Фенолы и хиноны: методы и задачи
Химия фенолов и хинонов дает неплохие и довольно оригинальные методы постороения новых C-C связей, и в этом разделе много такие реакций.
Главная черта химии фенолов состоит в том, что она замкнута в самой себе – в том смысле, что вы можете вводить самые разные заместители, даже очень сложные, в молекулы фенолов, но не можете никуда деть гидроксильную группу. В классической органической химии нет методов (практически нет, что для нас значит – нет совсем) замены фенольного гидроксила на что-либо еще с разрывом связи углерод-кислород. Ароматическую связь C-O невозможно разорвать средствами обычной органической химии (из этого общего правила есть несколько исключений, одно из которых вы знаете – это активированное нуклеофильное замещение в ароматических нитро-производных).
Стоит сказать, что современная химия, использующая комплексы переходных металлов, отлично справляется с этой задачей, и для нее фенолы – одна из любимых исходных структур для превращения в самые разные продукты. Но для нас это пока за пределами обсуждения. Созреем, обсудим.
Хиноны в этом смысле являются более гибким типом молекулы. Так как по многим свойствам это просто кетоны, то продукты превращения хинонов отлично можно уконтрапупить так, что исходный хинон невозможно или очень трудно будет узнать. При высокой способности хинонов вступать во всякие реакции с усложнением скелета (сопряженное присоединение, циклоприсоединение) с дальнейшим преобразованием, эта химия становится очень богатым источником в синтезе. Более того, с помощью хинонов можно и обойти нежелание фенолов расставаться со свои кислородом – фенолы довольно легко превращаются в хиноны, а продукты превращения хинонов вполне могут уже и не содержать взятого из фенола атома кислорода.
Иными словами, этот раздел довольно богат реакциями синтеза, но в качестве компенсации совсем не содержит никакой стереохимии. Впрочем, мы уже забыли, когда последний раз имели с ней дело. Терпение – уже в следующей теме она снова к нам заявится.
Новые C-C связи
Реакции фенолов
В молекулы фенолов ловольно просто вводить альдегидные группы (формилировать) в орто- или пара-положение в гидроксилу. Для этого существует не менее десятка реакций разной степени сложности. Но одна из них особенно интересна и необычна, хотя бы потому что использует совсем уж простые реагенты – хлороформ и щелочь. В хлороформе в этой роли ничего загадочного нет, потому что мы хорошо знаем что для формилирования нужны производные муравьиной кислоты, а хлороформ вполне таковым является.
Но механизм реакции очень далек от обычных реакций ацилирования, в которых мы берем производное кислоты, активируем его электрофильность обычно какой-нибудь кислотой, и далее наблюдаем ароматическое электрофильное замещение. В реакции Реймера-Тимана хлороформ в присутствии крепкого раствора щелочи дает дихлоркарбен точно так же, как он это делает в реакции двойной связи олефинов с образованием дихлорциклопропана.
Дихлоркарбен является карбеном (кто бы мог подумать!?) с выраженным электрофильным характером. Он реагирует с активированными ароматическими соединениями в основном через циклопропанирование по одной из связей цикла, а образующийся при этом дихлорциклопропаны в условиях реакции превращаются в другие продукты с раскрытием трехчленного кольца. Но с фенолятами дихлокарбен реагирует не так, как с другими ароматическими соединениями, не как карбен, а просто как электрофил. Напишем эту реакцию, точнее то, как она начинается, для орто-атаки дихлоркарбена на фенолят. Все, как обычно для фенолята – образуется σ-комплекс типа кето-форма фенола за счет присоединения электрофила к ароматической системе, но он получается немного необычный, потому что на углероде бывшего карбена возникает полноценный карбанион (потому что нейтральный углерод карбена при образовании связи получает пару электронов от фенолята).
И после этого должно произойти нечто головокружительное – кето-форма должна перейти в енол (фенол), но для этого нужно взять протон от углерода и перенести его на кислород. Все бы ничего и как обычно, но попробуйте-ка провернуть эту операцию под носом у живого карбаниона! Не забываем, что протоны сами по себе не скачут, а их кто-то переносит в прямом смясле этого слова – берет в одном месте и отдает в хорошие руки в другом. Этот кто-то – основание, а в этом случае просто гидроксид-ион. И вот, берет гидроксид-ион протон у кето-формы, и так, бочком-бочком, начинает пропихиваться молекулой воды к кислороду. Понятно, что это путешествие тут же и закончится – протон до кислорода не долетит, а тут же приклеится намертво к карбанионному центру, потому что просто сравните pK типичного карбаниона и фенольного гидроксила. И вообще все это можно было не писать, потому что реакция идет в воде, а в воде такие карбанионы долго не живут.
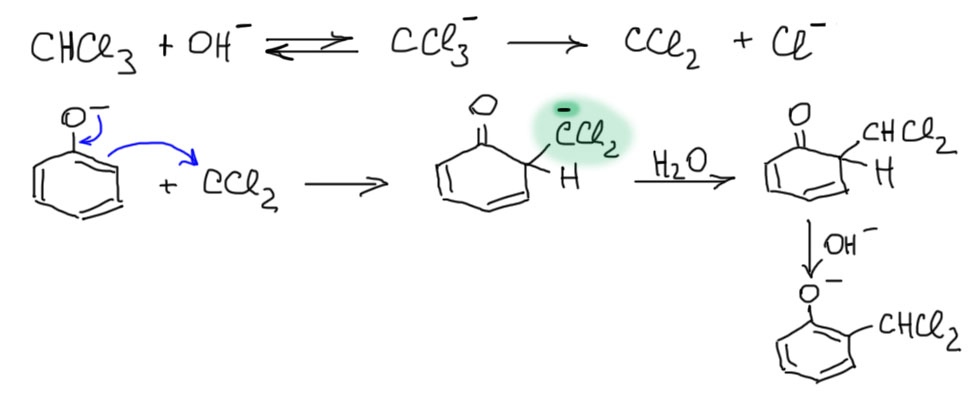
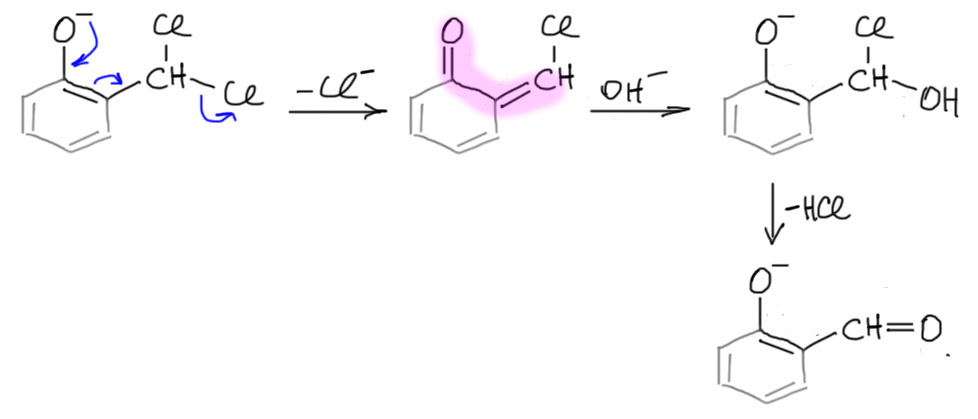
Дальше происходит нечто загадочное. Дихлорметильная группа количественно и быстро гидролизуется в альдегидную, настолько полно и быстро, что даже остатков дихлорметильного интермедиата найти не удается. Что же в этом загадочного? Только то, что обычно такие группы вполне устойчивы и для их гиролиза в альдегиды приходится применять весьма жесткие условия. А здесь все как-то само собой происходит, настолько легко, что даже начинаешь сомневаться в механизме. Но все, видимо, очень просто, как только мы еще раз вспомним про аналогию фенолят-енолят, и глубокое родство химии фенолов с химией карбонильных соединений. Нарисуем этот фенолят еще раз, для удобства выделив одну двойную связь, а все остальное маость притушив. Тогда мы ясно видим фенолят как енолят какого-то кетона, у которого на β-атоме углерода висит пара хлоров. О, да это же нам хорошо знакомо в химии кетонов с сопряженной двойной связью – мы так любим почем зря присоединять HCl или HBr к таким кетонам, чтобы получить “антимарковниковское” присоединение, а на самом деле просто 1,4-присоединение галогенида к сопряженному кетону. Но такое присоединение обратимо, и галогенид очень легко отщепляется с образованием как раз сопряженного кетона, а он, как ему и положено, легко берет другой нуклеофил их реакционной смеси, где у нас как раз полно гидроксида. Все получилось – как только на одном углероде окажется одновременно и хлор и гидроксил, от этого немедленно отвалится HCl и образуется альдегид. Вот почему гидролиз идет так легко – опять кето-енольное превращение помогло.
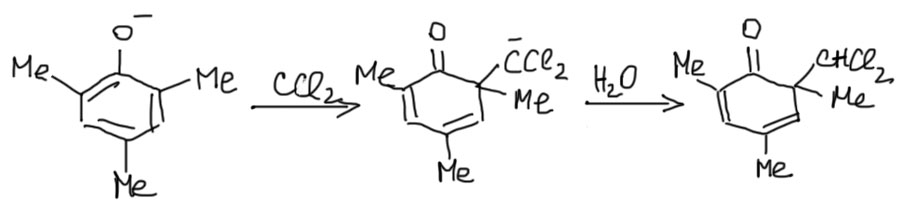
То, что это именно так, хорошо видно по результатам реакции фенолята с заместителями в орто-положениях. В этом случае из реакционной смеси выделяется как раз циклогексадиенон с дихлорметильной группой. Гидролиз не происходит, потому что кето-форма не может превратиться в (ф)енол, и дихлорметильная группа оказывается изолирована от карбонила. Продуктом реакции оказывается неароматическое соединение, циклогексадиенон. Превратиться ему некуда – в отличие от водорода, алкильные группы не умеют переползать на другое место. Как это не умеют – а перегруппировки карбокатионов? Да, но там алкильная группа переползает на соседний атом с 6 валентными электронами и со своими электронами. Здесь нет такой ситуации.
