Методы и задачи в химии соединений азота
Этот раздел – совершенно удивительная помойка. Раньше нам никогда не приходило в голову соединять в одной теме все соединения одного элемента. Представьте себе тему: соединения кислорода. Валим в одну кучу спирты, эфиры, альдегиды, кетоны, карбоновые кислоты и наслаждаемся произведённым результатом. После этого закрываем лавочку и расходимся – всё равно любой человек, который попробует в один присест загрузить это на свой чердак, либо отправится в дурдом, либо придёт к выводу о своей полной непригодности к изучению органической химии. Спокойно! Органическую химию изучить можно, но только небольшими кусочками, потихоньку. Тогда и чердак будет цел, и содержимое чердака будет интересно не только летучим мышам.
Но вот в этом разделе мы злобно нарушаем этот принцип. Неужели азот проще кислорода? Не проще, скорее сложнее – степеней окисления у кислорода от силы 4, с одной из которых мы никогда не встретимся. А у азота их в два раза больше, и ни одна не является экзотикой. Соотвественно и классов соединений у азота много больше. И с довольно представительным списком их представителей мы здесь встретимся, с большинством, впрочем, мимолётно.
Понятно, что у нас неожиданно возникли проблемы. Но вместе с проблемами – и уверенность в том, что мы с ними справимся. Если мы до этого момента не занимались просто, извините, тупым запоминанием реакций, а более-менее разобрались в том, как устроена органическая химия, то задача станет совсем небезнадёжной. Законы и закономерности хорошо работают во всех классах органических соединений, и мы и здесь будем видеть электрофилы и нуклеофилы, CH-кислотность, основность по Бренстеду-Лоури, замещение и присоединение, кинетический и термодинамический контроль, принцип Ле Шателье, обычные интермедиаты и механизмы, являющиеся разновидностями уже известных механизмов.
В этом разделе довольно мало новых методов образования C-C связей, а для синтеза соединений азота мы в основном используем то, что мы с блеском изучили в карбонильных соединениях и карбоновых кислотах.
И ещё здесь совсем нет стереохимии. В соединениях азота нет стереохимии??! Конечно есть! В химии азота стереохимии больше, чем где бы то ни было. Но мы просто её не изучаем, или делаем вид, что ничего нового относительно уже изученного здесь нет.
Последнее обновление:
20.03.2020 – исправлена ошибка на вкладке про расщепление по Гофману. И небольшой рестайлинг страницы с новым введением.
Новые C-C связи
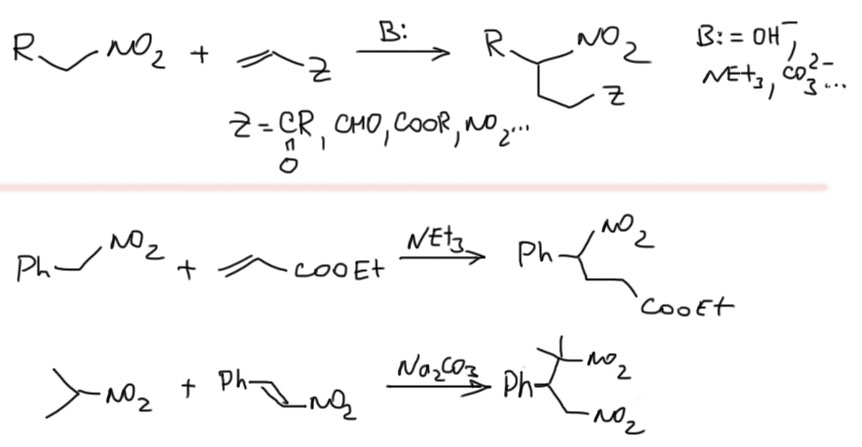
Второе важное обстоятельство – одна нитро-группа более мощный акцептор, чем два карбонила. Поэтому мононитросоединения обладают большей CH-кислотностью, чем малоновый или ацетоуксусный эфиры. Для депротонирования нитросоединений достаточно таких оснований как амины или карбонат, и не нужны не только LDA, но и этилат. Так как нитро-соединения еще и не дают самоконденсации, то реакции с ними проводят наиболее простым способом, добавляя основание к смеси нитросоединения и электрофила.
Конденсация с альдегидами и кетонами.
реакция Анри (или Генри, если французскую фамилию Henry читают как английское имя) – конденсация нитросоединений (метиленовая компонента) с альдегидами и иногда кетонами (карбонильная компонента). Сопряженное основание нитро-соединения (у этих анионов нет такого удобного названия как енолят для сопряженных оснований карбонильных соединений, их называют или нитронатами или солями аци-формы, и то, и другое не очень хорошо запоминается) – довольно слабый нуклеофил, что непосредственно следует из высокой стабильности этих анионов, поэтому реакции делают в основном с аальдегидами, а также самыми реакционноспособными кетонами (циклоалканонами, метилкетонами), так как реакции с более сложными кетонами обратимы и в равновесных смесях преобладают исходные. Конденсирующий агент – амины, карбонаты щелочным металлов, щелочи. Продукты – нитроспирты, если есть сопряжение с ароматическим кольцом – нитроолефины. Реакция с самым реакционноспособным альдегидом, формальдегидом, дает продукты полного замещения всех кислых протонов.

Нуклеофильности нитронатов не хватает для атаки на карбонильную группу сложных эфиров – аналога сложноэфирной конденсации в химии нитро-соединений нет.
Присоединение к электрофильным олефинам
После карбонильной группы следующим важным C-электрофилом является двойная связь, сопряженная с карбонилом или другим мезомерным акцептором. Мы хорошо знаем 1,4-присоединение разных нуклеофилов к таким связям (присоединение по Михаэлю). Анионы нитро-соединений не исключение. Это довольно полезная реакция. Кстати, в качестве активных олефинов могут выступать и нитро-олефины из реакции Анри.

Сами исходные гидразины получаются двумя путями. Симметричные – просто восстановлением нитросоединений цинковой пылью в щелочной среде.
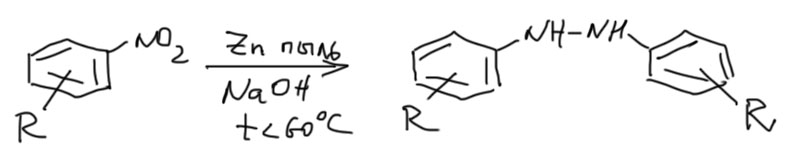
Несимметричные – гораздо более морочной процедурой. Для этого одна половина представлена соотвествующим анилином, вторая – нитрозобензолом. Нитрозосоединение пролучается в несколько стадий из нитросоединения, например, восстановлением до арилгидроксиламина, и окислением последнего, например, хлорным железом (еще годятся с десяток других окислителей, в частности надсерная кислота и трет-бутилгипохлорит, то есть так называемые переносчики положительного, в смысле электрофильного, кислорода). Полученные компоненты просто конденсируют в ледяной уксусной кислоте – это называется реакция Миллза, а азобензол восстанавливают цинком в щелочной среде.

