Алкины
Главная идея – алкины это не алкены. Это очень глубокая идея, которой нужно пропитаться. При общем поверхностном сходстве химия алкинов и химия алкенов очень сильно различаются. Тройная связь вообще очень необычная и странная штука. Если вам так не кажется, попробуйте представить ее форму. Вот двойная связь – это очень просто, – есть шесть атомов в плоскости, и над и под двумя центральными такие облака электронной плотности. Понятно, где у двойной связи бока, перед, зад, верх, низ; понятно, как к ней подбираются электрофилы и другие реагенты. А вот как выглядит тройная связь? Вроде есть такая ниточка из четырех атомов, и над, и под, и справа, и слева от двух центральных углеродов такие четыре облачка плотности, такие же как в олефинах, только четыре, а не два? Так это рисуют. Осталось понять, а где у ниточки верх и низ – она же со всех сторон одинаковая…
Как алкены называют олефинами, так и алкины еще скопом называют ацетиленами, то есть если увидите слово ацетилен во множественном числе, речь не идет о нескольких молекулах самого ацетилена C2H2, а о собирательном названии для любых соединений с тройными связями, в том числе и таких, которые имеют заместители любой сложности.
Обновления
6.01.2021 Добавлено про конкуренцию двойной и тройной связи в гидроборировании
Программа по разделу
1. Электронная природа тройной связи С≡С. Гибридизация атома углерода. Строение ацетилена (длины связей, углы).
2. ХИМИЧЕСКИЕ СВОЙСТВА. Гидрирование алкинов. Катализатор Линдлара и Р-2-Ni. Восстановление интернальных алкинов натрием в жидком аммиаке.
3. С-Н Кислотность алкинов. Ацетилениды натрия, лития и меди. Получение и использование в органическом синтезе. Особая роль ацетиленидов меди в превращениях производных ацетиленов. Кросс-сочетание ароматических иодпроизводных с ацетиленидами меди (некаталитическое – реакция Стефенса-Кастро, каталитическое – реакция Соногасиры, см. ниже).
4. Электрофильное присоединение к алкинам. Сравнение реакционной способности алкенов и алкинов. Общее представление о механизме реакций. Галогенирование и гидрогалогенирование алкинов.
5. Гидратация алкинов (реакция Кучерова).
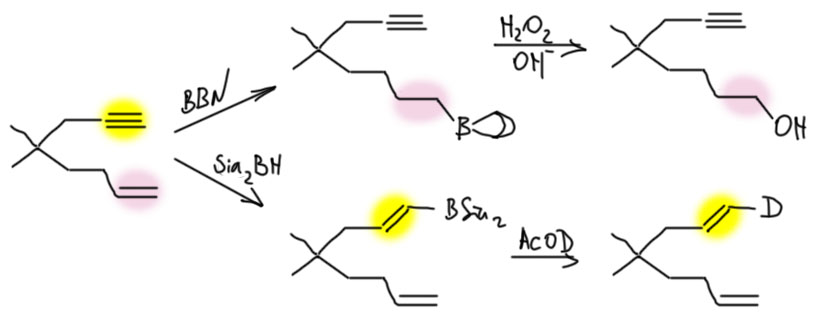
6. Регио- и стереоселективное присоединение гидридов бора. Гидроалюминирование алкинов с ДИБАЛ-Н. Селективное введение изотопов водорода как демонстрация возможностей метода. Региоспецифические гидроборирующие агенты (дисиамилборан, тексилборан, 9-ББН). Превращение борорганических производных в алкены, карбонильные соединения.
7. Ацетилен-алленовая изомеризация. Смещение тройной связи в терминальное положение (zipper-реакция). Реагенты для смещения тройной связи в терминальное положение.
8. Окисление алкинов с разрывом тройной связи.
9. Окислительная димеризация терминальных алкинов в присутствии соединений меди.
10. Нуклеофильное присоединение к алкинам. Почему алкины легче вступают в реакции нуклеофильного присоединения чем алкены. Синтез виниловых эфиров.
11. Три- и тетрамеризация ацетилена на Ni-катализаторах.
СИНТЕЗ АЛКИНОВ.
1. РЕАКЦИИ ЭЛИМИНИРОВАНИЯ.
а) дегидрогалогенирование вицинальных дигалогенидов под действием сильных оснований.
2. АЛКИЛИРОВАНИЕ АЦЕТИЛЕНИДОВ МЕТАЛЛОВ. (Только для первичных RHal. Плохо для втор., нельзя для трет. – закономерности определяются SN2-механизмом реакции, см. ниже)
3. СИНТЕЗЫ С ИСПОЛЬЗОВАНИЕМ СОЕДИНЕНИЙ Cu(I) КАК КАТАЛИЗАТОРОВ ИЛИ РЕАГЕНТОВ . (Реакции а) димеризации, б) окислительного сдваивания, в) Стефенса-Кастро, г) Соногасиры (Cu-Pd-катализ.)
4. АЦЕТИЛЕН-АЛЛЕНОВАЯ ПЕРЕНРУППИРОВКА (Фаворский). Синтез терминальных алкинов из интернальных (КАРА, LAЕA (LAРA)).
5. РЕАКЦИЯ ФАВОРСКОГО (КОН) –РЕППЕ (Cu2C2).
Структура и особенности ацетилена и алкинов
Так что же такое тройная связь и чем она отличается от двойной и, тем более, от простой. О, это весьма противоречивая и интересная штуковина!
В тройной связи углероды находятся с sp-гибридном состоянии, следовательно сама связь линейна вместе с двумя атомами, связанными с углеродами. Тройная связь короче двойной. Более короткая связь с очевидностью указывает на то, что тройная связь прочнее двойной и, тем более, одинарной. Но к этому высказыванию стоит относиться с осторожностью – имеется в виду тройная связь целиком. Нужно больше энергии для того, чтобы разорвать тройную связь пополам (если это произошло, образующиеся частицы CH или CR называются карбинами). С такими процессами мы точно не столкнемся, хотя с обратным процессом – образованием ацетилена димеризацией двух карбинов, мы неявно сталкиваемся, так как должны знать, что основной способ промышленного производства ацетилена – высокотемпературный пиролиз метана, происходящий при сгорании метана в кислородном пламени с соотношением метан:кислород, недостаточным для полного сгорания метана – часть метана горит и дает энергию для пиролиза других молекул метана. При температуре сильно выше 1000ºС связи C-H в метане рвутся одна за другой, пока не останется последняя, самая прочная (по мере отрыва атомов водорода каждый следующий отрывается тяжелее) образующиеся частицы, карбины рекомбинируют с образованием ацетилена. Горелку делают так, чтобы газ двигался через нее с большой скоростью, и чрезвычайно быстро проходит через область очень горячего пламени, после чего очень быстро охлаждается, а образовавшийся ацетилен поглощается растворителем. Получается, что при очень высоких температурах ацетилен более устойчив чем метан, этан и этилен (понятно, что по дороге к карбину должны образовываться метильный радикал и карбен, которые тоже могли бы димеризоваться в этан и этилен, но выживает в адском пламени только ацетилен).
В молекуле ацетилена только два типа связей – тройная и связь sp-C-H, которая тоже существенно прочнее родственников в алканах и алкенах (почему, выясним чуть позже). Этот эксперимент ясно дает понять, что молекула ацетилена чрезвычайно прочна и может переносить чрезвычайно жесткие условия, в которых большинство других органических молекул, включая заклейменные за инертность алканы, распадаются. Не зря ацетилен – одна из немногих органических молекул, существующих в межзвездном космическом пространстве. Вы скажете – какие проблемы, там же холодно и пусто, что за проблемы там существовать. Но чтобы существовать надо сначала образоваться, и обычно там встречаются такие частицы и молекулы, которые смогли образоваться и выжить в каком-нибудь жутком катаклизме. Да и образовавшись надо выживать дальше, ведь там может юыть и холодно, но излучения летают взад вперед вполне себе адские. И вот там и выживают только самые живучие молекулы типа ацетилена, цинистого водорода, а то и совсем кусочков типа карбена.
И при этом она, молекула ацетилена, нестабильна относительно распада на элементы, при котором выделяется много энергии. Точнее, метастабильна, то есть сама по себе вполне стабильна, может существовать бесконечно долго, если ни с чем другим не сталкивается. Это довольно распространенное, хотя и немного парадоксальное свойство многих молекул, которые, по расчету термодинамических параметров нестабильны относительно элементов, из которых сделаны. При этом химия так устроена, что реакция может быть сколько угодно выгодна, но если нет удобного пути, по которому реакция могда бы пойти – мы называем такие пути механизмами – ничего не произойдёт. Нет механизма – нет реакции. Но как только мы найдём механизм, возможно обходной, сначала превратив молекулы во что-то еще, и дальше маленькаим шагами – можем осуществить реакцию. Так и устроен катализ, и для этого и нужен катализ: катализом нельзя помочь сделать невыгодное превращение. Катализ может сдвинуть с места только выгодное превращение. Вот и ацетилен, когда молекулы одни в пространстве, в вакууме, они живут бесконечно долго. Но стоит им собраться в одном месте, в жидкости, даже не в жидком ацетилене, а в растворе под небольшим давлением, как самые разные молекулы оказываются способны помочь ацетилену развалиться на элементы. Даже сами молекулы ацетилена, когда сближены, умеют это делать. В одиночестве ацетилен живуч, в толпе себе подобных склонен к массовому самоубийству. Механизмы такого распада могут быть самыми разными, во многих случаях это даже толком и неизвестно, хотя в этом долго пытались разобраться, хотя бы для того чтобы понять, как безопасно работать с ацетиленом – зная механизм, можно придумать, как его заблокировать. Успехи в этом направлении были, и связаны они были с одним из весьма мрачных персонажей в истории химии, немцем Вальтером Реппе, работавшем в нацистской Германии не без использования тех дьявольских возможностей, которые предоставляла эта отмороженная страна. Понятно, что когда дым рассеялся, далеко не всеми достижениями Реппе удалось воспользоваться в нормальной жизни.
Поэтому, например, ацетилен никогда не хранят в сжиженном виде – в баллонах находится пористый материал пропитанный раствором ацетилена. И по той же причине ацетилен очень неохотно используют как сырье в химической промышленности несмотря на множество интересных и важных реакций – слишком опасно и дорого. Ведет себя так ацетилен потому что сама молекула хранит в себе солидный запас энергии – энтальпия образования из элементов сильно положительна, и при распаде на элементы эта энергия выделяется. В этом месте обязательно стоит подчеркнуть – такой характер (положительная энтальпия образования, формально означающая, что соединение неустойчиво по сравнению с элементами, из которого оно сделано, если это углеводород, то по сравнению с углеродом и водородом) свойствен всем ненасыщенным соединениям, в которых преобладают двойные или тройные C-C связи над простыми C-C связями: таковы и этилен, и аллен, и даже, о ужас, 1,3-бутадиен, и, совсем кошмар – бензол. Эталон стабильности, бензол, эндотермичен относительно элементов. Но у всех этих соединений, кроме ацетилена, эта эндотермичность не так велика, и движущая сила распада на элементы поэтому слишком мала, чтобы доставлять всем этим знаменитым углеводородам хоть какие-то неприятности. А вот у ацетилена эта эндотермичность уже весьма значительна, и это реально проявляется в его свойствах.
Причина этого, вероятно, довольно проста – тройная связь представляет собой, неаккуратно выражаясь, бочку с электронами. Шесть электронов (по паре на каждую черточку) обслуживают эту очень короткую, компактную и очень прочную связь – шесть электронов вынуждены обитать в очень тесном пространстве. Электроны не любят такой тесноты, как не разводи орбитали по перпендикулярным направлениям π-связей. Получается парадокс – очень прочная связь в потенциально нестабильной молекуле. Единственное, что удерживает молекулу ацетилена от немедленного распада на элементы – отсутствие мономолекулярного механизма для такого процесса. Молекула ацетилена мечтает о распаде, но не умеет распадаться сама, – ей требуется помощь или от других молекул ацетилена, либо от катализаторов, всяких переходных металлов. Именно поэтому в газовой фазе, а особенно при пониженном давлении ацетилен очень устойчив, но под давлением и в жидкой фазе ацетилен становится нехилой взрывчаткой. Это очень поучительная история – можно быть или казаться очень, даже невроятно прочным, но если знать знать тонкое место и вовремя ткнуть в него, то такую штуковину вмиг разносит в дым. Аналогии каждый да найдет сам.
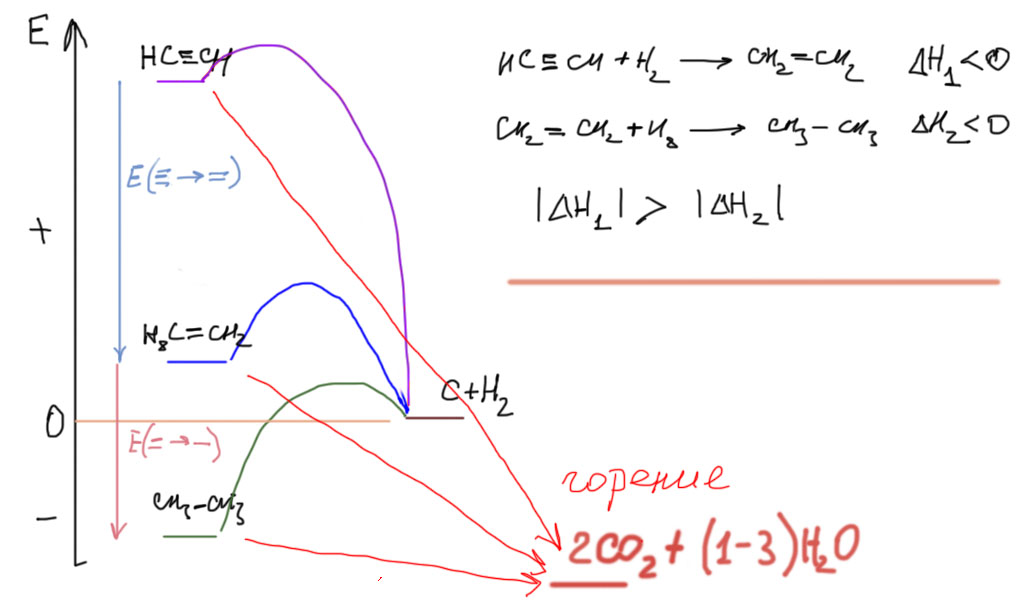
Избыточная энергия ацетилена имеет одно хорошо известное применение – горение ацетилена в кислороде дает чрезвычайно горячее пламя, используемое в ацетиленовой сварке. В наши дни ее активно вытесняет электросварка, но еще в недавнем прошлом на любой серьезной стройке можно было найти ряды голубых и белых баллонов с кислородом и ацетиленом. Ацетилен применяют в сварке даже несмотря на огромные неудобства, связанные с невозможностью хранить этот газ в сжиженном или сжатом виде – только в виде раствора, из-за чего емкость ацетиленовых баллонов существенно меньше таких же с любым другим простым углеводородом. Избыточная энергия, запасенная в молекуле ацетилена, при сгорании выделяется и дополнительно разогревает продукты сгорания, превращаясь в кинетическую энергию молекул двуокиси углерода и воды (справедливости ради заметим, что у ацетиленового пламени есть и второе слагаемое успеха – при сгорании ацетилена образуется сравнительно меньше воды чем при сгорании метана или этана, или этилена, а энтальпия образования воды существенно меньше энтальпии образования двуокиси углерода, поэтому уровень энергии продуктов сгорания еще и ниже всех – на картинке упрощения ради везде показан одинаковый).
