Фосфор в органической химии
Мы не изучаем химию фосфора в нашем курсе, но постоянно на неё натыкаемся. Полифосфорная кислота как кислотный катализатор. Орто-фосфорная тоже иногда используется. У нас есть реагенты для замены гидроксила на галогены, попроще типа галогенидов фосфора, и посложнее типа смесей трифенилфосфина и источников галогенов. У нас есть реакция Виттига и ее разновидности. Мельком поминается реакция Михаэлиса-Арбузова, фактически просто для того, чтобы делать реагенты для реакции Хорнера-Водсворса-Эммонса. Если доберемся до темы о применении комплексов переходных металлов в синтезе, там полно фосфиновых лигандов, они там просто царят, и ни о чём кроме фосфинов там никто и не говорит. Ну, вообще впечатляет. Получается, что это очень важный элемент, фосфор. А мы знаем о нём хоть что-нибудь? Нам кажется, что знаем, мы же привыкли так легко смотреть на Периодическую таблицу и считать, что раз мы можем найти там элемент, то в каждой группе всё одно и то же. Азот изучили, ну и все эти остальные фосфор, мышьяк, сурьма (правда элемент так смешно называется? – я всякий раз лезу в таблицу проверять), висмут – наверняка все одно и то же, тем более что вот с этими мы совсем почти не встречаемся, кроме фосфора. Нынче всю эту компанию под азотом принято называть пниктогенами.
Открою маленький секрет – они все разные. У каждого своя химия. Сходство есть, но различий едва ли не больше. И для всех элементов пятой-А (или пятнадцатой) группы, включая азот, ключевое слово – гипервалентность, но у каждого элемента группы это явление – выход за формальные валентные возможности по образованию ковалентных связей – проявляется по-своему. Для фосфора это явление настолько важно, что оно буквально определяет химию этого элемента.
Попробуем разобраться и тогда уже со знанием дела разберемся и в реакции или перегруппировке Михаэлиса-Арбузова и ее продолжениях.
Реакция Михаэлиса-Арбузова. Вступление.
Реакция Арбузова, а точнее, как её принято называть во всём мире, Михаэлиса-Арбузова – это важнейший прототип множества реакций с участием соединений фосфора, для органической химии фосфора это такая отправная точка. Как только появилась эта реакция и представление о том, что фосфор любит устраивать довольно удивительный трюк – менять степень окисления и валентность в процессе самого простого нуклеофильного замещения – химия фосфора поехала вперед очень быстро и реакции, построенные на этой способности фосфора, стали появляться сразу пачками. Трудно даже сказать, сколько всего существует таких реакций – там только именных не менее десятка.
Реакцию обнаружил немецкий химик Август Михаэлис (Карл Арнольд Август Михаэлис, не стоит путать с более молодым коллегой Леонором Михаэлисом, одним из авторов фундаментального в ферментативной кинетике, да и вообще в кинетике катализа уравнения Михаэлиса-Ментен) в 1898 году. Это довольно скромная единичная работа, – Август Михаэлис был не самым знаменитым химиком своего времени, он больше занимался преподавательской и методической работой, чем исследованиями. Но фосфорорганики считают его отцом-основателем своей химии, потому что он действительно опубликовал около двух десятков коротких работ в конце 19 века, в каждой из которых описываются одна-две типичные реакции соединений фосфора – так, что последующие исследователи обычно находят прототипы важнейших реакций фосфорорганических соединений в работах Михаэлиса. Вот и в этом случае Михаэлис строго говоря описывает не вполне то, что стало после знаменитой реакцией – работа описывает в общем довольно банальную кватернизацию фенильного эфира фосфористой кислоты парой простейших иодалканов с выделением фосфониевых солей и их последующим отдельным гидролизом при нагревании. Единственное, что здесь по настоящему интересно – почему при гидролизе легко уходит только одна группа и не уходят другие, почему вместо гидроксильной группы получается атом кислорода вроде как на двойной связи. Если мы уже знаем необычную химию фосфора, то понимаем, что это очень важная особенность, это неспроста. Но в те времена о таких вещах не знали ничего – получили, прогидролизовали, выделили, определили анализ, написали возможную формулу – всё, пожалте в вечность, вон местечко на 1325 ряду в 154 секторе свободно.
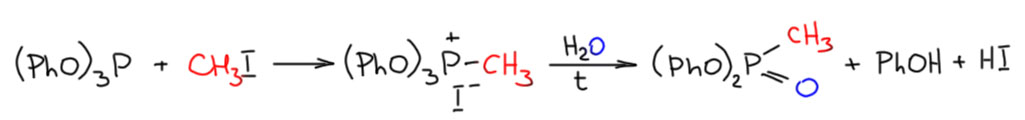
Строго говоря, в той же работе есть и реакция триэтилфосфита с иодистым метилом, которая привела к метилфосфоновой кислоте, но она сделана как-то совсем криво, почему-то при очень сильном нагревании, отчего там произошло еще и элиминирование, и где там что и в какой последовательности, Михаэлис со своим коллегой разбираться не стали.
Спустя несколько лет реакцией заинтересовался молодой русский химик Александр Ерминингельдович Арбузов, ученик знаменитого профессора Зайцева, того самого, который до сих пор командует нашими элиминированиями. И заинтересовался серьёзно, не просто чтоб статейку накропать, потому что положено, а для того чтобы разобраться в тогда малопонятной особенности химии фосфора – взаимопревращении 3-х и 5-тивалентных соединений. Арбузов проделал очень большую работу, разобрался, нашел именно то, что и называется реакцией Михаэлиса-Арбузова, а иногда и перегруппировкой Михаэлиса-Арбузова. Последнее – популярное, но неудачное обозначение; Михаэлиса еще можно считать открывателем прототипа реакции, но никак не перегруппировки. За пределами СССР Арбузовым довольно долго пренебрегали, реакцию просто называли по имени Михаэлиса, но это не какие-то происки злобных завистников и махровых антисоветчиков, а следствие того, что Арбузов опубликовал свои работы в русском журнале ЖРФХО, и на международном языке того времени вышел только коротенький реферат. А полностью исследования Арбузова вообще можно найти только в его диссертации, вышедшей уже во время Первой мировой, а потом сами знаете что было. В общем, просто время было такое, да и сам Арбузов что-то то ли забыл выучить немецкий, то ли заболел модной в то время германофобией – но вот так чуть было не упал по собственной воле с корабля истории. Почти все знаменитые русские химики тех времен отлично знали, что самые важные работы надо обязательно публиковать на немецком, и почти никто не забывал этого делать, в том числе учитель Арбузова Зайцев. К счастью, в том числе и усилиями Арбузова, в СССР сложилась очень мощная школа фосфорорганики, вклад которой в общую мировую фосфорорганику был столь велик, что со временем все во всем разобрались (в первую очередь с помощью человека с колоритной фамилией Косолапофф, о чем немного ниже), история области устаканилась и все заняли свои места. Сын Арбузова, тоже Арбузов, Борис Александрович, тоже большой ученый, однажды много позже немецкий таки выучил и уже в 1950-е написал про роль своего отца в становлении фосфорорганической химии в немецкий журнал Ангевандте, восстановив последовательность событий. Это было нелишне. потому что у фосфорорганики всегда и особенно в ранние годы ее истории был шлейф науки, с помощью которой делают что-то зловещее, секретное, особой государственной важности – из-за этого ученые, работавшие в этой области были невыездными, а статьи были нарочито невнятными, так что разбирать начальный этап фосфорорганики – занятие для хорошего расследователя скорее, чем для обычных ученых.
Пара слов про самого Арбузова и про его немного необычное отчество. Арбузов в СССР стал одним из самых статусных ученых с бесконечной чередой званий, наград, премий, должностей. Если мы посмотрим на официальную биографию Арбузова, с интересом обнаружим, что он родился в селе Арбузово-Баран, и ныне существующего в Татарстане. У нас обычно очень скупые биографии – будет сказано, что родился и всё – дальше уже идут достижения, но про Арбузова зачем-то написано еще, что в детстве он был на домашнем образовании. Так кем же он был – крестьянином этого села, фамилия по селу давалась вполне часто, но крестьян на дому учили разве что скотину доить. Нет, оказывается всё же можно найти, что семья Арбузова была из землевладельцев, помещиков, и село с землями вокруг было их собственностью; вот откуда и домашнее образование. В этом месте у советских биографий, тех, что всё же осмеливается упомянуть помещичье происхождение, начинаются совершенно комические колики – пишут, что помещики были мелкопоместные (как же, как же – село немаленькое, земли вокруг не могло быть мало, да и вообще это богатые места с отличной землёй, дававшей прекрасный доход) а в одном месте и вовсе написано, что отца Арбузова “любил народ”. Конечно, конечно – в тридцатые чекисты любили взять очередного классово чуждого, бывшего помещика, например, и обязательно спрашивали, не любил ли его народ. Если выясняли, что любил, да и помещик мелкопоместный, тут же не только отпускали, но и давали компенсацию за задержание и рекомендацию к приёму в партию; а вот если нет, не любил народ – тогда к стенке без промедления. А вообще упоминать про помещичье происхождение приходилось, потому что страшно хотелось рассказать, как батюшка ученого вместе с маленьким сыном ездили в соседнее поместье самого Бутлерова. О чём они там говорили сказать невозможно, но вряд ли о химии – Бутлеров в конце жизни это не тот Бутлеров, который в 1860-70-е действительно был одним из самых передовых мыслителей в химии, в конце жизни он увлекся мистикой, спиритизмом и прочим мракобесием, и только лениво отбивался от гневно обличавшего такое предательство науки Менделеева. По совсем странному совпадению скоро после визита Арбузовых, Бутлеров упал в библиотеке с лестницы, и очень быстро после этого скончался. Так что идея, что на путь служения науке юного Сашу наставил сам Бутлеров, который в советской научной мифологии занимал место одного из главных божеств, явно несостоятельна. Отца будущего основоположника фосфорорганики действительно звали Ерминингельд. Интересно, что если посмотреть известных родственников этой семьи, у всех не просто обычные, а самые распространенные русские имена, да и отчество у Ерминингельда было Владимирович. В русских святцах есть такой святой, мученик Ерминингельд, царевич Готфский, память на 14 ноября по старому стилю. Довольно экзотический святой, мученик 6-го века, из Испании, которую после римлян завоевали варвары, вестготы, которые приняли неправильную разновидность христианства, арианскую ересь, а некий Ерминингельд оказался диссидентом и захотел правильного христианства, за что от своих же и принял смерть. А отец Арбузова при чём? Можно подумать – не повезло, родился человек, надо крестить, а тут в святцах как назло одни раннехристианские мученики с именами одно другого заковыристей, такое бывает. Но нет, на тот же день приходятся и Кузьма, Демьян, Иван, Яков – выбирай не хочу и не выпендривайся. Но ведь нет – выбрали Ерминингельда, не догадывались, что сын его станет важной фигурой и потомкам придется выговаривать это Александр Ерминингельдович, и гадать, из каких заморских краёв прибыл к нам сей персонаж. Не из каких – простой русский помещик из Прикамья.
Так вот молодой Александр Ерминингельдович в начале 20 века взялся за исследование реакции, кратко описанной Михаэлисом, только сделал все аккуратно и по уму, как следует очистил все исходные, проводил реакции в мягких условиях, умно подбирал реагенты, тщательно характеризовал продукты. И обнаружил, что при действии иодистого метила триметилфосфит гладко превращается в диметиловый эфир метилфосфоновой кислоты. Да это же настоящая перегруппировка! Поэтому собственно реакцию эту очень часто называют перегруппировкой Михаэлиса-Арбузова.
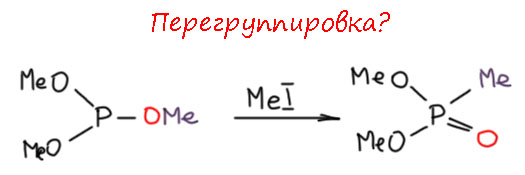
Поскольку хотя бы в общих чертах про эту реакцию знают все, могу легко предвидеть, что сейчас множество голосов сольется в порицании очевидной ошибки – метил-то в продукте не оттуда! Не торопитесь, это не ошибка, не всё так просто.
Надо сказать, что в органической химии есть очень странное поверие – все реакции одинаково интересны, но гроша ломаного не стоят рядом с самой простой перегруппировкой. Ученому повезло, если открыта новая реакция. Но если это перегруппировка, то уже при жизни является делегация уполномоченных божеств, документирует подвиг, и выписывает предписание о принятии открывателя в сонм небожителей. Даже подсознательно кажется, что это должно быть действительно круто – ведь если все на свете вещества созданы богами, то ученый, открывший способ взять такое творение, и просто поменяв части местами, сделать другое творение – становится равным или хотя бы полуравным богам химии – он как бы указывает им, что их трюки известны и могут быть повторены.
По-другому трудно к этому отнестись, ведь большинство перегруппировок – самые обычные реакции, а если удается разобрать их механизм, то многие оказываются еще и милым жульничеством – никакие это не перегруппировки, и в совпадения состава исходного и продукта часто чисто случайно. Вот так же и в этой реакции метил оказывется не из исходного фосфита, а из иодистого метила. Арбузов легко показал это, взяв разные алкилы. Придется тогда агкилгалогенид перенести со стрелки в уравнение реакции.
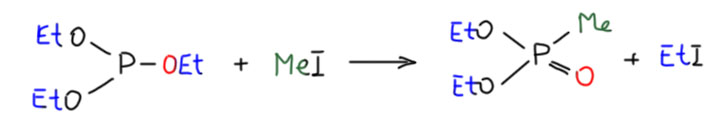
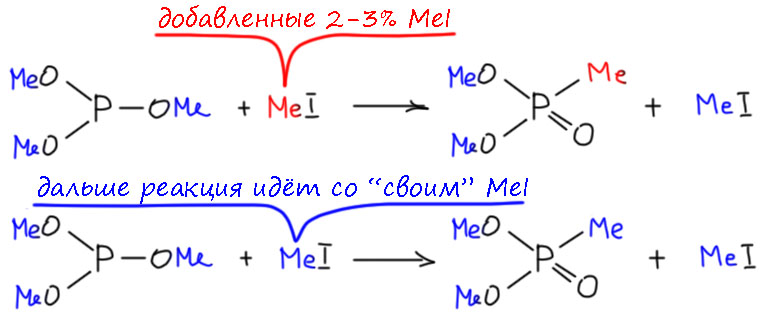
Получается, что это никакая не перегруппировка. Но нет, ведь есть и еще одна возможность сделать эту реакцию – можно использовать очень маленькое, каталитическое количество алкилгалогенида, того же иодистого метила, не более одной десятой эквивалента и даже еще меньше. В этом случае исходный иодистый метил инициирует реакцию, а дальше на фосфор пересаживается уже метил из исходного фосфита. И добавленный иодистый метил будет толко в первых процентах продукта. Мы могли бы применить изотопную метку, дейтерий в метиле или углерод-13 там же, и увидели бы это очень ясно. И тогда это уже во всех смыслах перегруппировка, и добавленный иодистый метил – просто катализатор этой реакции.
