Перед тем, как приступать к задачам, почитайте общие рекомендации, как это делать правильно и даже, в каком-то смысле, скучно.
Разложим карточки с методами и реакциями по алкенам, алкинам, диенам на те, с помощью которых делают новые углерод-углеродные связи, изменяют реакционные центры, создают стереоизомеры, и полезные дополнительные методы.
Новые C-C связи
Особое внимание! Это основные методы, которые вы будете использовать для получения более сложных молекул из более простых.
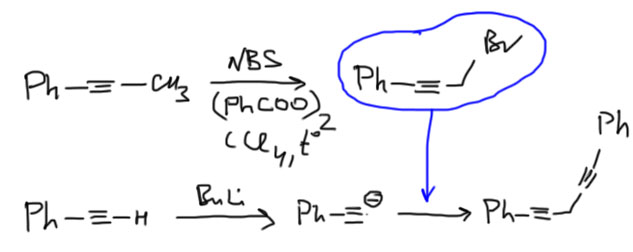
Основная реакция для C-C связей в этом разделе. Терминальный ацетилид превращают в карбанион действием очень сильного основания (бутиллития, амида натрия, гидрида калия), и затем добавляют алкилгалогенид. Механизм реакции – SN2, скоро изучим подробно и тогда поймем, откуда здесь всякие жесткие ограничения.
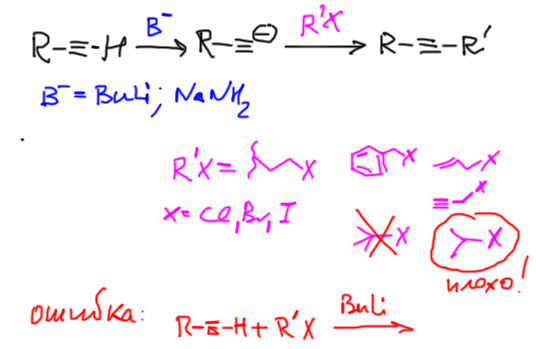
Алкилгалогениды это все галогенпроизводные, где галоген висит на sp3-гибридном атоме, галоген это хлор, бром или иод (иод мы пока вводить не умеем). Хороши для этой реакции метил, первичные алкилы, бензил, аллил, пропаргил (показаны на схеме). Нежелательны вторичные алкилы, но если деваться некуда – используйте. Третичные алкилы совершенно не годятся – использование таких алкилгалогенидов – фатальная ошибка. Не забывайте, что в реакции две стадии и их нужно выписать явно. Ошибка – месить все в одну стадию.
Примечание: для этой реакции хороши также магниевые производные ацетиленов, но пока это отложим.
Еще одно дополнение для любознательных. Сам ацетилен имеет два терминальных атома водорода, и может быть превращен в дианион. И это вполне может быть сделано для того, чтобы сразу сделать диалкилирование. Одна проблема – из-за низкой растворимости солей дианиона в обычных растворителях реакцию ведут в жидком аммиаке. В этом случае – пожалуйста.
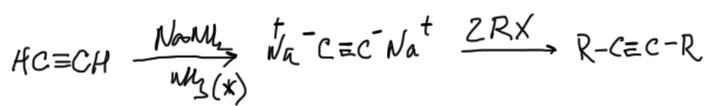
Но если вы хотите сделать только моноалкилирование, то задача становится не так проста. Проблема в том, что в этом случае трудно избежать побочного диалкилирования. Моноанион получить очень просто, использовав эквивалент амида натрия или другого очень сильного основания, потому что оторвать от него второй протон намного медленнее, pK по второму протону сильно выше чем pK по первому по очевидным причинам, но как только начнете прибавлять алкилгалогенид, возникнет равновесие между остающимся анионом незамещенного ацетилена и продуктом алкилирования. Кислотность ацетиленов приблизительно одинакова, а это значит, что после начала реакции (а реакция алкилирования не очень быстра даже с очень хорошими стубстратами), в реакционной смеси одновременно присутствует как исходный ацетиленид, так и получающийся замещенный ацетилен. Никто не может запретить этим двум веществам вступить в кислотно-основное равновесие и поменяться протонами – при этом в смеси образуется анион получающегося ацетилена – и неизбежно пойдет второе алкилирование. В результате получается смесь моно- и дизамещенного. Это не большая трагедия – при большой разнице в мол. массах эту смесь обычно нетрудно разделить даже перегонкой. Но мы хотим селективных реакций. Здесь применяют такой выход, обнаруженный экспериментально (Smith, Beumel, Synthesis 1974, 441). Ацетиленид лития образует комплекс с этилендиамином, и этот комплекс дает гораздо более чистое моноалкилирование. Более того, этот комплекс так устойчив, что его можно выделить и хранить, используя по мере необходимости, а саму реакцию алкилирования ведут в ДМСО (в следующей теме разберёмся, что это такое). Этот метод стал стандартным для моноалкилирования ацетилена, хотя встречаются и реакции, сделанные по старинке, в жидком аммиаке. При этом, кислотность ацетилена на много порядков выше кислотности амина, и отрыв протона от самого амина не является проблемой.
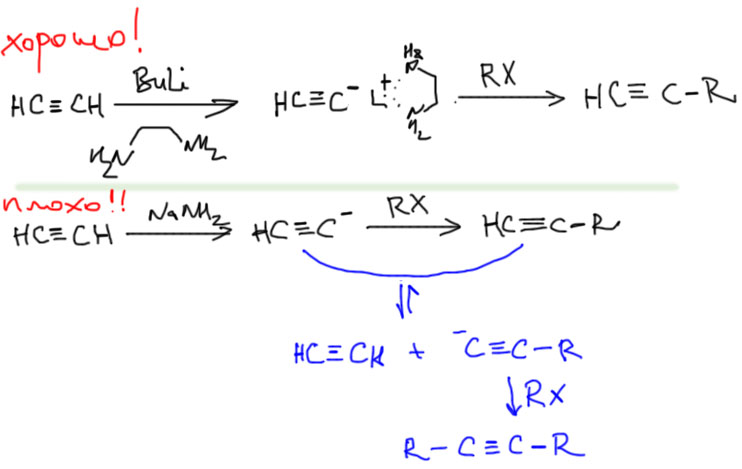
Вопрос, почему в этом комплексе не идет диалкилирование через обмен протоном, можно оставить без ответа – вот так найдено и это работает, и это главное. А можно и порассуждать о том, что возможно в комплексе не только катион лития связан с диаминовым лигандом, но и анион образует водородную связь в тем же лигандом, образуя такой комплекс катиона лития, в координационной сфере которого хелатный диаминовый лиганд удерживает и ацетиленид-анион. В такой конструкции анион будет иметь пониженную основность (будет закрыт водородной связью), и отрыв протона от образующегося ацетилена будет подавлен.
Это очень частная реакция с ограниченным применением. Димеризация эта не настоящая, а окислительная – в продукте не хватает двух атомов водорода из исходных, а это значит, что в реакции необходим окислитель. Носит разные названия – реакция Глазера (не Глейзера!, Карл Андреас Глазер – немец, весьма знаменитый химик второй половины 19 века), Эглинтона, Хэя, потому что ее открывали заново несколько раз, от Глазера до Хэя прошло почти ровно сто лет. Допускает широкую вариацию реагентов. Обязательно нужна соль меди (не важно Cu(I) или Cu(II)) типа ацетата Cu(OAc)2 или хлорида CuCl), окислитель (чаще всего просто кислород) и азотный лиганд (аммиак, пиридин, или что-нибудь посвежее типа тетраметилендиамина, сокращенно ТМЭДА). Вариант с двухвалентной медью может быть без кислорода – сама медь окислитель, но это детали невеликой важности. Распространённая ошибка с этой реакцией – нельзя так сшить два разных алкина, получите смесь всех возможных диинов. Для сшивания двух разных алкинов в современной химии есть мощные методы с использованием разных переходных металлов, но мы не будем это изучать и использовать.
