Вопросы
Здесь можно задать интересующий вас вопрос по органической химии. Вопросы анонимны, кто их задаёт мне совершенно безразлично. Можно задавать любые, но это не значит, что вы получите ответ. Вероятность есть, почему бы не попробовать. Ничего пока не точнее не знаю, это эксперимент. Логиниться тоже пока не будем, хотя это не исключено в будущем, поэтому заполнять поля с именем и адресом не обязательно.
С ответами пока не решил. Пока нет ни одного вопроса решить трудно. Скорее всего, я буду стараться отвечать на конкретные вопросы, на которые можно ответить коротко, небольшим текстом и одной-двумя схемами. Более общие вопросы задавать тоже можно – вообще можно задавать любые вопросы, касающиеся органической химии или даже химии в целом, если вам трудно решить, что касается органической химии, а что точно не касается – но я, скорее всего, буду расценивать таике вопросы как запрос на написание недостающих разделов, и буду соответственно составлять свои планы. Посмотрим, может даже введём что-то типа голосования, если будут многочисленные запросы на какие-то разделы и нужно будет понять приоритет.
Ответы будут публиковаться на отдельной странице.
Начинается новый год. Страничке с вопросами тоже исполнился год. Эксперимент вроде бы оказался удачным, поэтому продолжим. Всем задававшим вопросы – большое спасибо. Мы подняли занятные темы и кое-что важное узнали. Еще раз напоминаю, что глупых вопросов не бывает (глупые ответы бывают, и я не застрахован, но стараюсь попусту не влипать), поэтому не пытайтесь ранжировать свои вопросы сами – мне виднее, что заслуживает подробного ответа, а когда можно парой фраз отделаться. По-прежнему не обещаю оперативности. Как получится, так и получится. Минимум на один вопрос я до сих пор не ответил. Ответ получается таким объёмным, что допилить пока не выходит. Но рано или поздно это случится, тем более, что это пересекается с одной из лекций нового курса, которая тоже однажды будет сделана для сайта и выложена.
Добрый день!
Подскажите, а LDA может выступать литирующим агентом или он действует как основание в основном?
https://doi.org/10.1016/j.polymer.2018.11.018 в этой статье его использовали как основание, а бром остался не затронут, поэтому, собственно, и возник вопрос
АЧ: Добрый день. Ответил на страничке с краткими ответами.
Большое Вам спасибо за подробный ответ!
Руководитель дипломной дал задание сравнить химический состав растения (к примеру общий состав фенолов, протеина, токоферола листьев) из разных географических областей на основе существующих научных
статей. Предполагается, что на основе работы можно будет установить влияние окружающей среды на состав и определить лучшие области. Реально ли сделать достоверное сравнение, если в статьях отличаются методы сушки и методы экстракции?
АЧ: Вы опять не туда попали. И я никак не могу комментировать задание, данное кем-то, потому что не могу быть уверенным, что суть задания верно понята и передана.
Чисто из общих соображений задача сравнения химического состава растений из разных мест мне кажется бесполезной. Химимический состав растений зависит от случайного набора параметров, в которых местность играет далеко не первую роль. Важнее и состав почвы, и содержание питательных веществ, и конкретные погодные условия в сезон взятия проб, и соотношение частей растения в пробе, и так далее. Корректно все это учесть вряд ли возможно. И в статьях, естесвенно, все будет по-разному, в том числе и методы анализа.
Но диплом – это просто решение некоторой поставленной задачи. Поставили такую задачу – выполняйте. Соберите данные, сделайте корректную статистическую обработку, и она вам и покажет, насколько широки будут доверительные интервалы и все остальные статистические критерии. Но диплом можно написать всегда – кто мешает вам, например, сделать отрицательный вывод о достоверности.
Здравствуйте, Андрей Владимирович. Возник немного странный, наверное, вопрос. Имеет ли (или может иметь теоретически) какое-то применение в органической химии монооксид кремния? Просто вещество в принципе существует, но почти нигде и не упоминается. Хотя, его аналог, монооксид углерода в синтезе используется и вполне успешно. Может и SiO можно использовать для синтеза чего-нибудь кремнийорганического?
АЧ: Добрый вечер. СО в синтезе не просто используется, а можно сказать, является основой огромной области синтеза, как промышленного – а это самые крупнотоннажные каталитические процессы синтеза метанола, уксусной кислоты и многочисленных гидроформилирований, и не только. Это десятки миллионов тонн в год. В лабораторном синтезе, особенно с применением комплексов переходных металлов это многие десятки методов синтеза карбонильных соединений. Поэтому это одна из самых востребованных малых молекул.
Но кремний это не углерод. Во втором периоде мы пользуемся всем арсеналом, чёрт бы побрал эти милитаристские словечки, забыть бы их всех и навсегда – способов стабилизации необычных валентных состояний. Двухвалентный углерод стабилизируется мезомерией с соседним кислородом и так мы получаем стабильную, но очень реакционноспособную молекулу.
Но в третьем периоде и ниже ничего этого нет. Связи там длинные, гибридизация почти не проявляется, электронные оболочки пухлые, из-за чего при попытке приблизить атомы хоть немного поближе простой связи мы получаем отталкивание Паули, а это страшная сила. Поэтому мы даже не можем сделать нормальную двойную связь Si=O, не говоря уж о дальнейшей стабилизации секстетного кремния. Молекула получается дико нестабильная и ещё более дико реакционноспособная – надо же куда-то девать все эти свободные валентные возможности на обоих атомах. Образуется она только при адской температуре в высоком вакууме, и немедленно диспропорционирует при попутке вытащить её из ада в обычные условия. Есть еще какие-то попытки как-то это сделать при низкой температуре в матрицах, но тогда это должен быть очень странный полимер с двухвалетным дикоординированным кремнием, то есть такой силилен, а силилены это тоже очень особое состояние кремния, требующее особой стабилизации хорошими лигандами, и в простых соединениях не выживает. Я немного касался этого в своей лекции для Органики 21 века, про то, как p-элементы сражаются за место под солнцем с переходными металлами. Увы, она доступна только в видео- формате, я до сих пор не сделал для неё доступное слайд-шоу с комментарием. Сделаю в некоторой разумной перспективе.
Из этого конечно же следует, что оксосилилен, как наверное, стоит называть SiO, для органического синтеза абсолютно бесполезен. И в очередной раз изумиться, как повезло органикам с выбором углерода и вообще второго периода для своей химии, и славить богов за такой подарок.
Добрый день!
Не знаете ли вы способов превращения винилбромидов (скажем, они с одного конца дизамещённые, а с другой бром болтается) в просто олефин без использования металлов, их амальгам и металлоорганики (в интересующих меня молекулах есть кислые протоны и карбоксильные фрагменты)? Я порылся в литературе, ничего толкового кроме пары Zn-Ag не нашел.
АЧ: Это восстановительное дегалогенирование, соответственно, это или активная непереходная металлоорганика с протолизом. Или перреходные металлы, но тогда это не что иное как C-H кросс-сочетание, и в промежуточных комплексах будет гидридный, а это неизбежно приведет к конкурентному гидрометаллирвоанию, и дальше или к олигомеризации или к гидрированию.
Но всегда есть свободнорадикальное дегалогенирование, трибутилоловогидрид с инициатором и иногда с источником водорода. Лучше работают иодпроизводные, но и бромпроизводные тоже вполне подвержены. Конечно Bu3SnH это тоже металлоорганика, но в синтезе без металлоорганики, хоть какой-то продраться трудно. И с винилгалогенидом может быть проблема – радикальная олигомеризация уже дегалогенированного олефина, хотя возможно ее можно подавить просто избытком донора водорода.
Безусловно, в современной химии очень много попыток заметить гидрид оловяшки на что-то менее устрашающее. Но всё это в основном китайско-бумажная химия, а гидрид олова трудится более полувека в тысячах синтезов.
Добрый день!
Sn2 реакция при четвертичном атоме углерода с хорошей уходящей группой невозможна или все-таки иногда реализуема в зависимости от типа заместителей при этом атоме углерода?
АЧ: Ох, это чертовски непростой вопрос. Скорее всего да, но данные противоречивы, трудно исключить альтернативные механизмы, например, через перенос электрона. Что-то в третичном субстрате бензильного типа, но с акцепторами в кольце, чтобы убрать катионный путь. В эпоксидах тож. Здесь важно аккуратно выделить принципиальный вопрос – насколько точно мы можем идентифицировать переходной состояние SN2-типа. Без очень аккуратных расчетов не обойтись. Я понемногу собираю материал, у меня есть план на лекцию вокруг нуклеофильного замещения для нового сезона органики 21 века. Посмотрим. Пока отвечу уклончиво – скорее всего, если очень хорошо собрать воедино факторы, ускоряющие SN2, но блокирующие другие пути, то да.
АЧ: И как бы вдогонку. Поэтому мы безусловно правы, когда на 3-м курсе, когда речь заходит про SN2, начинаем истошно верещать как ворон у Эдгара Аллана По – никогда, никогда, никогда!
Безусловно если такие примеры есть, то это какая-то дико вымученная экзотика для любителей диковин.
К сожалению, в учебниках подают часто плохой пример, когда экстраполируют известный график скоростей SN2 на третичный субстрат, и получают какую-то мизерную, но ненулевую оценку скорости – в химии лучше никогда не экстраполировать, но только интерполировать. Нет таких данных, в оригинале у Ингольда и Хьюза этот график обрывается.
Добрый день.
У циклогексана есть конформация твист – она ниже по энергии, чем ванна, но выше, чем кресло. Подскажите, а какие атомы там “мешают” друг другу, и можно ли это хоть как-то представить при изображении структурной формулы?
АЧ: Добрый вечер. А зачем рисовать структурную формулу в твист-форме? Циклогексан имеет два очень хороших конформера типа кресло, находящихся в хороших, глубоких минимумах. Превращение одного кресла в другое идёт через барьер приблизительно в 10 ккал/моль – это соответствует очень быстрому процессу при комнатной температуре – и промежуточные конформеры типа твист, но они приблизительно на 5 ккал/моль выше кресел, а это значит, что в равновесной смеси конформеров их содержание меньше 1 процента. Твисты превращаются друг в друге с бешеной скоростью, потмоу что барьер всего 1 ккал/моль, что только чуть-чуть больше чем совсем ничего. Переходное состояние между твистами это и есть ванна – конформация, но не конформер, в ванне жизни нет. Напомню чем плоха ванна – прежде всего мерзкими заслоненными конфигурациями по нижним связям, и во-вторых, флагштоковым отталкиванием водородовю Эти взаимодействия дестабилизируют конфигурацию ванны. Быстрый уход от них понятен – скрутите связи, по которым была заслоненная конфигурация, чтобы от нее уйти, и как раз и попадете в эти мини-минимумы твистов.
Иными словами, твисты нужны только для того, чтобы корректно описать полный процесс инверсии кресел. ДЛя химии они не имеют значения, потому что время жизни в них ничтожно. Если бы нашлась реакция, для которой нужен именно твист, а такую реакцию не очень сложно себе представить – например, син-элиминирование – то твистами имело бы смысл заняться, потму что в таких случаях работает принцип Кертина-Гаммета, а все остальное идёт лесом.
Сделаем так. Вопрос, на самом деле, интересный и заслуживает иллюстрированного ответа. Сейчас я немного перегружен, но попробую на ближайших неделях покрутить циклогексан и сделать картинки. Тогда вернемся к этой истории.
Спасибо!
Андрей Владимирович, доброго времени суток!
Подскажите, почему при арилировании по Меервейну образуется радикал, сопряженный с электроноакцепторной группой исходного алкена, а не сопряженный с бензольным кольцом? Заранее спасибо за ответ.
АЧ: Возможно, тут какое-то недоразумение, но не вполне ясно, откуда взялась проблема со стабилизацией радикала-аддукта. Какой олефин брали, так и стабилизируется. Брали олефин с акцепторной группой, значит будет стабилизация сопряжением с этой группой. Брали стирол – тогда с фенилом. Или Вам хочется попробовать в качестве акцептора что-то типа омега-нитростирола или коричного нитрила?
Добрый день, вопрос по синтезу D3-тиализонов из Органикума т. 2, стр. 22 (или из ПОХа Б11.12).
https://doi.org/10.1002/ange.19670792202 – ссылка в Органикуме на статью по получению таких тиализонов.
Вопрос: а чем и как активируется сера в данном случае? Неужели обычный карбокатион может так просто раскрыть S8 или добавление амина (морфолина, пиперидина и т.д.) так способствует?
Я поинтересовался о таком с другой стороны: в одной статье (https://doi.org/10.1021/acs.orglett.7b00819) генерировали дихлоркарбен в присутствии трет-бутилата калия и серы, но выявить наличие тиофосгена у них не удалось, однако изоцианид присоединял серу, стало непонятно, почему изоцианид присоединяет серу, а дихлоркарбен – нет.
АЧ: Добрый день,
Это довольно простой кейс. Карбокатион тут ни при чём. Немного позже разрисую.
АЧ: Разрисовал на отдельной странице.
Спасибо!
Добрый день, Андрей Владимирович. А есть ли какое-либо четкое химико-физическое (возможно, квантово-механическое/химическое) определение ароматичности, её необходимые и достаточные условия? На данный момент все распространенные определения кажутся эвристическими и эмпирическими без какого-либо чисто физического представления, отсюда складывается впечатление, что понятие ароматичности, хоть и чуть ли не одно из фундаментальных в органической химии, не имеет четкого определения. А ведь сейчас ароматичность вышла за пределы органики, и можно встретить работы, описывающие ароматичность в кластерах d-металлов типа ртути.
Спасибо за ответ!
Добрый вечер!
Подскажите, почему соли диазония с противоионами BF4(-), PF6(-) в сухом виде можно хранить и о их взрывоопасности не говорят, а хлориды, гидросульфаты характеризуют как взрывоопасные? С чем связана взрывоопасность последних и меньше склонность к этому для первых?
АЧ: Это очень просто. Соли диазония неплохие окислители, и любят оторвать откуда-нибудь электрон. Как только оторвут, немедленно распадаются на азот и арильный радикал, радикалы быстро сдваиваются или находят атом водорода вокруг себя. Все это очень экзотермично и быстро. Очень быстрая экзотермичная ракцияя часто и воспринимается как взрыв. Тем более, что если соль хранилась в закрытой банке, выделяющийся нагретый азот занимает немаленький объём и её и разнесет.
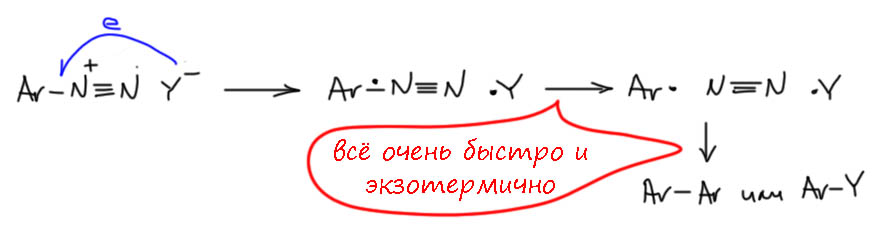
Откуда электрон? Например, от более-менее легкоокисляемого противоиона.
Хлорид окисляется довольно легко до атома хлора, и дальше часть схлопнется с теми же арильными радикалами (очень экзотермично), или друг с другом (тоже). А вот фторборат или фторфосфат окислить фактически невозможно – там нечего. Бисульфат кстати тоже окислить очень трудно (получился бы сульфоксильный радикал, но это непросто, поэтому такая реакция если и пойдёт, то только при сильном нагревании, но тогда скорее не перенос электрона, а прямая диссоциация соли диазония с образованием арильного катиона, который немедленно свяжет бисульфат). Бисульфаты солей диазония довольно устойчивы, если чистые, они вполне неплохо хранятся, ненамного хуже тетрафторборатов, то есть желательно в темноте и в холодильнике – темнота важнее, в промышленности соли диазония с устойчивыми противоионами хранят в самых обычных условиях и без проблем перевозят. Проблема с бисульфатом иногда бывает из-за низкого качества серной кислоты, используемой при диазотировании – там могут быть восстанавливающие примеси, которые и будут причиной разложения. Бывает, например, так, что по недосмотру в диазотирование вводили гидрохлорид анилина, но диазотировали в серной кислоте, получили бисульфат диазония, в котором осталось немного хлорида – этого может быть достаточно для неприятностей. Но если диазотирование сделано аккуратно и чисто, бисульфаты диазония вполне устойчивы.
В целом, соли диазония с неокисляемыми анионами очень устойчивы, хранятся годами без холодильника, и единственно, что нужно строго соблюдать – в темноте (обернуть фольгой, положить в картонный контейнер и т.п.)
Большое Вам спасибо!
Добрый день. Произошел залив кладовки из системы кондиционирования. Предположительно этиленгликолем или пропиленгликолем. Возможна ли очистить тканевые вещи (матрас, плед, подушки и т.д) от данного вещества? удаляется ли данное вещество полностью или же дальнейшее использование данных предметов опасно для здоровья?
АЧ: Ещё раз напоминаю, что это сайт для изучения науки химии, и бытовыми вопросами не занимается. Такие советы вообще лучше не давать, потому что нет никакой гарантии, что описанная суть проблемы верна. Что-то пролилось из системы кодиционирования. Да, это должен быть или пропиленгликоль, или этиленгликоль. Пропиленгликоль сам по себе вообще не токсичен, мы его каждый день едим из смых разных источников. Этиленгликоль токсичен, но только при приеме внутрь сразу немаленького количества. Надышаться этиленгликолем практически невозможно, хотя обязательно найдутся умельцы, у которых это получится. Но кто сказал, что это действительно так. Кто-то что-то туда залил, а кто-то другой что-то разбодяжил; гликолей не хватило, под руками была какая-то старая канистра чёрти откуда, может с полигона утилизации химоружия, может еще откуда-то, фломастером написано гликоль, вроде похоже, Ну залили, ну продали, вроде работало, потом вытекло, почему-то медная трубка с шипением растворилась. И так далее. Тут недавно люди пили сидр и все умерли.
Поэтому правило должно быть простое: пролилась любая химия на что-то бытовое – не надо разбирать что, надо устранить. Тряпки постирать хорошенько, то, что можно, проветрить пару месяцев. Что нельзя – выбросить. И если остается какой-то явно посторонний запах, повторять, пока не уйдёт.
Здравствуйте, Андрей Владимирович?
Почему в азулене электрофильное замещение происходит только в пятичленной части цикла? И возможно ли в азулене нуклеофиольное замещение?
АЧ: Как хорошо не знать, кто задаёт вопросы. Вот если бы я, например, знал что Вы случайно являетесь студентом нашего 3-го курса, то я бы ответил вопросом на вопрос: а Вы сами как думаете, ведь проанализировать электрофильное замещение в азулене нам должно быть по силам; мне даже кажется, что однажды у нас был такой вопрос на контрольной. А поскольку я не знаю, придётся немного разобраться. Пока кратко, но возможно в перспективе я сделаю ответ пополнее, тем более что азулен и правда – молекула предельно занимательная, взять хотя бы только то, что она демонстративно нарушает один из основных признаков ароматических соединений, при том что никто вроде бы не сомневается, что азулен это 10-электронная ароматическая система. Можно сказать, почти хрестоматийная, и в чём-то даже более бесспорная, чем изомерный нафталин. Отложим пока эти тонкости. Напомню общеизвестное: у азулена циклическое сопряжение с ароматическим счётом электронов, но которое можно представить через поляризацию системы так, чтобы один электрон переместился из 7-членного кольца в 5-членное: система, таким образом это конденсированный тропилий и циклопентадиенил-анион.
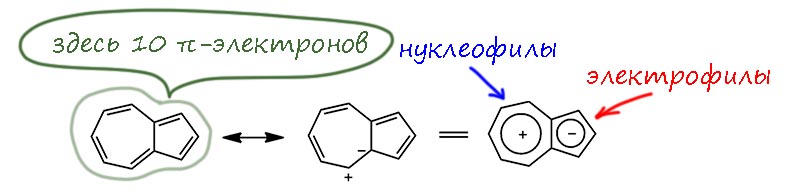
Такую поляризацию подтверждает значительный дипольный момент молекулы. Если вы (здесь и далее, вы это обращение к обобщённому читателю, а не только к автору письма) думаете, что это очевидно, то таки нет: почему бы азулену не быть просто [10]-аннуленом, для чего не нужна была бы никакая поляризация, вопрос не такой простой, но мы его здесь тоже оставим без ответа. Вообще, если вы всё же на 3-м курсе и пока не подобрали себе курсовую, очень советую сделать азулен: это несложно, есть именно 3-хстадийные синтезы из самых банальных исходных, а в конце счастье гарантировано – это реально невероятно красивое вещество.
Итак, приняли общеизвестное – поляризацию. Тогда сразу становится ясно, что азулен должен уметь реагировать как с электрофилами, так и с нуклеофилами, причём пятичленное кольцо электроноизбыточно (6 электронов на 5 углеродов), а значит активировано к электрофильному замещению. Это должно быть сильно похоже на 5-членные гетероциклы, в частности по реакционной способности и действительно это так: посмотрите как обращаются с пирролом, и получите довольно точное представление, какие реагенты лучше взять для азулена. Одна проблема – ориентация: в азулене два разных положения в 5-членном кольце. Не беда, рисуем сигма комплексы и анализируем стабилизацию ровно так как мы это должны делать всегда, когда нас спрашивают, куда то или иное ароматическое соединение согласно принять электрофил.
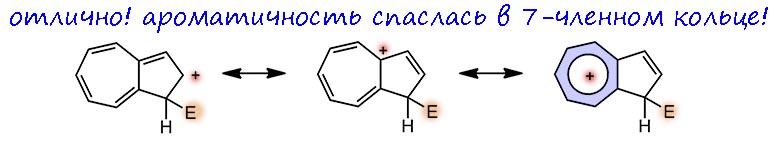
Рисуем сигма-комплекс по альфа положению. Рисуем мезомерные структуры и быстро видим, что одна из них как две капли воды похожа на настоящий тропилий – плюс, циклически делокализованный в 7-членном кольце. Отлично, это значит, что при образовании сигма-комплекса ароматичность не пропала, а нашла убежище, а это выгодно.
А вот если нарисовать сигма-комплекс по второму положению, то как ни гоняй плюс по системе, ароматичность не вырисовывается нигде. А это значит, что она потеряна, а это невыгодно. Поэтому можно считать, что направление ароматического замещения в азулене установлено, и это соответствует реальности.
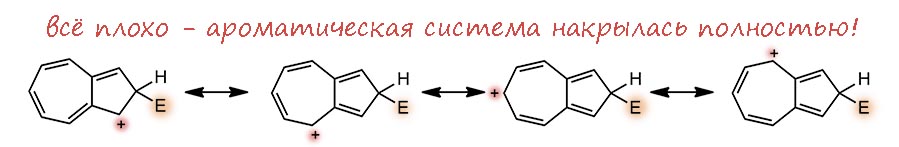
Второе кольцо азулена это фактически тропилий, электронодефицитная система: 6 электронов на 7 углеродов. Прямой аналогии мы не найдём, но если, например, в пиридине принять, что из-за большей электроотрицательности азота электронная плотность пи-системы смещена к этому атому, на углерода останется меньше, чем по одному электрону на рыло. И некоторое сходство усмотреть можно – они дезактивированы к электрофильному замещению, а в азулене это приговор, потому что под боком есть 5-членное кольцо, всегда готовое принять электрофил. И активированы к нуклеофильному замещению. Если там есть галоген или другая уходящая группа, то понтяно, что будет. Но можно подумать и про замещение водорода. Тогда опять рисуем сигма-комплексы, их тут будет три. И мы увидим, что в двух положениях, а это условно орто и пара, если принять циклопентадиенил за такой странный двухместный заместитель, минус может спрятаться в 5-членном кольце, принеся туда (а точнее, сохранив) ароматичность. Это выгодно.
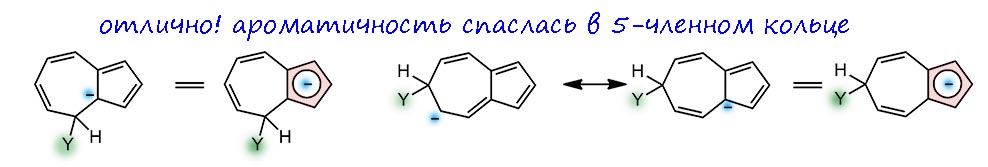
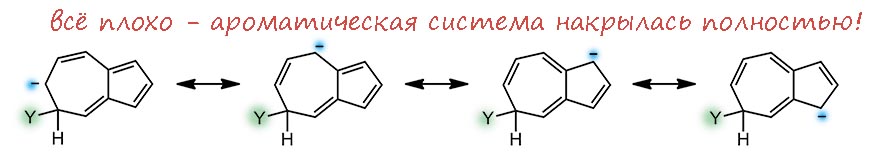
А вот в условном мета-положении опять минус будет метаться по всей системе без толку – ароматичность утрачена, и единственный способ ее вернуть – отправить нукелофил откуда пришёл.
Следовательно, нуклеофильная атака возможна по двум (а реально трём, потмоу что “орто”-положений два) положениям. Одна проблема – куда девать водород, ведь гидрид почти никогда не уходит. Ответ на это известен – окислить сигма комплекс, и для этого обычно применяют акцепторные хиноны. Подробности разбирать не будем.
Спасибо.
Андрей Владимирович, добрый вечер!
Подскажите, почему в ариновом механизме такой странный ряд подвижности галогенов: Br>I>Cl>F? Это можно объяснить без использования понятий о лимитирующих стадиях?
АЧ: Добрый вечер. А откуда у Вас этот ряд? В ариновом механизме очень трудно сделать нормальную кинетику, поэтому сведения о реакционной способности очень разнообразны и к одной схеме не сводятся. Вообще это интересная химия, и я её когда-нибудь с среднесрочной перспективе разберу – там много нового, механизм работающий и приносящий новые результаты.
В самом общем виде, чтобы генерировать арин нужно сильным основанием отнять протон рядом с галогеном, после чего происходит E1cb элиминирование. В отдельных случаях можно подумать и о согласованном E1cb-подобном E2 элиминировании. Галогенид уходит всегда не просто так, а с помощью катиона металла из реакционной смеси, обычно это противоион основания.
Вот и смотрите на факторы, влияющие на скорость этой реакции: во-первых, это CH-кислотность отщепляемого протона. На неё сильно влияет тот самый галоген, которому еще предстоит уйти, но часто и другие заместители. При прочих равных, вроде бы более электроотрицательный галоген дает большую кислотность и облегчает депротонирование. Если бы только этот фактор работал, то ряд был бы F > Cl > Br > I (это только по электроотрицательности, то есть по индуктивному эффекту, но там ещё есть гиперконъюгация, сигма-пи* сопряжение, с грубо обратным порядком, но пока пренебрежем, просто потому что этот фактор очень тяжело вытащить из всей каши факторов). Во-вторых, это собственно качество уходящей группы и здесь мы привыкли к тому, что ряд тоже I > Br > Cl >>>> F (фторид легко уходит только в механизме SNAr, где скоростьопределяющая стадия задана и это не уход галогенида). На этот ряд влияет эффект противоиона, но это тоже пока оставим.
Вот и получается, что скорость образования арина определяется взаимно противоположными рядами вляния, и в конкретных случаях игра эффектов может дать разные результаты. Фторид все равно в ариновых реакциях уходит редко, а остальные три галогена сложатся в ряд типа того, который Вы привели: бромид оказался компромиссом между легкостью отщепления протона и легкостью ухода (хлор дает большую кислотность, но хуже уходит; иод лучше уходит, но кислотность меньше).
Но еще раз подчеркну, в ариновых реакциях строит ряды занятие неблагодарное. Как у нас всегда бывает с такими рядами – кто-то 50 лет назад это опубликовал для конкретного случая, оттуда это попало в какой-то учебник и стало общей закономерностью, и теперь все должны это с важным видом хлебать.
Спасибо!
Ряд был в лекциях для третьего курса химфака, а в литературе встречались другие ряды, поэтому вопрос и появился…
АЧ: Ну да, так и есть. В преподавании обычно стараются давать какие-то однозначные схемы, чтобы удобнее было экзамены принимать. Но разобрав эту схему мы видим и возможности отклонений от неё – в элиминирвоании, а это частный случай бета-элиминирвоания, всегда есть вариативность механизма – что раньше, что позже, где переходное состояние – раньше или позже. А это все зависит от всего – заместителей, основания, уходящей группы, противоиона, растворителя, времени года и международной обстановки. Поэтому стоит гибко относится к всяким рядам и корреляциям для таких рассредоточенных механизмов – когда события реакции происходят не на одном реакционном центре, а в разных частях молекулы и разной степенью согласованности.
Здравствуйте, Андрей Владимирович. Хотелось бы узнать Ваше мнение по поводу квантовой химии. Насколько она нужна и важна для исследований в органической химии?
АЧ: Нет у меня сил подробно отвечать на этот вопрос. Тем более что и задан он в стиле старинного советского анекдота: Любите ли вы помидоры? – Есть люблю, а так нет
Моё мнение сугубо положительное, нужна и важна. Оставим сразу настоящих, профессональных квантовиков-затейников, которые считают, что это такая самостоятельная наука, которой по силам объяснить всё, не прикасаясь к колбам и прочей hardware. С упоением строчат они уравнения (моё любимое – уравнение Клопмана, своеобразный шедевр идиотизма от избытка ума), концепции, индексы и коэффициенты, призванные избавить химическое человечество от грязной работы – но это самое человечество почему-то в упор не замечает этих титанических усилий. Видимо, надо ещё поработать.
Но для обычной химии, в колбах, квантовая химия – исключительно важный инструмент. Я думаю, что уже в самом ближайшем будущем обходиться без него совсем станет неприлично. Как без спектров ЯМР в синтетической органической работе. Можно себе представить такую работу без ЯМР? Всего 60 лет назад и ранее так и было всегда, еще 40 лет назад как минимум часто.
