Карбоновые кислоты и их производные
Последнее обновление: 21.01.2020
В принципе это не самостоятельная тема, а просто продолжение карбонильных соединений. Карбоновые кислоты – типичные карбонильные соединения, и их свойства и реакции практически полностью определяются именно этим. Если мы разобрались в свойствах и реакциях альдегидов и кетонов, нам нужно будет просто понять, как все это применить к карбоновым кислотам и их производным. Берем каждую реакцию оттуда и транслируем на эти соединения. Проще не придумаешь.
Увы, химия, как всегда полна гадких сюрпризов. Процесс переноса знаний оттуда сюда не будет очень гладким, хотя бы потому, что под скромным названием “производные” не скрывается, как мы привыкли раньше, туповатый процесс замены “метила на этил”, и “хлора на бром”, ну в самом крайнем случае, “водорода на алкил или галоген”. Нет, здесь каждое производное – это фактически новый класс органических соединений, вполне самостоятельный. Именно поэтому в номенклатуре характеристические группы производных карбоновых кислот считаются главными группами, имеют очень высокое старшинство, определяются в конце названия. Поэтому для каждого из типов производных придется разобраться в специфике свойств, вносимых именно этой характеристической группой. Впрочем, не все так ужасно, общего у них не так мало, и даже сами различия вполне неплохо описываются как общие и характерные особенности.
Подробная программа раздела
Методы синтеза: окисление первичных спиртов и альдегидов, алкенов, алкилбензолов; гидролиз нитрилов и других производных карбоновых кислот; синтез на основе металлоорганических соединений; синтезы на основе малонового эфира. Получение муравьиной кислоты и уксусной кислот.
Строение карбоксильной группы. Физико-химические свойства кислот: ассоциация, диссоциация, влияние заместителей на кислотность. Галогенирование кислот по Гелю-Фольгарду-Зелинскому. Пиролитическая кетонизация, электролиз по Кольбе, декарбоксилирование по Хунсдиккеру.
Галогенангидриды. Получение с помощью галогенидов фосфора, тионилхлорида, бензоилхлорида. Свойства: взаимодействие с нуклеофильными реагентами (вода, спирты, аммиак, амины, гидразин, металлоорганические соединения). Восстановление до альдегидов – по Розенмунду и комплексными гидридами металлов. Взаимодействие диазометана с галогенангидридами карбоновых кислот (реакция Арндта-Эйстерта).
Ангидриды. Методы получения: дегидратация кислот с помощью Р2 О5 и фталевого ангидрида; ацилирование солей карбоновых кислот хлорангидридами. Реакции ангидридов кислот.
Кетен. Получение и свойства.
Сложные эфиры. Методы получения: этерификация карбоновых кислот (механизм), ацилирование спиртов и их алкоголятов ацилгалогенидами и ангидридами, алкилирование карбоксилат-ионов, реакции кислот с диазометаном. Методы синтеза циклических сложных эфиров – лактонов.
Реакции сложных эфиров: гидролиз (механизм кислотного и основного катализа), аммонолиз, переэтерификация; взаимодействие с магний- и литийорганическими соединениями, восстановление до спиртов и альдегидов комплексными гидридами металлов.
Сложноэфирная и ацилоиновая конденсации.
Ацетоуксусный эфир и его использование в синтезе. Кето-енольная таутомерия эфиров b -кетокислот, амбидентный характер енолят-иона.
Амиды. Методы получения: ацилирование аммиака и аминов, пиролиз карбоксилатов аммония. Синтез циклических амидов – лактамов. Свойства: гидролиз, восстановление до аминов, дегидратация амидов. Понятие о секстетных перегруппировках. Перегруппировки Гофмана, Курциуса.
Нитрилы. Методы получения: дегидратация амидов кислот (с помощью Р2 О5, SОCl2, РОCl3), алкилирование амбидентного цианид-иона. Свойства: гидролиз, аммонолиз, восстановление комплексными гидридами металлов до аминов, взаимодействие с магний- и литийорганическими соединениями.
Двухосновные кислоты. Методы синтеза: окислительное расщепление циклоолефинов и циклических кетонов, окисление полиалкилбензолов. Главные представители: щавелевая кислота, диэтилоксалат в сложноэфирной конденсации. Малоновая кислота: синтезы с малоновым эфиром, реакция Михаэля, конденсации с альдегидами (Кневенагель). Янтарная кислота, ее ангидрид, имид, N-бромсукцинимид. Адипиновая кислота. Конденсация Дикмана. Ацилоиновая конденсация эфиров дикарбоновых кислот как метод синтеза средних макроциклов.
Фталевая и терефталевая кислоты, промышленные методы получения. Фталевый ангидрид, фталимид и его использование в синтезе.
СПОСОБЫ СИНТЕЗА КАРБОН. КИСЛОТ.
1) ОКИСЛЕНИЕROH, RCHO, R-CH=CH2, R-CºCH.
2) ОКИСЛЕНИЕ алкилароматич. соед. (α-С)
3) ГИДРОЛИЗ R-CºN (Н+ или ОН-, очень жесткие условия)
4) RMgX или RLi + CO2
5) СИНТЕЗЫ С МАЛОНОВЫМ ЭФИРОМ.
6) ПРОМЫШЛ. спос. синтеза. (НСООН, АсОН)
7) Галоформная реакция
8) Р-ция Канниццаро
9) Р-ция Байера-Виллигера для нециклических кетонов (через сложные эфиры)
СПОСОБЫ СИНТЕЗА КИСЛОТ, КОТОРЫЕ БУДУТ ИЗВЕСТНЫ ИЗ ПОСЛЕДУЮЩИХ ЛЕКЦИЙ.
10) Гидролиз производных (хлорангидридов, ангидридов, эфиров, амидов).
11) Метод Арндта-Эйстерта.
12). СИНТЕЗЫ с АЦЕТОУКСУСНЫМ ЭФИРОМ (АУЭ).
ВАЖНЕЙШИЕ СВОЙСТВА КАРБОНОВЫХ КИСЛОТ.
1) кислотность 2) замещение ОН, синтез производных 3) восстановление
4) декарбоксилирование: а) термическое, б) по Кольбе, в) реакция Бородина-Хунсдиккера, г) пиролитическая кетонизация (тема – ” карбонильные соединения).
5) реакции по α-С-атому (Гелль-Фольгард-Зелинский). Радикальное галогенирование кислот.
Данные по кислотности карбоновых кислот.pK
AcOH (4.8), HCOOH (3.8), ClCH2COOH (2.8), Cl2CHCOOH (1.3), CCl3COOHи CF3COOHприближаются к сильным кислотам (pK 0.7 и 0.5 в воде).
RCH2COOH, где R = MeO (3.6), CN(2.5), PhCH2 (4.3).
CH2=CHCOOH (4.3).
α-, β-, и γ-хлормасляные (соотношение констант кислотности 140:9:3).
Производные карбоновых кислот (КК).
Галогенангидриды КК. Синтез ХА из кислот – как пример реакций замещения ОН группы карбоновых кислот. Механизм нуклеофильного замещения при sp2-углеродном атоме в С=О группе с разрывом связи С(О)-ОН. Создание хорошей УГ или протонирование С=О? Синтез с помощью PCl3, PCl5, SOCl2, хлорангидридов карбоновых кислот (напр. оксалилхлорида).
Взаимодействие с нуклеофильными реагентами и использование галогенангидридов в синтезе других производных КК. Восстановление ГКК, реакции с металлоорганическими соединениями.
Метод удлинения углеродной цепи по Арндту-Эйстерту. Механизм.
Синтез других пр-ных карбоновых кислот, не обязательно из кислот.
1. Ангидриды. Синтез. 3 группы методов синтеза и 8 примеров реакций.
2 Кетены как “внутренние” ангидриды карбоновых кислот. Синтез – 3 способа.
3. Синтез сложных эфиров: 7-8 способов.
СВОЙСТВА ПРОИЗВОДНЫХ КАРБОНОВЫХ КИСЛОТ. АЦИЛИРОВАНИЕ.
Ряд ацилирующих реагентов, по убыванию электрофильности.
Свойства ангидридов, кетенов (в основном ацилирование).
Свойства сложных эфиров. Гидролиз – 2 важнейших механизма (на 3 балла) и два второстепенных (на 5 или 5+).
Синтез амидов – 1) ацилирование NH3 и аминов с RCOX – т.е. с более сильными ацилирующими реагентами, чем амиды, и даже солями! (примеры). 2) Образование циклич. имидов (сукцин- и фталимид, их использование). 3) Частичный гидролиз нитрилов в щелочной среде с 10-15% Н2О2. 4) Что такое лактамы?
Гидролиз амидов (жесткие условия, сильно кислая или щелочная среда, знать механизм!).
Восстановление амидов: LiAlH4 – до аминов. Слабые восстанавливающие. агенты –LiAlH(OEt)3 или ДИБАЛ-Н – до альдегидов. Реакция RMgXc перв. или трет.-амидами. Реакция RMgX с ДМФ. Что получится? Дегидратация амидов с SOCl2 в ДМФ.