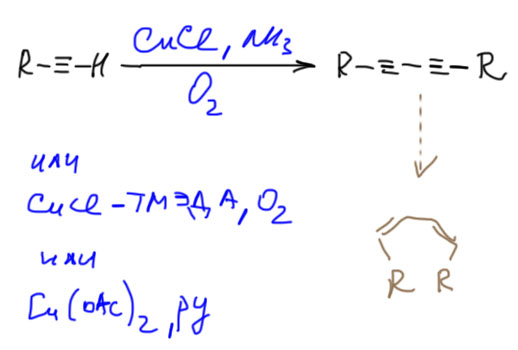
Единственное применение этой реакции – последующее гидрирование диина в диен. В реальности это не так просто, но в учебных целях годится. Я разберу это на вкладке про гидрирование алкинов в алкены.
Кроме этой весьма полезной реакции есть и настоящая димеризация ацетилена, без окислителя, при которой водород никуда не девается, он формально как будто переселяется с одной молекулы ацетилена на вторую – образуется винилацетилен. Это вещество, газ с ещё более капризным нравом, чем у самого ацетилена, легко взрывающийся при повышении давления, в начале 20 века пытались пристроить в промышленность в знаменитой американской компании Дюпон де Немур, но так же как с самим ацетиленом это не получилось, опасно слишком. Но от тех экспериментов и патентов пошло общепринятое сокращение для этого соединения – его называют МВА, то есть моновинилацетилен, просто потому что та же реакция дает и тример – дивинилацетилен (ДВА), а это тоже весьма норовистая вонючка. Впервые реакцию описал в 1931 году Ньюланд и др. (J. A. Nieuwland et al. J. Am. Chem. Soc. 1931, 53, 4197), и реакция эта выглядит совершенно невменяемо. Готовят суспензию CuCl в крепком водном расторе хлористого аммония, и в эту смесь булькают ацетилен пока растворяется, и через некоторое время нагреванием выгоняют оттуда смесь МВА и ДВА с побочными продуктами, выход МВА не превышает 4% (четырёх!) на исходный ацетилен и приблизительно столько же ДВА. В наши времена мы посмотрим на эту реакцию совсем по-другому, признав, что это каталитическая реакция с участием комплекса переходного металла, а такие реакции в последние 30-40 лет стали невероятно модными и популярными. Поэтому Ньюланд с сотрудниками фактически были одними из самых пионеристых пионеров этой ныне великой химии, хотя сами они об этом и не догадывались. Впоследствии реакцию много изучали химики-технологи и несколько улучшили результаты, использовав ту же CuCl, но в органическом растворителе типа ДМФА с добавкой солей всяких алифатических аминов. Выход МВА доходит до 20%, но и это не фонтан – ацетилен ведь штука недешёвая, на его производство тратится чёртова уйма энергии, и вот так просто превращать 80% этого сырья чёрт знает во что – занятие малопочтенное. Это сошло бы, если бы винилацетилен можно было бы применить в каком-нибудь синтезе чего-нибудь очень ценного, но ничего такого найти не удалось, хотя это вещество интересное и в лабораторном синтезе очень ценное.
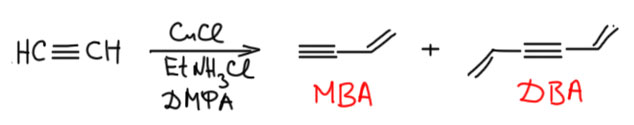
Надеюсь вы поняли, что нам эта реакция даром не нужна. Но она попала в некоторые учебники и иногда всплывает без каких-либо упоминаний о её проблемах. Более того, эта реакция попала в школьную химию, и ею важно трясут на олимпиадах и даже, насколько я слышал, чуть ли не на егэ. Удивительно, но это странный отголосок очень старой химии, похоже что ещё довоенной, когда таким веществам усердно пытались найти применение, кому-то показалось, что скоро найдут и нужно срочно пихать в учебники. Применения так и не нашли, а в учебниках осталось.
К тому же она, в ее оригинальном виде не применяется к замещённым ацетиленам – только к самому ацетилену. Но в современной химии реакция двух ацетиленов с образованием сопряжённого енина очень хорошо исследована и понята, но она требует современных методов с использованием весьма изощрённых катализаторов, комплексов переходных металлов, прежде всего родия, рутения, палладия. Я разберу ее в курсе про переходные металлы как-нибудь.
Если соединить эту реакцию с озонолизом можно получить остроумный метод быстрого синтеза весьма непростых молекул. Отработаем этот шаблон в задачах.
Изменения реакционных центров
Образование двойной связи
Скелет не меняем, делаем двойную связь в определённых местах.
Превращение алканов в алкены – промышленная реакция, в лаборатории не используется. И не используем. У нас тут университет, а не химзавод.
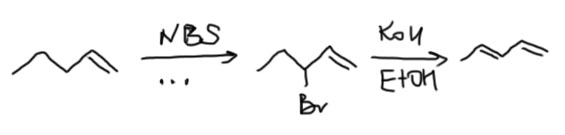
Превращение галогенпроизводных в алкены (отщепление, элиминирование) – очень важный метод, но подробно мы его рассмотрим позже. Сейчас только простой вариант отщепления HBr или HCl любым сильным основанием (KOH/EtOH). Не забываем, что получается самый стабильный олефин из возможных (см. Алкены, про стабильность).
Превращение спиртов в алкены. Кислотно-катализируемая дегидратация (реагент обозначается условно, например, H2SO4 кат.). Тоже получается самый устойчивый олефин. Реакция плохая, осложняется перегруппировками. Потом разберемся. Пока использовать только для простых случаев (2-пропанол в пропен, трет-бутанол в изобутилен, циклогексанол в циклогексен). Во всех остальных избегать.
Превращения двойной связи
- Выбираем...
- Двойная в простую
- Присоединение галогена
- Присоединение галогеноводорода
- Присоединение воды
- Присоединение солей ртути (сольвомеркурирование)
- Гидроборирование
Двойная связь очень часто превращается в разные заместители и группы. Важно разобраться, какие методы хороши, а какими не стоит пользоваться ни при каких обстоятельствах.
Используем самый стандартный катализатор – палладий, нанесенный на уголь, Pd/C. Не забываем про водород. Про все остальное лучше забудьте. Никель дешев и могуч, но с легкостью вызывает побочные реакции. Платина – нет смысла, никих преимуществ перед палладием, а стоит во много раз дороже. И умоляю, не используйте модные гомогенные катализаторы гидрирования, всякие комплексы Уилкинсона и прочее. Это очень сложно, требует точных знаний о том, что конкретно они гидрируют (далеко не все алкены, и у каждого такого комплекса свой норов). Нам это сейчас не поднять, а туманные знания часто бывают хуже полного неведения, так как создают иллюзии, заводящие в тупик.
Хлор присоединяем только, если просят. Хлор с водой не присоединяем – просто по аналогии с бромной водой это не получается. Похожая реакция действительно есть, но сделать ее непросто и потребовало бы использовать раствор готовой хлорноватистой кислоты HOCl.
Фтор и иод не присоединяем. Фтор – очень слабый электрофил, и очень сильный, как мы уже успели убедиться, свободнорадикальный агент, поэтому в реакции почти с любым алкеном будет много чего кроме присоединения. А с иодом все непросто, мы вернемся к этой проблеме, когда будем обсуждать реакции элиминирования (если не забудем). Сейчас не используем.
Присоединяем HBr и иногда HCl. Никогда не присоединяем HF – вообще-то это возможно, но очень непросто. Не присоединяем HI – хотя эта реакция происходит очень легко, легче чем присоединение всех остальных галогеноводородов, но образовавшийся алкилиодид очень легко восстанавливается иодистым водородом в алкан, и тогда нужно очень тщательно следить за условиями реакции.
Присоединение HBr идет по правилу Марковникова, а точнее, нужно анализировать стабильность карбокатионов и смотреть, куда присоединяется протон, и куда идет бромид.