Изменения реакционных центров
Синтез аминов
Общие замечания – удобный алгоритм построения почти любого амина
В аминах азот находится в самой низкой степени окисления -3, поэтому амины по определению являются продуктами восстановления. Это немного формальное обстоятельство стоит воспринимать совершенно буквально – основные методы получения аминов связаны с восстановительными реакциями. Восстановительные методы – первое, о чем стоит думать, когда нужно построить какой-то амин. На втором месте – полезные перегруппировки, которым нужно владеть очень хорошо, потому что они решают несколько специфических задач, которые почти невозможно решить по-другому. И только на третьем, а точнее, на тридцать третьем (места с 3 по 32 не заняты) всякое замещение, без которого, в принципе, можно совсем обойтись, как без хлеба в бессмертном учении Винни Пуха.
Второе обстоятельство связано с тем, что в аминах может быть до трех разных органических групп. Методы синтеза нужно подбирать в соответствии с природой этих групп. Можно воспользоваться вот таким несложным алгоритмом (это так красиво называется рецепт изготовления блюда, в данном случае блюдо – амин, а поскольку блюдо это по определению несъедобное, то пусть будет алгоритм), а расшифровка методов, на которые ссылается алгоритм, даны ниже на вкладках.
И ещё одна важная вещь. Чтобы успешно решать задачи органической химии нужно правильно думать. Когда мы вводим в органические соединения разные функциональные группы, мы думаем именно так: есть структура, в неё есть подходящие реакционные центры, нужно туда ввести требуемую группу. Ну и вводим. Но когда нам нужно получить амины, вся схема мышления переворачивается с головы на ноги: мы видим атом азота, на котором болтается одна, две или три группы. И вместо того, чтобы думать, как засунуть атом азота в одну из этих групп (какую? и что тогда делать с остальными?), мы, во-первых, сортируем эти группы приблизительно так, как это описано ниже, и дальше, одну за другой, грузим на азот в том порядке, который получился при сортировке групп.
Строим амин RN(R”)R’
0. Амин циклический (азот в цикле) или нециклический, но с характерной особенностью – имеет два заместителя, отличающихся только CH2-группой (R и RCH2). Такой амин строится перегруппировкой Бекмана, а при наличии еще и третьего заместителя у азота переходим к шагу 4 и вводим его в последнюю очередь. Дальше пойдем по порядку.
1. Есть ли среди заместителей третичный алкил? Если нет, переходим к шагу 2. Если есть, допустим это R”, получаем сначала R”NH2 с помощью перегруппировок (реакции Гофмана или Курциуса) после чего переходим к шагу 3. Внимание: случай, когда в амине есть одновременно третичный алкил и его гомолог, например tBu и tBuCH2 уже описан в шаге 0. Внимание: в амине не должно быть больше одного третичного алкила. Если есть два или три, переходим к шагу 10. Если в оставшейся части есть ароматический заместитель, переходим к шагу 11.
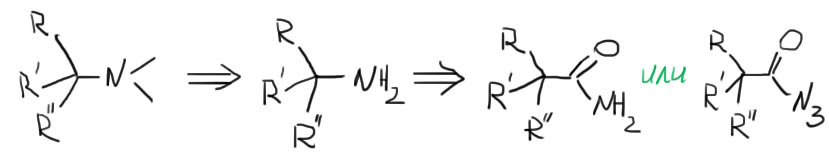
2. Есть ли среди заместителей фенил или замещенный фенил. Если есть, допустим это опять R”, получаем сначала R”NH2 с помощью реакций, описанных на вкладке Синтез анилинов. Потом переходим к шагу 3. Внимание: а амине не должно быть больше одного ароматического заместителя – если есть два или три, переходим к шагу 11.
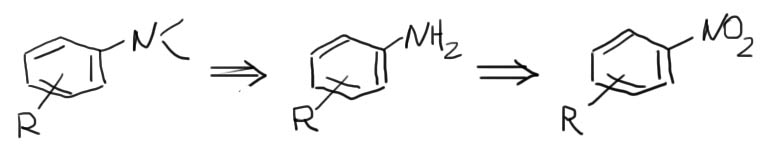
3. Внимательно осматриваем предложенный амин или остаток после шагов 1 и 2, а точнее каждый из оставшихся органических остатков. Что-то из них может оказаться сложным остатком, в котором после двух атомов углерода находится карбонил, карбоксил, амид, сложный эфир, нитрил, нитро-группа, или еще один амин. Это случай присоединения по Михаэлю. Таких остатков может быть до трех, и каждый навешиваем присоединением по Михаэлю. Внимание: выполнение нужно отложить до шага 6, а пока переходим к шагу 4.

4. Итак, в исходном амине не было ни третичного алкила, ни ароматического заместителя, или были, но мы их уже сделали. Продолжаем. Есть ли среди оставшихся заместителей вторичный алкил. Если есть, навешиваем с помощью восстановительного аминирования из соответствующего кетона. Если вторичных заместителей два или три, навешиваем в порядке уменьшения размера тем же способом. Если еще остались заместители, переходим к шагу 5.

5. Есть ли среди оставшихся заместителей первичные алкилы, но не метил. Если есть, навешиваем. Для этого используется много разных способов, смотрим их на вкладке Введение первичного алкила. Переходим к шагу 6.
6. Навешиваем сложные заместители присоединением по Михаэлю. Если необходимо, дополнительно их модифицируем. Переходим к шагу 7.
7. Остались, если остались, только метилы. Навешиваем метилы. Сравниваем результат с заданным амином. Если совпадает, радуемся. Если не совпадает, переходим к шагу 10.
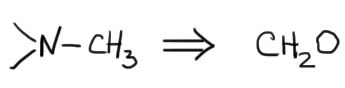
8-9. В резерве. В текущей версии алгоритма не используются.
10. Выбрасываем белый флаг и начинаем горько плакать, так как задачу решить не удалось, и никто нам не поможет. Это провал.
11. Выбрасываем черный флаг и начинаем бурно возмущаться, потому что нас явно спрашивают о том, чего нет в программе. Это провокация!
- выбираем соответствующий метод
- Перегруппировки в реакциях Гофмана и Курциуса
- Синтез анилинов
- Введение вторичного алкила
- Введение первичного алкила
- Перегруппировка Бекмана для синтеза аминов.
- Присоединение по Михаэлю в синтезе аминов
- Введение метила
- Что не нужно делать
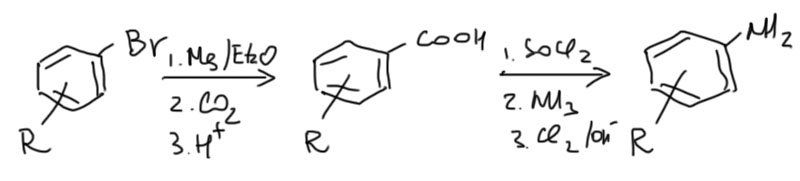
В принципе, это вполне общий метод синтеза аминов, но он совершенно незаменим только в одном случае – когда с азотом связан третичный алкил. Еще этом метод иногда применяют для синтеза ароматических аминов, но только в том случае, когда не срабатывают более простые методы. Оба метода используют одну и ту же секстетную перегруппировку и одно и то же исходное – карбоновую кислоту.