Добавление к ацетиленовому фрагменту алкилов стабилизирует молекулу. Уже метилацетилен намного стабильнее самого ацетилена, и отлично существует в сжиженном виде без опасности самопроизвольного взрыва. Так как и метилацетилен тоже обладает значительной избыточной энергией из-за наличия в нём тройной связи, этот углеводород стали активно применять в сварке – пламя горения метилацетилена хоть и немного холоднее ацетиленового (молекула всё же несколько стабильнее, да и воды получается больше, а вода забирает часть энергии, выделяющейся при полном окислении углеводорода из-за большей теплоёмкости в сравнении с теплоёмкостью совершенно жёсткой линейной молекулы диоксида углерода), но всё равно горячее пламени горения всего остального возможного углеводородного топлива, если не брать такую дорогую экзотику как циклопропаны). Безопасность и удобство использования в сжиженном виде в баллонах перевешивают небольшую потерю температуры пламени, которую к тому же можно довести до максимума инженерными решениями в конструкции горелки. А поскольку метилацетилен (в смеси с алленом – почему это так будет описано на отдельной вкладке) в огромных количествах получается при паровом крекинге пропана в производстве важнейшего мономера, пропилена, в качестве побочного продукта, и девать его в таких количествах принципиально некуда, то идея использовать его вместо ацетилена в сварке оказалась удачной. Такая смесь (метилацетилен-аллен, сокращённо MAPD) продаётся в больших количествах как горючее для сварки вместо опасного и неудобного ацетилена.
Если добавить алкилы ещё немного подлиннее, то алкины от 4 углеродов и более становятся совсем ручными и безопасными, если не считать, конечно, обычных проблем с легкостью воспламенения.
Следующий шаг такой. Мы помним рассуждения про стабильность двойной связи в алкенах. Помним, сколько шума нам удалось произвести, сравнивая двойные связи в разных олефинах. Так вот, если посмотреть на ацетилены не как на вещества с одной очень прочной тройной связью, а разложить тройную связь на одну сигма и две пи-связи, то тогда мы сможем попробовать рвать эти связи поочередно. И когда мы порвем первую пи-связь, то уже из картинки наверху видим, что энергии выделится больше, чем при разрыве второй пи-связи. Гидрирование ацетилена более экзотермично чем гидрирование этилена. Первая пи-связь в ацетилене менее устойчива чем пи-связь в этилене и других олефинах.
Из этого мы легко получаем практическое следствие – если мы хотим гидрировать ацетилен, то мы сможем рассчитывать на то, что сможем остановиться на этилене, так как ацетилен должен быть более реакционноспособен в реакции гидрирования чем этилен. Но в общем случае гидрирование алкина трудно остановить на алкене, – в большинстве реакций гидрирования с обычными катализаторами гидрирование будет полным с образованием алкана. Простые соображения в химии часто не работают, потому что нужно еще знать механизм конкретной реакции, а это не всегда можно легко выяснить. Для частичного гидрирования необходимо применять специальные катализаторы, которые часто по недоразумению называют отравленными, хотя на самом деле их особые свойства определяются особыми механизмами гидрирования. Для частичного гидрирования алкинов в цис-алкены применяют катализатор Линдлара – палладий, нанесенный на карбонат кальция, в присутствии ацетата свинца и хинолина. Почему этот катализатор останавливается на алкене мы разбирать не будем, так как это никому и не известно. Механизмы гетерогенных реакций вообще до сих пор толком не изучены. Работает, и хорошо! Не забывайте, что это только катализатор, и для гидрирования нужен водород. И отдельный вопрос – нужно ли выписывать все эти хинолин и ацетат свинца над стрелкой в схемах реакций? Не нужно. Это детали приготовления катализатора. Когда мы пишем Pd/CaCO3, мы уже имеем в виду катализатор Линдлара, а то, как точно его готовят, вы узнаете, когда будете готовить его сами. Возможно и не узнаете никогда, потому что готовый катализатор есть в продаже.
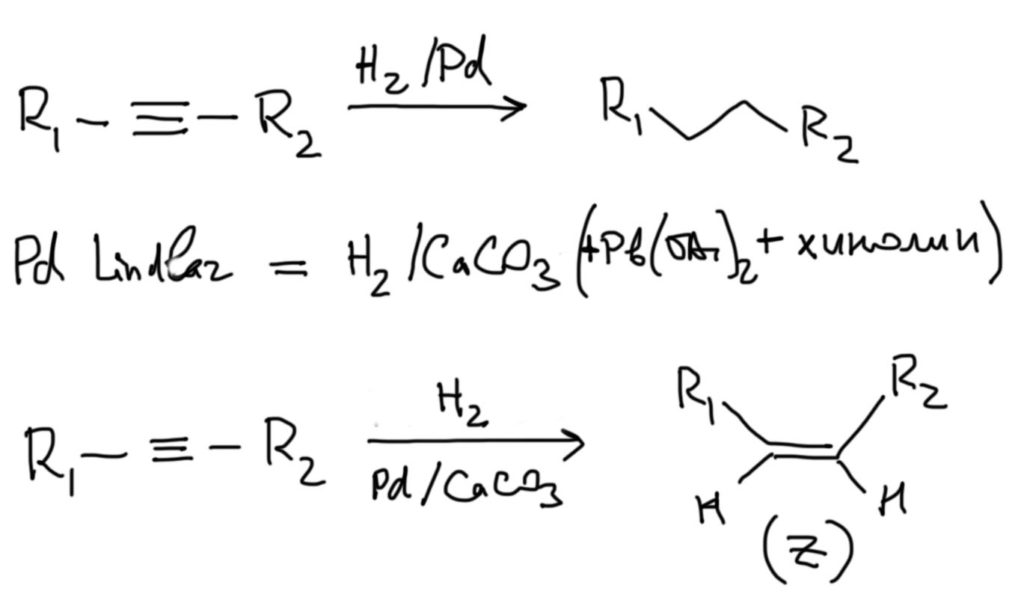
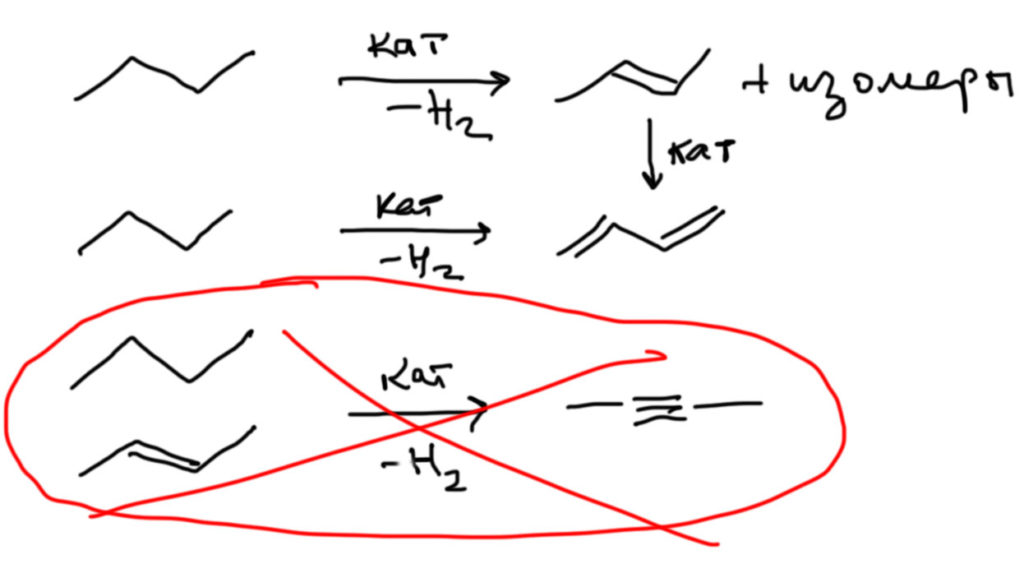
И еще одно практическое замечание относительно обратной реакции. Алкены часто, особенно в промышленности, получают каталитическим дегидрированием алканов, и это вполне осуществимая реакция несмотря на довольно существенную эндотермичность – катализаторы, переходные металлы и их производные, часто имеют огромное сродство к водороду и охотно отнимают его. Но следующий шаг сделать нельзя (или очень-очень трудно на грани с невозможностью) – нельзя дегидрировать алкен в алкин, вместо этого вы всегда будете получать гораздо более устойчивый диен. Просто посмотрите на верхнюю картинку, где показаны уровни энергии, соответствующие алкану, алкену и алкину, – и поймите, что дегидрировать алкан в алкен это как забросить мяч в кольцо, стоя на земле; а дегидрировать алкен в алкин – это все равно, что забросить мяч на крышу небоскреба, стоя на баскетбольном кольце.
Для дегидрирования алканов в алкены и диены используют всякие промышленные катализаторы, состав которых – коммерческая тайна компаний, которые разрабатывают и эксплуатируют такие процессы. Многие промышленные катализаторы являются сложными смесями каталитически активных компонентов, подложек и промоторов, и ничего конкретного про это сказать невозможно. В лаборатории эти реакции не делают. Поэтому, пожалуйста не устраивайте холивар про то, какой катализатор нужно использовать для дегидрирования алканов в алкены или диены. Множество переходных металлов и их производных (платиновые металлы, молибден, железо и др и пр., их оксиды) могут вызывать дегидрирование, а дальнейшая разработка катализатора – проблема более инженерная и экономическая, а не химическая. Нам это до лампочки. Мы изучаем лабораторную органическую химию. Вот те катализаторы, которые используют в лаборатории, например, палладий на угле или катализатор Линдлара для гидрирования мы должны знать, а катализаторы дегидрирования только в самых общих чертах, типа, такие есть.
Самые простые алкены типа этилена и пропилена получают ещё проще и без всякого катализатора – просто крекингом насыщенных углеводородов, получаемых или из природного газа после удаления из него основного компонента, метана (остается этан, пропан, бутан), или из низших фракций нефтепеработки. Такие углеводороды разбавляют водяным паром и подвергают кратковременному нагреванию до 700-850º (поэтому этот процесс называют паровым крекингом) с быстрым охлаждением: происходят свободнорадикальные реакции разрыва C-C связей с диспропорционированием образующихся радикалов, а также разрыва C-H связей, с образованием атомов водорода, которые отрывают другие атомы водорода. Водяной пар подавляет плохую реакцию глубокого распада углеводородов до элементарного углерода, забивающего реактор. Здеь нет никакой особой химии, разбавление инертным газом (а в этой реакции вода – инертный газ, потому что единственная возможная реакция – расщепление связи O-H – не идёт, потому что эта связь прочнее всех связей C-C и C-H) просто снижает вероятность контакта молекул углеводородов и стенок реактора (чисто вероятностный эффект – молекул больше, вероятность меньше), а именно такие соударения и приводят к более глубокому распаду молекул, заканчивающееся углеродом, сажей. Отложения сажи снижают эффективность теплопереноса, поэтому реактор приходится время от времени переводить в режим выжигания сажи, а в присутствии водяного пара такое происходит намного медленнее и производительность сильно увеличивается.
Поэтому паровой крекинг стал основным видом термического крекинга. Образуются водород, этилен, пропен, бутены, бутадиен. И немного ацетилена, который в этих промышленных процессах страшно всех раздражает, потому что является нежелательной и опасной примесью, его приходится отделять и обратно гидрировать в этилен-этан. Это лишний раз напоминает нам, что ацетилен довольно легко образуется при термическом распаде углеводородов при высокой температуре, но без участия катализаторов. И еще меньше смеси метилацетилен-аллен (смесь МАПД – метилацетилен – пропадиен), буквально доли процента. Но учитывая совершенно колоссальную производительность таких установок, даже доли процента – это десятки тонн. Эту смесь отделяют и в отличие от самого ацетилена находят ей применение – просто упаковывают и продают как газ для сварки, более безопасный чем сам ацетилен, МАПД можно сжижать и продавать в обычных баллонах. Это такая типичная промышленная проблема – найти оптимальный режим процесса, компромисс между производительностью и затратами. Если бы температура газов в крекинге была поменьше, ацетилен бы совсем почти не образовывался, можно было бы сэкономить на аппаратуре для его удаления, но и скорость крекинга уменьшилась бы, и общая производительность установки тоже, а затраты энергии на единицу продукции наоборот увеличились бы – большая часть энергии тратилась бы на тупое нагревание. Ну вот инженеры поэкспериментировали с режимами и остановились на таком решении. Установки парового крекинга дают достаточное количество простых алкенов от этилена до бутенов, а также бутадиен и обеспечивают сырьём процессы, использующие эти олефины и бутадиен, а это в основном полимеризация, но не только. Смеси газов крекинга разделяют на компоненты самым обычным способом – фракционной перегонкой, для промышленности перегонка при отрицательных температурах и повышенном давлении не проблема.
CH-Кислотность терминальных алкенов. Зависимость свойств анионов, катионов, радикалов от гибридизации углерода
Итак, задача – объяснить, почему водород в ацетилене легче снимается основаниями, чем водороды в алканах и алкенах. при поверхностном взгляде здесь вообще разница катастрофическая – с ацетилена протон снять можно, и мы знаем как, а про метан и этилен нам никто ничего не говорил – наверное, потому что ничего подобного сделать невозможно? Это не так. разница есть, но это не разница между “да” и “нет”, а количественная разница между константами кислотности: ацетилен кислотнее этилена где-то на 15 порядков (15 единиц pK), и приблизительно такая же разница между этиленом и этаном или метаном. Тенденция, однако.
Везде у нас связи C-H. Но это разные связи. Мы знаем, что в алканах гибридизация углерода sp3, и эта гибридизация распространяется и на связи C-C и на связи C-H. В этилене гибридизация sp2, и такими гибридными орбиталями привязаны с углероду атомы водорода. В ацетилене – sp. Это ясная и банальная вещь, но по какой-то малопонятной причине про это часто забывают, считая, что гибридизация говорит нам про простую, двойную, и тройную связь, – и только. Нет, и про все остальные связи тоже, и это важно.
Так, а чем они отличаются? Вот тут понадобится немного квантовой зауми, впрочем, в весьма упрощенном виде. Мы знаем про s и p-орбитали атомов, и про то, что они отличаются формой. Формой чего? Во-первых, соответствующих волновых функций (их графического образа), но это малопонятная заумь, а во-вторых распределения электронной плотности в пространстве. Тоже несладко, но хотя бы можно понять. К тому, что электрон как-то то ли движется так быстро, что не уследить, то ли вообще размазан (распределен) в пространстве, мы как-то уже привыкли. Электрон притягивается к ядру, и это хорошо – это просто. Чем ближе электрон к ядру, тем сильнее он притягивается, и тем сильнее нужно упираться рогом, чтобы его оттуда оторвать – это даже еще проще и понятнее. Проблема в том, что мы не знаем, где он находится, этот самый электрон. Но – из графика распределения электронной плотности – мы знаем где наибольшая вероятность его застать. Это как раз и есть максимум этой функции. Чем ближе максимум к ядру, тем ближе в среднем электрон к ядру, и тем прочнее он с этим ядром связан силами кулоновского притяжения. Здесь придется поверить в то, что в пределах валентной оболочки сферическая s-орбиталь в среднем более компактна чем p-орбиталь, состояшая из двух более протяженых и сильнее размазанных половинок. S-электроны в среднем ближе к ядру чем p-электроны. И держатся за ядро сильнее, и оторвать их сложнее. Теперь возьмем гибридные орбитали. Их вполне можно себе представлять как просто сумму: sp – сумму s и p; sp2– сумму s и двух p, и т.п. Вполне можно так и говорить – в sp-орбитали 50% s- и 50% p. В sp3– 25% s и 75% p. Прямо акционерное общество. И точно – с ограниченной ответственностью. Получается довольно простая закономерность – чем больше в гибридной орбитали s-участия, тем ближе в среднем электроны на этой орбитали к ядру. И наоборот.