Присоединение против правила Марковникова по Харашу с использованием свободнорадикальных инициаторов не используем – вместо этого идем через гидроборирование. Присоединение по Харашу – это классика, но это не препаративный метод, а просто наблюдение над механизмом реакции, и эта реакция на практике чрезвычайно ненадежна. Подробнее обсудим эту почти детективную историю на основной странице про алкены. Но – ее использование не является ошибкой, и если она вам так понравилась, то можете ее использовать на бумаге (но не в лаборатории!). И если уж пишете, не пишите глупостей типа инициирования реакции светом. Там нечего инициировать светом – все реагенты бесцветны. Пишите тогда перекись бензоила в качестве инициатора (см. Алканы. Инициирование)
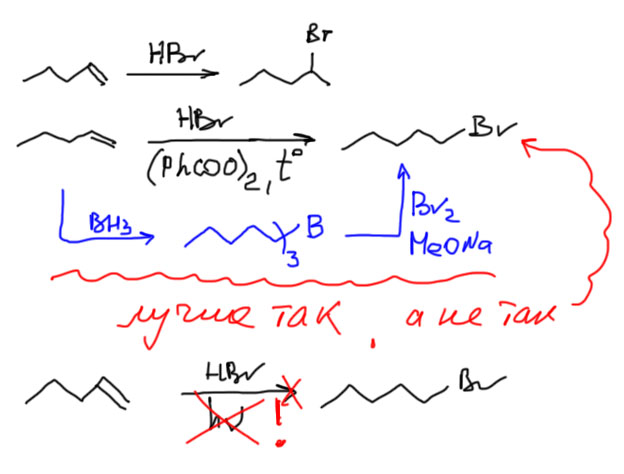
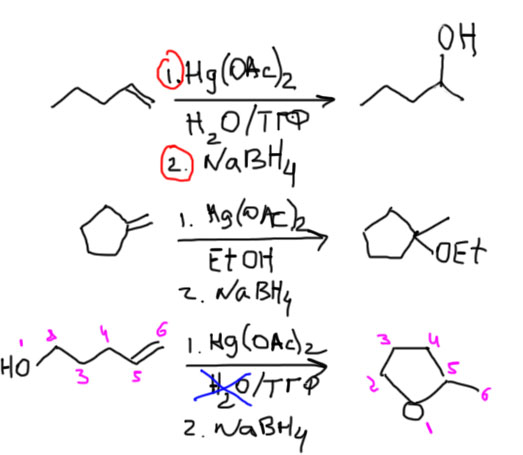
Мощная реакция. Рекомендуется использовать везде, где только можно. С этой реакцией есть несколько малоприятных недоразумений, но нам важно понять, что при гидроборировании алкенов бор присоединяется к менее замещенному атому углерода. В отличие от присоединение галогена, или протона мы не можем нарисовать карбокатион (потому что его там и нет – водород из борана садится на свое место одновременно с бором). Основная причина этого стерическая, но так как это формально совпадает с правилом Марковникова (атом бора всегда присоединяется против правила Марковникова), то можно просто это обстоятельство использовать как раз и навсгда установленное. Когда мы говорим о стерической причине, возможно очевидное возражение, что боран – очень маленькая молекула, и ей не должно так уж сильно важно, к какому атому подходить – менее замещенному или более замещенному. Но боран – не такая уж и маленькая молекула, так как в растворах она почти всегда находится в комплексе с основаниями Льюиса, например, теми же растворителями – гидроборирование почти всегда делают в эфирных растворителях (эфир, ТГФ и т.п.), которые всегда образуют комплексы с бораном. Фактически, в атаку на олефин боран идет в тесной связи с немаленькой молекулой ТГФ или эфира, и эта молекула, кроме того, что увеличивает стерический объем, еще и сильно снижает электрофильность борана – боран становится в таком комплексе более селективным. Используем всегда для последующего превращения в противо-марковниковский спирт или бромид, или как замену гидрированию, возможно с заменой на дейтерий. Реагенты – комплексы борана с основаниями Льюиса, эфиром, ТГФ, триэтиламином (это для нас одно и то же, поэтому выбирайте по вкусу). Обратите внимание, как удобно записывается продукт гидроборирования с тремя одинаковыми остатками.
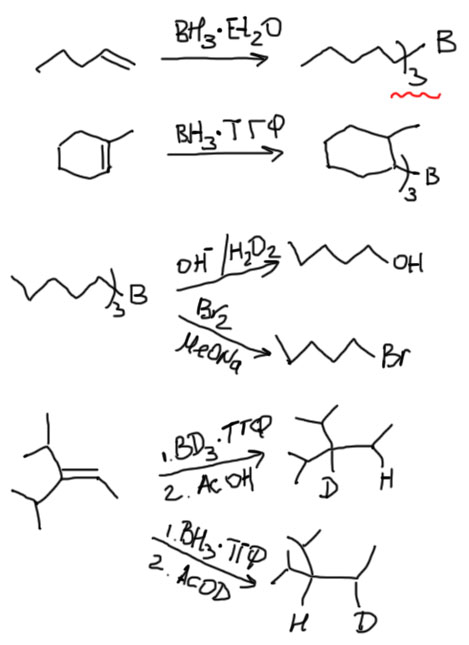
Для замещения борного остатка на водород или дейтерий используют слабую кислоту, обычно уксусную (или дейтероуксусную AcOD). Избегайте сильных кислот (типа HCl или DCl) для этой реакции – получите осложнения, связанные с конкурирующим электрофильным присоединением протона к двойной связи, что неизбежно приведет к потере стереохимической конфигурации или дейтериевой метки, а реакции гидроборирования почти всегда используют для селективных превращений с полным контролем стереохимии или положения изотопных меток. Вот это вы и потеряете.
Атом рядом с двойной связью (аллильное положение)
Всего одна реакция, но очень полезная
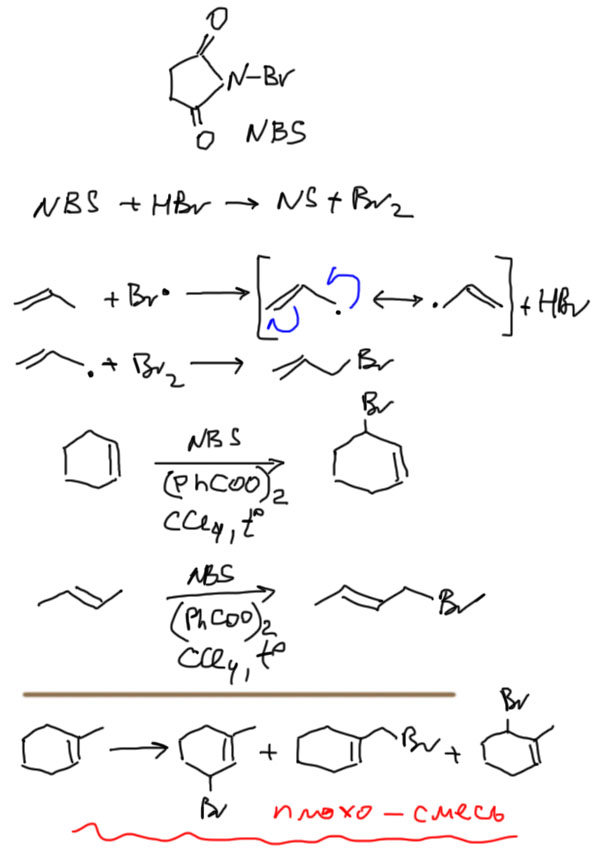
Кроме электрофильных реакций у алкенов есть и радикальные. Если вспомним свободнорадикальное бромирование алканов, радикал брома очень селективен и отнимает атом водорода от самой слабой связи. Там такой связью была третичная, но в алкенах есть и еще более слабая связь – аллильная. Аллильный радикал стабилизирован не жалкой гиперконъюгацией. а настоящим сопряжением. Но есть проблема – нельзя использовать сам бром, потому что он тут же присоединится к двойной связи. Поэтому используют специальный реагент – N-бромсукцинимид (NBS, а по-русски NБС), который является источником радикалов Br⋅ (механизм инициирования в этом случае непрост, и мы его как-нибудь отдельно обсудим), обязательно в неполярном растворителе CCl4, и желательно при дополнительном инициировании перекисью бензоила. И избегайте вводить в реакцию несимметричные алкены – получите смеси всех возможных продуктов (там внизу пример, но продукты перечислены не все). Это упрощённый подход, в реальности всё немного сложнее и интереснее, и про это можно почитать на странице про Алкены. Но для решения задач настоятельно рекомендую именно упрощённый подход, состоящий в том что а) если предлагаете эту реакцию сами, выбирайте только олефины с одинаковыми аллильными положениями и не волнуйтесь о возможном смещении двойной связи – пусть она остается там, где была; б) если олефин для аллильного бромирования вам подсунули и в нём разные аллильные положения, выбирайте по возможности вторичную группу (CH2) и тоже не волнуйтесь о смещении двойной связи – составители задач обычно сами про это забывают.