Но поскольку мы не разделяем поверия о величии перегруппировок, и ничего особенного в них вообще не видим (отчасти поэтому на сайте до сих пор нет отдельной странички про перегруппировки), у нас открыты глаза на то, чтобы увидеть в этой реакции много по настоящему интересного помимо слегка бессмысленного спора о том, перегруппировка это или просто реакция. На первый взгляд ведь это просто очередное SN2-замещение, ведь трудно не заметить у исходного фосфита неподеленной пары на фосфоре, это ведь не что иное как фосфин, триалкоксифосфин. Но мы видим, что просто замещением дело не ограничивается, ведь с нуклеофилом тоже что-то случилось – фосфор изменил степень окисления и валентность, стал представителем совсем другого ряда соединений. И почему так легко уходит один алкил. И чисто практически – у нас получилось фосфорорганическое соединение. Напомню, что элемент-органическими соединениями называются не любые соединения элемента с оранической частью – обязательно нужна хотя бы одна ковалентная связь элемент-углерод. В исходном ее не было, в продукте есть. Богатая реакция – в ней много фишек, в них надо разобраться и тогда мы, возможно, увидим, большую органическую химию фосфора. Смысл открытия Арбузова оказался именно в этом.
Но чтобы разобраться даже в самых простых особенностях этой химии, нам понадобится хотя бы немного углубиться в химию фосфора. А что за проблема с химией фосфора? Проблема в том, что мы её плохо знаем, и поэтому не вполне понимаем почему реакции производных фосфора идут так, как они идут.
Краткий курс химии фосфора
Мы в органической химии соединениями фосфора пользуемся весьма активно, но уделить им хоть немного внимания самим по себе не догадываемся. Вообще у нас часто возникает такой вульгарный периодизм – мы смотрим на элементы из 3-го периода, а еще реже из 4-го и 5-го, видим интересные реакции, но наше отношение такое – это пятая группа (или шестая, если нам попалась сера или даже селен), все одно и то же. Азот знаем. Амины знаем. Вот фосфины это амины из 3-го периода. Что там ещё? Фосфорная кислота? Ну это как азотная, только фосфорная почему-то орто-, а азотная какая? Пара или мета? А что это вообще значит?
Это очень большое заблуждение. Аналогии никто не забывает – это одна из основ периодичности. Но ведь это и называется не аналогичность, а периодичность именно потому, что закономерности намного сложнее и интереснее, чем просто повторение с небольшими количественными вариациями. Нет двух одинаковых элементов. Нет двух даже очень близких элементов. Каждый элемент – ярчайшая индивидуальность, и его положение в группе и периоде задает только самые общие тенденции. Поэтому фосфор это не азот, и забывать это нельзя. А если бы мы спустились на один элемент, то с удивлением увидели бы, что мышьяк это не фосфор, прямо таки совсем ни разу не фосфор. Но мы туда сегодня не полезем.
Фосфор – это неметалл, но с очень небольшой электроотрицательностью. Это важная особенность третьего периода, в котором под валентной оболочкой находится внутренняя полностью заполненная 2p-оболочка, очень компактная и поэтому отлично экранирующая ядро атомов, из-за чего электроны на валентной оболочки относительно слабо связаны, легко уходят и не очень охотно приходят. В третьем периоде очень популярны высшие степени окисления, и даже у хлора, настоящего галогена, вице-фтора, силенок не хвататет, чтобы удержать все семь валентных электронов, и это самая устойчивая степень окисления кроме, конечно, обязательной для галогена -1. Сера по электроотрицательности не очень далеко от углерода, и очень любит свою высшую валентность, а ведь сера – это элемент под кислородом. Ну и к фосфору мы доходим до настоящей электроотрицательной немощи. Такое впечатление, что у этого элемента одна цель в жизни – всеми способами избавиться от всех валентных электронов, сбагрить их хоть кому-нибудь. “Валентные электроны, настоящие, отдам в хорошие руки, в плохие тоже отдам” – кажется так прямо и написано в клеточке этого элемента в Периодической таблице. Для нас важно, что по электроотрицательности он уступает всем без исключения типичным элементам органики – всем галогенам, включая иод, кислороду и сере, азоту, углероду (да! и сильно) и даже водороду (да!да!да! – чуть-чуть, но чуть-чуть не считается, хоть на волосок, уступает и всё, извольте записать и пользоваться – это важно для определения электроотрицательностей, и здесь не должно быть никаких колебаний и разговоров про то, что фосфин образуется при гидролизе фосфидов – это не имеет ни малейшего отношения к электроотрицательности).
Фосфор – это элемент, для которого особо характерны всего две валентности – три и пять. В точном сответствии с номером группы: номер группы и восемь минус номер группы. И поскольку у нас такие дела с электроотрицательностью, это означает, что и по степени окисления фосфор небогат (сравните с азотом, только сами) – плюс три и плюс пять, остальное – экзотика.
Со стереохимической точки зрения фосфор тоже скуп невероятно. Маниакальная приверженность тетраэдру, иногда без одной вершины, которую занимает неподеленная пара. Можно сказать, что подавляющее большинство соединений фосфора имеет фосфор в тетраэдрическом окружении. Обе основные валентности фосфора довольствуются такой стереохимей и не жалуются, что вон даже у углерода насколько больше. Что значит “даже у углерода”!? Углерод для фосфора – настоящий электроотрицательный неметалл, и извольте подчиняться. И чтобы уж окончательно понять, куда мы попали, заметим, что у фосфора тетраэдр не имеет никакого отношения к sp3-гибридизации. А к какой имеет? Да в общем ни к какой. Вторая стереохимическая привязанность фосфора немного нам непривычна – это тригональная бипирамида, такой треугольник в плоскости и сверху и снизу над и под серединкой, то есть собственно атомом фосфора – еще по заместителю.
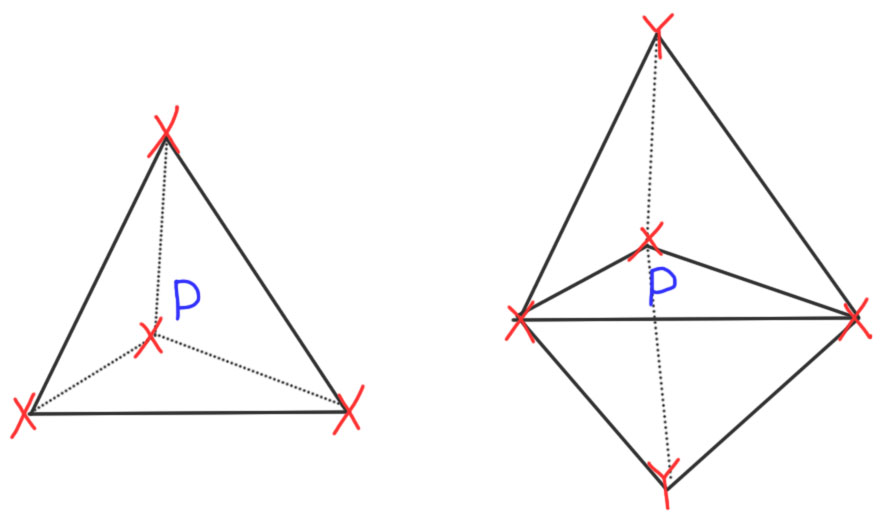
Следующая важнейшая и ярчайшая особенность фосфора – стремление к валентным состояниям, которые принято называть гипервалентностью. Фосфор – типичный p-элемент, имеет валентную оболочку из 3s и 3p-орбиталей, не имеет никакого доступа к 3d-оболочке, которая пока ещё очень высоко и пользоваться ей невозможно. Если бы было возможно, мы бы называли фосфор d-элементом, но такая мысль никому не приходит в голову, хотя по традиции даже в самых крутых учебниках типа Коттона-Уилкинсона любят писать, что расширение октета достигается за счет использования d-оболочки. Это старое заблуждение, давно преодоленное, но очень сильно въевшееся в учебную, да и научную литературу. Но надо понимать, что несмотря на это есть вполне устоявшийся консенсус относительно настоящей причины гипервалентности – и это довольно простая идея: при значительной разнице в электроотрицательностях ковалентная связь может получить настолько большую примесь ионности, что пять, шесть или даже семь таких связей у элементов конца периода, могут спокойно обслуживаться октетом электронов. И это не обычные связи, а гипервалентные.
Фосфор, как и любой другой р-элемент, где бы он ни находился, может образовать не более четырёх обычных ковалентных связей (обычных – значит двухцентровых двухэлектронных, что со времен Льюиса и Сиджвика является золотым стандартом химической связи, и никакая современная, современнейшая, наисовременнейшая, и т.д. теория структуры с этим не спорит, только предлагает свою интерпретацию этого понятия) и должен соблюдать правило октета Льюиса-Лангмюра. При этом, у фосфора пять валентных электронов, и для него особенно характерны валентность пять и степень окисления +5, потому что, как мы уже выяснили, это вообще очень свойственно именно элементам третьего периода, но фосфор еще и довольно далек от его конца, где все же эффект увеличивающегося заряда ядра начинает наконец хоть как-то удерживать валентные электроны.
О том, как это достигается при явном недостатке возможностей для ковалентного связывания, в химии давно есть консенсус. Этот консенсус можно выразить несколькими разными конкретными способами, но общий знаменатель у всех один – если мы рисуем пятую связь у фосфора, эта связь обслуживается за счет второго атома, не фосфора. Ну и на следующий вопрос – а как же может быть так, что связь есть, а фосфор вроде ни при чём, – ответ простой: не совсем ни при чём, но в химии есть такой тип взаимодействия, который может обходить ковалентные ограничения – этот тип называется ионностью, ионной связью, или ионным компонентом ковалентного связывания.
У гипервалентных состояний фосфора (других элементов тоже, но у фосфора это особенно ярко выражено) есть очень чёткая линия поведения – фосфор готов образовывать гипервалентные связи, и в общем может рассматривать любые серьёзные неметаллы как партнеров в таких связях, но если есть выбор – будет стремиться сделать такую связь с максимально электроотрицательным элементом в ассортименте. Это фтор, конечно, но мы почти не будем иметь дело с фторными соединениями фосфора (подальше от этого, подальше!), но следующий – кислород. Никаких слов не достаточно, чтобы выразить желание фосфора образовать гипервалентную связь с кислородом и ни с кем её не делить. Хотя если есть выбор и имеется фтор, то фосфор предпочтет именно фтор и тут же забудет про такой ещё недавно любезный кислород. Хорошо известно, например, что при действии фторид-ионов в водном растворе фосфаты превращаются в гексафторфосфаты (при избытке фторида) или промежуточные оксифториды, при недостатке фторида. Это производит впечатление, потому что кажется, что ничего на свете не может быть прочнее и устойчивее иона фосфата, но вот нет, конкуренция за атом пятивалентного фтора будет выиграна еще более электроотрицательным элементом в одну калитку.
Посмотрим, как образуется высшая валентность фосфора. Трехвалентный фосфор это тетраэдр, одна из вершин которого – неподеленная пара, или, если смотреть только по атомам, тригональная пирамида. Для дальнейшего расширения валентных возможностей фосфора приходится использовать эту пару. Из пары можно сделать ковалентную связь только одним способом – через донорно-акцепторное взаимодействие: пара от фосфора, свободная орбиталь от акцептора. Результат – тетраэдр, фосфор с формальным положительным зарядом (фосфоний). Фосфор переходит в степень окисления +5 (если придумаете акцептор из элемента менее электроотрицательного, чем фосфор, тогда степень окисления останется +3, но это будет непросто). Примеров полно – протонирование, кватернизация с галоидными алкилами, реакция с бромом или другими переносчиками электрофильных галогенов и т.п.
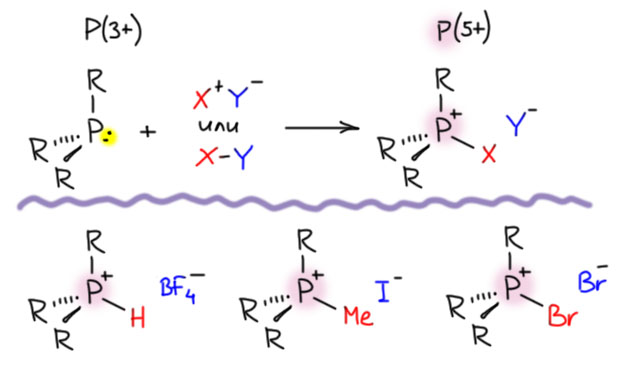
Вторая большая химия случается, когда пару фосфора использует на донорно-акцепторное взаимодействие не одновалентный, а двухвалентный акцептор. Это кто такой? Это тоже атом с вакантной орбиталью, но при этом и с неподеленной парой – сектетный атом кислорода, азота, или углерода в частицах, которые называют оксен, нитрен, карбен. Или, что чаще, при реакции с молекулой, у которой есть и такой атом-акцептор, и еще уходящая группа, и тогда вся реакция становится нуклеофильным замещением на атоме-акцепторе, но мы пока воздержимся от таких обобщений, а то закипят мозги, им и так в последнее время есть от чего кипеть. Тут надо понимать, что речь идет не обязательно о настоящей реакции, хотя точно можно сказать, что и сами такие реакции действительно есть. Чаще речь идет о формальной реакции – просто принципе образования таких молекул, производных пятивалентного фосфора, а которых на фосфоре тоже образуется формальный положительный заряд, а на бывшем двухвалентном акцепторе отрицательный. Таких молекул великое множество, фосфор очень любит этот тип структуры особенно когда акцептор – кислород. И это знаменитые молекулы, точнее, типы молекул.