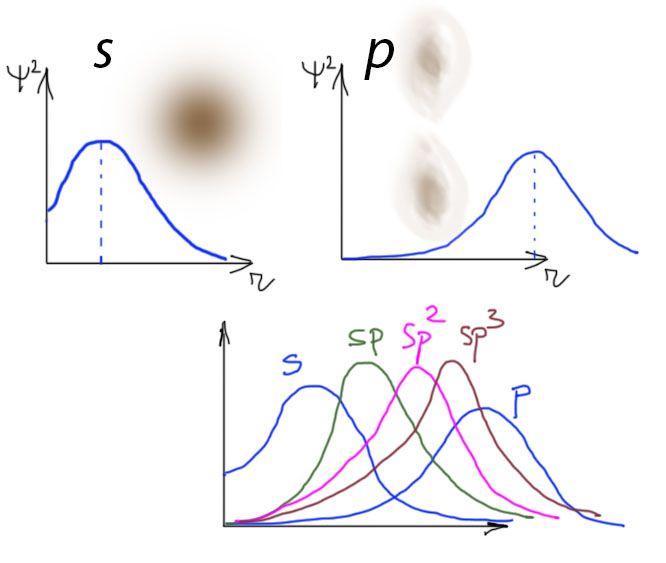
Вопрос скептика из зала – и что, это действительно так сильно влияет, какие-то картинки корявые??! Ответ скептику прост. Во-первых, отвали. Во-вторых, химия – наука экспериментальная. Если есть экспериментальные данные о том, что что-то как-то более-менее закономерно изменяется, то это очень важно, потому что это так в Природе установлено. Наше дело попытаться это объяснить, и наше объяснение должно объяснять почему то, что наблюдается действительно наблюдается. Мы не можем вместо этого искать способ доказать, что нам померещилось.
Итак, что мы видим из картинок. То, что чем больше s-вклад в орбиталь,
- тем она компактнее,
- тем короче связи, образованные с помощью этой орбитали,
- тем, следовательно, прочнее эти связи,
- тем сложнее забрать электрон с такой орбитали,
- и тем больше радости от того, что на эту орбиталь попал дополнительный электрон.
Чуть-чуть поясним. Что значит оторвать электрон и добавить еще один электрон. Каждому атому в Таблице Менделеева выдано определенное количество электронов, известная часть которых находится в валентной оболочке. У углерода валентных электрона 4. Частицей, в которой центральный атом имеет ровно столько электронов, сколько предписано, является радикал (на картинке у метильного радикала помечены электроны – свои синим, чужие красным). Если от радикала оторвать электрон, получим карбокатион (при этом нужно затратить энергию, это называется потенциал ионизации, IP). Если добавить – карбанион (при этом энергия выделяется – это называется сродство к электрону, EA). Так вот, чем больше нужно энергии, чтобы оторвать электрон, тем менее стабилен карбокатион (внимание – речь идет только о похожих частицах, широкие обобщения не приветствуются и могут оказаться неверными, но нам они и не нужны). И наоборот, чем больше энергии выделяется при образовании карбаниона, тем он стабильнее.

Теперь ясно, сколько всего мы можем сходу объяснить просто из относительного размера гибридных орбиталей и s-вклада. Мы вполне приближаемся к бессмертной славе Храброго Портняжки, замочившего одним ударом семерых. Итак, из относительного вклада s-орбиталей в гибридные орбитали мы видим, что в ряду алкан – алкен – алкин:
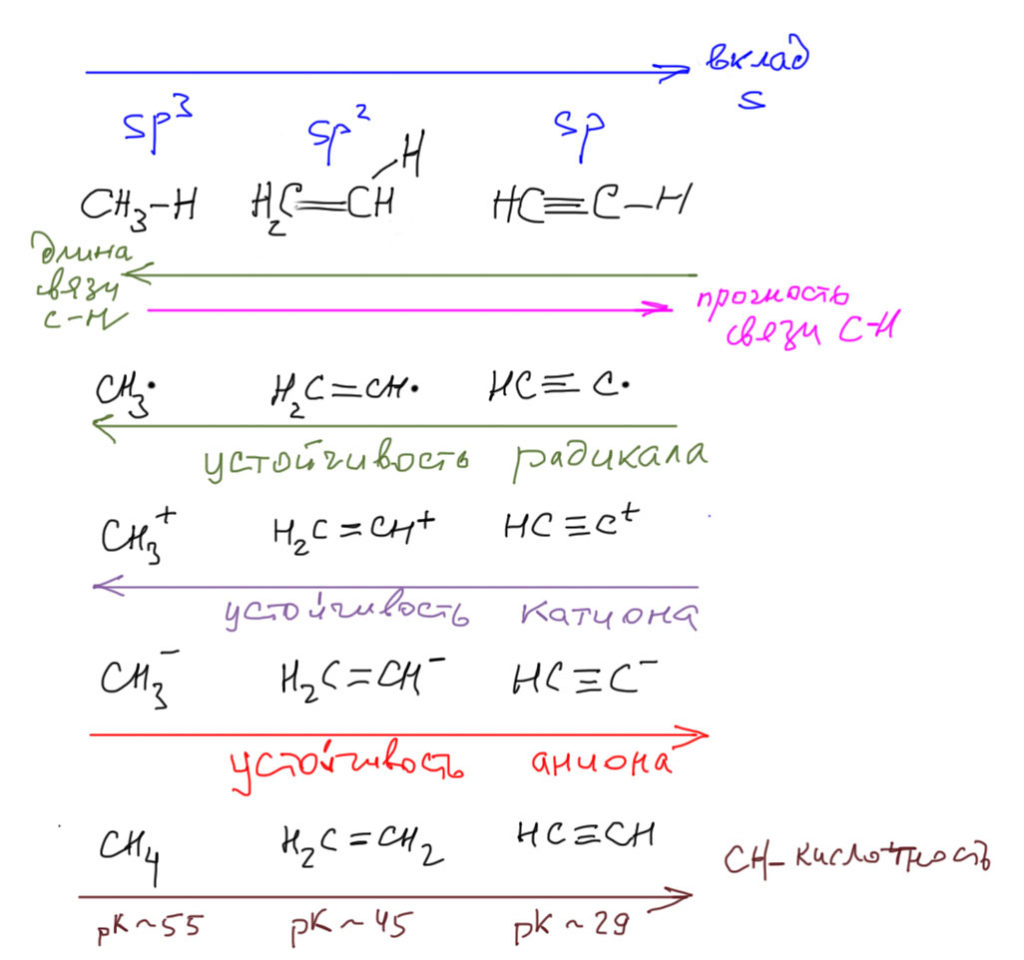
Вот так. Немного не хватило до подвига Храброго Портняжки. Впрочем, вы сами, может быть, что-нибудь еще заметите и добавите. Но если честно, то среди упомянутых некоторые свойства тесно связаны и должны рассматриваться как одно, например, энергия связей С-H и устойчивость радикалов. И в очередной раз напоминаю, что слово “устойчивость” здесь и почти везде используется в общехимическом смысле, а не в строгом термодинамическом. Это такая полукачественная мера трудности создания таких частиц и их относительной реакционной способности в ряду похожих частиц. Но это ровно то, что нужно при обсуждении механизмов реакций, в которых участвуют такие частицы.
Корректности ради заметим, что в ряду карбокатионов гибридизация не соответствует формально указанной, так как при образовании карбокатиона происходит изменение гибридизации. Например, алкильный катион не пирамидальный, а плоский (если не забыли правило Джиллеспи из курса неорганической химии, то оно здесь тоже работает), а следовательно гибридизация в алкильном катионе не sp3, а sp2. В алкенильном катионе – sp. Про алкинильный вообще лучше не думать. Собственно, в общих тенденциях это ничего не меняет, мы просто опускаемся на одну ступеньку вниз.
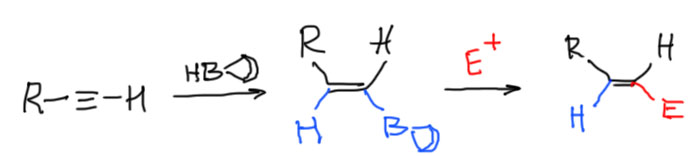
Электрофильное присоединение к алкинам
Невероятно мутная, как ни странно, тема. Очень простой, вроде бы, вопрос – что более реакционноспособно, алкены или алкины, в простейших реакциях присоединения брома, бромоводорода, боранов и других простых электрофилов. Экспериментальные данные-то наверняка есть, что тут обсуждать. тем не менее, по какой-то причине ясного ответа на этот вопрос найти непросто.
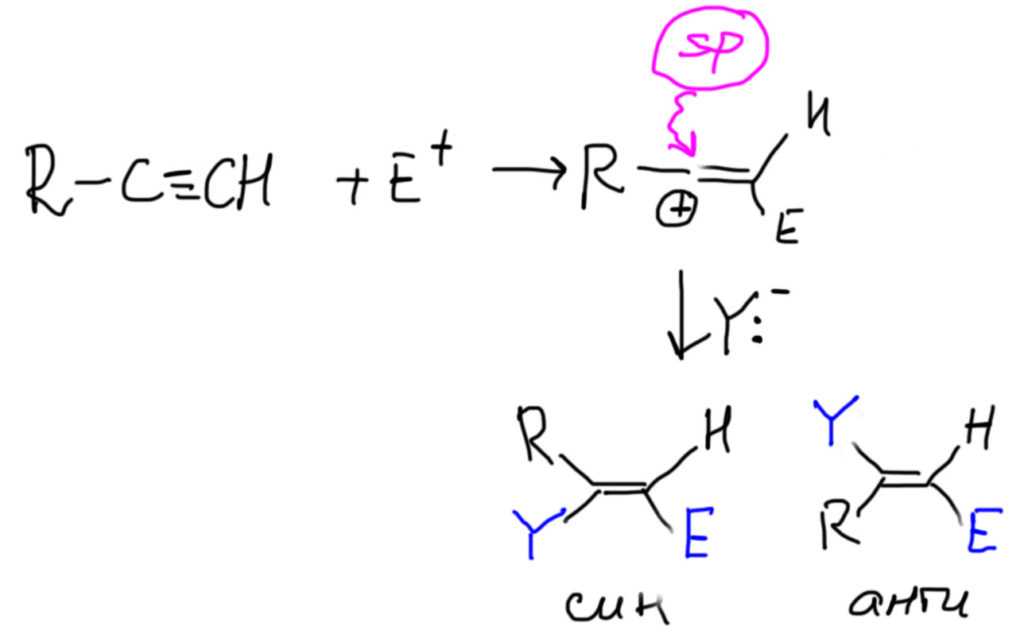 Ничего особенного в такой ситуации нет. Алкины, как мы уже успели заметить, довольно противоречивые соединения. В электрофильном присоединении это проявляется очень ярко. Попробуем разобраться.
Ничего особенного в такой ситуации нет. Алкины, как мы уже успели заметить, довольно противоречивые соединения. В электрофильном присоединении это проявляется очень ярко. Попробуем разобраться.
Если совсем коротко, то реакционную способность тройной связи по сравнению с двойной определяют несколько важных факторов, и проблема в том, что они действуют в разные стороны, поэтому алкины могут быть как менее, так и более реакционноспособными, чем алкены.
- в тех случаях, когда механизм присоединения предполагает образование карбкатиона или мостикового иона, алкины намного (часто на два-четыре порядка) хуже алкенов. Таковы, например все реакции галогенирования. Можете смело бромировать двойную связь в присутствии тройной, только не лейте избыток брома.
- а вот в согласованных механизмах на первый план выходит то, что первая π-связь в тройной связи намного менее стабильна (это обсуждалось выше), чем π-связь алкенов. В таких случаях тройная связь более реакционноспособна, иногда сильно более. Это особенно ярко проявляется в реакциях гидроборирования, за исключением реакции с 9-BBN, в которой дополнительно работает очень специфический фактор необычной формы самого реагента (об этом подробнее ниже на вкладке про гидроборирование). В современной химии таких реакций особенно много, из-за этого органический синтез любит тройные связи больше двойных. А вот реакции с протонными электрофилами, например, с галогеноводородами, идущими, скорее всего, по согласованному механизму типа AdE3, но с значительным смещением электронной плотности в переходном состоянии, напоминающем карбокатион, играют обе тенденции, съедая друг друга: в таких реакциях реакционная способность алкенов и алкинов очень близка, и определяется в основном третьим фактором, а именно:
- стерическим фактором. Тройная связь, даже внутренняя, намного доступнее для атаки электрофила, чем двойная, осоенно сильно замещенная. Но и концевая двойная связь тоже вполне открыта. Это создает возможности для мастеровитого синтетика тонко играть на таких различиях. Нам этого не поднять, но иметь представление о том, как искусно выбирают реакционные центры в сложных синтезах на головоломно сложных структурах, не помешает. Органический синтез – это искусство, но имеющее вполне рациональные основы, и не чурающееся расчетов и моделирования.
Если написать, как выглядит обычный механизм электрофильного присоединения в случае алкинов получим, что почти все то же самое, кроме карбокатиона (такой карбокатион принято называть винильным), в котором секстетный атом оказывается sp-гибридным. Выше мы разобрались, как гибридизация связана с устойчивостью всяких частиц, которые встречаются в механизмах, в том числе карбокатионов. Получается, что винильные карбокатионы чрезвычайно неустойчивы, а следовательно их образование очень энергозатратно, а следовательно реакционная способность алкинов с таким механизмом должна быть очень мала, и существенно ниже чем алкенов. Как проверить это предстказание. Очень просто – посмотрим, как к алкинам присоединяется самый простой электрофил, протон, для которого просто нет других возможностей. Для алкенов есть реакция гидратации в присутствии сильных кислот. А для алкинов – нет такой реакции! Мы же знаем, что гидратация алкинов не идет, пока не будет добавлена соль ртути (именно это, говорят, и открыл Кучеров, когда безуспешно пытался гидратировать ацетилен в растворе серной кислоты, пока случайно не разбил ртутный термометр – это легенда, но довольно правдоподобная). Что там делает ртуть, разберемся позднее, но факт – протон к ацетиленам почему-то присоединяться не торопится (если быть совсем корректным, то в более жестких условиях гидратация без солей ртути идет, но только с замещенными алкинами, когда катион может быть стабилизирован электронными донорными эффектами заместителя). Хорошо, а бромистый или хлористый водород уж наверняка присоединяется как положено? Хлористый винил, важнейший мономер, разве не так делают? Так, да не так. Если посмотреть на описанные в литературе методики присоединения гологеноводородов к ацетиленам, мы и там увидим катализ ртутью, а иногда и другими металлами, а вот просто присоединение HBr мы увидим только в одном случае – к фенилацетилену и похожим алкинам. Это исключение только подтверждает общую закономерность, так как в этом случае карбокатион стабилизирован сопряжением и его образование становится возможным.