Я бы это сформулировал как обращение к химику экспериментатору: знайте свои молекулы! Прежде чем что-то замышлять, посчитайте молекулярную и электронную структуру, посмотрите, как выглядит молекула в 3D (надежный рачет в этом смысле даже лучше, чем рентген, который все же дает молекулу в решетке, с геометрией, подходящей для упаковки, а это довольно часто сопряжено и с искажениями структуры, и с тем, что пакуется далеко не самый заселенный конформер) – будете намного лучше понимать стерику и пути подхода реагентов. Размышляя о механизме, посмотрите на распределение электронной плотности, орбитали, возможные напряжения – это часто трудно понять по рисунку на бумаге. Расчеты для таких целей сейчас делаются на самых обычных, хотя и приличных, домашних компах, за разумное время, с очень приличными уровнями теории, почти исключающими значимые ошибки для устойчивых молекул в основном состоянии. Поэтому пренебрегать такой возможностью лучше понимать свои реакции как минимум неразумно.
А вот все остальное – более сложные вещи (расчеты активных интермедиатов, необычных молекул, возбужденных состояний, и тем более переходных состояний и путей реакции, спектров, термодинамических функций, производных очень тяжёлых элементов со дна Периодической таблицы, поисков необычных связей с помощью модной теории Бейдера, и т.д.) лучше доверить хорошему коллаборатору из класса профессионалов (тех самых, которые затейники, пусть пользу приносят какую-то) и не ломиться в эти двери по принципу: нет таких рек, которые не могли бы повернуть большевики. Будет такой же конфуз как с реками, которые всё текут, куда текли, дошлифовывая истлевшие кости незадачливых большевиков. Там всё сложно, и нужно хорошо понимать, что и как делать, фильтруя большой объём литературы и свободно обращаясь с десятком пакетов с не самыми удобными интерфейсами. Сейчас в каждой второй статье усердно считают координаты реакции с переходными состояниями, и важно делают из этого выводы о механизмах, хотя подавляющее большинство таких расчетов и выводов из них совершенно смехотворны, и ничего не прибавляют к знаниям о механизмах – именно потому что это делают не профессионалы (которые скорее всего, от этого занятия отговорили бы), а сами экспериментальные химики, почему-то решившие, что это такая же рутина, как расчет устойчивой молекулы в основном состоянии.
Добрый день!
Касательно той лекции про фотохимию и разницу обычной колбы от флоуреактора: а что если светодиод (или много светодиодов) водрузить на кончик механической мешалки и включать/выключать светодиод на один оборот мешалки? Тогда и энергия выделяемая светодиодом будет уходить в среду, и выделение света будет в разных точках колбы, одно лишь понять, как эту гирлянду соорудить. Или с этим все равно будут неизбежно проблемы?
АЧ: Добрый вечер,
Немного не понял, Вы сами собираетесь заняться этими исследованиями? Ну так тогда и думайте над реализацией идеи. Если получится, с удовольствием включу получившуюся работу в какой-нибудь из будущих апдейтов по теме.
Иммерсивные источники света это не новость. Именно так делают в обычной фотохимии – там всегда источник внутри, в кварцевом или пирексном пальце, вокруг еще рубашка, потому что надо охлаждать. Очень громоздко, легко ломается, дико дорого, и при этом все равно производительность небольшая – работают те самые законы фотохимии и материальность света.
В фотокатализе будет то же самое, потому что относительно света это не катализ, света нужно много, если, конечно, хотите делать реакцию не на микромоли, а на что-то осязаемое. А светодиоды, даже очень мощные, сильно уступают по световому потоку хорошим УФ-лампам – просто прикиньте, откуда берутся фотоны там и здесь, и поймёте, что чудес не бывает.
Поэтому те, кто пытаются заставить служить фотокатализ каким-то практическим целям, а не только заполнению гламурных журналов мутными статьями, и городят эти якобы поточные реакторы (поток в них стоит сутками неподвижно), с десятками метров тонкой трубки, обложенной со всех сторон тысячами светодиодов – только так удается доставить до реакционной смеси какое-то разумное число фотонов.
Но наука тем и хороша, что ждет новых идей. Так что дерзайте. Успехов!
Добрый день! Благодарю за ваш труд.
Возник вопрос по R,S номенклатуре данного примера. По первому слою понятно – Cl>C>C, второй слой фенила CCC, а слой гидроксиметила OHH, по сумме выигрывает фенил, однако у Вас старшинство у него.
http://orgchem.avchem.ru/intro_concentr/stereochemistry/rsnomenclature/
АЧ: Добрый день и спасибо,
Но вопрос Ваш я не понял, а точнее, делаю вид, что не понял. Очень стараюсь, чтобы было правдоподобно: прочитал и не понял, что же тут написано. Про какую-то сумму, что за сумма, где сумма, куда сумма, что нужно суммировать и зачем. Не понял. Гидроксиметильная группа в номенклатуре КИП безусловно старше фенильной по второму слою, потому что кислород старше углерода, и всё. Один кислород старше ста углеродов, если бы мы могли представить себе такое количество углеродов в слое. Старше любого количества углеродов. Так устроена номенклатура и только так она работает однозначно в подавляющем большинстве случаев. За это в номенклатуре отвечал Владимир Прелог, нобелевский лауреат и один из самых дотошных учёных в истории химии – он специально придумывал все возможные сценарии, чтобы убедиться, что номенклатура с его именем будет работать всегда и однозначно.
Здравствуйте!
Можете добавить в главу/раздел Х Природные соединения – Алкалоиды и Пурины.
Здравствуйте!!
Ваше предложение очаровательно во всех отношениях! Могу ли я добавить? А где я её, главу или раздел, возьму? На этом сайте всё написано, записано, нарисовано и свёрстано мной. Новая глава по любой теме – три-четыре месяца работы в лучшем случае. Где я их возьму?
Поэтому, уж простите, но я сам буду решать, что мне важнее написать в первую очередь, а второй может и не быть. Я вот углеводы мечтаю доделать, а там процентов на 70 все готово, но нужно еще пару месяцев. Так и висит.
К тому же алкалоиды – понимаю интерес и сам когда-то с него начинал ещё в школьные годы – это безразмерная совершенно тема с современной химии, алкалоидов сейчас известны сотни только типов. Что из этого выбирать будем? Как сто лет назад, понятно что? Нет уж, увольте, я не хочу чтобы сайт этот прикрыл роскомхрензнаетчто за то, что здесь упомянуто нечто такое, что упоминать нельзя.
С пуринами ситуация проще конечно. Можно подумать, но ведь эти соединения и так описаны во всех мыслимых местах от химии до биохимии. Не знаю, может когда-нибудь я с ними попробую разобраться хотя бы в контексте ароматичности.
Андрей Владимирович, добрый вечер!
Почему при нитровании N-ацетиланилина в уксусном ангидриде возрастает доля орто-изомера? Орто-эффект начинает работать, или другая причина есть?
Заранее спасибо!
АЧ: Добрый вечер,
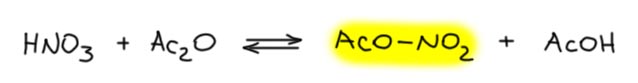
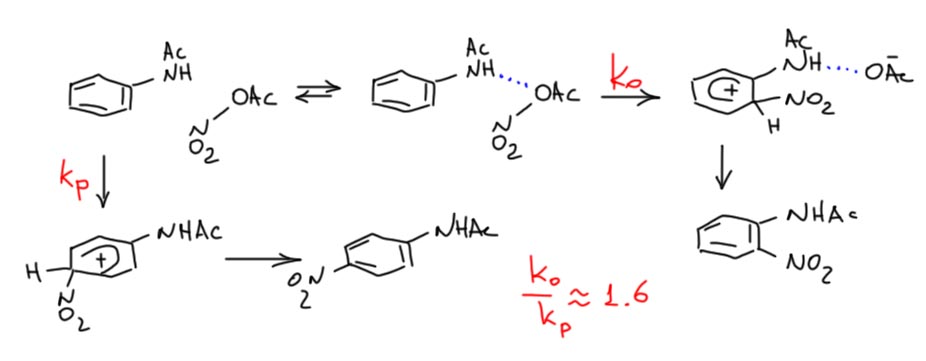
Вопрос Ваш касается одного из самых мутных разделов органической химии – электрофильного ароматического замещения. Нитрование открыли ещё в начале 19-го века, и вроде бы это одна из самых исследованных реакций, ведь Ингольд и Хьюз именно на неё переключились после великих свершений в алифатическом нуклеофильном земещении и эдиминировании. И с тех пор много кто туда совался, но тема как была мутной, так и осталась. Можно себя утешать, что в других ароматических замещениях – сульфировании, галогенировании, Фридель-Крафтсах всех мастей – дела ещё хуже, а хуже уже некуда.
Механизмы ароматического замещения описаны только в самых общих чертах, которые слабо помогают что-то осмысленно предсказывать. Поэтому в этой области так много мифов и откровенной лажи – никому не хочется все это разгребать, ведь это ужасно скучно, да строго говоря и инструментарий пока довольно примитивный, чтобы ожидать каких-то серьёзных прорывов.
Особенно это касается орто-пара соотношения при изменении реагента. Начнём с того, что термин “орто-эффект” никакого смысла не имеет, если не ясна природа этого эффекта, а она в разных реакциях разная. Это может быть обычная стерика, действие индуктивного или полевого эффекта заместителя, или тот или иной направляющий эффект (за счет координации через противоион, водородной связи или чего-то ещё).
Вот пример ацетанилида, хорошо известный. Обычной нитрующей смесью он нитруется преимущественно в пара положение (по разным данным и с разной нитрующей смесью орто изомера там от 5% до 10-15%. Это может означать, что в таких условиях ацетиламино-группа протонирована, причём не по азоту, а по карбонильному кислороду, и это деактивирует орто-положение к электрофильной атаке. Косвенно это подтверждается данными статьи Шофилда и сотр, откуда все эти результаты и идут.
Теперь смесь азотной кислоты и уксусного ангидрида (запрещён в Российской федерации). Во-первых, азотную кислоту для этой смеси всегда берут безводную, свежеперегнанную в вакууме с смеси с концентрированной серной. В такой смеси, как считают, обратимо образуется ацетилнитрат, являющийся нитрующим агентом. Это слабый нитрующий агент, но мы не можем быть уверены (вернее, можем быть уверены в обратном), что он образуется количественно, и что в смеси не остаётся азотная кислота, то есть более сильная протонная кислота, чем уксусная.
В реакции с ацетанилидом образуется (по данным статьи Шофилда) смесь 77% орто-изомера и 23% пара изомера. Причем реакция нитрования почти на пять порядков быстрее, чем нитрование в серной кислоте – это окончательно показывает, что там нитруется протонированная форма, а здесь непротонированная – здесь нет действительно сильной кислотности, азотная кислота намного слабее серной, а здесь ее еще и остатки. В этом месте мы в первую очередь задаем вопрос – можно ли называть такую реакцию обладающей значительной орто-селективностью. На мой взгляд, нет. Потому что если бы реакция была совсем неселективной, то орто-изомера получалось бы 66%, а пара – 33% (по числу положений, статистическое соотношение) – то есть 2:1. А в этой реакции 77:23 = 3.3, иными словами, селективность мизерная, перераспределилось всего 10% от пара к орто. Небольшое превышение орто над статистическим соотношением можно попробовать объяснить немного более выгодным путём, в котором между нитрующим агентом и заместителем возникает водородная связь – это чуть-чуть улучшает энергетику такого пути,
и надо весьма трезво понимать, что такой маленький эффект может вызвать совершенно ничтожный вклад – около 1 ккал/моль или даже меньше. Понятно, что как-то пытаться прояснить эту проблему означает пытаться что-то моделировать с точностью лучше 1 ккал/моль – совершенно безнадежная затея для достигнутого уровня теоретической химии. Я в таких случаях всегда говорю: приходите лет через 200, тогда что-то прояснится. Мораль отсюда простая – мы в химии любим давать красивые объяснение эффектам, которые нам кажутся значительными, а на самом деле весьма малы, почти ничтожны, забывая о том, что уровень нашего понимания химии до сих пор весьма приблизительный – как я люблю повторять по делу и без дела: химия – весьма молодая наука, и уровень понимания в ней вполне соответствует её нежному возрасту.
Спасибо!!
здравствуйте !задали кр по химии, не могу решить вопросы:
#1 кислотой не являются оба вещества
1) NaOH и HNO3
2) HNO3 и HCI
3) HCI и Fe2O3
4) NH3 и HCI
5) K2CO3 и HCIO3
#2 установите соответствие
А) NAHCO3
Б) H2SiO4
В) O3
Г) FeO
1) оксид
2) простое вещество
3) соль
4) кислота
АЧ: Дорогая Милана, добрый день и простите меня за то, что не смогу Вам помочь. Ваш вопрос по школьной химии, а это совершенно особый вид химии, в котором я совершенно не разбираюсь, даже не могу понять смысл приведенных Вами вопросов. Здесь мы занимаемся обычной химией, которую проходят в университетах. Школьная химия настолько далеко ушла за последние 10-15 лет от обычной химии, что мы, несчастные преподаватели университетов, совсем ее перестали понимать, и даже не пытаемся. Поэтому Вам стоит поискать специалиста именно по школьной химии. Я знаю, что такие есть и иногда им завидую.
Еще раз прошу меня извинить.
Добрый день!
А возможна ли реакция Манниха в таких условиях, где вместо вторичного амина берут замещённый амид?
АЧ: Реакция Манниха – это целый мир. В общем это азотистый аналог альдольной конденсации, и насколько легко обобщается альдольная конденсация, настолько же и еще гораздо шире – реакция Манниха. Поэтому вариантов реакции Манниха в современной химии бесчисленное количество. Приэтом мы должны понимать, что принципиально возможно, а что совсем нет. В реакции Манниха электрофилом всегда является иминиевый катион, образующийся из карбонильного соединения и азотистого нуклеофила, в классике вторичного амина, но возможны варианты. Иминиевая соль находит себе второй нуклеофил – обычно это донорный олефин (енол, енамин, эфир енола и т.п.) или сильнодонорная ароматика и дает продукт – основание Манниха, у которого дыльше бывает еще и своя весьма затейливая судьба. Получается такая могучая комбинаторика, которую и за двести лет не расхлебаешь к вящему удовольствию органиков – не реакция, а сокровищница.
Если мы полумаем, как можно в реакцию Манниха пристроить амид, то вариантов будет два – для образования иминиевого электрофила и как второй нуклеофил. Первый вариант кажется мне крайне маловероятным – амиды это крайне слабые нуклеофилы, хотя есть и способы их активировать, но иминиевая соль у которой на азте висит с одной стороны карбонил, а с другой фактически карбокатион (азакарбениевый ион) мне представляется весьма нестабильной и больших шансов на образование в реальных условиях не имеющий.
Другое дело если иминиевая соль, полученная обычным образом из вторичного амина и кабонильного соединения найдёт себе второй нуклеофил в амиде (или скорее его депротонированной или металлированной форме) – это легко себе представить и я думаю, что примеры в литературе найти можно. Образовываться должны при это гем-диамины, ацилированные по одному азоту (или же моноацилированные аминали) – ткие соединения известны, они весьма реакционноспособны, и могут иметь возможности дял продолжения реакции. В этом всегда и достоинство и сложность реакций Манниха – поди попробуй остановить их на конкретном продукте, им всё куда-то дальше хочется.
Копирую вопрос, который почему-то затерялся где-то ближе к концу потока.
“Здравствуйте! Подскажите, не знаете ли вы какого нибудь ресурса или чего-то, где можно почитать про осмоление? Чем может быть вызвано, как избежать, какое-то пояснение процессов?”
АЧ: Добрый день. Вопрос, конечно, интересный. Почти то же самое, что зайти на кулинарный ресурс высокой кухни и спросить, отчего вместо изысканых блюд по вашим рецептам выходит какая-то дрянь, подгорелая, пересоленная, воняет жженой резиной, вылили свиньям, свиньи пятачки воротят, объявили голодовку, требуют обратно чан любимой ботвиньи. В чём дело? Не знаете ли вы какого-нибудь ресурса или чего-нибудь ещё, где можно прочитать про несъедобную стряпню? И вообще – как отличить хороший вопрос от вопроса-ни-о-чём? Хороший вопрос был бы таким: вот есть такая-то конкретная реакция, там произошло осмоление, почему и как уменьшить. Вопрос-ни-о-чём ищет основы Общей Теории Осмоления (ОТО) и Специальной Теории Осмоления (СТО) специальный ресурс по осмолению (WikiTarring?), Энциклопедию Осмоления, Введение в Общее Осмоление, и так далее – не много не мало. Проще было проигнорировать, потому что химия конкретна и основана на эксперименте, а болтовня ни о чём в ней ничего не даёт. Но всё же – осмоление это действительно главная проблема химии. Химия вообще выросла из осмоления, и по статистике, валовой химический продукт на 90% состоит из продуктов осмоления. Давайте вкратце рассмотрим. Нальём воды на мельницу осмоления.
Конечно, в химии всё же проще, чем в кулинарии: там это искусство и мастерство, которые не пропьёшь, но и не купишь. В химии есть всякие закономерности, правила, и так далее, с помощью которых можно хотя бы приблизительно действовать целенаправленно и получать ожидаемое там, где это в принципе возможно. Мастерство тоже нужно, но не везде и не всегда, скорее опыт. А вот что точно нужно, так это знания.
Что такое осмоление? Это такое узаконенное словечко для обозначения побочных продуктов в реакциях. Обычно не просто побочных продуктов, а таких неприятных побочных продуктов, тёмно-окрашенных, не кристаллизующихся, трудноотделимых, часто являющихся признаком безнадёжно или частично испорченного синтеза. Такие свойства обычно свойственны органическим соединениям большой молекулярной массы с молекулами неопределенной формы и представляющими собой смеси похожих соединений с разной молярной массой – такие никогда не кристаллизуются, не перегоняются, не дают чётких пятен на хроматограммах, не разделяются на компоненты никаким способом. Такие свойства говорят, что скорее всего это так называемые олигомеры – продукты реакции исходных реагентов самих с собой или дополнительно ещё с чем-то. Во многих случаях образование таких пробуктов легко предсказать, если химик понимает химию процесса, и чем лучше понимает, тем скорее сможет справиться с осмолением. У нас на 3-м курсе в практикуме по органике в лабораторном журнале есть хорошее требование (увы, часто игнорируемое) расписывать не только механизм выполняемой реакции, но и прикинуть, какие побочные процессы могут вмешаться в выпоняемый синтез. Тогда мы можем действительно понять, что нас может ожидать, что может пойти не так, и что нужно сделать, чтобы этого было поменьше.
В органике крайне редко бывает так, что реагенты чисто и без альтернатив дают один продукт: захотели, например, присоединение брома к олефину, ожидаем дибромпроизводное, что может быть проще. Делаем реакцию, видим, что вроде всё нормально, бром обесцвечивается, но – что это за дымок? Это HBr. Откуда? Ответ – от верблюда – неверный. Придется серьезнее посмотреть на механизм, увидеть там возможность конкурентного элиминирования. Понять, что как только образовался HBr, немедленно возникла возможность кислотно-катализируемой олигомеризации. А значит и появления карбокатионов, а значит и гидридных сдвигов и перегруппирововок, и так далее – уже глаза разбегаются от множества путей. И когда реакция сделана, начинаем выделять и перегонять одидаемый дибромид, видим, что выход далёк от количественного, а в перегонной колбе остаётся немаленький неперегоняемый остаток, тёмная, мерзкая, дымящая зловонная жижа, и немало. А это элементарнейшая реакция, одна из древнейших, но в галогенировании очень много вариантов развития превращений. Так и в почти в любой другой реакции. Поэтому органическая химия совершенно одержима проблемами селективности и специфичности, и без устали предлагает усовершенствованные реагенты, альтернативные подходы и так далее, но ни один новый метод не решает проблему полностью. Так устроена химия соединений углерода.
Когда чаще всего бывает олигомеризация? Многие сценарии стандартны. Первый – реакции непредельных соединений с электрофилами – вместо присоединения нуклеофила промежуточные карбокатионы присоединяются к следующей двойной или реже тройной свяязи, и понеслась. Непредельные соединения могут быть в смеси изначально или образоваться на месте в результате элиминирования – напрмиер, во всяких дегидратациях. Всё это ещё осложняется скелетными перегруппировками, и получается совсем невозможная каша. Второй стандартный сценарий – реакции карбонильных соединений, все конденсации и так далее – они всегда рекурсивны, потому что на каждой стадии воспроизводятся реакционные центры и никто не мешает идти побочным самоконденсациям, Михаэлям, и свободной комбинации всего этого. Третий сценарий – реакции всякой донорной ароматики тоже с электрофилами, это похоже на реакции непредельных соединений, потому что донорная ароматика очень легко теряет ароматичность (деароматизуется) и дальше играет в обычные игры непредельных соединений. Но и акцепторная ароматика может подвести, например, пиридин и производные часто считают образцово дубовыми молекулами, с которыми можно делать что угодно и никуда они не денутся – но не дадут боги пиридин кватернизовать и поднести даже довольно слабый нуклеофил – кольцо раскроется и вы немедленно окажетесь в химии непредельных карбонильных соединений с множеством путей самоконденсациию Других сценариев тоже немало. Почти в каждой реакции есть какой-то выход на олигомеризации того или иного типа, и надо уметь их отлавливать. Поэтому знание механизма реакции в органике – это не блажь для коллоквиума, а совершенно необходимая вещь для осмысленной работы. В 19-м век механизмов не знали, работали вслепую, и в большинстве реакции получение какого-то желаемого продукта было скорее побочным процессом к общему осмолению реакционной массы – поэтому в ранней химии почти никогда не публиковали выходы. Многие старые работы того времени или вообще невоспроизводимы или воспроизводимы с очень низкими выходами. И как только представление о механизмах стало в химию проникать, появились и селективные реакции, и стали публиковать регулярные методики с выходами, и методики стали воспроизводимы. Только тогда возникает сложный многостадийный синтез, в котором нельзя допускать, чтобы на двадцать пятой стадии всё осмолилось.
Почему происходит осмоление? Есть две группы очевидных причин. Первая – такова реакция сама по себе, осмоление неизбежно. Очень многие реакции особенно ранней химии очень неселективны и дают желаемый продукт с небольшим выходом. В таких реакциях со сравнимыми скоростями идут множество конкурирующих процессов, и в смеси накапливаются продукты всех, в том числе и олигомеризации, которую мы называем осмолением. В таких реакциях обычно есть какой-то более или менеее удобный метод выделения целевого продукта, а всё остальное идёт на выброс. Так часто делают всякие исторические самоконденсации. Уж совсем яркий пример – получение мезитилена самоконденсацией ацетона, этой реакции уже почти 200 лет – добавляем к ацетону немного крепкой серной кислоты и ещё и немного греем – всё к чёрту осмоляется, потому что там много путей самоконденсаций, большая часть из них просто наращивает разветвлённую цепь, и даёт олигомер, смолу. Но небольшая часть превращается в мезитилен – маленькое симметричное летучее соединение, которое можно выгнать из мерзкой жижи и очистить аккуратной перегонкой. Тут именно мезитилен – побочный продукт осмоления. Есть и другие такие примеры, а много примеров не таких ярких, но всё равно похожих тем, что полезный продукт с небольшим выходом получается наряду с большим количеством побочных. Так делают, например, и такие красивые и важные вещества как порфирины – большая часть идет в олигомеризацию и смолу, меньшая в красивый макроцикл, который кристаллизуется или хроматографируется. По мере развития химии таким методам усердно ищут более селективные замены, часто находят, но иногда оказывается, что дешевле со смолой по-старому, чем без смолы по-новому. Тем не менее, современная промышленная химия просто жёстко требует сокращения отходов, и старые методы в ней императивно вытесняются новыми, иначе просто разоришься на утилизации отходов.
Кроме объективных причин (реакция такая, хоть тресни) есть и субъективные причины осмоления – ошибки экспериментаторов, обойдёмся без эмоциональных оценок. В органике вы или работаете над своими исследованиями и делаете что-то новое, в этом процессе без осмоления не обойтись – путь к новой химии всегда идёт через осмоление: пока достаточно всего не осмолите, не найдёте своего. Я думаю, что если бы можно было бы сложить вместе всю смолу, которую произвели органики за время развития органической химии, немного более 200 лет, то получился бы небольшой океан.
Но часто мы делаем какие-то соединения по опубликованным методикам: исходные всякие и так далее, никто не идет в свой путь прямо с самого начала, от элементов. Опубликованная методика должна воспроизводиться, конечно, не прямо точно, но в разумных пределах: если написан выход 80%, а вы получили 70, это нормально, а если 20%, то это плохо. Кто виноват? Почему низкий выход? Толковый химик всегда достает из реакционной смеси все, и взвешивает сумму продуктов, в том числе смолы, чтобы понять, не теряет ли просто в процессе выделения. Если не теряет, то смотрит, сколько вернулось исходного – если много, значит по какой-то причине реакция не пошла, надо задуматься по какой. Если исходное прореагировало, а продукта мало, значит много побочных, в том числе, возможно, смолы. Начинаем думать, в чём дело. Причина первая – плохая методика. Увы, такое бывает, и нередко, причем не застрахован никто. Есть, например, странное поверие, что старые немцы все делали отлично и никогда не лажали. Это заблуждение: немцы такие же люди как все, и в старых работах лажи очень много. То же касается всех остальных без исключения. С опытом приходит некоторое понимание того, где точно заминировано, а где почти точно нет. Любые методики вообще нужно прежде чем делать очень внимательно читать и продумывать – очень часто ошибки (просто забыли что-то очевидное) выявляются имено так. А самое лучшее – посмотреть, пользовался ли кто-то ещё этой методикой, пробив структуру по базам (оставим здесь вопрос, где взять доступ – хотите рабоатть в современной химии, просто ищите место работы, где такой доступ есть) – если пользовался и в работе есть ссылка – получено по работе такой-то – то это уже хорошо. Бывает, что и методику повторят и даже с улучшениями.
Если с методикой все нормально, то проблема у вас. Вы не смогли воспроизвести методику, и знаете об этом: например, написано, что медленно прикапывали три часа, а вы вылили струёй. Упустили температуру (написано держать до 5 градусов, у вас один раз всего выскочило к 20, и т.п.). У вас плохое оборудование, чаще всего плохая мешалка – реакционные смеси надо тщательно перемешивать, и без хорошей надёжной мешалки это невозможно. Мешалка должна перемешивать, а не просто крутиться внизу – вы должны видеть конус перемешивания. Слишком мелкая или гладкая мешалка часто перемешивает только жидкость вокруг себя, а всё остальное остается неподвижно. То же с вязкими реакционными смесями, их вообще трудно хорошо перемешивать. Или со смесями с осадком. Мешалка должна вращаться непрерывно, а не только тогда, когда вы на нее смотрите. Нужно отрегулировать скорость вращения так, чтобы перемешивание не срывалось. И так далее. Если перемешивание плохое, то в месте попадания прибавляемого реагента неизбежно будет его избыток и возможно локальное разогревание, которое вы не увидите, если термометр далеко от этого места – вот там и пойдёт осмоление. Другой источник проблем – качество реактивов и особенно растворителей. В органике обычно используют сухие растворители. Вода – очень плохая вещь во многих реакциях, очень частая причина осмоления: вода создаёт протонную кислотность в электрофильных реакциях, и вызывает полимеризацию. Вода снимает многие защиты, и этого тоже может быть достаточно для побочных реакций, даже если такая реакция пройдёт на несколько процентов. Вода – нуклеофил, создаёт побочные продукты с группами, способными вызвать олигомеризацию. В реакциях с металлоорганикой даже небольшая примесь воды создаст сильную альтернативную основность, которая направит реакцию не туда, куда хотелось. И так далее: мелкие и крупные пакости воды неисчислимы, и там, где есть такая опасность, реактивы и растворители должны быть сухими; а вода умеет прятаться, и ее много не нужно из-за маленькой молярной массы даже небольшая примесь воды в эквивалентах может оказаться сравнимой с загрузками реагентов.
В общем, если не искать теоретических основ осмоления, а просто подумать про механизмы органических реакций, и немного навести порядок в технике эксперимента, то и осмоление больше не покажется бичом богов, уничтожающим выходы.
Можно ли использовать многократно раствор пищевой соды с морской солью в теплой воде для ингаляций при простуде?
АЧ: А Вы что, их смешиваете, соль с содой? Там вроде карбонаты кальция и магния должны выпасть.
Здравствуйте!
Не без интереса прочитал вашу последнюю страницу, посвященную окислителям спиртов, и у меня возникло два вопроса.
Первый вопрос: а как делают TEMPO сейчас из TMP? Неужто и тут зеленая химия во все поля или все же надо брать какой-то окислитель повеселее и смириться с тем, от чего зеленая химия так стремится отказываться.
Второй вопрос: все эти методы замечательно работают, когда растворитель условно инертный, что-то типа дихлорметана (исходные и продукты растворимы, очень удобно), не найдется ли метода, который работает для таких органических веществ, которые растворимы в полярных растворителях типа воды, изопропанола (например, если надо окислить первичный спирт, а с вторичным реакция уже будет идти медленнее)? Ведь даже если, допустим, исходное вещество хоть немного растворимо в дихлорметане и будет реагировать с имеющимся массивом окислителей, вдруг и его производное, тоже имеет плохую растворимость (вследствие разных факторов в уже исходной молекуле) – тогда у нас будут проблемы с выделением. Нет ли?
АЧ: Добрый день. С интересом прочитал Ваш вопрос. По поводу TEMPO. Во-первых, не бывает ничего совершенно зелёного. Курс на озеленение не значит, что мы от незеленого скачком окажемся в зеленом. Предлагается медленно, но верно двигаться в этом направлении, оценивая степень озеленения с помощью разных метрик. То есть противопоставляется не зелёное-незеленое, а менее зелёное и более зеленое. Чем зеленее тем лучше. И так далее. И в этом смысле TEMPO действительно без вопросов выигрывает у остальных методов. Это каталитический метод, и TEMPO используют в количестве нескольких мольных процентов, а стехиометрический окислитель можно подбирать и здесь есть простор для дальнейшего озеленения. Для окисления TMP в TEMPO используют разные перекисные окислители, а они всегда восходят к перекиси водорода, а это один из самых зеленых реагентов по всем метрикам. Плюс ещё есть возможность использовать иммобилизованные версии, над этим работал один из отцов зеленой химии Роджер Шелдон, и там в совершенно буквальном смысле всё схвачено. В общем, это перспективная химия, хотя и довольно капризная и сырая.