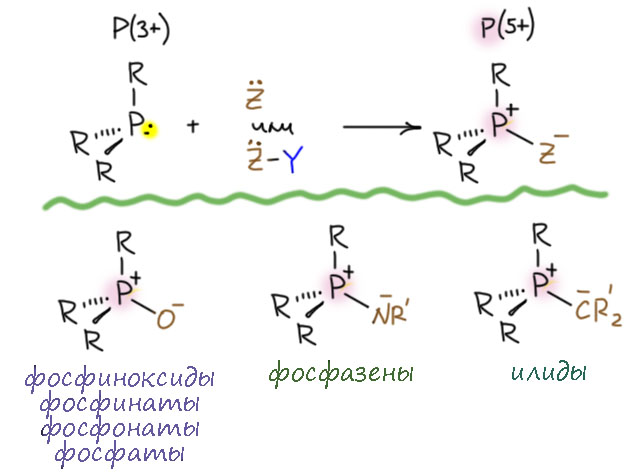
Остановимся на этом типе структуры подробнее, потому что это одна из тех вещей, которая делает химию фосфора очень сложной и нелогичной. И всё потому что сложилась определенная традиция, от которой никто не хочет отходить, хотя более-менее понятно многим, что тут всё очень запущено и это не метафора, а предельно точная констатация факта.
Дело в том, что особенно в случае кислорода такую связь почти всегда рисуют как двойную. В современной литературе вообще произошло нечто странное – одинаковую связь стали рисовать по разному. Фосфиноксиды и илиды стали почти всегда рисовать одинарной связью с зарядами, а все остальные молекулы (фосфаты, фосфонаты, соответствующие кислоты и производные, фосфазены) – с двойной связью. Почему? А чёрт его знает! Так получилось – но тип связи во всех этих случаях один и тот же.
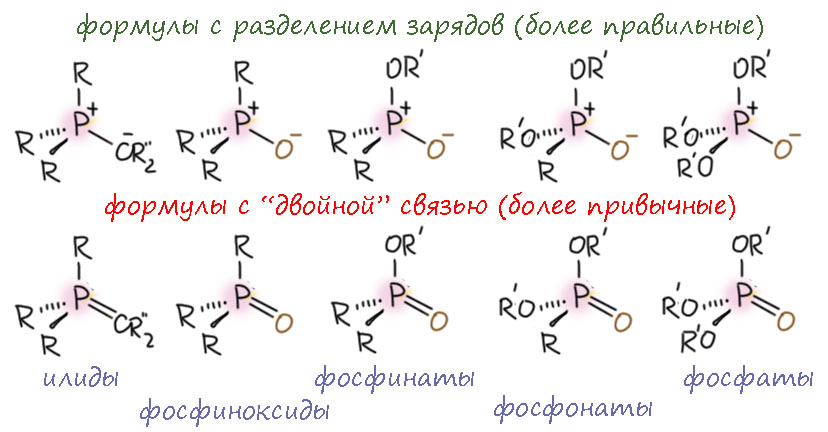
Более того, именно так эту связь рисовать велит ИЮПАК (не только ее, но и все прочие гипервалентные связи, сделанные по принципу простая ковалентная плюс ионная), правда в явном виде оговариваясь, что корень этой рекомендации – эстетика формул и только эстетика, и что авторы рекомендаций вполне в курсе проблем с валентностью и нарушением октета. Вот эстеты чёртовы на нашу голову нашлись! Ведь это не должно быть произвольным выбором – типа, так более красивше, ну и рисуем так. А это правда двойная связь? Это совершенно потрясающая ловушка для всех, кто занимается химией фосфора не постоянно, и иногда, проездом. Если мы рисуем эту связь обычной двойной, то сразу возникает непреодолимое желание обращаться с этим фрагментом, как с карбонильной группой, например, присоединять к нему нуклеофилы, использовать похожие на карбонильную химию механизмы нуклеофильного замещения на фосфоре. И рассматривать группы с такой связью – фосфиноксиды, фосфонаты и т.п. – как обычные мезомерные акцепторы, тем более, что такие группы точно являются акцепторами и довольно серьёзными.
С этой связью много проблем, которые легко сбивают с толку. Во-первых, можно спросить, а каков порядок этой связи? Порядок связи определяется подсчетом количества электронов на орбиталях, обслуживающих связывание в конкретной паре атомов, эту величину легко найти в выдачах квантово-химических расчетов, когда считают всякие обычнные параметры, связанные с распределением электронной плотности. И вполне неудивительно, что этот порядок оказывается равен двум – то есть это вполне официально двойная связь. Двойка эта получилась вполне очевидным способом – связь действительно обслуживается 4-мя электронами, так как ионный компонент этой связи очевидным образом локазизован на занятой орбитали атома кислорода. И это очень короткая связь, она существенно короче обычной связи P-O, когда на кислороде есть еще один атом (у “двойной” связи длина 1.46-1.47 Å, у простой 1.60 Å). Иными словами, есть очень существенные основания для того чтобы рисовать такие связи двумя черточками. Это не преступление – рисовать эту связь двойной. Все, кто так делают, не должны немедленно раскаяться, порвать всё уже написанное и впредь не грешить. Рисуйте на здоровье, это не ошибка. Но я попробую доказать, что это путь к ошибкам и альтернативный способ, действительно немного громоздкий, более адекватен и позволяет многое понять в реакциях соединений фосфора.
Ведь, с другой стороны, мы научены органической химией вкладывать в понятие двойной связи более точное содержание, чем просто формальный счет электронов, участвующих в связывании. Эрих Хюккель (да, тот самый, из ароматичности, но его первой работой в органической химии был анализ структуры двойной связи, и это именно он придумал разделить ее на сигма- и пи-составляющие) научил нас видеть в двойной связи очень характерный элемент структуры, для образования которого требуются орбитали π-типа, то есть орбитали, область связевого перекрывания которых находится не на оси связывания – и даже более того, эта ось должна попасть на узловую поверхность π-орбиталей. Мы отлично знаем как классно это работает в основной органике, где такие связи легко образуют три основных элемента-органогена – углерод, азот, кислород. И с этим связана вся необъятная химия непредельных соединений, карбонильных соединений и множества всего вокруг. И нам кажется, что совершенно естественно, что в этой химии смогут участвовать и другие элементы, особенно ближайшие аналоги органогенов – сера, фосфор. Но это заблуждение, причем чрезвычайно глубоко укоренившееся, но при этом, кажется никогда в явном виде не изложенное. Как можно излагать заблуждение? Элементарно, ведь заблуждающиеся не знают, что они заблуждаются.
Так вот, в соединениях фосфора, где рисуют двойную связь, просто не может быть π-связей. Это просто как божий день: для π-связей нужны π-орбитали, а это орбитали, свободные от σ-связывания. Но в соединениях фосфора, в основном имеющих тетраэдрическую геометрию вокруг атома фосфора, просто нет таких – всё, что есть, уже задействовано в построении четырёх связей тетраэдра. Некоторые скажут – почему обязательно свободные от σ-связывания – а как же гибридные орбитали участвуют в гиперконъюгации, в σ*-π-сопряжении, в σ-σ*-сопряжении? Да, но гиперконъюгацию никогда не считают основным компонентом связей, это просто такой подстроечный эффект, вносящий небольшой вклад в уже имеющиеся связи в молекулах. Такого и у фосфора очень много.
Итак, мы имеем этот характернейший элемент структурной химии фосфора – образование в высшей степени окисления чрезвычайно прочной и короткой связи фосфор-кислород, которая вяглядит как обычная двойная связь, но в отличие от настоящих двойных связей, состоящих из сигма и пи-ковалентных компонентов, связь фосфор кислород состоит из сигма-ковалентного и ионного взаимодействия. Когда говорят про ионное взаимодействие, обычно возражают – ну, это слабо, электростатика это ерунда по сравнению с настоящей ковалентной связью. Но это, во-первых, основано на неверном обобщении наблюдений за ионными соединениями в растворах, где растворитель помогает растаскивать заряды. Без помощи растворителя взаимодейсвтие в ионных парах особенно когда ионы небольшие по размеру, очень даже немаленькое. Но здесь фосфор и кислород уже связаны ковалентной связью, и это сближает атомы кислорода и фосфора на еще более короткое расстояние, чем то, которое достижимо при чисто ионном взаимодействии. А сила притяжения зарядов очень быстро возрастает на коротких расстояниях – такова природа кулоновского взаимодействия, так что есть все основания считать, что в этой связи этот компонент может ничем не уступать чисто ковалентному. Поэтому мы получаем очень прочную и короткую как будто двойную связь, которая на самом деле является гипервалентной (потому что сделана из пары валентных электронов фосфора и вакантной орбитали атома акцептора) связью, состоящей из двух компонент – обычной двухцентровой двухэлектронной простой ковалентной связи, и ионной связи.
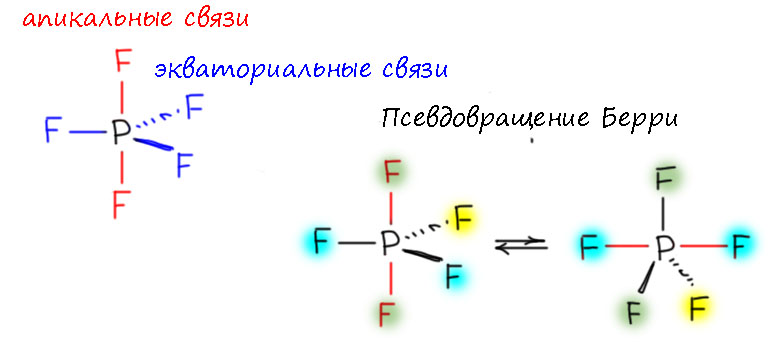
Второй важнейший структурный тип у пятивалентного фосфора – тригональная бипирамида. Соединение-прототип для этого типа – пентафторид фосфора, знаменитая молекула. Геометрически у этой молекулы два типа связей P-F – те, что в плоскости, экваториальные. И те, что сверху и снизу, апикальные (или аксиальные – оба термина используются равноправно). Апикальные связи заметно длиннее экваториальных. И из структуры совершенно явно следует, что в нем два типа фторов. Но в ЯМР фтор ровно один. Это наблюдение в 1960 году натолкнуло Стефена Берри на гипотезу о спонтанном превращении, которое во времени эффективно усредняет атомы фтора в этой молекуле. Это превращение, точнее, внутримолекулярное движение, с тех пор называют псевдовращением Берри, и это второе по важности и популярности конформационное превращение в химии самых разных молекул после заторможеннного вращения. В химии фосфора оно совершенно вездесуще. Оно отвечает за взаимопревращения конформации 5-координированных молекул, а сами эти конформации образуются из-за того, что природа экваториальных и апикальных связей радикально отличается. Это движение за один ход меняет местами две пары атомов – два апикальных и два экваториальных, при этом один атом остается на месте, поэтому само движение можно при достаточном воображении действительно представить, как такое вращение вокруг одной из связей, но с деформацией. Множество таких дижений и усредняют во времени (не в пространстве) все пять связей. Это очень быстрые движения, ведь барьер активации для них приблизительно такой же по величине, как и барьер во внутреннем вращении простых связей, как мы изучаем в конформационных превращениях соединений углерода.
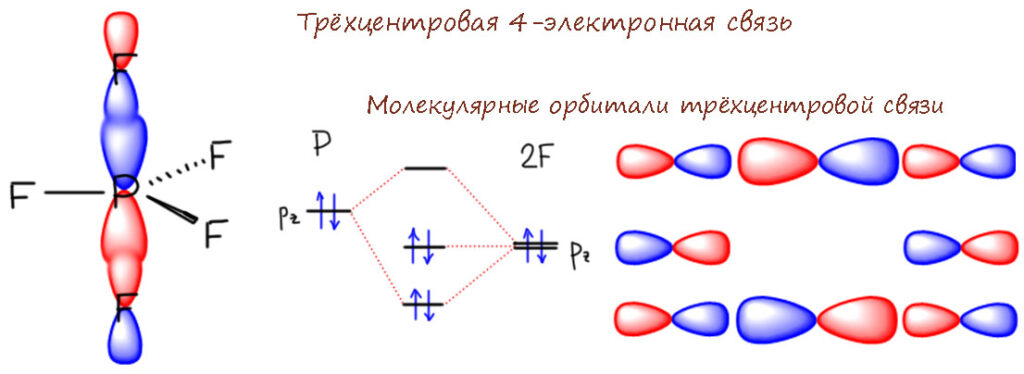
Фосфор, еще раз напоминаю, не может образовать пять нормальных ковалентных связей. Нормальная ковалентная связь по определению двухэлектронная двухцентровая (часто обозначают как 2c-2e), и большинство молекул образовано именно такими связями. На это иногда возражают – но теория МО показывает, что связывающие молекулярные орбитали делокализованы. Вот, например, в метане молекулярные орбитали есть даже и пятицентровые. Это так, но не нужно путать связывающие молекулярные орбитали и сами связи. Теория МО выдает нам целую пачку молекулярных орбиталей на каждую молекулу или молекулярнй фрагмент. Но если мы аккуратно посчитаем, сколько занятых МО обслуживают систему из n связей (например, четыре связи в метане), мы обнаружим в подавляющем большинстве случаев n связывающих МО (например, четыре – одну снизу и три вырожденных повыше – в метане), каждая из которых в молекуле будет иметь два электрона. В теории МО атомные орбитали, взаимодействуя попарно в соответствии с симметрией системы, образуют новую пару молекулярных – одну связывающую, и одну антисвязывающую, из которых почти всегда запонена будет только связывающая. Но сами орбитали, являющиеся результатом решения квантово-химической задачи об электронной структуре электронов в поле нескольких ядер, будут соответствовать симметрии молекулы, и в соответствии с этой симметрией смешивать разные атомные орбитали. Количество занятых МО при этом будет точно таким же, как если бы орбитали были двухцентровыми и связывание атомов было бы попарным.
В тригональной бипирамиде апикальные связи устроены не так. Это не двухцентровые, а трехцентровые связи. Они образованы не двумя, а тремя орбиталями – одной от фосфора, и двумя от фторов, от фосфора мы берем пару электронов и по одному от фторов, итого 4 электрона. Иными словами это как будто не две связи, а одна связь ее так и обозначают – трехцентровая четырехэлектронная, 3c-4e. При этом, поскольку со стороны фосфора участвует только одна атомная орбиталь, мы учитываем в валентной оболочке фосфора только ее формальное содержимое, а атомная орбиталь не может содержать больше пары электронов – поэтому правило октета не нарушается – три пары от экваториальных связей и одна пара от трехцентровой апикальной. Остальные два электрона отогнаны на атомы фтора, что создает ситуацию значительного вклада ионности. В этом смысле два атома фтора выполяют ту же роль, что и один атом кислорода в уже рассмотренной связи, но то, что может сделать один двухвалентный кислород, здесь на пару делают два одновалентных фтора.