Метод Гофмана отличается от Курциуса простотой (азид натрия есть не у всех, и желание получать азид и нагревать его с потенциальным риском досрочно отправиться на встречу с Курциусом, есть у еще меньшего количества разумных людей), и еще тем, что реакция заведомо идет в присутствии воды и в результате сразу получается искомый амин. А в Курциусе продуктом является не амин, а изоцианат, который нужно дополнительно гидролизовать. В остальном это одно и то же.
Реакцию используют для синтеза аминов через кислоты, например, через Гриньяр из третичного углерода (третичные алкильные Гриньяры вполне хорошо получаются обычным способом, хотя и труднее чем менее стерически затрудненные).


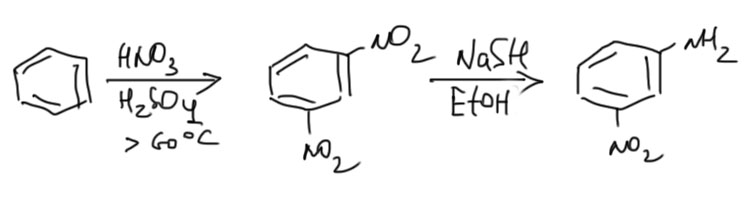
Есть еще одна важная реакция – частичное восстановление одной нитро-группы из нескольких. Для этого используют особый восстановитель – NaSH в спирте. При этом, реальное применение у этой реакции очень узкое – только такие молекулы, у которых не могут получиться разные продукты при восстановлении одной ниро-группы из нескольких. наиболее часто реакцию применяют для синтеза очень полезного в синтезе м-нитроанилина.
В редких случаях, когда по какой-то причине доступна бензойная кислота, можно получать анилины реакцией Гофмана. Например, так иногда бывает проще превратить ароматическое галогенпроизводное в анилин, так как нуклеофильное ароматическое замещение (освежите в памяти) – реакция непростая и часто осложняемая всякими странными процесами типа смещения входящей группы от места нахождения уходящей.
Кроме этого, неплохо не забывать про нуклеофильное ароматическое замещение.
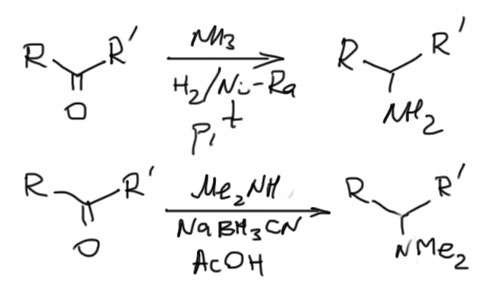
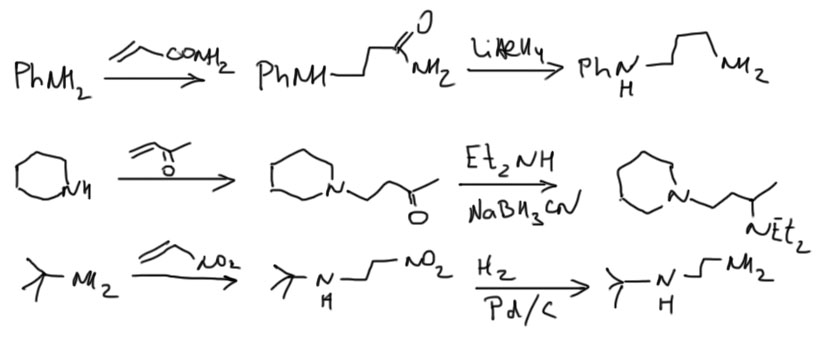
Для восстановительного аминирования используют множество восстановителей, но нам можно ограничиться двумя. В простых случаях (когда вторичные алкилы суть что-то банальное типа изопропила, циклогексила и т.п.) это гетерогенное гидрирование водородом над никелем Ренея, в более сложных (это когда вторичный алкил может иметь дополнительные двойные связи или какие-то функциональные группы – спец. восстановитель цианоборгидрид натрия в слабокислой среде.
Если кетон енолизуемый, и вы еще не забыли, что такое енамин и можете его получить, то енамины отлично восстанавливаются обычным боргидридом натрия, но в этом случае заместитель будет последним, так как енамины образуются из вторичных аминов. Вот пример такого синтеза. Без этого подхода можно вполне обойтись, и все это сделать так, как описано выше без выделения енамина.

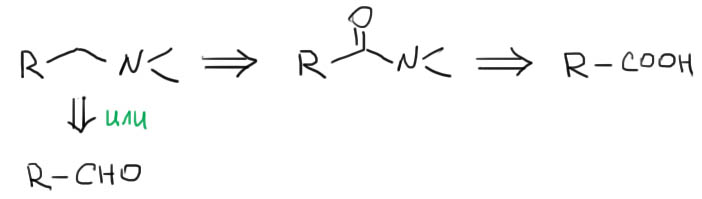
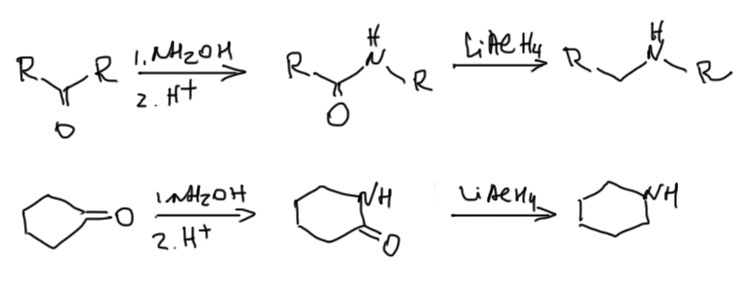
На первом месте востановление амидов карбоновых кислот или нитрилов алюмогидридом лития. Амиды легко получаются, при этом в амине могут уже быть другие заместители, нитрилы дают только амины с NH2-группой. Это очень надежный способ с огромным количеством реальных примеров применения.

Восстановление амидов используется и в подходе с использованием перегруппировки Бекмана на отдельной вкладке.
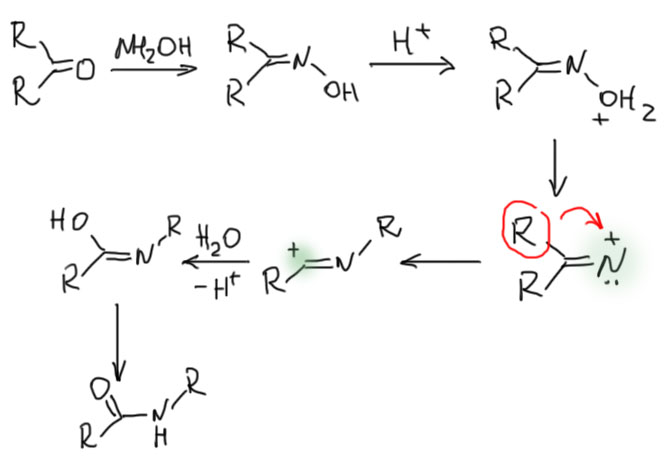
На втором месте восстановительное аминирование альдегидов, которое делается или так же, как и восстановительное аминирование кетонов, или, если в амине уже есть один заместитель, то получают основание Шиффа, и его восстанавливают боргидридом натрия.