Секстетные перегруппировки – Гофмана, Курциуса, (Шмидта – на 5+), механизм, нитрены. Можно ли использ. для амидов ароматических или пространственно затруднённых. кислот? Как выделить изоцианаты? Секстетные перегруппировки ведут к укорочению цепи
Нитрилы. Синтез. 1) Амбидентная природа СN– аниона. Когда получаются изонитрилы? 2) синтез из амидов. Свойства. 1) гидролиз (в какой среде? Легко или нет?) 2) кислотность α-СН. Конденсация Торпа-Циглера (аналогичная Дикману) – будет в теме “Циклоалканы” 3) Алкоголиз нитрилов. Имидаты, амидины, ДБН и ДБУ – тоже амидины. 4) Восстановление нитрилов (когда в амины, а когда в альдегиды?). 5) реакция Риттера (трет- амины.)
Изонитрилы – понятие, синтез из цианидов и дихлоркарбена. Св-ва. Аналогия с СО, р-ции с галогенами, серой, водой (формамиды), аминами (амидины). Реакция с RLi .
Непредельные к-ты. Синтез – Кневенагель, Виттиг, Перкин, Хек, из β-оксикислот. Св-ва, особенности свойств в зависимости от положения С=С: 1) Присоединение НВг, нуклеофилов, 2) миграция С=С 3) образование лактонов (аллилуксусная к-та).
Жирные кислоты. Образование в организме – из ацетил-КоА в малонил-КоА и т.д. Особенности жирных кислотв организме – прямая цепь, четное число атомов С, нет сопряжения С=С, Z-конфигурация С=С. С12- лауриновая, С16-пальмитиновая, С18- стеариновая.
Непредельные жирные кислоты – С18, D9 – олеиновая, D9,12 – линолевая, D9,12,15 – линоленовая (на 5), С20 D5,8, 11,14 – арахидоновая (на 5+). Все цис! Последние три – незаменимые. Из них образуются простагландины.
Двухосновные к-ты. Тривиальные названия и способы синтеза С2, С3, С4, С6 дикарбоновых кислот, поведение при нагревании. Диэтилоксалат в синтезе. Малоновый эфир в синтезе, в реакции Михаэля и Кневенагеля. Янтарная кислота, её ангидрид, имид, N-бромсукцинимид. Адипиновая кислота. Конденсация Дикмана. Ацилоиновая конденсация эфиров дикарбоновых кислот как метод синтеза средних и макроциклов. Фталевая и терефталевая кислоты, получение в промышленности. Фталимид и его использование в синтезе. Фумаровая и малеиновая кислоты, их эфиры и использование в синтезе (все это уже было раньше).
Рассмотрим некоторые важные вещи подробнее.
Что такое карбоновая кислота?
Иногда, то есть почти всегда говорят так – это соединение, содержащее карбоксильную группу, COOH. Строго говоря, это неверно, и более того, проблема не в названии – ещё великий Дэн Сяопин не зря ведь учил, что неважно какого цвета кошка, если она умеет ловить мышей – в этом смысле любые соединения с карбоксильной группой являются кислотами (умеют ловить мышей), следовательно так ли важно, как их называть? Ну, называйте кислотами, но зачем именно карбоновыми? Ужас в том, что проблема эта есть только в русском языке. Если вы спросите себя, как эти кислоты называются по-английски (а также испански, французски, итальянски и т.д.), то получите нечто более определённое – carboxylic acids. Английский язык (и все романские) совершенно точно называет кислоты именно по карбоксильной группе – мы могли бы тоже использовать называние карбоксильные кислоты, но у нас уже есть своё слово “карбоновые”. Нам до лампочки, как это у них там называется, тем более, что в английском это термин взялся не от большого и мудрого размышления о будущем использовании термина, а просто потому, что слово carbonic acid у них давно занято – это просто угольная кислота (в самом деле, как иначе, carbon – уголь, carbonic – угольная, точно так же как sulfur – sulfuric и т.п.). А как это будет в самом главном языке с начала органической химии – немецком? Совсем просто – Karbonsaüren – просто углеродные кислоты, немцы оказались самыми простыми и вообще не стали париться – кислоты, производные элемента углерода.
Получилось так, что в русский язык термин “карбоновые кислоты” попал очень давно, и, скорее всего, просто был заимствован из немецкого, и тогда, конечно никто не мог заметить, что то, что у них просто входит в корень слова, в русском неявно, а потом и явно стало суффиксом -он. А у этого суффикса есть очень точное значение в том, что касается как раз кислот, причём значение совсем не только русское, а вполне международное. И очень точно формулируемое. Если у элемента есть кислородная кислота в высшей степени окисления, и у этой кислоты в структуре больше одного гидроксила (иными словами, кислота не менее чем двухосновная), то замена одного гидроксила на органический остаток (алкил, арил и т.п., но подразумевается связь углерод-элемент) даёт соединения которые и называют -оновыми кислотами. Вот, например, несколько таких пар, но такие кислоты есть практически у всех непереходных элементов, металлов и металлоидов. 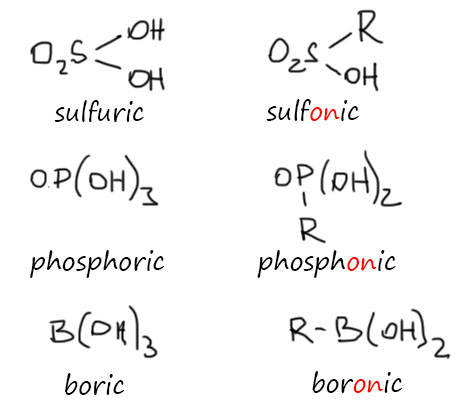
Когда мы переносим эту номенклатуру в русский язык, используем наши названия для родоначальных кислот, но вполне систематические -оновые – для органических производных кислот: серная – сульфоновые, фосфорная – фосфоновые, борная – бороновые и т.п. И с карбоновой кислотой мы точно так сюда же и попадаем: угольная – карбоновые. Получается, что карбоновыми кислотами можно в этом смысле называть только такие, в которых карбоксил связан с углеродным остатком. И ни угольная кислота, ни её родственники хлоругольная кислота, карбаминовая кислота и т.п. – карбоновыми кислотами не являются. Но самое страшное – и муравьиная кислота не должна считаться карбоновой, ведь у неё карбоксил связан с водородом, а водород это не углерод, один родил воду, другой – уголь. А вот это уже настоящий кошмар – поколениями муравьиную кислоту считали родоначальником класса карбоновые кислоты. Может ли родоначальником быть не родственник, или родственник, но очень далёкий? Что делать?
Ничего особенного. Ломать точно ничего не будем. В конце концов, эта странная проблема есть только в русском языке, и возникла она довольно случайно – сначала просто заимствовали термин из немецкого, а много после с ним влипли в какую-то номенклатуру. Я вам таких ситуаций в органике ещё с дюжину найду, не меньше. Будем же следовать заветам великого Дэна и ценить кошек за ловлю мышей, а карбоновые кислоты – за их восхитительные свойства и производные, а не за случайный суффикс. Впрочем, в этой формальной схеме есть толика существенного. Нельзя не заметить, что при некотором сходстве, угольная кислота и её родственники во многом другом ведут себя иначе и не замечать этого глупо. И даже муравьиная кислота имеет совершенно особенные свойства и реакции – строго говоря, она образует отдельный класс органических соединений, состоящий из одного единственного соединения, настолько отлично ее поведение и поведение ее производных от всех остальных карбоновых кислот. Мы много раз с этим столкнёмся, а то, с чем мы не столкнёмся, ещё более обширно и интересно. Так бывает со всеми классификациями, как их ни крои, а что-нибудь да вывалится.
И ещё одно существенное следствие этой бури в пробирке. Всё это можно легко формализовать, если использовать не очень популярную в органике степень окисления. Тогда получится, что в угольной кислоте и ее родственниках углерод имеет степень окисления +4. В настоящих карбоновых +3. А в муравьиной – только +2, столько же, сколько у карбонильного углерода в кетонах. И если мы возьмёмся расположить все органические классы по степени окисления углерода в том месте молекулы, которое этот класс и определяет, все эти кислоты расположатся на разных ступенях – настоящие карбоновые между муравьиной и угольной. А степень окисления – вполне рабочая характеристика, ее мощно используют в классификациях реакций, да и просто для того, чтобы уравнять окислительно-восстановительные реакции.
И это вполне подтверждает то, что под классификациями, как бы формальны они ни были, обычно есть нечто существенное. И ещё то, что система терминов в русском языке иногда бывает точнее и содержательнее, чем системы терминов в других языках. Увы, далеко не всегда.
Почему карбоновые кислоты такие кислые?
То, что карбоновые кислоты кислые, знают все. Уксус, лимонная кислота, другие “фруктовые кислоты” в незрелых фруктах – вырви глаз! Наверное, это сильные кислоты, такая кислятина? pH разбавленных растворов уксусной кислоты между 2 и 3, это приблизительно то же самое, что раствор капли соляной кислоты в стакане воды.