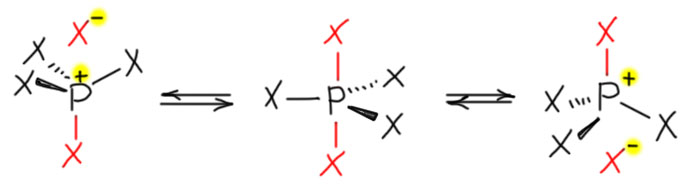
Следующий момент такой. Связь трехцентровая, и тогда вопрос, а может только один из атомов уйти из этой связи, или иными словами, может ли трехцентровая связь порваться как обычная – по паре атомов. Ну конечно, а как по-другому это может быть? Не фосфор же разрывать пополам. Уходит один из атомов (или групп) и это очень важное второе свойство такого типа фосфора – взаимопревращение между пятикоординированной и фосфониевой формой в ионной паре с анионным лигандом. Группы на атоме фосфора могут быть как одинаковые, так и разные, если они разные, то псевдовращение Берри переставляет их, но если они разные по электроотрицательности, в апикальные положения предпочитают уходить атомы с большей электроотрицательностью.
Немного вернемся к “двойной” связи фосфор-кислород. Итак, допустим мы согласились, что у фосфора нет двойных связей. А что нам это даёт? Может это такая схоластика – согласились, выучили, забыли. Мало ли чего там понапридумывали. Но нет, с этим очень прочно связана химия фосфора, в которой очень много необычного, если мы привыкаем переносить представления, полученные в химии элементов 2-го периода. Вот, например, фосфоновые кислоты очень вроде бы похожи на карбоновые – так и кажется, что вся разница – одна лишняя гидроксильная группа. У фосфоновых кислот есть сложные эфира точно так же как у карбоновых. Эфиры карбоновых кислот исключительно легко гидролизуются в условиях основного катализа, потому что идет легкая реакция замещения на карбонильной группе именно за счет присоединения нуклеофилов к двойной связи C=O с образованием тетраэдрического интермедиата и последующим расщеплением с восстановлением карбонильной группы. Реакции гидролиза (щелочь в воде, а еще лучше в спиртах) расщепляет сложные эфиры карбоновых кислот практически моментально. А что эфиры фосфоновых кислот – похожи на эфиры карбоновых? Так же легко гидролизуются? Если мы остаемся во власти представлений о тенденциях в химии элементов 2-го периода, мы можем вспомнить о большей силе фосфорных кисло по сравнению с карбоновыми и предположим, что щелочной гидролиз должен быть или таким же легким, или еще более легким. На деле всё не так просто: гидролиз идёт, но точно медленнее, и щелочной гидролиз идет довольно медленно и почти всегда останавливается на омылении только одной группы OR. Поэтому в синтезе гидролиз эфиров фосфоновых кислот почти всегда делают в условиях кислотного катализа – либо используют галогеноводороды при повышенной температуре, либо, что является стандартным приёмом – используют кремниевый (силильный) катализ.
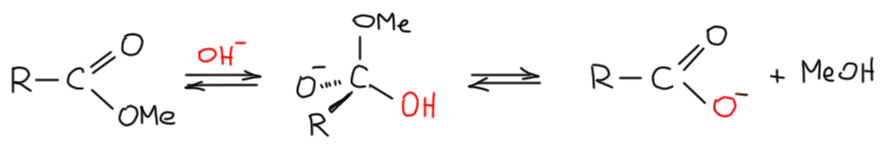
Видимо, там всё же что-то не так с механизмом. Как же идет замещение на атоме фосфора максимальной валентности – хоть здесь есть аналогии с химией углерода? Есть, и очень близкие. Но сразу приходится сказать, что в отличие от химии углерода, где стараниями Ингольда и Хьюза и их последователей выстроена хорошо знакомая нам и весьма стройная теория механизмов, в химии фосфора ничего подобного нет. Механизмы есть, но чтобы найти и изучить их потребуется некоторая доля любопытства. Плата за это велика: я наверняка всех достал рассказами о том, что если бы в органике не было теории механизмов, мы до сих пор перебирали бы сотни и тысячи реакций вместе с условиями, выходами и прочим, не зная как упорядочить материал. Вот если вы когда-нибудь видели обзоры и книжки по органической химии фосфора, они ровно так и выглядят – это описание десятков и сотен очень похожих реакций, со всеми условиями, температурами, выходами. Очень тоскливое зрелище. А почему они не используют механизмы для систематизации знаний? А потому, что с механизмами там, прямо скажем, некоторая фигня приключилась. Это вообще тема для отдельного разговора, а здесь только намекну – исследователи в начале решили не париться, и прямо целиком перенести всю систему механизмов из химии карбоновых кислот и их производных на фосфорные и фосфоновые кислоты и их производные, только заменив слово “ацил” на слово “фосфорил”, и вот так все это и пытались сделать, но как-то через некоторое время поняли, что аналогия сильно хромает. Напомню, что в карбоновых кислотах (возьмём, например, катализируемый основаниями гидролиз сложных эфиров, и давайте не заморачиваться алкилом в эфире) реакция идет через тетраэдрический интермедиат:
 А теперь посмотрим на аналогичную реакцию фосфоната (могли бы взять и фосфат и фосфинат). И напишем две версии. Одну для полноты аналогии с двойной связью. Присоединяем гидроксид, вроде видим пока всё отлично, только интермедиат не тетраэдрический, а тригонально-бипирамидальный. Ну, ничего страшного, группа пятая, но вяглядит как аналог. Но! Мы уже знаем, что это гипервалентное производное, фосфоран, и апикальная связь у него трёхцентровая. И продолжения аналогичного нет, потому что нет никакого смысла смещать электронную плотность к фосфору, ей гораздо лучше на кислороде. Аналогия разрушается.
А теперь посмотрим на аналогичную реакцию фосфоната (могли бы взять и фосфат и фосфинат). И напишем две версии. Одну для полноты аналогии с двойной связью. Присоединяем гидроксид, вроде видим пока всё отлично, только интермедиат не тетраэдрический, а тригонально-бипирамидальный. Ну, ничего страшного, группа пятая, но вяглядит как аналог. Но! Мы уже знаем, что это гипервалентное производное, фосфоран, и апикальная связь у него трёхцентровая. И продолжения аналогичного нет, потому что нет никакого смысла смещать электронную плотность к фосфору, ей гораздо лучше на кислороде. Аналогия разрушается.
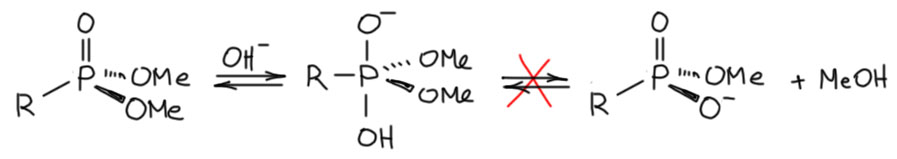
Но поскольку мы уже знаем химию фосфоранов, мы видим другое продолжение. Знаем мы то, что уходящая группа должна попасть в апикальное положение, тогда она без проблем обратимо отвалит. Дорисуем продолжение. Псевдовращение переставляет группы на фосфоре, и один из вариантов ставит уходящую группу в апикальное положение. Мы видим фосфоран и уже знаем, что происходит с такими молекулами – обратимая диссоциация одной из апикальных групп с образованием фосфониевого центра. Проделав эту нехитрую операцию, и перебросив очевидно более кислый протон на более сильное основание, мы видим – что? – дианион? – нет, моноанион кислого эфира фосфоновой кислоты, и он сразу получился в форме с разделением зарядов, и это правильно, потому что так устроено обратимое образование и распад пятикоординированного интермедиата, фосфорана. При желании мы можем перерисовать то же самое с привычной “двойной” связью, так почти все и делают, и вы делайте, если хотите, но, надеюсь, я хотя бы кого-то убедил, что это лишняя суета (новый мем в стиле реплики на расстрел герцога Энгиенского – это меньше чем преступление, это суета).
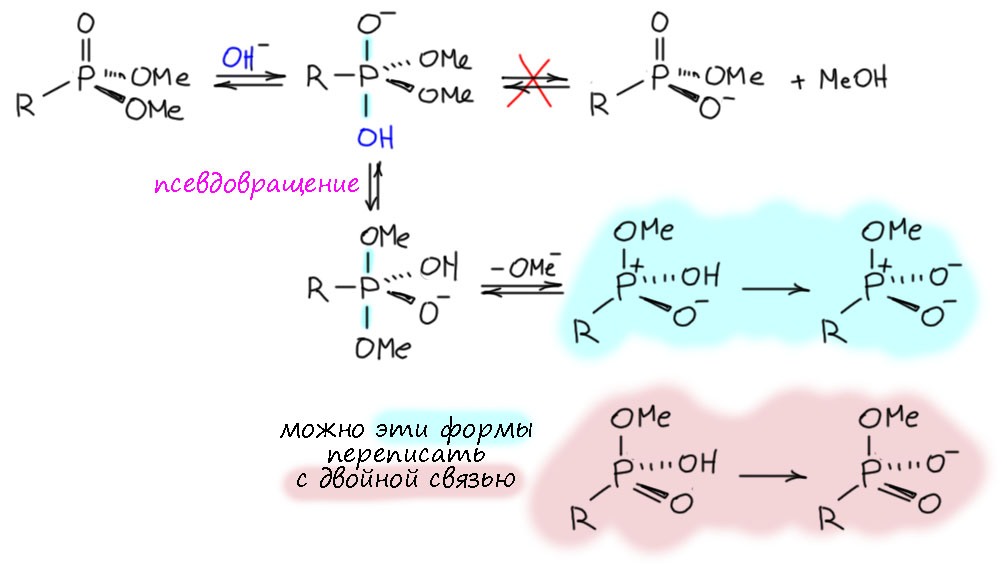
Ведь давайте быстренько перерисуем схему, и исходное изобразив без “двойной связи”. Что изменилось? Схема стала логичнее: мы видим очень характерную для фосфора(V) апикальную атаку нуклеофила на фосфониевый центр с образованием фосфорана. Дальше мы всё уже сделали, но схема становитсяя полностью симметричной, и это хорошо, потмоу что все реакции такого типа принципиально обратимы и должны иметь одинаковый механизм для прямой и обратной реакции. Заодно мы видим, что в результате получается в самом конце анион (два минуса один плюс), а это в значительной степени блокирует обратную реакцию, так как анионные нуклеофилы очень не любят наезжать на анионные электрофилы – отталкиваются они, никак не могут сблизиться на расстояние взаимодействия.
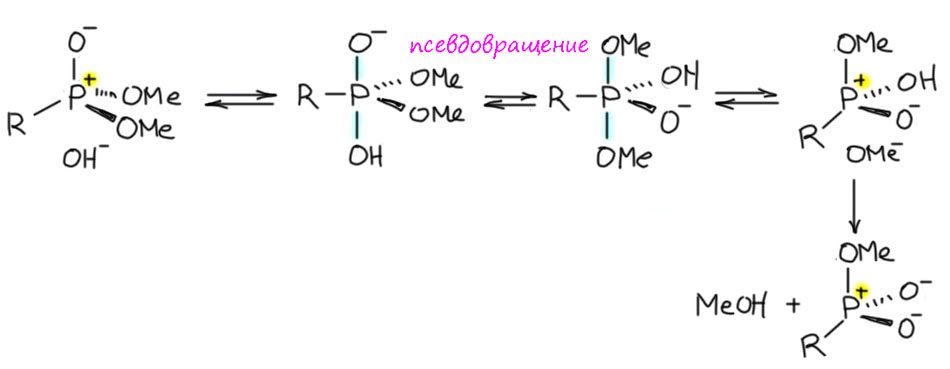
Ещё одно достоинство представления “двойной” связи в виде гипервалентной с разделением зарядов состоит в том, что мы получаем для фосфора единый механизм для замещения на P(V). Такое замещение иногда бывает в фосфониевых производных, имеющих уходящие группы – галогены, алкокси и т.п. и мы используем такие реакции, например, для превращения спиртов в галогенпроизводные с помощью фосфорных реагентов типа трифенилфосфин-бром, можете посмотреть, где-то то рядом это обсуждается. Замещение идет как апикальная атака на тетраэдрический фосфоний, только в этих случаях нет “двойной” связи фосфор-кислород, а все связи чисто одинарные ковалентные. Образуется фосфоран, и дальше, если есть во втором апикальном положении есть уходящая группа, она уходит. Если уходящая группа не в апикальном положении, потребуется небольшая затрата энергии на псевдовращение.
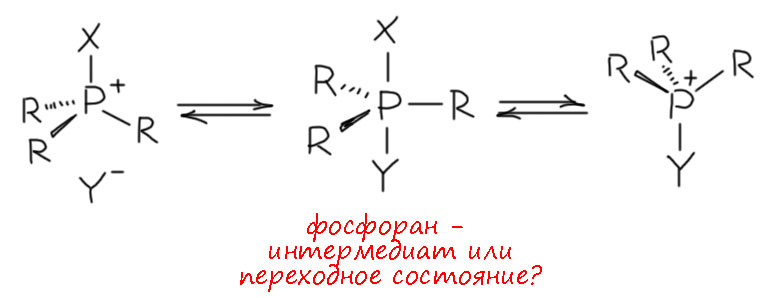
Такой механизм можно назвать SN2 на фосфоре. И уже очень давно поставлен вопрос – это согласованный механизм как в химии углерода, или это стадийный механизм, потому что в отличие от химии углерода, где нет и не может быть пятикоординированных соединений углерода, в химии фосфора такие соединения есть – фосфораны. Но при этом, несмотря на очень большой интерес и весьма длительную историю исследований, до конца не ясно, насколько устойчивы такие интермедиаты – данные противоречивы. Впечатление такое – или совсем неустойчивы, или очень неустойчивы; исключений мало, как-то поймать что-то похожее удается редко, почти никогда, и только для каких-то моделей, где удается зафиксировать фосфоран, например, с помощью хелатных циклов. Тем не менее, время жизни такой штуки может быть достаточно хотя бы для одного псевдовращения, если нужно переставить уходящую группу в апикальное положение. Если уходящей группы не находится, интермедиат просто диссоциирует обратно.
Напоследок еще раз вернёмся к замещению в фосфатах или фосфонатах, и доведем параллель до конца, чтобы показать, что использование не “двойных” связей, а связи с разделением зарядов, позволяет и замещение в таких соединениях рассматривать как SN2 на фосфоре. Сравним. Поскольку всё уже сказано, можно без дополнительных комментариев.