Что касается воды. Здесь всё не очень весело. Причина понятна – большинство методов окисления спиртов предполагают электрофильную активацию, а вода или другой нуклеофильный растворитель банально будут конкурировать со спиртом за реагент. А поскольку растворителя по молям всегда на порядки больше чем растворенного вещества, конкуренция будет невыгодна для спирта, даже если он может предъявить большую нуклеофильность. Изопропанол как растворитель очевидно будет такой же проблемой. Именно поэтому большинство методов окисления действительно использует инертные ненуклеофильные или малонуклеофильные растворители типа дихлорметана, бензола, ацетона, ну или хотя бы ацетонитрила.
Но в общем дело не безнадёжное. Можно окислять и в воде, и в достаточно нуклеофильных растворителях. ТОт же метод Мофата может использовать не только ДЦК, но и водорастворимый карбодиимид, очень популярный в сшивке пептидов. Из хромовых окислителей, Джонс фактически работает в водной среде – там сначала ацетон, но он быстро становится водным. Дихромат пиридиния работает в ДМФА, правда медленно, но с этим будет проблема в полярных нуклеофильных растворителях всегда. Предшественник Десса-Мартина, IBX прямо специально применяют в полярных раствоителях, потму что в неполярных он не растворяется, и реакции очень часто ведут в ДМСО. Те же методики с TEMPO тоже обычно водные, хотя они двухфазные и субстрат находится в органике, но там нет принципиальных проблем, и есть место для творчества. Есть еще всякая каталитическая рутениевая химия, я ее здесь не разбирал, это скорее для курса по переходным металлам, я планирую туда однажды добавить лекцию про каталитическое окисление, но это точно не в этом году.
В общем, варианты есть.
Здравствуйте!
Я как аспирант не могу не поинтересоваться: а к тем слайдам, которые вы выложили на главной странице, не будет какой-то записи лекций? Ведь смысл смотреть на картинки без интерактивного пояснения. Смысл же так теряется, кмк.
АЧ: Если Вы аспирант, и Вам так интересно, что было в этих лекциях, могли бы их и посетить, и послушать. Но я вообще не уверен, что Вы имеете в виду мои лекции, потому что никак не могу понять, что за слайды и на какой главной странице я выложил. Поскольку я на главной странице точно ничего не выкладывал, то видимо это не я, и страница какая-то другая. Потрудитесь объяснить, что конкретно Вам хочется, и какой смысл Вы опасаетесь потерять. И тон ещё желательно менее императивный выбрать для общения, это так принято в тех местах, где ещё не успели потерять здравый смысл, а это единственный вид смысла, потерять который действительно чрезвычайно опасно.
>И тон ещё желательно менее императивный выбрать для общения
Не имел в виду ничего плохого, извините.
Я про http://orgchem.avchem.ru/organic-chemistry-21st-century/
АЧ: Да, хорошо, понятно, что на этой странице слайды не выложены, а ссылки на лекции со слайдами есть в меню сверху. Записи лекций выкладывает компания Teach-in у себя и на ютубе. Всего лекций было десять, и они еще не все выложены ни слайдами, ни видео. Надеюсь, что они доделают остальные в январе, а я тоже буду стараться довыложить и слайды.
Громадное спасибо! С большим интересом сейчас буду смотреть эти лекции и ждать продолжения так же, как и ответы на многие вопросы здесь…
С праздниками!
АЧ: Хорошо. Спасибо за интерес. Я задерживаю последние три лекции, потому что хочу еще сделать краткое описание слайдов текстом, а это быстро не получается. Но обязательно получится.
А, всё, нашёл ссылку. Извините за беспокойство.
Здравствуйте, Андрей Владимирович, подскажите, пожалуйста, есть мезо-2,3-дибромбутан при его обработке йодидом калия в ацетоне получается транс-бутен-2, а если взять мезо-1,2-дидейтеро-1,2-дибромэтан, то в результате получим цис продукт. Как такое происходит учитывая, что на ключевой стадии отличий нет?
АЧ: Добрый вечер, Кирилл,
Спасибо за вопрос. Это очень интересная история, такая классика, работы Сола Уинстейна, но с продолжением. Постараюсь в ближайшие дни разрисовать, что там происходит, на страничке Ответы.
АЧ: Выложил ответ на страничке Ответы.
Спасибо вам большое, очень познавательно!
АЧ: Да, хорошо что спросили. Занятная химия, стоило немного разобраться.
Доброго времени суток!
Подскажите, почему график зависимости потенциальной энергии (например, бутана) от величины торсионного угла выходит не из начала координат? И в целом он смещен относительно оси абсцис.
Это какой-то минимум внутренней энергии?
АЧ: Добрый день,
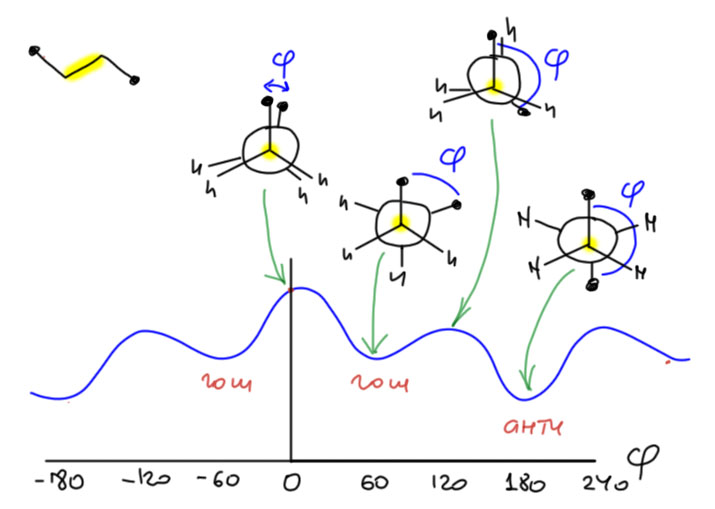
Не вполне понятно, о каком графике идёт речь. Если рассматривать конформационный анализ бутана, то речь может идти о зависимости внутренней энергии конформаций от торсионного угла по связям, допускающим внутреннее вращение. В бутане таких связей три, но интерес представляет только средняя связь С-С, потому что только по ней образуются разные конформеры, которые и определяют стереохимическое поведение бутана. Обычно это изображают в виде графика, по абсциссе как раз этот самый двугранный угол, который изменяется от 0 до 360, или от -180 до 180 с обычной для угловых величин периодичностью. Угол ноль соответствует (обычно так, но любой может выбрать другое начало координат) заслоненной конформации с двумя метилами друг напротив друга – это самая невыгодная конфигурация, и ей соответствует максимум. То есть это еще и переходное состояние для перехода из одного гош-конформера в другой. Дальше сканируем угол, и получаем при 60, 180, 300 заторможенные конформации, которым соответствуют гош, анти, гош конформеры. Это минимумы, и это реальные молекулы, находящиеся в тепловом равновесии в реальном веществе бутане. Соответственно при 0, 120,240 имеем заслоненные конформации с максимумами энергии – это переходные состояния для превращения гош-анти, анти-гош, гош-гош.
Вот собственно и всё. Отсюда и имеем понимание того, что реальный бутан это равновесная смесь гош и анти конформеров, в которой преобладает анти. Чтобы знать константы равновесия и соответственно доли конформеров, нужно знать конкретные величины свободных энергий, и это несколько сложнее, чем просто внутренние энергии, но конкретные величины мало кому нужны.
В связи с этим не вполне понятно, о каком смещении относительно оси абсцисс идет речь. Нас интересуют энергии, они по ординате, и энергии это всегда относительные величины, а выбор уровня всегда за вами, какой хотите, такой и выбирайте, потому что реально вы всегда имеете дело с разностями энергий.
Имелось в виду, что в минимуме, который соответствует анти-конформации, потенциальная энергия не равна 0, а имеет какой то запас энергии, из-за чего, видимо, график находится несколько выше горизонтальной оси
АЧ: А с чего энергия должна быть равна нулю и что это вообще значит? Энергии всегда относительны, могут быть хоть отрицательны, хоть положительны, и значение имеют только разности энергий. В конформационном анализе нас интересуют разности энергий конформеров, из этого мы поймем их соотношение в равновесной смеси, или же по другому заселенности конформеров, хотя для этого нужно было бы иметь не оценки внутренних энергий, а термодинамические функции энергий Гиббса, но для оценки сойдут и энергии, в этих вещах всё весьма приблизительно.Ещё нам могут понадобиться величины барьеров превращений конформеров, для оценки скорости превращания, и здесь уже все очень приблизительно. Сами же величины энергий будут разными и сильно в зависимости от метода расчета или экспериментального определения, потому что в работе с энергиями всегда есть произвол выбора точки отсчета, и это ровным счетом никого не волнует, потому что не принято сводить на одной энергетической диаграмме (или в термохимическом цикле) величины, полученные разными методами, а когда поневоле так делать приходится, то сначала правдами и неправдами сводят их к одному уровню отсчета. В термохимии это едва ли не главная проблема – как это сделать корректно – из-за которой многие вещи так и не получается свести вместе. Но в конформационном анализе такой проблемы обычно нет, потому что профили энергии берут из расчета молекулярной механикой или квантовой химией, и в процессе расчета осуществляют сведение к одному уровню отсчета. .
Понятно, большое спасибо!
Обычно считается, что метансульфоновая и трифторметансульфоновая кислоты не проявляют ни свойств окислителей, ни свойств электрофилов. Тем не менее, в реакциях, с которыми мне приходится иметь дело, это не совсем так. Мочевина при нагревании с сульфоновыми кислотами даёт много весёлой чёрной смолы, в то время как трифторуксусная кислота к такому не приводит. Однако, ключевой стадией исследуемого мной процесса является кислотно-катализируемое превращение, и сила кислоты играет решающую роль. В частности, в классическом литературном варианте реакцию проводят в токе сухого газообразного хлороводорода. Однако, не менее важен и температурный режим, поскольку необходимый мне продукт получается только при температурах выше 100 °С, а трифторуксусная кислота при атмосферном давлении кипит при 72 °С.
В связи с этими обстоятельствами у меня возникает вопрос прикладного характера. Какие есть высококипящие сильные кислоты, не способные проявлять свойств окислителей или электрофилов? Желательно, чтобы они были сильнее трифторуксусной хотя бы по константе кислотности в растворе, но можно и как среду рассматривать. Ещё желательно, чтобы можно было с ними работать в стекле, но это уже в некотором роде мечты.
АЧ: Здесь, возможно, есть некоторое, как минимум, терминологическое неоразумение. Высокая кислотность это и есть один из источников электрофильности. Если в молекуле есть пи-связи, то их протонирование приведет к появлению электрофильных центров карбокатионного типа, более или менее стабилизированных, и это дальше ведет к олигомеризации, что и воспринимается как осмоление. Можно сделать вывод, что в Вашем случае это проявляется при увеличении кислотности. MeSO3H и тем более TfOH кислоты довольно сильные, даже в растворе. Как среда TfOH – суперкислота, MeSO3H малость недотягивает, но это очень сильная кислота. Трифторуксусная кислота сильной не является, она протонирует пи-связи только если образующийся карбокатион очень хорошо стабилизирован, иначе мы имеем общий кислотный катализ, которого для некоторых реакций недостаточно. Так что ничего необычного в описанных Вами явлениях нет, и никакой специальной электрофильности от сульфокислот не требует. Да у них и нет ничего. Сульфонилирование может быть электрофильной реакцией, но без дополнительной активации это неактуально.
Если дело в температуре кипения, попробуйте проводить реакции в закрытом сосуде. Маленькие пузырьки хорошего качества, к таких сейчас любят ставить реакции, можно перегревать на сорок-пятьдесят градусов выше температуры кипения. Для загрузок поольше есть такие фирменные толстостенные колбы с завинчивающейся тефлоновой крышкой – тоже можно перегревать на несколько десятков градусов (только экран поставить надо, они иногда взрываются).
Или можно поискать перфторпропионовую кислоту, или даже масляную, но это та еще дрянь. Если реакция терпит воду, то добавлене пнебольшого количества воды к трифторуктусной дает азеотроп, который кипит при 105 градусах.
Если не хватает кислотности, к трифоторуксусной можно добавить немного сильной кислоты, даже серной, или MsOH/TsOH/TfOH. Посредине между трифторуксусной и сульфоновыми все равно нет по кислотности ничего доступного.
Продолжая тему антиароматичности. На мой взгляд, было бы неплохо дополнить рассуждения об “антиароматичности” малеимида ещё и комментарием по поводу “антиароматичности” парабановых кислот (1,3-имидазолидин-2,4,5-трионов). В них, как ни считай электроны, всё равно получится 4n, а значит антиароматика. Но они плоские и дубовые. Понятно, что опять кросс-сопряжение всё рушит, никакой циклической сопряжённой системы там нет, но как пример для образовательных целей – куда более простая и понятная система, чем малеимид.
АЧ: Добрый день, Олег,
А что нового мы узнаем, если рассмотрим парабановую кислоту? Я напомню, что проблемы антиароматичности в контексте малеинового ангидрида нет как таковой – экзоциклические пи-связи, хотя бы одна, уничтожают саму основу ароматичности или антиароматичности: электронное строение таких молекул не имеет отношения к аннуленам Хюккеля, и говорить не о чем. Но есть некая производная проблема – а что если в делокализации можно усмотреть формально ароматические или антиароматические граничные структуры – не влияет ли это как-то на свойства таких молекул. Именно это я разбирал на страничке про тропон и малеимид. Малеимид – очень удобное соединение для такого разговора, потому что имеет хорошо известную и вроде бы аномально высокую реакционную способность в реакции Дильса-Альдера – не объясняется ли это участием антиароматической дестабилизации? Я разобрал тот кейс и показал, что есть более простое объяснение – циклическое напряжение. ДЛя меня тема снята, пока не будут новые данные, например, кто-нибудь умудрится оценить вклад напряжения и покажет, что этого недостаточно для объяснения. Не знаю как, но в принципе, такое исследование можно было бы ожидать. Выйдет, разберёмся.
Парабановая кислота – штука занятная. Конечно, у нее есть 4е граничная структура. Но что в этой молекуле аномального? Относительно высокая кислотность? Но это наверняка можно объяснить просто удачной делокализацией. И что ещё? Думаю, что ничего особенно обобенного там нет. И вообще, этих молекул, у которых найдётся 4е-граничная структура десятки. ДЛя того, чтобы получился полезный разбор нужны более веские основания – всё же что-то сильно необычное в структуре или реакционной способности.
Андрей Владимирович, здравствуйте!
Подскажите, каков механизм декарбонилирования, когда мы в сложноэфирной конденсации с эфиром щавелевой кислоты продукт нагреваем с каталитическим количеством кислоты?
АЧ: Добрый вечер. Ох, противная это история, хотя забавная, и немного забытая. Кислота там не нужна. Декарбонилирование происходит просто при нагревании – это старая фишка ещё со времён главного классика кетоэфирной химии Вислиценуса-отца. Тогда все перегоняли без вакуума, и проcто нашли, что кетоэфиры из конденсации с диэтилоксалатом, уже выделенные и очищенные от кислот и оснований, при перегонке чисто выбрасывают CO где-то выше 150 градусов. Это получается удобнее, чем конденсировать с диэтилкарбонатом, потому что диэтилоксалат намного электрофильнее.
Это просто термическое разложение, причем однозначно доказано, что карбонил выламывается из карбоксила, а не кето-группы, а алкокси переезжает на карбонил. Сейчас мне лень, завтра может схему нарисую.
Спасибо, по возможности, нарисуйте, пожалуйста, схему…
АЧ: Ответил на странице Ответы.
Второй вопрос: насколько сильно кросссопряжение может мешать протеканию реакции Манниха, например, если взять такой дизамещённый алкен, чтобы с одной стороны был метилен, с другой одновременно и донорная группа (амин какой-нибудь или этокси-группа), а с другой акцепторная (карбоксил, карбонил и т.д.)? Можно ли как-то нивелировать действие одной из этих групп (например, донорной, чтобы реакция Манниха шла)?
АЧ:
Простите, но я ни черта не понял. Ещё раз привлекаю внимание к тому, что химия конкретна. Ни одна просто реакция не мешает другой просто реакции. Если есть проблема хемоселективности, надо ее решать конкретно. И что за кросс-сочетание?? Что с чем сочетается? Олефин это не вполне субстрат для кросс-сочетания, нужна или уходящая группа, или магний, бор, олово, цинк. Все это вполне совместимо с кратными связями, как и уходящие группы разные. Манних – это фактически та же альдольная конденсация, только вместо карбонила имин или иминий – опять непонятно, что тут за проблема с кросс-сочетанием. Реакция Манниха, если это не классический кейс с иминиевой солью формальдегида, вообще реакция весьма сложная, с кучей альтернативных путей, там каждый случай надо отдельно разбирать. В общем, если есть конкретный кейс, можете мне на почту прислать структуру, вызывающую восросы, тогда посмотрим.По позднему времени мне померещилось кросс-сочетание вместо кросс-сопряжения. Извиняюсь. Но я пытаюсь придумать случай, где в Маннихе может попасться кросс-сопряжение, но что-то не получается. Со стороны какого из реагентов – нуклеофила (а это в Маннихе или енол, или енамин, хотя возможны более экзотические случаи) или электрофила (а это в Маннихе всегда иминиевая соль или активированный водородными связями или координацией по кислоте Льюиса имин)? Право, поточнее опишите проблему, а я постараюсь впредь быть внимательнее.
Чёрт, я тоже ошибся и перепутал Манниха с Михаэлем. В общем присоединение к 1,4-непредельному карбонилу, только так, чтобы в 3 положении еще донор торчал
АЧ: Ну и пусть себе торчит. Положение 3 – это бета? Уточнение нелишнее, потому то у многих акцепторов Михаэля акцепторы неуглеродные – нитро, сульфонил и т.п. Если это бета, то есть тот углерод, по которому идёт атака нуклеофила, то во-первых, здесь нет кросс-сопряжения. Если там мезомерный донор, то получается идеальная цепь сопряжения донор-акцептор, стабилизирующая исходный олефин, а любая стабилизация исходного уменьшает реакционную способность. Вообще, реакция Михаэля очень непроста, и в ней часто случаются всякие неожиданности. Нужно очень внимательно всегда выписывать механизм и смотреть, хорошо ли стабилизированы интермедиаты, и нет ли альтернативных путей. Поэтому вы нечасто найдёте замещённые хоть по альфа, хоть по бета-положению акцепторы Михаэля в реальных реакциях. Найдёте, безусловно, и есть очень важные частные случаи, но в большинстве работ все будет крутиться вокруг простых винилов с одним акцептором. Еще очень существенную роль играет стерика, на бета-углероде особенно, потому что присоединение нуклеофила обратимо, и в случае стерических препятствий, а уже один метил не подарок, константа равновесия становится плохой, и если нет быстрого продолжения, выводящего интермедиат из равновесия, то всё будет скверно с реакцией Михаэля. Реакция Михаэля это ведь такой общий шаблон реакции, под которые можно подобрать практически бесконечное количество реальных пар реагентов. У нас есть только самые общие идеи, как идёт реакция – мы понимаем обратимость первой стадии, закономерности стабилизации интермедиата, видим продолжение, которое выведет продукт из равновесия, и вот должны в каждом конктретном случае все это собрать и призадуматься – что тут важнее. В наше время очень нелишне было бы посчитать структуры исходных (это даст представление о масштабе стерических проблем) и интермедиата (это даст лучшее понимание стабилизации – вдруг мы что-то на бумаге неправильно оцениваем), тогда будет немного легче понять, отчего это задуманная реакция, вроде вполне банальная, ну никак идти не хочет или идёт с выходом 4 процента
годовых. Химия конкретна.Хорошо, если конкретнее, то вот:
https://doi.org/10.1016/j.tet.2011.09.034
5-метилиденгидантоин, по одну сторону двойной связи слабенький акцептор, по другую слабенький донор.
Реакция Михаэля не идет просто так и только добавление кислоты Льюиса в отдельных случаях исправляет ситуацию. Получается, что можно взять уксусную кислоту или добавить какую-нибудь конкретную кислоту Льюиса типа бромида меди и она поможет присоединить нуклеофил, стягивая на себя электронную плотность, так?
АЧ: В реакциях Михаэля часто применяют активацию и кислотами Бренстеда и кислотами Льюиса. В данной статье я вижу только один пример сопряжённого присоединения – цианид. Реакцию катализируют модным трифлатом меди в водной среде, и все идет неплохо, причем обращаем внимание на то, что енолят, образовавшийся при присоединении цианида успевает присоединиться к второй молекуле метиленгидантоина. То есть не такая уж плохая реакционная способность. Может быть не выдающаяся, как у всяких заправских акцепторов Михаэля, но и неплохая. И в чём тогда проблема?
И кстати CuBr2 лучше не применять. Это неплохой бромирующий агент, и если не сам исходный гидантоин, то промежуточный енолят он пробромирует с удовольствием. Берите инертные кислоты Льюиса.
Здравствуйте! У меня сразу два вопроса. Второй задам отдельно.
Используется ли озонолиз сейчас или это такая же старая химия как и действие на спирты йодоводородной кислотой? Насколько сложно проводить данную реакцию, препаративна ли она?
АЧ: Озонолиз это не старая химия, это в основном химия послевоенная, когда Криге, чудом уцелевший на Восточном фронте, взялся за исследваоние механизма и разобрал там всё. Потрясающе интересно. Я давно мечтаю найти на неё время и аккуратно разобрать. И это невероятно популярная реакция в современном синтезе, потому что она удивительно селективна и можно прогрызть правильную двойную связь в очень сложной молекуле.
Оставим пока, но я поставлю себе в планы с серьёзным приоритетом.
Здравствуйте я меня такой вопрос , какая химя может оставить на метале разводы несмываемые, я переделываю старую дверь и за этого задаю вопрос.
АЧ: Обращаю внимание задающих вопросы, что это образовательный сайт, и вопросы принимаются только имеющие отношение к химии как науке и предмете изучения. Я не против корректных бытовых вопросов, но только в том случае, если можно понять, что конкретно вызвало проблему. Что за дверь, из какого металла (может быть она у Вас титановая или золотая), и что может на неё попасть.
Здравствуйте, Андрей Владимирович!
Почему цианоборгидрид натрия селективно восстанавливает имины, но плохо восстанавливает альдегиды и кетоны?
АЧ: Приветствую Вас,
Это отличный вопрос. Если говорить коротко, то конечно восстанавливает и обычную карбонильную группу, в условиях общего кислотного катализа – этот реагент не расщепляется в слабокислой среде при рН около 3, что и позволяет использовать мягкую кислотную активацию восстанавливаемой группы. Поскольку реагент намного более слабый донор гидрида по сравнению с просто борогидридом, для восстановления очень важна такая активация. И тогда понятно, что именно иминная группа, которая из-за большей основности более склонна к такой активации, получает преимущество перед просто карбонильной. Но вы селективно не восстановите имин в присутствии альдегида, а с кетонами ситуация проще, потому что они часто сильно уступают в электрофильности.
Пока так кратко, но я поставлю себе в очередь более подробный разговор про гидридные восстановители. Окисление вот допилю, там просто немного увлёкся, а времени мало.
Спасибо!
Андрей Владимирович, здравствуйте! Вопрос по образованию лактонов, если взять циклогексанон и подействовать на него CF3CO3H, то образуется семичленный цикл – лактон, а можно ли получить лактон, когда в альфа положении от карбонильного углерода находятся заместители? Спасибо!
АЧ: Приветствую Вас,
Это реакция Байера-Виллигера, довольно типичная секстетная перегруппировка. Заместители ей не мешают. Там другая проблема: если замещение несимметричное, то мигрировать могут обе стороны, соответственно получаются два лактона. Часто это совсем неселективно, но иногда бывает только один продукт. Предсказать направление довольно трудно. Это в основном вопрос стереохимии. Нужно сделать конформационный анализ исходного кетона и интермедиата, и постараться увидеть там правильную конфигурацию для миграции, которая идет всегда с тыла относительно уходящей группы. Ну или литературу поискать конкретно по интересующему кетону. Реакция Байера-Виллигера довольно популярна в синтезе, и примеров можно найти очень много.
Добрый вечер, Андрей Владимирович! Возникла необходимость синтеза интермедиатов гликолиза. Требуется субстраты для определения активности ферментов. Подскажите пожалуйста, как синтезировать глюкозо-1,6-бифосфат или фрутозо-1,6-бифосфат из глюкозы/фруктозы? Как селективно присоединить остатки фосфорной кислоты к гидроксильным группам? И как далее окислить фруктозо-1,6-бифосфат до фосфоглицеринового альдегида? Как возможно получить дальнейшие интермедиаты гликолиза: 1,3 – бисфосфоглицерат, фосфоенолпируват. Заранее спасибо!
АЧ: Добрый вечер, Руслан,
Воспрос Ваш – совсем перебор. Помощь в научных исследованиях надо искать у своего научного руководителя, а если Вы и сам такой, то у себя самого, и у коллег, специализирующихся в той же области химии.
Из общих соображений могу сказать, что обычная препаративная химия вряд ли может давать незащищённые фосфорилированные углеводы. Таеие вещи должны делаться ферментативным катализом, используя те ферменты, которые и работают в клетке для селективного фосфорилирования. Все это наверняка есть в продаже, и соответствующие методики в соответствующей химии наверняка есть. То же касается и других фосфорилированных интермедиатов. Это либо покупается, либо делается с помощью ферментов.
Добрый вечер!
Подскажите, почему при получении термодинамических енолятов используют протонные растворители и в основном нуклеофильные основания?
АЧ: В химии очень важна корректность. И очень важно понимать, что вот эти два термина: “кинетический” и “термодинамический” енолят – это такой жаргон, используемый, чтобы сокращённо выражать более сложные и длинные понятия. И если “кинетический” енолят действительно можно получить в прямом смысле этого слова хотя бы в растворе, то под термодинамическим енолятом мы понимаем енолят, образующийся в равновесии при переносе протона от енолизуемого карбонильного соединения как CH-кислоты к основанию сравнимой силы в растворе, обычно в низкой концентрации (ее можно оценить для конкретных оснований по величинам кислотности/основности) только в качестве реакционноспособного интермедиата в реакциях типа альдольной конденсации. Слово “получить” здесь неприемлемо, потому что вы даже не можете говорить о растворе енолята – в таких растворах это только один из компонентов, тем более что немедленно начинаются реакции самоконденсации или конденсации. Вы просто обсуждаете механизмы этих реакций, используя представление об образовании равновесного (равновесие и термодинамика это практически синонимы) енолята. Основания для этого годятся любые, подходящие по силе, а нуклеофильность не имеет значения, потому что это просто будет еще одно равновесие присоединения-отщепления нуклеофила.
Есть и другая проблема – в случае несимметричных кетонов, енолизуемых в обе стороны. Тогда термин “кинетический енолят” мы применяем к менее замещенному, и можем его конкретно получить количественным депротонированием кетона стерически затруднённым сильным основанием типа LDA.
Термодинамическим енолятом в этом случае жаргонно называют второй енолят – более замещённый (ещё иногда возникает проблема диастереомерии, но сейчас это не важно). Такой енолят тоже можно именно получить количественно, но не депротонированием, а например, расщеплением силилового или ацетильного эфира такого енолята. Такой способ не является кислотно-основным равновесием и никак не относится к терминологии депротонирования, а эпитет “термодинамический” применяют только чтобы показать, что такой енолят имеет такую же структуру, что и тот, который образуется в равновесии.
Что касается протонных растворителей для равновесного депротонирования, то это не обязательно. Обычно в таких случаях берут в качестве основания алкоголяты, а их удобно брать в растворе соответствующего спирта. Но с тем же успехом можете взять и другой растворитель, в котором растворяется основание, а сопряжённая кислота основания будет исполнять роль перносчика протона.
Спасибо!!
Здравствуйте!
А обработка иодоводородной кислотой спиртов с получением алканов – это хороший препаративный метод или тут есть свои подводные камни?
АЧ: Это история химии, глубокая архаика, традиционная ценность. Если вам дороги экспонаты из музея истории химии, то вперед. Старые химики очень ценили эту реакцию, потому что она во многих случаях прямо проявляет скелет сложного органического вещества, обрывая с него гидроксилы, метоксилы, и часто даже и другие группы. Это сначала нуклеофильное замещение, а дальше свободнорадикальная реакция по механизму. Результаты почти непредсказуемы Но выходы мизерные, всегда смеси, условия жесткие. Препаративно реакция бесполезна. В последние сто лет вы её не увидите в работах ни по синтезу. ни по исследованию строения, потому что появились нормальные методы и того и другого. Если вам нужно отчекрыжить гидроксил, есть куча методов, через ионное гидрирование, гидрогенолиз, реакцию Бартона-МакКомби, замещения на галоген и гриньяр с протолизом и хрен знает что ещё, всё селективно, если нужно и стереоселективно.
Спасибо!
Доброго времени суток, Андрей Владимирович?
Возможно, немного странный вопрос и не совсем связанный с химией, но все же: почему кротилы и кротоны так называются?
АЧ: Добрый вечер. Посмотрите на страничке про альдольную конденсацию внизу на вкладке про альдольно-кротоновую самоконденсацию. Все эти названия (кротоновые альдегид и кислота, кротонат, кротиловый спирт и радикал кротил) происходят от одного корня, а он из кротонового масла, и далее к растениям рода кротон.
А вообще история довольно запутанная. Кротоновое масло никакой кротоновой кислоты не содержит – это была ошибка фармацевтов из первой половины 19 века, тогда было очень модно пытаться выделять вещества из лекарственных растений, но техника эксперимента и анализа была весьма примитивна. Ни одно более-менее современное исследование кротонового масла не находит в нём кротоновой кислоты или ее производных. Весьма известный немецкий химик Гойтер в 1870-х с немного уже более продвинутой техникой эксперимента обнаружил в кротоновом масле гомолог кротоновой кислоты, которую назвал тиглиновой (латинское название Кротона слабительного Croton tiglium L).