Посмотрим на практические вещи.
- Что нужно для реакции Реймера-Тимана? Фенол в растворе хлороформа, и к нему прикапывают водный раствор NaOH с хорошей концентрацией. Смесь нагревают несколько часов. Потом нейтрализуют и выделяют продукты. Если будете делать, не пугайтесь того, что реакционная смесь выглядит жутковато – все сильно темнеет и превращается в массу странного вида, граница раздела фаз не видна. Так всегда выглядят сильно щелочные водно-органические смеси. Образуется куча свяких побочных непонятных веществ с выраженными поверхностно-активными свойствами.
- Хороша ли реакция? Стоит ли ее использовать? Нет, реакция очень “грязная” – много побочных, осмоления. Выходы целевых продуктов небольшие, редко выше 20%. Но она очень простая и дешевая и для орто-гидроксиальдегидов, а это очень полезные продукты, она дает самый простой и удобный метод синтеза в одну стадию. И еще она очень популярна как метод синтеза вот этих дихорметилциклогексадиенонов, у которых своя, богатая и интересная химия.
- Это метод селективного получения орто-формилфенолов (орто-гидроксиальдегидов), которые часто называют по простейшему представителю ряда салициловыми альдегидами? Там что, происходит что-то подобное на карбоксилирование по Кольбе-Шмитту? Не совсем. Катион натрия может удерживать диоксид углерода, координируясь по кислороду, но в дихлоркарбене ему зацепиться не за что. Да и вода кругом, полностью забивающая все окрестности катиона. Поэтому в реакции всегда получаются и орто- и пара-изомеры приблизительно в одинаковых количествах. Фокус в том, что орто-изомеры легко отгоняются из смеси продуктов с водяным паром (это старинный метод выделения некоторых органических соединений), и оттуда – из отгона – очень просто выделяются в чистом виде. А пара-изомеры остаются в остатке после перегонки, и это нужно, как обычно, выделять утомительной экстракцией, отмывкой, упариванием, перегонкой или даже хроматографией.
C реакцией карбоксилирования – введения карбоксильной группы в молекулу – мы очень хорошо знакомы и подробно разбирали ее в теме Карбоновые кислоты. Проблема здесь простая: диоксид углерода – это очень слабый электрофил. Исключительно интересен вопрос почему, ведь диокид углерода – довольно типичное карбонильное соединение, и на атоме углерода у него висят аж два атома кислорода, что должно, по идее, наводить на углерод нехилый плюс и делать его весьма электрофильным. Наводить наводит, но не делает. И еще более странно – диокисид углерода по структуре мало чем отличается от молекулы кетена, строение собственно карбонильной группы точно такое же, а электрофильность должна быть даже выше, ведь единственное, чем диоксид углерода действительно отличается от кетена это замена второго кислорода метиленовой группой. Кетен – очень активный электрофил, жадно взаимодействует даже со слабыми нуклеофилами в низких концентрациях. А диоксид углерода – электрофил настолько ленивый, что до сих пор нам удавалось его ввести в реакцию только с самыми зверскими нуклеофилами типа литий или магнийорганики. Почему же он такой слабый? Заинтриговал – и брошу. Здесь не будет ответа на этот непростой вопрос, но я постараюсь раскрыть эту проблему позже и в другом месте. Это ведь очень важная проблема. Задача научиться делать из диоксида углерода что-то полезное весьма остро стоит в современной химии, на исследования в этой области тратятся колоссальные средства. Понятно почему – это не просто чрезвычайно дешевый реагент, к которому совершенно буквально применимо определение “дешевле грязи”, но есть даже обоснованная надежда, что за хороший способ использования диоксида углерода еще и доплачивать будут, ведь накопление CO2 в атмосфере – главная причина катастрофического потепления земной атмосферы, и любой способ сделать так, что его там станет меньше, бурно приветствуется.
Выдающаяся реакционная способность фенолятов в реакциях ароматического электрофильного замещения делает именно их способными реагировать с таким немощным электрофилом, как диоксид углерода.
Одна из древнейших реакций диоксида углерода в органической химии, открытая еще в 1860, реакция Кольбе-Шмитта или карбоксилирование по Кольбе-Шмитту эксплуатирует именно это свойство фенолятов. Херманн Кольбе обнаружил эту реакцию в 1860, но в те времена техника эксперимента была еще очень примитивной, и выход продукта был очень мал. И только в конце 19 века, когда Шмитт сделал реакцию под давлением и получил хороший выход продукта. Но и здесь не без проблем – реакция обратима, а так как один из реагентов – газ, то единственный способ погнать реакцию сперед, к продуктам, как учит великий Ле Шателье, – использовать повышенное давление газообразного реагента. Реакция идет с расплавленным фенолятом натрия или еще лучше, с растертым в тонкий порошок сухим фенолятом натрия при высокой температуре и серьезном давлении CO2 до 100 атм. Так как реакция все равно медленная, требуется довольно высокая температура. Обратим внимание на то, что для обратимых реакций что-то плюс газ равно продукт температура, как учит великий Ле Шателье, смещает равновесие влево, в сторону исходных. Но без повышенной температуры не идет реакция. Приходится противодействовать температуре давлением. Поэтому такое высокое давление. Из этого мы понимаем. что это скорее промышленная реакция чем лабораторная. И еще у нее довольно узкий диапазон – замещенные фенолы в ней участвуют плохо или вообще не участвуют. Годятся только простые производные фенола типа метилфенолов (крезолов). А, скажем, дезактивированные фенолы, например, нитрофенолы даже не стоит пытаться карбоксилировать, хотя в экстремально жестких условиях при 260º и давлении 1200 атм (! это не ошибка) небольшой выход нитросалицилата все таки образуется.
Образуется почти исключительно орто-изомер продукта карбоксилирования – натриевой соли салициловой кислоты. С этим собстченно и связана важность этой реакции – салициловая кислота и ее сложный эфир по фенольной группе, О-ацетилсалициловая кислота или аспирин – одни из самых старых, но до сих пор широко применяемых фармацевтических препаратов. C внедрения и начала производства аспирина фактически начинается фармацевтическая промышленность в современном смысле этого слова, а компания, развернувшая это производство, Байер (Bayer) из Леверкузена, до сих пор является одной из крупнейших химических и фармацевтических компаний мира.
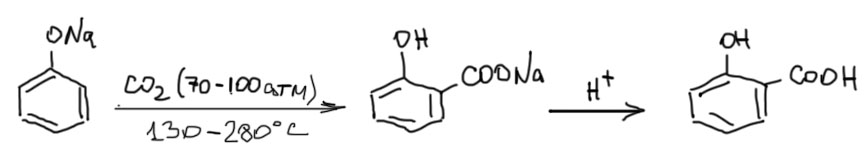
Образование орто-изомера обычно объясняют тем, что катион натрия является хоть и слабой, но вполне явной кислотой Льюиса, образующей слабые координационные связи с кислородами и фенолята и CO2. С литием происходит то же самое, но работать с фенолятом лития гораздо менее удобно. Именно поэтому в реакции Кольбе-Шмитта тщательно исключают воду – вода мешает катиону натрия взаимодействовать с кислородами фенолята и CO2, вытесняя их из окрестностей катиона натрия. Поэтому в этой реакции для образования фенолята нельзя использовать NaOH или Na2CO3 – при этом выделяется вода. В принципе, можно даже вполне обойтись без координационных связей, а просто считать взаимодействия ионными – катион притягивает молекулы реагентов за места с самым отрицательным зарядом, те же кислороды по странному совпадению. А как правильно? Как всегда правда состоит в том, что на такие вопросы нельзя дать однозначного ответа. Да, катионы щелочных металлов, особенно самые маленькие, литий и натрий, способны образовывать координационные связи с хорошими лигандами. И да, эти связи не совсем классические координационные (то есть пара от основания, пустая орбиталь от кислоты), а имеют очень большую степень ионности. Без ионности не обойтись, потому что катион s-элемента натрия, вся валентная оболочка которого состоит из одной s-орбитали, все равно может образовать не более одной настоящей координационной связи, а нам нужно было бы минимум две. И даже если мы исподтишка притащим в валентную оболочку металла пустые p-орбитали, это не очень много даст, так как они все равно пустые и высокие по энергии, и дадут вклад скорее в ионный характер связей, чем в ковалентно-координационный. В общем, если эта казуистика кажется вам пустым словоблудием, просто согласитесь с тем, что мы имеем право писать такие взаимодействия, которые удерживают реагенты рядом в переходном состоянии электрофильной атаки CO2 на орто-положение фенолята.
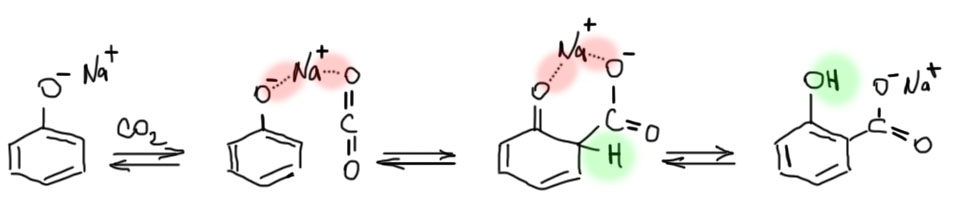
То, что катион натрия действительно выполняет очень важную роль в этой реакции, становится очевидным, когда его заменяют на катион калия. Катионы щелочных металлов часто, а точнее почти всегда считают такими невинными свидетелями большой химии – предполагается, что у каждого элемента Периодической таблицы есть своя, богатая и уникальная химия. Кроме щелочных металлов, которым обычно выделяют роль балласта, неизбежного просто для баланса зарядов в реакциях и более ни для чего не нужного, просто шариков с положительным зарядом. Но это большое заблуждение. Катионы щелочных металлов очень сильно различаются между собой. Катионы лития и натрия имеют вполне определенные свойства кислот Льюиса, образующих настоящие комплексные соединения с разнообразными лигандами, и у каждого есть свои предпочтения в выборе лигандов. Три тяжелых щелочных металла – калий, рубидий и цезий – в принципе, гораздо больше похожи на просто положительно заряженные шарики с увеличивающимся размером. Их поведение ближе похоже на просто положительный заряд, противоион. Они мало с чем взаимодействуют специфически, не образуют связей, похожих на координационные, и удерживаются вблизи отрицательных ионов за счет чистой электростатики. Взаимодействие больших щелочных катионов с незаряженными (хотя и полярными) молекулами совсем слабое, настолько слабое, что этим обычно можно пренебречь.