Аррениусовская кислотность уксусной кислоты в воде измеряется как pK 4.76. Судя по этой величине это все же довольно слабая кислота. И это неудивительно. Карбоновые кислоты относятся к кислородным кислотам, и такие есть у каждого неметалла, даже если ограничится высшими степенями окисления. Строго говоря в этом смысле мы должны начинать рассуждение не с уксусной, а с угольной кислоты. Угольная кислота неустойчива и не дает растворов с концентрацией выше 0.0001 М, если не использовать повышенное давление диоксида углерода, поэтому мы еле-еле чувствуем кислый вкус даже у воды, насыщенной диоксидом углерода. Если бы удалось создать большую концентрацию, то почувствовали бы и не хуже, чем с уксусом.
Угольная кислота – типичная кислородная кислота. В своем периоде у нее есть только один аналог – азотная кислота, считающаяся сильной кислотой. Борная кислота не считается, она не является протонной, это кислота Льюиса. Если опуститься на следующий период, то там будет ряд пошире – хлорная кислота (очень очень сильная), серная кислота (сильная), орто-фосфорная кислота (слабая, но в ряду слабых ближе к сильному концу с pK по первой ступени 2.2), кремневая кислота (точно очень слабая, хотя полного аналога угольной у кремния нет, и реальная химия кремнекислот весьма непроста). Тенденции в рядах очевидны – все определяет электроотрицательность элемента.
Поскольку мы занимаемся органической химией, то вместо обычных кислородных кислот возьмем их органические аналоги – одну из гидроксильных групп обычной кислородной кислоты заменим на органический остаток, и если гидроксилов в исходной молекуле больше одного, то в результате мы будем иметь тоже протонные кислоты. Такие кислоты всегда называются -оновыми: сульфоновыми RSO2OH, фосфоновыми RP(O)(OH)2, карбоновыми RCOOH, и так далее -селеноновыми, арсоновыми… Общие тенденции в этих рядах тоже просты – поскольку органический остаток это либо слабый индуктивный донор, либо слабый индуктивный акцептор (ключевое слово – слабый), то кислотность -оновых кислот близка к таковой у кислоты-родоначальника. Все сульфоновые кислоты, например, сильные, как серная. Именно поэтому мы используем, например, толуолсульфоновую кислоту в качестве кислотного катализатора, она почти такая же сильная, как серная, но при этом удобна в работе, она кристаллическая, и не является окислителем. Далее, фосфоновые кислоты недалеко ушли от фосфорной. А карбоновые – от угольной, а это уровень pK 4-5 (есть несколько оценок кислотности угольной кислоты, различающиеся тем, что используется либо равновесие с растворенным диоксидом углерода, либо вводится поправка на реальную концентрацию H2CO3, но нам эти детали не очень важны).
Довольно часто задают другой вопрос – почему кислотность карбоновых кислот существенно больше, чем кислотность спиртов. Почему спиртов? По очевидному сопоставлению OH-кислот, например, поставив рядом этанол и уксусную кислоту:
CH3CH2OH CH3COOH
Сходство молекул очевидно. Как очевидно и различие, и это различие очень существенное, из разряда по настоящему драматических. На место слабого индуктивного донора поставлена карбонильная группа, обладающая сильным акцепторным эффектом, причем сразу обоих типов – и индуктивным, и мезомерным. Работают оба и оба в одну сторону, увеличивая кислотность. Как мы уже договорились, при анализе кислотности и основности стараемся смотреть прежде всего на влияние на стабильность заряженных частиц, в данном случае анионов в равновесии. Поскольку пока что мы имели в виду именно кислотность по Аррениусу (диссоциация кислоты в разбавленном водном растворе, и напомню, что в воде мы пишем протон, подразумевая гидратированные формы протона типа гидроксоний-катиона), так и запишем:
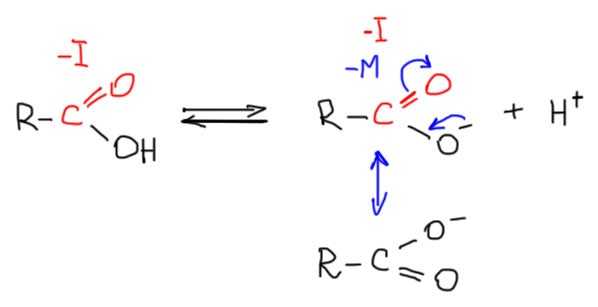
Видим, что отрицательный заряд на кислороде делокализован как за счет акцепторного индуктивного эффекта, то есть просто за счет поляризации связей и смещения электронной плотности. Так и за счет мезомерной стабилизации – карбоксилат-анион представляет собой пример идеальной делокализации с двумя совершенно одинаковыми граничными структурами.
А какой фактор важнее – мезомерия или индуктивный эффект? В принципе, это довольно сомнительный вопрос: разделить действие двух факторов, действующих в одну сторону, очень трудно, во многих случаях получается совершенно бессмысленное препирательство. Но иногда это все же полезно. Общий подход состоит в том, что мезомерный эффект обычно сильнее индуктивного. Это хорошо, но мы уже видели, что бывают исключения из этого неизвестно кем и когда установленного, но очень популярного правила. На следующих вкладках эта проблема будет возникать еще не раз. В данном случае сомнения могут быть связаны, например, со случаем кислотности самой угольной кислоты. Если посмотреть диссоциацию по второй ступени, то мы получим даже не две, а все три совершенно одинаковые мезомерные формы.
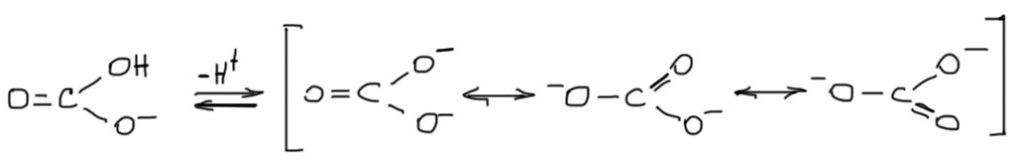 Если аргумент об определяющем эффекте мезомерии верен, то угольная кислота должна иметь очень высокую кислотность по второй ступени. Очень высокую, естественно, но не выше кислотности по первой ступени – более высокая кислотность двухосновной кислоты по второй ступени это чрезвычайно редкое явление. Близкую к ней или что-то типа этого, что-то такое, что нельзя не заметить. Но нет, ничего подобного, pK по второй ступени целых 10.3 – целая пропасть разделяет первую и вторую ступень, кстати это гораздо большая разница чем то, что наблюдается у двухосновных карбоновых кислот типа щавелевой. И мы это отлично знаем и пользуемся этим очень часто, выбирая между карбонатом и бикарбонатом, всегда помним, что основность первого значительно выше основности второго. Видимо, причина большого ослабления кислотности по второй ступени то, что мезомерный эффект в этом случае явно не смог перебороть индуктивный, а в бикарбонате эти эффекты не сонаправлены, а противоположны, и вместо индуктивного акцептора карбонила находится индуктивный донор отрицательно заряженный карбоксилат COO—.
Если аргумент об определяющем эффекте мезомерии верен, то угольная кислота должна иметь очень высокую кислотность по второй ступени. Очень высокую, естественно, но не выше кислотности по первой ступени – более высокая кислотность двухосновной кислоты по второй ступени это чрезвычайно редкое явление. Близкую к ней или что-то типа этого, что-то такое, что нельзя не заметить. Но нет, ничего подобного, pK по второй ступени целых 10.3 – целая пропасть разделяет первую и вторую ступень, кстати это гораздо большая разница чем то, что наблюдается у двухосновных карбоновых кислот типа щавелевой. И мы это отлично знаем и пользуемся этим очень часто, выбирая между карбонатом и бикарбонатом, всегда помним, что основность первого значительно выше основности второго. Видимо, причина большого ослабления кислотности по второй ступени то, что мезомерный эффект в этом случае явно не смог перебороть индуктивный, а в бикарбонате эти эффекты не сонаправлены, а противоположны, и вместо индуктивного акцептора карбонила находится индуктивный донор отрицательно заряженный карбоксилат COO—.
Есть и второй аргумент. Он связан с тем, что под кислотностью частно понимают не одно, а несколько связанных друг с другом свойств. Мы пока что занимались кислотность по Аррениусу – в воде. Поговорим ниже еще и кислотности в смысле Бренстеда-Лоури. Оба этих типа кислотности связаны с равновесием сопряженной кислоты и основания, и для карбоновых кислот обязательно подразумевают образование аниона. Но у кислот, и мы это уже видели в кислотном катализе реакций карбонильных соединений, есть еще и функция образования водородной связи со всякими атомами с неподеленной парой. В таком типе кислотности кислоту называют донором водородной связи. И в водородной связи протон остается на своем доноре, и образования аниона не происходит, соотвественно, не работает и мезомерия, по крайней мере, в том ярком виде двух одинаковых граничных структур. И если бы именно мезомерия была главной причиной относительно высокой кислотности карбоновых кислот, то кислотность, связанная с образованием водородной связи была бы существенно ниже. Но нет, ничего подобного, такую кислотность легко измеряют, и величины доступны для очень большого количества соединений. Правило здесь простое – если причины, определяющие кислотность по Аррениусу или Бренстеду-Лоури с одной стороны, и водородной связи, с другой стороны, одинаковы, то эти величины отлично коррелируют друг с другом. А если разные и существенно разные, то не коррелируют. Подробнее останавливаться не будем, но просто скажем – для карбоновых кислот обе кислотности отлично коррелируют, а следовательно и причина у них в основном одинакова. И такой причиной, одной для двух типов кислотности может быть только очень сильный акцепторный индуктивный эффект карбонила.