Забавно, что и само название кротон тоже результат путаницы. Кротоновое масло часто путали с касторовым – оба невероятно горькие и имеют слабительное действие, чем пользовались итальянские фашисты, которые любили издеваться над оппонентами, предлагая тем выпить за здоровье дуче стаканчик касторового масла (пересмотрите Амаркорд, там есть такая сцена). Оба растения попали в Европу из дальних стран, поэтому их путали. Кротон назвали кротоном потому что спутали семена этого растения с семенами клещевины, из которой добывали касторовое масло, а семена клещевины немного смахивают по форме на клещей, по-гречески как раз кротос. Название клещевины по-лытыни Ricinus – а это как раз клещ, только по-латински, ну и собственно русское название клещевина это просто перевод латинского. фактически получается что оба растения названы одинаково, только одно использует греческий, а другое – латинский корень. В ботанике таких фокусов немало. Так это подобрал Линней, когда делал ботаническую номенклатуру вслед на зоологической, а к растениям в отличие от животных Линней относился довольно прохладно и напорол в названиях множество ошибок и путаницы. Название закрепилось, никто не стал пересматривать, так мы и живем с растением, которое перепутали с другим, из которого якобы выделили кислоту, которую перепутали с другой. А мы ещё к этому добавляем путаницы, называя кротоном очень популярное комнатное растение, которое тоже не кротон, а кодиеум, правда ботанически растения близки, и некогда название кротон использовалось как синоним.
Нельзя не заметить, что и действие касторового и кротонового масла только поверхностно похоже, а действующие вещества в них разные, и довольно сложные.
Но кротоновое все же содержит хотя бы тиглиновую кислоту. Но и здесь есть вопрос – скорее всего сами семена кротона и её не содержат, а такие низкомолекулярные компоненты появляются в результате выдерживания семян на воздухе (ферментации) и тиглиновая кислота это просто результат разложения и окисления весьма непростых компонентов семян. Так или иначе, кротоновое масло обладает ясно выраженным раздражающим и даже нарывным эффектом, и это очень легко отнести как раз к действию сопряженных непредельных кислот, акцепторов Михаэля – все активные акцепторы Михаэля имеют слезоточивое, раздражающее, нарывное действие, и хуже того, они канцерогенны. По этой причине кротоновое масло давно не используется в медицине.
Путаницу усугубляет и еще одно обстоятельство. В литературе кротоновым маслом называют не только настоящее масло, выжатое или экстрагированное из семян, но и эфирное масло, точнее масла растений рода кротон, а их десятки или даже сотни в тропиках. И в этих эфирных маслах как положено, одни терпены и никаких следов никаких непредельных кислот.
Большое Вам спасибо за подробный и интересный ответ!
АЧ: Не за что. Я люблю копаться в названиях соединений, растений, улиц, городов и т.п. Очень неожиданные бывают повороты.
Здравствуйте, Андрей Владимирович!
Хотел бы вновь обратиться к вопросу региоселективности замещения в ароматике.
Стоит ли, на ваш взгляд, удивляться замещению водорода именно в пара положении к карбонильной группе в 2,5-диметоксибензальдегиде в реакции с йодом и нитратом серебра.
На первый взгляд, если собрать ориентирующие эффектызаместителей, можно было бы ожидать замещения в 3, а не 4 положении, а тут вон как!)
«Увидено» в этой статье: 10.1039/C4OB00493K
Благодарю за возможность задавать вопросы на вашем сайте.
АЧ: Добрый вечер, Герман,
А я благодарю за вопросы, хотя не всегда успеваю отвечать, особенно когда ответ получается немаленький, как с предыдущим. Вопросы это отличный способ узнать, что кажется проблемой, а это всегда интересно и часто выводит на всякие обобщения, иногда неожиданные.
Ваш вопрос проще и отвечаю сразу. Во-первых, в электрофильном ароматическом замещении не надо собирать эффекты ориентации. Практически всегда ориентацию определяет доминирующий ориентант. Если есть мезомерный донор, то он. Если есть несколько мезомерных доноров то фенолят перешибет любого амина, который перешибет алкокси, который перешибет любого галогена, а из галогенов самый сильный ориентант фтор, который перешибет другие галогены. Соответственно мезомерный донор перешибет индуктивного донора. А донор перешибёт акцептора.
В данном случае есть метокси, который перешибет альдегида. Поскольку метокси два в пара-положении, то ориентация будет в первом приближении орто к любой из метокси. Выписываем возможные продукты:
Теперь дополнительные соображения, при прочих равных заместители будут стараться избегать становиться в ряд по трое и совсем плохо по четверо. Это отлично работает для крупных входящих групп типа брома или иода (с хлором работает плохо) – это банальная стерика и больше ничего. С вторым кандидатом есть ещё отягчающее обстоятельство – акцептор в орто-положении. Это довольно слабый фактор, который выплывает только при прочих равных, чоответственно здесь его можно в расчет не принимать, и так все плохо. Рассматриваем продукты и убираем плохие. Остается ровно то, что получилось.
Эта схема очень хорошо работает в огромном количестве случаев, хотя с химией всегда могут быть исключения. Но это будет большой редкостью, и имело бы какое-то объяснение.
Итого: получили ровно то, что ожидалось.
Большое спасибо за ответ!
Андрей Владимирович, здравствуйте!
Почему реактивы Коллинза, Кори (CrO3 в пиридине и т.п.) не затрагивают при окислении другие функциональные группы (в частности, кратные связи)? С чем связана такая избирательность?
Заранее спасибо за ответ!
АЧ: Это общий вопрос к тому, почему реагенты селективны – в данном случае хемоселективны – затрагивают только определенные реакционные центры. Это всегда определяется механизмом реакции. В частности, реагенты 6-тивалентного хрома это, с точки зрения органики, что-то типа ацилирующих реагентов (здесь это можно назвать хромилирующими, но так не говорят) – они образуют смешанные эфиры хромовых кислот со спиртовыми группами, и дальше эти производные претерпевают внутримолекулярную реакцию, в которой водород из-под гидроксила переносится на хромовый остаток. А с двойной связью такие производные хрома просто не реагируют – механизма нет. Вас какие-то конкретные реагенты интересуют или вообще?
АЧ: Если вы ждёте более подробного ответа на этот вопрос, наберитесь немного терпения, ответ будет, в перспективе пары недель, сочиняю.
АЧ: Пара недель превратилась в несколько месяцев, но зато страничка получилась вполне информативная, я туда запихал много всего полезного и занятного.
Здравствуйте, Андрей Владимирович!
Большое спасибо за ваш сайт, он очень полезен даже нам, занимающимся комплексами металлов и металлоорганикой.
Вопрос про номенклатуру: если нет главной цепи, а, скажем, лиганд образован циклами и гетероциклами, как выбирать, кто главный и с чего начать нумерацию? Например, у нас в ходу вот такой силандиамин Me2Si(NHPbt)2, где Pbt — это 2-фенил-1,3-бензотиазол; азот связан с положением 2 фенильного заместителя (если считать, что с положением 1 связан бензотиазол). Как правильно назвать вот этот заместитель Pbt? Должны ли мы при выборе главного (гетеро)цикла заместителя выбирать тот, который связан с аминогруппой в данном случае (то есть, тот, который -ил)?
Коллеги предлагали варианты для Pbt = 2-(фен-2’-ил)-1,3-бензотиазол или 2-(1,3-бензотиазол-2-ил)фенил ; ChemDraw для этого заместителя даёт название 2-(benzo[d]thiazol-2-yl)phenyl.
Тут же вопрос, для таких тиазолов нужно ли обязательно указывать что они 1,3-тиазолы, или в этом нет необходимости (как делает ChemDraw); а если указывать, что они 1,3-, то нужно ли обозначать место сочленения с бензольным кольцом [d], ведь другого варианта для 1,3-тиазолов нет?
АЧ: Добрый вечер, Николай, спасибо за добрые слова. Делать что-то полезное очень оптимистическое занятие. Не могу себе отказать в этом удовольствии. Кстати о комплексах – у меня ведь есть и второй сайт, как раз про комплексы, но правда в контексте катализа.
Ваш лиганд выглядит так?
Тогда это, во-первых, силан – именно это основа. Всё остальное заместители. Называем сложный заместитель: это 2-замещенный фениламино, заместитель просто бензтиазол-2-ил, если хотите указать гетероатомы, это можно, хотя ИЮПАК сохраняет основные традиционные названия, в том числе тиазол это 1,3-тиазол по умолчанию. Но пусть будет так, тогда собираем название
бис(2-(бенз-1,3-тиазол-2-ил)фениламино)диметилсилан. Указывать связь, по которой конденсированы кольца не надо, потому что это и так однозначно, а ИЮПАК не требует избыточных локантов.
ИЮПАК допускает и альтернативные названия, но они все будут более громоздкие и неуклюжие.
Здравствуйте, Андрей Владимирович! Как объяснить, что замещенный ацетилен при гидрировании в жидком аммиаке в присутствии натрия дает транс-алкен? Спасибо огромное за ваш труд!
АЧ: Ой, а разве это не описано? Я точно это расписывал во всех подробностях. Надо поискать.
АЧ: Проклятье! Простите, пожалуйста. Давно написано, но застряло в черновиках. Вообще давно почти готова новая версия страницы про ацетилены, но чтобы допилить нужна еще неделя-другая, а взять негде. Поэтому много материала зависло. Но я на днях материал по гидрированию выложу на старой странице. Не сегодня только.
АЧ: Выложил развёрнутый ответ на странице про Алкины – внизу появился раздел про гидрирование.
Есть у Хараша реакция с хитрым пероксидом и бромидом меди (I), можно ли её применять для енолизуемых альдегидов и кетонов или с этим будут сложности (может какие-то сложности в плане стабилизации получаемого радикала)?
https://doi.org/10.1021/ja01536a062
АЧ: В этой реакции нет ничего хитрого кроме того, что это нечто историческое, из тех времен, когда плохо понимали, как идут реакции, и что там могут делать соединения переходных металлов. Вообще для начала удивляет заявка на стереоспецифичность реакции, в которой нет никакой стереохимии, из чего мы узнаем, что в 1958 году в джаксе редактор не в курсе, что такое стереоспецифичность, как впрочем и сам Хараш и Джордж Сосновски. Имеется в виду, видимо, региоселективность. Вообще эта статья имеет отношение только к истории химии, ничего полезного для современной работы в ней нет. Химия не стоит на месте, и как бы ни был велик Хараш, а это так и есть, но это химик первой половины прошлого века, наделавший много работ, которые давно потеряли актуальность. Свободнорадикальное замещение, в том числе и в присутствии соединений меди, конечно, известно, но сейчас это делается немного на другом уровне и эксперимента, и понимания.
Здравствуйте! Известно, что циклопентадиенильный анион ароматичен, там это достигается засчёт того, что образуется ионная связь. А возможны ли случаи, когда ароматичность достигается вследствие ковалентной связи? Моё предположение лишь таково, что для этого нужно иметь какую-то длинную, малоэнергетичную связь, так ещё и сильно исказить привычную геометрию (чтобы эта связь была орбитальна в пи-кольцевом сопряжении), но тогда другая связь при этом атоме, например, углерода, должна быть ей перпендикулярна, получается.
АЧ: Эти рассуждения мне вообще не очень понятны. Циклопентадиенильный анион ароматичен именно как анион, и это не имеет отношения к противоионам – что там за катион, натрий, литий, калий, цезий, кальций, и т.п., хотя ряд невелик, потому что скоро начнутся уже кординационные соединения сендвичевого типа, которые тоже считаются ароматическими с точки зрения циклопентадиенильного лиганда. Причём тут ионная связь и с кем эта связь?
А начиная со второй строчки вопроса я вообще не в состоянии интерпретировать текст. Как Вы себе представляете ароматичность, достигаемую вследствие ковалентной связи? Какой ковалентной связи, с чем, причём тут ароматичность и вообще я не вполне уверен, что Вы достаточно хорошо разобрались с тем, что такое ароматичность. Или формулируйте вопросы корректнее, или мы вряд ли сможем достичь взаимопонимания, необходимого для того, чтобы я смог понять, что требуется объяснить.
Забыл саму статью прикрепить, извините за оплошность:
doi.org/10.1021/jo00834a051
АЧ: На этот вопрос отвечу несколько позже и в более развёрнутом виде на странице Ответы, так как нужно сделать несколько рисунков.
АЧ: Развернутый ответ выложил на странице Ответы.
Огромное спасибо!
И ещё один вопрос:
есть статья, в которой упоминаются дегидрогидантоины и получение одного из них; так вот, там брали N-хлорогидантоин и элиминировали HCl триэтиламином. Какова природа такого элиминирования, разве поляризация связи не смещена в сторону азота в данном случае, давая на азоте условный дельта-минус?
АЧ: Вопросы простые, но отвечу позже, завтра-послезавтра, сейчас совсем нет времени, извините.
Может ли енол какого-нибудь карбонильного соединения присоединять HBr в уксусной кислоте?
АЧ: Конечно да, и мы это отлично знаем. Енол – это сильнодонорный олефин, активированный к электрофильному присоединению. Поэтому что HBr, что любая другая более-менее сильная кислота протонируют двойную связь с образованием мезомерно-стабилизированного катиона, обратимое депротонирование которого дает кето-форму.
ТО есть мы имеем дело с кислотным катализом кето-енольной таутомерии, и совершенно один черт какую кислоту использовать. Хотите HBr – ради богов. У меня на сайте немало написано про кето-енольную таутомерию, в том числе и с кислотным катализом.
Я понимаю, что процесс вполне себе равновесный, но что если оставить какое-нибудь енолизующееся карбонильное соединение надолго в смеси HBr-AcOH? Я вполне могу наивно предположить следующее: а не может ли HBr присоединиться к этому сильнодонорному олефину? Причем можно предположить два варианта (если исключить самоконденсацию енола с кетоном): продукт присоединения по Харашу – не всегда же мы проводим кислотный катализ, избавляясь от вообще всех перекисей, понимая под перекисью здесь вполне доступный кислород воздуха; даже если и избавиться от кислорода, вот равновесно присоединится HBr по правилу Марковникова, а затем в кислой среде на получившуюся гидроксильную группу сядет протон, отщепится вода, произойдет Е1-элиминирование. Причем взять специально какой-нибудь 4-метокси-ацетофенон, чтобы самоконденсация шла медленно. Возможно ли такое?
АЧ: А что, простите, вы ожидаете получить при присоединении HBr к енолу? Вы не пробовали не словами это описывать, а просто схему реакции написать – что получится, устойчиво ли это, нужно ли это и так далее. А что касается свободнорадикального присоединения и эффекта Хараша, то он наблюдается только тогда, когда скорость электрофильного присоединения меньше скорости радикального. Это ровно то, что происходит с терминальными алкенами, но уже с дизамещенными алкенами обычное электрофильное присоединение быстрее и Хараш просто не работает. Тем более это будет с енолами, которые настолько сильно активированы к электрофильному присоединению, что радикальное там рядом не стояло.
Здравствуйте!
Задам несколько вопросов по отдельности.
Расскажите, пожалуйста, возможно ли, а если возможно, то хорошо ли протекает реакция Sn2 в тех случаях, когда на одном атоме углерода есть две уходящих группы (например, дибромметан)?
АЧ: Химия конкретна, поэтому надо обсуждать конкретные соединения или хотя бы родственные соединения. Две уходящие группы на одном углероде – это галогены. Все остальное или неустойчиво или очень непросто сделать (непросто не значит невозможно, но нужно думать про такие случаи отдельно). А два галогена – хлора, брома или иода – пожалуйста, вполне такие соединения реагируют в SN2 с разными нуклеофилами, и так собственно и получают разные дизамещенные метаны (или гомологи), причем второе замещение часто идет легче первого, так что остановиться на монозамещенном не очень просто. Используйте обычные способы активации анионных нуклеофилов – полярные растворители, межфазный катализ. Неплохо заходит и старинный способ – использование серебряных солей нуклеофилов. Если галоген фтор, то просто так реакции не идут, но как ни странно два фтора заместить проще чем один, если субстрат выдерживает сильные кислоты – в таких условиях можно, например, делать гидролиз гем-дифторзамещенных – уходящий фторид протонируется и отваливает в виде HF. Этот немного странный метод иногда всплывает в синтезах разных кетонов (нам он не понадобится, потому что мы всё равно не сможем делать дифторпроизводные).
Здравствуйте!
А будет ли пост/лекция про нобелевку по химии за этот год? За прошлый вы так, к сожалению, и не выложили, очень хотелось бы послушать. 🙁
АЧ: Добрый вечер,
Нет, про это не будет. Я про органику, а не вообще про всё, а квантовые точки это чистая неорганика с точки зрения материала, и фотофизика во всём остальном. Более того, я довольно долго занимался всякими органическими флуорофорами и оптическими материалами, и вечно была эта проблема – что ни напишешь, кто-то из рефери обязательно заметит: Нафига вы этой ерундой занимаетесь, ведь есть же квантовые точки, они умеют всё. Приходилось каждый раз терпеливо прояснять, что нет, далеко не всё, и есть ещё место и другим материалам. Так что я ненавижу квантовые точки. Но это действительно очень крутая вещь, и нобелевка за дело. И хотя наука – дело совершенно общечеловеческое, но всё же здорово, что в химии у нас наконец второй лауреат. Только вот у кого у нас. Вопрос непраздный, но всё же есть повод так сказать. А это хорошо, потому что говорит нам одну простую вещь: в любой жизни есть место нобелевке, поэтому может даже и в нашей есть, но это мы узнаем сильно нескоро.
Насчёт прошлого года и правда получилось скверно. Я просто не справился с обработкой тех лекций для выкладки, не успел, других дел полно. Но я восполню это в этом году, лекция ещё раз будет прочитана и теперь уж точно выложена; собственно я ровно сейчас и занят вёрсткой этой страницы.
Андрей Владимирович, здравствуйте!
Иногда в реакциях с кратными связями (которые, по сути, являются избытком электронной плотности) происходит взаимодействие с анионами или другими электроноизбыточными частицами. Почему они вступают в реакцию, а не отталкиваются друг от друга (ведь электростатику никто не отменяет…)?
Заранее спасибо за ответ и за ваш превосходный сайт!
АЧ: Добрый день и спасибо за добрые слова.
Увы, я не совсем понял Ваш вопрос. Химия конкретна. С какими кратными связями, какие электроноизбыточные частицы?
В общем виде, во всех реакциях схема простая. При сближении частиц возможность реакции определяется балансом и электростатики, и ковалентных взаимодествий. Оба этих типа взаимодействия довольно короткие, если только речь не идет о двух настоящих ионах, когда электростатика сильна и начинает работать сильно раньше ковалентных взаимодействий. Если речь не идет о ионах (оба реагента), а ион только один реагент, или ни одного, тогда это намного более слабые ион-дипольные или диполь-дипольные взаимодействия с очень крутой зависимостью от расстояния, очень быстрым спадом. Поэтому если есть потенциал ковалентного взаимодействия, то он успевает зацепиться и начать развиваться, а энергии в ковалентных взаимодействиях обычно больше, что и позволяет молекулам начать процесс перестройки связей, то есть собственно реакцию. Реакции обычно идут в растворах, то есть в жидкой фазе, где молекулы и так сталкиваются во всех возможных ориентациях, то есть достигают близкого контакта, преодолевая электростатическое отталкивание, и те взаимные ориентации, которые соответствуют орбитальным взамиодействиям, и развиваются дальше уже как реакция.
А потенциал ковалентного взаимодействия мы и оцениваем с помощью волшебных слов нуклеофил-электрофил, в разных вариантах, в том числе с помощью понятий о граничных орбиталях (нуклеофил = ВЗМО – электрофил = НСМО) и их взаимодействиях. И когда анализируем возможность какой-то реакции, оцениваем электрофильность и нуклеофильность разными качественными и полуколичественными способами, в том числе и по ВЗМО/НСМО и тогда и приходим к выводам, возможна или нет реакция, и даже оцениваем, хорошо ли она пойдёт.
Вот так это выглядит, если кратко, и в самом общем виде. Но химия конкретна и надо разбирать конкретные реакции.
Спасибо!
Если на конкретных примерах, то почему можно присоединять, например, спирты к алкинам? Ведь у алкинов тройная связь, где достаточно высокая электронная плотность, а у спиртов – неполноценные пары электронов на кислороде, тоже “концентрация минуса” большая. Как спирту удается прорваться через этот “тор” из двух пи-орбиталей двойной связи, а не оттолкнуться от нее?
Или, например, присоединение MnO4(-) к алкенам.
АЧ: Сейчас на странице Ответы появится развёрнутый ответ на этот вопрос.
Большое Вам спасибо за подробный ответ!
Здравствуйте!
А Вы не скажите, пожалуйста, почему при реакции кокосульфата натрия с лимонной кислотой выделяется углекислый газ?
АЧ: Ну, это дерьмище какое-то, а не кокосульфат, видимо, импортозамещение. При производстве кокосульфата после сульфирования спиртов из кокосового масла, образующиеся эфиры серной кислоты тщательно нейтрализуют, скорее всего бикарбонатом натрия. В продукте хорошего качества гидрокарбоната оставаться не должно. А если это дерьмо кое-как сделано, то может остаться и будет шипеть при добавлении кислот. Но тогда уже вопросы более серьёзные возникнут к качеству – если уж нейтрализовать нормально не смогли, то и сам процесс сульфирования мог быть сделан кое-как. А ведь это ингредиент косметики.
Здравствуйте, почему трет-бутильный радикал стабильнее, чем фенил-радикал? В трет-бутиле третичный атом стабилизируется за счёт +I эффекта трёх групп СН3, а в фениле возможна делокализация электрона по кольцу. Как понять, кто более стабилен в этом случае?
АЧ: Добрый вечер,
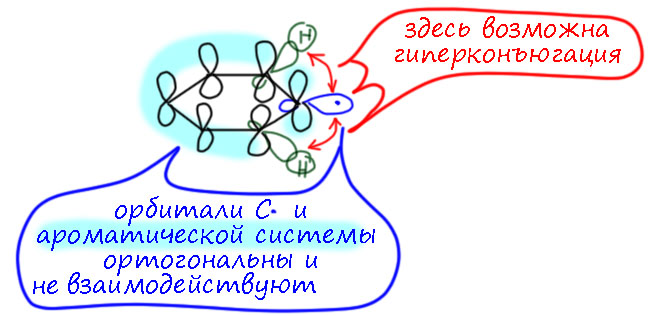
В Вашем вопросе немало путаницы. Во-первых, индуктивный эффект не имеет никакого отношения к стабилизации радикалов, потому что радикалы это электронейтральные частицы, в которых атом с неподеленной парой не имеет ни избытка, ни недостатка электронной плотности. И индуктивный эффект ничем его порадовать не может – не нужно этому атому просто смещения электронной плотности за счет поляризации. Есть только один случай, когда полярные эффекты (а так называют любые эффекты, связанные со смещением плотности и перераспределением заряда в частице; индуктивный эффект – типичный полярный) стабилизируют радикалы – это так называемый каптодативный эффект, когда на атоме с неспаренным электроном есть сразу и донорный и акцепторный заместитель. Но это редкий случай, и там много своих заморочек и непоняток. Оставим это.
В большинстве случаев радикальные частицы стабилизируются за счет делокализации или гиперконъюгацией (сопряжением пи и сигма-орбиталей), или обычным сопряжением (пи-пи делокализация). Трет-бутильный радикал стабилизирован гиперконъюгацией – сопряжением пи-орбитали с неспаренным электроном и связывающих гибридных сигма-орбиталей C-H связей.
Фенильный радикал не имеет очевидных путей делокализации. Обычное сопряжение с ароматической пи-системой не работает, потому что орбиталь, на которой находится неспаренный электрон (часто коротко говорят – спин, просто имея в виду, что главной характеристикой неспаренного электрона является спин; поэтому когда мы коротко говорим – в такой-то частице делокализован спин, то имеем в виду именно делокализацию неспаренного электрона) находится в плоскости кольца, и перпендикулярна (или ортогональна, что одно и то же; первое слово обычно относят к очевидной геометрии, а второе еще имеет более строгий математический смысл ортогональности векторов в общем смысле этого термина). Ортогональные орбитали не взаимодействуют по определению. В фенильном радикале можно увидеть возможность для гиперконъюгации с C-H связями – орбитали коллинеарны, и вполне могут взаимодействовать, но геометрически они смотрят немного в разные стороны, поэтому хоть и взаимодействуют, но это довольно слабое взаимодействие.
Поэтому даже с точки зрения стабилизации радикала выигрывает трет-бутильный – у него больше возможностей для гиперконъюгации – целых девять CH-связей, хотя нужно учитывать вращение вокруг связей CC и взаимное расположение в конформерах трет=бутильного радикала, но всё равно возможностей больше, чем в фенильном.
Но это еще не всё. Когда говорят о стабильности радикалов, имеют в виду не некоторую абсолютную стабильность с строгим термодинамическим определением, потому что это просто бесполезно. В химии, и это относится не только к радикалам, под стабильностью часто имеют в виду немного другое. Это чертовски тонкий момент, который путает многих и является причиной противоречий – стабильность всегда относительна, и для оценки стабильности используют какую-нибудь простую реакцию. Стабильность радикалов обычно оценивают по переносу атома водорода (часто используют аббревиатуру HAT – hydrogen atom transfer) от молекулы с C-H связью к радикалу. Такой процесс по определению обратим, но в большинстве случаев идет в одну сторону – и тот радикал, который образуется в такой реакции и является более стабильным. И вот в реакции фенильного радикала с изо-бутаном образуется бензол и трет-бутильный радикал, и это в этом направлении экзотермическая реакция. А в обратном эндотермическая и в заметной степени не идущая.
Из этого мы делаем вывод, основанный на эксперименте, – трет-бутильный радикал более стабилен, чем фенильный. И из того же делаем и второй вывод – связь CH в бензоле более прочная, чем связь CH в изобутане. И даже есть повод для обобщения: связь sp2-гибридного углерода с водородом обычно прочнее, чем связь sp3-гибридного атома углерода с водородом. Подтверждение этому можно увидеть и том, что связи с sp2-C немного короче, чем связи с sp3-C.
В общих чертах такая схема работает и позволяет удовлетворительно предсказывать, какой радикал отщепит какой-то атом водорода в другой молекуле. Или наоборот не отщепит.
Увы или слава богам, но химия сложнее таких схем. Скептики сразу начинают ехидно спрашивать, а почему мы оцениваем стабильность углеродных радикалов именно относительно переноса атома водорода. А что будет, если использовать другие элементы. И так далее. Но нам на 3 курсе это незачем, потому что мы практически исключительно в радикальных реакциях рвем именно связи C-H.
Пойдет ли гидролиз крахмала добавлением азотной кислоты вместо серной?
АЧ: Ну и вопросы у вас, милостивые государи! В химии важно понимать а) свойства веществ и б) природу реакций. ТОгда сами ответите на такие вопросы за десять секунд.
Гидролиз крахмала это реакция полисахарида с водой. Чтобы реакция шла за разумное время, ей нужен катализ. Самый полезный катализ из простых, неэнзиматических, в таких случаях – кислотный, катализ протонными кислотами. Поскольку речь идет о водных средах, то можно сказать, что протоном (точнее, ионами гидрония или по- старому, гидроксония), то есть нас интересует только рН среды и больше ничего (растворы разбавленные, поэтому рН является адекватной мерой кислотности). Чем больше кислотность (меньше рН), тем быстрее. Но – в сильнокислой среде уже глюкоза неустойчива, как и другие углеводы, происходит и эпимеризация и дегидратация в фураны. Поэтому нельзя перебрать с рН. Гидролизуют очень аккуратно, держат рН около 3, даже выше, а низкую скорость при такой слабокислой среде компенсируют быстрым нагреванием. В общем получается все равно не очень чистая глюкоза, поэтому давным-давно перешли на энзиматический гидролиз, очень мягкий и аккуратный.
Но если держаться кислотного гидролиза, то нужна любая сильная кислота, для доведения рН до 3-4. Можете сами прикинуть концентрацию. Будет немного, около 1% или даже меньше.
Азотная кислота в разбавленном водном растворе это просто еще одна сильная кислота. И если соблюсти рН, то действовать будет так же. Но – азотная кислота это довольно серьезный окислитель. Углеводы (альдозы) окисляются азоткой при нагревании до альдаровых (сахарных) кислот – это когда и верх и низ карбоксилы. Это старая и очень полезная химия. Но для окисления концентрация должна быть повыше. Иными словами приблизительно 1%-ной азоткой при нагревании гидролиз пойдет так же как другими сильными кислотами сравнимой молярной концентрации (серная – сильная кислота только по первой ступени, поэтому рН разбавленного раствора одинаковой молярной концентрации серной и азотной кислоты будет приблизительно одинаков). И всё же азоткой гидролизовать глюкозу не будут, все равно побоятся окисления, хотя если не ошибиться с концентрацией, то окислением можно пренебречь.
Еще раз здравствуйте!
Как работают и как работать с ионообменными смолами?
АЧ: Ещё раз здравствую, да и Вы тоже не хворайте, а здравствуйте,
Но здравие здравием, а скоро придется ввести ограничение: вопрос должен быть хотя бы минимально конкретным. «Расскажите обо всём или хотя бы о чём-нибудь» – это не ко мне. Любителей попусту болтать на любые темы нынче легионы, вот к ним и обращайтесь. Ионообменные смолы в общем это бесконечная тема.
Поэтому просто кратко. Ионообменные смолы – это полимеры, практически всегда сшитые, в молекулы которых введены ионные группы. Если анионные, например, сульфонатные SO3–, то это катиониты (катионо-обменные смолы). Если катионные, например, четвертичные аммонии, то это аниониты (анионо-обменные смолы). Самый распространённый полимер для таких смол это сополимер стирола и дивинилбензола (ДВБ). Стирол полимеризуется в линейный полимер (там есть всякие дефекты, но это неважно), а линейные полимеры растворяются в подходящих растворителях. Если в полимер стирола ввести ионные группы, такой полимер будет, как и почти любое ионное вещество, медленно, но верно растворяться в воде, и просто не будет работать как стационарная неуносимая фаза. Поэтому добавляют к стиролу немного дивинилбензола, он встраивается в цепочки, но из-за второй винильной группы цепляет другую растущую цепочку, так получаются сшивки между цепочками, а полимер (крупиинка полимера, гранула полимера) фактически становится одной огромной молекулой (реально всё же не одной, но это тоже неважно, в любом случае молекулы огромны и перепутаны друг с другом). Смеху ради можно сказать на такие гранулы – вот молекулы лежат, щас я чайную ложку молекул возьму и прямо их все пощщитаю, типа, на счётах. Такой полимер не может ни в чём раствориться (в физическом смысле, безусловно, можно найти какую-нибудь агрессивную жидкость, которая этот полимер расщепит или окислит в маленькие молекулы). Вместо этого он вбирает некоторое количество растворителя внутрь, так как цепочки лежат неплотно, растворитель немного всё это распирает, всё это хозяйство увеличивается в объёме, – набухает. Это рабочее состояние всех таких полимеров – набухший гель. Если в сухом состоянии это были такие крупинки, гранулки, то в набухшем частички увеличиваются в объёме, и становятся упругими, мягкими, но не слипшимися, и растворитель свободно проходит между ними.