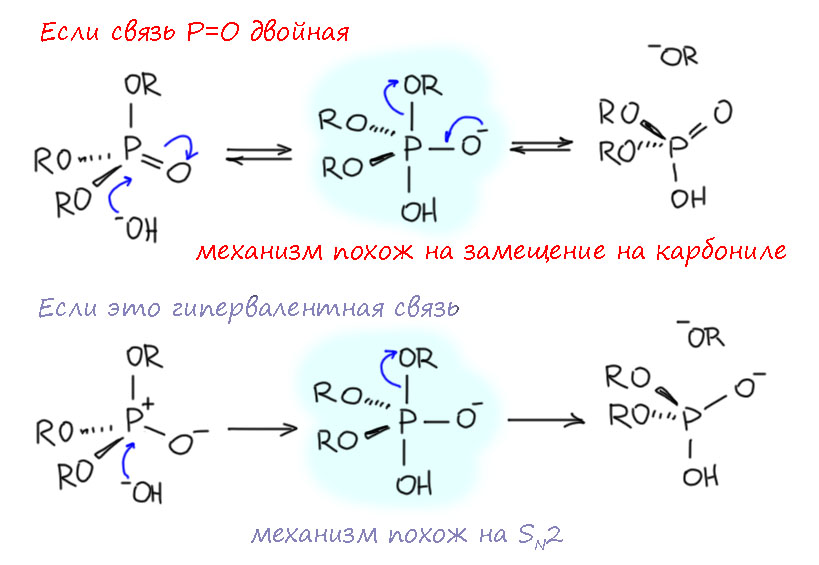
Для таких фосфоранов получается в разных подходах один и тот же вывод: в собственно процессе замещения участвует как раз апикальная трехцентровая связь. Её особая природа дает ей высокую лабильность, а псевдовращение Берри поможет переместить в апикальное положение уходящую группу. Очень важно понимать и то, что эта особая, гипервалентная природа обусловливает важность того, чтобы в такой связи участвовали более электроотрицательные атомы, те же кислороды.
А теперь подробнее
Теперь применим знания в химии фосфора к пониманию того, почему реакции соединений фосфора происходят неким особенным образом.
Кислородные кислоты фосфора(V)
Возьмём трехвалентный фосфор и кислоту трехвалентного фосфора, фосфористую. Если это производное именно трехвалентного фосфора, это просто тригидроксифосфин. Очевидно, что это основание и нуклеофил, как и все фосфины. Но это и протонная кислота. А не может ли она протонировать сама себя? Запишем.
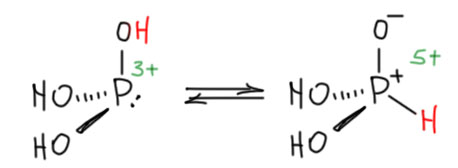
Ой, это что же произошло! Я же просто перетащил протон от атома кислорода к атому фосфора. Но фосфор стал фосфониевым, а на кислороде возник отрицательный заряд – а это разве не та самая гипервалентная связь, которую принято рисовать двойной, но мы решили придерживаться форме с разделением зарядов. Да, конечно, она самая – ведь в ней нет никакой мистики, это и правда ровно оно – ковалентная простая связь плюс ионная. Если хотите, можно ее нарисовать двойной, и тогда мы увидим стандартную формулу для хорошо известного таутомера фосфористой кислоты. И лишний раз убедимся – в этой форме фосфор пятивалентный и имеет степень окисления +5.
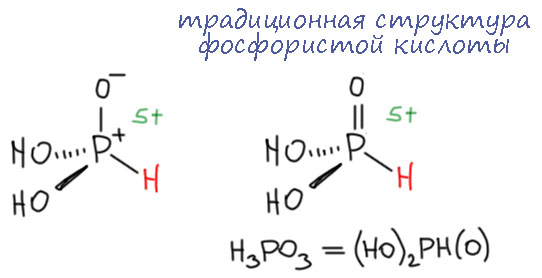
Иными словами, решив записать совершенно невинную вещь – просто протон перместить от одного атома к другому мы нечаянно попали в старую проблему химии фосфора – в каком виде существует фосфористая кислота. Эту проблему обнаружили еще в 19 веке, и как раз А.Е.Арбузов и обнаружил свою реакцию, когда захотел наконец разобраться в этом. Ведь кислота такая есть, она хорошо известна, устойчива, вполне индивидуальна. И получается совершенно очевидным образом, например, просто гидролизом треххлористого фосфора (трихлорфосфина). Здравый смысл и химия остальных элементов говорит, что это должна быть трехосновная кислота, тригидроксифосфин. Но все измерения свойств говорили, что это не то. Кислота, например, очевидно двухосновная, а не трехосновная, и соли у нее с двумя эквивалентами одновалентных металлов, а не с тремя. И тогда мы вспоминаем еще одну вещь – хорошо нам известную кето-енольную таутомерию. Есть две молекулы, одна из них CH-кислота, другая OH-кислота. И тогда мы говорим, что в смеси таутомеров преобладает тот, у которого меньше кислотность. Это так очевидно, что кажется издевательским трюизмом – протон предпочитает сидеть на кислотном центре с меньшей кислотностью (или, что то же самое – переезжать на основный центр с большей основностью). Так устроена вся химия протонных кислот – более сильная кислота протонирует более сильное основание.
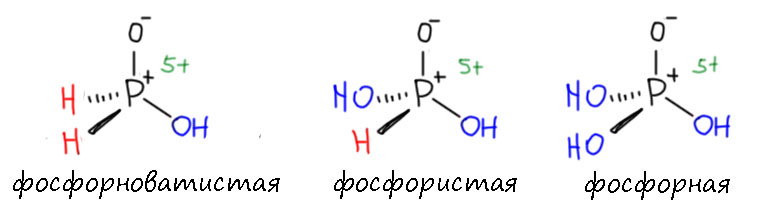
Ну а здесь? В исходной форме это OH-кислота. Мы не знаем ее кислотности, но просто по аналогиям понимаем, что это кислота слабая, но не слишком – сделаем очень широкую оценку: pK не меньше единицы (это приблизительно кислотность большинства кислородных кислот фосфора по первой ступени) и вряд ли больше 10 (у фосфорных кислот по вторым-третьим ступениям кислотность не бывает сильно ниже). А протон на фосфоре – это PH-кислота. Знаем мы что-нибудь про кислотность фосфинов? Немного. Но мы знаем, что связь P-H практически неполярна, а если судить по электроотрицательностям, даже слегка поляризована в сторону водорода, водород там скорее гидридный, и это имеет смысл – фосфины в химии являются гидридными восстановителями. Если порыться по химии фосфинов и о том, как их можно депротонировать, увидим, что кислотность очень мала. Она побольше, чем у аминов – у аммиака и простых аминов pK около 40 и даже еще побольше. У фосфинов это, видимо, где-то в районе 30, может чуть меньше. Такая грубая, но разумная оценка говорит нам, что протон от гидроксильной группы должен ласточкой перелететь на фосфор, и там и остаться, а в в равновесии двух форм фосфористой кислоты, трехвалентной фосфиновой формы должно быть очень мало. Все опубликованные данные говорят, что это так и есть: фосфористая кислота это производное пятивалентного фосфора с одним водородом фосфинового типа, и это кислота, очень похожая на фосфорную, по первой ступени кислотности этих двух кислот похожи с очень большой точностью – ну так у них и структуры почти одинаковые. Более того, мы отлично знаем, что у фосфора есть третья кислота, где уже два водорода висят на фосфоре – мы с удовольствием используем эту кислоту как восстановитель для солей диазония. Вот три этих кислоты рядом, у всех трех фосфор имеет валентность пять и степень окисления +5. Все эти три кислоты очень похожи друг на друга, например, это довольно сильные слабые кислоты, с практически одинаковым pK по первой ступени в районе 1.5-1.7, но первая кислота однооосновная, вторая двухосновная, третья – трёхосновная, и каждая следующая ступень значительно слабее первой., что неудивительно, так как протону приходится отщепляться от частицы со значительным суммарным отрицательным зарядом. Названия этих кислот остались с тех пор, когда в их структуре не разобрались, и думали, что это кислоты меньших степеней окисления, +1 и +3. Когда разобрались, их стали рассматривать как родоначальников рядов кислот при замещении водорода на органический остаток, и первые кислоты стали называть фосфиновыми, а вторые фосфоновыми.
Два ряда эфиров фосфористой кислоты
Вернёмся к фосфористой кислоте. Фосфористая кислота существует как фосфоновая, но вот эфиры есть отдельно у каждой, и это тоже очень старая загадка. Эфиры фосфористой кислоты стали пробовать получать еще в середине 19 века реакцией треххлористого фосфора с спиртами, и тогда получили нечто, что долго считали триалкилфосфитом, но потом стали все делать лучше и убедились, что иногда треххлористый фосфор реагирует с тремя эквивалентами спирта, а иногда только с двумя. Чистые триалкоксифосфины, они же триалкилфосфиты образуются только если спирт реагирует в присутствии основания (пиридина, диметиланилина, диэтиланилина), причем надо строжайше следить, чтобы в процессе прибавления было очень хорошее перемешивание и никогда не образовывались в реакционной смеси непромешиваемые места, где выделяющийся хлористый водород не успевает связаться с основанием. Тогда получается чистый триалкилфосфит, совершенно устойчивые обычно жидкие вещества с неприятным запахом – запах, очевидно, выдает то, что фосфор в них трехвалентный и это фосфины, летучие фосфины – штука вонючая.
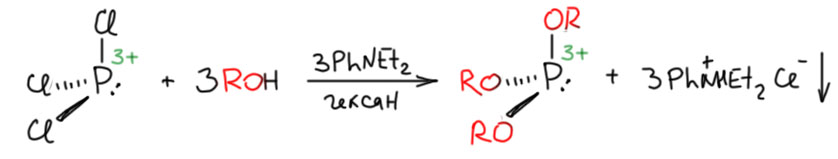
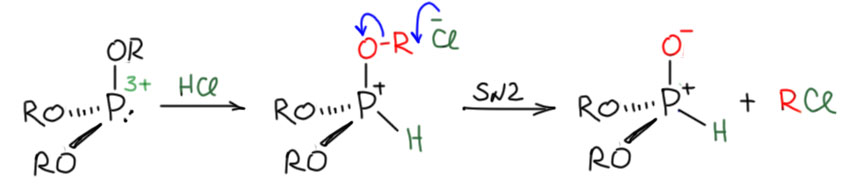
Ну и почему надо так бояться свободной сильной кислоты, я думаю, уже ясно. Вместо триалкилфосфита, получите диалкилфосфит, он же диалкилфосфонат, а механизм мы уже знаем, ну, почти. Понятно, что происходит протонирование, и пятивалентный фосфор в фосфониевой соли ищет, как бы ему обзавестись его любимой гипервалентной связью с кислородом. Сделать это уже не так просто, как в самой исходной кислоте, и приходится призывать на помощь SN2-замещение, чтобы выделяющийся хлорид снёс один из алкилов. Он это делает с удовольствием, и это значит, что образование гипервалентной связи очень выгодно, что и делает такой фрагмент превосходной уходящей группой. Здесь мы очень ясно видим эту общую тенденцию в химии фосфора – если на 5-валентном фосфоре висит одновалентный кислородный лиганд (типа OR), очень выгодно превратить его в двухвалентный, создав на кислороде минус, и второй, ионный компонент в связь фосфор-кислород. Этот выигрыш в энергии и создает хорошую уходящую группу.
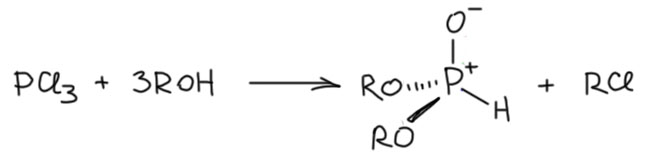
И чтобы прямо получить диалкилфосфит (диалкилфосфонат) из треххлористого фосфора, просто делают реакцию со спиртами, но без добавления оснований. Обратите внимание, что спирта все равно берут три эквивалента.
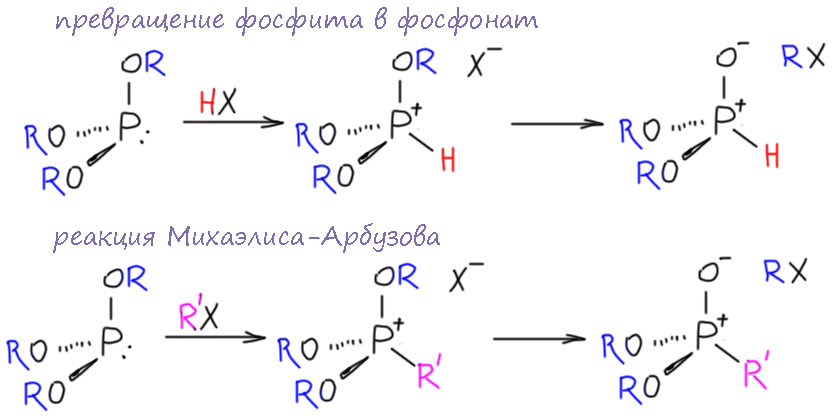
Так можно получить оба типа эфиров. Еще раз напомню, что они долго считались эфирами фосфористой кислоты, и поэтому так и называются до сих пор, название диалкилфосфит вместо диалкилфосфонат встречается очень часто, ведь его просто считали кислым эфиром фосфористой кислоты. Но мы теперь отлично понимаем, что никакого кислого эфира фосфористой кислоты нет, он спонтанно перегруппировывается в диалкилфосфонат. И пока во всем этом разбирались, была проблема получить чистые тралкилфосфиты. Вот и у Михаэлиса в той исторической работе триэтилфосфит, видимо, был грязным, откуда и двусмысленные результаты. А вот у Арбузова все было чисто, эфиры фосфористой кислоты хорошо очищены, и ему поэтому удалось однозначно увидеть реакцию с алкилиодидом.
Реакция Михаэлиса-Арбузова
И мы уже понимаем, что реакция Михаэлиса-Арбузова очень похожа на превращение триалкилфосфита в диалкилфосфонат, только вместо протонирования фосфора, здесь происходит кватернизация, но природа процесса та же самая – пятивалетный фосфор хочет получить полноценную гипервалентную связь с кислородом, потому что это очень выгодно.