Желательно этими методами и пользоваться. Но для аминов с незамещенной аминогруппой можно использовать еще и заместительные методы, в которых связь C-N образуется в результате SN2-замещения. В старинном методе Габриэля нуклеофилом является фталимид. Калийная соль фталимида – очень дешевый и доступный реактив, не требующий никаких специальных приемов работы. Но это очень скверный нуклеофил, поэтому реакции ведут, даже с самыми реакционноспособными бензилгалогенидами, в жестких условиях – при нагревании без растворителя. Получающийся N-алкилфталимид разлагают гидразином. Другой метод наоборот использует исключительно мощный нуклеофил – азид-ион, один из самых лучших нуклеофилов, известных человечеству. Реакции с азидом идут быстро даже с менее реакционноспособными алкилгалогенидами. Пока никто не видит, намекну, что он быстро реагирует даже с вторичными алкилгалогенидами. Получающийся азид разлагают трифенилфосфином (метод Штаудингера). Это – очень хороший и довольно универсальный, но дорогой и немного опасный (азиды очень токсичны) метод. Если хотите, используйте, но не забывайте проверять субстрат на пригодность для SN2.

Как и в реакции Байера-Виллигера, и здесь лучше использовать симметричные кетоны, и по той же причине. Получающиеся амиды и сами по себе неплохие вещества, но их еще можно восстановить алюмогидридом лития до аминов. Амины при этом всегда получаются однотипные, и по этому признаку этот метод легко угадывается.
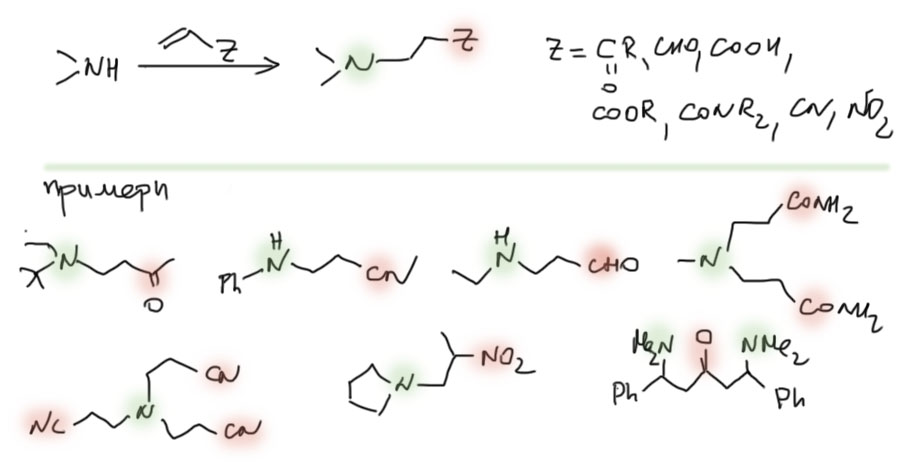
Но в полученных молекулах карбонилы, амиды, нитрилы, нитро-группы могут быть превращены в амины уже описанными методами. Получаются диамины или даже полиамины, у которых между аминами как правило три углерода, или два углерода, если была нитро-группа.
Восстановительное аминирование формальдегида в присутствии муравьиной кислоты называется реакцией Эшвайлера-Кларка. Он никогда не дает четвертичных солей, как заместительное метилирование метилиодидом или диметилсульфатом, но все водороды будут заменены на метилы. Пока сделаем вид, что нам не приходит в голову, что может возникнуть задача заменить только один водород в NH2-группе на метил. 
Те редкие случаи, когда заместительные реакции работают, перечислены на вкладках. Старайтесь не выходить за пределы рекомендованных методов.
Реакции солей диазония
- Выбираем...
- Термическое разложение солей диазония
- реакции солей диазония с восстановителями
- Реакции солей диазония с солями меди(1+) - реакция Зандмейера
Реакции солей диазония многочисленны. Но очень хорошо распределяются на три группы.
- Превращение солей диазония в фторпроизводные и фенолы – термическое разложение
- … в иодпроизводные, а также уничтожение аминогруппы через диазотирование – восстановительные реакции
- … во все остальное (хлор и бромпроизводные, нитрилы и т.п.) – реакцией с солями меди.
Выбирайте соответствующую вкладку
Соль диазония – соединение достаточно склонное к разложению, так как в нем присутствует лучшая в мире уходящая группа – молекулярный азот. Но после ухода азота остается карбокатион, в котором плюс находится на sp2-гибридном атоме и никак не стабилизирован (пустая орбиталь находится в плоскости ароматического кольца), поэтому такие катионы чрезвычайно неустойчивы и, если образуются, немедленно схватываю первый попавшийся нуклеофил, вообще не разбирая, хорош он или плох. В этом как раз и заключается проблема – если в растворе несколько нуклеофилов, то будет смесь продуктов. Например, если нагревать диазониевую соль, полученную диазотированием в соляной кислоте, то нуклеофилами будут хлорид-ион и вода, и мы получим смесь хлорпроизводного и фенола. Поэтому для таких реакций подбирают условия с одним нуклеофилом. Таких реакций известно несколько, но самых важных две:
Получение фенолов
Анион серной кислоты, бисульфат-ион, чрезвычайно слабый нуклеофил, как и положено аниону сильной кислоте. Поэтому в диазорастворе, полученном диазотированием с серной кислотой, нет другого нуклеофила кроме воды. В этом случае получаются фенолы. Диазораствор просто нагревают и выдерживают при кипении до полного разложения соли диазония. Чтобы в этом убедится, время от времени делают пробу на образование азокрасителя. Это – самый универсальный метод синтеза фенолов из доступных нам, хотя реакция довольно грязная и дает невысокие выходы просто потому, что соль диазония разлагается медленно, и по дороге начинает кусать образующийся фенол с образованием окрашенных смол.