Образование тройной связи
Тройная связь делается только из двойной
Прямого способа делать тройную из двойной нет – посмотрите еще раз почему (Алкины. Структура и особенности…), и никогда не пытайтесь дегидрировать алкены – это ошибка.
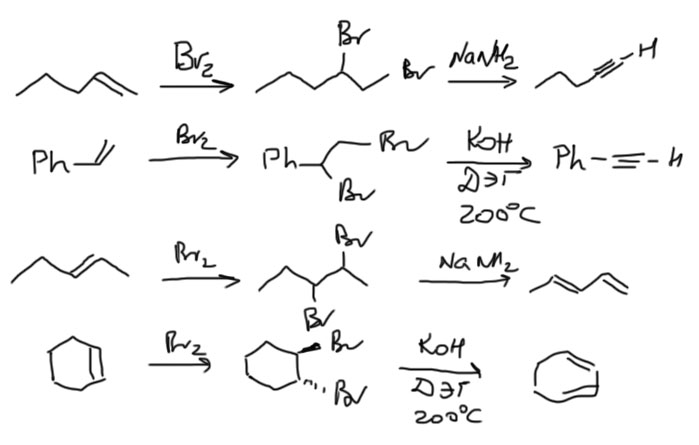
Но можно присоединить бром и отщепить два HBr сильным основанием. Внимание – это надежно работает только, если связи – и двойная и тройная – концевые. Если связь внутри, то вместо алкина часто получается диен. И ни в коем случае не делайте это в циклоалкенах. Еще одна важная вещь – используйте достаточно сильное основание. Обычная спиртовая щелочь, которая работает для получения двойной связи, здесь недостаточна. Используйте амид натрия, или щелочь (обязательно KOH – потом разберемся почему) в растворителе, обладающем свойствами образовывать комплекс с катионами калия – это диэтиленгликоль или триэтиленгликоль, при высокой температуре. Также годится трет-бутилат калия.
Превращения тройной связи
- Выбираем...
- Электрофильное присоединение
- Реакция Кучерова
- Гидроборирование
- Альдегиды из алкинов
- Гидрирование
- Концевой водород в терминальных алкинах
- А аллильное замещение в алкинах есть?
Тройная связь даже еще более полезна, чем двойная – ее легко превратить во множество других заместителей и групп.
Самая полезная для нас сейчас реакция, потому что с ее помощью легко осуществляется селективное превращение тройной связи в альдегиды (здесь такая же история как с реакцией Кучерова, но завершить цепочку синтеза альдегидом не откажется ни один вменяемый составитель задач). Не забываем про то, что гидроборирование и реакция Кучерова взаимно дополняют друг друга. Для химии “на бумаге” выбор между дисиамилбораном и ББН по вкусу.
Вторая важная схема – введение дейтерия в стереохимически точные положения через гидроборирование и разложение борана уксусной кислотой, при том, что дейтерий может быть в любой из этих молекул, или в обеих сразу. Для тренировки решите задачи в теме Алкины. Там же более подробное обсуждение гидроборирования алкинов.
Ещё раз повторим – эта задача решается гидроборированием и более никак. Для гидроборирования обязательно используем стерически затруднённые бораны: дисиамилборан или BBN.
- Если у вас одна концевая тройная связь, то выбор не важен – используйте любой, например:
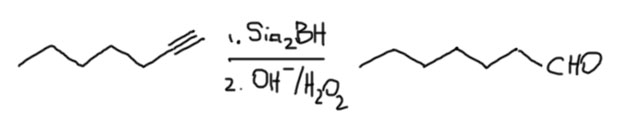
- Если кроме концевой тройной есть внутренняя двойная, выбор за дисиамилбораном, например:
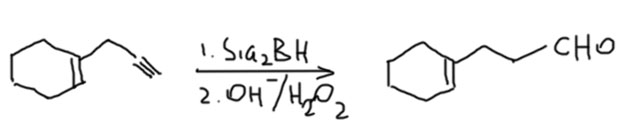
- Если есть и концевая и внутренняя тройные связи – BBN, например:
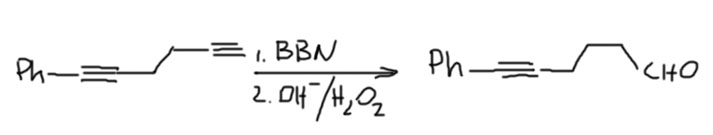
- Если и тройная и двойная концевые, только дисиамилборан, и в этом случае нужно очень точно дозировать боран – любой избыток начнет реагировать с двойной связью.
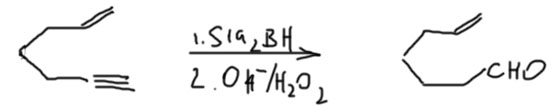
- Ну и наконец, хотя это не вполне вписывается в этот раздел, но для полноты картины, но в том же случае, если вам захотелось превратить в альдегид именно двойную связь, то можно использовать BBN, только спирт придётся доокислить, например:
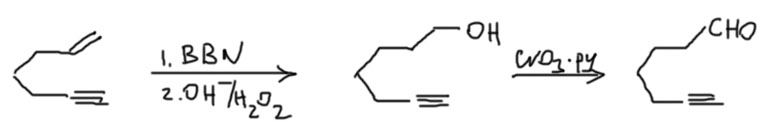
Старайтесь решать задачи с помощью реакции гидроборирования, не стесняйтесь этой реакции. Это великая реакция, она очень хорошо изучена, её реально применяли во многих тысячах (скорее даже десятках тысяч) синтезов. Это очень важное преимущество – иметь реакцию, про которую много известно. Если придётся применятьее на практике, с хорошей долей вероятности найдёте в литературе готовый пример применения к чему-то очень похожему.
Неудачная замена. Замену такой мощной реакции найти непросто. В учебнике Курца, описана другая реакция, и некоторые любопытные там ее находят и пытаются применить. Суть ее в том, что к алкину свободнорадикально присоединяют тиол, а образующийся виниловый тиоэфир гидролизуют в присутствии солей ртути, очень похоже на то, что мы скоро будем делать с производными 1,3-дитиана. Как-то так, только я немного подправлю условия этой реакции на более реалистичные, использовав стандартный инициатор радикальных реакций AIBN, вместо показанного в учебнике УФ-света, практически несовмстимого с большинством органических соединений – получите совсем другую фотохимию. Синим цветом показано, как тиол присоединяется к тройной связи.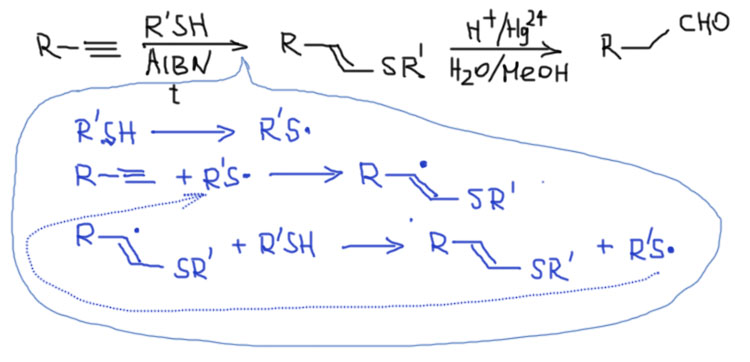
Здесь всё вроде бы разумно, кроме чисто практических причин – чудовищная вонь сернистых соединений плюс огромные количества соли ртути. Проблема в том, что у этой реакции почти нет никакой истории. Она восходит к одной старой статье довольно крупного немецкого химика Берингера, но в дальнейшем следы ее реального применения можно найти в 2-3 не очень внятных работах, и всё. Никто этой реакцией не заинтересовался, никто ее не применял и не исследовал. Насколько можно понять из условий, она не была бы селективна, имела бы массу проблем с побочными реакциями, ну а о ее практическом применении в лаборатории всё уже сказано. С вонью сернистых соединений все уже свыклись – с хорошими тягами и аккуратной работой это не проблема, сернистые соединения очень популярны в современном синтезе. А вот ртуть в стехимометрических количествах – это почти приговор, использовать это будут, только если реакция не имеет конкурентов. Эта, как мы знаем, имеет. Не надо её использовать.