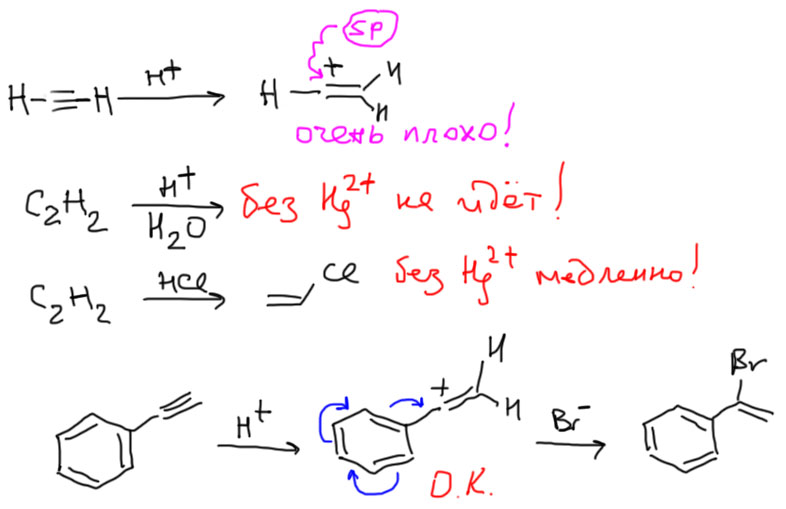
А бромирование что, тоже без ртути не идет? Идет, но довольно медленно. Бромирование алкенов гораздо быстрее. По этой причине при присоединении брома к ацетиленам непросто остановиться на дибромпроизводном, реакция пролетает дальше, и даже если мы берем соотношение реагентов 1:1, мы получаем тетрабромпроизводное и остается непрореагировавший алкин. Обсуждать присоединение галогена более подробно поэтому не очень интересно. Можно только задать вопрос про возможность образования мостикового иона. Хорошего ответа на это нет. Может есть такой тон, а может нет. Экспериментальные данные не дают хороших поводов обсуждать это. Одно можно сказать более-менее надежно. Если такой ион и образуется, то он должен быть очень неустойчив, потому что структура этого иона – трехчленный цикл с двойной связью. Очень трудно так сильно согнуть связи – нормальный угол в sp2-гибридном углероде 120º, а согнуть нужно до 60º. Мы позднее обсудим эту проблему, которая называется напряжением. Сейчас это не так важно. Просто признаем, что простой перенос механизмов электрофильного присоединения из химии алкенов в химию алкинов приводит к очень неустойчивым частицам – винильному катиону или очень напряженному мостиковому иону.
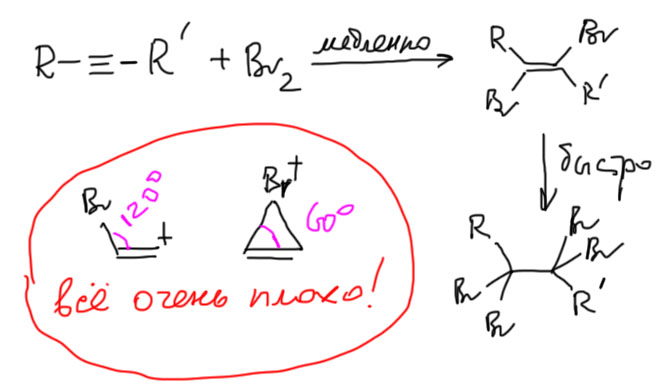
Резюмируем: в реакциях электрофильного присоединения алкины обычно намного менее реакционноспособны чем алкены, если механизм реакции предполагает образование карбокатионов или мостиковых ионов.
Но есть еще гидроборирование или реакции с участием ионов ртути. А что там?
Что делает ртуть в реакции Кучерова?
Итак, ацетилены обычно намного менее реакционноспособны чем алкены в “обычных” реакциях электрофильного присоединения. Под “обычными” имеются ввиду реакции, условия которых приблизительно одинаковы для алкенов и алкинов, что позволяет предположить, что и механизм должен в общем быть одним и тем же. Так и с гидратацией. Для алкенов мы это на соответствующей вкладке посмотрели – реакция идёт в присутствии сильных кислот, хотя и осложняется перегруппировками, олигомеризацией и прочими радостями, связанными с буйным нравом карбокатионов. А что с алкинами? Все знают еще со шкоы реакцию Кучерова, как минимум из патриотических соображений, а почему такая разница в способах гидратации, не вполне понятно, и почти никто это не объясняет.
На самом деле алкины (не сам незамещённый ацетилен!), имеющие алкил или фенил на одном или обоих углеродах вполне можно гидратировать практически в тех же условиях, что и алкены. Это открыл еще знаменитый французский химик Марслен Бертло аж в 1862 году, но применения эта реакция не нашла из-за жёсткости условий – действия концентрированной серной кислоты с последующим выливанием смеси в воду (видите, уже Бертло знал, что серную кислоту льют в воду, а не наоборот!), а как мы скоро узнаем в химии кетонов, продукт такой реакции, виниловый спирт немедленно перегруппировывается в кетон, а он в таких условиях просто и немедленно вступает в реакцию альдольно-кротоновой самоконденсации с образованием целой кучи всяких продуктов. Осмоляется всё, проще говоря. Зачем такая реакция. 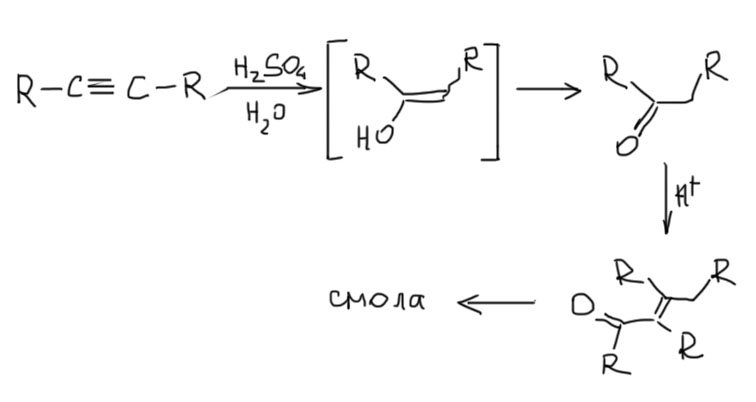
Решение всего через 20 лет нашел Михаил Григорьевич Кучеров, случайно или нет, достоверно неизвестно и не важно – большинство серьезных находок в химии сделаны немного или совсем случайно. Мощь учёного в том и состоит, чтобы не упустить интересный результат, разобраться в том, что произвошло, и хорошенько это описать, чтобы и другие подивились и порадовались. Кучеров к тому же не был профессиональным химиком, не получил химического образования, был в прямом смысле этого слова дилетантом. Это не страшное слово, а по своему первому смыслу очень лестное. По-итальянски так называют людей, занимающихся чем-то не заработка ради, а для развлечения, удовольствия. Вот Вивальди, например, был композитором-дилетантом. И Бородин наш тоже. Бородин вообще уникум, он еще и как химик тоже был дилетантом. Будьте такими, как они – будьте дилетантами! Слава и признательность потомков найдёт вас. Почему в русский язык это слово вошло с таким негативным смыслом – видимо, потому, что у нас не принято получать удовольствия от дела, которым занимаешься.
Напомню, что в отличие от алкенов, гидратация которых происходит в присутствии сильных кислот, алкины еще требуют немного соли двухвалентной ртути. Часто возникает вопрос – а что происходит с алкенами в таких условиях. Ответ простой – если проводить реакцию аккуратно, то ничего. Реакция Кучерова селективна по отношению к алкинам, и алкены не затрагивает. Примеры можно дюжинами обнаружить в литературе, от самых простых до очень сложных (вот, например, пример из синтеза стероидного лекарственного препарата, где Кучеров не затрагивает аж три разные двойные связи).
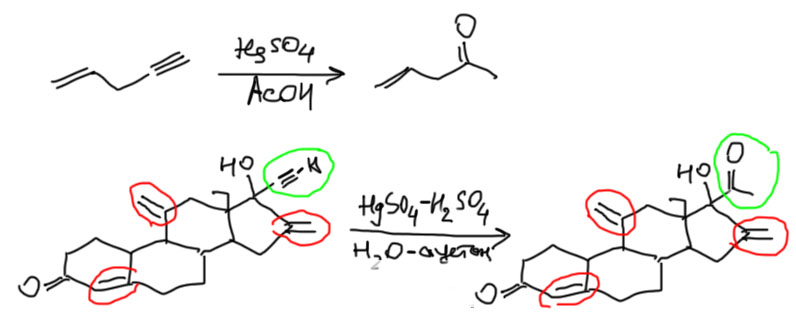
Слово “аккуратно” означает, что в таких случаях необходимо тщательно подбирать условия реакции и использовать небольшие количества кислот и соли ртути, так как в более жестких условиях двойные связи будут затронуты хотя бы обычной гидратацией. Но химия вся об этом, не зря ее часто сравнивают с поварским искусством – пересолил, переперчил, добавил не ту пряность, – и вместо изысканного блюда получаем мерзкое несъедобное варево.
Тем не менее, совершенно очевидно, что в присутствии ртути тройная связь реагирует намного легче двойной. В чем тут дело, и что там делает ртуть? На это есть два ответа, простой и сложный. Начнем с простого.
То, что двухвалентная ртуть электрофил, хорошо известно – мы уже применяли ее к алкенам и получали весьма полезную реакцию селективной гидратации по Марковникову. Правда там ртуть используется не в каталитическом, а в стехиометрическом количестве, и в конце ее нужно специально отгрызать от продукта присоединения восстановлением боргидридом натрия.
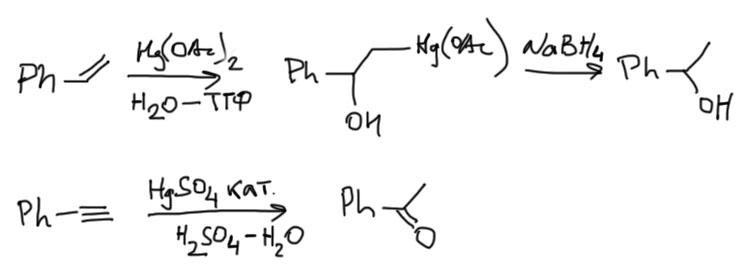
Ртуть – очень большой катион, а связи Hg-C имеют относительно очень большую длину. Поэтому, если мы захотим нарисовать механизм электрофильного присоединения катиона ртути к тройной связи, использовав идею о мостиковом ионе, то получим вполне симпатичную картинку, внешне очень похожую на механизм того же бромирования с участием мостикового иона. Этот ион, как положено, заберет из реакционной смеси нуклеофил, воду. Дальше все довольно просто, если знать, что спирты с гидроксилом на двойной связи (енолы) немедленно самопроизвольно перегруппировываются в карбонильные соединения. Последняя стадия требует уже некоторого интимного знания о том, что ртутьорганические соединения с карбонильной группой рядом легко расщепляются кислотами (обычные ртутьорганические соединения типа того, что появляется в реакции с алкеном, так себя не ведут), образуется конечный продукт, а ртуть возвращается в реакцию, являясь в данном случае фактически катализатором гидратации. Почему в данном случае тройная связь более реакционноспособна чем двойная становится ясно, если учесть, что в данном случае нет винильного катиона, а в мостиковом ионе металл так далеко от углеродов, что эта структура больше не напоминает насильно согнутый треугольник, а скорее мирно присоседившийся к двум углеродам атом ртути. Получается, что тех факторов, что ограничивают обычные реакции электрофилов с тройной связью больше нет, а в остатке мы имеем только то, что и так знаем и уже обсудили – существенно меньшую устойчивость пи-связи в тройной связи по сравнению с пи-связью в двойной связи (прошу прощения за это неуклюжее нагромождение связей, но попробуйте сами это выразить более изящно).
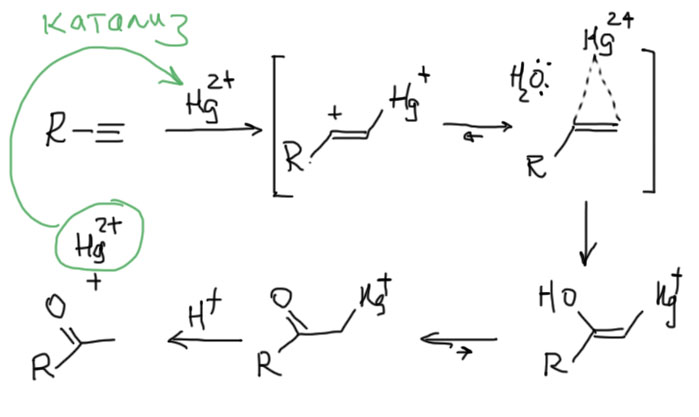
На этом вполне можно остановиться. Но если совершенно случайно стало интересно, то можно посмотреть на проблему немного с другого угла. Даже с другой стороны. А что если это не “закрытый, а …открытый перелом”? Не электрофильное, а … нуклеофильное присоединение. Именно так и смотрит на эту реакцию, точнее, на эти реакции современная химия (есть и такая, …ой, а какую тогда собcтвенно мы изучаем?!). Так вот, в современной органической химии реакций, похожих на реакцию Кучерова, воз и маленькая тележка, и называются они угрожающе реакциями нуклеометаллирования. Устроены они все одинаково. Берется какой-нибудь металл в виде соли или комплекса (в современной химии нет разницы между этими понятиями, поэтому всегда используют более общее слово “комплекс”), обладающий свойствами кислоты Льюиса, а такими свойствами обладают почти все производные металлов, переходных или непереходных в положительных степенях окисления (а что, у металлов бывают отрицательные? – бывают, но не будем пока о таких грустных вещах). Дают ему алкин. Образуется комплекс, в котором алкин выполняет роль лиганда, образующего координационную связь с металлом за счет пары электронов пи-связи и пустой орбитали где-нибудь в валентной оболочке металла. Комплексы такого типа очень хорошо известны для множества металлов, многие из них вполне стабильны, могут быть выделены, положены в баночку и поставлены на полку. Другие образуются в реакционных смесях и видны во всяких спектрах. В таком комплексе электронная плотность с пи-связи частично съезжает в сторону металла. И тогда вдруг оказывается, что алкин в виде лиганда становится способен реагировать с нуклеофилами, даже весьма слабыми. Нуклеофил буквально присоединяется к алкину-лиганду, и тогда связь с металлом переходит из π-типа в обычную ковалентную связь (σ-связь). Дальше происходит простая вещь – σ-связи углерод-металл обычно очень слабы и разрываются любыми источниками протонов, даже слабыми кислотами. Все, новый механизм готов. Это именно нуклеофильное присоединение к алкину, активированному π-связью с металлом. И металл (исходный комплекс металла) играет здесь роль катализатора. Посмотрим на общий механизм (M+ здесь просто обозначает металл в положительной степени окисления со своими лигандами). И не обращайте внимание на стереохимию, здесь все не так просто.