Карбоновые кислоты, в первую очередь, уксусная кислота являются отличными донорами водородной связи, и это свойство очень часто используют в реакциях, требующих кислотного катализа. Напомню только одну реакцию из химии карбонильных соединений, требующих кислотного катализа для кето-енольного равновесия: бромирование в уксусной кислоте. Ни при каких обстоятельствах нельзя рассматривать протонирование карбонильной группы уксусной кислотой – только водородную связь. Катализ кето-енольного превращения в присутствии карбоновых кислот – одно из такиз превращений, а вообще их сотни. Еще раз – не пишите протонирование очень слабых оснований типа карбонильного кислорода слабыми кислотами типа карбоновых. Это совершенно некорректно, так как разница в pK в этих случаях составляет много, иногда десятки порядков. В присутствии сильных кислот – специфический кислотный катализ через протонирование и превращение сопряженной кислоты субстрата. В присутствии (а часто и в растворе – высокая концентрация компенсирует более слабый эффект) слабых кислот – общий кислотный катализ через образование водородной связи, приводящего к смещению электронной плотности в том же направлении, что и при протонировании.
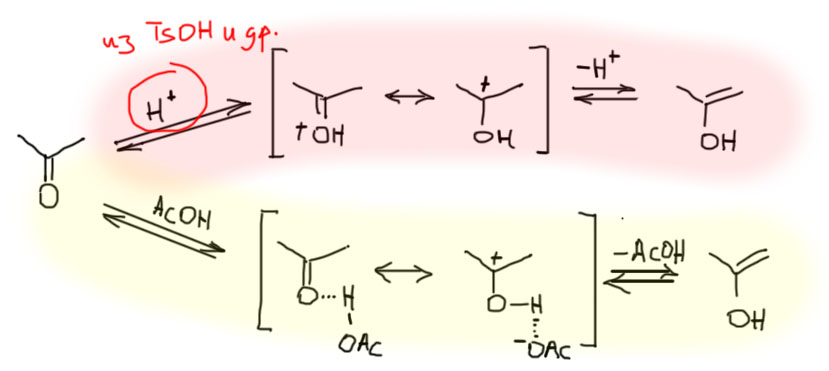
Кислотность по Бренстеду-Лоури и нуклеофильность анионов карбоновых кислот
В органических растворителях карбоновые кислоты также являются слабыми. Особенно сильно, как мы знаем (вспомните рассуждения об основности и нуклеофильности в SN2-реакциях), влияют и на то, и на другое полярные апротонные растворители, такие как ДМСО, в которых и нуклеофильность и основность всегда значительно возрастает.
Основность ацетат-иона в ДМСО оценивается величиной pK около 12. Напомню, что шкалы кислотности-основности Бренстеда-Лоури, в отличие от шкал кислотности-основности по Аррениусу, относительные и плавающие, но известная шкала основности (шкала Бордуелла) в ДМСО устроена так, что величины в pK в ДМСО можно непосредственно сравнивать с величинами pK в воде (аррениусовской кислотностью-основностью) – как это сделано, можете еще раз вспомнить на страничке про кислотность-основность. Поэтому основность ацетата в ДМСО (pK 12.6) на 8 порядков выше основности ацетата в воде.
Напомню, зачем это нужно знать. Ацетат и другие анионы карбоновых кислот (карбоксилаты) – нуклеофилы. Для нуклеофилов из неметаллов 2 периода действует качественное правило – нуклеофильность определяется основностью. В воде и других протонных растворителях ацетат и другие карбоксилаты – слабые основания (pK в районе 4-5 это очень мало, например, в водном растворе pH даже не будет на щелочной стороне шкалы), а значит и слабые нуклеофилы. И это действительно так – реакции SN2-замещения даже самых серьезных субстратов типа метильных, бензильных и т.п. а обычных растворителях ну просто очень медленные. А все остальное совсем не реагирует. А реакция весьма полезная – хорошо бы уметь получать сложные эфиры реакцией солей карбоновых кислот с алкилгалогенидами и тозилатами.
И это один из тех случаев, когда специальные растворители типа ДМСО и ДМФА или методы межфазного переноса играют решающую роль. Повышение основности на 8 порядков отражается в столь же впечатляющем увеличении нуклеофильности. При этом очень важно то, что pK 12.6 это все еще на настолько много, чтобы опасаться конкуренции элиминирования. Подробнее про сами реакции, использующие этот подход, можно прочитать на страничке методов.
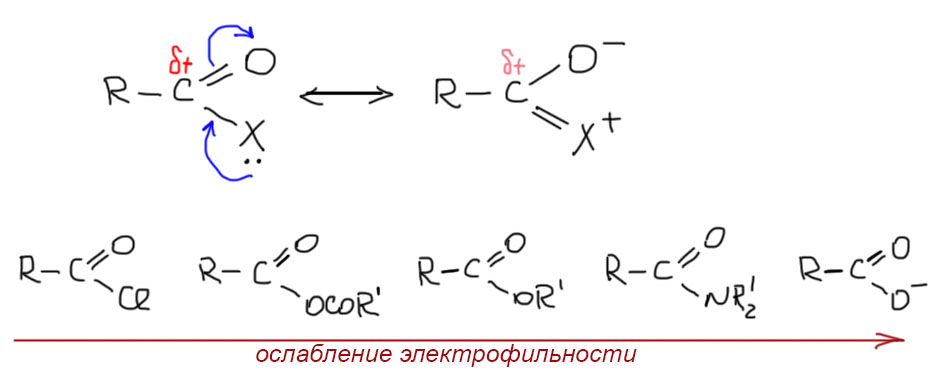
Чем отличается карбонильная группа в карбоновых кислотах и производных от карбонильной группы альдегидов и кетонов
На первый взгляд вообще ничем. На второй мы обращаем внимание, что в тех производных, где есть карбонильная группа, например, галогенангидридах, ангидридах, сложных эфирах, амидах, а также в самих карбоновых кислотах на карбонильном углероде всегда есть еще один заместитель, и у него есть неподеленная пара, а следовательно и донорный мезомерный эффект. Тот же заместитель одновременно имеет еще и акцепторный индуктивный эффект. Единственное исключение – соли карбоновых кислот, в которых этот заместитель донорный (кислород с отрицательным зарядом – донор и в мезомерном и в индуктивном смысле). Влияние мезомерного донора на карбонильную группу вполне описывается мезомерным эффектом. Электронная плотность смещается в сторону кабонильной группы, что приводит к двум серьезным последствиям:
- положительный заряд на карбонильном углероде уменьшается (но всегда все равно остается положительным, даже в суммарно отрицательной соли), а значит снижается электрофильность;
- связь С=O ослабевает, удлиняется, становится не полностью двойной – этот эффект хорошо виден в ИК-спектрах, но не так важен для химических свойств.
С другой стороны, в галогенангидридах преобладет не донорный мезомерный, а акцепторный индуктивный эффект, и заряд на углероде наоборот выше, чем в альдегидах и кетонах, и электрофильность тоже сильно выше. Тот же эффект, но в меньшей степени проявляется в ангидридах. Почему в ангидридах? – ведь там тоже атом кислорода сидит на карбониле, как в сложных эфирах? Такой же, но с другой стороны у него такой же ацил, и его мезомерная донорность расползается в равной (если ангидрид симметричный) или неравной (если несимметричный) на две стороны, а следовательно ослабевает, а индуктивный эффект, наоборот усиливается, так как этот атом кислорода к собственному индуктивному эффекту кислорода, добавляется индуктивный акцепторный эффект второго ацила.
Картинка получается очень красивая, но, похоже, далеко не полная, и даже не совсем точная. Очень важную информацию мы можем получить из уже упомянутых ИК-спектров. Мы как-то позабыли этот важнейший метод, который еще недавно был основной спектроскопией в органике, а ныне почти полностью заменился ЯМРом. Для изучения карбонила во всех мыслимых формах ИК совершенно незаменим. Напомню, что ИК (точнее, так называемые валентные колебания в ИК) показывает нам не атомы и не группы атомов как ЯМР, а целые связи. А связи в колебательных спектрах в ИК-диапазоне – это просто пружинки такие, и поведение этих пружинок отлично описыватся простым законом Гука. Пружинки любят колебаться – туда-сюда, туда-сюда – и частота этих колебаний тем выше, чем а) пружинка жестче; б) грузики на концах (то есть атомы) легче. Когда мы изучаем одну и ту же связь, как в случае карбонила, то грузики одинаковы, и все определяется жесткостью связи. А жесткость вполне однозначно связана с порядком связи – чем он меньше, тем связь мягче, и частота колебаний меньше.