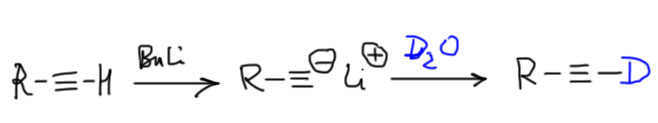
Больше мы пока с анионом ничего не сделаем. Присоединение к альдегидам и кетонам отложим до них самих. Можно еще получить галогенпроизводные с хлором, бромом или иодом на конце молекулы – алкинил галиды, это очень необычные и интересные вещества, но нам они не понадобятся.
Кроме солей с щелочными металлами можно получить производные других металлов, и это уже не соли, а настоящие координационные и металлоорганические соединения. Чаще всего мелькают производные меди, которые очень легко получаются. Химия у них очень богатая, но это не столько органическая химия, сколько металлоорганическая. С одной их реакцией мы знакомы – это именно они окисляются кислородом в диацетилены (см. вверху). Все остальное оставим в покое.
Есть. Но только на бумаге. На страничке про алкины разберем эту проблему подробнее, чтобы разобраться, почему такая простая аналогия реально не работает. Но на бумаге применять эту реакцию можно, и примеры можно найти в задачах. На бумаге это делается точно так же, как аллильное бромирование. В реальности это вообще не делается, но мы договоримся не обращать на это внимание. Аналог аллила с тройной связью называется пропаргилом. Пропаргилбромиды применяются так же как и аллильные, например в синтезе с ацетиленид-анионами, можем получить несопряженные диины. В реальности и с этой реакцией есть сложности, но мы не будем обращать на них внимания.
Диены
- Выбираем...
- Диены из алкенов
- Диены из алкенов через аллильное бромирование
- Диены из алкинов
- Реакции присоединения к диенам - обновлено 13.11.2021
Диенов у нас мало, потому что химия у них сложнее и запутаннее. Особое внимание на реакцию Дильса-Альдера.
Это мы уже рассмотрели в самом верху – через димеризацию алкина в диин, с последующим гидрированием. Здесь есть один серьёзный затык. Одиночную тройную связь мы умеем селективно гидрировать и цис и транс. Но в диине так свободно орудовать не получится. Особенно сложно гидрировать в транс-транс. Мы даже не будем пытаться. Натрий не даст такого результата, потому что механизм этого восстановления, работающий через перенос электрона, для диина даст совсем другой результат.
А вот в цис-цис восстановить можно, и это довольно часто используют в реальных синтезах. Мы можем написать гидрирование над катализатором Линдлара, но в реальности обычно выполняют гидроборирование стерически затруднёнными боранами, у нас это дисиамилборан.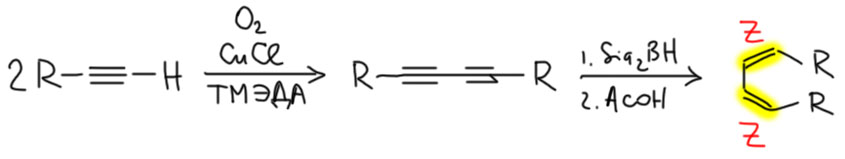
Только реакция Дильса-Альдера. Все остальное – только для дискуссий о механизмах. Старайтесь избегать всяких присоединений, потому что там так много фокусов, что на 3 курсе разобрать это невозможно. Впрочем, может такое быть, что в какой-то задаче используется присоединение к диену. Тогда действуйте по обстоятельствам и по условию задачи попытайтесь понять, что от вас требуется.
Во всех случаях избегайте 1,2-присоединения брома или бромистого водорода. Эта проблема подробно разобрана на основной старничке про диены. В учебниках к этой проблеме часто подходят совершенно халтурно, навязывая идею, что если присоединение вести при охлаждении получается 1,2-аддукт, а если при повышенной температуре, то 1,4-адукт. Это настолько далеко от реальности, что не годится даже как упрощённая картина. В реальности вы никогда не получите 1,2-аддукт просто потому, что все такие соединения перегруппировываются в равновесную смесь 1,4- и 1,2-аддукта с весьма значительной скоростью уже при комнатной температуре. Кинетический контроль, это замечательно, но вы не можете заморозить термодинамику. Вернее, можете, только для того чтобы снять спектр, или аналитическую хроматограмму. Но реакции ставят обычно дял того, чтобы получить продукт. Поэтому даже если реакцию проводить очень быстро при отрицательной температуре, чтобы максимально ближе подойти к кинетическому контролю, то как вы собираетесь такой продукт выделять из реакционной смеси. Все эти манипуляции, которые мы делаем в практикуме, и которые являются стандартом обработки реакционных смесей в органической химии – это называется по-английски aqueous work-up, хорошего короткого перевода на русский, увы нет – выливание реакции в воду, иногда на лёд, иногда с добавлением нейтральзующих реагентов типа бикарбоната, потом экстракция оргеническим растворителем, промывание органической фазы, сушка, упаривание растворителя – делаются при комнатной температуре. Можете попробовать поработать при нуле градусов, поищите где-нибудь на кафедрах, связанных с биохимией, у них бывают холодные комнаты, убедитесь, что это удовольствие не для органики. А пока вы копаетесь со всем этим, пройдет пара часов, и равновесие установится, результатом будет смесь с преобладанием 1,4-продукта. А если учесть, что продукт надо еще как-то очистить – перегнать, перекристаллизовать, хроматографировать – то задача станет совсем невыполнимой.
А вот 1,4-присоединение брома или бромоводорода уже вполне можно сделать. Во многих случаях это вполне практическая реакция, которую и правда можно сделать. И чем более замещён диен алкилами или фенилами, тем лучше, тем чище идёт именно 1,4-присоединение. Для этого не нужна никакая повышенная температура, в большинстве случаев реакция отлично идёт при комнатной, а часто даже при охлаждении. Например, 2,3-диметилбутадиен чисто присоединяет и бром и хлор, и HBr уже при 0ºС. В некоторых методиках бром прибавляют даже при -78ºС, но после прибавления брома реакционную смесь нагревают до 0ºС и выдерживают при этой температуре пару часов для завершения установления равновесия. И никогда не нагревают реакционную смесь выше комнатной температуре потому что продукты присоединения – бромпроизводные аллильного типа, очень быстро осмоляются при повышенной температуре, да это и не нужно, потому что присоединение идет с большой скоростью уже при охлаждении. 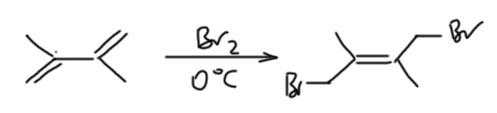
Ещё один вопрос – какой диастереомер получается, E или Z. Поскольку реакция определяется равновесием, то продукт это и есть равновесная смесь, и это касается не только преобладания 1,4-аддукта над 1,2-аддуктом, но и диастереоселективности. Может образоваться и E-изомер и Z-изомер. И нам нужно оценить, кто из них стабильнее (обладает меньшей ΔGº образования). Чаще всего по разным причинам этот вопрос возникает при присоединении к 2,3-дизамещенным диенам. И результат, конечно, будет зависеть от конкретных заместителей R, но в большинстве случаев E-изомер стабильнее, потому что обычно отталкивание заместитетелей R немного больше, чем отталкивание бромметилной группы от заместителя (бром – довольно большая плюха, но она довольно далеко, ее можно отогнуть в сторону, а если заместитель R – это вторичный алкил или третичный алкил или фенил, то их стерический объём больше). Поэтому E-изомер – основной продукт реакции.