Механизм реакции Михаэлиса-Арбузова конечно же установили позднее, когда возникла теория механизмов. Общепризнанный механизм – это две последовательные стадии SN2-замещения. С этим довольно долго копались, и не сам Арбузов и его школа – увы, в СССР органика очень долго развивалась как такое продолжение чисто экспериментальной химии 19 века – делаем реакции, выделяем продукты, делаем элементный анализ, алавим, перегоняем, все это описываем. Никаких попыток как-то это осмыслить не было и потому что советским химикам так было удобнее, они сложились в классической школе и не собирались изменяться. Но даже если бы они захотели, это могло плохо кончиться – совершенно невежественные и необразованные советские вожди не допускали даже мысли, что может быть какая-то другая теория кроме тупой демагогии марксизма-ленинизма. И все достижения электронной теории, квантовой физики и химии, все новые воззрения на эффекты заместителей, и все остальное фактически было под запретом. А чтобы никто и не смел никуда соваться, над научным сообществом были поставлены в основном престарелые академики, которые некогда, обычно до революции были учеными, даже очень крупными и знаменитыми, но с тех пор превратились в свадебных генералов, надсмотрщиков над научным сообществом и истуканов для навешивания орденов, званий, премий, должностей, привилегий и прочего. И если мы последим по публикациям работы таких академиков, то удивимся невероятному постоянству – десятилетиями не меняется тематика исследований, как будто химия остановилась, все в ней сделано, и надо просто терпеливо перебирать все возможные комбинации заместителей. В результате в стране образовалось множество профессиональных химиков, которые умели делать реакции и получать новые соединения в ошеломляющих количествах. Но уровень осмысления остался в 19 веке. Здесь надо еще заметить, что исследования фосфорорганики шли очень активно во всех химических державах довоенного и послевоенного мира – потенциал фосфорорганики в поиске физиологически активных веществ был осознан очень быстро и фактически началась общемировая гонка – кто получит больше разных типов таких соединений. Большая химия пестицидов начинается именно с фосфорорганики, а это время химизации сельского хозяйства, когда основную надежду на увеличение урожаев связывали с тотальной борьбой с вредителями, и все агрохимические исследовательские центры фактически занимались поисками оружия массового поражения вредителей. А за кулисами шли очень похожие исследования, где под вредителями понимались совсем другие организмы – все это шло параллельно как минимум потому что основной механизм действия фософорорганики на все разнообразие животных от насекомых до млекопитающих один и тот же – это ингибиторы холинэстераз. Поскольку в те времена поиск шел чисто перебором огромных количеств однотипных соединений, вот их и получали в невероятных количествах, сбрасывая в открытую печать всё, что не показало никаких привлекательных характеристик, а таких соединений всегда в любом поиске 99.99% минимум – но из-за этого лихорадочного случайного поиска в химии фосфорорганики случилось колоссальное перепроизводство экспериментальных результатов. Может сложиться впечатление, что в этой химии перед химиками была задача – поставить все возможные реакции и выяснить как реагирует каждый фосфорный реагент с каждым моно- или полифункциональным органическим соединением, и все это опубликовать. Ни в одной другой значительной области органики и элементооорганики так не работают – делают только обоснованные эксперименты, активно используют разные способы осмысления и предсказания, ставят только те реакции и синтезы, которые действительно могут привести к чему-то интересному. И вот это лихорадочное накопление результатов в фосфорорганике делает эту область такой огромной и плохо разобранной помойкой из новых соединений, похожих друг на друга структур и реакций, колоссальных таблиц, перебирающих все возможные заместители. И при этом – крайне слабый уровень осмысления тенденций в изменении свойств и реакционной способности.
А исследования механизма реакции Михаэлиса-Арбузова и других реакций в химии фосфора переехали в основном в США. Там в это время (в середине 20 века) работал очень занятный персонаж Дженнэйди Майкл Косолапофф (возможно, Геннадий Михайлович, но следов этого человека в СССР я не знаю, так что возможно, он перебрался в Штаты очень давно, и уже стал просто американским химиком; сын его точно с таким же именем – американский подполковник, ветеран вьетнамской войны). Так вот этот Косолапофф для химии фосфорорганики стал просто легендарным основоположником; хотя он сам сделал не очень много собственных работ, но написал совершенно удивительный по объёму и подробности труд Фосфорорганические соединения (вышел в 1951 году на Wiley), где собрал просто все, что было известно к этому времени, в том числе совершенно все советские работы, выходившие в малодоступных на западе советских жарналах по-русски, и таких работ оказались сотни – казанская школа фосфорорганики была невероятно плодовита и фактически на первом этапе развития этой науки забила всех остальных с разгромным счетом. И все фосфорорганики, как только возникали вопросы по химии, всегда сразу же спрашивали – а что Косолапофф, а как у Косолапоффа. В общем, Косолапофф это была такая библия фосфорорганики, Ветхий завет. Потом было уже очень много новых книг по фосфорорганике.
В общем, начиная с 1940-х в этой реакции стали разбираться с точки зрения механизма и реакционной способности. И увидели, что она довольно типична для SN2-замещения – например, в реакции участвуют в основном первичные алкилгалогениды и все те субстраты, которые мы знаем как лучшие для механизма SN2. Вторичные алкилгалогениды реагировали намного медленнее первичных, и только самые маленькие типа изопропил или циклогексилиодида, а обычно реакция с вторичными галогенпроизводными дает элиминирование – хотя фосфиты это весьма слабые основания, но реакцию Арбузова традиционно делали в довольно жестких условиях при длительном нагревании часто выше 150ºС. Про третичные галогенпроизводные и говорить нечего. Из галогенпроизводных иодпроизводные реагируют быстрее бромпроизводных, и тем более хлорпроизводных. И в реакции происходит обращение конфигурации.
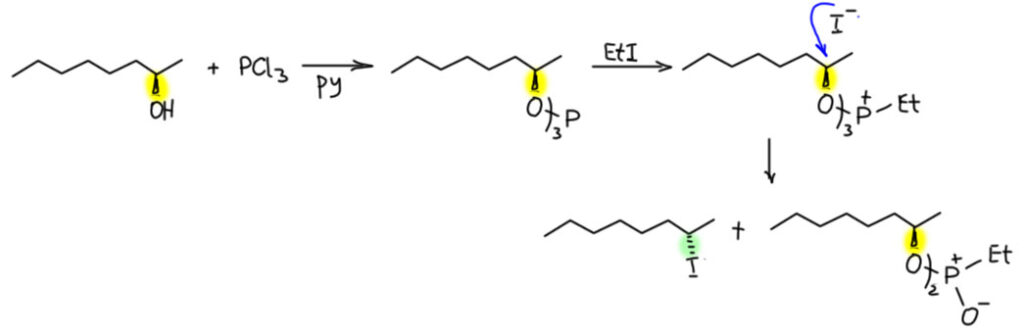
Из механизма стало понятно, как работает каталитический вариант реакции, выглядящий как настоящая перегруппировка. Исходный алкилгалогенид действует как инициатор, а дальше реакцию ведет уже образующийся алкилгалогенид, то есть фактически катализатором является именно галогенид при очевидной необходимости в самом начале иметь откуда угодно хоть совсем немного алкилгалогенида для алкилирования фосфора.
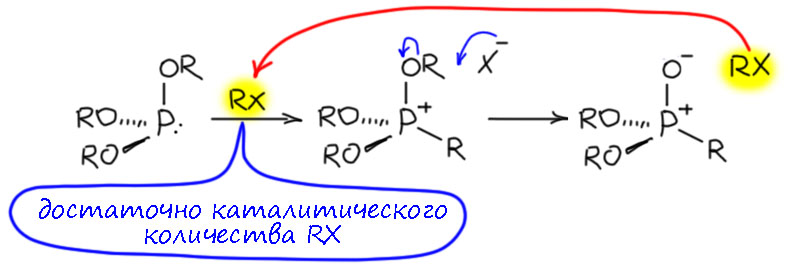
Реакция Михаэлиса-Арбузова не отличается высокой селективностью. Проблема в том, что выделяющийся алкилгалогенид точно так же может участвовать в реакции, образуя побочный продукт. Этого не происходит, если реакционная способность нужного галогенпроизводного намного выше, например, если мы используем бензилгалогенид, а уходит этил – бензил намного активнее этила. Но если у нас и там, и там первичные, то образования смеси не избежать, и приходится использовать либо избыток, либо, как рекомендовал Косолапофф просто отгонять этилгалогенид из реакционной смеси. По той же причине почти никогда не берут триметилфосфит – метилирование, как мы знаем, в SN2-замещении идёт очень быстро.
Можно ли использовать тозилаты вместо галогенпроизводных? Если не забывать, что галогенид из галогенпроизводного участвует в реакции как нуклеофил на стадии второго SN2-замещения, то тозилат, очень слабый нуклеофил, должен быть бесполезен. Тем не менее есть минимум одна старая статья (Myers, T. C., Preis, S., Jensen, E. V. J. Am. Chem. Soc. 1954, 76, 4172), в которой утверждают, что толилаты работают и дают ровно то же самое. Но эксперимент там реально описан только один и он по-настоящему драконовский – смесь триэтилфосфита и этилтозилата греют 2 часа при 150ºС и потом еще всю ночь, то есть часов десять при 120ºС – выход диэтилового эфира этилфосфоновой кислоты получается почти количественный и трудно этому не поверить, но не оставляет впечатление, что там что-то все же напортачили, например, откуда-то взялась небольшая примесь галогенида, может из фосфита (недочистили от соли в синтезе) или из тозилата (его получают из тозилхлорида, и каждый, кто работал с этим соединением знает, как трудно бывает очитстить продукт реакции от примеси хлористого тозила. Маленькая примесь галогенида вполне могла дать реакцию Михаэлиса-Арбузова, а поскольку там один алкил, то достаточно каталитического количества. Во всяком случае больше я нигде не видел, чтобы тозилаты реально применяли в этой реакции. Всякое бывает в химии, ошибки не исключены, в старых журналах еще не было хорошего рецензирования, и написанное было некому поправить. Есть правда и еще одна статья (J. Org. Chem. 1967, 32, 2172), в которой тоже показано, что когда алкил в тозилате и фосфите одинаковый (метил или этил) то реакция действительно идет и довольно бурно, но если они разные, то в результате получаются малопонятные смеси. Из этого можно сделать вывод, что алкилирование фосфита конечно происходит, но дальше этот тозилат фосфония ведет себя очень странно, и не исключены или какие-то другие механизмы, или действительно работали примеси.
В этом месте так и хочется задать вопрос – а почему не использовать все эти замечательные способы ускорения SN2-замещения, которые мы знаем – все эти замечательные полярные растворители, межфазный катализ и всё такое. Ну, просто вспомним – все это хорошо работает для анионных нуклеофилов, и то в основном для сильно сольватируемых. И совершенно бесполезны для нейтральных нуклеофилов. А фосфит – это как раз и есть нейтральный нуклеофил, и поэтому ничего серьёзного с ним сделать нельзя, и все что остается, это побольше концентрацию (поэтому реакцию любят делать вообще без растворителя) да повыше температуру, и здесь возникают всякие неприятные вещи типа того, а что делать, если алкилгалогенид летуч. Тогда реакцию делают в закрытом сосуде под давлением (в запаянной ампуле, в специальных толстостенных пробирках, в автоклавах) – но тогда невозможность отгонять образующееся галогенпроизводное и избежать самоалкилирования. В общем, эту реакцию как делали при А.Е.Арбузове в начале 20 века, так почти без изменений делают и сейчас, и либо получают что-то полезное и умудряются это выделить из реакционной смеси, либо не получают. Если получают, всё решает крайняя простота этой реакции, именно это делает ее невероятно привлекательной в синтезе самых разнообразных производных фосфоновых и фосфиновых кислот, а также фосфиноксидов. Смешал исходный алкоксифосфин с электрофилом, и дальше просто погрел как следует образовавшуюся фосфониевую соль – и либо все накрылось, либо получилось новое соединение. Поэтому с реакции Михаэлиса-Арбузова часто начинают проработку синтетических подходов к очередной разновидности фосфорорганики. Это такой общий способ синтеза соединений P(5+) из соединений P(3+) с введением нового органического заместителя на фосфор. Метод не очень надежный, очень ограниченный, мало пригодный для дополнительной оптимизации, но очень простой и максимально прямой. В этом состоит и историческое и актуальное значение этой реакции.
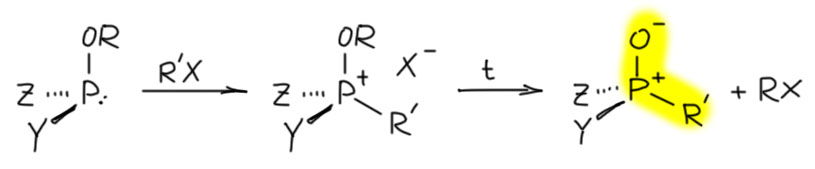
Отчасти поэтому в современном синтезе при необходимости вводить в молекулы посложнее фосфонатные группы вместо реакции Арбузова предпочитают просто алкилировать соли диалкилфосфитов – это часто называют реакцией Михаэлиса-Беккера по небольшой работе, опубликованной в 1897 году (Michaelis A, Becker T.H. Chem. Ber. 1897, 30, 1003). Этот Михаэлис, возможно, должен быть признан рекордсменом по признанию заслуг перед человечеством – попробуйте найти второго ученого, который остался в истории двумя именными реакциями, опубликовав ровно три немудрёных опыта. Впрочем, как уже было замечено Михаэлиса почитают именно как пионера фосфорорганической химии, такого легендарного Отца-основателя, который сам бы был немало удивлен, если бы к нему в иной мир явилась делегация благодарного потомства чествовать за заслуги, и стал лопотать бы что-то типа того, что мы там со студентами ставили какие-то опыты, учились статьи писать в Берихьте, ну что-то вроде получилось, спасибо, спасибо, я очень тронут, цветы отдайте ангелам(бесам), а вот бутылочки грушевого шнапса не захватили случайно…
У диалкилфосфитов, которые, как мы знаем, то же самое, что фосфонаты есть этот атом водорода на фосфоре – кислотность у него очень мала, но она есть, и этот протон можно отодрать сильными основаниями типа трет-бутилата калия или гидрида натрия. Обратим внимание на то, что такой анион чем-то неуловимо похож на енолят: там тоже у двух таутомеров одно сопряженное основание.