Получают такие смолы (смола, по-английски resin – это вполне допустимое короткое название для полимерных материалов любого типа, хотя многие из них ни на какие смолы в бытовом смысле близко не похожи; а говорить на полимеры – смола – модно и выдаёт в вас большого специалиста, а вовсе не братца Кролика) или прямо какой-то химией с уже готовым полимером. Например, тот самый сополимер стирола и дивинилбензола (это невероятно популярный полимер с такой кучей разных применений, что диву даёшься), содержит много бензольных колец, которые можно просульфировать (скорее сульфохлорировать и потом гидролизовать, это более аккуратная реакция) и получить сульфированный полимер. Конечно, количество и положения сульфогрупп в нем будут совершенно случайны, но это не всегда плохо. А можно пронитровать, потом восстановить и кватернизовать и получить четвертичные аммонии. Но если есть желание получить более понятный материал с известным количеством и положениями групп, то лучше к смеси стирола и ДВБ добавить замещённый стирол (их много есть просто в продаже, например, сульфированнй стирол). Совсем стирол заменять на замещённый не надо – будет бессмысленный неработающий избыток ионных групп, и работать такой материал будет плохо, да и с полимеризацией будут проблемы.
И вот, такие смолы с ионными группами и есть ионообменная смола. Чаще всего применяют сульфированную, это катионит, есть куча марок, и наших (обычно КУ) и фирменных, особо собаку съела на ионитах фирма Дау (Dow Chemical Co) из США, которая их выпускает уже лет сто, настолько, что фирменные названия типа дауэкса и амберлиста стали нарицательными, а всяких марок и типов с разными группами и структурами там бесчисленное количество на все применения.
Любой ионит перед применением подвергают набуханию в растворителе задуманной работы (обычно в воде), и зарядке нужными ионами. Катионит, напрмиер, часто заряжают ионами натрия, для чего просто выдерживают с водными раствором NaCl по инструкции. Это можно сделать и в колонке типа хроматографической, или просто в колбе.
Для чего используют? Поскольку я отвечаю за органическую химию, забудем про водоподготовку и умягчение воды (катионит в натриевой форме связывает ионы магния и кальция, замещая их в потоке на ионы натрия, когда емкость исчерпывается, катионит снова обрабатывают раствором NaCl и так много раз, в промышленных установках такие циклы повторяют автоматически), или ионообменную хроматографию для разделения ионов. В органике ионообменные смолы чаще всего используют как катализаторы, чаще всего кислотные, для чего берут сульфированный катионит и заряжают его протонами, просто промывая раствором сильной кислоты. Такие гранулы обладают кислотностью и катализируют обычные кислотно-катализируемые реакции, например, этерификацию. После реакции катионит просто отфильтровывают, и возможно используют много раз, хотя это не так просто, особено если реакции разные, ведь смола сорбирует немало органики, которая будет пачкать следующие реакции. Удобно? Не то слово! Когда это работает, это обожают технологи, потому что не образуется кислых жидких отходов, а это большая проблема. Всегда ли можно заменить обычный кислотный катализатор (серную, толуолсульфоновую, трифторметансульфоновую и т.п. кислоту) катионитом в H-форме? Увы, нет. У таких реакций часто бывает проблема с кинетикой, потому что каталитические центры локализованы на поверхности гранул, их не так много, к ним молекулы еще должны подплыть диффузией, и это весьма дорого обходится в смысле кинетических параметров. Если реакция не очень быстрая в растворе, то с катионитом она вообще может не пойти. К тому же не стоит преувеличивать кислотность таких пришитых кислот, хоть это и сульфокислоты. Впрочем, великий химик Джордж Ола в своё время придумал катионит, в котором сульфогруппы приделаны к перфторированному полимеру, назвал это Nafion-H, и это весьма сильная кислота (но не суперкислота!), которая катализирует в том числе и перегруппировки, и реакции Фриделя-Крафтса и многое другое. Дорогая только чертовски, впрочем ее можно регенерировать и использовать много раз.
А основной катализ можно? Можно, если уже анионит зарядить гидроксид-ионами или каким-то другим анионным основанием. Но это работает плохо, используется редко, в работе капризно, и случаев реального применения мало.
Вот собственно кратко всё. Просто от себя добавлю, что катализ катионитами вещь полезная, но в целом это удивительно скучная химия, в основном паханая-перепаханная, хотя, конечно, сейчас полимерная химия набрала совершенно удивительные обороты и фигачит новые полимеры, в том числе и ионообменные, со страшной скоростью. Там точно есть что-то новое и интересное. Как-то точно надо решать проблему плохой кинетики, диффузии, ведь группы внутри гранул обычного полимера не работают, и нужно работать над пористостью и прочими интригующими вещами.
Спасибо!
Здравствуйте!
Зачем после синтеза метилиодида из диметилсульфата с иодидами нужно промыть раствор содой? Видно образование пузырьков, значит выделяется CO2, но каким образом в перегонный куб попадает кислота? Неужели HI образуется?
АЧ: Приветствую.
Самое общее правило в органике в препаративной работе – вы при обработке реакционной смеси и экстракции продукта в органическую фазу (иногда продукт сам отделяется и образует органическую фазу), должны отмыть этот органический раствор от кислот или оснований, которые в ней могут быть растворены. Если этого не сделать, то при дальнейших процедурах выделения продукта (упаривание растворителя, перегонка) кислота или основние сконцентрируются и могут быть причиной разложения продукта, особенно во время нагревания во время перегонки. А иногда кислоты или основания бывают летучими, перегоняются незаметно вместе с продуктом, и дальше при хранении становятся причиной разложения. Получили бесцветную жидкость “перегонялась в точке, показатель преломления как в таблице”, запаяли в ампулу, положили на полку, через месяц достаём, а там чёрная смола. Это обычная история с образцами, полученными в практикумах.
Как понять, кислота или основание могут образоваться в продукте во время обработки. Смотрим на конкретную реакцию и думаем.
Реакция диметилсульфата с иодистым натрием. Образовался иодистый метил и метилсульфат натрия (натриевая соль полуэфира серной кислоты). Ни то ни другое не кислота и не основание. Но диметилсульфат вряд ли израсходовался на 100%. В органике крайне редко делают реакции с чистой стехиометрией (всегда что-то в избытке, а кроме того, в органике чистота реактивов редко бывает выше 95%, чаще меньше), и реакции не идут со 100%-ной конверсией, скорости реакций сильно замедляются ближе к концу и их просто прерывают раньше, чтобы не ждать сто лет ради ещё пяти процентов.
Итак, в смеси почти наверняка остался диметилсульфат. Как только в смеси появится вода, а она появится, потому что вам надо отмыть продукт от солей, этот диметилсульфат начнет гидролизоваться. Гидролизуется он медленно, но верно. Результат – сильная кислота (метилсульфокислота или уже даже серная, а поскольку там еще мог остаться и иодид, то и иодистоводородная, которая как раз летучая). Если не отмоете их и будете перегонять, с хорошей вероятностью кислота попадет в продукт, и если это HI, начнется свободнорадикальное разложение. Для подстраховки, кстати, метилиодид при хранении стабилизируют чем-то типа серебряной проволочки (у вас есть?).
Но есть и еще проще проблема. Диметилсульфат у вас прямо из магазина Aldrich-Fluka-Merck? Свежайший? Вряд ли. Скорее всего, это нечто, что стояло под тягой полсотни лет и немного заплесневело. Так там уже и без всякой реакции полно всякой кислой дряни, и все это вы потащите в реакцию.
Поэтому моем от кислоты, тщательно и правильно. Я надеюсь, ваши преподаватели научили вас это делать так как это нужно делать, потому что иначе промывка ничего не даёт. Моем, сушим, перегоняем, храним в холодном месте без доступа света, если есть серебряная проволочка, добавляем.
И во всякой следующей препаративной реакции думаем, что может остаться в продукте, кислота или основание, и тщательно отмываем соответственно.
Это общее правило каждый добросовестный препаративный химик соблюдает неукоснительно, просто на автомате.
Здравствуйте!
Не подскажете, почему ксантан, хотя растворим в воде, собирается в комки, а если всыпать ксантан в глицерин, то он хорошо распределится?
АЧ: Доброй ночи,
Это не такой простой вопрос, как может показаться. И в воде ксантан растворим не вполне, по крайней мере то, что получается, нельзя назвать истинным раствором – это не жидкость, в которой плавают молекулы растворенного вещества.
Ксантан, или ксантановая смола это полисахарид, и очень интересный. В некотором смысле это модифицированная целлюлоза. Целлюлоза, как известно, в воде нерастворима вообще, потому что представляет собой линейные цепи полимера глюкозы, склеенные друг с другом водородными связями. В молекулах ксантана к целлюлозе сбоку пришиты короткие кусочки небольших олигосахаридов, часть из которых несет карбоксильные группы. Поэтому это больше не ниточки, это трехмерная структура, сшитый полимер, боковые ответвления не дают целллозным цепочкам склеиваться друг с другом. Молекулы огромны и перейти в раствор в полном смысле этого слова не могут. Но поскольку там чёртова тьма гидроксильных групп, с молекулами воды это взаимодействует отлично. И именно вода входит в эти молекулы, а не молекулы в воду. И войдя, распирает эту сетку, расталкивает цепочки и сшивки – это называется набуханием. В результате образуется система, которую принято называть гелем – масса молекул сшитого полимера, распертого молекулами воды. Структура полимера такова, что туда может поместиться очень много воды, и в конце вы получите систему, в которой воды по массе больше чем полимера, она образует частично внутри области вполне жидкой фазы, все это гибкое, находится в непрерывном тепловом движении, и может с виду показаться раствором. Но это не раствор, а именно гель, но при большом количестве воды он приобретает текучесть, становится похож на жидкость, только вязкую. Поэтому ксантан используют как загуститель, когда надо у водной системы создать вязкость, тягучую консистенцию, заставить, например, какие-то легкие штучки типа блесток парить в толще, не падая на дно – ведь им для этого придется не молекулы жидкости расталкивать, а буквально пропихиваться через набухшую сетку полимера.
Чем глицерин отличается от воды? Глицерин это полярная жидкость, довольно вязкая сама по себе, и тоже с сильно ассоциированными друг с другом молекулами – работают динамические водородные связи, которые относительно легко рвутся и вновь образуются. Для ксантанового полимера глицерин – очень удачная жидкость, она точно так же как вода проникает в промежутки между углеводными звеньями, образуя водородные связи. Полимер точно так же набухает. Но происходи это быстрее, чем с водой. Глицерин по свойства молекул ближе к углеводам (в общем он и есть углевод, спирт от любой из двух треоз) и быстрее заполняет пространство внутри полимера. Вода особенно сначала делает это медленно, поэтому вначале и образуются такие склизкие комки – когда гидратация прошла только с поверхности частички полимера. В глицерине ксантан быстрее набухает, а благодаря близости показателей преломления (опять – здесь важна природа молекул), кажется, что частички становятся прозрачными, исчезают. Но это все равно гель, а не раствор. И тоже вязкий.
АЧ: С утра посмотрел работы по структуре ксантана, и должен внести несколько уточнений. На основные выводы ответа они не влияют, но точность всегда лучше неточности. Первое – молекулы ксантана это разветвлённый, но не сшитый полимер. Это действительно по структуре модифицированные цепочки целлюлозы. Модификация боковыми олигосахаридами (поскольку ксантан это продукт биосинтеза, а не случайной полимеризации, его структура довольно регулярна, боковые цепочки одинаковы, хотя степень полимеризации у отдельных молекул разная) препятствует слипанию цепочек в пачки, как у обычной целлюлозы. Тем не менее, есть возможность слипания двух таких цепей, если боковые цепи отгибаются в сторону – получается такая “толстая шерстяная нитка” (метафора моя), димерный или двухниточный (double-stranded – термин взят из структуры ДНК) от которой во все стороны торчат волоски боковых углеводов.
То что ксантан несшитый полимер безусловно помогает его молекулам отделятся друг от друга, если жидкость предоставляет им возможность водородных связей (и вода, и глицерин годятся). Можно ли считать такую систему раствором? Полимерные химики используют это понятие, если молекулы полимера отделены друг от друга в жидкости, чем напоминают молекулы обычного растворенного вещества, или как говорят, силы адгезии сильней сил когезии – взаимодействие отдельных молекул с растворителем суммарно сильнее взаимодействия молекул растворителя друг с другом и молекул полимера друг с другом. Здесь надо понимать еще и то, что вода в этом смысле хуже как растворитель чем глицерин, потому что у воды очень велика сила когезии, молекулы воды очень сильно взаимодействуют друг с другом и не любят, когда в эту гармонию кто-то лезет. Поэтому вода медленнее взаимодействует с ксантаном, чем глицерин – это более сложный процесс.
И не стоит забывать ещё и про размер. Средний размер молекул ксантана около 100 нанометров. Каждая облеплена молекулами воды или глицерина, образующими непрерывную фазу с соседними молекулами растворителя. Такая штука условно говоря плавает, подвержена тепловому движению. Но размер таков, что это больше напоминает не раствор, а дисперсную систему. В хороших коллоидных растворах (напрмиер, хрестоматийном коллоиде золота) размер частичек всего несколько нанометров. Если несколько десятков, это уже считается крупным и не очень устойчивым золем. Сотня нанометров – это уже микросуспензия, потому что частички такого размера склонны самопроизвольно падать на дно, седиментировать. Безусловно, углевод это не металл. Плотность частичек почти не отличается от макроскопической плотности жидкости, поэтому седиментировать им смысла нет. Здесь самое главное – можно ли считать такую систему гелем? Гель – это скорее намного более крупная и трехмерно сшитая молекула полимера. Целый макроскопический кусок (гранула) такого полимера может быть одной молекулой (примеры нам отлично известны, это, например, сшитые полиакриламидные гели, используемые для поглощения воды, и собственно это не что иное, как органическое подражание трехмерной сетке диоксида кремния, силикаГЕЛЯ). Поэтому там не может быть ничего кроме набухания, раствор, даже такой, как у несшитых полимеров, невозможен. В случае ксантана, если концентрация побольше, система будет себя вести как гель, потому что отдельные набухшие молекулы будут сцепляться друг с другом водородными связями. Но если это очень сильно разбавить, молекулы разойдутся. Скорее всего, это должно сопровождаться значительным снижением вязкости и увеличением текучести, но тут надо искать данные по реологии системы ксантан-вода и ксантан-глицерин и много всего ещё.
Всё остальное, написаннное ночью, вполне адекватно и изменений не требует. Ксантан – очень интересная и необычная штука, неслучайно ее так любят исследовать и применять.
Спасибо!
Подскажите, каким образом триметиламин реагирует с раствором хлорида натрия.
АЧ: Вот и я тоже думаю – каким же образом триметиламин может взаимодействовать с раствором хлорида натрия? И кто вообще это придумал? Очевидно, что триметиламин растворяется в воде, и будучи неплохим основанием, частично и равновесно даёт ион триметиламмония и смещает рН в щелочную область. Ионы натрия и хлорида благополучно остаются в том же растворе, и в первом приближении никак не влияют на поведение триметиламина. А если покопаться чуть поточнее, то выяснится, что растворимость триметиламина в присутствии сильного электролита снижается, но собственно и все – немного смещается кислотно-основное равновесие и больше ничего.
Право, это из той же оперы, что и странная идея про экстракцию каких-то комплексов. Откуда вы это берете? Источники информации должны быть понадёжнее, что всегда можно проверить проверив информацию по другим источникам. Хорошая информация всегда есть в разных местах. Сомнительная – только в одном. В каком?
Здравствуйте!
Скажите, пожалуйста, как так получается, что изоамиловый спирт может экстрагировать йодидные комплексы серебра, ведь одно – органическое, а другое – неорганическое?
АЧ: Прямо скажем, странный вопрос. Во-первых, что такое “иодидные комплексы серебра”, и откуда известно, что они экстрагируются изоамиловым спиртом?
Во-вторых, что бы это ни было, почему бы ему не экстрагироваться? Противопоставлять органику и неорганику – дело неблагодарное. Это очень формальная характеристика вещества, вообще скорее филологическая, чем сущностно химическая. И органических и неорганических веществ необозримые количества, и свойства их изменяются настолько широко, что всегда можно найти самые диковинные сближения между с виду совсем никак не связанными веществами.
Толковый химик вообще никогда не будет зацикливаться на том, органическое вещество или неорганическое. Нужно разбирать структуру, а для анализа растворимости понимать, как устроено твердое вещество (кристаллическая решетка, если она есть) и какие взаимодействия можно ожидать при переходе молекул в раствор (и ещё не всегда очевидно, что образуется именно раствор, а не что-то упорядоченное).
Алифатические спирты – очень хорошие растворители для множества самых разных веществ, в том числе неорганических. Они довольно полярны, имеют гидроксилы, способные быть и донорами, и акцепторами водородной связи, и хорошо взаимодействовать по нескольким разным механизмам. Если у серебра действительно есть более-менее устойчивые иодидные комплексы, то у таких комплексов будет а) значительная координационная ненасыщенность, то есть они смогут принимать дополнительные донорные лиганды, и почему бы и не те же спирты; б) иодиды способны взаимодействовать и через водородные связи (слабые, но все же, для растворимости даже слабые взаимодействия вполне работают из-за огромного избытка растворителя над растворенным веществом) и через так называемые галогенные связи с теми же атомами кислорода. Возможны и еще более непростые эффекты, потому что иодид серебра это очень непростое вещество, тут даже комплексы никакие не нужны. Обсуждать это стоило бы серьёзно, если бы были надежные данные по такой растворимости, но я их что-то не вижу.
Неорганика вообще часто неплохо растворяется в самой разной органике. Даже с виду очень простая. Например, самые простые соли лития типа хлорида отлично растворяются и экстрагируются из водного раствора в самые разные органические растворители, включая те же бутиловый или изоамиловый спирт. Этим очень активно пользуются при отделении катионов лития от натрия и калия и в анализе, и в промышленном выделении лития. С виду хлорид лития – самое простое неорганическое вещество, какое только можно придумать. Но на деле всё сложнее, происходит очень сложная агрегация и такие агрегаты уже переходят в органику, взаимодействуя и как кислоты Льюиса и через другие взаимодействия. И так далее.
К слову об ортоэфирах, какая история у приставки орто- в этих эфирах? Всё ли гладко в получении этих эфиров реакцией Вильямсона, ведь наверняка в таких условиях может получаться карбен :CCl2?
АЧ: Историю проследить будет непросто. Я попробую на досуге. Но из общих соображений это в ранней химии была такая идея. На современный лад я бы ее переиначил вот как: если есть два ряда кислородных кислот, то тот, которая соответствует более полной степени гидратации соответствующего ангидрида называется орто- – эта приставка несет смысл “правильная, соответствующая ожиданиям”. Бывает и второй ряд – неполная гидратация, как бы мы сейчас сказали – с координационно ненасыщенным элементом. Такую кислоту называют с приставкой мета – то есть просто другая, следующая в ряду, менее ожидаемая.
В ранней химии еще совершенно не понимали закономерностей структуры и просто смотрели валентность и стехиометрию, и когда реально обнаруживали два ряда кислот у элемента (не только кислот, но и солей или производных типа эфиров) – радовались как дети, и давали им эти приставки. С химией углерода понятно получилось так, что сначала обнаружили угольную кислоту и карбоновые кислоты – соответствующие неполной гидратации. Их можно было бы называть мета-, но это не прижилось. Зато когда нашли второй ряд, соответствующий полной гидратации, с удовольствием назвали его орто-. И не стоит забывать, что уже с 1860-х стали здорово смотреть и сравнивать элементы одной группы, и бывало так, что у более нижнего элемента преобладала орто-форма, а у второго периода – мета-форма.
Что касается карбена, то и хорошо, что образуется, наверняка образуется. Синглетные карбены с огромной скоростью реагируют со спиртами, это вообще одна из самых быстрых реакций, поэтому наверняка это и есть механизм алкоголиза хлороформа.
Здравствуйте!
Расскажите, пожалуйста, про реакцию Бодру-Чичибабина. Можно ли этой реакцией получать кетоны из триэтилортацетата или других ортоэфиров карбоновых кислот?
АЧ: На страничке про карбоновые кислоты: методы и задачи на вкладке альдегиды из производных муравьиной кислоты можете уже прочитать про реакцию Чичибабина-Бодру.
Это старая реакция, довольно малополезная в наше время. хотя для простых альдегидов ее иногда применяют. Сейчас все же слишком много других методов синтеза альдегидов, куда более надежных. Скорее это такая историческая реакция, одна из первых появившихся для только что открытых гриньяров. Распространять ее на другие ортоэфиры – занятие довольно бессмысленное – там реакции быдут заведомо намнного медленнее, а сами рто-эфиры не так уж доступны, в отличие от триэтилортоформиата, который является весьма дешевым и легкодоступным реактивом, очень широко применяемым в синтезе.
Здравствуйте!
А как протекает реакция этилхлорформиата с фенолом в присутствии пиридина и что это за реакция?
АЧ: Как обычная этерификация. Хлороформат – это производное угольной кислоты, наполовину хлорангидрид, наполовину эфир. Это такое историческое название, не без путаницы. По-русски кислоту называют хлоругольной, а её эфиры хлорформиатами. Сама хлоугольная кислота неустойчива, её эфиры вполне устойчивы, хотя и отвратительно агрессивны (если с маленькими заместителями и летучи), напоминают фосген и по запаху, и по действию. Этерификация хлорангидридом требует добавления основания, а на самом деле нуклеофильного катализатора, пиридина – посмотрите на страничке про методы в карбоновых кислотах.
Так получают смешанные эфиры тех же фенолов, и например, когда исходный эфир не этиловый, а третбутиловый, это называется Boc. А если бензиловый, то Cbz, бывают и другие. Это такие защитные группы для гидроксила, довольно хлипкие впрочем.
Не может ли в этой конкретной реакции с фенолом образовываться продукт орто-присоединения?
АЧ: Это маловероятно, хотя в количестве нескольких процентов не исключено, так, что обнаружить это сможет только кто-то сильно озабоченный. Не бывает органических реакций без побочных. Но вообще орто-замещение в фенолах это почти всегда вторичный процесс, происходящий, ели атака электрофила по кислороду обратима в условиях реакции. Тогда атака по углероду, которая необратима в тех же условиях понемногу перетягивает продукты в эту сторону. В данном случае сложные эфиры фенолов в очень мягких условиях ацилирования вполне устойчивы, ацилирование в этих условиях можно считать необратимым. Поэтому можно не париться про орто-ацилирование (здесь – карбоксилирование).
Здравствуйте, Андрей Владимирович! У меня несколько вопросов:
1) Почему перед использованием сорбент с С18 цепочками должен быть подготовлен и увлажнен с помощью смеси органического растворителя с водой?
2) Для чего нужен сополимер полидивинилбензола с N-винилпирролидоном (сорбент Oasis HLB), если он удерживает и полярные, и неполярные молекулы?
3) Почему после сорбции бисфенола из воды на этом сорбенте бисфенол смывают смесью метанола с водой, а не, например, просто метанолом? А другие органические соединения ведь тоже попадут вместе с бисфенолом в раствор? Получается, получиться отделиться только от полярных молекул?
АЧ: Милостивый государь, доброй ночи! У меня встречный вопрос – Вы меня ни с кем не перепутали? Тут об органической химии идёт речь, а не о хроматографии. Некоторые простые соображения, возможно, завтра-послезавтра напишу, потому что я некогда встречался с сорбентами подобного типа, но вообще в любом деле надо искать специалиста, а не общие соображения, которыми вымощена дорога на свалку истории.
АЧ: Найдём 10 минут на эту совершенно постороннюю тему. Хроматография впрочем чрезвычайно важна для органической химии, и она не стоит на месте. Сначала несколько общих вещей.
Здесь почти все вопросы связаны с обращённо-фазовой хроматографией – это когда сорбент из высокополярного и снабжённого кислотными и/или основными центрами вещества (силикагеля, окиси алюминия и т.п. – это нормально-фазовая хроматография, к которой мы привыкли) превращают наоборот в нечто малополярное. Обычные (нормально-фазовые) сорбенты сильнее удерживают полярные вещества (а также те, у которых тоже есть кислотные или основные центры). Элюируют обычные сорбенты органическими растворителями, с очень простой закономерностью, грубо говоря, чем полярнее тем сильнее (а точнее есть общеизвестный элюотропный ряд растворителей по силе элюирования). Часто применяют градиентное элюирование – от плохого элюента к лучшим. Плохими элюентами в обычной хроматографии считаются неполярные углеводороды. Соответственно и элюирование происходит от менее полярным к более полярным (а также содержащим кислотные центры, кислые протоны и т.п.)
В обращенно-фазовой хроматографии берут сорбент с малополярной поверхностью. Но прямо такой сорбент найти трудно, поэтому берут хроматографический силикагель и пришивают к поверхности длинные углеводородные хвосты. Стандарт – октадецилсилил (ODS, C18). Элюируют высокополярными жидкостями, обычно метанолом или ацетонитрилом. В градиентном элюировании портят их не малополярными жидкостями, а водой. В этом типе хроматографии плохой элюент – вода (иногда буфер). Порядок элюирования обратный – сначала более полярные, потом менее. Совсем малополярные вещества выходят последними, когда элюируют уже почти чистым растворителем без воды.
Первый вопрос – зачем при подготовке сорбента используют водный растворитель. Подготовка сорбента – приведение его в максимально равновесное состояние для работы с элюентом. Поскольку элюент почти всегда содержит воду, сорбент необходимо «приучить» и к воде тоже. Сначала сорбент замачивают в основном элюенте – это приводит к тому, что хвосты С18 максимально распрямляются, и образуют такую плотную щётку, которая работает как жидкая неполярная фаза – она должна быть насыщена основным элюентом. Но никакое пришивание хвостов полностью не закрывает саму поверхность, а это силикагель (иногда частично прикрытый – endcapped – маленькими группами типа триметилсилилила), и эта остаточная поверхность должна быть гидратирована – достигнуто равновесие с водой. Иначе при движении элюента свойсва сор,ента будут меняться, а это всегда ухудшает эффективность разделения (пики хвостят и уширяются, плывут времена удержания).
Второй вопрос про особую фазу, не модифицированный силикагель, а полимер, на основе малополярной ароматической и полярной гидрофильной компонент. Здесь это фирменный сорбент Оазис ГЛБ. Такой сорбент применяют не для хроматографии, а для подготовки проб. Реальные пробы – всякие жидкости типа сточных вод или биологических жидкостей содержат много всякой дряни, от солей до биополимеров. И в этих жидкостях надо определять какую-нибудь органику типа лекарств или органических поллютантов. Если сразу все это дерьмо колоть в хроматограф, дорогущие колонки придется менять каждый день, да и точность определения из-за забивания сорбента дерьмом будет невелика. Поэтому пробы готовят, обычно с помощью твердофазной экстракции на какой-нибудь обращённо-фазовый сорбент, вот как раз типа этого сополимера. Такие сорбенты не всегда имеют неполярную фазу из насыщенных алкилов, но часто это например, полимер, содержащий ароматические кольца. Такой полимер будет иметь сродство не просто к малополярным веществам, а скорее к малополярным, но ароматическим или ненасыщенным – это дает некоторую долю более специфической сорбции, например, тоже ароматических молекул. То есть это скорее не просто обращённая фаза, а фаза со специфическим сродством к более узким классам соединений.
Пропускают через слой сорбента анализируемую жидкость – органика сорбируется, а всякую дрянь просто отмывают водой или сильноводным элюентом – органику это не смывает. Заодно происходит относительное концентрирование. Потому все сорбированное вымывают тем элюентом, который пойдет в хроматограф (или делают промежуточный смыв с концентрированием). Поскольку сорбент тоже обращённофазовый, его элюируют типичными растворителями для такой хроматографии, то есть тем же метанолом с добавлением воды. Количество воды подбирают по анализируемому веществу – смывание должно быть количественным, но есть возможность небольшого фракционирования, похожего на градиентное элюирование – чтобы смыть не все, а что-то малополярное осталось на сорбенте. Чем меньше растворенных веществ в пробе, тем больше точность определения нужного. Но надо понимать, что на этой стадии (пробоподготовки, твердофазной экстракции) нет задачи разделения – эту задачу будет решать хроматография, а сейчас важно максимально упростить задачу хроматографии, убрать всякую дрянь, повысить концентрацию определяемого вещества и т.п. Поэтому конечно будет смывать не только определяемое вещество, но и другие компоненты. А сотав элюента подбирают разработчики методики разделения и определения чисто экспериментально.
Здравствуйте!
Стоит ли какая-то история за реакцией Нефа? Неочевидно с первого взгляда, что при подкислении соли нитросоединения получится карбонильное соединение.
АЧ: Конечно, это очень интересная реакция. И что занятно – она у меня давно написана, года два назад. Я планировал страничку про алифатические нитросоединения и довольно далеко в ней продвинулся, но не доделал и отложил. Так и сидит в черновиках.
Я на днях посмотрю этот черновик и решу, выложить ли только Нефа в ответах, или может быть все же страничку в том виде как она есть. Там нет Анри, собственно поэтому я ее и отложил, это скучно, и дело завяло, что-то интереснее всплыло.
АЧ: Выложил страничку про алифатические нитросоединения, где есть и реакция Нефа.
Спасибо!
А про реакцию Анри отдельную страничку будете делать?
АЧ: Когда-нибудь обязательно. Но прогноз неутешительный. Очередь длинна, жизнь коротка. Посмотрим.
какая степень окисления углерода в циклопентене?
АЧ: А самим слабо прикинуть? Посмотрите на страничке про валентность в конце есть описание, как считать степени окисления в органике. Это не просто, а очень просто.
У циклопентена (а также у всех остальных циклоалкенов есть sp3-углероды, на каждом по два водорода, значит -2. И sp2-углероды, на каждом по одному водороду, значит -1.
Здравствуйте!
Может ли металлил хлорид реагировать с нуклеофилами не только через реакцию Sn2, а, например, через двойную связь с последующим отщеплением хлора и образованием более термодинамически выгодного алкена? Если да, то насколько это выгоднее?