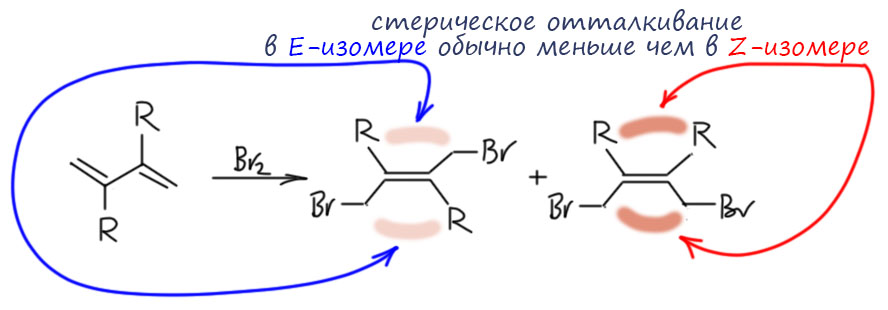
Но разница обычно невелика. Например, если R – фенил, то разница всего около 1 ккал/моль (оценка из моего собственного расчета, просто для информации методом DFT, B3LYP/6-311G(d,p), не важно, что это значит, важно, что это хорошо согласуется с экспериментальным соотношением продуктов около 90:10, Dodson, R.M., et al J. Org.Chem. 1972, 37, 2367-2372). Можно спросить – ну и что, какое нам дело до изомера, содержание которого меньше 10%. А равновесие? Равновесие, как мы уже не раз говорили – страшная сила. Ле Шателье не даст соврать. В равновесии находится не только 1,2- и 1, 4-аддукт, но и диастереомеры. Механизм равновесия тот же самый – ионизация аллильного бромида. В делокализованном аллильном катионе вращение облегчено, и обратимая диссоциация и возврат бромида дают равновесие между двумя диастереомерами. Это равновесие то же самое, что и равновесие между 1,2 и 1,4-аддуктом, потому что основная стадия – ионизация и образвоание катиона – одна и та же.

Представьте теперь себе реакцию, которая идет только с Z-изомером, например, нуклеофильное замещение, приводящее к циклизации. Если бы равновесия не было или оно было бы медленным, то прореагировал бы этот изомер, и реакция бы остановилась на мизерном выходе. Но поскольку есть быстрое равновесие, по мере выбывания одного изомера, равновесие быстро заполняет дефицит. И так реакция идет с большим выходом, причем совершенно неважно, что взять в начале – чистый Z-изомер, чистый E-изомер, или их смесь.
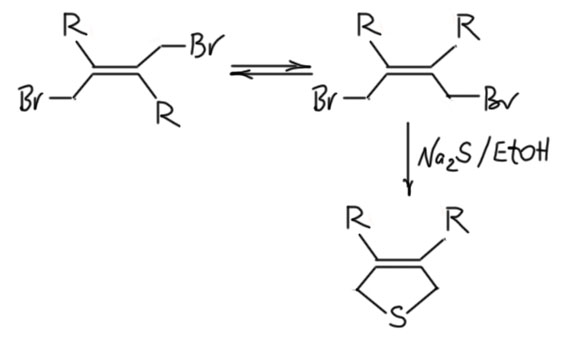
Эта химия дает еще один пример того, как важны в органической химии равновесия, и как важно их уметь оценивать и учитывать.
Что еще можно сделать с диенами в смысле присоединения?
Гидроборирование? Если диен несопряжённый, гидроборируйте на здоровье, в реакцию будет вступать
менее замещённая связь. Для надёжности используйте большой боран, дисиамилборан или BBN, например: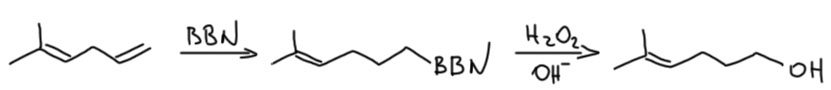
Если обе двойные связи доступны для гидроборирования, иногда может происходить двойное гидроборирование, причём во второй стадии может участвовать боран, образовавшийся на первой стадии. Самый яркий пример такой реакции мы знаем – это образование BBN из циклооктадиена. Другие примеры такой реакции придётся искать очень долго, но желающие найдут.
А вот сопряжённые диены гидроборировать не рекомендуется. Даже так – не рекомендует. Кто не рекомендует? Кто посмел?? Сам Герберт Браун, не много не мало. На это должна бы последовать слегка непристойная реплика из старинного советского анекдота про Петьку и Василия Иваныча. В общем, если сам Браун не рекомендует, то выпендриваться смысла нет. А в чём проблема? В том, что реакция приводит к образованию совершенно неудобоваримых смесей разных продуктов. Если использовать просто боран, то получается совсем нехорошая вещь – нерастворимые полимеры, в которых боран беспорядочно цепляется за двойные связи молекул диенов, сшивая всё это в хаотический трёхмерный полимер. В этом пытались разобраться ещё совсем недавно, но большого успеха не достигли кроме опубликования статьи в новом модном журнале (O. A. Scherman. Chem. Sci. 2015, 6, 6262). Здесь оставим эту проблему, а на основной страничке может однажды разберём её подробнее.
Стереоселективные реакции
- Выбираем...
- Присоединение к алкенам
- Присоединение к алкинам
- Циклоприсоединение
- Стереоселективное гидрирование алкинов в алкены
- Under construction
Очень важный раздел. Разберитесь в стереохимических схемах (шаблонах) и доведите их использование до автоматизма.
- присоединение галогена, в том числе с чужим нуклеофилом (водой и т.п.) – анти-присоединение
- гидроборирование с последующей реакцией борана – син-присоединение
- дигидроксилирование тетраоксидом осмия или перманганатом калия – син-присоединение
- дигидроксилирование через раскрытие эпоксида – анти-присоединение
Все остальное, в том числе присоединение протона (гидратация, присоединение галогенводородов), сольвомеркурирование с разложением боргидридом натрия, гидрирование водородом над всякими катализаторами нам не годятся, так как предсказать стереоселективность в конкретных реакциях невозможно.
Результат стереоселективной реакции легко предсказать и записать хоть проекцией Фишера, хоть естественной проекцией. Во-первых, нам нужен олефин как минимум виц-дизамещенный. Возьмем анти-присоединение и любой годный олефин. Отметим заместители в олефине, находящиеся в транс-расположении друг относительно друга. Рисуем Фишера, в котором эти заместители находятся на концах вертикали или естественную проекцию, в которой они на концах основного зигзага. Тогда в Фишере вошедшие заместители будут по одну сторону от вертикали, а в естественной проекции, наоборот, по разные. Это – эритро-конфигурация.
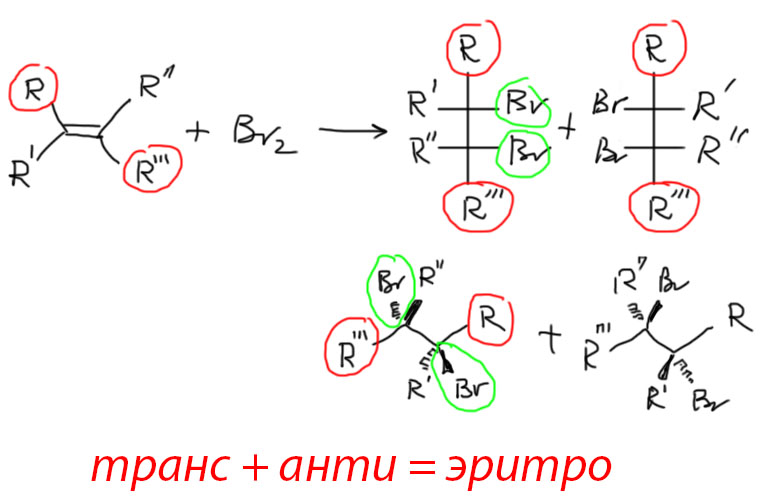
Обратите внимание, что продукты получаются всегда в виде рацемической смеси двух энантиомеров. Для корректности изображают оба. В дальнейшем, когда идея о том, что в реакциях оптически неактивных исходных не может получиться один энантиомер, а получается рацемат, войдет в привычку, можно будет не рисовать второй энантиомер, так как это очевидно.