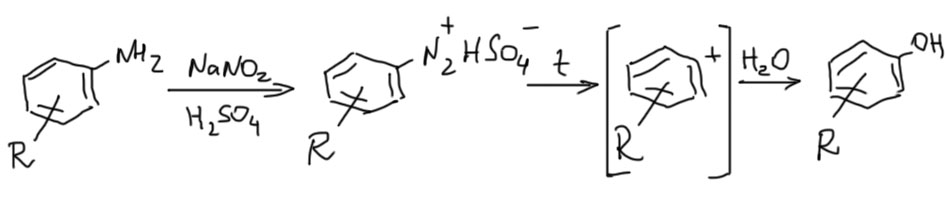
Получение фторпроизводных
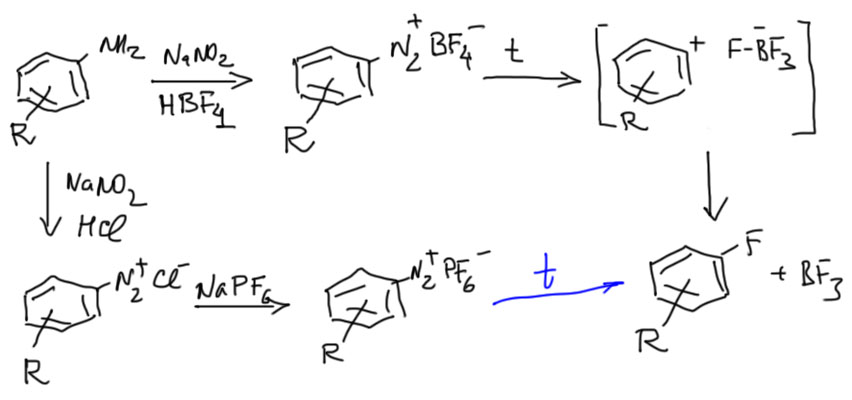
Фторид в качестве нуклеофила – штука крайне капризная и непростая. Можно было бы просто диазотировать в фтористоводородной кислоте, но она в водном растворе слабая, а с безводным фтористым водородом охотников работать немного. Поэтому пришлось использовать довольно неожиданный метод. Диазотируют в присутствии тетрафторборной кислоты – это легко доступный и дешевый реактив и получают тетрафторборат диазония. Альтернатива – диазотировать в соляной кислоте, и потом из водного раствора осаждать тетрафторборат солью тетрафторборной кислоты – немного дешевле, но и опаснее, потому что в такой соли диазония остаются ионы хлорида, что иногда приводит к взрывам при разложении. Вместо тетрафторборной кислоты или ее соли используют еще гексафторфосфорную кислоту, но это чисто косетическое, непринципиальное улучшение.
Соль выделяют и сушат и разлагают в твердом виде, просто нагревая смесь с чем-нибудь инертным, например, чистым отмытым и сухим песком (песок должен быть чистым! песок прямо из песочницы во дворе с хорошей долей вероятности сделает этот эксперимент последним в вашей карьере). Тогда арильному катиону не остается ничего, кроме как выдрать фторид из тетрафторбората. Это очень трудно и красноречивее любых слов говорит о чудовищной реакционной способности катиона. Этот метод – называют реакцией Бальца-Шимана – и это самый универсальный способ введения фтора в ароматическое кольцо. Если видите фтор в ароматическом кольце – будьте уверены, что он там по вине Бальца и Шимана. В нашем курсе вариантов нет. Вы, конечно, можете вспомнить нуклеофильное ароматическое замещение, которое мы проходили в 1 семестре и уже намертво забыли, и там есть возможность ввести фтор вместо хлора, но только при наличии нитро-групп, желательно двух или трёх в орто- и пара-положениях, относительно обмениваемого галогена. Здесь нам эта химия вряд ли встретится, потому что не очень легко понять, что делать с этими нитро-группами. И, безусловно, это не касается задач, в которых фтор уже присутствует в исходных.
Другой важный способ превращать соли диазония во что-то полезное – реакция с восстановителями. Перенос электрона от восстановителя на соль диазония приводит к ее моментальному разложению с образованием того же азота и фенильного радикала. Радикал намного более стабильная (точнее, менее нестабильная) частица, чем катион, и такие реакции происходят намного легче термического разложения. Образующийся радикал быстро с чем-нибудь рекомбинирует, или выдирает атом водорода, откуда полегче. Таких реакций очень много, но мы возьмем самые важные.
Получение иодпроизводных
Иодид-ион – самый сильный восстановитель из галогенидов, и это всем известно. Поэтому реакция иодидов с солями диазония идёт совсем не так, как реакции с остальными галогенидами. Во-первых, в этой реакции не нужна медь ни в каком виде. Используют просто раствор иодида калия или натрия. Когда вы прибавляете этот раствор к раствору соли диазония, и если всё пока ещё, как положено, холодное, вы можете увидеть, как сначала выпадает обильный тёмно-красный или коричневый осадок. Это иодид соли диазония. Этот осадок очень быстро начинает пузыриться. В этот момент стакан с диазораствором ставят в холодную водяную баню. Запомните, что все реакции диазотирования и разложения солей диазония всегда делают в стаканах, лучше с большим запасом объёма – если у вас 100 мл реакционной смеси, стакан берите минимум поллитровый, а лучше литровый. В стакан можно положить магнитную мешалку, но это не принципиально – это тот случай, когда реакционная смесь отлично перемешивает сама себя. Ни в коем случае не берите маленьких стаканов без запаса, и тем более, колб с мешалками. Если возьмёте, то будете иметь шанс устроить любительскую инсценировку сказки братьев Гримм про горшочек каши. Горшочек, то есть ваш стакан с реакцией вдруг начнёт варить сам по себе, не надо ничего и говорить, как вы увидите неуклонно поднимающуюся в нём чёрную пену, полминуты, и она уже прёт наружу, стекает по стенкам стакана, мерзко пузырясь и смердя, стекает на пол, и ручьями устремляется к выходу из практикума. И бесполезно тогда кричать: “Раз, два, три, горшочек, больше не вари…”. Он вас уже не послушает.
Вот чтобы этого не было, берите стакан побольше, и нагревайте смесь очень медленно через водяную баню, сначала холодную, сказав, для порядка: “Раз, два, три, горшочек, вари…”. Тогда обычно пена на успевает подняться до жерла стакана, и всё проходит спокойно.
И обязательно нагрейте содержимое стакана почти до кипения, градусов до 80-90, и подержите в этом состоянии полчаса. Это обязательно, если этого не сделать, то у вас может остаться неразложившаяся соль диазония, которая обязательно напомнит о себе при выделении продукта реакции хорошим бамсом. Если смесь хорошо прогреть, вы будете застрахованы от этой неприятности. И еще – не нужно греть смесь до хорошего кипения – очень многие иодпроизводные великолепно летят с водяным паром, и могут отправиться вместе с ним в тягу. После этого в окрестностях химфака выпадет иодбензольный дождь, сильно озадачив Роскомгидромет, где, вероятно, решат, что загрязнение окружающей среды приняло угрожающие размеры.
В методиках про этом синтез часто пишут, что его нужно после добавления раствора иодида оставить на ночь. Постарайтесь этого не делать, так как это бесполезно, а если вы забудете прогреть смесь, то и опасно – за ночь просто при комнатной температуре соль диазония разложится не полностью, и на следующий день с удовольствием преподнесёт вам гадкий сюрприз. Смесь после прогрева можно оставить и на ночь, и на неделю. Иодпроизводные все очень тяжелы, и скапливаются на дне стакана тяжёлым чёрным маслом, или мерзкими чёрными блямбами. Не пугайтесь, так и должно быть. Все реакции солей диазония очень грязные (об этом как-нибудь отдельно), к тому же там всегда есть много иода, образовавшегося из иодида реакцией с почти всегда остающимся избытком азотистой кислоты, как её ни гаси мочевиной.