АЧ: Вопрос довольно туманно сформулирован. Могут ли аллильные галогениды реагировать не по обычному SN2, то есть с тыловой атакой нуклеофила на тот углерод, который несет уходящую группу? Да, конечно, есть альтернативный путь реакции – атака на дальний от этого центра атом углерода двойной связи, при этом двойная связь перемещается на соседнее положение с одновременным уходом уходящей группы. Происходит аллильная перегруппировка, и образуется изомер нормального продукта. Такой механизм обычно называют SN2 штрих, тоже считают согласованным (и конечно есть и SN1-версия с образованием делокализованного аллильного катиона, который конкурентно связывает нуклеофил на один из концевых атомов аллильной системы – именно этот механизм лежит в основе конкуренции 1,2- и 1,4-присоединения электрофилов к диенам).
Аллильная перегруппировка (или аллильный сдвиг, allylic shift) в нуклеофильном замещении аллильных субстратов это совершенно обычное осложнение. И его очень трудно нормально описать а тем более предсказать, поэтому мы на 3-м курсе на этом не останавливаемся, иначе просто закопались бы без шансов выплыть. Хороших закономерностей там нет. Обычно, поскольку это согласованный механизм, управляет стерика, и это легко понять. Но, увы, это не так просто, потому что стереохимия двух центров принципиально разная: один самый обычный тетраэдрический углерод, но второй это плоский тригональный углерод с пи-системой и подходом сверху или снизу. Поэтому тут нельзя просто сравнить количество заместителей, и сказать – вот тут меньше, туда идём. Результат – это дико мутная область, которую обычно описывают как сумму накопленного опыта. Если вам это нужно по работе – разберетесь, или просто эксперимент сделаете. А так просто это разбирать – смысла не очень много.
Не знаю, специально ли Вы взяли металлилхлорид, или случайно попался, но в этом случае аллильная система симметрична, и различить по продуктам механизм не получится, пока не введете какую-нибудь изотопную метку. Соответственно термодинамика к этому отношения вовсе не имеет никакого.
Если Вы имели в виду что-то другое, уточните вопрос.
Хорошо, с металлил хлоридом опешил. Что если 3-хлор-бутен-1 ввести в реакцию Sn2, какое примерно соотношение продуктов можно ожидать? Можно ли как-то реакцию сместить в одну или другую сторону?
АЧ: Да какая разница? Повторю ещё раз – аллильные системы чрезвычайно сложны для какого-то разумного обобщения. Это же будет зависеть и от нуклеофила и от условий. В общем случае будет конкуренция SN2/SN2’/SN1/SN1′, а для SN2/SN2′ будете еще иметь альтернативу путей реакции с ранним и поздним переходными состояниями. В одном будет доминировать стерика, которую придется реально моделировать, во втором – добавится еще термодинамика. Поэтому Вы нигде не найдете обобщающих обзоров или книг по региоселективности аллильного замещения. Когда-то этим пытались заниматься, но быстро завяли.
Это химия. Если в конкретной работе есть такой вопрос, то пути два. Или подробно изучить прецеденты по литературе. В нормальном мире для этого есть реаксис или сцыфиндер. В мире традиционных ценностей двигайте в библиотеку, как мы сорок лет назад, там стоят еще наверняка тома бейльштейна и кемикал абстрактс. Учитесь пользоваться, и если не задохнётесь от пыли, то наберете за пару месяцев некоторое количество ссылок. Изучив их Вы поймёте, что потратили время и надышались пылью зря, потому что ссылки как-то совсем беспардонно противоречат друг другу. Но зато приобщитесь к традиционным ценностям, одной из которых и является хорошая пыль.
Тогда просто начнете сами делать реакции в своих условиях и со своим нуклеофилом, поняв простую вещь – в случае, когда продукта только два, реакцию проще сделать, чем гадать, как она пойдёт. Сейчас ведь ещё ЯМР есть, хотя это и не совсем традиционная ценность, так что скорее всего, через некоторое время придется вернуться к старой доброй фракционной перегонке и старому доброму показателю преломления.
Другой органической химии у меня для Вас нет. Может у кого-то есть, поищите.
Здравствуйте!
Можно ли как-то и, если можно, то как, прогнозировать растворимость тех или иных веществ в разных растворителях, отталкиваясь от структуры этих веществ?
АЧ: Конечно можно. И это делает любой профессиональный химик, работающий руками в препаративной химии, постоянно и почти инстинктивно – всё время нужно принимать решения – из чего перекристаллизовать, чем экстрагировать, полностью ли перешло вещество, какой растворитель брать для реакции, чем элюировать, чем высаживать и т.д.
Если коротко, то дя этого нужно представлять себе основные взаимодействия между веществами, и подбирать растворитель так, чтобы взаимодействия соответствовали (или наоборот, не соответствовали – для перекристаллизации и высаживания). Взаимодействий основных немного: водородные связи, полярность, гидрофобность, ароматический стекинг, галогенная связь, основность, кислотность.
По этим признакам и потому что важнейшей жидкостью в химии является вода, вы сначала делите вещества на гидрофобные и гидрофильные, а гидрофильные – на легкорастворимые в воде (или смешивающиеся с водой) и немного растворимые с водой (частично смешивающиеся). Легкорастворимые обычно содержат ионные группы (прямо соли) или очень полярные группировки с высокой степенью разделения заряда. Просто растворимые – группы с кислыми протонами, способные хорошо образовывать водородные связи (гидроксильные группы, особенно в виде вицинальных диолов, которые по влиянию на гидрофильность почти эквивалентны настоящим ионным группам). Дальше вы смотрите на молекулу и оцениваете размер ее гидрофобной части (самое гидрофобное – это насыщенные углеводородные части; умеренно гидрофобны ненасыщенные фрагменты и ароматические кольца, галогены, двухвалентная сера) и соотношение гидрофильной и гидрофобной части. Если гидрофильного много, а гидрофобного мало, вещество скорее всего легкорастворимо в воде. Если наоборот – оно сохраняет умеренную растворимость и это говорит о том, что например, его можно перекристаллизовать из водного растворителя, или наоборот, нужно быть особенно тщательным при экстракции из воды – вместо обычных трех раз может потребоваться пять-шесть и более полярным растворителем.
Дальше начинаются более тонкие вещи. Ионные вещества (настоящие соли), например, почти всегда совсем нерастворимы в эфирах и кетонах (эфир, ацетон).
Гидрофильные вещества, даже умеренно – нерастворимы в насыщенных углеводородах (пентан, гексан, петролей), но растворимы почти во всем остальном (ароматика, хлорорганика, эфиры, кетоны, нитрилы и т.д.).
Ароматика (бензол, толуол, хлорбензол, нитробензол) обладает способностью специально растворять большие полициклические ароматические соединения, которые почти нерастворимы во всем остальном.
Хлорорганика несмотря на собственную малополярность, но из-за так называемой галогенной связи обладает способностью растворять, хотя бы умеренно, даже сильнополярные (и часто даже и ионные) вещества, поэтому ее (дихлорметан, хлороформ, дихлорэтан, трихлорэтилен) так любят для экстракции – она почти никогда не подводит, хотя для сильнополярных веществ экстрагировать нужно долго,а лучше всего даже непрерывно. Она же, из-за той же связи галоген-пи-система хорошо тянет ароматику.
Вещества, обладающие некоторой основностью (азотистые и т.п.) хорошо относятся к растворителям с кислыми протонами (спирты, карбоновые кислоты).
Если в соединении есть металл, ионный или ковалентный, нужно брать кординирующие растворители, обладающие льюисовой основностью – эфиры (эфир, ТГФ, диметоксиэтан, диглим, диоксан и т.п.), нитрилы (ацетонитрил, или более высокие нитрилы, если там еще есть большая гидрофобная малополярная часть).
Есть и всякие специальные растворители с особыми свойствами. Например, нитрометан и другие небольшие нитроалканы потрясающе растворяют полярные и ионные вещества и при этом не смешиваются с водой.
Диметилсульфоксид хотя бы немного растворяет практически всё – от ионных веществ до совсем гидрофобных – поэтому его так любят для массового тестирования веществ и для ЯМР, но смешивается с водой и непригоден для экстракции.
Ну а дальше идет личный опыт каждого практикующего химика. Чем дольше работаешь, тем больше понимаешь, что в чём растворяется. И что обязательно найдется вещество, которое сможет удивить, отказавшись следовать любым рецептам. В химии ни одно правило не бывает совсем универсальным, потому что возможности вариации структуры молекул и их взаимодейтсвий бесконечны.
Как пользоваться правилами Вудварда-Хоффмана?
АЧ: Ой, только не это! Рисовать надо много, толку мало. Это же везде разобрано во всех видах, что я могу тут еще добавить. Я положу себе закладку, если в каком-то будущем найдётся свободное время, может быть. Но у меня гора недоделанных страниц в очень высокой степени готовности, но на которые времени не находится, что лучше я одну-две доделаю, чем эту общеизвестную систему буду в сотый раз описывать.
Почему тропон и трополон ароматичны, а малеимид неароматичен, хотя по той же логике должен быть антиароматичным?
АЧ: Ну простите, это очевидные вещи, которые разбирают буквально везде. А на второй вопрос Вы найдёте ответ, если просто прочитаете мою страничку про ароматичность или ответ на вопрос, почему антиароматичность можно ожидать только у малых циклов.
Если Вас не устраивают обычные объяснения, напишите, что конкретно Вам кажется сомнительным.
АЧ: И ещё – про малеимид, а заодно малеиновый ангидрид, бензохинон и т.п. Все эти вещи не могут быть ароматическими и антиароматическими в прямом смысле этих слов, так как не имеют циклической делокализации (по причине наличия экзоциклических кратных связей, которые мешают перемещать кратные связи в цикле в смысле структур Кекуле – а из этого следует, что все кратные связи в таких циклах локализованы). Другое дело, что некоторые граничные структуры (с поляризацией карбонилов) имеют признаки антиароматической делокализации. Это не антиароматичность в прямом смысле слова, а некоторый, довльно слабый дестабилизирующий фактор. Возможно, именно этим объясняется аномально высокая реакционная способность всех этих соединений, например, в реакции Дильса-Альдера и не только. Но это утверждение не имеет прямых доказательств.
Спасибо за ответ!
1. Если было бы поле для рисования, было бы удобнее.
2. Вот как я понимаю свой вопрос: у тропона есть граничная структура, где есть поляризация карбонила, из-за чего эта структура ароматична (6 электронов, плоская, связи усреднены); не может ли быть у малеимида (и подобных соединений) такой же граничной структуры, где одновременно оба карбонила поляризованы, либо один карбонил поляризован, а другой карбонил находится в виде “енола” [HO-C=N] (не могу вспомнить, как такие фрагменты называются)? Может ли малеимид приобрести структуру пиррола с двумя гидроксилами?
АЧ: Поля для рисования не будет. Можете письмо обычное написать, если хотите приложить что-то графическое. Вопрос я понял, и скажу по секрету, немного уже начал набрасывать ответ, потому что увидел в вопросе одну вещь, которую действительно стоит обсудить. Найду еще немного времени на днях, закончу.
Здравствуйте!
Почему в реакции Тиффено-Демьянова протекает с миграцией C-C связи, а не с образованием эпоксида? Или всё же можно сместить как-то эту реакцию в сторону образования последнего?
АЧ: Потому что это не что иное как разновидность пинакон-пинаколиновой перегруппировки. Почему при дегидратации диола не получается эпоксид? Потому же и при дезаминировании аминоспирта не получается эпоксид. Это одна и та же реакция, идущая через перегруппировку на оксокарбениевый ион. Это намного выгоднее, чем образовать напряжённый эпоксид. И сам эпоксид кислотами немедленно раскрывается ровно так же.
Здравствуйте!
Вписывается ли гибридизация орбиталей (метод валентных связей) в теорию МО или где-то есть расхождения?
Спасибо за ответы на предыдущие вопросы, очень интересно было читать.
АЧ: С таким же успехом можно спросить, вписывается ли санный спорт в чемпионат мира по футболу. Ответ простой – не вписывается, это разные виды спорта.
Так и тут. Во-первых, нет никакой теории МО, а гибридизация орбиталей относится к методу валентных связей как бублик к выхлопной трубе. Надеть можно, только непонятно зачем.
МО – это метод приближенного решения уравнения Шрёдингера, вполне хорошо реализованный. ВС – то же самое, только плохо реализованный и в общем не нужный никому, кроме нескольких сильно упорных деятелей, которые десятилетиями пытаются из этого что-то выдоить (некоторое количество статей и диссертаций выдоить получилось, а больше вроде бы ничего).
В методе МО и всех его надстройках, а также в методе функционала плотности вы получаете в принципе одно и то же – набор молекулярных орбиталей. Вот их вы и используете. Это довольно неприятное занятие для молекул хоть немного сложнее метана и бензола, потому что МО обычно включают в себя кучу атомных орбиталей с совсем периферийных атомов, все это дико захламляет картинку и мешает её разглядывать. Поэтому существуют чисто математические фокусы, позволяющие преобразовывать оригинальный набор МО в орбитали, более аккуратно локализованные на привычных нам фрагментах – такие орбитали проще разглядывать и они даже неплохо напоминают что-то такое, что мы из общих соображений рисуем.
Что касается гибридизации, то это просто совершенно тупая сумма атомных орбиталей – за гибридизацией не стоит ничего осмысленного ни в какой теории или методе. Если в какой-то МО на каком-то атоме оказались одновременно вклады и s и p -, а это случается нередко – вон посмотрите, что я тут на днях написал про нитрование на страничке Ответы – там есть такой пример – то вы и увидите нечто очень похожее на восьмерку с разными лопастями, как мы обычно гибридную орбиталь и рисуем. В электронной структуре всегда рулит симметрия, и для сигма-связей, а также МО, которые их обслуживают, s-АО и p-АО вдоль связи соответствуют симметрии и поэтому практически всегда входят в соотвесттвующие МО, создавая впечатление гибридной орбитали. Но – это только для второго периода. В периодах ниже s- и p-орбитали сильно разнятся и по энергии и по протяженности (а также внутренней начинке), поэтому там они обычно не входят в состав одной МО, и впечатление гибридизации пропадает.
АЧ: Забавно, я пролетел глазом по страничке и сразу несколько раз наткнулся на словосочестание “теория МО”. Это то, что называется устойчивым словосочетанием, въелось. В общем-то это не важно, теория так теория, но уместно спросить – а в чём теория? Просто привыкли мы так это называть. Ещё раз повторю – это не теория, а приближенный математический метод решения уравнения. Который дает приближенное представление о реальной вещи – уровнях энергии электронов в молекуле. И мы как-то довольно ловко насобачились этими приближенными решениями пользоваться в мирных целях – чтобы судить о других реальных вещах, свойствах молекул и реакционной способности. Обратите внимание здесь на главную засаду – мы не можем в полном смысле этого слова рассчитать эти свойства и параметры реакционной способности, а можем только получить некоторое приближенное представление о них. В принципе, именно это можно было бы назвать теорией – вот такое утверждение: знание МО (а есть некоторый объективный критерий предела этого метода, поэтому МО не являются произвольным следствием конкретного метода расчета, а в некотором смысле этого слова вполне объективны) дает нам хорошее приближение к свойствам и поведению вещества, составленного из молекул, подвергшихся вычислениям. Теорией это высказывание делает то, что мы можем как подтверждать, так и опровергать его, делая расчеты и сравнивая их с тем, что мы пытаемся объяснить.
Спасибо!
АЧ: Да, пожалуйста, извините за несколько задорный тон ответа. Это не значит что вопрос какой-то не такой, вопрос отличный и совершенно понятно, откуда идущий. К сожалению, в учебной литературе отношение к всем этим квантовым наворотам, типа МО и ВС, очень неадекватное. Всё это относится к дискуссиям более чем полувековой давности, которые в корректной литературе давно преодолены как неактуальные, но продолжают переписываться без какой-либо попытки осмысления в разных вторичных пособиях. Придется как-то с этим жить, понемногу самостоятельно разбираясь в том, что этого заслуживает. Я буду в дальнейшем стараться на новых страничках больше уделять внимания разбору МО и всего, что касается строения и электронной структуры, отделяя всякие глупости от содержательных вещей.
Здравствуйте!
Можно ли как-то увидеть разницу между ковалентной и ионной связью с помощью диаграмм молекулярных орбиталей?
Почему TiCl4 и SnCl4 – жидкости?
АЧ: Разделю на две части. По поводу МО ионных и ковалентных связей. Здесь есть одно досадное противоречие, которое несколько усложняет задачу. Чисто теоретически ничего не может быть проще. Ионная связь – это не химия, а физика, просто электростатическое притяжение зарядов. Поэтому у настоящей ионной связи нет представления в МО. Порядок ионной связи равен нулю. Порядок связи очень просто выясняется из молекулярных орбиталей – просто находите все орбитали, отвечающие за связывание между интересующими атомами (орбитали должны быть занятыми, а между атомами должна быть одинаковая фаза, а не узел), суммируете заселенности и получаете порядок связи. Программы расчетов делают это автоматически. Он вполне может быть дробным, может быть меньше единицы, но он не должен быть равен нулю. Вообще, если у нас ионная пара, то не должно быть МО, одновременно включающих атомы разных ионов. Допольнительно можно еще прикинуть расстояния между атомами. Ионная связь – это сумма ионных радиусов или больше. Если меньше, то либо есть вклад ковалентности, либо у нас проблемы с величинами ионных радиусов.
В реальности все это работает плохо. Причин тому миллион. Поэтому вы можете, например, озадачиться таким вопросом – существует ли молекула NaCl, чего проще. Понятно, что речь идет о газовой фазе, вакууме. В конденсированных фазах такой молекулы нет. В вакууме всяческие спектроскопии показывают наличие таких частиц. Но что это – молекула или просто ионная пара, слипшиеся ионы? Можете попробовать поискать литературу по этой проблеме. Убедитесь, что однозначный ответ дать чрезвычайно сложно – требуются расчеты очень высокого уровня, сильно за пределами того, что мы называем теорией МО и даже всякими надстройками, которые все же позволяют с кучей оговорок рассуждать о МО. В конце концов, обрушив на несчастную “молекулу” всю вычислительную мощь, вроде бы получается, что это ионная пара, а не молекула. Но сомнение остается. Хотя бы потому что атомы и ионы это не жесткие шарики, и не существует поверхности атома – плотность нигде не кончается, в том числе она продолжается и за пределами ионного радиуса. Безусловно, реальным химикам до таких проблем дела нет, но для теоретической химии вопросы остаются.
С ионами сложнее проблем будет еще больше. Большиснтво расчетных методов имеют мерзкую привычку завышать ковалентность. Опять потребуются сильно выходящие за уровни МО уровни теории. Где к чёрту летит всякая наглядность результатов.
Еще одна проблема уже более практическая. В химии мы имеем дело обычно с довольно сложными системами – с растворами или чем-то еще того сложнее. И если у нас, например, возникает вопрос типа такого – ацетат кальция построен как взаимодейсвующие ионы, или это все же настоящий комплекс с координационными связями и т.д. Но ясно, что нас интересует не абстрактное нечто гордо бороздящее межзвёздное пространство, а вещество в растворе – нас интересует, напрмиер, реакционная способность, потому что мы, например, решили, что это хороший источник ацетат-ионов или кислотный катализатор или еще что-то. Но как только мы этот ацетат кальция положим в раствор, мы быдем вынуждены считаться с тем, как это переходит в раствор, как взаимодействует с растворителем, с другими компонентами реакционной системы, образует сложные агрегаты и т.д. В этом месте вы потеряете всякую надежду докопаться до отдельных взаимодействий на уровне ионный/ковалентный.
Это большая проблема в химии, на самом деле. Хороших и простых ответов почти никогда нет. Поэтому мы сплошь и рядом видим ситуации, когда люди просто не заморачиваются ионностью/ковалентностью, и например, рисуют в явном виде связи даже со щелочными металлами (например, BuLi рисуют как Bu-Li, а не Bu(-)Li(+)), потому что для их конкретных целей это просто не важно. Химия намного сложнее, чем иногда кажется.
АЧ: Про жидкие тетрахлориды олова и титана. Это любопытная история, совсем не ограничиваюшаяся этими соединениями. Скажем так: почему галогенпроизводные элементов в высоких степенях окисления (от 4 и выше) очень часто бывают не кристаллическими, а жидкими,а иногда и газообразными; и даже если они твердые, то легкоплавкие и летучие. Уж совсем знаменитый, в том числе и печально, пример – шестифтористый уран, который многие считают даже газом, но это всё же кристаллическое вещество, но невероятно летучее.
При этом это часто молекулы с большой молярной массой, с очень сильно поляризованными (но не ионными) связями, и что самое страшное – в серединке у таких молекул почти всегда находится чрезвычайно реакционноспособный, сильно координационно ненасыщенный атом с немаленьким зарядом.
Но чудес не бывает. Когда мы спрашиваем себя, почему некое вещество не хочет кристаллизоваться, или кристаллизуется, но очень легко плавится, и очень легко летит это однозначно свидетельствует о том, что у молекул этого вещества нет возможности образовать более-менее прочные межмолекулярные взаимодействия с такими же молекулами. Вроде должны же быть? Спрашиваем себя – какие? Центральный атом почти всегда кислота Льюиса и очень часто сильная (современная химия разрешила нам использовать это прилагательное с кислотами Льюиса), и это почти всегда ярко отражается на свойствах таких соединений – они жадно связывают другие лиганды, например, воду – многие дымят на воздухе как ненормальные. Причина разная – у соединений переходных металлов это очевидная координационная ненасыщенность (у тетрахлорида титана счет электронов – восемь, у титана минимум два свободных координационных места (формально даже пять, но у атома титана нет столько места вокруг). У соединений главных групп – гипервалентность, острое желание добить к.ч. до октаэдра.
Но в чистом веществе нет других лигандов. Есть другие молекулы того же. И чтобы взаимодействовать нужно образовывать мостиковые связи – трёхцентровые. Они всегда слабее обычных ковалетных. Возникает обычная термодинамическая проблема – что выгоднее, оставить только двухцентровые ковалентные связи, или часть их ослабить переведя в трехцентровые мостиковые, и получить еще некоторую выгоду на увеличении координационного числа. Это всегда вопрос баланса. Есть молекулы, для которых это невыгодно, а есть те, для которых это выгодно. Возьмите какую-нибудь мощную книгу по химии элементов, того же Гринвуда-Ёрншоу или Виберга-Виберга-Холлемана, где аккуратно сведены свойства и структуры соединений, и прогуляйтесь по высоковалентным фторидам и хлоридам элементов – увидите оба варианта, часто даже в одной группе, причем ниже будут чаще встречаться димеры и олигомеры с мостиками (летучие твердые вещества), а выше будут чаще попадаться летучие жидкости.
Никаких других опций для галогенпроизводных нет – либо мономер, либо олигомеризация через мостики. У высоковалентных соединений не бывает ионных связей даже с галогенами, по очевидной причине – вы ободрали атом и увеличили очень сильно эффективную электроотрицательность, поэтому даже фторид не сможет рядом с таким атомом удержать свои электроны и противостоять обратному смещению электронной плотности, то есть ковалентности. Есть еще один способ – передать один или два галогенида такой же молекуле, насытив ее координационную сферу, и оставшись катионом или дикатионом – тогда мы получим ионное соединение, кристаллическое. Но это редко бывает потому что тот, у кого отнимают, становится еще более ненасыщенным и жадным и требует свое обратно. В кристалле иногда бывает, что можно получить компенсацию за счет энергии ионной решетки. Я думаю, что такие примеры есть, но их мало, и что-то я сейчас ничего вспомнить не могу.
И еще одно важное обстоятельство. Допустим, у нас не получается образовать мостики – невыгодно. А почему сами такие молекулы не пакуются в кристалл – они же часто весьма симметричные, тяжёлые. А потому что кристаллы образуются за счет взаимодействий, отвечающих за притяжение (это или разные виды электростатики, в том числе всякие модные сигма-дырки, если у нас нет никакой ковалентности. а ее уже нет, мы уже от нее отказались). Но такие молекулы всегда устроены одинаково – в середине атом с возможностями, но мы (мы – это другие молекулы) его не видим, потому что он окружен атомами галогенов,, которые очень хорошо экранируют серединку. Нам кажется. что четырех атомов для этого мало. Посмотрите. как это будет выглядеть, если представить атомы ван-дер-ваальсовыми радиусами – и увидите. что серединка в лучшем случае просвечивает в щёлочки между внешними шарами – и кто в эту щёлочку протиснется? А нам и некому – есть только такие же молекулы. Экранирование работает великолепно особенно в верхних периодах даже маленькими фторами. Есть совершенно поразительная молекула гексафторида серы, где добраться до атома серы просто невозможно – этой газ, которым можно дышать и который спокойно булькает через горячую щёлочь. Другие молекулы этого типа не такие поразительные, но тоже экранированием защищают себя хотя бы от димеризации.
Здравствуйте, Андрей Владимирович!
Не так давно в книге Ингольда «Теоретические основы органической химии» увидел данные о том, что коричная кислота и 2-фенилнитроэтилен при нитровании (вроде бы в условиях ничего необычного не было, ссылку на работу, цитируемую в книге прикреплю) дают очень малое количество продукта мета-замещения (~2%).
В связи с этим вопрос: разве этиленовый фрагмент не будет выступать эдаким мостом для переноса мезомерного акцепторного влияния карбоксильной и нитро-группы?
Или не стоит под таким углом эти системы рассматривать и дело в совершенно других эффектах?
Отдельное спасибо за вашу работу над сайтами!
J. Am. Chem. Soc. 1948, 70, 3, 1191–1193
Вот ссылка на оригинальную работу.
Спасибо за добрые слова. Да, это любопытная история, и вполне достоверная. Есть еще несколько работ, в которых этот эффект воспроизводится. У меня есть одна гипотеза, почему это работает именно так. Собственно я всегда это говорю, выражая в виде такой максимы: мезомерный эффект не может быть дестабилизирующим. Сопряжение может и часто бывает дестабилизирующим, а мезомерный эффект нет.
Но чтобы не быть голословным, мне надо посчитать несколько структур. Некоторое время это займет. Тогда напишу.
Спасибо!
АЧ: Ответил весьма подробно, потому что с запасом и на вырост, на страничке Ответы.
Спасибо за очень подробный разбор этого интересного эффекта!
Здравствуйте!
Объясните, пожалуйста, наглядно, как работает уравнение Гаммета(-Тафта), и зачем (кому?) оно вообще нужно?
АЧ: Простите, а наглядно это как? Вот же в тупик поставили. Наглядно объяснить, как работает уравнение. Которое на деле вовсе никакое не уравнение.
Вообще это дикая тоска, хоть наглядно, хоть ненаглядно. Давно надо было что-то написать по этому поводу, потому что это очень полезная штука. И столь же бесполезная. Это великий прорыв в органической химии. Который стал причиной одного из величайших провалов, просто эпического масштаба.
Я подумаю. Быстро не обещаю, а что самое главное, это точно будет коротко. Но и вас прошу всё же объяснить мне, что значит “наглядно”. А то может и браться не стоит.
Подразумевалось спросить у вас, чтобы вы на каких-то примерах показали, как это все работает.
Хорошо, я понял. Наберитесь тогда терпения, возможно, и правда настало время уделить этому внимание. Попробую в некоторой перспективе найти несколько дней для этой темы.
Здравствуйте!
Почему при образовании “кротона”, который протекает в основных условиях (Е1cb механизм) из “альдоля”, наиболее кислым оказывается протон, из которого получается карбанион, а не протон на спиртовой группе альдоля? Неужели и тут термодинамика решает, что ей лучше пойти именно по пути образования карбаниона, а не просто образовать соль, или мы просто греем посильнее в данном случае, чтобы утащить из реакционной сферы воду, чтобы сместить равновесие? Обьясните, пожалуйста.
АЧ: Приветствую! Кротон – это растение семейства молочайных, даже целый род. Кротоновая кислота оттуда.
Про элиминирование и место депротонирования. Еще раз повторяю – это уже в куче мест было – протон отщепляется из всех мест с хотя бы минимально сравнимой кислотностью. OH-кислотность от CH-кислотности в енолизуемом положении отличается не более чем на пять единиц рК. Да даже и десять не трагедия. Это просто значит что в равновесии енолята на пять-да даже пусть десять порядков меньше чем алкоголята. Число Авогадро видели когда-нибудь? Поделите-ка его на десять миллиардов. Сколько осталось? Сдается мне что дофига по сравнению с этим числом – сущий мизер.
Поэтому у вас в системе есть енолят и есть алкоголят. Алкоголят ни во что не превращается, просто находится в равновесии. А енолят вступает в элиминирование и уходит из своего равновесия. Это выгодно, только тогда когда при этом получается сопряженная с карбонилом и еще с чем-нибудь типа фенила двойная связь – цепь сопряжения. Енолят выходит из своего равновесия депротонирования. Равновесие подкачивает еще – и это туда же. Возникает канал выхода из равновесия в конкретный продукт – дегидратации альдоля. Все туда в конце концов и свалится.
Если альдоль простой, и двойная связь будет сопряжена только с карбонилом, это обычно не работает. Выгода элиминирования меньше, скорость реакции сильно меньше, а при очень малом содержании енолята в системе это дает пренебрежимо малую скорость реакции.
Вывод: во все случаях конкурентного депротонирования из нескольких мест, может выиграть то, которое имеет продолжение в виде выгодной и формально необратимой реакции, даже если разность в кислотностях составляет много единиц рК (все равно разумное количество, так как скорости реакция всегда зависят от концентраций, и если концентрация слишком мала, никакая выгодность не вытащит такой путь.
АЧ: Более подробный ответ на странице Ответы.
Здравствуйте!
Можно ли как-то повысить нуклеофильную способность у амидов (первичных/вторичных), имидов и мочевин? Синтез Габриэля наталкивает на мысль, что как нуклеофил фталимид калия неплох, ещё вы говорили про диформиламид натрия, но тут речь про соли этих соединений; легко ли, например, снять протон с мочевины и пустить получившуюся соль в реакцию SN2 с чем-нибудь?