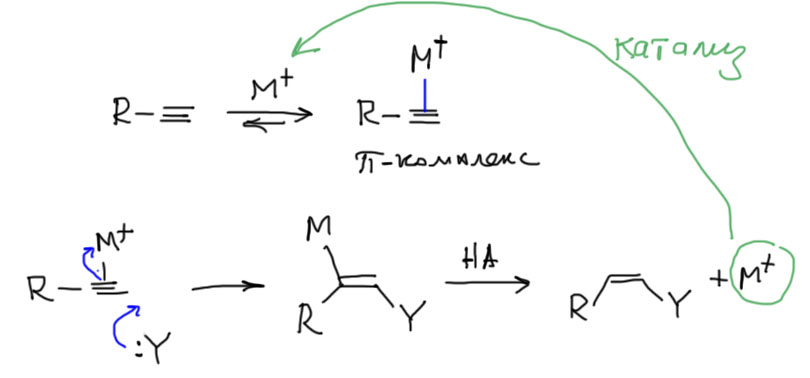
Когда такие реакции стали исследовать серьезно, немедленно выяснилось, что ртуть не лучший металл для этих целей (токсичен, тяжел, да и связи слишком прочные), а лучше подходят соседи ртути по Таблице Менделеева медь, серебро и золото, а потом в ход пошли платиновые металлы типа рутения, родия, иридия, да и многие другие тоже как-то отметились. И нуклеофилом может быть не только вода, но и карбоновые кислоты, амины, спирты, и много чего еще. Вот пара примеров: современная реакция Кучерова с комплексом серебра, и присоединение карбоновой кислоты с комплексом рутения.
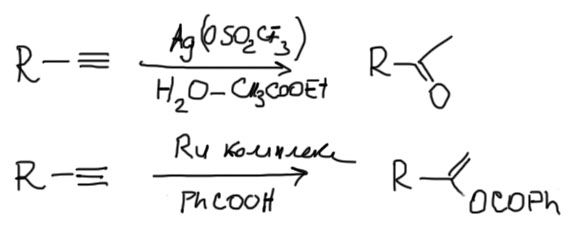
А у алкенов есть такие реакции? Безусловно есть, и ничем существенно они не отличаются. Но реакционная способность алкинов в таких реакциях гораздо выше. Классическая реакция Кучерова, открытая 130 с лишним лет назад, фактически первый пример таких реакций, и теперь окончательно становится ясно, почему она предпочитает алкины алкенам.
Гидроборирование алкинов
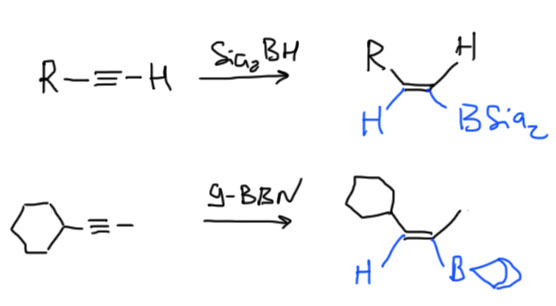
Гидроборирование алкенов – очень важная реакция (нобелевки просто так не дают), но гидроборирование алкинов не менее важно и интересно. Алкины обладают высокой реакционной способностью в реакциях гидроборирования – это пример электрофильной реакции, в которой реакционная способность тройной связи может быть выше чем двойной. Понятно почему. В обычных электрофильных реакциях с промежуточным образованием карбокатиона или мостикового иона главный фактор, определяющий реакционную способность – стабильность этих промежуточных частиц. В случае ацетиленов здесь все очень плохо – карбокатионы винильного типа чрезвычайно невыгодны, и мостиковые иона – трехчленные циклы с оставшейся от ацетилена двойной связью, – тоже очень напряжены и невыгодны. В гидроборировании нет ни невыгодного винильного карбокатиона, ни напряженного циклического иона, а обе половинки реагента, электрофильная и нуклеофильная, присоединяются одновременно. Как мы выяснили, механизм гидроборирования – согласованная реакция. Для согласованных реакций основную роль играют не электронные донорные или акцепторные эффекты заместителей, а стерические факторы – чем проще реагенту подобраться к месту реакции, тем лучше. В этом смысле тройная связь лучше двойной – она вся такая кругленькая и в стороны ничего не торчит. Боран может подобраться с любой стороны (в плоском олефине – только с двух сторон) и заместитель на тройной связи мешает гораздо меньше. Впрочем, это не только достоинство, но и недостаток, но об этом ниже.
Второй очень важный фактор, определяющий скорость согласованной реакции с бораном – относительная устойчивость кратной связи, к которой присоединяется боран – чем она меньше, тем быстрее присоединяется боран (почему объяснено в основной вкладке про гидроборирование в алкенах). Но мы уже выяснили, что пи-связь тройной связи менее стабильна – намного менее стабильна – чем пи-связь двойной связи. И это не менее важный фактор – алкины гидроборируются легче алкенов. Единственное исключение среди тех боранов, которые мы разобрали – BBN, но об этом ниже.
Высокая реакционная способность алкинов и очень маленькие стерические препятствия создают много проблем. Первая – низкая региоселективность при использовании обычного борана, в большинстве случаев просто неприемлемая – образуется смесь марковниковского и антимарковниковского продукта, во многих случаях не очень далекая от 1:1. Это неприемлемо.
Вторая проблема – двойное гидроборирование. Вернее, это не проблема, а осложнение. Не забывайте, что на боре остаются гидриды, и они тоже пойдут в дело – попробуйте нарисовать, что получится. И не забывайте, что первый боран в значительной степени присоединяется по Марковникову, поэтому продуктов получается немало. Это тем более неприемлемо.
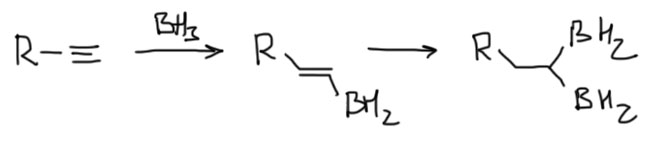
Поэтому для алкинов лучше совсем не использовать простой боран. Лучше ограничиться моно-гидроборированием, а для этого нужно сильно увеличить стерические препятствия, чтобы вторая молекула борана просто не влезла. Использование стерически затрудненных боранов, дисиамилборана и BBN, в этом случае почти обязательно.
Гидроборирование такими реагентами идет точно так же как обычным бораном, но “лезет” только одна молекула, поэтому нарисовать очень просто. Дисиамилборан намного более реакционноспособен чем 9-ББН, но иногда это даже плохо, поэтому под рукой держат оба (и еще десяток других). Стереохимия, как и положено в гидроборировании – син, то есть обе части реагента присоединяются с одной стороны. Обратите внимание, что направление гидроборирования определяется стерическими факторами, а не электронными (карбокатиона нет, а значит и стабилизировать нечего). Чаще всего поэтому гидроборируют терминальные алкины, в которых бор всегда идет к первому углероду. Если тройная связь внутри, образуются оба изомера, что плохо, поэтому предпочитают брать симметричные молекулы. Селективность еще неплохо проявляется, если с одной стороны маленький метил, а с другой что-нибудь явно побольше. 9-ББН при этом селективнее дисиамилборана, здесь вполне работает, если не забыли, лозунг “более реакционноспособный реагент менее селективен”.
А вот где ББН действительно блещет так это в способности выбирать концевую кратную связь в присутствии внутренней, причем это работает как для тройной, так и для двойной связи. Из этого иногда делают ошибочный вывод о преимуществе двойной связи перед тройной, но дело, как мы видим, не в типе связи, а в том, что громоздкий ББН просто не лезет в середину молекулы.
BBN отличается от других боранов тем, что он труднее реагирует даже с концевой тройной связью, чем с двойной. Очень вероятно, что это связано с необычной формой молекулы – атом бора с своим водородом в ней расположен как ручка у такой своеобразной корзинки. И чтобы произошло гидроборирование концевой тройной связи, она должна так изящно вдвинуться в середину этой корзинки водородом вперед – прямо внутрь корзинки. Беда в том, что корзинка непуста – в ней “лежат атомы”. Строго говоря, не лежат, а сама корзинка сделана из этих самых атомов, она очень толстая – атом водорода борана будет тыкать прямо в атомы корзинки, возникнет стерическое препятствие на пути к переходному состоянию гидроборирования. Поскольку водород все же невелик и запас некоторый там есть, то это не фатально – взаимодействие осуществиться, но вся эта толкотня сделает его более высоким по энергии, менее выгодным, а скорость гидроборирования будет меньше. Особенность BBN – именно в том, что у него жесткая структура, это такой каркас, где каждый атом находится на своем месте, и нет никакой повижности. Вот как это выглядит, если не учитывать некоторые важные вещи. Возникает область стерического отталкивания. 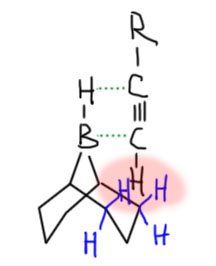
Эта картинка, с одной стороны, очень хорошо показывает суть проблем, которые называют стерическими препятствиями. С другой стороны, она немного нас дезинформирует. Если ей поверить совсем, то станет совершенно очевидно, что алкины с тройной связью внутри цепи вообще не должны реагировать с BBN – если уж водород там мешает, то что будет делать хотя бы метил – вообще рогом упрется в корзинку!? Ничего подобного, реагируют внутренние алкины с BBN, да, медленнее, чем концевые, но реагируют вполне, как например, следует из приведенного примера гидроборирования циклогексилпропина. В чем фокус? В том, что нельзя считать атомы точками, соединенными отрезками связей. Атомы имеют размер, и все разный. Если прикинуть молекулы BBN с размерами атомов (это те размеры, которые приблизительно показывают, насколько должны сблизиться молекулы, чтобы началось что-то, похожее на реакцию), то увидим более реалистическую картинку. Вот как это приблизительно выглядит с использованием реальной структуры BBN из данных рентгеноструктурного анализа всяких производных BBN. Сравнение с привычной нам структурой может показаться шокирующим – вместо такого проволочного каркаса с рогами видим такую пухленькую кругленькую фиговину, и не поймешь где там эти кресла. Да, это так и выглядит – именно так “видят” эту молекулу другие молекулы. Важно то, что мы в обычных структурах не совсем правильно представляем себе бор, он нам кажется таким маленьким, один из первых элементов. Бор, как элемент, близкий к началу периода, весьма объемист и пузат (розовый шарик), а водород, который мне пришлось пририсовать такой кипой на лысине бора (с приблизительным соблюдением длин связей и радиусов) весьма невелик. Поэтому ацетилен не будет подходить вертикально (как следует из проволочной структуры), а с некоторым углом, который выведет концевой атом водорода, ослабив стерические препятствия (но не убрав их совсем). Понятно, что на месте водорода вполне может быть и нечто более объемистое. 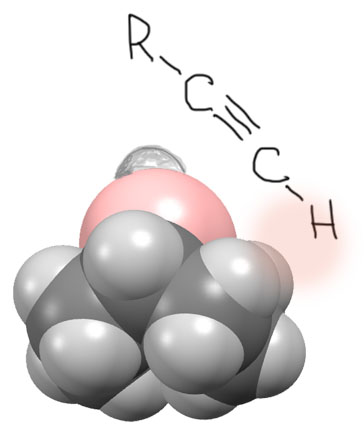
Гидроборирование алкинов - использование
Как и в случае алкенов продукты гидроборирование алкинов используются дальше в разнообразных синтезах. Связь C-B достаточно легко разрывается, причем из-за относительно большей электроотрицательности углерода эта связь поляризована так, что отрицательный заряд находится на этом атоме. Следовательно, бороорганические соединения – нуклеофилы, и подвергаются замещению борного остатка на электрофилы – это электрофильное замещение. Электрофильное замещение почти всегда происходит с сохранением стереохимической конфигурации, так как электрофил почти всегда подходит с той же стороны, где находится уходящая группа, в данном случае это бор вместе с теми заместителями, которые на нем остаются. Это очень удобно, ведь и гидроборирование – стереохимически селективная реакция.
Электрофилов, умеющих замещать бор в органоборанах пруд пруди, но нам стоит ограничиться тремя – электрофильным кислородом, галогеном, и протоном. Электрофильный кислород – это перекись водорода (как-нибудь отдельно разберем почему), бром – это бром, и с протоном тоже проблем вроде нет. В реальности проблемы есть, потому что нужно еще и подумать о судьбе борного остатка, который освобождает мсто для входящего эоектрофила. Если ни о чем не думать, то получится, что бор уходит в виде катиона. Но это страшно невыгодно. Вспомните, что трехвалентный бром – уже секстетный атом, а у катиона вообще было бы всего 4 валентных электрона – это слишком мало, атомы неметаллов, а бор все же какой-никакой, а неметалл, – не выносят такого унижения. Это у металлов вообще можно ободрать валентную оболочку до голого остова внутренних оболочек, а у неметаллов собственная гордость, валентные электроны в полном комплекте у них – как удостоверение личности неметалла. Самое большее (точнее, меньшее), на что они согласны, это 6 электронов, секстет, и то это всегда признак высокой реакционной способности и электрофильности. Итак, у бора проблемы. Решить их можно довольно просто – дать бору на время подержать еще один лиганд со своей парой, тогда электронов временно станет все 8, а после ухода восстановится шестерка, положенная бору по Таблице Менделеева, и все будут довольны. Поэтому в реакционных смесях всегда есть лиганды-основания: гидроксид, метоксид, или анион слабой кислоты.
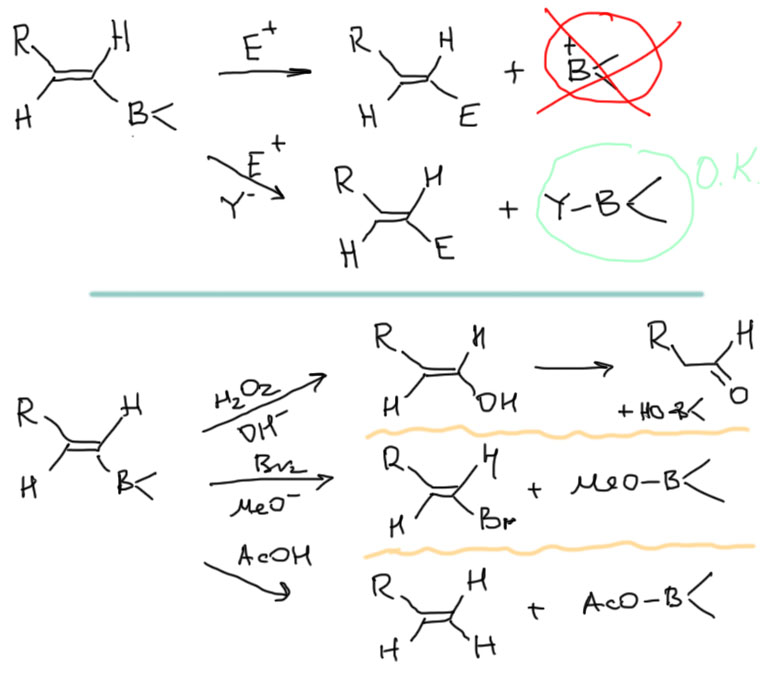
Обратите внимание, что в случае, когда электрофил из перекиси водорода (формально это HO+), первоначально образующийся непредельный спирт (енол) перегруппировывается в карбонильное соединение – альдегид (это мог бы быть и кетон, если бы мы гидроборировали не концевой алкин, но, как мы уже убедились, это редко делают из-за низкой селективности гидроборирования). Ещё обращу внимание на то, что во второй реакции бор замещается на галоген бром. Эта реакция здесь приведена как дань немного странной традиции использовать эту реакцию в учебных синтезах, хотя в реальности она идёт крайне плохо и разделить замещение бор на бром и присоединение брома к кратной связи невозможно. Тем не менее так получилось, что она часто используется в учебных задачах, и мы её пока оставим, потому что ничего криминального в ней нет, и более того, аналогичное превращение гораздо лучше можно сделать с иодом ровно по тому же механизму, а осложнений там меньше. Когда-нибудь я допилю до ума новую редакцию страницы про алкины и там получше разберу эту проблему ближе к реальности.
