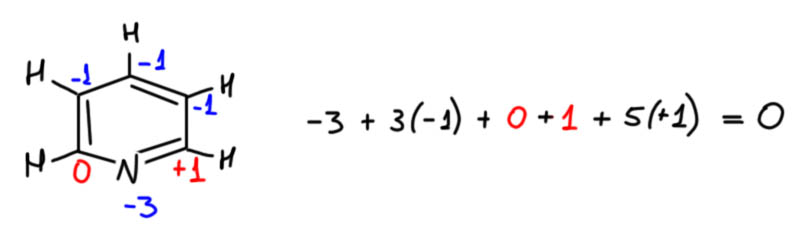
Ну и ничего нам не остается, кроме деления пополам. Получается дробная степень окисления +1/2. Сумма ноль. Все в порядке. Но придется смириться с дробной степенью окисления. ИЮПАК не возражает и ничего другого не предлагает.
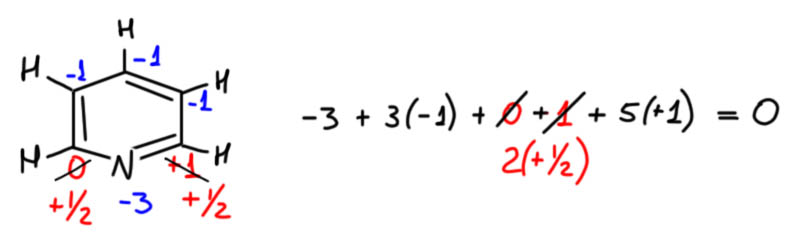
А теперь возьмем несимметричную молекулу, да хоть просто заместитель воткнем в тот же пиридин. Вот что получится. Здесь у двух углеродов будут разные степени окисления в двух граничных структурах. Выписываем все, убеждаемся, что сумма равна нулю и там, и там. Поскольку структуры в равной степени правильные, всё, что на м остается, это для каждого углерода взять среднее. Получим дробные степени окисления, не забудем проверить, что сумма осталась равна нулю. Не пренебрегайте этой проверкой – чаще всего ошибки мы делаем в самых примитивных расчетах, когда делим две морковки и три яблока на белочку, зайчика и ёжика (прим. – ёжику всё это на фиг не сдалось).
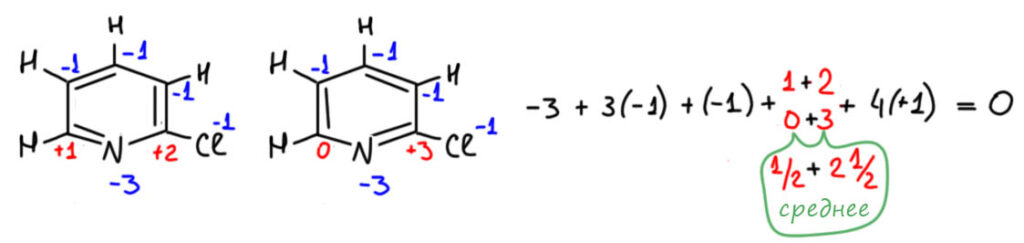
В молекулах, для изображения которых необходима мезомерия, всегда проверяйте по каждой граничной структуре степени окисления. Вот наша любимая молекула оксида углерода (напоминаю, что валентность углерода равна 2). Стпени окисления в обоих граничных структурах получаются одинаковые (во второй связь тройная, но надо учесть заряды)
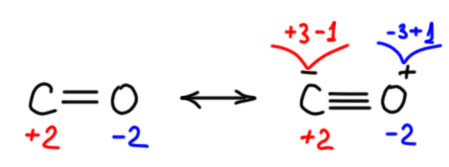
Так будет не всегда. Вот, например, аллильный анион. Две одинаковые граничные структуры, но в каждой одинаковые углероды будут иметь разные степени окисления. Очевидно, что так оставлять нельзя, это ни в какие ворота не лезет – мы точно знаем, что аллильный анион совершенно симметричен, и это как раз и отображается с помощью граничных структур. Во-первых, проверяем, что сумма степеней окисления равна – в данном случае не нолю, а заряду аллильного аниона, -1. Все правильно, и тогда мы просто берем среднее и получаем в качестве степени окисления крайних углеродов минус два с половиной, сумма при этом остается той же минус единицей. 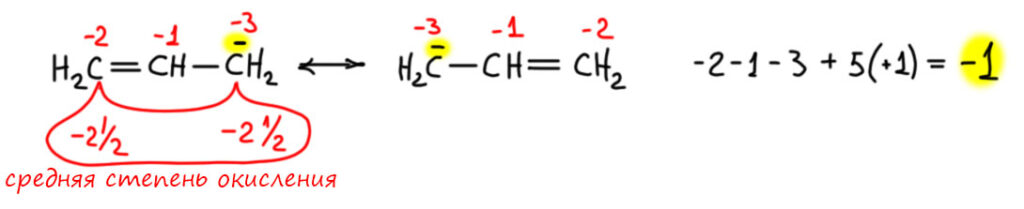
В общем, получается, что определить степени окисления элементов не просто, а очень просто. Давайте напоследок посмотрим, какими степенями окисления могут похвастаться другие важные элементы в органике. После углерода самый богатый ряд у азота – от -3 до +5.
У кислорода почти везде степень окисленния -2. Есть еще -1 в пероксидах всевозможных типах. Ноль в самом кислороде и гипофторидах R-OF, эти соединения иногда используют для фторирования. Положительные степени окисления у кислорода – большая редкость: плюс один у центрального кислорода в озоне (у крайних кислородов минус половинка). Плюс два у фторида кислорода.