Так вот, если мы берем фенолят не натрия, а калия (ну и само собой понятно, рубидия или цезия, но какой смысл попусту деньги тратить на эти дорогущие элементы, если калий уже неплохо работает) и запускаем его в реакцию с диокидом углерода, то, во-первых, реакция становится медленнее, и требует еще более жестких условий (температура от 220º), а во-вторых, образуется почти исключительно пара-гидроксибензойная кислота, точнее ее калийная соль. Более того, если взять калийную соль салициловой кислоты и нагревать ее в замкнутом сосуде желательно в атмосфере CO2, выше 200º, то происходит перегруппировка в калийную соль пара-гидроксибензойной кислоты.
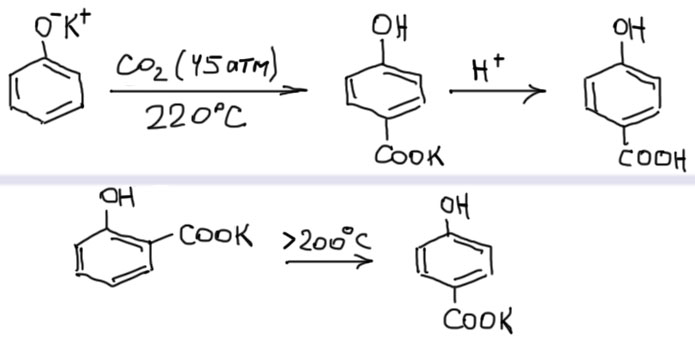
Обратное неверно – сколько бы мы не грели пара-гидроксибензоат натрия, в салицилат натрия он не превратится. Это означает, как обычно в таких ситуациях, что орто-карбоксилирование в присутствии катиона натрия – это кинетически контролируемая реакция, потому что катион натрия, как показано на схеме выше, активирует молекулу CO2 и делает реакцию быстрее, работая фактически как кислотный катализатор карбоксилирования. А карбоксилирование фенолят иона без содействия катиона с образованием пара-гидроксибензоата – термодинамически контролируемая реакция, то есть это результат установившегося равновесия.
Пара-гидроксибензойная кислота также производится в промышленных масштабах в огромных количествах. Ее сложные эфиры под названиями парабены являются одними из самых популярных консервантов (веществ, препятствующих размножению бактерий) в производстве пищевых продуктов и особенно косметики. Можете сами заняться ловлей этих веществ по этикеткам, где их часто обозначают международными E-индексами: в диапазоне E214-E219 сидят метиловый (метилпарабен), этиловый (этилпарабен), пропиловый и бутиловый эфиры и их натриевые соли. Можете и не заниматься, но в последние несколько лет общественность сильно настроилась против парабенов из-за всяких сообщений об их вредности и даже канцерогенности (объективного подтверждения этим страхам нет, но это уже мало на что влияет), и их стали понемногу заменять другими консервантами.
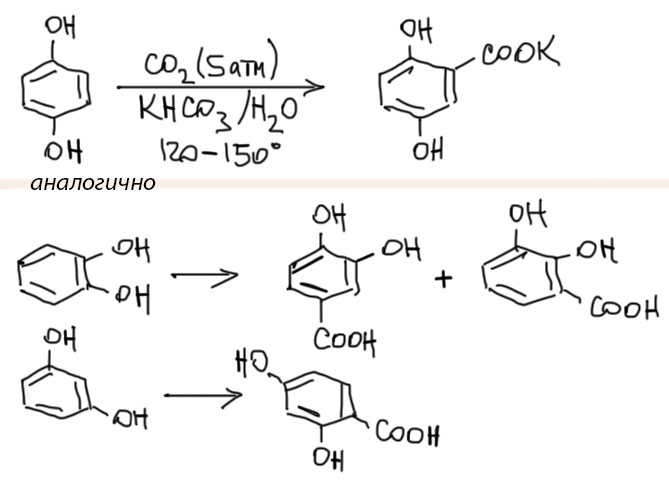
Карбоксилирование дигидрокси- и тригидроксибензолов.
В этих случаях реакция происходит намного легче, и для карбоксилирования сами эти соединения (орто-дигидроксибензол – пирокатехин, мета – резорцин, пара – гидрохинон; 1,3,5-тригидроксибензол – флороглюцин) нагревают в водном растворе бикарбоната калия в замкнутом сосуде в атмосфере CO2, но при небольшом давлении. Кислотность всех этих соединений выше, чем кислотность фенола, поэтому основности бикарбоната вполне хватает для депротонирования, и фактически реакция идет не с анионом бикарбоната, а в растовренным CO2.
Мощнейшая и невероятно гибкая реакция, имеющая колоссальное применение в сложном органическом синтезе, и продолжающая развиваться. Она входит в целое семейство родственных реакций, не менее дюжины из которых имеют имена.
Строго говоря, перегруппировка Кляйзена является частным случаем более общей перегруппировки Коупа, хотя последняя была открыта на 28 лет позже в 1940. Перегруппировка Коупа – это самопроизвольное превращение друг в друга двух молекул, содержащих две двойные связи (которые, как часто бывает в органической химии, могут быть и тройными), разделенные тремя простыми. 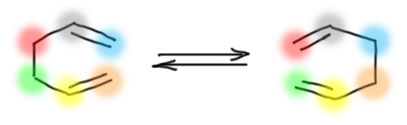 Вот так это обозначим, использовав цвета вместо обозначения элементов, потому что атомы могут быть разными, не обязательно углеродами. На каждом из атомов могут быть заместители, связи могут находиться в циклах, но обязательно так, чтобы вся эта штуковина могла выгнуться так, чтобы крайние атомы могли сблизиться, образовав нечто похожее на 6-членный цикл. В самом общем случае это равновесие, и перегруппировка может происходить как справа налево, так и обратно, что вполне понятно, потому что и слева, и справа система имеет одинаковое расположение связей – двойная–простая-простая-простая–двойная. В реальных случаях слева и справа могут оказаться существенно различные молекулы, одна из которых может быть или намного стабильнее, или быстро превращаться во что-то третье, в этом случае равновесие, как учит великий Ле Шателье, смещается, возможно даже целиком, именно в эту сторону.
Вот так это обозначим, использовав цвета вместо обозначения элементов, потому что атомы могут быть разными, не обязательно углеродами. На каждом из атомов могут быть заместители, связи могут находиться в циклах, но обязательно так, чтобы вся эта штуковина могла выгнуться так, чтобы крайние атомы могли сблизиться, образовав нечто похожее на 6-членный цикл. В самом общем случае это равновесие, и перегруппировка может происходить как справа налево, так и обратно, что вполне понятно, потому что и слева, и справа система имеет одинаковое расположение связей – двойная–простая-простая-простая–двойная. В реальных случаях слева и справа могут оказаться существенно различные молекулы, одна из которых может быть или намного стабильнее, или быстро превращаться во что-то третье, в этом случае равновесие, как учит великий Ле Шателье, смещается, возможно даже целиком, именно в эту сторону.
Перегруппировка Кляйзена – это перегруппировка Коупа в случае, когда слева один из атомов, третьих с конца (красненький или зелененький, возьмем для определенности красненький) – это кислород, а все остальные углероды. 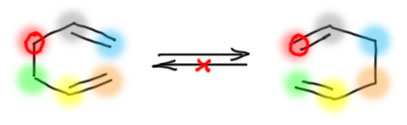 В этом случае слева и справа оказываются совершенно разные молекулы: слева простой эфир (аллилвиниловый), а справа что-то карбонильное, которое может быстро еще куда-нибудь превратиться. Поэтому перегруппировка Кляйзена, в отличие от общей перегруппировки Коупа, как правило, необратимо, и идет слева направо.
В этом случае слева и справа оказываются совершенно разные молекулы: слева простой эфир (аллилвиниловый), а справа что-то карбонильное, которое может быстро еще куда-нибудь превратиться. Поэтому перегруппировка Кляйзена, в отличие от общей перегруппировки Коупа, как правило, необратимо, и идет слева направо.
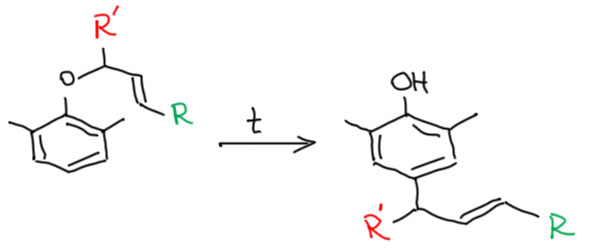
И хотя такая перегруппировка Кляйзена, когда на кислороде висят такие нециклические, неароматические фрагменты, гораздо чаще встречается в реальной органической химии, мы ограничимся более частным случаем перегруппировки Кляйзена, в которой один из фрагментов – бензольное кольцо. Получается перегруппировка аллильного эфира фенола в – сначала, как мы уже привыкли в химии фенолов, в кето-форму, – которая немедленно перегруппировывается в енольную форму – орто-аллилфенол. Обратите внимание – образовалась новая углерод-углеродная связь, а это событие мы уже научились ценить превыше всего на свете, потому что это усложняет структуру, а это – главная цель органического синтеза.