Вот, берем карбонил, и считаем, что идеальный карбонил в самых простых карбонильных соединениях, альдегидах. Смотрим в ИК, в ту часть спектра, где поглощают как раз связи, колеблющиеся, как пружинки (это называется валентными колебаниями, а по-английски точнее и понятнее – stretching vibrations, то есть растягивающие колебания). Где виден карбонил у альдегидов – у 1725 см-1 (это не частота, а волновое число, но это неважно, так как обе величины пропорциональны, фактически одно и то же, просто в разных единицах измерения). Возьмем эту величину, как характеристику настоящей двойной связи C=O, такой эталонной пружинки, с которой мы будем сравнивать все остальное.
ОК, а где тогда кетоны? У 1715 см-1 – о, почти то же самое, но немного меньше, потому что индуктивный донор немного электронную плотность от себя отгоняет, что вызывает небольшое смещение плотности к кислороду, а это то же самое, что небольшое уменьшение порядка связи – пружинка стала чуть мягче.
А амиды? – между 1630 и 1680 см-1. О, так и должно быть, мезомерный донор дал серьезный вклад второй граничной структуры с одинарной связью C-O, и порядок связи естественно уменьшился гораздо сильнее. Видим из этих тенденций, что ИК действительно отлично отражает изменения в структуре и действие электронных эффектов. А в солях кислот совсем стала пружинка мягкой – 1550-1610 см-1 – прямо так и должно быть, ведь здесь обе граничные структуры одинаковы и мы совершенно точно можем сказать насколько уменьшился порядок связи – с двух до полутора (одна двойная связь поровну делится на две, добавляя по половинке к простой σ-связи C-O).
Посмотрим теперь с другой стороны. Хлорангидриды: 1810-1775 см-1 – связь стала сильно жестче. Почему? Граничные структуры ничего такого не говорят – как вообще связь может быть больше чем двойной? Это понять сложнее без помощи настоящей квантовой химии, но чисто приблизительно считаем, что индуктивный акцептор откачивает плотность от углерода на себя, что приводит к тому, что карбонильный кислород своей неподеленной парой немного компенсирует это смещение. Углерод пожалел! Фокус в том, что от углерода плотность скачивают через σ-связь, а кислород компенсирует это смещение через π-связь. Можно даже это выразить мезомерией.
Эта мезомерия не для слабонервных. Если она вам кажется странной, просто пропустите, ничего не потеряете. Но все же, если мы хотим изобразить мезомерию и граничную структуру для случая, когда индуктивный акцептор скачивает плотность с углерода по σ-связи, а кислород компенсирует это смещение как π-донор, нам придется во второй граничной структуре показать тройную связь на карбониле, и чисто ионную связь с акцептором X. Мы не имеем права оставлять черточку связи к X, потому что граничные структуры должны соблюдать правило октета Льюиса – не более 4 черточек на атоме неметалла, p-элемента. Вклад этой странной, но вполне легитимной граничной структуры в реальную структуру производного невелик, но она объясняет, каким образом акцепторный индуктивный эффект группы X может приводить к упрочению, увеличению порядка связи выше чисто двойной, связи CO в карбониле. Некоторые наблюдательные читатели могут спросить, а с какого … вдруг мезомерия стала отображать индуктивный эффект, ведь договаривались же, что индуктивный и мезомерный эфекты принципиально различны, и отображаются по разному. Вопрос правильный, но не по адресу – в этом случае мезомерия не отображает индуктивный эффект, а отображает существеную роль граничной структуры с тройной связью углерод-кислород (а ее можно нарисовать в любом случае, даже для альдегида или кетона, только вклад ее там ничтожен) в том частном случае, когда индуктивный эффект уже привел к смещению электронной плотности.
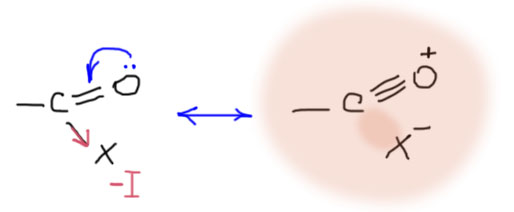
С хлорангидридами все ясно. Ангидриды имеют частоту валентного колебания карбонила приблизительно там же, но с этим мы уже разобрались, и просто принимаем, что картина такая же как в хлорангидридах, но с немного меньшим вкладом граничной структуры с тройной связью.
А вот что со сложными эфирами? Из того, что мы узнали до этого можно предположить, что это будет как в амидах, но поменьше – просто потому что мы знаем, что алкокси-группа – это мезомерный донор, как и амино-группа, но только послабее. И мы привыкли, что для алкокси и гидрокси-групп мезомерный донорный эффект сильнее акцепторного индуктивного, и в этом проявляется их отличие от галогенов. Сюрприз: в сложных эфирах карбонил колеблется при 1735 см-1, то есть он почти такой же как в альдегидах, но все же немного жестче. Оказывается, сложные эфиры находятся в ряду со стороны хлорангидридов и ангидридов, а не амидов! Это буквально означает, что акцепторный индуктивный эффект алкокси-группы в этом случае преобладает над мезомерным донорным. Как же так, мы же договорились что +M > -I !?
Очень хорошо, что предоставилась возможность прояснить эту важную проблему. Индуктивный эффект определяется разностью электроотрицательностей атомов, и поэтому приблизительно одинаков в разных ситуациях. Мезомерный эффект устроен намного сложнее, это такое отображение электронных, орбитальных взаимодействий в молекулах, зависящих от конкретной структуры молекулы, взаимного расположения взаимодействующих групп и т.д. Наиболее сильно мезомерный эффект проявляется, когда
а) он приводит к стабилизации молекулы или иона;
б) когда есть цепь сопряжения, на концах которой находятся донор и акцептор (что это такое можно вспомнить вот здесь).
В таких случаях действительно всегда алкокси-группа и другие кислорожные заместители, действующие как донор, обладают очень сильным донорным эффектом, стабилизирующим, например, карбокатионы и другие акцепторные центры, особенно секстетного типа.
В других случаях, а именно когда
а) эффект дестабилизирующий;
б) донорность и акцепторность в цепи сопряжения выражены неявно, например, мы имеем дело с устойчивой нейтральной молекулой, у которой и так все хорошо;
в) мы имеем дело не с одной цепью сопряжения, а с несколькими пересекающимися, и т.п.
– мезомерный эффект может быть довольно слаб и вполне может уступать другим эффектам, если их действие противоположно. Может уступать, а может и не уступать – в этих случаях баланс всегда очень тонок и довольно непредсказуем. Именно поэтому мы еще в самом начале говорили, что анализировать действие электронных эффектов всегда проще и однозначнее во всяких заряженных частицах, а не в нейтральных молекулах. Вот в этом конкретном случае мезомерный эффект алкоксигруппы на карбонил как раз имеет дестабилизирующий характер. Как это работает можно посмотреть на вкладке ниже, посвященной енолятам производных карбоновых кислот, но в отличие от енолятов, где мы имеем дело с заряженной частицей, и вклад мезомерного эффекта весьма существен, здесь у нас нейтральная стабильная молекула, в которой мезомерия работает слабо.
Теперь уже не будет неожиданным увидеть, что у самих кислот карбонильная группа даже немного жестче, чем у сложных эфиров – индуктивный эффект OH безусловно немного больше чем у OR. Но это для нашей химии совсем неважно, потому что кислоты слишком легко депротонируются и в равновесиях участвуют не в виде нейтральной молекулы RCOOH, а в виде аниона, с которым мы уже разобрались.
Итак, порядок электрофильности, приведенный в начале таким и остался, но если мы к нему добавим альдегиды и кетоны, то получим, что они встраиваются в серединку, между сложными эфирами и амидами. С альдегидами там не совсем все ясно, потому что разница очень невелика, и реальный порядок электрофильности будет сильно зависеть еще и от эффектов группы, висящей на карбониле. Но нам это не так важно – мы никогда не будем устраивать соревнования в электрофильности между альдегидами и сложными эфирами. Но с кетонами это гораздо более однозначно, да к тому же не забудем, что реакционная способность карбонила в кетонах дополнительно ослабляется по стерическими причинам – мы это разбирали в теме про альдегиды и кетоны. Карбонил в сложных эфирах более электрофилен, чем в кетонах, и это очень важно, потому что как раз в этом случае соревнования в электрофильности устраивают довольно часто, например, в реакции конденсации енолизуемых кетонов с сложными эфирами, енолизуемыми или неенолизуемыми. В смеси енолизуемого сложного эфира и енолизуемого кетона мы можем в равновесных ненаправленных условиях селективно получать продукт ацилирования кетона – кетон выступает в виде метиленовой компоненты (а у него еще и CH-кислотность выше), а сложный эфир в виде карбонильной, то есть электрофильной компоненты.