Конкуренция двойных и тройных связей в гидроборировании
Двойные и тройные связи имеют разную реакционную способность в гидроборировании, причём разница эта достаточно велика, чтобы было возможно в соединениях, содержащих и то, и это селективно добраться до одной из них. Безусловно, это не стопроцентно работающий подход, в химии вообще таких не бывает, и скорее надо сказать так – возможность есть, но насколько она реализуема, зависит от конкретной молекулы. Попробуем немного разобраться в проблеме.
Сразу скажем, что связи не должны быть в сопряжении. Гидроборирование вообще очень плохо работает для сопряженных кратных связей, что двойных, что тройных. Поэтому никогда не берите сопряженные диены и енины. Впрочем, сопряжённые диины вполне нормально гидроборируются большими боранами. Это очень специфическая химия, точно не для нашего курса.
1. Сам незамещённый боран в любом виде не годится для этой цели, он малоселективен и будет давать смеси продуктов почти во всех случаях.
2. Нам понадобятся стерически затруднённые, большие бораны. Можно ограничиться двумя – дисиамилбораном и BBN. Общая идея проста – большинство боранов предпочитает тройные связи двойным, а BBN – двойные тройным. Это не железный закон, а общая тенденция, со своими исключениями и заморочками. В общем случае нельзя сказать, что BBN будет цепляться за любую двойную связь, не замечая тройной, а дисиамилборан – наоборот.
3. Любое гидроборирование предпочитает терминальные кратные связи внутренним. Почти всегда терминальная связь имеет преимущество перед внутренней. Гидроборирование вообще в основном управляется стерикой, и только во вторую, даже в третью очередь – электронными эффектами. На втором месте среди факторов находится стабильность связи, а точнее даже нестабильноть – всё, что связь дестабилизирует. Дестабилизируют кратные связи та же стерика (поэтому цис-алкен обычно более реакционноспособен, чем транс), напряжение в циклах (это связано с размером и стереохимией циклов, пока оставим до темы Циклоалканы), сильнодонорные заместители.
Теперь посмотрим на те случаи, где у нас есть шанс селективности.
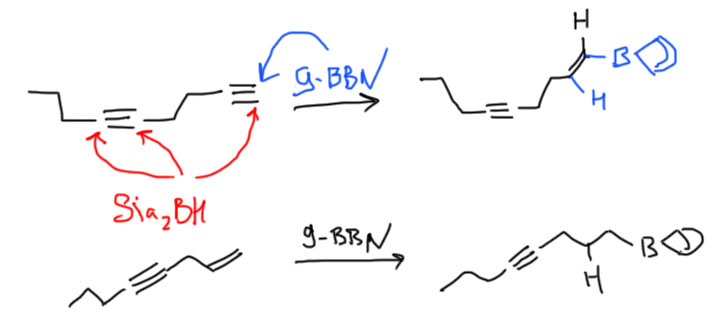
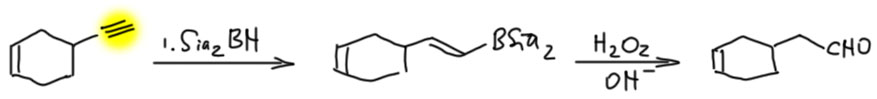
- Есть терминальная тройная связь и еще что-то внутри молекулы (двойная или тройная). Не проблема – берём один эквивалент дисиамилборана и гидроборируем тройную. Например,
- Есть терминальная двойная связь и ещё что-то внутри молекулы (двойная или тройная). Не проблема – берём один экв. BBN и гидроборируем терминальную двойную. Например,
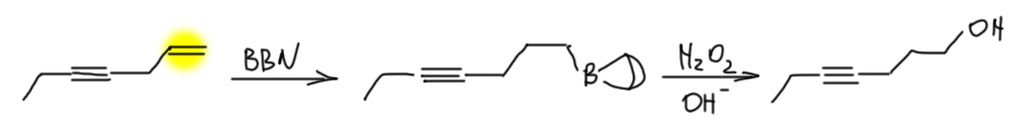
- Есть терминальная двойная и терминальная тройная связи. Здесь можно достать любую – двойную BBN-ом, тройную дисиамилбораном. Например,
- Есть внутренняя тройная связь и внутренняя двойная. До тройной можно добраться дисиамилбораном, до двойной – никак, но зато она останется незатронутой и сохранит всё самое ценное, что у неё было – положение и стереохимию. Единственная проблема – тройная связь будет гидроборироваться нерегиоселективно (если только там рядом нет с одной стороны какой-нибудь раскоряки, которая направит боран на дальний от себя углерод), поэтому единственное, для чего это можно использовать – это син-гидрирование тройной).
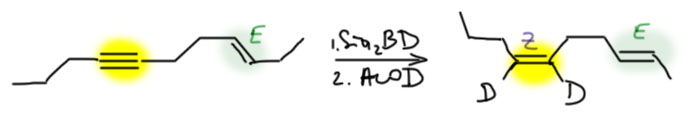
селективное гидрирование
Алкины можно и нужно уметь гидрировать до алкенов, причем чисто и стереоспецифично до цис или транс, если алкин дизамещенный. Это важнейшая реакция для целенаправленного синтеза чистых стереоизомеров алкенов. Более того, в нашей химии это единственный способ. И если в задаче видите, что от вас хотят определеные E/Z двойные связи, сразу прибегайте к этой реакции и ни к чему более. Никаких альтернатив у нее нет. В этом смысле это даже хорошо – думать не надо, только вспомнить. Но и в серьёзном синтезе и самом современном это очень востребованная реакция, позволяющая надёжно делать изомеры алкенов, поэтому её хорошо исследовали, для каждого типа восстановления есть несколько альтернативных методов.
Транс-гидрирование
Восстановление растворами щелочных металлов в аммиаке
Транс-гидрирование почти всегда делается одним единственным способом, восстановлением растворами щелочных металлов в жидком аммиаке. Все щелочные металлы, от лития до калия (последние два – перебор, хотя они тоже растворяются точно так же) растворяются в жидком аммиаке с образованием очень тёмных, почти черных, но на самом деле темно-синих растворов.
Что такое раствор щелочного металла в аммиаке
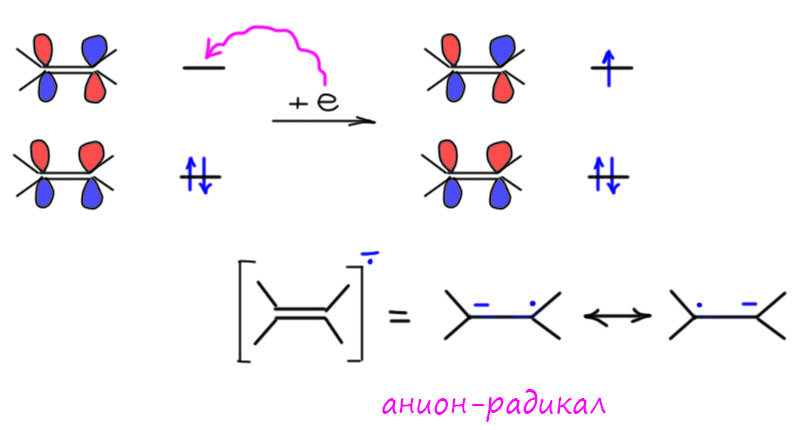
Растворы эти содержат сольватированные катионы металлов и электроны. Звучит малость дико, но это действительно так, и дело в том, что электроны по разному взаимодействуют с разными молекулами. Чаще всего лишние электроны прилипают, попадая на нижнюю свободную орбиталь, и образуя анион-радикалы. К многим другим молекулам электроны прилипают, но анион-радикалы часто неустойчивы и распадаются по слабым связям на радикал и анион, а они уже реагируют дальше. Так происходит, например, с водой – мы знаем, что бывает, когда щелочные металлы попадают в воду: на самом деле они просто отдают электроны, электроны садятся на связи O-H и разваливают их на анион на кислороде и радикал на водороде, радикалы сдваиваются в молекулу водорода. Но есть такие молекулы, к которым электроны не клеятся вообще никак – про такие молекулы говорят, что у них сродство к электрону равно нулю. Вот одной из таких молекул является аммиак. Но аммиак как молекула это одно дело, а аммиак как жидкость немного другое. В жидком аммиаке электроны могут существовать в поле многих молекул и ионов, они практически свободны, не связаны химически ни с какими молекулами, но несвободны в том смысле, что они связаны со всем раствором, потому что нужно сохранять общую электронейтральность, там же катионы щелочного металла. В этом смысле когда говорят, что растворение щелочных металлов в аммиаке даёт раствор электронов в аммиаке, это не совсем корректно, потому что на самом деле это раствор электронов в растворе катионов щелочного металла в аммиаке. Не было бы там катионов щелочного металла, не было бы и раствора. Не верите, найдите какой-нибудь источник электронов, например, катод, или вообще найдите источник электронного пучка, и опустите его в жидкий аммиак, попробуйте налить чистых электронов из катода в аммиак, увидите, что ни черта не выйдет, не пойдут. Это безусловно троллинг, но не совсем тупой – иногда полезно проделывать такие мысленные эксперименты. Катионы нужны.
Вообще, если подумать, что это очень интересная система получилась, немного похожая на сам металл. При этом при увеличении концентрации, а щелочные (и щелочноземельные металлы) отлично растворимы в аммиаке, с большим тепловым эффектом, появляется у раствора металлический блеск, обычно золотой, и возникает настоящая проводимость металлического типа – если приложить напряжение к электродам, то потечет ток, и носителями заряда будут электроны (катионы перемещаются намного медленнее, и этой составляющей тока можно пренебречь). При растворении металла аммиак кипит – будете сами делать, не торопитесь, это может быть не менее опасно, чем растворение щелочного металла в воде. Если бросить кусочек побольше, то недалеко до проблем, иногда это так бурно, что даже кажется, что вспыхивают огоньки, и только немаленькая теплоемкость аммиака и то, что реакция идет при минус 33 градусах, делают этот процесс все же менее опасным, чем растворение в воде, что совсем невозможно сделать без эксцессов. Значительный тепловой эффект как раз и говорит, что процесс выгодный – металлу выгодно отдавать электроны, превращаясь в сольватированный катион. На досуге можете подумать о таком парадоксе: при растворении металла в аммиаке металл окисляется, образуется катион. Но с аммиаком ничего не происходит (кроме того, что часть молекул становятся лигандами, но от этого степени окисления не изменяются). А кто тогда окислил металл? Неужели мы нашли окислительно-восстановительную реакцию, у которой есть восстановитель (металл), но нет окислителя! А теперь напишите ту же реакцию в обратную сторону – получится восстановление катиона металла электронами, или что то же самое, электрофхимическое восстановление, причём только полуреакция. Чудны дела!
И как это восстанавливает?
И вот, если в такую удивительную систему добавлять разные органические соединения, то у электронов появится возможность переехать от сольватированных катионов металла, с которыми они связаны только электростатически, на настоящие молекулы, где для электронов может найтись более достойная работа – делать новые ковалентные связи. В самый первый момент этот процесс очень похож на электрохимическое восстановление – электрон попадает на низшую свободную орбиталь молекул, у кого какая есть. Есть только одна разница с электрохимическим восстановлением – на катоде можно создать любой отрицательный потенциал и вколотить электроны в любую молекулу, которая принципиально может принять электроны (напомню, что не любая молекула может принять электроны, а только такая, у которой положительно сродство к электрону; например, тот же аммиак, алканы, алифатические амины электронов не принимают). А раствор щелочных металлов в аммиаке похож на катод с определенным, заранее заданным и неизменным потеницалом, этот потенциал значителен, но ограничен. Поэтому в этой сиуации восстанавливаться будут только те молекулы, потенциал восстановления которых меньше (по абсолютной величине) некоторой величины. Цифрами бросаться не будем, они нам ничего не скажут, просто поймем качественно, как это устроено.
Тройная связь – один из зачётных объектов для восстановления в этой системе. Мы уже выяснили, что увеличение порядка связи от одинарной йк двойной и дальше к тройной сильно дестабилизирует связь. Еще раз вернитесь к обсуждению этого парадокса – тройная связь прочнее всех как целое (относительно полного разрыва пополам), но при этом она крайне неустойчива относительно превращения в двойную – это всегда сильно экзотермический процесс и только отсутствие подходящего механизма спасает тройную связь от того, чтобы реагировать с первой попавшейся молекулой. Я в сотый раз это повторю, потому то это важнейшая идея, которой нужно пропитаться так, как настоящая ромовая баба, какие бывают только в Неаполе, пропитана ромовым сиропом, – чтоб аж текло: молекула может быть сколь угодно неустойчива в термодинамическом смысле, но если нет механизма ее реакции с другими молекулами в данной системе, то она будет жить вечно, или пока в систему не запустят другие молекулы, у которых есть механизм. Механизмы реакций – важнейшее, что только есть в химии. Химия была поваренной книгой, пока в ней не появилась теория механизмов. Хотите понимать химию, а не только запоминать, изучайте механизмы, разбирайтесь в них, добивайтесь понимания, как они работают.
Это я все время повторяю, потому что сколько раз это не повторяй, обязательно найдется некто, кто изумленно спросит: “Вот, некоторые говорят, что тройная связь неустойчива. А почему же у меня на полке стоит баночка гексина (пропаргилового спирта, ацетилендикарбоновой кислоты и т.п.) и не разлагается? А почему у меня под окном стоит баллон ацетилена и не взрывается?”
Двойная связь тоже неустойчива и тоже с удовольствием вступает в многочисленные реакции присоединения, но не все – опять нужен механизм. И неустойчивость двойной связи даже близко не напоминает неустойчивость тройной. Посмотрим, что будет, если на двойную и на тройную связь попадает электрон, в чём разница. Формально, разницы нет, и там и там электрон попадает на разрыхляющую орбиталь, а это у π-связей всегда одно и то же – две p-орбитали в противофазе. То, что получается, называется анион-радикалом. Такие двойные слова через дефис обычно означают нечто такое, то не является ни одним из слов по отдельности: генерал-майор, например, это и не генерал, и не майор, а кресло-качалка это и не кресло, и не качалка. И анион-радикалы у многих молекул это тоже нечто особенное, некая частица с дополнительным электроном, который может быть как-нибудь затейливо делокализован. Когда дойдет до восстановления ароматических соединений в той же системе, увидим такой случай. Но здесь у нас нет делокализации, восстановлению подвергается одна связь, и результат попадания электрона это действительно одновременно и радикал, такую вещь можно по-разному себе представить: если воображение хорошее можете подумать о слабой трехэлектронной связи – это та же связь, что и была (потому что пара связевых электронов на связывающей орбитали никуда не делась), но она сильно ослаблена, потому что еще один электрон на разрыхляющей орбитали значительно усиливает отталкивание (любая связь это компромисс притяжения и отталкивания, в котором преобладает притяжение) – связь удлиняется, ослабляется. Вполне уместно рисовать для такой связи мезомерию между двумя анионами и радикалами, и это правильно, потому что мы не знаем, где у нас анион и где радикал, и одна формула была бы недостаточна. В общем, можно сказать, что π-связь рвётся и дальше с анион-радикалом что-то происходит, и это что-то можно предсказать потому что мы знаем химию и радикалов и анионов – будет много интересного, скорее всего неселективного, возможно, просто полимеризация. Но нам это не нужно, потому что электрон можно загнать на обычную двойную связь (не сопряженную) только электрохимически, а раствор щелочного металла в аммиаке не может этого сделать, не хватает восстановительного потенциала.