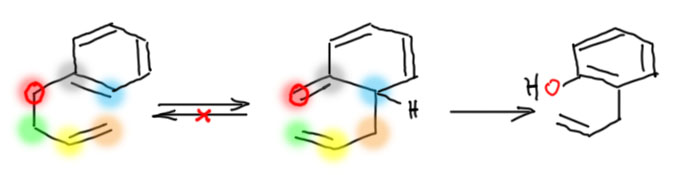
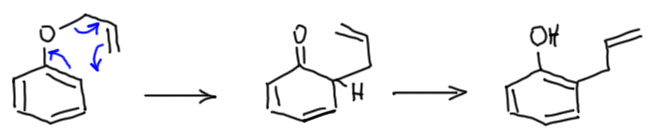
Перевернем теперь для удобства все это хозяйство. Механизм у этой реакции какой? Это нечто очень похожее на то, что мы видели в реакции Дильса-Альдера: механизма как такового у таких реакций нет, просто исходные превращаются в конечные без каких-либо остановок по дороге, то есть это еще один пример согласованных реакций, в которых практически одновременно, но не обязательно прямо совсем-совсем одновременно, но без остановок и задержек рвутся связи и образуются связи, в чем участвуют сразу три пары электронов (каждую, как обычно, изображает кривая стрелочка), всего шесть штук электронов, перераспределяясь в циклическом переходном состоянии.
Обратим внимание на то, что в реакции участвуют не только двойные связи, как в Дильсе-Альдере, но еще и две простые (сигма) связи, одна из них рвется, другая образуется. Такие реакции – какие такие? – во-первых, согласованные; во-вторых, с участием сигма-связей, которые как будто переезжают на другое место – называются сигматропными. Конкретно в перегруппировках Коупа и Кляйзена, рвущаяся и образующаяся связи разделены 3 атомами с каждого из концов – поэтому реакция называется [3,3]-сигматропной перегруппировкой. Еще раз – если мы посмотрим, как устроена система атомов, в которой происходит эта перегруппировка, то мы увидим два трехатомных фрагмента, в которых разрываются-образуются только π-связи, а между этими фрагментами есть в начале одна σ-связь, связывающая условно первые атомы каждого из фрагментов, эта связь в процессе реакции рвется, и возникает новая σ-связь, но уже между третьими атомами фрагментов: связь как будто переезжает с первых атомов на третьи. 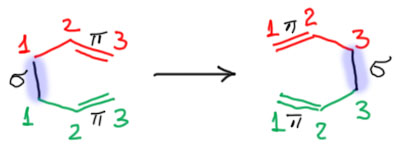 Поэтому и сигма-тропизм, от многозначного греческого корня -троп-, обозначающего изменение, переход, движуху всякую (слов с таким корнем полно, например, тропосфера – часть атмосферы, где происходят всякие климатические процессы, а мы много обсуждали прототропную таутомерию, то есть процесс перемещения протона с дного атома на другой).
Поэтому и сигма-тропизм, от многозначного греческого корня -троп-, обозначающего изменение, переход, движуху всякую (слов с таким корнем полно, например, тропосфера – часть атмосферы, где происходят всякие климатические процессы, а мы много обсуждали прототропную таутомерию, то есть процесс перемещения протона с дного атома на другой).
Видели ли мы в нашем курсе другие сигматропные реакции? Видели, но не обратили внимания. Оставим пока это, когда-нибудь еще вернемся. Кето-енольная таутомерия похожа на сигматропную реакцию, но не является такой, потому что не является согласованной, а требует катализа и включает промежуточные стадии.
Перейдем от общих вещей к конкретным.
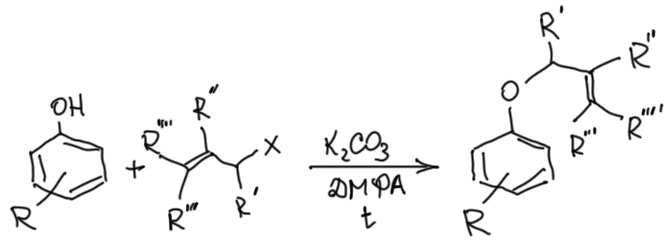
- Что нужно для перегруппировки Кляйзена? Ничего, кроме исходного эфира фенола с группой аллильного типа – связанной с кислородом насыщенным углеродом, и двойной С=С-связью на следующем атоме. И еще, это должно быть способно выгнуться в 6-членный цикл. Остальное на ваше усмотрение, любые заместители и на ароматическом кольце, и на аллиле, и даже еще могут быть двойные связи в других местах молекулы. Заместители – донорные или акцепторные – влияют на скорость реакции, но слишком слабо, чтобы нам это обсуждать. Безусловно, в реальности может помешать, например, стерика, какой-нибудь растопыр на дороге. Но это учесть можно даже на глазок, приблизительно так, как мы это делали в реакции Дильса-Альдера.
- Как вызвать перегруппировку Кляйзена? Просто нагреванием, иногда довольно сильным, до 200-240º в каком-нибудь высококипящем растворителе, или в специальном закрытом сосуде с толстыми стенками, если мы хотим использовать растворитель с меньшей температурой кипения (хоть это и очевидно, все же напомню, что нельзя в открытом на атмосферу сосуде нагреть жидкость выше температуры кипения содержимого, а если это смесь, то выше температуры кипения самой легкокипящей жидкости в смеси). В современной химии для ускорения реакции иногда используют катализ кислотами Льюиса и другими реагентами, но мы не будем этого делать.
- А откуда берутся аллиловые эфиры фенолов для перегруппировки? А вот это совсем просто, потому что феноляты – это нуклеофилы, а галогенпроизводные и тозилаты аллильного типа – превосходные субстраты для SN2, даже если галоген висит на вторичном углероде. А аллильные бромпроизводные легко получаются и аллильным бромированием, и еще кучей всяких реакций, например, селективным восстановлением карбонила продуктов кротоновой конденсации получаем аллильные спирты, которые легко переводим в бромиды или тозилаты. И если у нас есть аллильный бромид или тозилат, то эфир фенола получается совершенно элементарно просто нагреванием фенола и аллильного бромида или тозилата в присутствии основания, как правило просто безводного поташа, в подходящем растворителе типа ацетона, ацетонитрила или ДМФА. В реальной химии это не всегда бывает совсем гладко, потому что есть такое явление, как аллильная перегруппировка при замещении, но мы пренебрежем этими осложнениями и всегда будем считать, что все ОК.
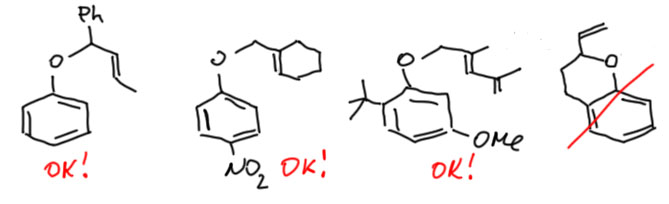
- А не мешают все эти заместители на аллиле? Мешают, конечно, но не так сильно, как во многих других реакциях, чувствительных к стерическим препятствиям. Заместители на конце аллила могли бы мешать, если бы фрагмент, в котором происходит перемещение связей, был плоским – тогда эта рогатина упиралась бы в ароматическое кольцо и не давала бы образоваться новой сигма-связи. Но фрагмент не плоский, а конец аллильной системы наезжает на кольцо фенола немного сверху (или снизу), поэтому эти заместители непосредственно ни во что не упираются. Впрочем, реальных примеров перегруппировок, когда есть оба заместителя на конце (на схеме R”’ и R””), очень мало (я не знаю ни одной, но не могу утверждать, что нет вообще). Но с одним таким заместителем примеров полно.
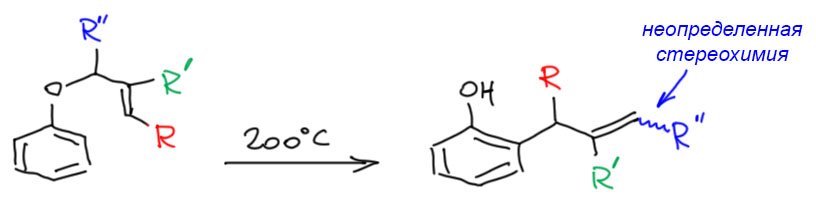
- Есть ли стереохимия у перегруппировки Кляйзена? Есть, и очень затейливая, поэтому эту реакцию так любят в современном синтезе. Но мы не будем с этим разбираться. Если у вас в каком-то примере возникает вопрос, цис- или транс-двойная связь образуется в продукте (это значит, что образуестя дизамещенная двойная связь), рисуйте транс просто потому что температура используется немаленькая, и при таком нагревании цис- все равно изомеризуется в транс. Но если заместителей больше, и двойная связь три- или тетразамещенная, то стереохимия не определена, как в следующем примере.
- Не забываем, что аллил в процессе (это такое сальто прогнувшись) переворачивается и начало становится концом, а конец началом.
- А может ли быть перегруппировка на нескольких аллилфенольных фрагментах? Вполне, и так делают довольно симпатичные синтезы из очень простых исходных. Учтите только, что желательно подбирать исходное так, чтобы у аллилов было только одно место для перемещения, иначе получите смеси.
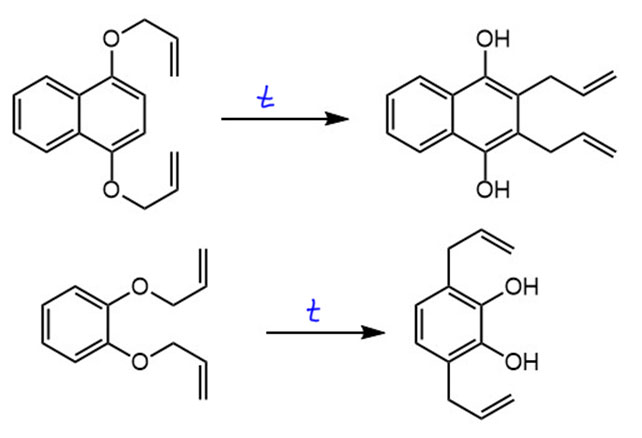
- В фенольной части двойная связь берется из ароматического кольца – а в аллильной части такое может быть? Нет. Так не получается, видимо, потому что в этом случае оба ароматических кольца временно теряли бы ароматичность. Это слишком невыгодно. Нет такой реакции. Попробуйте сами написать, как это могло бы выглядеть и оцените ущерб.