Реакции с нуклеофилами
Карбонил, он и на Луне карбонил, и основная его реакция всегда – присоединение нуклеофилов с образованием тетраэдрического аддукта. Все, что по этому поводу можно сказать, уже сказано в Альдегидах и кетонах. Присоединяет. Реакционная способность рассмотрена на предыдущей вкладке – хлорангидриды, ангидриды и сложные эфиры более электрофильны чем альдегиды и кетоны, амиды менее. Сложные эфиры очень близки к альдегидам, из-за этого кроме электрофильности в реакционную способость дает большой вклад и второй фактор – стерический. Поскольку в альдегидах карбонильный углерод всегда более доступен чем в сложных эфирах похожей структуры (вместо водорода алкокси-группа), реальная реакционная способность альдегидов в реакциях с нуклеофилами будет выше. Про формальдегид мы естественно не говорим – его реакционная способность столь велика, что он, наверное, мог бы и с ангидридами поспорить… если бы такой спор когда-либо реально состоялся бы, то есть была бы какая-то реакция, в которой формальдегид и какой-нибудь ангидрид конкурировали бы за нуклеофил. Мне ничего подобного в голову не приходит. А если нет такой реакции, то эта проблема, видимо, относится к той же области, как и исход знаменитой битвы слона с китом, или жаркого спора, у кого больше атомных боеголовок.
Где в этой схеме сами кислоты? Это непростой вопрос, так как в реакциях с нуклеофилами, которые очень часто являются основаниями, кислоты переходят в соли, а с солями все ясно – наименее реакционноспособны из всех форм карбонила. Анионы карбоновых кислот, карбоксилаты, реагируют только с самыми реакционноспособными нуклеофилами типа нестабилизированных карбанионов в простых литийорганических соединениях, или комплексными гидридами металлов, и то далеко не со всеми. Это неплохо, потому что позволяет селективно превращать соединения с несколькими функциональными группами. Но в условиях кислотного катализа, когда анион не образуется, кислоты вполне реакционноспособны, и, вероятно, не сильно отличаются от эфиров. Косвенным подтверждением этого является то, что взаимопревращение кислоты и сложного эфира в условиях кислотного катализа – очень подвижное равновесие, которое легко смещается хоть в одну, хоть в другую сторону.
Реакция с нуклеофилами сначала дает обычный тетраэдрический интермедиат, который отличается от таких же интермедиатов у альдегидов и кетонов,
- во-первых, тем, что он всегда неустойчив и никогда не становится конечным продуктом реакции (там таких случаев немало);
- во-вторых, тем что практически всегда дальше превращается по одному и тому же сценарию, а именно, происходит уход той группы, которая была в производном на карбонильном углероде и вновь образуется полноценный карбонил.
Получается очень простая картинка. Для корректности напишем два равновесия, с основной и кислотной формой нуклеофила. Разницы мало. 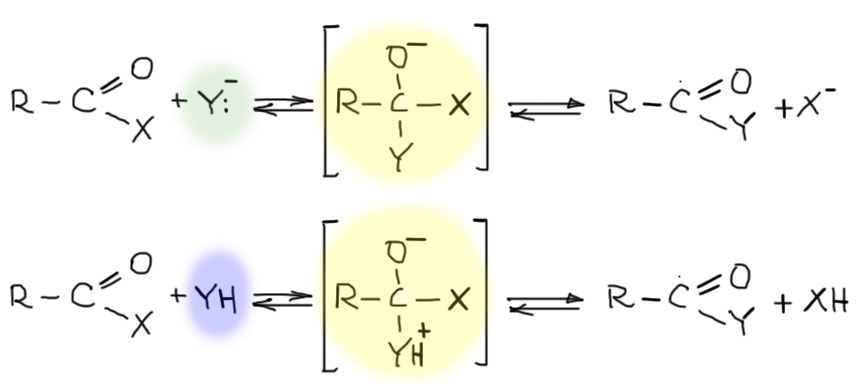
Если посмотреть на эту реакцию не с точки зрения механизма, а с точки зрения результата, то ее суть заключается в том, что производное карбоновой кислоты отдает нуклеофилу остаток карбоновой кислоты, который называется в общем случае ацилом. Поэтому эту общую реакцию, точнее общий тип реакций называют ацилированием нуклеофилов (ацилированием аминов, спиртов, солей карбоновых кислот, тиолов, и т.д., и т.п). С этим термином мы уже сталкивались в ароматических соединениях, где было ацилирование бензола и его производных по Фриделю-Крафтсу. Строго говоря, это просто частный случай того, что мы рассматриваем здесь: бензол и другие ароматические соединения в этой реакции это нуклеофилы, очень слабые, поэтому трубующие катализа (активации ацилирующего реагента) хорошей кислотой Льюиса. И мы помним, что ароматические соединения можно ацилировать хлорангидридами, ангидридами, самими кислотами, амидами, а сложными эфирами нельзя только потому что кроме ацилирования получится еще продукты алкилирования спиртовой частью эфира. И если дальше мы начнем копаться в том, что мы уже знаем, и что можем еще узнать, то реакция ацилирования окажется очень распространенной и очень востребованной. Практически любой нуклеофил можно проацилировать, только придется, как обычно, подумать о реакционной способности и способов ее раскочегаривания.
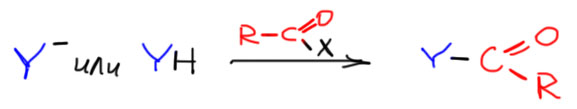 Реакции ацилирования, используемые для синтеза. поэтому обычно записывают, исходя из нуклеофила, если только он не совсем простой, а ацилирующие агенты и все остальное пишут над стрелкой.
Реакции ацилирования, используемые для синтеза. поэтому обычно записывают, исходя из нуклеофила, если только он не совсем простой, а ацилирующие агенты и все остальное пишут над стрелкой.
Частные случаи реакции ацилирования называют по конкретным ацилам, когда их названия общеизвестны, а слово получается удобопроизносимое. Остаток муравьиной кислоты – формил, формилирование. Уксусной – ацетил, ацетилирование. Бензойной – бензоил, бензоилирование. Но, например, пропионовой – пропионил, но “пропионилирование” не говорят. Масляной – бутирил, но “бутирилирование” не говорят. И так далее. В общем случае ацилы называют по ИЮПАК как кислоты, но вместо “-овая кислота” заканчивают на “-оил” с сохранением всего остального: от декановой кислоты, например, ацил называется деканоил, а ацилирование – деканоилированием. И так всегда, если мы знаем название кислоты, то знаем и ацил и ацилирование, даже, возможно, не догадываясь о структуре самой кислоты. Была бы, например, цапцараповая кислота, то ацил от неё назывался бы цапцарапоил. И реакцию можно было бы назвать цапцарапоилирование, а если бы это слово показалось громоздким и неуклюжим, то говорили бы – ацилирование цапцарапоил хлоридом. И так далее…
А енолы у всего этого есть?
То, что с енолами у карбоновых кислот и их производных все не так просто можно очень просто убедиться, написав структуру енольной формы самой простой енолизуемой кислоты – уксусной кислоты. Обратим внимание на то, что слово “енолизуемый”, как и изображение стрелки равновесия, не означает того, что енол действительно существует в измеримых количествах, – это просто констатация принципиальной возможности енолизации из-за наличия α-протонов. Итак, если мы напишем енол у уксусной кислоты, то обнаружим довольно страшную молекулу, 1,1-дигидроксиэтен, фактически являющийся продуктом гидратации – присоединения воды к карбонильной группе – кетена. Кетена??! С кетеном мы пока еще не познакомились, но более-менее все знают, что это бешено реакционноспособное соединение, бурно реагирующее даже со следами воды с образованием … уксусной кислоты. Еще раз – енол уксусной кислоты это гидрат кетена, но продукт гидратации кетена – это уксусная кислота. Таким образом мы чисто случайно обнаружили вероятный механизм реакции кетена с водой – при этом сначала должен образоваться енол уксусной кислоты, который перегруппировывается, или другими словами, претерпевает кето-енольную таутомеризацию в обычную (кетонную) форму уксусной кислоты. 
Хорошо, может быть, а сколько там этой енольной формы. Про простые альдегиды и кетоны мы знаем, что содержание енола составляет тысячные и десятитысячные доли процента. что соотвествует константам равновесия кетон-енол в диапазоне 10-4 – 10-6. Содержание енола в растворе уксусной кислоты в сравнении с этими величинами по различным оценкам составляет абсурдно малую величину порядка 10-20. Когда мы имеем дело с такими малыми величинами всегда полезно сравнивать их с числом молекул в моле, числом Авогадро. Тогда мы поймем очень ясно, что 10-4 – 10-6 – это не так уж и плохо, это огромное количество молекул, вполне измеримое множеством современных методов, и вполне могущее играть какую-то серьезную роль в реакциях как реакционноспособный интермедиат. Мы это и видели в химии енолизуемых альдегидов и кетонов – множество важнейших реакций зависят от малого содержания равновесного енола. В сравнении с этим константа в 10-20 – это буквально считанные молекулы, это уже больше на гомеопатию похоже, чем на серьезную науку. Таике количества вещества не определяются в растворах никакими реальными спектральными методами (в газовой фазе определяются масс-спектрометрически, но это не имеет прямого отношения к тому, что происходит в обычных реакциях). И никакой реальной роли в реакциях соединение, присутствующее в такой концентрации, иметь не может.