Соответственно, если в другом примере что-то одно изменится (заместители цис, а не транс, или син-присоединение, а не анти, то получится продукт с входящими группами по разные стороны в Фишере (или по одну сторону в естественной проекции) – это трео-изомер. Если изменились оба исходных обстоятельства (цис-заместители и син-присоединение, то результат будет опять эритро. Третьего не дано.
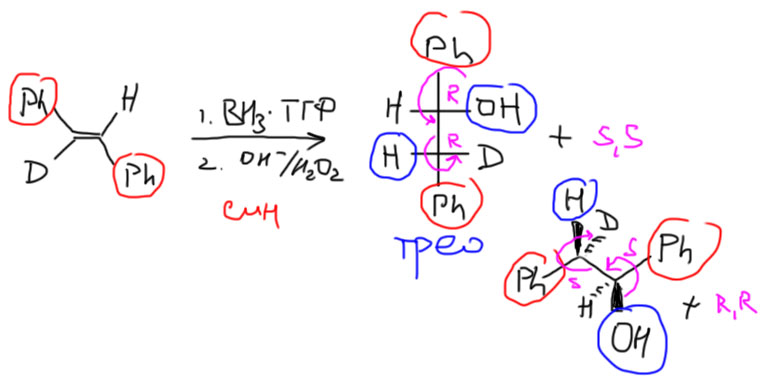
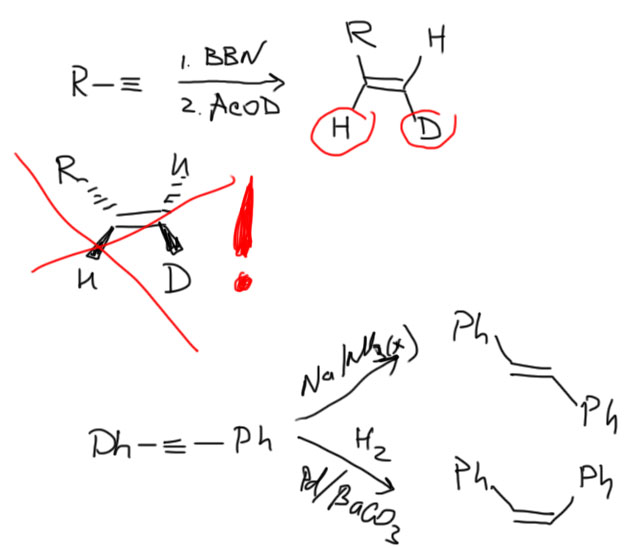
Из-за недостатка заместителей в реакциях алкинов в стереоселективных реакциях часто используют дейтерий в разных положениях. Освежите этот прием в памяти, решив задачу на странице Алкины.
В задачах часто соединяют присоединение к тройной связи с последующим присоединением уже к стереохимически определенному алкену. Потренируемся в задачах.
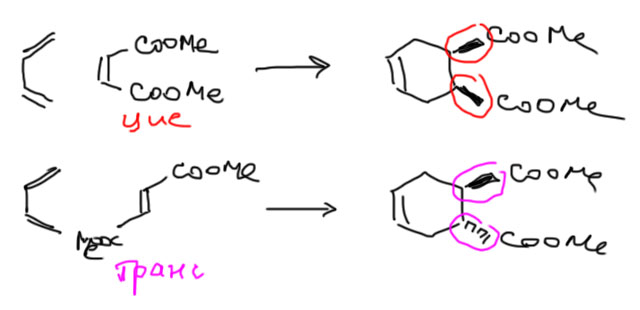
Про эндо и экзо-продукты можно вспомнить на странице Диены.
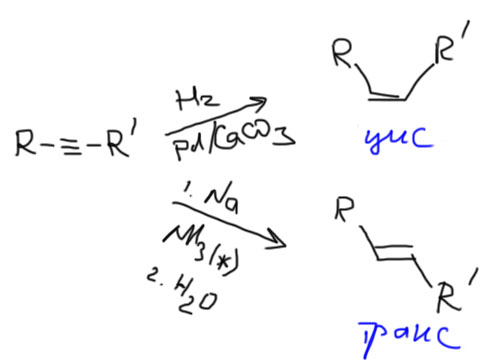
Важнейшая реакция, точнее, несколько реакций, позволяющих получать чистые диастереомеры (цис или транс) дизамещенных алкенов из дизамещенных алкинов. В нашем курсе никаких других способов достичь такого результата нет и не будет. А поскольку чистые цис- или транс-изомеры алкенов очень часто используются для стереоселективного присоединения, что является одним из самых распространенных приемов в задачах на стереохимию, реакции гидрирования алкинов особенно важны.
Для получения чистых транс-изомеров используем восстановление металлическим натрием или литием в жидком аммиаке в присутствии слабой кислоты Бренстеда, этанола или трет-бутанола. Как работает эта удивительная реакция – фактически восстановлением свободными электронами, посмотрите в теме Ароматические соединения, восстановление по Бёрчу. ![]()
Разные вспомогательные методы
Особое внимание на озонолиз – он часто используется в задачах
Сильные основания вызывают превращение алкинов, имеющих рядом с тройной связью хотя бы одну, а возможно и сколько угодно связанных друг с другом в непрерывную цепь CH2-групп, в равновесную изомерную смесь всех возможных алкинов и алленов. Теорию этой перегруппировки, которую называют ацетилен-алленовой или алкин-алленовой, а могли бы назвать перегруппировкой Фаворского, если бы это название не было уже заиграно другой реакцией, разберем на отдельной странице. Это интересно с теоретической точки зрения, но практически не имеет смысла – зачем портить вещество. Есть возможность практически превращать концевой алкин в практически чистый 2-алкин, и это тоже там разобрано, но мы не будем это использовать в задачах.
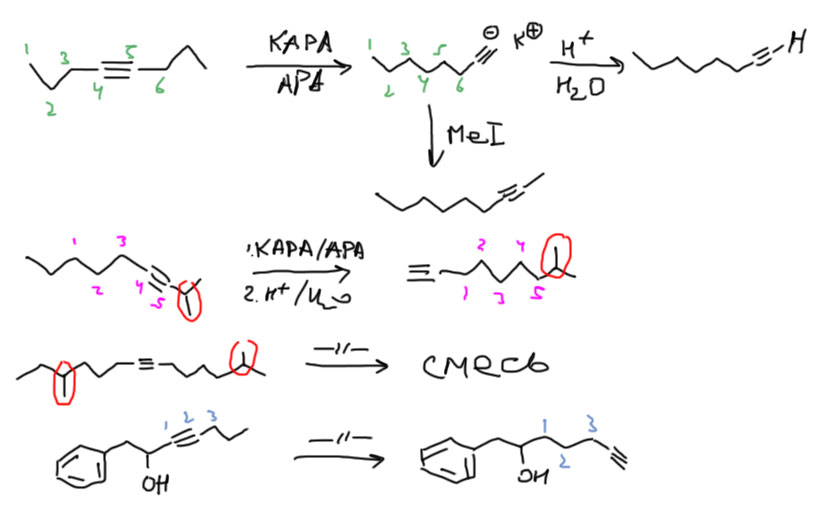
Но, если подобрать такой растворитель, в котором самая стабильная в этой системе соль терминального алкина выпадает в осадок, по закону Ле Шателье, равновесие полностью смещается в сторону такой соли (это распространённое объяснение на самом деле не совсем верно – подробнее настоящая причина смещения связи разобрана на той же странице). Если подобрать систему, в которой депротонирование и протонирование происходят с близкими скоростями, а основность достаточно высока, то появляется возможность наоборот смещать тройную связь из глубины цепи в конец. Такой системой является раствор калийной соли 1,3-диаминопропана в самом этом диамине (1,3-пропандиамин, которые часто называют для красоты сокращения немного неправильно 3-аминопропиламином, сокращенно АПА (APA), но сокращение ДАП (DAP) от 1,3-диаминопропана, что тоже не совсем точно соответствует номенклатуре ИЮПАК, не менее популярно). Калийную соль этого амина делают прямо перед реакцией, добавляя гидрид калия в АПА (это чрезвычайно опасный реагент, самовоспламеняющийся и даже взрывающийся от контактов с влажным воздухом и, тем более, водой!). Калийная соль АПА, игриво сокращаемая в КАПА , оказалась удивительно полезным реагентом для смещения тройной связи “в торец” – реакция проходит при 0ºС за несколько секунд даже если тройной связи нужно преодолеть с десяток связей по дороге, а продукт либо гидролизуют в 1-алкин, либо используют сразу как ацетиленидный карбанион в синтезе. У реагента есть еще натриевый (НАПА) и литиевый аналог (ЛАПА), но они намного менее эффективны, особенно ЛАПА, впрочем, и тот и другой легко активировать с помощью добавления трет-бутилата калия, и такая смесь работает почти так же как КАПА, хотя и медленнее, реакция требует несколько часов перемешивания при комнатной температуре. Условие – двойная связь бежит только по неразветвленной и незамещенной углеродной цепи, любое препятствие ее останавливает, но если с одной стороны путь открыт, то реакции не мешают некоторые простые заместители типа гидрокси и алкокси, но не галогены или любые более сложные группы (карбонилы, карбоксилы, нитро, и т.п.). Другие кратные связи тоже нежелательны.