- А если орто-положение в исходном эфире занято? Одно – пожалуйста, реакция пойдет в другое. А если оба? Тогда происходит очень интересная вещь – аллил делает второе сальто и становится в пара-положение. А почему не в соседнее? Потому что именно при перемещении из орто в пара происходит нормальная перегруппировка Коупа, хотя всю эту реакцию – миграцию аллила из аллилового эфира 2,6-дизамещенного фенола в пара-положение чаще называют пара-перегруппировкой Кляйзена, но фактически эта перегруппировка – это последовательность обычной перегруппировки Кляйзена и обычной перегруппировки Коупа. И мы каждый раз видим эту ситуацию со связями – двойная-простая-простая-простая-двойная.
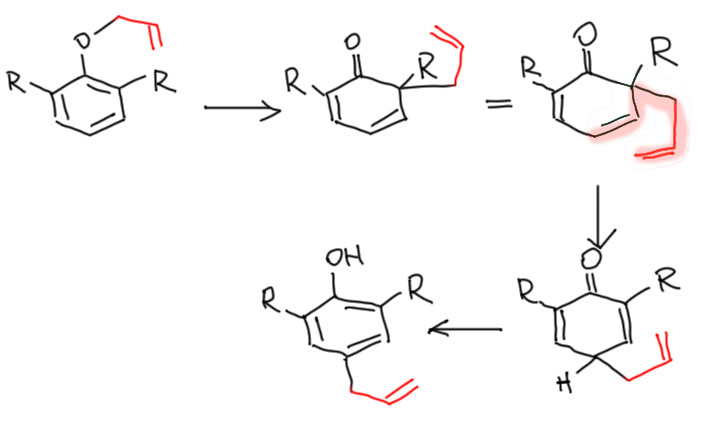
- В пара-Кляйзене аллил делает двойное сальто прогнувшись и встает на место, сохранив тот вид, который он имел в исходном эфире фенола. Этим пара-Кляйзен отличается от обычного Кляйзена.
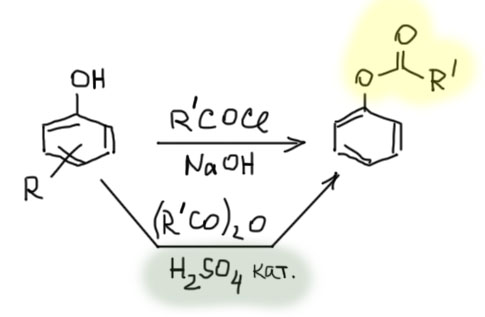
Можно ли фенолы ацилировать? Хотелось бы, потому что ароматические кетоны с фенольными заместителями – очень полезные и нужные соединения.
Но прямо взять фенол и ацилировать его по Фриделю-Крафтсу не получится. Фенолы очень легко и быстро взаимодействуют с обычными ацилирующими агентами – хлорангидридами и ангидридами, образуя сложные эфиры – O-ацилфенолы. Если мы хотим сделать именно это, то лучше действовать аккуратнее. Для хлорангидридов хорош метод Шоттен-Баумана, использующий водную щелочь как основание (почему это работает, а не происходит банальный гидролиз хлорангидрида, можно вспомнить в разделе про Карбоновые кислоты). Для ангидридов предпочитают кислотный катализ – одну-две капельки сильной кислоты типа серной.
Полученные ацилированные фенолы нагревают в присутствии эквимольного количества безводного хлористого алюминия иногда вообще без растворителя, иногда в сильнополярных растворителях типа нитробензола. Сразу замечу, что это очень неудобный в работе растворитель, его трудно удалять после реакции, так как он очень высоко кипит и его невозможно отогнать на роторном испарителе, к тому же он имеет очень сильный запах и высокую токсичность; а если он еще и плохо очищен и содержит м-динитробенозол, то работа с ним становится протсо опасна – и при этом свойства нитробензола чрезвычайно хорошо подходят для реакций с участием безводного хлорида алюминия и часто его нечем заменить.
После реакции смесь как обычно обрабатывают льдом и большим количеством соляной кислоты до полного растворения солей алюминия. И выделяют ацилированные фенолы, при этом преобладают орто-изомеры. Гидрокси-группа – сильнейший орто/пара-ориентант в реакциях электрофильного замещения, но то, что в этой реакции получается преимущественно орто-изомер, наталкивает на мысль, что это действительно перегруппировка, происходящая внутримолекулярно. Увы, это вряд ли так. Вполне очевидно, что кислота Льюиса садится на атом кислорода эфира фенола (может сесть на любой из двух, рисуют и так и так, но это не принципиально; скорее всего оба комплекса находятся в равновесии). Карбонильный кислород при этом становится более электрофильным. Но вот что дальше? Внутримолекулярная атака этого углерода на орто-положение вряд ли возможна, потому что там было бы 4-членное переходное состояние, а это невыгодно. Иногда рисуют диссоциацию комплекса с образованием ацильного катиона, сильнейшего электрофила, который немедленно атакует ближайшее положение ароматического кольца. Это такое химическое выражение принципа: “Где родился, там и пригодился”. А именно, если в каком-то процессе образуется высокореакционоспособная частица, то она с большей вероятностью вступит в реакцию как можно ближе к тому месту, где она образовалась. И вот как раз если реакцию вести не просто так, а в растворе нитробензола, продукт пара-ацилирования тоже образуется, и иногда даже становится основным. Дело в том, что нитробензол очень полярен, и содержит нитро-группу, в которой есть два атома кислорода с значительным отрицательным зарядом. В таком растворителе ацильный катион может иметь большее время жизни, и имеет больше шансов “доплыть” до пара-положения. Я все же нарисовал более тривиальный механизм, похожий на обычный механизм ацилирования Фриделя-Крафтса, но с учетом специфики фенола, σ-комплексы которого фактически являются нейтральной молекулой циклогексадиенона, то есть кето-формой собственно фенола.
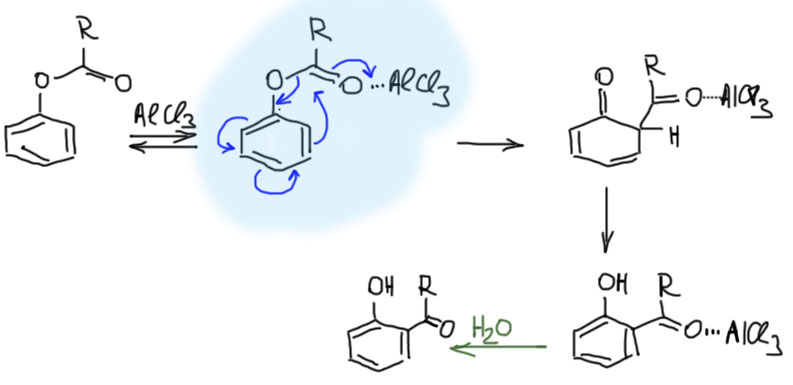
Итак, механизм реакции малость туманен, но сама реакция очень полезна.
Реакции хинонов
Довольно популярное верование состоит в том, что пара-хиноны реагируют с реактивами Гриньяра так же, как и с другими нуклеофилами (см. вкладку ниже) по схеме 1,4-присоединения, что должно давать 2-замещенные гидрохиноны.
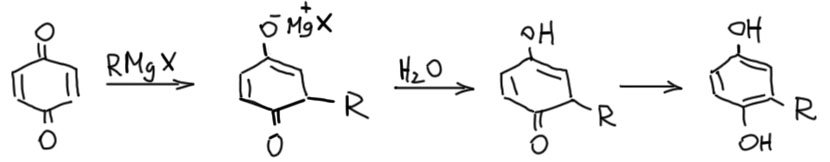
Это верование восходит к работам начала 20 века, не нашедшим подтверждения в последующих исследованиях. В реальности реакция гриньяров с хинонами дает смеси продуктов 1,2- и 1,4-присоединения, и еще много побочных продуктов. Сосбтвенно, в этом нет никакого сюрприза – вспомним, что и в реакциях с обычными ненасыщенными кетонами (енонами) гриньяры дают кашу и не рекомендованы. Там рекомендованы литийорганические соединения для чистого 1,2-присоединения. С хинонами происходит то же самое. Получающиеся продукты обычно называют хинолами. Нам они не очень нужны, но все же для полноты картины напишем:
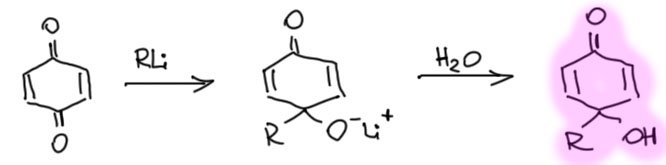
В этом месте многие наверняка вспомнят купраты, которые успешно решили эту проблему для обычных сопряженных ненасыщенных кетонов, но здесь им ничего не светит, потому что хиноны их просто окисляют без какого-либо полезного результата.
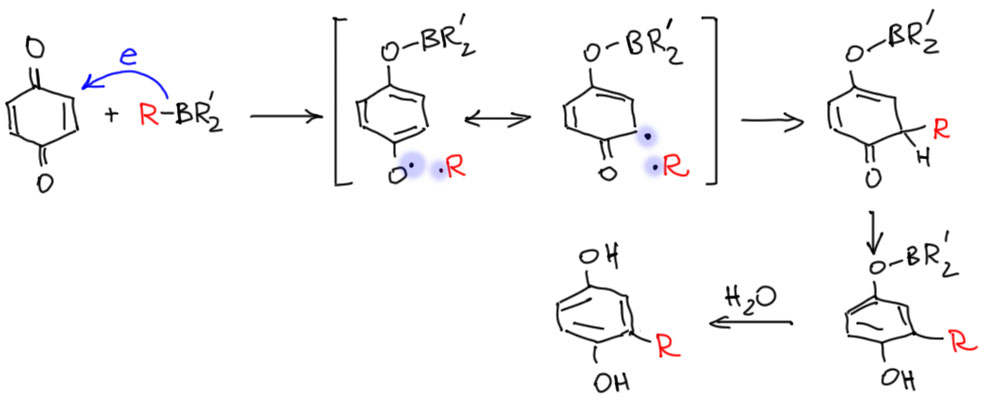
Если нам действительно нужно получить то, что мы ожидаем от присоединения гриньяров, есть вполне проверенная и надежная реакция с боранами, например, продуктами гидроборирования. По механизму это не нуклеофильная, а свободнорадикальняа реакция – хинон окисляет боран, он разваливается на радикал R• и борный остаток, который садится на кислород, и происходит рекомбинация радикалов по месту наибольшей спиновой плотности. Кстати, бораны точно так же реагируют с обычными енонами.