Чтобы это быстро очистить вчерне, предпочитают перегонку с паром. Большинство простых ароматических галогенпроизводных легко летят с водяным паром, оставляя в колбе всякую окрашенную дрянь. Для надёжности перед перегонкой с паром к смеси добавляют раствор щёлочи до щелочной реакции, но делая это, будьте осторожны, ведь для диазотирования вы могли взять очень большой избыток кислоты (это придётся проверить, посмотрев в методику и свои записи о ходе эксперимента и загрузках реагентов). Если избыток был очень большой, щёлочь лучше не добавлять. Зачем щёлочь? – для того чтобы перевести побочно образующийся фенол в нелетучий фенолят. Фенол, скорее всего, полетит вместе с продуктом, но вы его потом отмоете раствором щелочи. Ещё обязательно полетит иод – конденсат будет розовым, и иод растворится в иодбензольном слое, опять окрасив его. Вам может показаться, что очистка не получилась, но это не так – от окрашенного органического дерьма вы точно избавились. После перегонки к отгону ( а это очень много тёплой воды, на дне которой тёмно-розовое масло или кристаллические шматки) добавляют немного хлороформа, переливают в делительную воронку, и последовательно встряхивают а) с раствором некрепкой щёлочи; б) с раствором тиосульфата натрия (не сульфита! и не гидросульфита) до полного ухода окраски иода.
кликните, чтобы узнать почему не надо заменять тиосульфат бисульфитом или метабисульфитом
Причина проста – нам надо быстро и количественно убрать иод, потому что даже следы иода окрашивают органику в розовый цвет и отлично перегоняются вместе с органикой хоть в вакууме, хоть просто так, так что перегонкой от иода не избавиться. Поэтому нам нужна быстрая количественная реакция, восстанавливающая иод до иодида, иодид-ион уходит в воду, и больше никому не мешает. Самая известная быстрая и количественная реакция с иодом это реакция с тиосульфатом – недаром ее используют для иодометрического тирования, настолько она аккуратная и быстрая. Встряхивание органического слоя, содержащего иод, убирает его моментально. Важно только чтобы а) вы умели обращаться с делительной воронкой – надо уметь очень хорошо встряхивать, так как будто вам самого/саму просто трясет от ненависти к этому иоду и вы готовы на очень серьёзную встряску, чтобы его убрать. Проблема в том, что иод у вас в органике, а тиосульфат в воде, и реагируют они только на границе раздела фаз и трясти надо так, чтобы жидкость разбилась (делительная воронка и надежды при этом разбиться не должны) на мельчайшие капельки, только тогда реакция произойдёт быстро, прямо у вас на глазах. Кроме этого б) тиосульфата должно быть достаточно для количественного восстановления иода. Если тиосульфата недостаточно, иод останется и это очень хорошо видно по цвету водного слоя – он будет не бесцветный, а желтый. Тогда смело добавляйте еще раствор тиосульфата (раствор! не кристаллы – никогда не сыпьте в делительную воронку кристаллические соли!) и еще раз встряхивайте – водный раствор должен оставатьсяс бесцветным. Это поможет вам убедиться в том, что вы убрали весь иод даже если раствор органики густо окращен всякими органическими примесями, которые часто образуются в диазотировании, и на этом фоне чёрт поймёт, есть там иод или нет – но именно встряхивание с тиосульфатом скажет вам это – если иод есть, водный слой сначала окрасится в желтый цвет, и если тиосульфата достаточно, быстро обесцветится. Если вместо тиосульфата взять бисульфит или метабисульфит, или просто сульфит иод тоже восстанавливается, но намного медленнее, особенно если иода мало – эти анионы гораздо хуже, чем бисульфит действуют в двухфазной водно-органической системе. Поэтому вы иод просто, скорее всего, нацело не отмоете, и все у вас будет розовым. Берите тиосульфат, и если все делаете правильно, результат гарантирован.
Трясти надо усердно, взяв нетекущую делительную воронку. После, как обычно, сушат, отгоняют на роторе хлороформ, и перегоняют остаток в вакууме.
Что происходит в реакции. Иодид-ион восстанавливает соль диазония, радикал разваливается, азот улетает, и два радикала немедленно рекомбинируют в иодпроизводное. И всё. В учебнике Курца про эту реакцию написано, что у неё SRN1-механизм. Это странная и для меня необъяснимая фантазия автора учебника, не имеющая никакого отношения к реальности. В мезанизме SRN1 связь C-I получиться просто не может.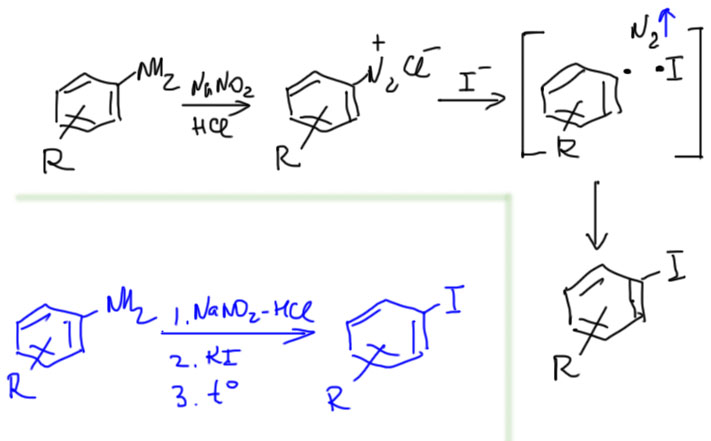
Уничтожение амино-группы
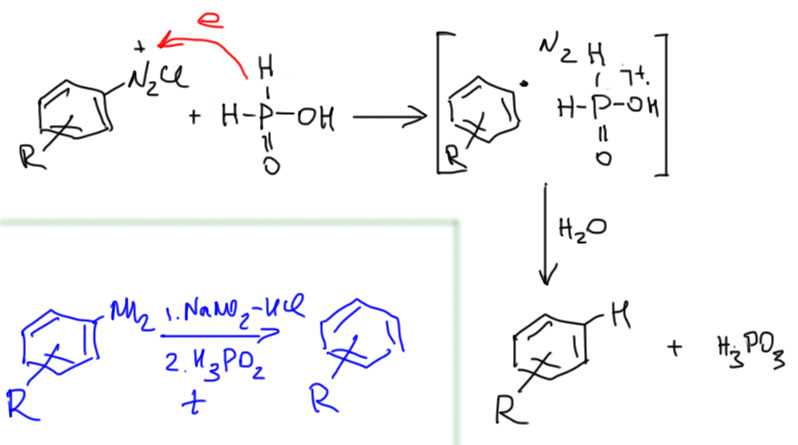
Это очень важная реакция в много стадийных синтезах, когда амино-группа используется как мощный ориентант, а затем удаляется. Такое превращение можно вызвать множеством реагентов, которые могут тыть источниками атома водорода – это и спирты, и боргидрид натрия, и т.п. – но самый надежный и универсальный – гипофосфористая кислота, которая по своей природе является фосфином, то есть содержит очень слабую связь P-H. Она же и восстановитель.