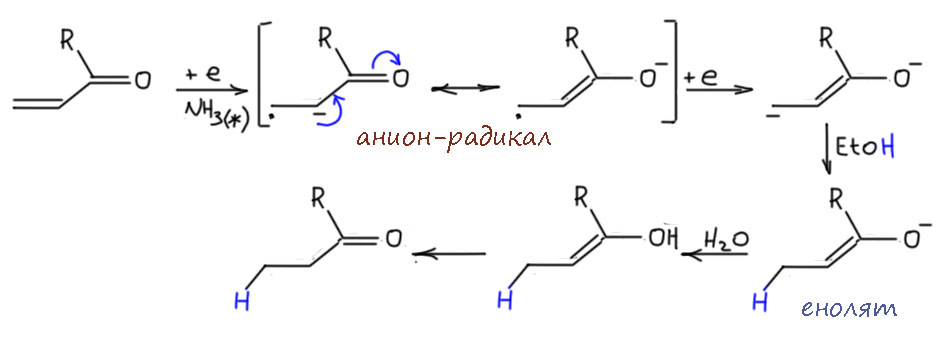
Но если двойная связь сопряжена с хорошей акцепторной группой, напрмиер, карбонилом, то перенос электрона становится выгодным, потмоу что анион-радикал стабилизирован сопряжением, минус уходит в акцепторную группу, а радикал принимает ещё один электрон – это хорошо известная реакция сопряженного восстановления непредельных карбонильных соединений, и мы ее еще раз изучим и будем много применять, когда доберёмся до этих производных.
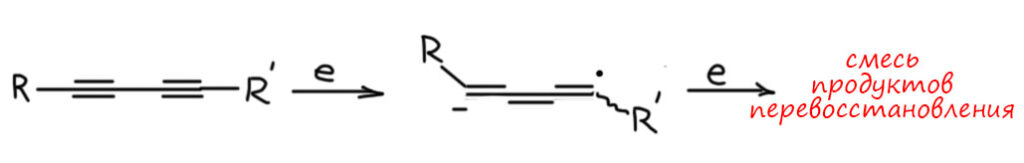
Если вместо алкена взять алкин, в общих чертах будет то же самое, но в деталях – другое. Детали важны. Во-первых, у алкина две орбитали соответствующие π-связям, вернее две связывающие и две разрыхляющие. Каждая пара имеет одинаковые энергии, они вырожденные, и это очень понятно, потому что мы не найдем ни одной причины, чтобы они могли быть разными. Можно использовать симметрию, а если не хочется, то просто подумать о том, что заместители на тройной связи находятся на прямой линии связи и совершенно одинаково относительно орбиталей π-связей. Фокус в том, что электрон попадает на одну из них, и как только попадает, возникает проблема. Орбитали остаются одинаковыми или нет? Если мы наивно считаем орбитали такими полочками для электронов, прибитыми гвоздями к оси энергий, то конечно нет – полочка же не сдвигается, если на неё поставить горшок с цветком. Но орбитали – не полочки, это уровни энергии электронов в молекуле, и раз молекула изменилась (был алкин, стал анион-радикал алкина), то изменились и уровни энергии. И конечно, стали разными, ведь на одном есть электрон, а на другом нет. Есть другой подход к тому же самому: если бы уровни не изменились, то у нас получились бы вырожденные орбитали с одним электроном – и на какую его поставить. Квантовая наука не любит таких ситуаций, очень не любит. Молекула будет стремиться изменить геометрию так, чтобы вырождение оказалось снято и уровни энергии (орбитали) разведены, электрон окажется на нижнем, и все успокоится. В случае тройной связи самое простое изменение состоит в том, что заместители уйдут с прямой, а это то же самое, что мы называем изменением гибридизации. Было sp- станет sp2.
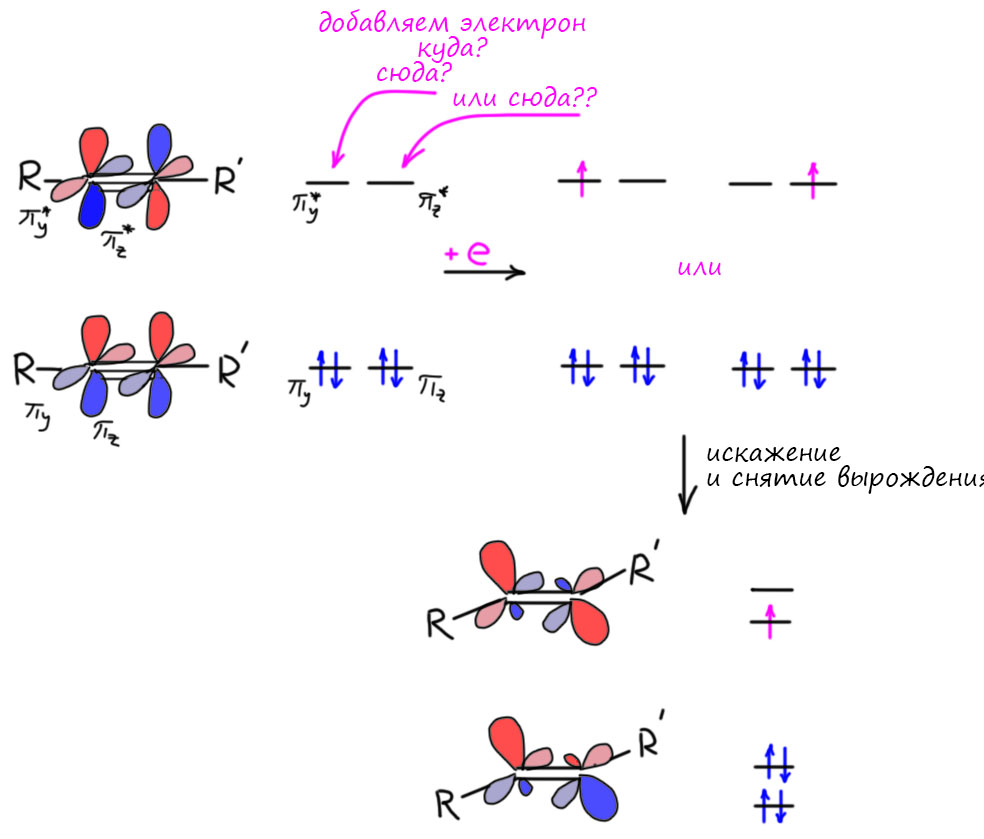 Исказится-то она исказится, но в какую сторону. Можно придумать два способа: в одну сторону, син-, или в разные – анти-. Выбрать легко, в таких случаях почти всегда побеждает самое простое – отталкивание, и в смысле стерического отталкивания заместителей на тройной связи, и в смысле отталкивания электронов на новых, гибридных орбиталях – там их, электронов всего три, но электроны одного спина отталкиваются, и этого достаточно. Стерика впрочем не менее важна – мы отлично знаем по химии алкенов, что цис-алкены менее стабильны чем транс-алкены из-за взаимного отталкивания заместителей. Если заместители маленькие, это очень маленькая величина, не более 1 ккал/моль, но и это важно. А если побольше что-то, то разница становится значительной.
Исказится-то она исказится, но в какую сторону. Можно придумать два способа: в одну сторону, син-, или в разные – анти-. Выбрать легко, в таких случаях почти всегда побеждает самое простое – отталкивание, и в смысле стерического отталкивания заместителей на тройной связи, и в смысле отталкивания электронов на новых, гибридных орбиталях – там их, электронов всего три, но электроны одного спина отталкиваются, и этого достаточно. Стерика впрочем не менее важна – мы отлично знаем по химии алкенов, что цис-алкены менее стабильны чем транс-алкены из-за взаимного отталкивания заместителей. Если заместители маленькие, это очень маленькая величина, не более 1 ккал/моль, но и это важно. А если побольше что-то, то разница становится значительной.
![]()
Возможно еще и второе восстановление ещё одним электроном уже в дианион, радикалы обычно очень легко восстанавливаются в анионы (это то же самое, что с обратным знаком взятая энергия удаления одного электрона из аниона на углероде, а это очень небольшая энергия по определению, ведь углерод – далеко не самый электроотрицатеотный неметалл), но в простом несопряжённом алкене вторе восстановление приведет к дианиону на соседних атомах, а это очень невыгодно. Вот в уже рассмотренном сопряженном кетоне первый минус уходит далеко, и радикал восстанавливается легко.
Каждый анион забирает протон от источника протонов, кислоты Бренстеда-Лоури с достаточной кислотностью. Оценим её. Образующиеся анионы имеют минус на sp2-гибридном углероде, а это мы уже разобрали на вкладке про зависимость разных свойств от гибридизации углерода, и увидели там, что CH-кислотность на sp2-гибридном углероде находится посредине между весьма немаленькой кислотностью терминального ацетилена (sp-гибридный углерод) и чрезвычайно слабой кислотностью на насыщенном sp3-гибридном атоме. Итого, это что-то в окрестностях рК 35-37. Аммиак недостаточно кислотен для протонирования такого карбаниона (рК аммиака это что-то в районе 43-44, не путайте, имеется в виду кислотность, а не основность). Вот почему в такие смеси всегда добавдяют два эквивалента или чуть-чуть больше спиртов, обычно или этанола, или трет-бутанола, которые достаточно кислотны для протонирования карбанионов. Итого, в результате переноса двух электронов из раствора и протонирования образующихся карбанионов спиртом мы получаем почти всегда чистейший транс-изомер алкена. Это очень надежный, хотя и довольно хлопотный способ восстановления.
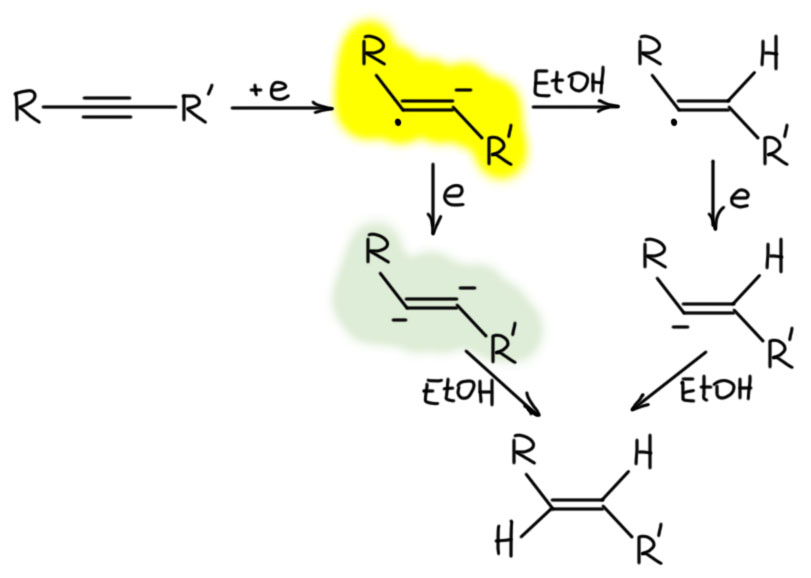
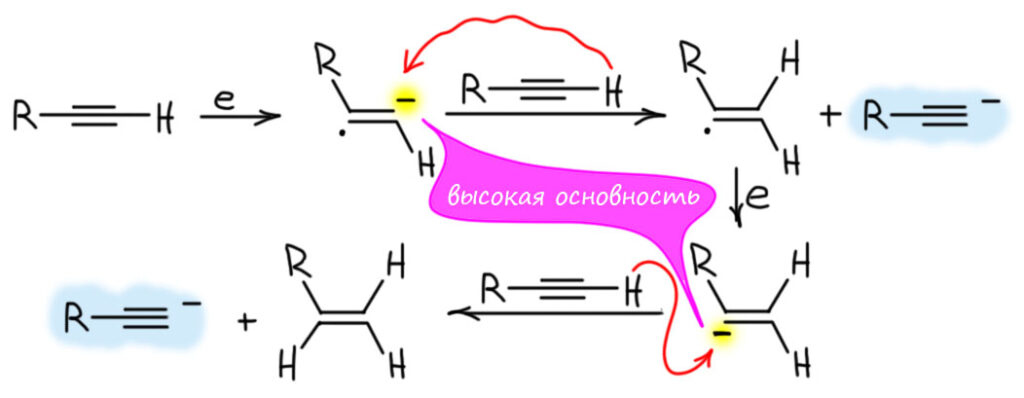
К сожалению, это способ применим только к дизамещенным ацетиленам. Ни сам ацетилен, ни монозамещенные не восстанавливаются в заметной степени. Дело, видимо, в том, что восстановление начинается, и карбанионы винильного типа образуются, но дальше они просто забирают протоны от исходного ацетилена. Таким образом, алкин в основном превращается в ацетиленид, который не принимает еще электроны, пскольку и так уже имеет отрицательный заряд. Небольшая часть исходного алкина, конечно, восстановится, но выход даже теоретически не может быть больше 33%, а в реальности гораздо меньше, что делает процесс бессмысленным. Напрашивается вопрос – а почему протон забирают от ацетилена, а не от спирта, ведь спирт – более сильная кислота Бренстеда-Лоури, чем терминальный ацетилен. Это типичное заблуждение, происходящее от того, что бы берем неправильные величины. Когда говорят о кислотности спиртов, обычно выплывают величины рК в районе 16-17, что конечно, намного лучше чем кислотность ацетиленов. Увы, эти величины для спиртов получены для раствора в воде. Как в воде?! Нельзя же получить алкоголяты в водном расторе. Алкоголяты получить нельзя, а кислотность оценить можно. Вообще, мы всегда преувеличиваем, когда говорим, что кислотность воды намного больше кислотности спиртов, и алкоголяты полностью гидролизованы. Это не совсем так, там разница не больше 2 единиц рК, что соврешенно не мешает равновесной оценке кислотности. Но кислотность спиртов в неводных, а особенно апротонных растворителях намного меньше, на много единиц рК, в ДМСО она достигает 30-31 и сильно опережает кислотность ацетиленов. Ну а в аммиаке? Точно это не известно, никто не мерил, но из общих соображений можно понять, что это хоть и не ДМСО, но тоже растворитель апротонный (более апротонный, чем ДМСО, у которого кислотность намного выше, чем у аммиака – и обратите в этом месте внимание, что апротонность растворителя понятие относительное – относительное чего? – относительное основности содержащихся в конкретной реакционной смеси оснований) и умеренно полярный, поэтому кислотность спиртов в нем тоже сильно понижена – для протонирования винильных анионов хватает с запасом, ацетиленидов – нет.