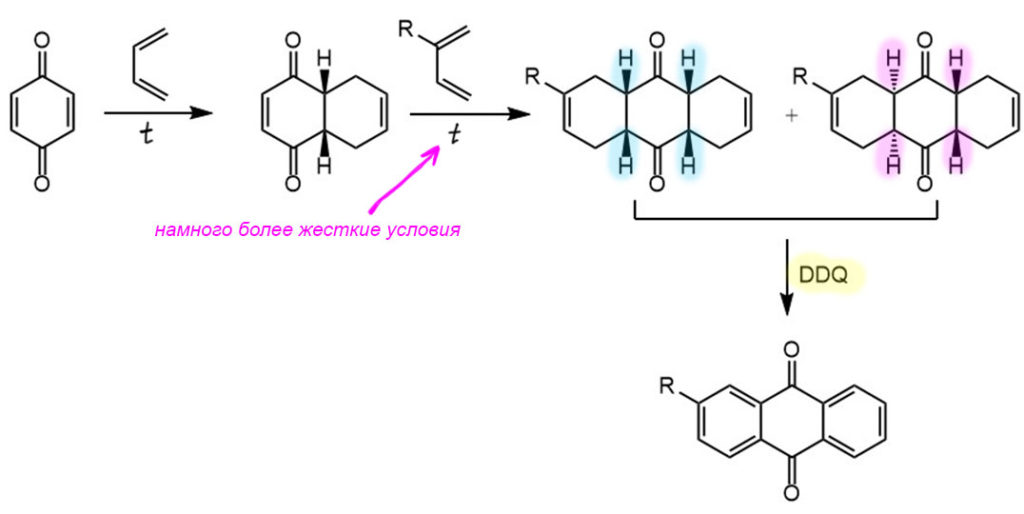
Пара-бензохинон и его производные, и 1,4-нафтохинон тоже – отличные диенофилы в реакции Дильса-Альдера. Очень много интересных синтезов делается с помощью этой реакции.
Сам бензохинон очень хорошо реагирует с одной молекулой диена. Образующийся аддукт вполне стабилен, может быть выделен и использован, – это непредельный дикетон, с которым можно сделать много разных реакций. Но чаще его подвергают кето-енольному превращению. Само собой это не происходит, нужен кислотный катализ, и чаще всего его просто нагревают с несколькими каплями сильной кислоты, хотя бы соляной.
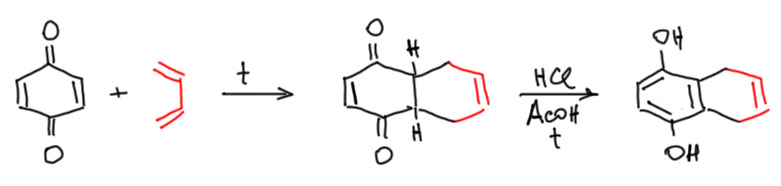
Это очень интересный случай. Мы, пожалуй, впервые сталкиваемся с ситуацией, когда и кето-форма, и енольная форма совершенно устойчивы и могут жить неопределенно долго, если не озаботиться катализом. При этом енольная форма намного устойчивее (она ароматическая) и поэтому превращение идет в одну сторону (точнее, обратимая реакция имеет очень большую константу равновесия), и обратно в кето-форму перейти нельзя. Контанта равновесия велика, а вот константа скорости превращения кето-форма в енол без катализа очень мала.
При желании можно присоединить вторую молекулу диена, но реакционная способность моно-аддукта намного меньше, чем у исходного хинона, и реакция требует длительного нагревания. Из этого, в частности, следует, что диен для второго присоединения можно заменить на другой. Конечный дикетон уже не может дать ароматический енол, и поэтому ни во что не превращается. Есть, впрочем, одна засада. У аддукта сочленение с новым кольцом имеет цис-конфигурацию, и поэтому у бис-аддукта сочленения могут быть в одну сторону и в разные стороны, но это разные молекулы – разные диастереомеры. И если получается смесь, а она получается, то их нужно делить, а это непросто. Но реакцию двольно часто используют для синтеза антраценов и антрахинонов, и тогда эти кольца просто ароматизуют (про DDQ и ароматизацию вспомните в методах в теме про ароматичность), и не заморачиваются с разделением диастереомеров.
Изменения реакционных центров
Электрофильное замещение в фенолах
Сопряженное присоединение к 1,4-хинонам
Это очень важная и одновременно очень капризная реакция. Общая схема состоит в том, что происходит обычное для ненасыщенных кетонов (енонов) 1,4-присоединение, при этом образуется циклогексендионы, а это кето-форма (енолизуемый кетон по обоим карбонилам), очень легко превращающаяся в енольную форму, имеющую ароматическую структуру. Эта енольная форма – пара-дигидроксибензол, который принято называть тривиальным именем гидрохинон. Итак, первичный продукт реакции с нуклеофилами– замещенный гидрохинон.
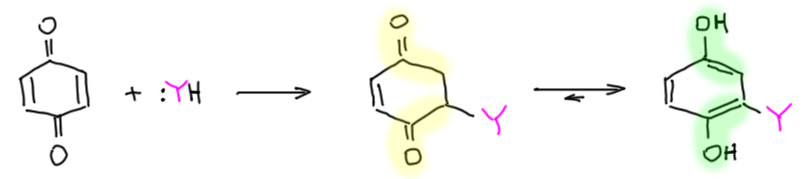
Именно так присоединяются к хинонам амины и многие другие азотсодержащие нуклеофилы, а также галогенид-ионы в галогенводородных кислотах.
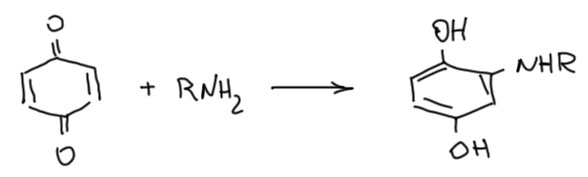
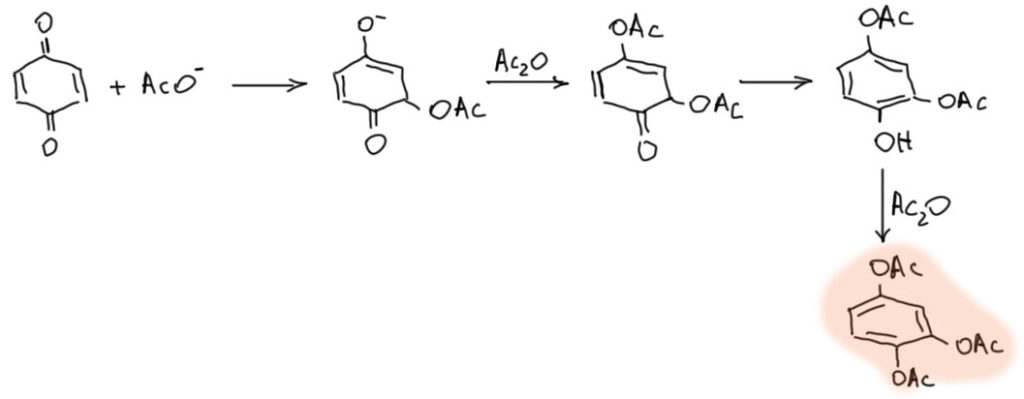
Очень популярный вариант этой реакции, имеющий даже имя – реакция Тиле – взаимодействие с солью карбоновой кислоты в присутствии ее ангидрида. При этом происходит не только присоединение нуклеофила, но и ацилирование гидрохинона и получается триэфир 1,2,4-тригидроксибензола. Особенно часто реакцию делают с уксусным ангидридом (запрещен в Российской федерации).
К сожалению или к счастью, реакция часто идет не совсем так. Проблема в том, что образующийся гидрохинон может окисляться исходным хиноном в замещенный хинон (обратите, кстати, внимание, как записывают реакцию вспомогалельного реагента, если хотят указать не только его самого, но и во что он превращается – такой пересекающейся реакцией с касательной стрелкой) и это часто происходит, потому что реакция с нуклеофилом довольно медленная, и исходный хинон расходуетсся небыстро, и очень долго остается в смеси с образующимся продуктом. Так вот, поскольку нуклеофил еще не израсходовался, то он присоединяется к монозамещенному хинону с образованием дизамещенного гидрохинона. Понятно, что выход продукта в этих случаях, считая на введенный в реакцию хинон, не может быть больше 50%. Интересно, что второе присоединение можем дать 4 разных продукта, но по разным причинам, в том числе стерическим, как правило в основном получается 2,5-дизамещенный. Так присоединяются спирты, фенолы, сернистые нуклеофилы, и много что еще, так что можно считать, что именно такой путь является самым распространенным. В реальности, во многих случаях получаются смеси моно- и дизамещения.