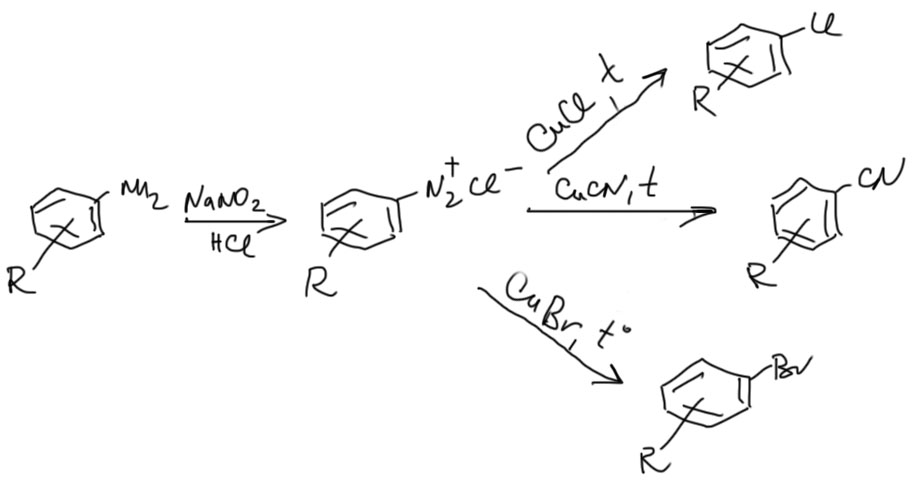
Но еще больше реакций солей диазония можно описать одним шаблоном – взаимодействие с производными одновалентной меди. При этом получаются производные с заменой диазогруппы на то, что было у меди. Механизмы этих реакций до сих пор толком неизвестны, но в любом случае это нечто для нас необычное, требующее рассуждений о реакциях в координационной сфере металла. Скорее всего, эти реакции, так же как и реакции с восстановителями, включают распад соли диазония до радикала, вполне вероятно за счет восстановления одновалентной медью, но дальше радикал не рекомбинирует, а получает один из лигандов с атома меди, который при этом переходит в Cu(2+). Даже писать это не будем.
Все эти реакции скопом называют реакцией Зандмейера. Так получают, среди прочего, хло-, бромпроизводные, и нитрилы.
Очень важно
Реакция диазотирования и превращения солей диазония дают исключительно мощный инструмент синтеза производных бензолов. В принципе, использовать эти реакции можно в самых разных комбинациях и исходя из самых разных исходных совершенно произвольным образом. Но многие разумные люди не хотят тратить свое время на фантазии и предпочитают более ясные рекомендации. В данном случае такой фундаментальный рецепт есть. Так как он действительно очень строго и определен, он носит очень серьезное название “Теорема о трех нитроанилинах”.
Эта теорема до сих пор не доказана. Даже Великую теорему Ферма уже доказали, а Великую Теорему о Трех Нитроанилинах нет. Но ее никто и не опроверг. Опровергнуть ее можно, предложив такое производное бензола, которое нельзя получить из трех нитроанилинов. Может быть у вас получится? Если получится, держите это в тайне, потому что Великой Теореме не понравится, что кто-то её пытается опровергнуть, а жрецы культа Великой Теоремы сделают так, что вы больше не сможете правильно решить ни одной задачи.
Если коротко, то действовать нужно так.
1. Берем один из трех нитроанилинов.
2. Используя невероятную орто-пара-ориентирующую мощь аминогруппы (возможно огромную реакционную способность придется даже немного притушить ацетилированием) производят электрофильное замещение в свободные орто и пара положения относительно амина.
3. Повторяют электрофильное замещение, если остались свободные места.
4. Аминогруппу диазотируют, и или замещают, или вовсе убирают, как описано на карточках про соли диазония.
5. Нитро-группу восстанавливают до амина
6. Используя невероятную орто-пара-ориентирующую мощь аминогруппы (возможно огромную реакционную способность придется даже немного притушить ацетилированием) производят электрофильное замещение в свободные орто и пара положения относительно этого нового амина.
7. Повторяют электрофильное замещение, если остались свободные места.
8. Аминогруппу диазотируют, и или замещают, или вовсе убирают, как описано на карточках про соли диазония.
9. Радуются очередному триумфу Великой теоремы. В сэкономленное от ненужных раздумий время веселятся и развлекаются, прославляя Великого Создателя Теоремы (это не я).
С этим следствием все очень просто, так как любой из трех нитроанилинов можно получить из бензола. Следовательно, это высказывание сводится к Великой теореме и может быть или доказано или опровергнуто только вместе с ней.
Напомню, как это делается. Орто- и пара-нитроанилины получаем из анилина, который получаем из бензола.
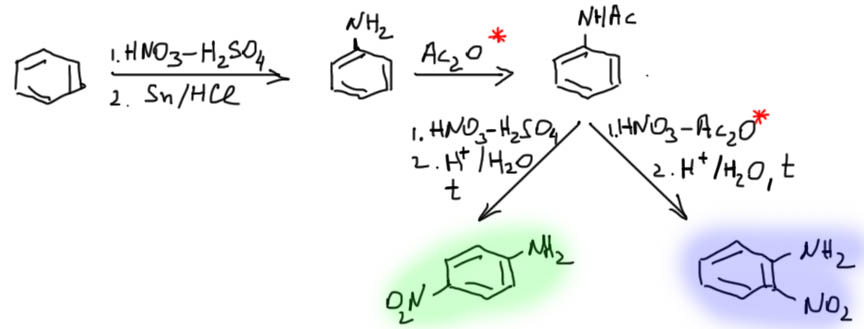
(* запрещен в Российской Федерации)
Мета-нитроанилин получаем парциальным восстановлением м-динитробензола
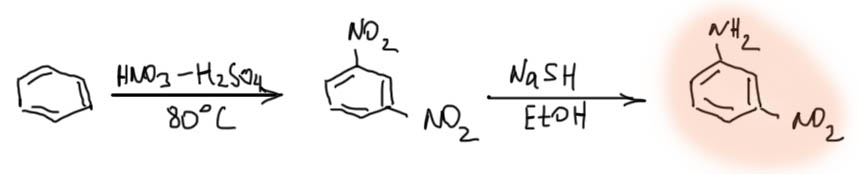
Далее действуем согласно Теореме.
Это следствие можно было бы сформулировать как отдельную теорему, но на самом деле это просто разновидность Великой теоремы. Его применяют гораздо реже самой Великой теоремы и обычно только тогда, когда в исходных даны как раз одна из амино- или нитробензойных кислот, или их очевидных предшественников, например, толуола. Из толуола можно получить все три аминобензойные кислоты.
Действуют точно так же, как в самой Великой теореме, но в тех местах, где там делают восстановление нитро-группы в амино-группу здесь применяю превращение карбоксила в амин с помощью перегруппировок Гофмана или Курциуса (см. выше Алгоритм).