АЧ: Во-первых, как нуклеофил фталимид калия очень плох – он сильно делокализован, и даже по критерию основность-нуклеофильность не проходит; NH-кислотность фталимида весьма высока, фталимиды натрия и калия получаются просто действием щёлочи на фталимид, и даже водой толком не гидролизуются. Метод Габриэля – старинная химия 1895-го года использует жестокий способ делать реакцию – без растворителя смешивают фталимид калия и галогенпроизводное и просто жучат всё это пламенем горелки пока дым не пойдёт. И используются только самые активные в SN2 субстраты типа бензилгалогенидов и галогенкарбоновых кислот. Большого значения метод Габриэля давно не имеет, но его бережно сохраняют ради нескольких удобных методик. Никаких особенно более культурных методов делать эту реакцию нет, апротонные полярные растворители ее несильно ускоряют, так как анион сильно делокализован и от сольватации зависит слабо. С диформиламидом похожая история – опять только активные субстраты, и реакция в смеси формамида-диметилформамида с хорошим и длительным нагреванием.
Амиды, имиды, мочевины – очень слабые нуклеофилы. Алкилировать можно только через депротонирование, и это несложно, потому что кислотность довольно приличная. Эти реакции малопопулярны, потому что опять сильно ограничены субстратами, опять тот же набор в основном. Методик и протоколов опубликовано немало. Обычно это что-то типа межфазных условий, щелочь или более сильные основания, растворители типа ДМСО, нагревание. Выходы скромные, всегда есть опасность разложения амида или мочевины сильным основанием. В этой химии в общем предпочитают сразу сделать нужный амид или мочевину, тем более что методы отлично отработаны и исходных полно. Есть обширная химия не алкилирования, а арилирования амидов и мочевин, с помощью комплексом переходных металлов, палладия и меди, но алкилирование так не делается.
Доброго времени суток;
спасибо, было очень интересно про амбидентные нуклеофилы!
>В конкурсе на самую идиотскую статью всех времен и народов это, я уверен, один из самых достойных претендентов на второе место (почему на второе – потому что первое занято).
А кем занято первое?
И как скоро можно ожидать лекцию по нобелевке 2022?
АЧ: Спасибо за высокую оценку странички. Я давно собирался, но откладывал, потому что радости мало в этом вздоре копаться. Но как всегда бывает и для себя кое-что занятное нашёл, в основном из серии “как тесен мир, а химия ещё теснее”. Да уж теперь и отделался, что не может не радовать. Так что спасибо, что подвигли меня на это.
Насчет первого месте, во-первых, в этом конкурсе как на международной олимпиаде по химии семьдесят три первых места, сто тридцать вторых, а сколько третьих до сих пор сосчитать не могут. Придет время расскажу о некоторых кандидатах.
Над лекцией работаю. Закопался, потому что параллельно несколько вещей всплыло и вообще у меня новая идея появилась. Пока не скажу какая. Но больше никаких прогнозов по времени, но точно работаю, и точно время выхода неумолимо приближается.
Добрый день, Андрей Владимирович.
Почему в органической химии принято писать именно схемы реакций, а не их уравнения?
АЧ: В Москве сейчас полвторого, поэтому Доброй ночи. Что значит принято и кем принято. Если речь идет о профессиональных химиках, занимающихся исследованиями или разработкой промышленных процессов, то пишут и схемы и уравнения. Схемы реакций пишут, когда нужно придумать или коротко описать путь синтеза и вас интересует, какие соединения и какими способами превращаются друг в друга. Вас интересует исходное и продукт, и над стрелкой условное обозначение основных реагентов, иногда с важными деталями (соотношения, загрузки катализаторов, растворитель, основные условия) – если вы уже знаете эти детали; или без них, если только начинаете планировать и пока ничего не знаете, кроме того, что и из чего хотите получить.
Уравнения обязательно пишут, когда приступают к реальному выполнению синтеза, потому что вам нужно посчитать загрузки реагентов, избытки, и т.п. Хороший химик обязательно сам проверяет весь расчет по стехиометрии (которая берется из уравнения), даже если методика взята из лучшего препаративного мануала типа СОПа, или статьи какого-нибудь солнцеликого светилы, типа Вудварда. Ошибки есть всегда и запоротый синтез из-за опечатки – ваша проблема, а не Вудварда. Себя самого надо проверять, что уж там.
В органике правда есть малоприятная проблема того, что вы не всегда знаете стехиометрию. Вы точно знаете исходное и конечное, а вот сколько нужно реагентов знаете не всегда. Вы не можете рассчитать количества катализаторов и вспомогательных веществ. Да и в окислительно-восстановительных реакциях в органике стехиометрия известна не всегда. В таких случаях уравнение это исходное = конечное, а все остальное берется из методики и просто масштабируется по загрузке исходного.
Расскажите, пожалуйста, про амбидентантные нуклеофилы, и можно ли, скажем, получать реакцией SN2 (или каким-то более изощренным способом) изонитрилы и изоцианаты, используя цианиды и цианаты металлов?
АЧ: Сколько лет я откладывал эту дурацкую тему. Догнали. Но может и неплохо, проще отделаться и забыть. Вот, целую страницу новую накатал. Читать только если правда интересно. В меню, где нуклеофильность. Ссылку наверх ставить не буду. Потому что чем меньше будут забивать себе голову этой мутью, тем лучше.
Андрей Владимирович, вот есть много всяких разных слабо или ненуклеофильных именно органических оснований, от DIPEA, HDMS до DBU и протонных губок, а в чем собственно отличие, где эти штуки хороши, а где можно и триэтиламином/пиридином обойтись?
АЧ: Это интересная тема, но пока отложу более подробный рассказ. Очень кратко. Во-первых, основания в органке бывают анионные и неионные. Про анионные более-менее всё понятно – движемся от кислородных (предел это трет-алкоголяты в ДМСО), к азотным (амиды, где-то до pK44), куда-то сюда вклинивается гидрид калия (но не натрия) для шибко смелых, и углеродным – литийорганика, литийорганика с комплекссобразователями, дальше чем ближе к свободному карбаниону тем сильнее, но совсем близко подобраться невозможно. Где-то на полтиннике дело заканчивается. Алканы и циклоалканы, кроме малых, напрямую депротонировать невозможно и это вроде бы непреодолимо на Земле в колбах.
Неионные. В основном это производные азота, хотя есть и другие атомы с хорошей основностью, но это экзотика. Неионные производные углерода – это карбены, там хороший диапазон основности, но это немного экзотика.
Азотные от слабых совсем типа пиридина (pK 5). 2,6-дизамещенные пиридины и диалкиламинопиридины – до 10. Третичные алифатические амины до 12-13. Еще чуть больше всякие каркасы типа хинуклидина. Стерически затрудненные третичные амины типа Хюнига (DIPEA) это там же. Дальше дырка, а после 20-22 начинаются амидины и гуанидины. Самое популярное основание здесь DBU (около 24). На этом обычные основания заканчиваются и начинаются супероснования – Веркаде (фосфорное основание), фосфазены (азотные) и всякие аналоги – pK можно довести почти до 40, но кровавыми трудами и очень большими и труднодоступными молекулами. Эта химия впечатляет тем, что про нее хочется читать, но самому делать не хочется.
Ионные отличаются от неионных тем, что а) у них есть противоион, и это иногда мешает; б) они растворимы только в полярных или координирующих растворителях, а если учесть ограничения по собственному депротонированию растворителя, то диапазон неприятно быстро упирается в эфиры (ТГФ, ДМЭ, диглим и т.п.) и работать приходится быстро и при минусах. в) преимущество в том, что можно подраскачать апротонным полярным растворителем типа ДМСО, но это только до рК 30-32, дальше ДМСО сам превращается в сильное основание, и работает как ограничитель где-то на уровне 34.
Неионные отличаются от ионных удобством работы. Но на рК не влияет растворитель (только надо учитывать, что шкалы рК в разных растворителях разные, и например, шкала в ацетонитриле почти на 5 единиц больше, что иногда вызывает недоразумения, когда сравнивают рК ионного основания в воде или ДМСО и неионного в ацетонитриле – кажется, что неионные сильно сильнее, и ща мы ацетиленовый протон снесем – чёрта с два! Зато диапазон растворителей очень большой, часто годятся даже углеводороды, ацетонитрил очень удобная штука, совсем непригодная для ионных; даже банальный дихлорметан имеет немаленький диапазон устойчивости к основаниям, потому что альфа-элиминирование довольно далеко.
Основания нужны для а) количественного депротонирования кислот, и для этого надо хорошо понимать величины рК, птому что понадобится запас минимум в 5 единиц и в данной среде;
б) для основного катализа – и это безбрежное море, здесь рК должно быть грубо сравнимо с тем, что нужно раскачать, можно даже меньше, но максимум на 4-5 единиц, а результат будет сильно зависеть от кинетики.
в) для E2-элиминирования, и здесь основность не помешает, поэтому, например, DBU так популярно в элиминировании – ещё и нагреть хорошо можно, DBU малолетуче.
Во многих случаях для основания важно подавлять нуклеофильность. Нуклеофильность дает другие реакции – замещение, реакции по карбонилу, другие присоединения. Если есть проблема с такими побочными, а она есть в каждой второй реакции, нужно предпочесть стерически затрудненное основание – лутидин или ди-тБу-пиридин вместо пиридина; основание Хюнига или тетраметилпиперидин (ТМП) вместо триэтиламина. DBU сам считается достаточно малонуклеофильным, хотя там бывают проблемы, но можно поднагреть, что часто смещает равновесия в сторону самого тупого – депротонирования.
Упоминалась еще протонная губка. Это скорее диковина, чем нечто полезное. Основность протонной губки невелика, только чуть выше триэтиламина. У нее есть одно класное свойство, которое иногда очень может пригодиться – она и правда сжирает протон. Дело в том, что большинство оснований в протонированной форме очень здорово делятся этим протоном водородными связями с другими реагентами. Как правило это ерунда, но бывает, что ищут ситуации, когда водородные связи надо максимально убрать. В протонированной губке протон закрыт с двух сторон азотами, и у него больше нет возможностей участвовать в зругих водородных связях. В общем, именно поэтому это соединение (при совсем невыдающейся основности) снискало такую славу – Протонная Губка(трейдмарк) – не в стоге нашли, передний край. Вообще, протонных губок очень много, это такой класс неионных оснований, эта, которая с большой буквы, просто первая и самая знаменитая. Везде надо быть первым, даже в губках.
Пока всё. Однажды напишу подробнее, но не очень скоро, но скорее всего в этом году.
АЧ: У меня большая просьба не задавать вопросы в виде уточнения каких-то очень старых вопросов. Чёрта-с два найдёшь, где это.
АЧ: Вот последний вопрос. Я его выудил с какого-то дна.
— Андрей Владимирович, есть ли какой-то доступный и несложный способ сделать NaHMDS, чтоб прям в дырявом ведерке и с протекающими капельными воронками (я утрирую)? —
Отвечаю. Нет, ни в разбитом корыте, ни в утятнице, ни в старой перечнице сделать силазид натрия не получится. Я вообще его никогда не делал, но делал литиевый, причем в 1980-х, то есть почти в ржавом ведре. Получилось неплохо. Красивые и необычные кристаллы, очень удобные в работе. Но литий это не натрий, литиевый силазид делается с помощью обычной литийорганики, не помню, что это было, були или фели. И это обычная работа, не намного сложнее хорошего гриньяра.
Но для натриевого силазида металлирующий реагент подобрать сложнее. Скорее всего это амид натрия, все остальные содирующие (это термин такой, вполне теперь встречается) реагенты труднодоступны, потому что нужно очень сильное основание, гидрид не годится, нужны или чистый RNa (основания Лохманна-Шлоссера не годятся) или действительно амид. Возможно, хотя я сильно не уверен, можно попробовать in situ натрийорганику, которая образуется при действии натрия на что-то типа стирола. Но хорошо ли отделится силазид от олигомеров, которые при этом получаются, я не знаю. Достоинством (и проблемой) силазидов является их легкая растворимость почти во всем, включая углеводороды, отмыть поэтому невозможно.
С амидом натрия, даже если это культурная суспензия в минеральном масле, работать трудно и опасно. Никаких вольностей он не простит, посуда и реактивы должны быть отличного качества.
Проблема еще в том, что последние лет тридцать-сорок щелочные силазиды никто не получает – их просто покупают готовые, они очень удобны, или кристаллические, или растворы. В нормальном мире это вообще не проблема. Их намного проще транспортировать, чем литийорганику – никаких проблем с пирофорностью, а в твердом виде вообще не опаснее соды. Они еще и отлично хранятся, и при необходимости их даже можно перегнать (но нужен хороший вакуум). Поэтому никто, скорее всего, я не проверял, не озаботился улучшением и упрощением методики (оригинальная методика 1961 года). Для оборудованной лаборатории с хорошей сухой камерой в производстве реактивов амид натрия – удобный и дешевый реагент.
Спасибо!
С запихиванием вопроса в реплаи вышло случайно, т.к. писать с телефона неудобно, форма для вопросов находится в самом конце страницы, и пришлось листать в самый низ, а я видимо задел “Уточнить вопрос”, и форма внизу пропала/заменилась на форму для реплая. Я это понял вот только как вы ответили.
АЧ: Я добавил сверху кнопу, которая будет быстро переносить на форму вопросов. Посимпатичнее потом сделаю, сейчас лень. Вообще эту страничку хорошо бы переверстать, но у меня нет хороших идей, и я нигде не видел прототип, которому хотелось бы подражать.
Андрей Владимирович, с новым учебным годом! Скажите, возможно ли проводить реакцию Sn2 с такими субстратами, где есть две хороших уходящих группы при одном атоме (дихлорметан, например, или этиловый эфир дибромуксусной кислоты)? Можно ли проворачивать Sn2 с субстратами, где есть хорошая уходящая группа при двойной связи (винилтозилат, например)?
АЧ: С новым учебным годом лучше поздравлять тех, кто не имеет к учебному году, ни новому, ни старому никакого отношения. В противном случае, это все равно что поздравлять лошадь с тем, что карета подана.
На второй вопрос сразу: ни в коем случае мы не говорим про SN2, если углерод не sp3. У винильных, фенильных, ацетиленильных производных тоже есть нуклеофильное замещение, но по совершенно другим механизмам и в совершенно других условиях. Теория SN2-замещения там не работает. Так что если у вас есть винилтозилат, а это довольно труднодоступное соединение, то делать с ним замещение можно, но не так как мы это делаем с алкилтозилатами. И это будет очень плохой субстрат, склонный к конкурентному замещению по атому серы. Так что мороки много, толку мало.
На первый вопрос – да, конечно, и таких реакций немало. Но надо очень хорошо понимать, что получается в результате замещения, и какие у этого свойства и вообще устойчиво ли это. К тому же дигалогензамещенные могут реагировать с теми нуклеофилами, которые являются еще и сильными основаниями, через альфа-элиминирование и образование карбена, и его дальнейшие реакции. В общем, здесь потребуется нечто большее, чем просто общие соображения. Но никаких принципиальных запретов нет, хотя это не самые лучшие субстраты в основном по стерическим причинам.
Спасибо!
И ещё вопрос – выложите ли вы лекцию про прошлогоднюю нобелевку?
АЧ: Да, надеюсь до конца января. Застрял немного.
АЧ: У меня вопрос к последнему задавшему вопрос – вот этот:
Здравствуйте. После прочтения статьи про резонансные структуры захотелось задать соответствующий вопрос. У Вас там сказано: (цитирую) “Результат будет противоположным – в реальной структуре будет преобладать вклад аниона с минусом на менее замещённом атоме, потому что во второй структуре индуктивные доноры дестабилизируют минус.”. Но разве, когда на углероде будет заряд (-), он не будет, наоборот, отдавать электронную плотность, так, что метильные группы будет акцепторами?
А как Вы умудрились запихать его куда-то в конец этого треда, да ещё и зарыть в реплаи?
АЧ: Отвечаю. Вопрос странный. Я советую хорошенько ещё раз ознакомиться с электронными эффектами. Важно понять, что они просто описывают те реальные смещения электронной плотности, которые уже произошли в молекуле, можно сказать, в момент ее появления на свет. Дальше там уже ничего не смещается, как это можно понять по Вашему недоумению, Вы мыслите это как будто так, что плотность сначала куда-то сместилась, а потому окружающие группы почесали репы, решили, что как-то нехорошо получилось, и стали её, плотность дополнительно между собой тасовать. Чем-то этот процесс выборы напоминает в одной незадачливой стране.
Но нет, смещение происходит один раз с учетом баланса эффектов. Этот баланс важно уметь оценивать (на глазок, как всё в химии). Когда речь заходит про мезомерию, мы считаем это более сильным способом смещения плотности, рисуем все возможные граничные структуры, и дальше на глазок оцениваем их вклад в реальную. Наибольший вклад дают те структуры, которые имеют больше причин для стабилизации зарядов на атомах, а это очень легко оценивается во-первых, по электроотрицательности (минусу лучше на более электроотрицательном атоме, а плюсу – на более электроположительном). И во-вторых, по дополнительным немезомерным эффектам, а мы собственно ничего кроме индуктивного больше не знаем. Индуктивные доноры дестабилизируют минус и стабилизируют плюс, и наоборот. Это простая и довольно эффективная схема, которая позволяет на глазок оценить вклады граничных структур.
Метильные группы являются индуктивными донорами. Они дестабилизируют минус, как и положено донорам.
Возможно, Вы где-то нашли рассуждения о том, что иногда метилы могут делокализовать избыток электронной плотности по механизму поляруемости. Это крайне спорная вещь, которая проявляется только как очень тонкий эффект там, где всё остальное отыграно. Да и обычно явления, которые пытаются объяснить таким способом, легче объясняются чем-то другим (гиперконъюгацией, регибридизацией, сольватацией, стерикой). Для оценки вклада граничных структур это совершенно бесполезный эффект, так как его заведомо не будет видно (даже если он есть) на фоне куда более сильного смещения за счёт мезомерии.
Добрый вечер, уважаемый Андрей Владимирович! Уже несколько моих писем Вам осталось без ответа. Может быть, они попадают в спам? Воспользовался этой страничкой, чтобы связаться с Вами и прояснить ситуацию.
Вопрос, возможно, не по адресу, но я его задам: есть банка с H3PO3 (фосфористой кислотой), USSR grade, даже не обводненной вроде. Как бы вы её (кислоту) чистили от примесей (меня пугает неиллюзорное наличие H3PO4 там)? Дальше я буду из него варить всякие фосфорорганические штуки, не хочется получить 0.0% продукта на первой же стадии…
АЧ: Старинная фосфористая кислота почти наверняка содержит фосфорную. Проще всего было бы ее выкинуть и найти свежую, какого-нибудь более вменяемого grade. Не думаю, что её легко очистить, потому что она по свойствам не сильно от фосфорной отличается. Вроде бы её можно перекристаллизовать, но это наверняка то ещё занятие, и точную методику искать придётся долго. Впрочем, я бы посоветовал поискатьво всех издания Брауэра и Неорганических синтезов. Если там есть методика получения фосфористой кислоты, то может быть и процедура очистки.
Я бы вообще сначала посоветовал сделать пробный синтез с этой и посмотреть, что получится. Если в одной реакции ничего не выйдет, это, право, не трагедия. В химии неудачи случаются нередко, и если Вас это пугает, то возможно, Вы ещё не поняли, что это за профессия. К тому же, мне немного странно, что за производные получают из самой фосфористой кислоты, обычно берут ее эфиры обеих форм, которые легко найти чистыми, да и очистить можно, или тригалогениды фосфора.
В любом случае, если хотите что-то конкретное делать в препаративной работе и возникают сложности, всегда ищите людей, у которых есть конкретный опыт работы именно с этим веществом. В химии плохо работают так называемые общие соображения, потому что у каждого вещества свой норов, и кто-то уже, возможно, с этим справился своими руками, осталось только найти кто.
Доброго дня!
Что, кстати, думаете насчет недавней нобелевки по химии? Есть ли что интересное поведать на этот раз?
АЧ: Добрый вечер. Хорошая нобелевка, своевременная. У меня было заблуждение – я считал, что в одни руки двух не дают, и это якобы прописано в завещании Нобеля. Но это оказалось неверно, и тогда проблем нет. Тем более, что остряки намекают, что Шарплессу дали не две нобелевки, а две полу-нобелевки, а значит одну полную, и тем более проблем нет.
Писать ничего не буду. Я недавно на эту тему выдал лекцию, и когда этот курс лекций закончится (где-то в середине декабря), я просто выложу ту лекцию на сайт, сделаю страничку и выложу. Придется набраться терпения, если это интересно.
Здравствуйте! Как вообще реакция превращения терминального алкина в интернальный работает (и наоборот)? Неужели у терминального протона алкина pKa больше, чем у протона в по соседству с алкильной группой?
АЧ: Нет, конечно, кислотность терминального протона намного больше, чем пропаргильных. Но так работает равновесие. Вы сами можете это понять, если просто представите себе равновесие между ацетиленами – териминальным и интернальными (и еще алленами), а не между карбанионами. У меня где-то это очень подробно расписано, скорее всего, где-то в черновикахю На днях посмотрю, насколько это готово, и может быть выложу, хотя бы частично.
“более редкие случаи пока не обсуждали:
3) Y– + RX+, например Вг– + R4N+ = RBr + R3N;
4) Y: + RX+, например R3N + R’SR”2 = R3NR’+ + SR”2”
Доброго времени суток, расскажите, пожалуйста, всегда ли четвертичные аммониевые соли будут вступать в реакции SN2 или тут тоже есть какие-то ограничения? Вообще расскажите, пожалуйста, когда эти типы реакций выше можно использовать. Спасибо!
АЧ: Нет, конечно, никто не отменял того, что амин – плохая уходящая группа, поэтому просто так четвертичные амонийные соли в качестве субстратов для замещения не используют. Такая группа отлично замещается, только если субстрат особо активированный к замещению, а это все в основном в химии гетероциклов (пирролов, индолов и т.п.) или переходной металлоорганики типа ферроцена. Здесь мы это не будем рассматривать.
В обычной химии скорее иногда можно встретить побочные реакции, в которых расщепляются четвертичные аммонийные соли, например, использованные в виде межфазных переносчиков. Вот, к примеру, старая проблема – почему нельзя получить чистый фторид тетрабутиламмония (продажная соль всегда содержит пару молекул гидратной воды) или гидроксид тетрабутиламмония (только водный раствор): при полном удалении воды что фторид, что гидроксид фактически в условиях межфазного переноса становятся сильными нуклеофилами и основаниями и расщепляют тетрабутиламмоний и в замещении, и в элиминировании. Ну и невозможно не заметить, что если не в замещении, то в конкурирующем элиминировании кватернизованные амины очень полезны, потому что в этом суть элиминирования по Гофману в методе исчерпывающего метилирования (это когда амины метилируют иодистым метилом и затем расщепляют нагреванием с влажной окисью серебра (если что, напоминаю, что представление о том, что гидроксид серебра – сильное основние – типичный и очень заразный миф, это не так). В теме Амины у меня эта реакция рассмотрена и мы ее применяем, а лет сто назад она была совершенно невероятно популярна.
Другое дело кватернизованный сульфоний: сульфид замещается довольно легко, как минимум потому что связь углерод-сера намного слабее связей между элементами второго периода. Замещение в кватернизованных сульфониях встречается не просто часто, а очень часто, особенно в современной химии. Реакций и методов таких пропасть. Например, синтез эпоксидов реакцией илида из кватернизованного ДМСО с кетонами. Эта реакция завершается внутримолекулярным SN2 замещением диметилсульфида.
Пока подробнее писать не буду, но, возможно, сильно позже, подсоберу фактуры и напишу с реакциями.
Доброго времени суток! Как наглядно понять что “Гриньяр завёлся”, кроме как наблюдая за градусником, погруженным в раствор?
Заранее спасибо за ответ.
АЧ: Во-первых, на химфаке запрещено произносить слово “градусник”. Это не градусник, а специальное термометрическое оборудование, сокращённо термометр. И не перепутали ли Вы доктора Гриньяра и доктора Айболита, который “и ставит, и ставит им грудусники”. Гриньярам ставить градусник, то есть термометр совершенно бесполезно. Когда гриньяр заводится, эфир почти моментально начинает кипеть, и это видно без всякого термометра.
Признаки запуска гриньяра очевидны – начинает шевелиться магний (я часто советую в момент запуска не включать мешалку – это создает локальную высокую концентрацию алкилгалогенида, в этом месте гриньяр заводится, и тогда его уже е остановить), и появляются пузырьки – локальное разогревание на поверхности стружинок нагревает эфир. Если использовали иод для активации, в момент запуска рыжая окраска раствора на глазах уходит, обесцвечивается. И эфир начинает кипеть. В этот момент надо быть готовым даже немного охладить смесь, если перестает справляться холодильник (ставьте хороший обратный холодильник, не “три шарика”). Если справляется – пусть кипит, но следующую порцию галогенпроизводного добавляйте только когда немного уймётся кипение. Обращайте снимание на цвет раствора над магнием – у алифатических гриньяров серовытый оттенок, у ароматических – коричневый. Это проявляется почти сразу, как только начинает образовываться гриньяр.
Если не заводится, обратите внимание не появилась ли белая взвесь – это признак сильно мокрого эфира, в котором, скорее всего, ничего запустить не получится.Если взвеси нет, но гриньяр всё равно не заводится, значит вы взяли слишком много эфира – в некоторых дурацких методиках его льют в диких количествах. Когда эфир совершенно сухой и все собрано тщательно, посуда сухая, прожаренная в сушильном шкафу – гриньяры можно запустить в разбавленном растворе и это даже хорошо. Но если эфир не идеальный, а посуда была сыроватой, гриньяр так не запустите. Лучше взять меньше эфира вначале, чтобы получить в момент запуска концентрации побольше. А разбавите потом, добавляя последние порции галогенида.
Хорошо, а с ранее упомянутым реактивом Нормана дела так же обстоят или все немножко хитрее?
В некотором смысле даже проще. Но ТГФ должен быть реально сух, и надо умудриться при переливании и загрузке реагентов не промочить его. Если ТГФ хорош, то гриньяры завариваются проще, чем в эфире, хотя бы потому что в ТГФ вы можете слегка подогреть смесь для запуска (феном лучше всего, если нет фена, то тёплой банькой – не включайте нагрев у мешалки!). И если не запускается, добавьте немного, процентов пять иодистого метила. Если и тогда не запускается, у Вас не ТГФ, а помои.
Доброго времени суток. Вот есть винилбромид, из которого можно сварить Гриньяр. Однако вот незадача, в ампуле, в которой он запаян, видны большие куски белой массы, вероятно, продуктов полимеризации. Температура кипения этого вещества 16 градусов. Как можно очистить это вещество, не взорвав/сжигая лабораторию? Как вообще работают с такого рода низкотемпературными ЛВЖ?
АЧ: Добрый вечер. Мне кажется, у Вас проблема. Если стоит задача взорвать или сжечь лабораторию, а под руками только ампула протухшего винилбромида, то задача кажется невыполнимой. А чем Вам так лаборатория насолила? Может, пусть живёт.
Из винилбромида, начнём с того, делают не гриньяр, а реактив Нормана. В нашей стране (и нигде больше) так называют гриньяры из винилгалогенидов, потому что их не могли получить ни сам Виктор Гриньяр, ни другие химики почти полвека, пока другой французский учёный Анри Норман не догадался в 1954 году заварить не в эфире, а в ТГФ – этот растворитель как раз тогда входил в моду среди быстро размножавшихся в это время как кролики металлооргаников – ТГФ оказался просто уникальным растворителем для этой науки. В этом растворителе гриньяры (или реактивы Нормана) завариваются очень легко, с небольшой активацией кристалликом иода, и иогда с добавлением небольшого количества метилиодида. Как работать с ТГФ, как его чистить и как сделать так, чтобы он не нахватал обратно всю ту воду, которую из него несколько дней удаляли, и как не взорваться на перекиси – надеюсь, Вам известно. Если нет, обязательно проконсультируйтесь с теми, кто это делать умеет.
Про винилбромид и как с ним работать. Это зависит от того, что точно требуется сделать. Если нужен просто винильный гриньяр для какой-то препаративной реакции, и в этой реакции можно использовать избыток гриньяра, тогда просто декантируйте жидкость с этой белой дряни – это, видимо, действительно полимер, хотя не исключено, что тот, кто его запаивал, добавил туда что-нибудь для защиты или доосушивания. Работайте быстро – легколетучие жидкости кипят, быстро охлажадют все вокруг себя, и туда быстро намораживается влага из воздуха. То есть нужно очень хорошо продумать прибор, все акуратно собрать, и продумать свои действия, чтобы всё было под рукой. После немного охлаждаете ампулу, и – обязательно в очках! – быстро ее вскрываете. Дозировать легколетучую жидкость сложно, поэтому советую приготовить раствор в растворителе реакции с некоторым запасом, просто взвешивая колбы до и после. Тогда прикинете объём этого раствора и спокойно его отмерите обычными способами.
Хуже если гриньяр нужен более чистый и в точном количестве. С количество все относительно просто – гриньяры титруют, легко найдёте методики, все более-менее одинаково. Тогда его заваривают с запасом и отбирают нужное количество.
Если винилбромид все же нужно очистить, его придется перегнать. Проще всего это сделать на вакуумной линии – это не совсем перегонка, а скорее перемораживание, но когда вещество очень летуче, а все примеси намного менее летучи (а это ровно этот случай), то это вполне адекватная процедура очистки. От воды она тоже очищает – для гриньяра более чем достаточно.