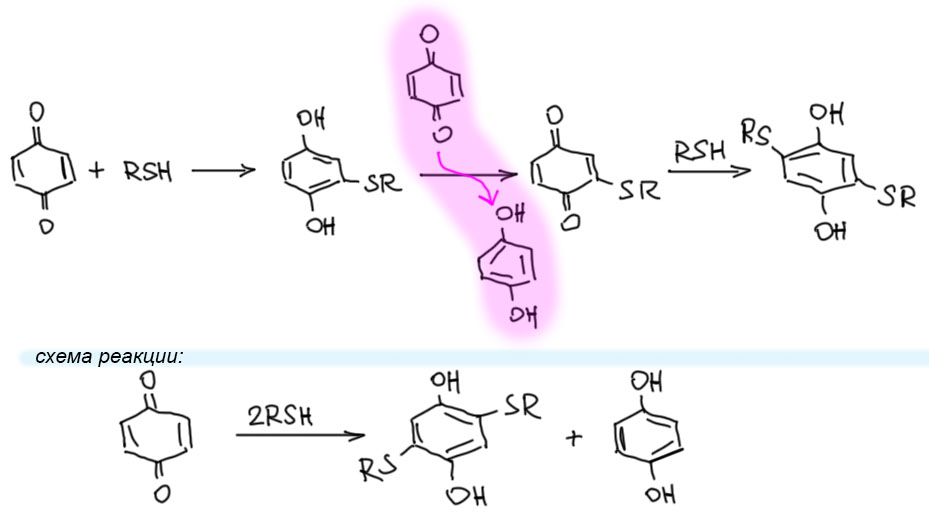
Хиноны из …
Окислять фенолы в хиноны, как ни странно, намного сложнее, чем многие другие исходные, даже анилины. Фенолы очень легко окисляются, и первое, что с ними происходит – образование ароксильных радикалов, которые хорошо делокализованы по кольцу и поэтому достаточно устойчивы, чтобы образовываться в достаточно больших количествах для того, чтобы стала возможной бимолекулярная реакция сдваивания. Вот они и сдваиваются, продукт снова окисляется, и все это бесконтрольное спаривание приводит к образованию олигомеров и осмолению. Большинство окислителей к этому и приводит. При очень тщательном подборе окислителя можно остановиться на сдваивании и образовании димеров – это очень важная реакция, но не в разделе про хиноны.
Чтобы получить хиноны, нужно умудриться остановиться раньше, чем реакция уйдет слишком далеко, и научиться эффективно перехватывать ароксильные радикалы раньше, чем они успеют накопиться для встречи друг с другом. Перехватывать радикал лучше всего другим радикалом – следовательно нужен радикал, концентрация которого в реакционной смеси достаточно высока, чтобы вероятность встречи этого радикала с возникающим ароксильным радикалом в очень низкой концентрации была велика. Следовательно, нужен стабильный радикал. Сразу приходит в голову просто кислород, но он дает гидроперекиси, которые дальше превращаются не в хиноны (есть одна очень интересная реакция, которую в промышленности используют для получения перекиси водорода, основанная на такой реакции одного своеобразного фенола с кислородом, но об этом где-нибудь отдельно).
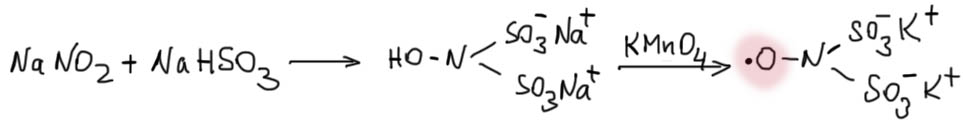
В общем, проблема. К счастью, есть одно соединение, которое решает эту проблему, стабильный свободный радикал с радикальным центром как раз на кислороде. Это неорганическая соль, нитрозодисульфонат калия, очень легко получаемая из нитрита и бисульфита с последующим окислением. Поскольку это очень необычное соединение, неудивительно, что оно носит имя открывателя – его открыл французский химик Эдмон Фреми в очень далеком 1845 году, то есть тогда, когда даже мысли никакой не могло никому в голову прийти, как она в действительности устроена, поскольку ни о каких электронах и свободных спинах было неизвестно ровным счетом ничего. Просто поняли, что соль какая-то странная, сделанная из самых банальных элементов, азота, серы, кислорода, калия, и при этом интенсивно окрашенная, и тоже как-то странно – кристаллы оранжевые, а водный раствор фиолетовый – и очень легко вступающая в разные реакции. Вот Фреми это чудо открыл, пусть и отвечает своим именем в веках. Если кто-нибудь справделиво возмутится, как так получилось, что из натриевой соли получилась в конце калиевая, в этом нет ничего удивительного, калий взялся из перманганата, а количественная замена натрия на калий происходит потому что соли калия с крупными анионами в воде почти всегда растворимы хуже солей натрия, и из растворов выкристаллизовываются первыми.
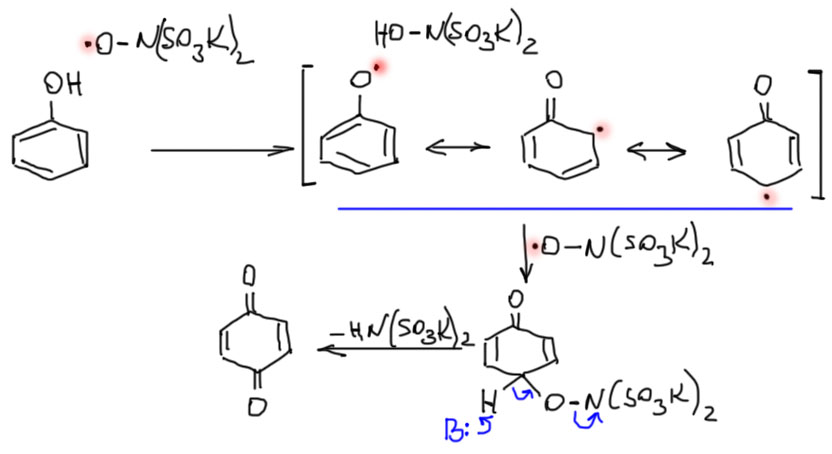
Соль Фреми окисляет фенолы в хиноны (это называется реакцией Тойбера) по очень простому и очевидному механизму. Первый эквивалент отрывает атом водорода от гидроксила, образуется ароксильный радикал, в котором неспаренный электрон делокализован по орто- и пара-положениям. Второй эквивалент рекомбинирует с радикальным центром в ароксильном радикале, c образованием типичного для химии фенолов циклогексадиенона с своеобразным заместителем, произошедшим из соли Фреми. Дальше происходит нечто, приводящее к уходу иминодисульфоната, вполне приличной уходящей группы из-за того, что нуклеофильность азота в ней подавлена акцепторным эффектом двух сульфонатов. Скорее всего, это просто хорошо нам знакомое β-элиминирование под действием основания, которое снимает протон, с одновременным уходом уходящей группы. Образуется хинон, второй кислород которого пришел из соли Фреми (это было подтверждено изотопной меткой). На схеме показан только пара-хинон, образование орто-хинона происходит точно так же.
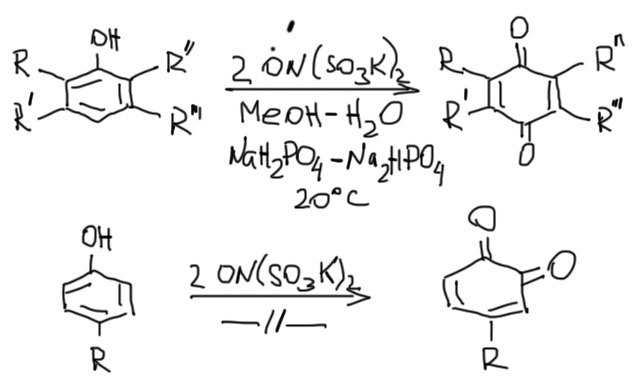
Реакцию с фенолами (реакцию Тойбера) проводят в смеси вода-метанол, вместо чистой воды используют фосфатный буфер, чтобы не допустить закисления реакционной смеси – соль Фреми этого не любит. К тому же, скорее всего для последней стадии, β-элиминирования, нужна некоторая концентрация основания, хотя бы гидроксид-иона, и ее и обеспечивает буфер, поддерживающий приблизительно нейтральную среду. Если пара-положение свободно или занято чем-нибудь легко уходящим, то окисление дает пара-хиноны, а если занято, то в орто-хиноны. В ряду нафтолов α-нафтол дает 1,4-нафтохинон, а β-нафтол – смесь 1,4 и 1,2-нафтохинонов.
Анилины вроде бы дальше от хинонов, но окислять их в хиноны легче, чем фенолы. Причина этого проста – почти любой окислитель берет анилин за неподеленную пару на азоте, а не за атом водорода, как у фенолов. Поэтому с самого начала взаимоотношений анилинов с окислителями возникает не радикал, склонный к сдваиванию, а положительно заряженный и делокализованный в кольцо интермедиат, которому проще схватить нуклеофил из реакционной смеси, например ту же воду. И тогда мы оказываемся сразу очень близко от конечного хинона. В реальности, конечно, все не так изящно. Анилины не любят окислителей, и от малейшего окислителя немедленно окисляются, все страшно темнеет и мутнеет, и приобретает вид мерзкой жижи, годной только для помойки. Но если это перегнать с паром, то из жижи замечательно отгоняются хиноны, обычно в виде желтых кристаллов, плавающих в воде, то есть они еще и перекристаллизовываются из горячей воды, когда пар с примесью хинона конденсируется. Хиноны вообще, как правило, очень легко перегоняются с водяным паром. Отфильтровал, посушил, и готово. Выходы не бывают очень велики, но это очень просто и дешево, поэтому анилины часто используют для получения несложных хинонов. Чаще всего окисляют обычной хромовой смесью. Орто-хиноны так не получают, но пара-хиноны – пожалуйста. Поэтому пара-положение в анилине должно быть свободно.
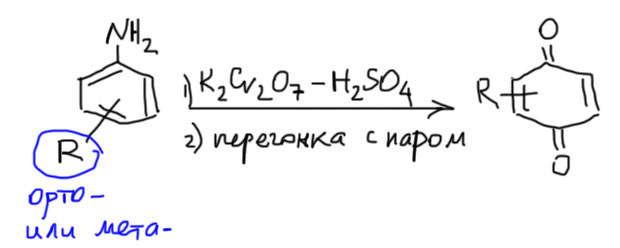
Гидрохиноны очень легко окисляются в пара-хиноны, ровно как и хиноны очень легко восстанавливаются в гидрохиноны. И то, и другое очень легко сделать практически. Если у вас есть гидрохинон, то он окисляется в хинон таким слабым окислителем, как хлорид железа(III). А обратно, из хинона в гидрохинон – раствором бисульфита натрия.