Разные вспомогательные методы
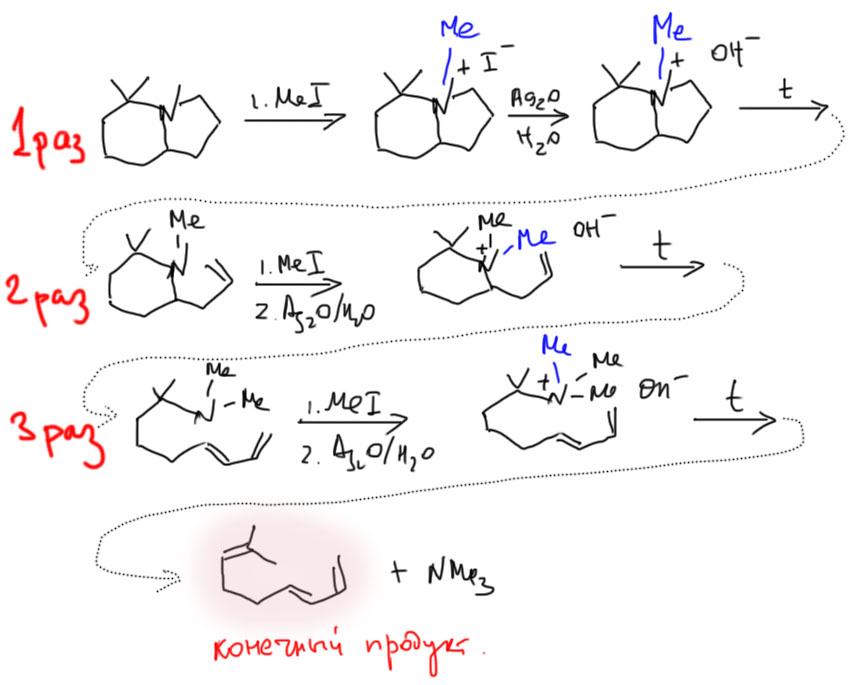
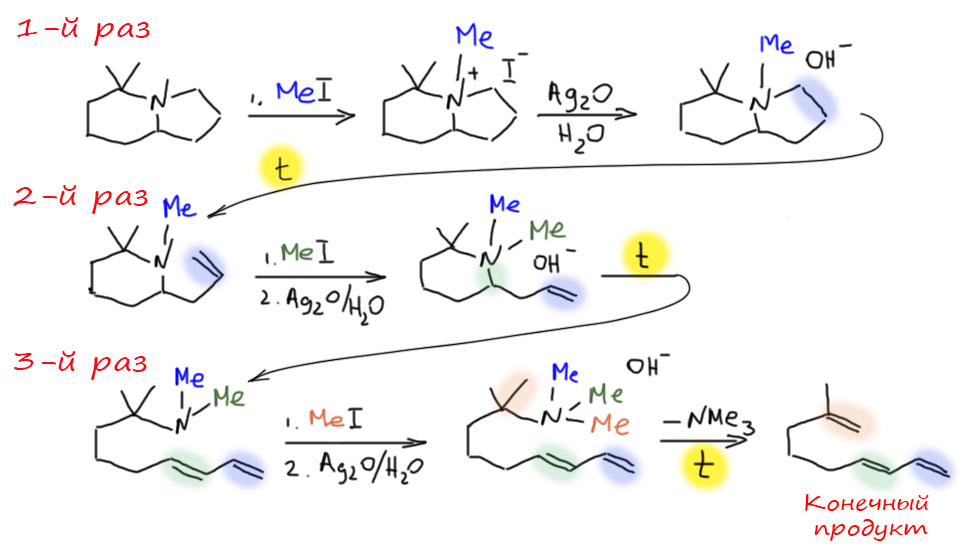
Практически чистый путь элиминирования предложил еще в 19 веке как раз Гофман, ничего не зная ни про E2-механим, ни даже про основность. Но смысл его решения такой – нужно взять очень плохую уходящую группу, тогда и с основанием можно особенно не заморачиваться. В качестве уходящей группы используют триметиламин, а вся процедура выглядит как раз на 19 век: берем амин, обрабатываем его избытком иодистого метила – происходит три подряд SN2-замещения и образуется четвертичный аммоний с противоионом иодида. Обрабатываем это влажной окисью серебра, осаждается иодид серебра и получается гидроксид четвертичного аммония. Дальше просто греем и происходит E2-элиминирование. Если немного подумать, то можно придти к выводу, что гидроксид четвертичного аммония – это просто идеальные условия того, что мы сейчас называем межфазным переносом. И вспомнить. что в этих условиях гидроксид становится сильнейшим основанием, ничем не хуже трет-бутилата. Так что заодно мы выяснили, что Гофман со своим методом фактически предугадали межфазный катализ.
Так как уходящая группа очень плоха, но и работает как индуктивный акцептор, отщепление протона опережает уход плохой уходящей группы, и тогда важно, какой из имеющихся протонов более кислый – а это именно протоны на менее замещенном атоме углерода.

Этот метод очень активно использовали в доспектральные времена для установления структуры аминов. Проводили в несколько стадий полную деструкцию исходного амина, после чего исследовали полученное непредельное соединение. На каждой из стадий образуется наименее замещенный олефин из возможных. Так как основным приемом метода была обработка избытком иодистого метила, метод называли исчерпывающим метилированием. Вот пример применения метода – обратите внимание, в каком порядке образуются двойные связи из исходной структуры.
Внимание! В этой схеме есть глупая ошибка, на которую обратил моё внимание г-н Хайкин. Я не буду её уничтожать, потому что исправление ошибок – лучшая тренировка знаний, а любые возможности надо использовать. Рассмотрите схему, найдите ошибку, и только после сравните с исправленной схемой.
Алифатические нитро-соединения
- Из акролеина и нитрометана получите 1,5-динитропентан-2-ол
- Из бензальдегида и фенилнитрометана получите 1,3-динитро-1,2,3-трифенилпропан
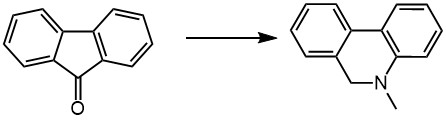
Перегруппировка Бекмана и др.
2. Из дибензилкетона и ацетона получите бензил-изопропил-(2-фенилэтил)амин
Перегруппировки Гофмана, Курциуса и др.
2. Из циклогексанона, метилиодида и формальдегида получите 1-(N,N-диметиламино)-1-метилциклогексан
Присоединение по Михаэлю и др.
2. Из малоновой кислоты, бензальдегида, циклопентанона и диазометана получите соединение B (структура внизу)
3. Из акролеина и изобутилена получите диамин C.
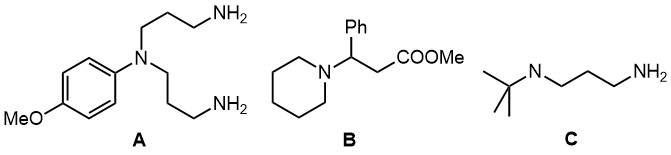
Ароматические амины, восстановительное аминирование и др.
2. Из анилина, ацетона, формальдегида получите диамин B (структура внизу)
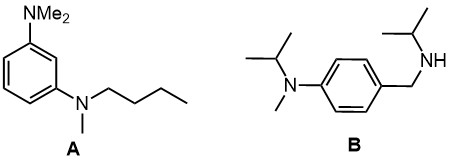
Диазотирование
1. Из п-нитроанилина получите 2,6-дихлор-3,5-дибромбензойную кислоту
2. Из 2,6-дидейтеробромбензола получите 3,5-дидейтеробромбензол
3. Из о-нитроанилина получите 1-фтор-3-хлор-4,6-дибромбензол
Диазотирование и синтез аминов
1. Из о-нитроанилина получите N-(2-фторбензил)-o-диаминобензола
Теорема о трех нитроанилинах
Получите из бензола и днр не менее пяти разных изомерных фторхлориодбензойных кислот на ваше усмотрение. Заодно выясним, может быть их и нет столько.