Вывод простой – в енолизуемых (еще раз: мы не перестаем называть их енолизуемыми, даже зная наперед, чем закончится это предложение) карбоновых кислотах содержание енола пренебрежимо мало, и на течение реакций влияния не оказывает. То же самое относится к сложным эфирам и амидам. Практически полная неспособность карбоновых кислот, сложных эфиров и амидов образовывать значимые количества енолов имеет совершенно ясные и определенные последствия для химии этих соединений – у них просто нет тех реакций с участием енолов, которые есть у енолизуемых альдегидов и кетонов, например, кислотно-катализируемого бромирования, нитрозирования, и тем более, кислотно-катализируемой конденсации по типу альдольной.
Но совсем на енолах в химии карбоновых кислот крест ставить не стоит. Если вспомнить, какие факторы способствуют повышенному содержанию енола в химии карбонильных соединений, то есть еще CH-кислотность – чем она выше, тем, при прочих равных условиях, выше содержание енола. В химии карбоновых кислот наивысшей CH-кислотностью обладают галогенангидриды, к которым примыкают еще и просто ангидриды. Именно эти типы соединений могут проявлять склонность к образованию значимых количеств енолов и использовать эти формы для каких-то реакций. Действительно, как минимум одна такая реакция очень хорошо известна – бромирование по Геллю-Фольгарду-Зелинскому. В этой реакции используют то, что галогенангидриды карбоновых кислот реагируют с бромом или хлором непосредственно без использования каких-либо дополнительных реагентов. Фактически это та же самая реакция, как и бромирование енолизуемых кетонов и альдегидов. С бромом или хлором реагирует галогенангидрид, почти наверняка в енольной форме. Здесь нехорошо только то, что это всего лишь гипотеза, потому что ни относительное количество енола в галогенангидридах определено не было, ни сами эти енолы не ловились каким-либо спектральным или другим методом. Слишком это неудобные для исследования, высокореакционноспособные соединения. Но гипотеза разумная, а других все равно нет. Сначала напишем саму реакцию галогенангидрида с галогеном, без деталей, которые можно вспомнить в разделе про бромирование кетонов.
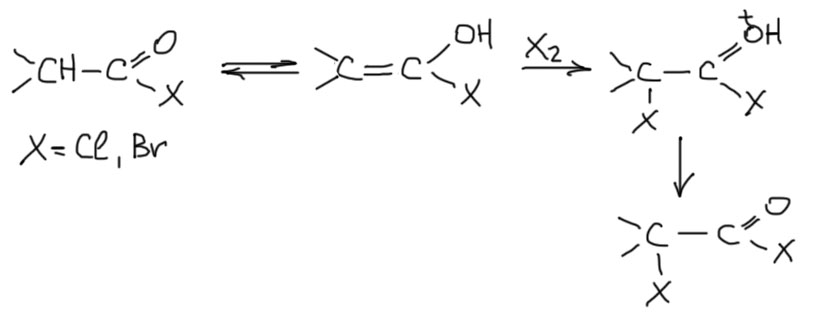
Галогенированный галогенангидрид можно гидролизовать и получить галогенированную кислоту. Это хорошо, но галогенировать хочется не галогенангидриды, а сами кислоты. Метод Гелля-Фольгарда-Зелинского решает эту проблему, используя кислоту, бром и небольшое количество фосфора. Прямо в реакционной смеси образуется PBr3, который реагирует с кислотой, образуя бромангидрид, который реагирует с бромом. Образующийся бромангидрид бромированной кислоты в равновесии с исходной кислотой дает новый бромангидрид и бромированную кислоту (это равновесие, и оно не сдвинуто в одной сторону на 100%, но это и не нужно. Так понемногу бромируется большая часть исходной кислоты. В конце всю смесь гидролизуют, и бромированную кислоту выделяют кристаллизацией или перегонкой.

А еноляты?
Еноляты у производных карбоновых кислот и даже у самих карбоновых кислот есть, но все не так просто. Это важная история, а в учебниках она рассмотрена удивительно невнятно, потратим на нее немного времени.
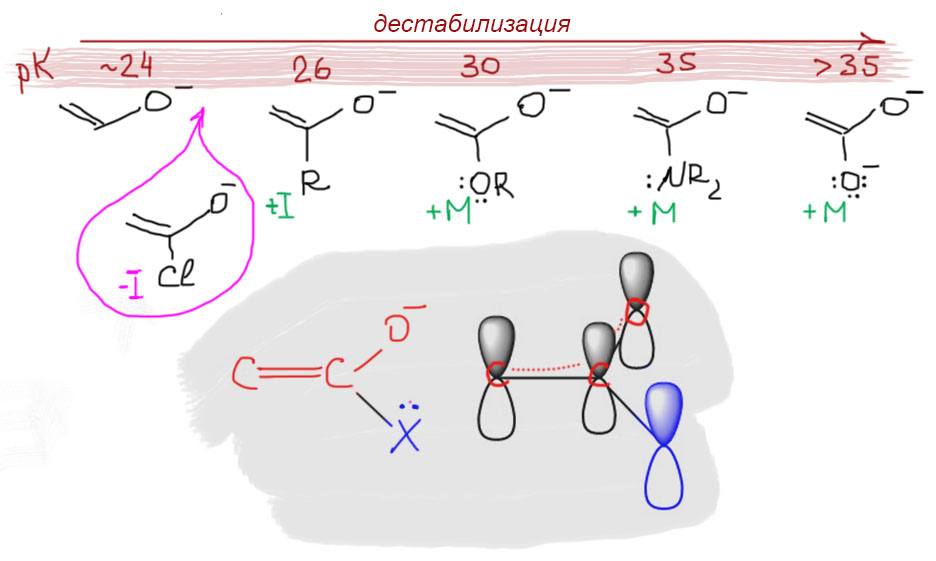
Стабилизация и дестабилизация енолятов.
Во-первых, кислотность α-протонов у производных кислот очень сильно зависит от того, какое производное мы хотим попробовать депротонировать. У всех производных кроме хлорангидридов (или галогенангидридов) кислотность сильно ниже чем у кетонов с таким же строением группы, присоединенной к карбонилу. Причину понять просто, но не очень. Возьмем, например, сложный эфир, оторвем протон, и подумаем, как стабилизирован получившийся анион. Заметим, что уже сам сложный эфир имеет мезомерную делокализацию, которая снижает положительный заряд на карбонильном углероде, уменьшая тем самым акцепторный индуктивный эффект всей группы COOR по сравнению с кетонной группой. Мезомерная форма аниона будет с виду такой-же как у кетона, поэтому это и называют также енолятом. Но сопряженная система здесь более сложная, потому что в нее включен еще и атом кислорода со своей парой, так что можно написать и третью граничную структуру. В этом месте у многих людей может начаться совершенно закономерная ломка – много раз ведь говорили, что чем больше граничных структур, тем лучше. Три же лучше чем две? Не всегда. Мы часто забываем вторую часть этого принципа мезомерной стабилизации – нужно рассматривать не только число граничных структур, но и их вклад в результирующую, реальную структуру молекулы или иона. Формы с малым вкладом либо ничего не вносят, либо могут даже оказывать отрицательный эффект. Плохими всегда являются формы, увеличивающие степень разделения зарядов. Очевидно, что это как раз этот случай – вместо одного минуса, два минуса и плюс. Это плохая форма. Но обычная енолятная форма есть, а значит вроде бы все должно быть точно так же, как у обычного кетона или альдегида. Но нет, CH-кислотность сложных эфиров меньше, значит что-то этот анион все таки дестабилизирует. Нам будет непросто понять что и как, так как для этого очень пригодилась бы квантовая наука.
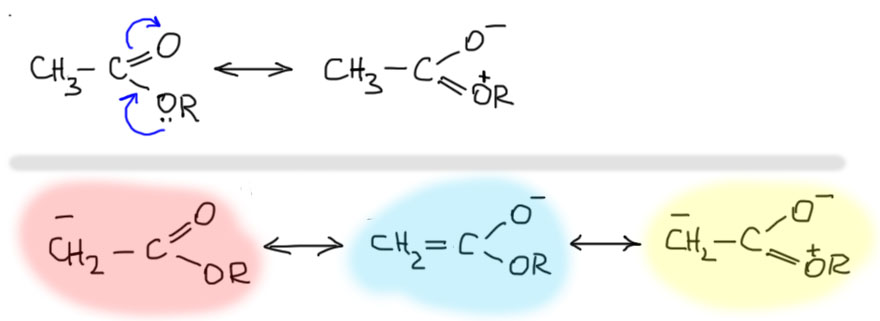
Проблемы у нас даже еще более серьезные. Я не хочу никого пугать, но посмотрим, например, на енолят, который мы получили бы из аниона карбоновой кислоты. Здесь три совершенно нормальные граничные структуры, две из которы заведомо одинаковые. И это должно быть хорошо, очень хорошо, точно так как учит нас теория резонанса – чем больше одинаковых граничных структур, тем лучше, тем больше стабилизация. Если мы ограничимся только этим, то неизбежно придем к очень странному выводу – CH-кислотность аниона карбоновой кислоты должна быть точно выше CH-кислотности, например, сложного эфира (там нормальных структур две, а здесь целых три!!!). Но нет, ничего не получается.