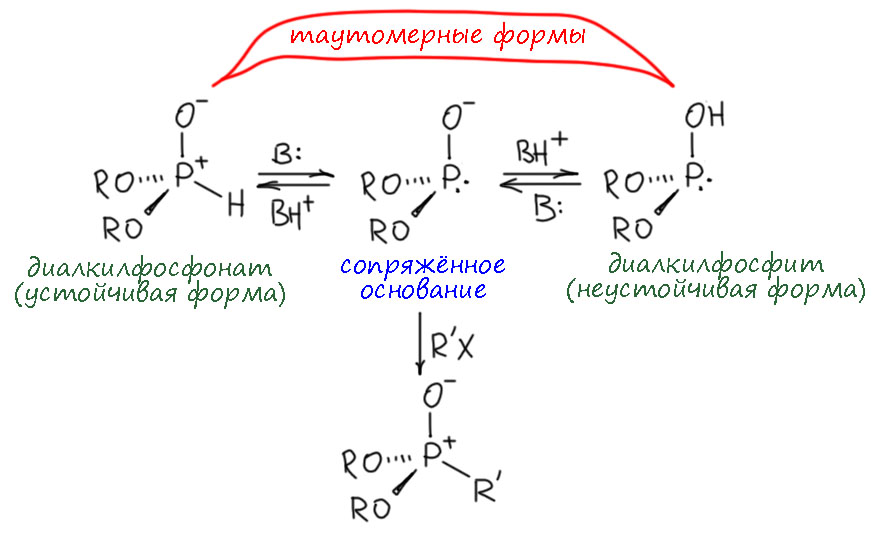
Так и здесь у диалкилфосфоната и гипотетического диалкилфосфита одно единственное сопряженное основание и это фосфорный анионный нуклеофил, соответственно в SN2-замещении дает намного более быстрые и чистые реакции, здесь нет проблем с побочным алкилированием потому что в реакции не образуется второй алкилгалогенид. Вот в этой реакции, поскольку нуклеофил анионный можно применять все способы увеличения реакционной способности – современные растворители, межфазный перенос и т.п. И понятно, что тозилаты спиртов в такой реакции ничем не хуже галогенпроизводных. Оборотной стороной является весьма высокая основность таких анионов, а это может приводить к своим побочным реакциям. Поэтому в химии фосфорорганических соединений реакции Михаэлиса-Арбузова и Михаэлиса-Беккера используют как два варианта, и в конкретных случаях выбирают метод синтеза на основе конкретных экспериментов по оптимизации.

Другие реакции по шаблону реакции Михаэлиса-Арбузова
От реакции Михаэлиса-Арбузова производится невероятное количество других реакций, сделанных по одному принципу: фосфор из фосфита (или другого алкоксифосфина) нуклеофильно атакует некий электрофильный центр, происходит замещение или присоединение, образуется алкоксифосфоний, дальше в смеси находится анионный нуклеофил, который расщепляет алкоксифосфоний с образованием пятивалентного фосфора с гипервалентной “двойной” связью фосфор-кислород, а это какое-то новое фосфорорганическое соединение, которое немедленно тащут на испытания биологической активности.
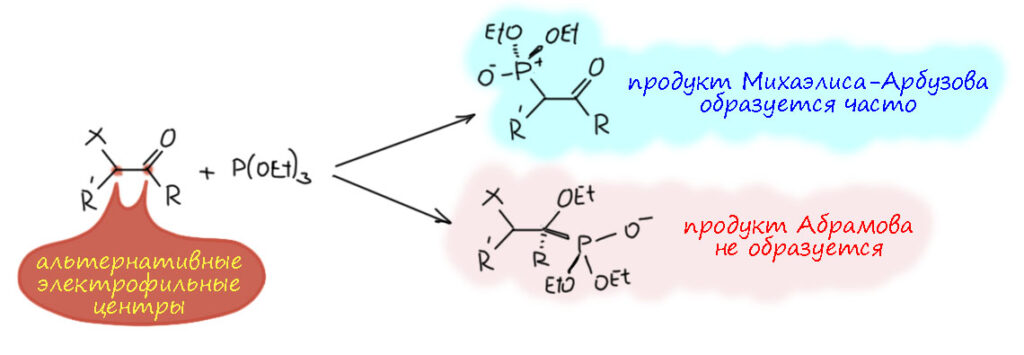
Логика в химии довольно хорошо задана. Ищите электрофилы. Сначала, конечно, просто перебирайте стандартные углеродный – карбонильная группа, акцепторы Михаэля. Карбонильную группу пробуйте всех основных типов – альдегиды, кетоны, производные кислот. С альдегидами и активными кетонами нуклеофильный фосфор присоединяется к карбонилу и дальше надо понять, что произойдёт с тетраэдрическим аддуктом. То, что получается, принято называть реакцией Абрамова, так как ее в начале исследовал Василий Абрамов, ученик Арбузова (). В своем оригинальном варианте реакция должна заключаться в присоединении фосфита к карбонильной группе с последующей нуклеофильном внутримолекулярном замещении – результатом был бы α-алкоксизамещённый фосфонат. Но в реальности эта реакция редко идет так гладко, потому что там есть другие пути превращений, и с разными альдегидами и кетонами получаются разные продукты или смеси продуктов.
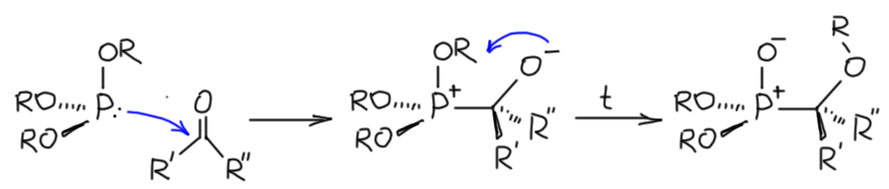
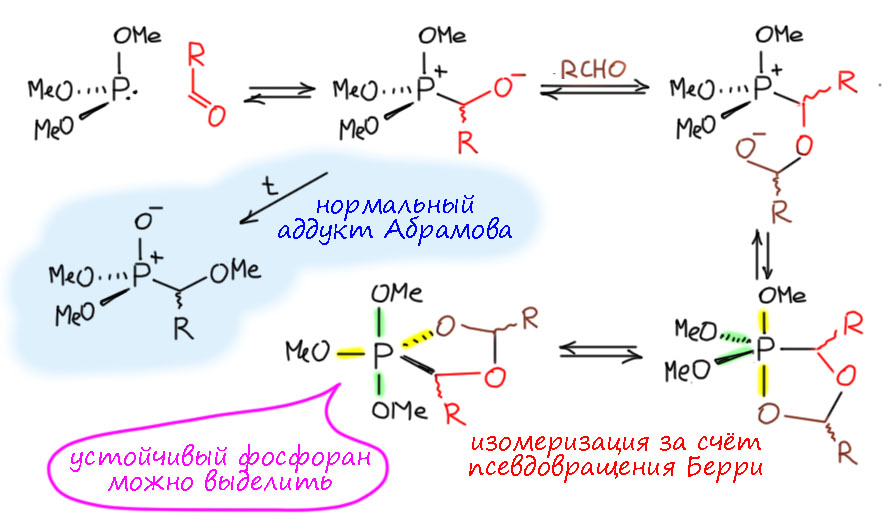
Рамирес с сотрудниками с помощью тогда только появившегося ЯМР на ядрах фосфора показали (Ramirez, F., Patwardhan, A. V., Heller, S. R. J. Am.Chem.Soc., 1964, 86, 514), что само присоединение фосфита к карбонильной группе альдегидов происходит очень легко в мягких условиях, при этом к первому аддукту обратимо присоединяется вторая молекула альдегида, и такой аддукт модет замкнуть пятичленный цикл присоединением кислородного центра к фосфониевому атому фосфора с образованием пятикоординированного фосфорана. Атака нуклеофила на фосфониевый фосфор всегда происходит с апикального положения чтобы образовалась трехцентровая гипервалентная связь, но первый аддукт претерпевает изомеризацию псевдовращением Берри (меняются местами апикальные и экваториальные связи помеченные желстым и зеленым), так чтобы в апикальные положения встали два метоксила – это выгоднее, потому что на гипервалентной трехцентровой связи всегда должны висеть наиболее электроотрицательные группы из возможных в фосфоране и псевдовращение постарается их туда отправить, если сможет по стерическим причинам. Здесь – может. Образуется устойчивый фосфоран с пятичленным циклом (гетероцикл – фосфадиоксалан), можно получить ЯМР – только не надо преувеличивать устойчивость таких гипервалентных соединений фосфора, она невелика, самое большее, что можно сделать это в мягких условиях зафиксировать образование спектроскопически, слово “выделить” здесь не означает – достать из реакционной смеси, почистить и положить в банку. А когда, как в условиях Абрамова, смесь сильно греют, фосфоран не образуется, он очень слабый, гипервалентная трехцентровая связь слаба, и при нагревании все равновесия смещаются в сторону моноаддукта и происходит деметилирование с образованием продукта Абрамова.
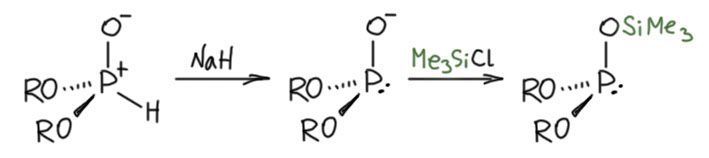
Реакцию Абрамова удалось сделать вполне предсказуемой и селективной очень простым приёмом – одна из алкокси-групп была заменена на силилокси. Это очень просто – мы уже умеем делать моноанион фосфита депротонированием диалкилфосфоната, этот моноанион вляется ссильным нуклеофилом и алкилгалогенидами алкилируется по фосфору (реакция Михаэлиса-Беккера), но хлорсилан это жесткий электрофил, который предпочитает кислород углероду, и тот же анион силилирует по кислороду. При этом получается искомый моносилилированный фосфит.
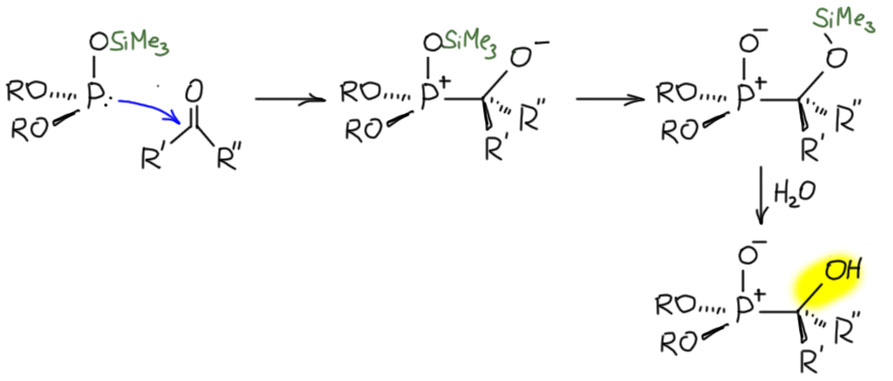
Этот фосфит используем в реакции Абрамова. Всё идет так же, но силильная группа гладко и мягко пересаживается на кислород бывшей карбонильной группы. Понятно почему – кремнию все равно на каком кислороде сидеть, а фосфору расчистили его любимую гипервалентную связь, что, как мы знаем, даёт огромный выигрыш в энергии. Дальше это просто гидролизуют, кремний слетает и образуется очень полезные соединения α-гидроксифосфонаты, а это такие аналоги гидроксикарбоновых кислот. Здесь уже можно подумать и об энантиоселективной реакции и о многих путях продолжения синтеза.
Теперь, как обычно, посмотрим на акцепторы Михаэля, на непредельные альдегиды и кетоны. Здесь, как и в любой другой химии с участием нуклеофилов мы увидим конкуренцию 1,2- и 1,4-присоединения. И еще раз убедимся насколько глупа концепция ЖМКО, ведь по ней уж фосфорный-то нуклеофил должен быть без вопросов мягким и ничего кроме сопряженного присоединения не давать. Но в реальной химии работают совсем другие законы – законы равновесий. И реакции фосфитов тоже выбирают между двумя реакционными центрами очень тщательно, сначала атакуя более реакционноспособный, но в результате равновесий в конце концов образуя просто самый устойчивый продукт. Сопряженные альдегиды почти всегда дают 1,2-присоединение, а здесь это просто продукт реакции Абрамова. И в этих случаях тоже удобно использовать моносилилированный фосфит, условия реакции намного мягче.
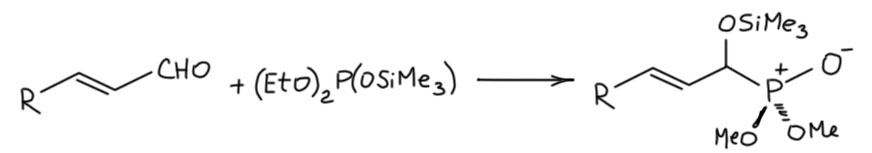
А еноны дают и 1,2- и 1,4-присоединение в зависимости от структуры – фактически важнее всего заместители, которые регулируют возможность довольно крупного реагента, фосфита подобраться к одному или другому электрофильному центру. !.4-присоединение тоже иногда называют реакцией Абрамова, ну или Михаэля-Абрамова (Михаэль это не Михаэлис!!!)
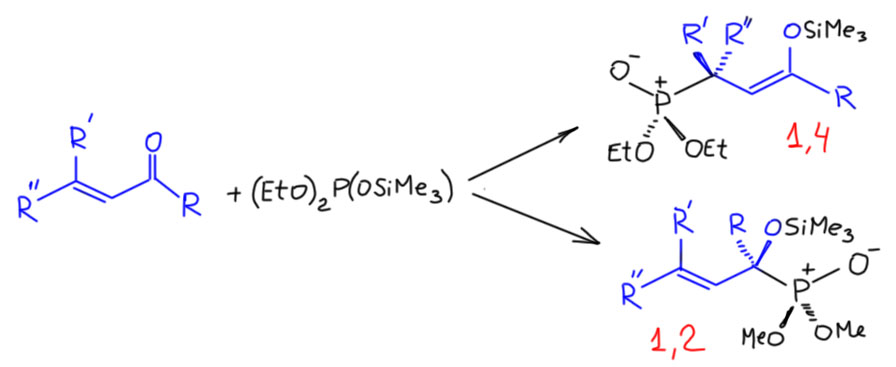
Небольшие, например, циклические еноны довольно чисто дают продукт сопряжённого присоединения. Классные продукты – это силиловые эфиры енолов, дальше можно делать конденсацию Мукайямы или много чего ещё.