Если вакуумной линии нет, то можно перегнать с самом обычном приборе для перегонки, но в холодильник придется пустить ледяную воду, а приемник охлаждать чем-нибудь еще похолоднее (смесь лед-соль вполне достаточна, переохлаждать всякими сухими льдами или азотами не нужно – мороки будет больше, а не меньше). Прибор придется собрать очень аккуратно, шлифы должны быть надежными, все соединения закрепить кекам или пружинками – через все неплотности будет намораживаться влага. Очень полезно на вход подать через силиконовую септу очень слабый ток азота или аргона, а на выходе повесить на другой септе через иголочку и тонкую трубочку маленькую барботерку с силиконовым маслом – она будет показывать, что в приборе есть положительное давление (пузырьки выходят, а не входят, но очень медленно два-три в минуту, иначе будет уносить вещество) и не происходит подсос влажного воздуха. И перегоняйте спокойно как любую другую жидкость, подставив еле тёпленькую баньку. В приемник добавьте чуть-чуть любого ингибитора свободнорадикальной полимеризации (гидрохинона, трет-бутилпирокатехина, и т.п.), но если вещество не планируете хранить, а сразу используете после перегонки, можно этого не делать. Перегоняйте медленно, потому что выход идет через маленькое сечение, и любое ускорение чревато вскипанием и вышибанием того, что слабее всего закреплено. После того, как перегонка закончена, быстро снимаете приемник, закрываете его свежей септой и ставите в морозилку. Ничего особенно в самом соединении нет – главная проблема во всех манипуляциях состоитв том. чтобы не наморозить туда влагу. Ни в коем случае поэтому нельзя оставлять даже ненадолго незакупоренный сосуд с таким веществом – оно кипит, охлаждается и намораживает в себя влагу. С точки зрения опасности ничего особенного в жидкостях, кипящих ниже комнатной температуры и даже при небольшом минусе нет – они закипают и сами себя охлаждают. Когда прибор закрыт септой с иголочкой, этого достаточно, чтобы вода туда не намораживалась – диффузия в узком канале против давоения очень медленная. У винилбромида еще и очень тяжелые пары – по мол массе это почти как хлороформ, такие пары сами себя запирают, и если нет перегрева, ведут себя совсем спокойно. Поэтому не опасайтесь, что это вскипит и все разнесет, в том числе ненавистную лабораторию.
Спасибо большое, очень познавательно!
Добрый вечер, Андрей Владимирович. Как определить абсолютную конфигурацию стереоцетров в молекуле Д-гюкозы?( последовательность конфигураций R/S). Когда в в молекуле один хиральный центр, то все понятно, что энантиомеру присваивается R или S конфигурация в зависимости от взаимного расположения четырех заместителей вокруг хирального центра. Для каждого из заместителей вначале определяется старшинство по правилу Кана-Ингольда-Прелога, затем молекулу ориентируют так, чтобы младший заместитель был направлен в сторону от наблюдателя, и устанавливают направление падения старшинства остальных трех заместителей. Это все хорошо. Как применяется это правило, если в молекуле имеется несколько хиральных атомов?
Спасибо за ёмкий ответ на предыдущие вопросы, жду с нетерпением почитать про консервацию альдегидов/кетонов. Пока читал, вспомнил ещё кое-что: ДМСО перегоняют под хорошим вакуумом, но во всех прописях (емнип) сказано гнать ещё неприменно, пропуская в вакуум, инертный газ, для чего такая трата аргона или азота вообще необходима? ДМСО там разлагается что ли по пути или боятся этого остаточного давления или ещё что?
АЧ: Написал я про бисульфитные производные – на страничке ответов. Довольно длинно, но это занятная химия, пусть будет.
Что касается ДМСО, то право, вопрос Ваш странный. Если Вы решили заниматься химией и серьёзно, придётся научиться не бояться трат. Химия – наука дорогая, и сделать её хорошо, особенно в 21 веке можно только если делать основательно и как следует, используя лучшие реактивы и стекло. Иначе будете просто производить грязь и ненадёжные результаты и однажды с этим крупно влипнете.
ДМСО перегоняют в вакууме, как и любые растворители, которые при атмосферном давлении кипят выше 150 градусов. Это делается, чтобы избежать разложения, и чтобы перегнанный растворитель не оказался хуже исходного. Перегонять лучше при таком вакууме, чтобы температура кипения не стала слишком низкой. ДЛя ДМСО это 10-20 мм рт ст. Если ниже – растворитель полетит ниже 70 градусов и очистка достигнута не будет. Прибор придется защитить от влаги, особенно если гоните на водоструе. При перегонке растворителей начисто лучше всегда подавать в прибор через капилляр азот или аргон. Истратите совсем немного, не бойтесь. Зачем? Кислород воздуха всегда плохая вещь, особенно когда булькает через нагретую жидкость. Во-первых, он мокрый – ведь его берут не из баллона, а из атмосферы. Сушили-сушили – и сами же пускаете туда водяной пар из воздуха, а его там немало. Не забывайте, что мы живем не в пустыне (пока не живём), и влажность воздуха редко бывает меньше 70%. Прикиньте сами, сколько вы туда нагоните воды, если будете в капилляр подавать воздух. А ДМСО никогда не гонят над осушителями. Его сушат отдельно, и в колбу для перегонки переносят, оставив осушитель на выброс. Ну и кислород – а это автоокисление. ДМСО не так к этому чувствителен, но многие другие органические жидкости – очень. Поэтому перегонять с поддувом инертным газом – просто хорошая практика.
Добрый день, у меня три разных вопроса, очень интересно зачем и почему:
1) крупицу йода добавляют при изготовлении Гриньяра
2) почему не стоит использовать бисульфит/метабисульфит натрия, а стоит брать тиосульфат при приготовлении йодсодержащей ароматики (вы про это писали как-то)
3) как грамотно подойти к “консервированию” альдегидов метабисульфитом/гидросульфитом натрия (деталь: как-то пробовал бутаналь так спасти, в итоге по ямр получился какой-то неразделимый неприятно пахнущий осадок моего вещества непонятно с чем)
АЧ: 1) Это очень просто. Магний, как и все активные металлы, сверху вседа покрыт окисленным слоем, защищающим металл от дальнейшего окисления. Магний сам по себе – металл очень активный, он самопроизвольно растворяется в спиртах (это метод осушки метанола, но не этанола), а от такого же растворения в воде его защищает только образование слоя гидроксида на поверхности. Соответственно, если мы хотим, чтобы магний реагировал с галогенпроизводными, обычно это защитный слой надо расшевелить, чтобы хотя бы частично открылась поверхность. В этом месте начнется взаимодействие, и дальше дело пойдёт. Когда мы делаем гриньяры, всегда очень хорошо видно, что реакция сразу не начинается, и ее надо расшевелить. У каждого практикующего химика, хотя бы иногда заваривающего гриньяры, есть свои приёмчики. Некоторые самые активные галогенпроизводные типа нормальных алкилиодидов и бромидов, особенно те, что поменьше (MeI, EtBr) сами справляются с этой задачей – мы чуть-чуть подогреваем смесь рукой или тёплой банькой (никогда не включайте нагрев на мешалке, когда завариваете любой гриньяр – с гарантией будете ловить его по всей лаборатории). Более ленивые галогенпроизводные (вторичные, третичные алкилы, ароматические галогенпроизводные, хлорпроизводные) просто так расшевелить очень трудно или почти невозможно. Ещё одна проблема – не совсем сухой эфир. Для гриньяров нужен абсолютный эфир – то есть сначала очищенный от перекиси, подсушенный хрористым кальцием, и свежеперегнанный со всеми предосторожностями, обычно над натриевой проволокой (просто бросить кусочек натрия в эфир и с важным видом перегнатьв приборе со старой хлоркальциевой трубкой – это бессмысленная халтура). В практикумах такого просто никто делать не будет и слава богам – это очень опасно, и мы не хотим, чтобы наши лаборанты, которые и так на вес золота, рисковали жизнью и здоровьем. В практикуме эфир просто сушат хорошей дозой прокаленного хлористого кальция. В таком эфире много воды, но гриньяр в нем запустить можно. ДЛя активации магния используют кучу разных приемов, обысно это чуть-чуть чего-то очень реакционноспособного. Кристаллик иода, капля брома, немного активного маленького галогенпроизводного (иодистого метила, бромистого этила, дибромэтана), отлично работают. У магния защитный слой не такой прочный, как, например, у алюминия, и активирующий реагент диффундирует через слой к поверхности, там начинается экзотермическая локальная реакция, и слой просто ломается, предоставляя доступ основного реагента к поверхности. Дальше дело идет само – мы видим, как магний начинает шевелиться, тепло реакции разогревает эфир до кипения, и дальше только держись – неопытные химики, в первый раз заваривая приличный гриньяр на полмоля-моль, почти всегда отправляют его в потолок – настолько неожиданно ленивая реакция разгоняется до состояния чернобыльского реактора.
2) Про тиосульфат для очистки от иода я дописал примечание в раздел про получение иодпроизводных из солей диазония.
3) Про бисульфитные производные альдегидов постараюсь на днях написать подробнее. Это интересная история, которой по разным причинам брезгуют, как каким-то замшелым старьём, где всё очевидно. Вот вовсе там не всё очевидно, а сама реакция очень полезна в синтезе. Кратко только замечу, что там очень много зависит от бисульфита. Бисульфит из банки не пригоден ни на что, кроме обесцвечивания избытка брома после реакции бромирования. Ни одна серьёзная реакция (нуклеофильное замещение, получение бисульфитных производных, синтез арилгидразинов по Фишеру, реакция Бухерера и т.п.) с бисульфитом “из банки” не идёт. Нужен насыщенный раствор бисульфита натрия, который нельзя получит просто растворением в воде реактивов из банок с наклейкой “бисульфит” или “метабисульфит”. Раствор бисульфита используют еще биохимики, и у них есть пропись, как получить раствор бисульфита из твердого метабисульфита – можете легко найти этот метод в сети, но для целей органики он не работает – мала концетрация. В недружественных странах можно купить готовый именно раствор (40%) бисульфита натрия, но он, во-первых, долго не хранится, а во-вторых, нам прописано импортозамещение – мы не ищем лёгких путей (знать бы ещё куда). Поэтому раствор бисульфита, пригодный для всех этих знаменитых реакций, придется готовить самим, усердно пропуская до полного насыщения диоксид серы в крепкие растворы щелочи или соды, причём придется сильно озаботиться количествами – в таких случаях рекомендуют пропускать сернистый ангидрид, периодически взвешивая колбу, чтобы добиться расчетного привеса. Некоторые старинные умельцы умудрялись делать такие реакции с колбой, сразу установленной на грубые технические механические весы, поставив на вторую чашку гирьку нужной массы.
Подробнее про реакцию напишу, скорее всего на страничке про методы в альдегидах-кетонах.
Добрый день, вопрос, возможно, глупый, но что будет, если попытаться перегнать ДМФА над натрием?
АЧ: Добрый день. Во-первых, прекратите называть вопросы глупыми. Это какая-то странная манера унижаться, видимо, из серии традиционных ценностей и прочих скрепок, когда надо кланяться, бить челом, называть себя презренным рабом или хотя бы покорным слугой. Если вам кажется, что ваш вопрос действительно глупый, зачем его задаете, а если задаете вопрос, значит вам хочется узнать на него ответ, и тогда что в этом глупого?
Ничего хорошего не будет. Я не уверен, что это кто-то реально пытался делать. Вообще, когда что-то нагревают с натрием, просто надо понимать, что натрий это весьма сильный одноэлектронный восстановитель, который с удовольствием сдаст свои электроны всем связям, которые готовы их принять – а это, например, связи с достаточно кислыми атомами водорода, связи углерод-галоген, и всевозможные кратные связи кроме уж совсем простых олефинов. Все карбонильные соединения охотно принимают электрон, одразуются анион-радикалы, которые дальше как-то реагируют или расщепляются. В амидах это произойдет обязательно, дальше все просто развалится, и даже возможно экзотермично, то есть с сопровождением фонтанами, летающими термометрами, и прочими эффектами. С диметилформамидом вообще шутить н принято, иначе вы его никогда не очистите – он сам собой немного разлагается при температуре кипения, особенно если в нем есть что-то кислотное или основное. Поэтому процедуры очистки ДМФА довольно занудные и медленные, например, его перегоняют только в вакууме, и для этого нужно уметь делать “плохой” вакуум – это очень просто если есть маностат, а если его нет, это непросто. Я обычно втыкал тонкую иголку в вакуумный шланг или септу. И гонят его над самыми простыми и инертными осушителями, чтобы не вызвать разложения – иначе перегнанный ДМФА будет хуже чем исходный. Чуть не сказал, что поэтому проще купить готовый сухой и чистый ДМФА, но вспомнил, что в ближайшие полвека нам это не светит.
Кстати, само требование перегонять в легком вакууме, чтобы температура кипения была не больше 100 градусов, накладывает ограничения на перегонку над натрием – попробуйте перенать что-нибудь в вакууме над натрием. Вернее, точно не пробуйте – жидкость тут же вскипит и перебросит – следы воды есть везде, пойдет струйка водорода и при вакуумировании эти пузырьки расширятся и выкинут всю жидкость к черту. Есть одна мерзкая жидкость. которую перегоняют в плохом вакууме над натрием – диглим, и это то еще занятие, не один раз приведшее к плохим последствиям, если это делать хотя бы немного неаккуратно и невнимательно и не продумав все от начала до конца. Другие вещи над натрием при нагревании и в вакууме почти никогда не перегоняют. А без вакуума и при сильном нагревании с натрием вообще шутить нельзя – восстановительное разложение модет случится при повышенной температуре даже с тем вещами, которые так просто с натрием вообще не реагируют.
Для ДМФА вы сами легко напишете, что может быть, если на эту молекулу приклеится электрон – анион радикал расщепится, получится диметиламид натрия и формильный радикал, он оторвет водород, образовавшийся формальдегид еще раз возьмет электрон от натрия и т.д. – в общем получится много молекул с ионными связями, это всегда выгодно, поэтому экзотермичность процесса гарантирована.
Добрый день, Андрей Владимирович.
А откуда идут истоки теоремы о трёх нитроанилинах?
АЧ: Добрый вечер. Никто не знает. Почти наверняка в глубокую древность, к трудам Теофраста, а другие говорят – Плиния Старшего. Правда соответствующие части их сочинений утрачены. Вроде бы Плиний писал этот папирус как раз накануне извержения Везувия, и перечитывал его в самый момент первого взрыва, находясь на Мизенском мысе, и ударной волной рукопись вырвало из его рук и она унеслась в пучины Неаполитанского залива.
В новейшие времена это учение само собой возродилось в думах ведущих органических мыслителей и обрело форму Великой Теоремы о Трёх Нитроанилинах и следствий из неё, которой я с вами и поделился. Руководствуясь Великой Теоремой в последние 120 лет было получено бесчисленное количество производных бензола, что и подтвердило законное место Великой Теоремы среди величайших образцов человеческого гения.
Доброго времени суток, Андрей Владимирович. Вот есть реакция Штрекера, почему там гидролиз так легко протекает из нитрила в кислоту, хотя мы знаем, что гидролиз нитрилов в кислой среде протекает в довольно тяжелых условиях. Можно было бы подумать, что аминогруппа этому как-то способствует, но она даст соль. Может здесь контранион этой соли как-то участвует в процессе гидролиза?
АЧ: Легко гидролизуются в условиях кислотного катализа не только аддукты Штрекера, но и циангидрины. Точная причина этого, насколько я знаю, никогда не изучалась, скорее всего, никому в голову не пришло, что это представляет интерес. Причины может быть две: просто индуктивный эффект акцептора (кислород гидроксила или даже аммоний), увеличивающий электрофильность нитрильного углерода. Или всё-таки содействие гидролизу. Что-то внутримолекулярное придумать можно, причём и нукелеофильное, и электрофильное. Для нуклеофильного содействия – трёхчленные циклы вполне функциональны, а протонирование амино-группы не будет количественным, основность амина рядом с нитрильной группой понижена. Второй вариант содействия – электрофильный, водородная связь на азот от гидроксила или аммония, тоже будет увеличивать реакционную способность. Чтобы судить точнее, кто-то должен это попробовать исследовать.
Доброго времени суток, Андрей Владимирович!
Почему реакции Петасиса нет ни в курсе, ни в методичках, вроде же полезная химия?
АЧ: Ну мало ли полезных реакций в химии? Реакция Петасиса на 3 курсе совершенно лишняя, она требует понимания химии переходных металлов. И ничего по методологии сверх классического Виттига не даёт. Если интересен Петасис, посмотрите на моем втором сайте про переходные металлы, там это разобрано.
Простите, уточню, я имел в виду не реакции с реагентом Петасиса, что, безусловно, лишнее для третьего курса, а реакцию борных кислот с альдегидами в присутствии аминов (https://en.m.wikipedia.org/wiki/Petasis_reaction), там ведь не используются никакие металлоорганические реактивы, разве что борную кислоту из Гриньяра сделать…
АЧ: О, подходящие боги! Ну это совсем экзотика. Вообще это плохая идея тащить всё подряд в базовый курс органики только потому что это где-то успешно применяется. В современной органике только именных реакции уже многие сотни ближе к тысяче и сверх того, а уж просто методов вообще не счесть. И те, кто занимается каким-то методом, всегда уверены, что ничего важнее уже упомянутые подходящие боги не создали и это должен знать каждый. Но мы так не будем делать. Важно понимать концептуальные основы химии, а дальше уж сами наполняйте, чем хотите. Реакция Петасиса в смысле понимания химии мало что даёт – это очередная разновидность нуклеофильного присоединения к карбонильной группе или ее аналогу, иминиевой соли, например. Она неплохо смотрится в контексте модного органокатализа, но там такого многие десятки только основных разновидностей. Закладочку я тем не менее положу, может в некоторой перспективе найду ей место в каком-то более общем контексте.
Спасибо!
Здравствуйте, Андрей Владимирович. Почему в алкилировании или аминировании для синтеза сульфонов, сульфамидов используют только сульфохлориды? Тут такая же аналогия. как и с галогенангидридами, то есть увеличивается δ+ на атоме серы и влияние -I, +M галогена; ? В учебниках как то этот момент особо не проговаривается. Заранее благодарю за ответ.
АЧ: Встречный вопрос – а какая альтернатива? Что ещё Вы могли бы использовать? Пожалуйста, проясните, чтобы я точнее понял суть вопроса.
Например, сульфоновую кислоту или ее соль (или вообще сложный эфир). Тут вылезает подвижный водород, из аминирования получится соль, но возможна ли последующая атака серы… А алкилирование… полагаю, пойдет, подкислим соль/кислоту и сложный эфир (с гидролизом), думаю, без проблем. провожу полную аналогию с карбоновыми
АЧ: К сожалению или к счастью, аналогия с карбоновыми кислотами неверна, не работает. Я приблизительно это описал тут в ответе про химию фосфора – в третьем периоде химия другая. К сере это относится в полной мере. В химии серы и фосфора нет аналогов тетраэдрических аддуктов химии карбонильной группы. Замещение на шестивалентной сере или пятивалентном фосфоре не идет как присоединение-отщепление, как на карбониле. Поэтому вам обязательно нужна приличная уходящая группа на атоме серы или фосфора, чтобы осуществлялось именно замещение. И ничего проще галогена не придумать, хотя при желании что-то придумать другое, например, смешанный ангидрид, можно найти замену.
Добрый вечер,не подскажете где можно посмотреть ваши видео лекции,видел 2 ваши лекции в teach-in, очень понравились?
АЧ: Задайте запрос в яндекс. Есть точно ещё пара лекций, и они где-то выложены. Больше нет. Был один год, когда все лекции на химфаке записывали, и что тогда записалось, то записалось. По переходным металлам я сам аудио-лекции записыываю и они размещены на сайте transmet.avchem.ru. Там только голос, без морды, но зато слайды получше сделаны.
Спасибо большое за ответ!
Доброго времени суток, у̶в̶а̶ж̶а̶е̶м̶а̶я̶ ̶р̶е̶д̶а̶к̶ц̶и̶я̶ Андрей Владимирович! Спасибо за ответ про Лёйкарта и Валлаха!
На одной из страниц вы разбирали основность и нуклеофильность. И вот сказали, что мол есть хорошие нуклеофилы, но при этом они слабые основания, и наоборот, есть отличные основания, но при этом они плохие нуклеофилы.
Вопрос: почему так? Что обуславливает способность отдельных групп частиц быть только хорошими нуклеофилами, а других только хорошими основаниями.
Еще вопрос по той же теме: есть гидрид-ион, почему NaH мы используем зачастую как основание, может ли он быть нуклеофилом при этом? Ведь, как мы знаем, есть так много различных реагентов с гидридом, те же ДИБАЛ-H и NaBH3CN, которые уже вроде как и нуклеофилы, а о их способности к основности немного то ли умалчивается, то ли сознательно не говорится (на третьем курсе).
Здравствуйте. После прочтения статьи про резонансные структуры захотелось задать соответствующий вопрос. У Вас там сказано: (цитирую) “Результат будет противоположным – в реальной структуре будет преобладать вклад аниона с минусом на менее замещённом атоме, потому что во второй структуре индуктивные доноры дестабилизируют минус.”. Но разве, когда на углероде будет заряд (-), он не будет, наоборот, отдавать электронную плотность, так, что метильные группы будет акцепторами?
Доброго времени суток, дорогая редакция. Не без удовольствия прочитал ваш ответ на вопрос про фосфорорганические соединения и их применение, спасибо!
Вопрос: можно ли в реакции Лейкарта-Уоллоха заменить формамид на ДМФА или его замещенные аналогии? А в реакции Эшвейлера-Кларка формальдегид на какое-нибудь другое карбонильное соединение, чтобы вместо метила получить амин с другими заместителями? Есть ли способы повесить лишь одну метильную группу?
Заранее спасибо!
АЧ: Рад, что доставил Вам удовольствие. А Вы всё редакцию ищете? Ищите уж тогда сразу издательский дом. На главной странице сайта написано, кто делает этот сайт, и это полная и исчерпывающая правда.
Реакция Лёйкарта-Валлаха это старинный способ восстановительного аминирования. Метод Эшвайлера-Кларка это просто частный случай реакции Лёйкарта-Валлаха, хотя и с другим диапазоном применения. В обоих методах восстановительным агентом является муравьиная кислота или ее анион, формиат, являющиеся гидридными переносчиками. В общей реакции Лёйкарта-Валлаха используются разные карбонильные соединения, обычно альдегиды, но и активные кетоны тоже. В методе Эшвайлера-Кларка используют только один альдегид – формальдегид. Источники азота тоже разные. В классике Лёйкарта-Валлаха это просто ион аммония или формамид, но в расширенном толковании этой реакции берут разные амины, или такие исочники аминов как диметилформамид (получаются диметиламины). В методе Эшвайлера-Кларка источник азота – любые первичные или вторичные амины. Но поскольку альдегид всегда один и тот же, в этом методе вводят только метилы. Прежде всего потому что если возьмете другие альдегиды, то это будет уже не Эшвайлер-Кларк, а Лёйкарт-Валлах.
Немного позже постараюсь написать более подробный ответ.
Андрей Владимирович, есть ли какой-то доступный и несложный способ сделать NaHMDS, чтоб прям в дырявом ведерке и с протекающими капельными воронками (я утрирую)?
Здравствуйте, Андрей Владимирович. Спасибо за все, что Вы делаете, Ваш сайт и лекции мне очень помогли.
Собственно вопрос: как можно синтезировать высшие циклоалканы, в которых в состав цикла входят 20,30,40,50 и т.д. атомов углерода? Наверняка это имеет смысл, так как что-то подобное могут использовать как молекулярные узлы, в составе ротаксанов.
Заранее благодарю.
АЧ: Спасибо за добрые слова. А почему именно циклоалканы? Но вопрос интересный, постараюсь кратко ответить в ближайщие дни, а себе поставлю в план вообще обсуждение макроциклизации.
Спасибо большое за ответ, было очень интересно
АЧ: Да, мне тоже. Вопрос неожиданно оказался весьма многогранным.
Доброго времени суток. Возможно немного вопрос в сторону, но хочется узнать чего-то и про фосфороорганику, благо и ей есть широкое применение, а говорят о ней не так много. Например, есть такие две реакции – Арбузова и Абрамова, – можно ли где-то о них по-подробнее прочитать? В учебниках о них написано вскользь, а хочется знать, почему у этих реакций не всегда хорошие выходы и какие у этих реакций могут быть подводные камни.
АЧ: Ох, чую, следующий вопрос будет “а можно ли что-то узнать про химию вообще”. Фосфорорганика – огромная наука, очень быстро развивающаяся, там просто невероятно много чудес, самых настоящих, потому что фосфор – это вам не азот (у азота тоже очень увлекательная химия), это элемент из 3-го периода с совершенно другими валентными возможностями. Там вообще всё не так, и лезть туда со знаниями по 2-му периоду всё равно, что научившись играть с котёнком, смело отправиться в вольер со львами, типа, тоже кошки и молоко, наверное, любят, справимся.
Я бы очень хотел однажды сделать небольшой обзор по особенностям химии фосфора, но это как-то совсем из 3-го курса выходит. Надеюсь, до этого дело когда-нибудь дойдёт, материалы собираются понемногу, концепция вырисовывается. Поэтому, если есть совсем конкретные вопросы, задавайте, я подумаю. Но в наше время во-первых, по любой химии есть что почитать, целые полки литературы по любой области. По-английски, конечно, читать надо. Про фосфор это, конечно, не полки, а книжные шкафы, и у некоторых уже ножки подгибаются. И, простите, у любых реакций, даже у реакции нейтрализации, не всегда хорошие выходы, а подводных камней в химии столько, что не устаешь удивляться, где там вообще вода помещается.
Хорошо, можете тогда, пожалуйста, вкратце (или не совсем) рассказать про реакцию Арбузова, зачем она нужна с̶т̶у̶д̶е̶н̶т̶а̶м̶ ̶т̶р̶е̶т̶ь̶е̶г̶о̶ ̶к̶у̶р̶с̶а̶ кроме как для дальнейшего использования в реакции Хорнера-Уодсворта-Эммонса; если этот процесс термодинамический, может ли побочно при присоединении триалкилфосфита к RX от фосфора отвалиться алкоксильный фрагмент с получением трехвалентного фосфора и дальнейшими нуклеофильными превращениями (иначе говоря, почему (RO)3R’P+ X- вроде как стабилен и его можно выделить, хотя может это и отдельный вопрос, не знаю)? Почему для этой реакции обычно берут какие-то галогенпроизводные (бром, хлор, а можно ли взять тозилат, например)? И немного в сторону (не обижусь, если не ответите, просто интересно): работает ли похожая реакция Перкова с производными карбоновых кислот, например, со сложными эфирами (иначе говоря, почему триэтилфосфит с этил хлорацетатом дает продукт реакции Арбузова, а не продукт реакции Перкова или что-то еще сложнее, типа (RO)2P(=O)-C(=O)-CH2Cl) )?
Заранее спасибо!
АЧ: Лучше задавайте вопросы на верхнем уровне, а не в ответах на ответы. И не грузите меня тем, обидитесь Вы или нет, это Ваши проблемы. Чтобы серьёзно написать про химию фосфора, мне нужно сначала доделать страничку про гипервалентность, потому что именно здесь ключ ко всей этой химии, начинающейся с реакции Михаэлиса-Арбузова, и далее к целому кусту родственных реакций, включая реакцию Перкова (это немец, если что, видимо, пруссак, там полно славянских фамилий). Та страничка готова процентов на 80, но увы, я ее не выпущу пока не доделаю полностью, поэтому это все время откладывается. По поводу Вашего вопроса, я подумаю, насколько можно из него достать чисто практические вещи, а общее оставить на потом.
АЧ: Два одинаковых вопроса, один убираю.
Ответ на вопрос про антиароматичность готовится, но может быть с большой задержкой. Скорее всего будет добавлен материал на страничку про ароматичность.
АЧ: А почему Sn – это же будет электрофильное замещение, а не нуклеофильное?
Тогда Se1, и вправду. Бывает ли или такое положение вещей бессмысленно рассматривать как общий случай (м.б. это частный случай чего-то еще?)?
АЧ:Ну хорошо, посмотрим. Но сами посудите, для такого механизма понадобится нечто, что самопроизвольно ионизуется с образованием карбаниона. Карбанион понадобится суперстабилизированный. Или очень выгодный распад. В принципе, попробуйте расписать галоформную реакцию и увидите ровно это – скоростьопределяющий распад тетраэдрического аддукта на CCl3-анион, который быстро подхватит электрофил, протон.
Доброго времени суток. Бывает ли Sn1cb механизм для органич. химии или что-то подобное с уходом “электрофуга” (уж даже не знаю, как это правильно обозначить), образованием карбаниона и присоединением электрофила?
Когда же все таки идет Ade2 (с образованием КК, со всеми перегруппировками и олигомеризацией. Ведь существуют данные о выходе продуктов, где четко видно, что перегруппировка КК есть), а когда согласованный Ade3? Или это зависит от подбора среды для проведения конкретной реакции? Или вообще это два разных подхода к рассмотрению механизма? Естественно, если мы не говорим об образовании ониевых ионов. Да, тема подробно раскрыта на сайте, но мне все же этот момент не совсем ясен.
Заранее )) благодарю за ответ.
АЧ: КК – это карбокатион? Если да, не отвечайте.
Справедливо ли суждение: Если нет betta протона (например, в хлористом метиле, бромистом неопентиле или в бензилбромиде), то засунув сильное основание и слабый нуклеофил (необязательно) , такое как LDA или трет-бутилат калия, пойдет альфа-элиминирование с образованием карбена? Зачем вообще нужны карбены?
За ранее благодарю за ответ. Еще хочу выразить огромную благодарность за то, что вы делаете и сайты, и лекции. Ваше изложение темы , это что то феноменальное! Это очень помогает разобраться в непонятных темах, а также черпнуть много нового. Очень ждем ваши следующие лекции. Правда, большое спасибо, вы делаете очень важное дело.
АЧ: Спасибо за высокую оценку, но “заранее” пишется вместе. Про карбены не обрадую, это огромная тема, я давно хочу сделать отдельную страницу про карбены, но пока это в перспективе. Но на Ваш вопрос кратко на днях отвечу.
Доброго времени суток, дорогая редакция. В теме про ароматичность вы упомянули, что молекула с маленьким циклом от 3 до 5 и замкнутой сопряженной системой, имеющая 4n электронов, называется антиароматической. Вот этот диапазон от 3 до 5 вообще условен, или, если это так, где можно прочертить условную грань между антиароматичностью и неароматичностью.
АЧ: Редакцией меня ещё никто не называл. Ну тогда я и главный редактор и просто редактор, и могу, в случае чего, сам на себя наорать, если допущу какой-нибудь позорный ляп. Удобно.
Мы хорошо знаем про присоединение HBr против Марковникова к олефинам. А к ацетиленам так присоединять бромистый водород можно?
Здравствуйте! Подскажите, не знаете ли вы какого нибудь ресурса или чего-то, где можно почитать про осмоление? Чем может быть вызвано, как избежать, какое-то пояснение процессов?
Добрый вечер, Андрей Владимирович. Возможно не заметил. Тема спирты, галогенирование. Почему при использовании PPh3 и Br2, по сравнению с другими способами) не происходит перегруппировок в случае неопентилового спирта? И вообще, почему эта реакция более стереоселективна? (возможно сморозил глупость по поводу стереоселективности)