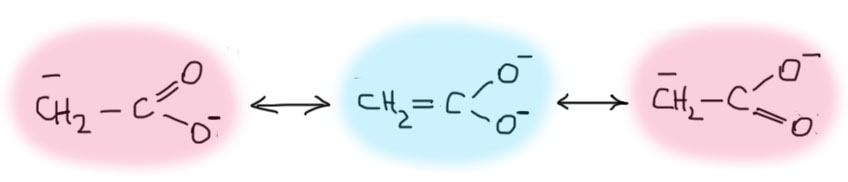
Этот поучительный случай показывает, что на теорию резонанса или мезомерной стабилизации слишком сильно полагаться не стоит. Просто посчитать количество граничных структур и считать, что это все и определяет, не получается. Каждый раз нужно думать и о том, как устроены эти граничные структуры, и действительно ли они дают вклад в стабилизацию. Попробуем рассмотреть проблему немного по-другому. Когда анализируют электронные эффекты, важно определить основную структуру, влияние на которую мы будем рассматривать, и дальше пытаться понять, как на нее влияют эффекты. Буквально это значит, что мы не будем смотреть на еноляты сложных эфиров и всего прочего, как каждый раз на новый тип частицы, определяя его стабилизацию или дестабилизацию заново, а просто посмотрим, как влияют заместители на одну единственную систему – енолят ацетальдегида. Это и будет точка отсчета. И увидим мы очень простую и понятную картинку – заместители с донорными эффектами дестабилизируют сопряженную систему енолята. Причина этого точно такая же, как и дестабилизация отрицательного заряда, локализованного на одном атоме – отрицательный заряд это избыток электронной плотности, электроны отталкиваются и вообще не любят быть рядом друг с другом в ограниченном объеме пространства, а донор еще добавляет к этому. Индуктивный донор типа алкила действует слабее – поэтому в альдегидах CH-кислотность α-протонов больше чем в кетонах. Еще сильнее действует донор с неподеленной парой, и эффект этот очень похож на известный нам α-эффект – отталкивание электронных пар на соседних атомах. Вон, на сереньком фоне нарисовано, как лежат орбитали в системе, похожей на сложный эфир, амид или соль (это не молекулярная орбиталь!, а просто атомные орбитали π-типа, отвечающие за сопряженную систему енолята и, синим, +М-заместителя). Хорошо видно, что они сонаправлены и отталкиваются, потому что в сумме в этой системе многовато электронов (4 штуки на еноляте плюс пара на заместителе). Результат – дестабилизация, отталкивание. Как всегда учитываем и донорность этого заместителя: нейтральный кислород в эфире слабее атота в амиде, а он слабее отрицательного кислорода в соли. Известные величины pK (в ДМСО, для альдегида величина экстраполирована, потому что экспериментально не измерена) вполне подтверждают этот эффект. А вот у галогена в галогенангидриде, как и в других случаях, акцепторный индуктивный эффект преобладает, и происходит, наоборот, стабилизация. Увы, pK в этом случае неизвестно потому что такие еноляты неустойчивы (см. ниже).
Существуют ли реально эти самые еноляты?
Во-вторых, нельзя забывать, что там есть еще уходящая группа, которая определяет особенности этих типов карбонильных соединений в реакциях по карбонильной группе. При попытке получить енолят эти уходящие группы тоже могут преподнести сюрприз. В общем виде можно написать что-то типа такой схемы. Отщепление α-протона дает мезомерно-делокализованный карбанион. Но если уходящая группа согласится на уход, то вместо мезомерной делокализации может произойти просто элиминирование с образованием своеобразного непредельного карбонильного соединения – кетена. Если рассмотреть этот процесс внимательно, то увидим, что это просто частный случай E1cb-механизма β-элиминирования. Тот же продукт, кетен, может получиться и прямо из исходного производного в результате самого обычного E2-элиминирования.
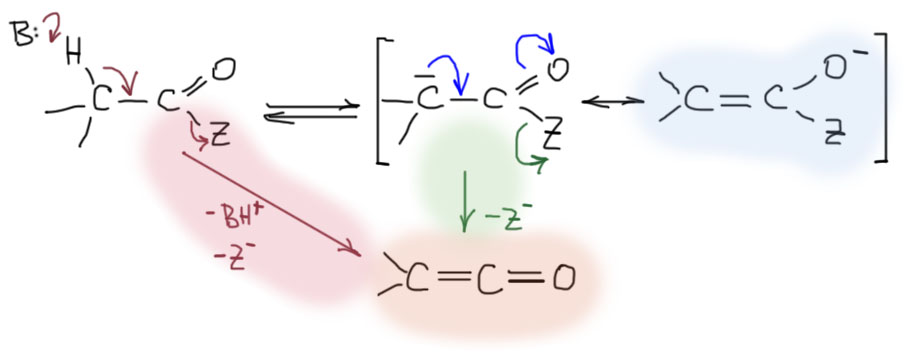
Ну и что же происходит в разных случаях. Все более-менее просто. Хорошие уходящие группы типа галогенов легко уходят, поэтому из галогенангидридов образуются кетены. Из ангидридов тоже, но так делают очень редко.
В сложных эфирах, амидах, самих кислотах хороших уходящих групп нет и поэтому образуются еноляты. Но есть всякие простые нюансы. Со сложными эфирами все просто – еноляты. Поэтому их чаще всего применяют, когда требуются еноляты.

В амидах нужно следить за тем, чтобы на азоте не было своих водородов – они обычно более кислые, и отщепляются в первую очередь. В результате будет не самая тривиальная химия, и мы ее пока отложим в сторону. Но даже если водородов на азоте нет, все равно возможны побочные реакции, потому что кислотность α-водородов в амидах существенно ниже, чем кислотность таких протонов в сложных эфирах, и иногда бывает проще оторвать протон от групп, которые висят на атоме азота. Разобраться во всем этом сложно, и поэтому не будем пытаться. В синтезе такие реакции используют, но очень редко.
В кислотах тоже есть нюанс. Хотелось бы и кислоты отложить в сторону, но нам они конкретно и очень скоро понадобятся именно в этом контексте. Понятно, что протон карбоксильной группы более кислый и отщепляется первым. И дальше протон придется отщеплять от соли кислоты, образуя дианион – и да, это тоже енолят, можно даже сказать ендиолят, но так не говорят. Пугаться этой формы не нужно – она нормальная. Но нужно помнить, что потребуется не один, а два эквивалента сильного основания.
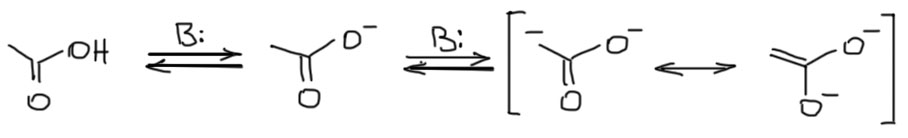
Тем не менее мы никогда не будем делать еноляты из сложных эфиров простых карбоновых кислот, и потому что основание потребуется для этого очень сильное – кислотность здесь еще ниже чем в амидах, и потому что в этом нет смысла – сложные эфиры депротонировать гораздо проще. Но в химии двухосновных кислот, прежде всего малоновой, необходимость в этой реакции есть. Так будет почти всегда, но не всегда. Бывают случаи, когда CH-кислотность сильнее кислотности карбоксила, например, при отщеплении протонов от малоновой кислоты. Оставим это до обсуждения малоновой кислоты и ее реакций.
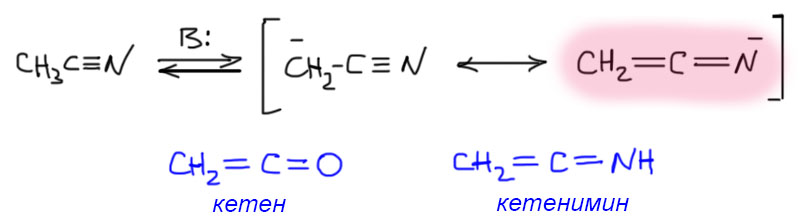
Так, с енолятами разобрались. А от нитрилов можно оторвать протон? Отлично можно, CH-кислотность нитрилов только немного меньше кислотности сложных эфиров той же кислоты (pK этилацетата в ДМСО 29.5 можно сравнить с кислотностью ацетониорила с pK 31.3). При отрыве протона от нитрила получается карбанион, мезомерно стабилизированный с второй граничной структурой с минусом на атоме азота. Немного необычно выглядит, но никаких конкретных претензий к такой форме предъявить нельзя. Судя по pK она ничем не хуже (почти ничем – один неполный порядок константы говорит о небольшой разнице) чем енолят из сложного эфира. Если эту граничную структуру рассмотреть внимательно, бросается в глаза сходство с молекулой кетена, и действительно это не что иное, как сопряженное основание азотного аналога кетена – кетенимина. А сам кетенимин – это таутомерная форма нитрила, намного менее устойчивая, чем сам нитрил, и поэтому в нитрилах практически нет в измеримых количесвах этого таутомера (как впрочем и енола в сложных эфирах). Но отрыв протона дает сопряженное основание нитрила, которое гораздо ближе к кетениминной форме, чем к форме с нитрильной группой. И такой анион отлично участвует в типичных реакциях енолятов, например, в аналоге сложноэфирной конденсации. Но об этом – в другой раз.