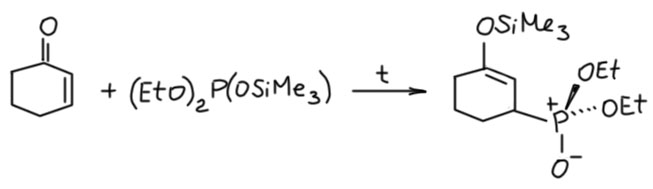
Следующая проблема тоже касается хемоселективности, когда в субстрате есть два электрофильных центра и фосфит решает, на какой ему наехать, а может быть и оба попробовать, а там по ходу дела разберёмся, вот как в только что разобранной химии с непредельными кетонами. Теперь разберемся с другим стандартным субстратом такого типа – α-галогензамещенными карбонильными соединениями. Нуклеофил может либо присоединяться к карбонильной группе, либо замещать галоген – и это очень хороший галоген, как мы знаем из теории SN2-замещения. И действительно, в реакции фосфитов в полной мере проявляется эта конкуренция. А что получается? По идее, либо продукт реакции Михаэлиса-Арбузова, либо Абрамова, или их смеси, если реакция неселективна. Но здесь возникает еще одна сложность – реакция Абрамова с такими субстратами идет необычно и дает совсем неожиданный продукт, в котором никак нельзя узнать возможный продукт реакции Абрамова. Собственно, Арбузов с сотрудниками несколько раз делали реакции с галогенкетонами и галогенальдегидами и описывали образование продуктов необычной природы, но понять структуру не могли, и это не удивительно, в то время структуру устанавливали по аналогии, по дополнительным реакциям с понятным результатом – но для этого случая старые методы классической органической химии не работали. Разобраться удалось только в 1952, и и это стало новой именной реакцией – реакцией Перкова, по имени немецкого промышленного химика Вернера Перкова. Такая совершенно славянская фамилия не должна удивлять – в Германии, особенно в Пруссии, очень много славянских фамилий, потому что Восточная Пруссия веками была местом теснейшего соприкосновения трех этнических групп – славян, балтов и германцев, и там исторически сложился такой сплав трех этнических субстратов, и среди прусской знати было полно славянских фамилий.
Перков, правда, тоже химик старой школы и ничего кроме реакций не использует, никаких спектров, хотя в это время уже точно есть ИК и здесь бы оно помогло. Но Перков ничего этого не знает, да и дело происходит в послевоенной Германии, пока еще не полностью оправившейся от разрухи, и никаких модных приборов там просто нет. Перков сделал реакцию триэтилфосфита с трихлоацетальдегидом (хлоралем), получил новый продукт и серией простых реакций – гидролиза до и после хлорирования – выяснил его структуру. Всё, достаточно, новая реакция свершилась.
Сам Перков – чистый прикладник, он изучал химию пестицидов и написал один из первых справочников по химическим средствам защиты растений. Ну и реакцию открыл очень полезную, с ее помощью делали минимум два очень популярных в прошлом пестицида – хлорофос и дихлофос, которые применяли во всем мире в колоссальных количества и загадили ими всю планету. Оба давно запрещены во всех более-менее вменяемых странах, но в окружающей среде остатки этих пестицидов еще долго будут себя проявлять. Перков открыл эту реакцию, как водится, случайно, и не будь оно просто прикладным химиком, которому было важно получить что-то и тут же бежать пробовать на тараканах, он мог бы заинтересоваться интригующей химией этого процесса и натолкнуться на многие интересные продолжения. Но он оставил все это потомкам, а как я уже замечал, в фосфорорганической химии вообще было засилье именно практиков, так что разбирать механизм реакции стали много времени спустя.
В реакции Перкова первоначально фосфит присоединяется к карбонильной группе как в реакции Абрамова, образуя обычный фосфониевый интермедиат. Но дальше его судьба другая, куда более необычная. Сказывается то, что рядом с фосфонием образуется алкоксидный кислород, хороший нуклеофил, и такая пространственная сближенность фосфониевого электрофила и кислородного нуклеофила. В этом месте можно сделать глубокий вдох и вспомнить реакцию Виттига – там ведь нечто очень похожее, только отрицательный кислород возникает в бета-положении – и там происходит образование гипервалентного пятикоординированного фосфора в четырехчленном цикле – оксафосфетане. Теория и практика циклизации говорит нам, что трехчленные циклы образуются легче четырехчленных – тогда и здесь мы должны ожидать образования пятикоординированного гипервалентного фосфора с трехчленным циклом – делаем усилие, вспоминая номенклатуру гетероциклов – э-э-э… – трехчленный насыщенный цикл – -иран – оксафосфирана. А вся вместе эта структура с пятивалентным пятикоординированным фосфором это фосфоран. Фосфораны, как мы уже неоднократно замечали, во-первых, почти всегда неустойчивы, и являются не продуктами реакций, а интермедиатами, а иногда даже только переходными состояниями. Фосфоран тем относительно устойчивее, чем более электроотрицательны элементы, находящиеся в апикальных положениях. А ще лучше, когда во всех пяти. Почему? Тоже уже проходили – фосфораны находятся в непрерывном конформационном взаимопревращении форм через псевдовращение Берри, поэтому все группы вокруг фосфора бывают в апикальных положениях. Еще один момент – группы в апикальных положениях висят на более слабой трехцентровой связи, с очень значительной долей ионности, причем отрицательные концы связи – на атомах, связанных с фосфором.
ОК. Рисуем фосфоран с трехчленным циклом оксафосфирана. Еще раз споминаем, что фосфораны образуются из тетраэдрических фосфониев так, что входящий нуклеофил встает в апикальное положение. Прикидываем, что будет при псевдовращении Берри, и видим, что один из вариантов – уже углеродная часть трехчленного цикла встает в апикальное положение. Возникает вопрос – а трехчленный цикл не мешает псевдовращению? Нет, потому что там ничего реально не вращается, а просто пары связей совершают короткое движение (колебание) навстречу друг другу, и трехчленный цикл никуда не девается, просто два ребра (связи) немного изменяют длину. Как только углерод попадает в апикальное положение, он становится сильно похож на карбанион, а в бета-положении у него галоген – и это не что иное как E1cb-элиминирование. Галогенид элиминирует, оставив на фосфоре очень интересный остаток – это енолят. В конце происходит то же самое, что в реакции Михаэлиса-Арбузова, галогенид снимает алкил и возвращает фосфору его любимую гипервалентную связь с кислородом, за которую фосфор готов отдать всё на свете. Результатом реакции является интереснейшая штука – фосфорный эфир енола.
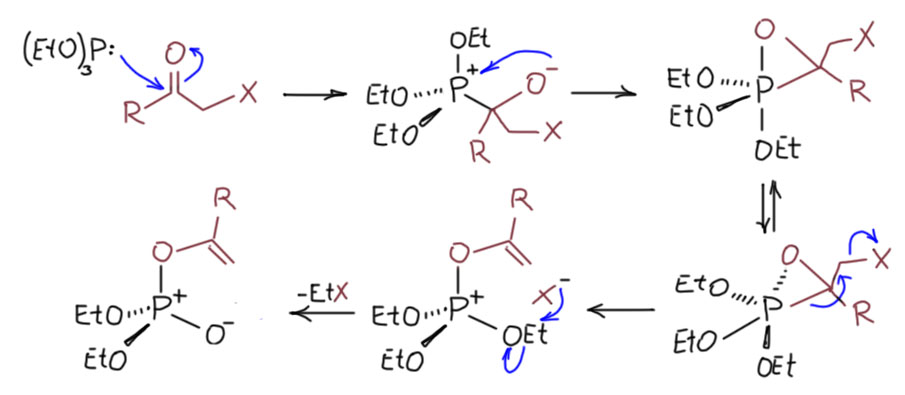
Вернемся на минуту к самой первой реакции, сделанной Перковом – триметилфосфит с хлоралем. В зависимости от условий в этой реакции получаются два разных продукта. Если реакция идет в присутствии воды, а это самый очевидный выбор так как хлораль обычно существует в виде гидрата, хлоральгидрата – то аддукт по карбонилу немедленно протонируется и выходит из реакции – и это продукт реакции Абрамова. Вопрос: а откуда там хлорид для последней стадии? Он просто есть в реакционной смеси, в промышленнности вообше используют не готовый триметилфосфит, а треххлористый фосфор, который сначала в том же реакторе обрабатывают метанолом. Это знаменитый пестицид, в СССР известный как хлорофос, а в других странах как метрифонат или трихлорфон, впервые был запатентован Байером в 1955, но химия настолько простая, что во многих странах плевать хотели на этот патент и делали свой хлорофос по десятками разных названий.
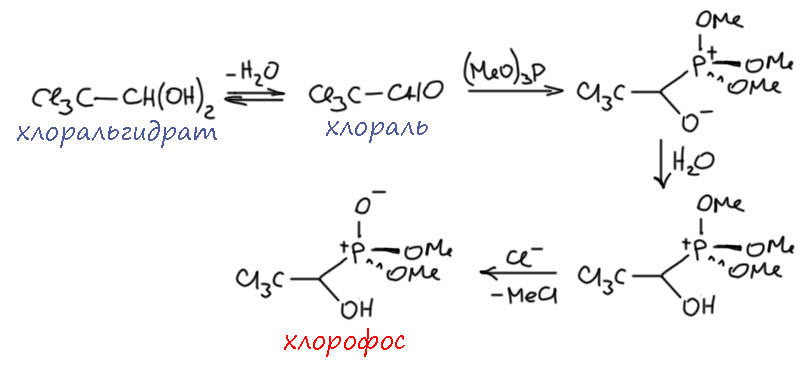
А если реакцию делать без источника протонов просто нагреванием хлораля с триметилфосфитом то образуется то, что и описал Перков – и это не менее знаменитый пестицид, дихлофос в СССР или дихлорвос в других странах (есть еще туча других торговых марок потому что это простые вещества, которые мог производить под своей маркой любой – проще химии не придумать).
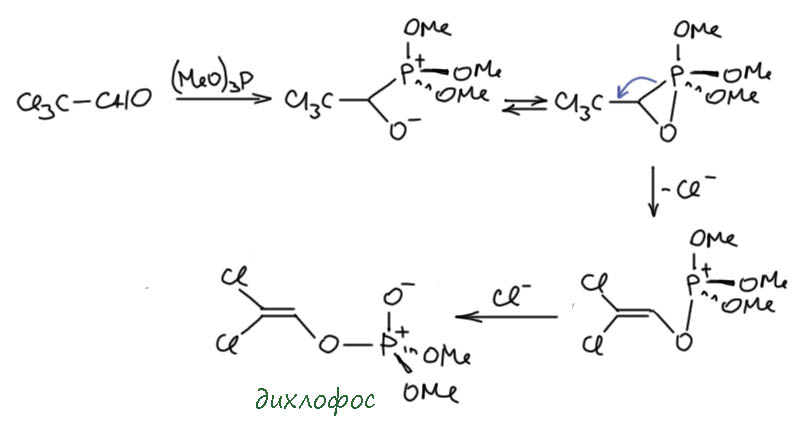
Оба этих инсектицида по простоте получения, дешевизне, эффективности почти не имеют себе равных. Оба являются ингибиторами холинэстеразы, поэтому действуют на всех животных, но не в одинаковой степени – холинэстеразы у разных животных немного разные, да и всякие сопуствующие факторы тоже работают – до холинэстеразы насекомых проще добраться, чем до холинэстеразы человека, поэтому те концентрации, которые используют в обработке, не убъют и даже не сильно отравят человека, а ингибирование холинэстеразы большинством таких соединений обратимо и быстро проходит само (мы теперь знаем примеры фосфорорганики, которая вызывает необратимое ингибирование холинэстераз человека, и надо хорошо понимать, что вся эта химия принципиально одинакова). Интересно, что хлорофос, попав в организм действует не сам, он подвергается медленному превращению в дихлофос (то есть, реакция Перкова может происходить и с уже готовым аддуктом Абрамова), который и работает. Поэтому дихлофос намного эффективнее и требуется в намного меньших количествах. Но у обоих пестицидов есть и масса других действий на организм человека, кроме того, особенно при производстве в недостаточно развитых странах, в них много опасных примесей; оба очень надолго остаются в окружающей среде, проникают в питьевую воду и сельскохозяйственную продукцию, создавая такой постоянный фон воздействия. За оба пестицида долго и мощно дрались производители пестицидов, уверяя, что без них не обойтись, особенно в теплых странах, где насекомые разносят опасные болезни, а денег на массовое применение более современных и дорогих препаратов нет. Оба пестицида в основном применяли не для обработки полей, а для унижтожения насекомых в закрытых помещениях – жилье, школах, хранилищах зерна, и т.п. Но всё же страна за страной за последние 20-25 лет отказывались и запрещали. Хлорофос в 2020 году наконец запретили даже в Индии, где из-за огромной плоности населения и климата проблема насекомых-переносчиков особенно остра.
Галогенкетоны и галогенальдегиды реагируют с фосфитами конкурентно по реакции Михаэлиса-Арбузова, Абрамова или Перкова в зависимости от условий. Не будем это подробно разбирать, это как всегда зависит от условий и особенно от природы карбонильного соединения, так как выбор предпочтительного места атаки в основном регулируется стерикой.
Галогензамещенные сложные эфиры реагируют только по Михаэлису-Арбузову по причине меньшей реакционной способности карбонильной группы в производных карбоновых кислот. Так получают фосфоновоуксусный эфир, применяемый в очень полезном варианте реакции Виттига – реакции Хорнера-Водсворса-Эммонса. Эта реакция рассмотрена на страничке про медоды в химии карбоновых кислот.
Есть впрочем одно производное карбоновых кислот, карбонильная группа которого достаточно реакционноспособна, чтобы быть единственным реакционным центром – это хлорангидриды. Реакция идет строго по пути Михаэлиса-Арбузова и получаются интересные соединения – ацилфосфонаты.
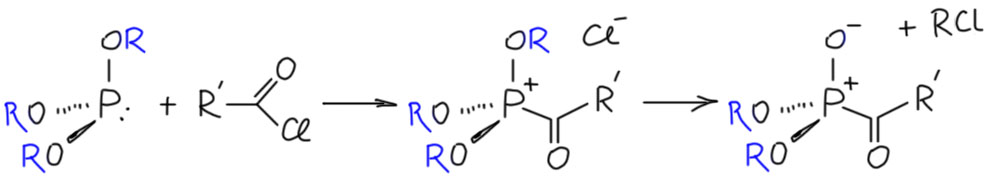
ок