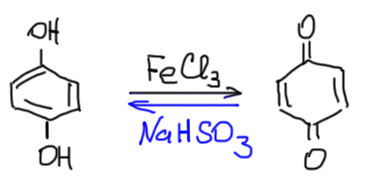
Если немного выйти за пределы органической химии, в смысле получения одних органических соединений из других, то здесь много интересного. Легкость преращения хинона и гидрохинона друг в друга ясно видна из того, что хинон и соответствующий гидрохинон образуют обратимую пару окислитель-восстановитель (редокс-пару):

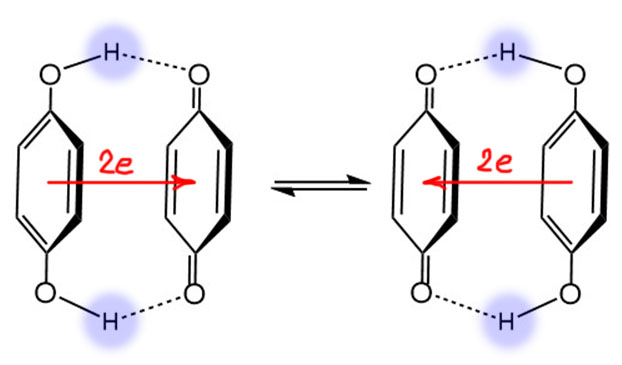
Эта пара настолько хорошо работает, что ее используют не только в органической химии, но и в прикладной электрохимии – потенциал платиновой проволочки, находящейся в растворе, где есть находящиеся в равновесии хинон и гидрохинон зависит, как ясно следует из равновесия, от концентрации ионов водорода. Такой электрод очень долго использовали в pH-метрах для измерения pH. Оставим электрохимию в стороне, но для нас интересно вещество, которое обеспечивает гарантированное соотношение хинон-гидрохинон в растворе. Это хингидрон – 1:1 комплекс хинона и гидрохинона. Это совершенно поразительная вещь, хочется сказать, молекула, но это не молекула, а молекулярный комплекс, комплекс двух полноценных молекул. Фокус этого комплеска в том, что это с одной стороны, донорно-акцепторный комплекс – хинон в нем акцептор, а гидрохинон – донор. Ароматическая и неароматическая молекулы связаны в комплекс кольцо-над-кольцом. Но связь двух молекул обеспечивается не донорно-акцепторным взаимодействием, а водородными связями. Красиво, – безусловно, но не только в этом дело. Попробуйте теперь мысленно в донорно-акцепторной паре перенести два электрона с гидрохинона на хинон. Это создало бы заряды на каждой из молекул, но зарядов не будет, так как вслед за переносом электронов (вслед, а не одновременно, протоны тяжелее и медленнее электронов) в том же направлении смещаются два протона – плюсы следуют за минусами. Хинон становится гидрохиноном, а гидрохинон хиноном. Как это работает в реальности (например, озадачившись вопросом, а что будет, если мы сдлеаем комплекс из изотопно-меченого хинона и немеченого гидрохинона – сможем ли мы разобрать, где будет метка) мы рассматривать не будем – кому станет любопытно и так найдет.
Бензол и замещенные бензолы в хиноны не окисляются, так как при это пришлось бы полностью пожертвовать ароматической стабилизацией. Но конденсированные углеводороды, начиная уже с нафталина, окисляются в соответствующие хиноны очень легко, хотя и достаточно сильными окислителями. Обычно берут шестивалентный хром, например, хромовый ангидрид в уксусной кислоте. При таких окислениях ароматичность не пропадает, а уползает в остающиеся бензольные кольца, где ей хорошо и уютно. Почему это так происходит, мы уже обсуждали в теме Ароматические соединения, где есть вкладка про специфику конденсированных углеводородов.
Нафталин окисляется в нафтохинон при комнатной температуре. Для нас это особенно важно, так как этот хинон действительно хинон, и его можно использовать в реакциях сопряженного присоединения и Дильса-Альдера, получая много интересных продуктов. 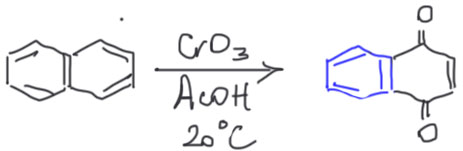 Антрацен очень легко окисляется в 9,10-антрахинон, получая даже больше ароматичности чем было, потому что два настоящих бензольных кольца это лучше, чем размазанные на три кольца 14 электронов. С антрахиноном мы почти ничего дальше сделать не сможем, потому что это с точки зрения реакций просто дезактивированное производное бензола, а в реальности дело обстоит еще хуже – антрахинон даже бромировать нормально не получается, настолько онинертен в реакциях электрофильного характера.
Антрацен очень легко окисляется в 9,10-антрахинон, получая даже больше ароматичности чем было, потому что два настоящих бензольных кольца это лучше, чем размазанные на три кольца 14 электронов. С антрахиноном мы почти ничего дальше сделать не сможем, потому что это с точки зрения реакций просто дезактивированное производное бензола, а в реальности дело обстоит еще хуже – антрахинон даже бромировать нормально не получается, настолько онинертен в реакциях электрофильного характера.
У антрахинона, как у всякого бензола с акцепторными заместителями (здесь их два на каждое кольцо, и это не шутка) конечно, есть нуклеофильная химия, но она очень необычная, и разобраться в ней непросто. Среди производных антрахинона много очень красивых и стойких красителей, поэтому эта химия очень хорошо проработана, но в промышленных реакциях, а они довольно сильно отличаются от того, что мы привыкли делать в колбах. Например, антрахинон при сплавлении (сплавлении!) с щелочью на воздухе (для этого потребуется железная сковорода, так как стекло с расплавленой щелочью отлично реагирует, просто растворяется в расплаве) дает ни много ни мало 1,2-дигидроксиантрахинон, хорошо известный как карминово-красный краситель ализарин. Эта странная реакция – просто нуклеофильное замещение водорода гидроксид-ионом, причем выделяется водород, а это опасно и приводит к частичному восстановлению исходного антрахинона, поэтому в расплав добавляют окислители типа селитры или хлората натрия – тогда водород не выделяется и улучшается выход ализарина. Когда мы обсуждали нуклеофильное замещение в ароматическом ряду, мы никогда не замещали водород, всегда галоген, именно потому что из σ-комплексов нуклеофильного замещения при атаке нуклеофила на незамещенный атом нужно удалять гидрид-ион, а это само-собой не происходит, гидрид не умеет уходить, в каком-то смысле это самая плохая уходящая группа в нуклеофильном замещении – вообще не уходит ни при каких условиях. Но этот комплекс можно окислить, и тогда вместо гидрида уйдет или атом водорода, или протон, а это уже возможно. В ряду производных бензола создать такие условия можно, но непросто. А в ряду антрахинона, исключительно стабильного и стойкого к действию кислот, электрофилов и окислителей, можно просто не церемониться и создать адски жесткие условия – ну вот типа сплавления со щелочью и селитрой. 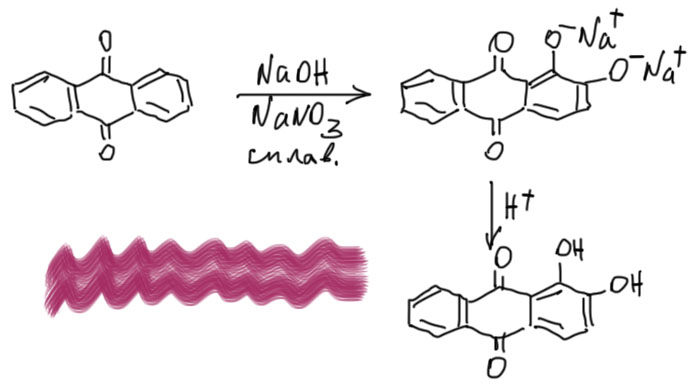
Ализарин – очень красивый краситель, известный с древности, потому что он содержится в одном растении (марена красильная), и его оттуда выделяли сплоть до конца 19 века, когда появилась промышленная химия. Из-за наличия двух гидроксильных групп он еще легко образует комплексы со всякими металлами типа магния или алюминия. Если вы когда-нибудь баловались любыми красками, акварелью, акрилом или маслом, наверняка знаете вишневую краску крапп-лак, вот это и есть комплекс ализарина с кальцием и алюминием.
Разные вспомогательные методы
Пока не из чего
Теорема о трех нитроанилинах, фенолы из аминов
1. Получите из анилина 5-фторсалициловую кислоту (в салициловой кислоте нумерация идет от карбоксила).
2. Получите из анилина 3-иодсалициловый альдегид
Реакции фенолов и простых эфиров фенолов
Получите из фенола 3-метил-4-метоксибензальдегид (в этой задаче была ошибка – раньше был 4-метил-5-метоксибензальдегид)
Дильс-Альдер с хинонами
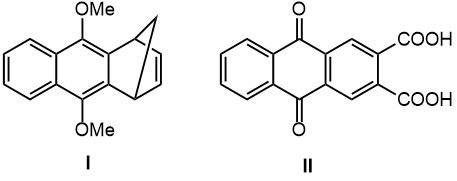
1. Из циклопентадиена и нафталина получите соединение I.
2. Из пара-хинона, бута-1,3-диена и 2,3-диметилбута-1,3-диена получите 9,10-антрахинон-2,3-дикарбоновую кислоту II.
Дильс-Альдер и присоединение к хинонам
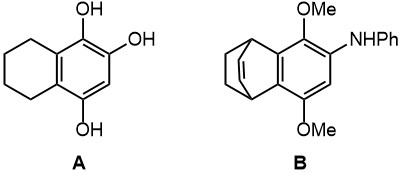
1. Из анилина и бутадиена получите трехатомный фенол A.
2. Из фенола, анилина и 1,3-циклогексадиена получите амин B.
Перегруппировка Кляйзена
1. Из п-толуидина и циклогексена получите 2-(циклогекс-2-енил)-4-метилфенол
2. Из бензальдегида, малоновой кислоты и фенола получите 2-(1-фенилпроп-2-енил)-фенол.
3. Из о-толуидина и ацетальдегида получите 2-втор-бутил-4-н-бутил-6-метилфенол.
Все вместе: фенолы и хиноны
1. Из пара-хинона, 2,3-диметилбутадиена и аллилового спирта получите 1,4-диметокси-2,3-диаллил-6,7-диметилнафталин