За бешеную легкость и полезность этого метода не только реагент, но и саму реакцию обозвали метафорически зиппер-реакцией (зиппер – это застежка типа молния). На схемах нужно указывать и реагент и растворитель, то есть KAPA/APA. Частая ошибка – потерять углероды в продукте. Чтобы не терять, не ленитесь пронумеровать ту часть цепи, по которой будет путешествовать тройная связь.
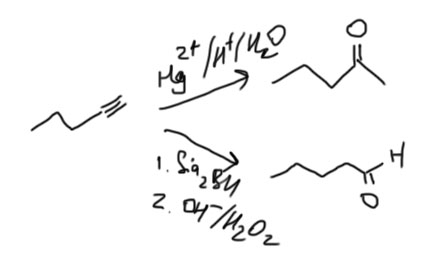
Озон очень легко разрывает двойные (и тройные тоже, но к алкинам реакцию редко применяют – смысла нет) связи, сначала образуется озонид, который не выделяют, а разлагают либо восстановителями (исторически это цинк, в современных методиках предпочитают более аккуратный диметилсульфид или трифенилфосфин)), или окислителями (перекисью водорода). Результат почти одинаковый, но из монозамещенного атома в восстановительном варианте получается альдегид, в окислительном – кислота.
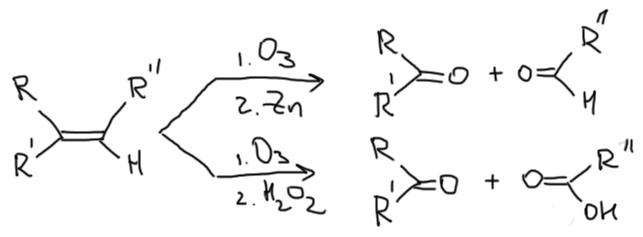
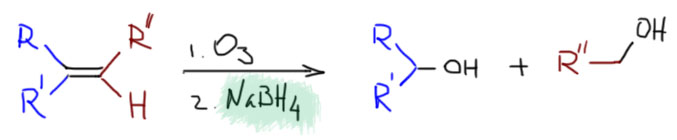
Есть ещё вариант восстановительного озонолиза, в котором применяют более сильный восстановитель – боргидрид натрия. В этом случае получаются первичные и вторичные спирты.
Особенно часто озонолиз используют для определения структуры олефина по двум продуктам, если двойная связь в открытой цепи, или по одному – если она была в цикле. Если связей было несколько, проблем будет больше, но и встречаться с такими случаями мы вряд ли будем. Бензольное кольцо озонолиз не берет (точнее, берет, но в таких жестких условиях, которые просто не используют для озонолиза двойной связи, которое обычно делают при -78ºС в хлороформе – и это очень мягкая и селективная реакция, не трогающая больше почти ничего).
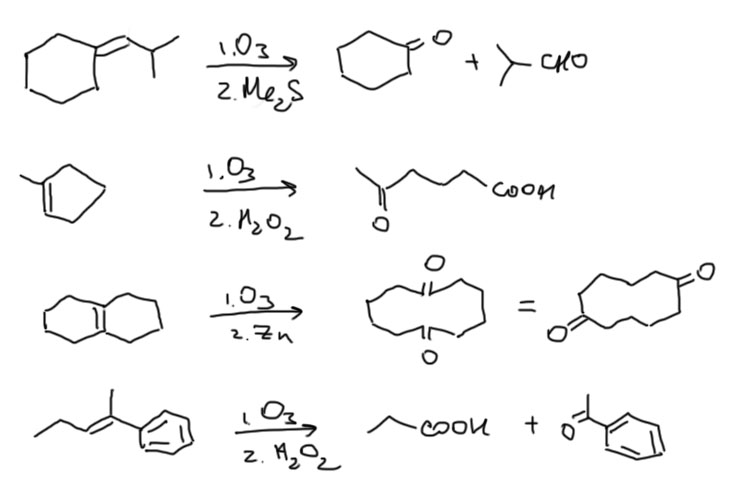
Алкилирование карбаниона, зиппер, реакции тройной связи
- Из пропина, 1-бромпентана получите октаналь и октанон-2. Нарисуйте схему ретро-синтеза и основанную на ней схему синтеза с указанием реагентов и условий реакций.
Димеризация ацетиленов
- Из фенилацетилена получите (Z,Z)-1,4-дифенилбутадиен-1,3. Будет ли этот диен легко реагировать с диенофилами по реакции Дильса-Альдера?
- Из 3,3-диметилбут-1-ена получите 2,2,7,7-тетраметилоктан
Стереоселективный синтез алкенов, стереохимия присоединения
- Из фенилацетилена, бромметана получите эритро-изомер 1,2,3-трибром-1-фенилпропана. Нарисуйте стереохимические проекции продукта (Фишера и естественную), разметьте стереоцентры по R/S-правилу.
Цепь с несколькими двойными связями с определённой стереохимической конфигурацией
1. Из изопропилацетилена и хлористого аллила получите (Е)-7-бром-2-метилгепт-3-ен
2. Из продукта предыдущей задачи и ацетилена получите (3E,8Z,10Z,15E)-2,17-диметилоктадека-3,8,10,15-тетраен.
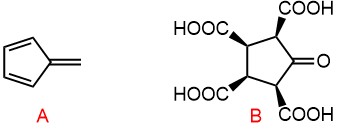
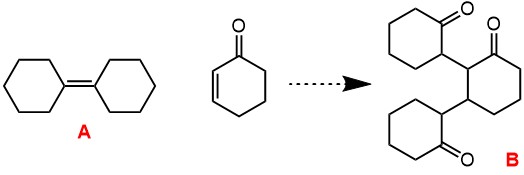
Реакция Дильса-Альдера, озонолиз
- Из 2,3-диметилбут-2-ена и бутендиовой кислоты получите трео-изомер 4,5-дикарбоксиоктан-2,7-диона (название не соответствует номенклатуре, но вполне годится, чтобы нарисовать структуру). Разрисуйте схему ретро-синтеза, раскройте ее в схеме синтеза. Нарисуйте стереохимическую проекцию продукта и дайте ему правильное название по ИЮПАК. В синтезе использована бутендиовая кислота, но существует два диастереомера этого соединения – малеиновая кислота (цис) и фумаровая кислота (транс). Какой из них использован в синтезе?
Алкилирование ацетиленид-аниона, аллильное бромирование
- Из пропена получите (E)-1,4-гексадиен
Дильс-Альдер и др.
Гидроборирование и озонолиз
Дильс-Альдер и озонолиз