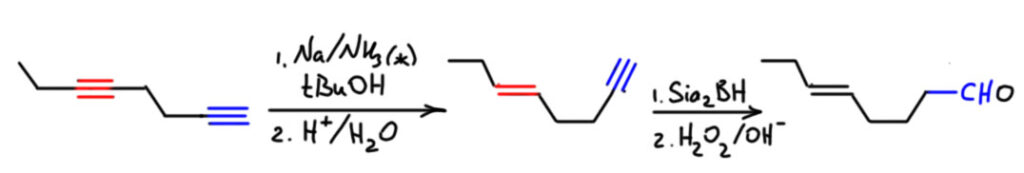
Если в соединении несколько тройных связей, то а) востанавливаются только внутренние, и это неплохо – позволяет оставить нетронутыми терминальные, и дальше развить на них синтез, как в этом примере, где терминальную тройную связь дальше превращают в альдегид:
Еще одна особенность этого метода – им нельзя восстанавливать сопряжённые диины. А так хочется – ведь нам нужны транс-транс-диены, например для реакции Дильса-Альдера. И это даже иногда попадается в задачах. Здесь фокус такой – если вы примените это на бумаге, почти никто ругаться не будет (это плохой совет – кто-нибудь обязательно будет и вам не повезёт, но от безысходности лучше так, чем никак). Но в реальности это не работает, и хорошо понятно почему – две сопряженные тройные связи образуют единую систему, электроны попадают на свободные орбитали этой системы,, и это, по идее, должно приводить к образованию трех двойных связей подряд (как у аллена, но с еще одной двойной связью – такие системыназываются кумуленами). Такое нагромождение двойных связей неустойчиво, а это значит, что оно предпочтет взять еще электроны и восстановиться дальше. В результате будет несколько продуктов перевосстановления, и ни о какой селективности, не то что стерео, но и просто относительно степени восстановления мечтать не приходится, а делить такие смеси тоже желающих не находится. Поэтому метод просто не применяют.
Резюмируя заметим, что для транс-гидрирования алкинов ничего лучше никто так и не придумал, поэтому эту реакцию продолжают делать, когда такая потребность есть несмотря ни на какие сложности. В реальной жизни аммиачная методика имеет еще один недостаток – аммиак не самый лучший растворитель для органики, потому что это растворитель, отдаленно напоминающий воду, состояший из полярных молекул, связанных водородными связями (всё намного слабее, чем в воде, поэтому жидкий аммиак растворяет органику, если в ней не очень много углеродов и она не слишком неполярна). Плохая растворимость органических соединений побольше размером вызывает необходимость использовать очень большие объёмы аммиака, потому что если вещество не растворилось и вы этого не заметили, то после реакции вы обнаружите очень большое количество непрореагировавшего, и куча аммиака, лития или натрия, времени и сил пойдет коту под хвост. Мне один раз пришлось так восстанавливать сопряженную двойную связь в одном стероиде, а стероиды это обычно малополярные гидрофобные вещества, и на 1 грамм исходного пришлось брать поллитра жидкого аммиака, проблема еще в том, что в очень тёмном растворе вообще ни чёрта не видно, и растворилось там вещество или нет приходится гадать, и перестраховываться).
Восстановление алюмогидридом лития
Кроме растворов щелочных металлов в жидком аммиаке для транс-гидрирования алкинов иногда применяют алюмогидрид лития. Это очень странная реакция, которая толком не описана нигде, и в большинстве книг по синтезу даже не упоминается. Но она есть, и ее эпизодически применяли в реальных синтезах. Самое приятное ее отличие от восстановления в аммиаке это пригодность для восстановдения сопряженных диинов, которое собственно и оправдывает наш интерес к ней. Без этого смысла упоминать ее не было бы никакого, так как аммиачная методика намного надежнее.
Поскольку эту реакцию толком никто не описывал и не исследовал, остается гадать, как она идет. Проблема очевидна – алюмогидрид это вроде бы гидрид алюминия, похож на гидриды бора, и по всем признакам такие реагенты должны присоединяться син-, а не анти. Но, видимо, фокус в том, что это не столько гидрид элемента, сколько анионный гидридный комплекс, и он не присоединяется так же как боран (или алан), согласованно, в четырехчленном переходном состоянии по очень простой причине – в анионных комплексах нет электрофильного атома бора или алюминия. Нейтральные бораны и аланы – реагенты электрофильные, они цепляются за кратную связь электронодефицитным секстетным атомом бора или алюминия, а дальше реакция уже развивается как согласованный процесс.
В анионном комплексе вообще нет электрофильного атома, и это нуклеофильный реагент, донор гидрид-иона. Проблема только в том, что в тройную связь гидрид просто так не идет, невыгодно.
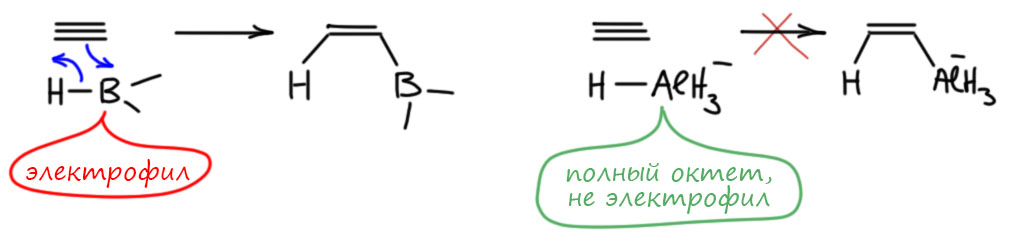
Поэтому и нет гидридных восстановителей для тройной связи (мы обсуждаем реагенты – доноры гидрида в теме про восстановление карбонильной группы). Вот только алюмогидрид, но и здесь реакция требует весьма жестких условий, обычно это нагревание до 140-150º в растворе диглима. Диглим – это диметиловый эфир диэтиленгликоля. Дело в том, что алюмогидрид лития – твердое кристаллическое вещество, растворимое только в эфирных растворителях (эфире, ТГФ и т.п. – простых эфирах) за счет сильного взаимодействия катиона лития с неподеленными парами кислорода. Сольватированный катион лития идет в раствор, ну и за ним тянется анион.

Диглим берут тогда, когда нужно реакцию посильнее нагреть, до 150º и даже чуть выше (температура кипения 162º). Если будете работать с этим растворитетем, учтите, что он очень опасен. Он точно так же как эфир и ТГФ дает перекиси при стоянии с доступом воздуха, но если низкокипящие эфир и ТГФ могут вас иногда простить, если вы их забудете очистить от перекиси перед использованием (а когда вы привыкнете к халтуре, вот тогда и жахнет), то нагревание до 150º диглима даже с маленькой примесью перекиси это почти то же самое, что поджаривание на гриле гранаты – разложение перекиси вызовет выброс сильно нагретой реакционной смеси, и это даже если не взорвется, воспламенится и гореть будет славно. Диглим перед использованием обязательно проверяют и тщательно очищают, нужно посмотреть конкретные методики в руководствах по очистке растворителей.
Итак, почему работает алюмогидрид, и почему транс. Моя гипотеза такова: в условиях реакции образуется небольшое количество алана, и он активирует тройную связь к присоединению гидрида, обычному нуклеофильному, а не согласованному. Образующийся при этом винильный карбанион подхватывает алан, образуя анионный комплекс, который же и живет до конца реакции, пока смесь не гидролизуют. Алан может образоваться как при термической диссоциации комплекса, так и при действии примесей. Интересно то, что в литературе эту реакцию чаще всего описывают, как идущую хорошо только для алкинов, в молекуле которых есть гидроксильная группа, причем не очень важно, в каком положении, рядом, через углерод, а иногда и очень далеко, до 7-8 атомов углерода (Rossi R., Carpita, A. Synthesis 1977, 561), что полностью исключает участие такой группы в внутримолекулярном направлении реагента на тройную связь. Странно, что до сих пор никто не догадался просто добавить в реакцию с простыми алкинами чуть-чуть любого спирта. Спирты вообще взаимодействуют с алюмогидридом с замещением гидридных лигандов на алкоксидные – так получают более селективные восстановители типа трис-третбутоксиалюмогидрида или Red-Al, но при высокой температуре реакция может быть обратима. Гидроксильная группа может как раз способствовать образованию небольшого количества алана. То есть, что-то типа такого:
![]()
Ещё раз предупреждаю, что никаких доказательств такого механизма нет, это моя гипотеза. Почему транс? Это обычная стереохимия, когда нуклеофил (здесь гидрид) присоединяется к двойной или тройной связи, на которой висит кислота Льюиса (здесь алан), и в результате и нуклеофил и кислота Льюиса садятся на порванную связь с двух сторон. В современной органике несметное количество таких реакций. Почему алан сам не присоединяется как боран? Хорошо известно, что у аланов реакционная способность ниже, поэтому реакции гидроалюминирования далеко не так полезны как реакции гидроборирования; к тому же реакция идет при нагревании, часто сильном, а в таких условиях даже гидроборирование смещено в стороны исходных (напоминаю про обратимость гидроборирования, и про то, что это очень важное явление в химии гидроборирования).
Повторю, что особое значение этой реакции придает то, что ее реально применяют для транс-транс-гидрирования сопряженных диацетиленов. В подавляющем количестве описанных реакций этого типа где-то в молекуле есть гидроксил, хотя бы один, хотя бы далеко. Но нам можно применять эту реакцию, когда нужно иметь транс-транс-изомер диена для реакции Дильса-Альдера. Здесь очень простое соображение – если такая реаккция попалась в задачах, значит составитель задач допустил лажу, такое бывает, составители живые люди, у них бывают провалы в памяти, да и голова иногда болит (по себе знаю) – и что вам тогда делать, соединение нужно по цепи синтеза. Но мы всё же надеемся, что составители страдают всем этим не очень часто, а значит такое может случиться очень редко.
Напомню, что цис-цис обычно вообще не реагирует по стерическим причинам в циклоприсоединении. В большом синтезе диены с конкретной стереохимией уже давно не получают гидрированием алкинов, там для этого есть кросс-сочетание, но нам делать нечего, для нас это полезная реакция.
![]()
Никаких других методов транс-гидрирования нет. В самой современной химии есть новые реакции, использующие катализ комплексами переходных металлов, но мы это использовать не будем, там все очень сложно и с катализаторами и с механизмами.
Цис-гидрирование по Линдлару
В отличие от транс-гидрирование, где есть полтора метода, и самый главный метод не работает в двух очень важных случаях – с терминальными ацетиленами и сопряжёнными диинами, а второй работает от случая к случаю и никогда толком не исследовался, для цис-гидрирования методов напридумывали с десяток. И они гораздо гибче, и с этими двумя частными случаями справляются играючи. Они очень популярны в синтезе, и это оправдано – дело в том, что и транс-олефины и транс-транс-диены легко получить другими способами, особенно современным кросс-сочетанием из доступных исходных. Кросс-сочетанием можно получить любые диены, но для цис-цис исходные гораздо менее доступны, а диины как раз очень легкодоступны, поэтому такое восстановление имеет большой смысл. Такая же история и с цис-олефинами.
Гидрирование по Линдлару
Это первый (описан в 1952 году) и самый популярный до сих пор метод цис-гидрирования алкинов до цис-алкенов. Обычные катализаторы гидрирования типа палладия на угле и катализаторов на основе платины и никеля гидрируют алкины со свистом до алканов, остановиться на алкенах невозможно. Более того, алкены гидрируются намного быстрее алкинов. Даже если отмерить количество водорода, соответствующее одному эквиваленту, получите смесь алкана и непрореагировавшего алкина, так что смысла в этом нет. В промышленности такое частичное гидрирование делается довольно легко, потому что в промышленных реакторах можно регулировать время контакта реакционной смеси и катализатора, но в обычной лаборатории это очень сложно.
Герберт Линдлар (английский промышленный химик, проработавший всю жизнь в Швейцарии на знаменитой фарм-компании Офман Ля Рош, прожил ровно сто лет, 1909-2009, органики часто живут долго, но до ста лет всё же далеко не все, хотя другие примеры есть) предложил решение. Это очень важная работа, потому что химия алкинов и возможность получения чистых стереоизомеров алкенов очень много применяются в синтезе при разработке лекарств, не зря и работа появилась у одной из самых передовых в смысле разработки и синтеза новых субстанций фармацевтической компании. Мы не зря так подробно это изучаем и используем в задачах – это очень полезная химия даже до сих пор, хотя сейчас очень много других методов. Линдлар за свою долгую деятельность ничем больше не прославился, но этой разработки вполне достаточно для того, чтобы занять очень почетное место и оставить своё имя потомкам. Цис-гидрирование алкинов применяли в тысячах синтезов, количество примеров реального использования совершенно бесконечно. После открытия Линдлара предложено очень много других методов, очень хороших, часть мы разберем ниже, но даже самые недавние обзоры утверждают, что за методом Линдлара до сих пор безусловное первенство. Вот что значит в химии хорошая идея – пара опытов и Олимп ваш, устраивайтесь поудобнее рядом с другими богами, впереди вечность. Тоска какая, чёрт возьми, слава богам мне никогда ничего подобного в голову не приходило.
Решение было очень похоже на катализатор Розенмунда, предложенный в 1918 для селективного гидрирования хлорангидридов в альдегиды учеником Дильса Карлом Розенмундом. Обычно это решение описывают как частичное отравление катализатора палладиевого катализатора гидрирования. Но это очень странное описание. Отравление – это порча . А здесь, и в случае Розенмунда и в случае Линдлара никто ничего не портил, катализаторы получились отличные, очень активные, но более селективные – не вызывают перевосстановления и других побочных, точнее, сводят всё это к разумному минимуму – не бывает абсолютно селективных решений, а здесь задача очень непроста – как изменить катализатор так, чтобы противопоставить что-то скорости реакции – алкены все равно гидрируются быстрее алкинов, значит действовать надо не через скорости реакций. Изобретение высокоселктивных реагентов, которые вызывают только желаемое превращение и не безобразничают, одна из главнейших задач органического синтеза, и странно считать, что эта задача решается отравлением. Типа, травили-травили, но не помер, а стал круче прежнего. Да, бывает, знаем, но не в химии.
Оба этих катализатора используют один и тот же приём – палладий наносится на очень “плохой” носитель. Знаменитые гетерогенные катализаторы гидрирования, да и многих других реакций, получаются нанесением активных частиц на материалы с очень большой и тоже активной поверхностью – силикагель, окись алюминия, алюмосиликатные минералы, цеолиты, углерод, и так далее. На таких носителях на поверхности есть множество активных групп и центров, с которым химически связываются частички катализатора, и при последующей активации это дает множество активных центров с очень высокой дисперсностью, да и сама поверхность подложки не инертна, а часто помогает реакциям кислотным со-катализом, активной сорбцией, или еще как-то. И Розенмунд и Линдлар пошли другим путем – они взяли в качестве носителей простые малорастворимые соли, по всем параметрам совсем неподходящие, просто дрянь какую-то. Даже как-то неудобно за крупных химиков, зачем они так… Постойте браниться, у них же всё получилось, сто лет пользуемся.
Розенмунд взял свежеосажденный сульфат бария, Линдлар – такой же карбонат кальция. Эти осадки считаются очень мелкими, недаром есть особая категория самых плотных фильтров для фильтрования осадков сульфата бария. Но и то, и то – кристаллики, барита или кальцита, мелкие, но это нельзя называть мелкодисперсной фазой с большой поверхностью. Поверхность невелика, а кроме того, она инертна и частички палладия держит скорее за счет физических сил, чем химических связей-якорей. Когда палладий наносят, а это делают очень просто, добавляя раствор подходящей формы, хлорида палладия в случае Розенмунда, или гидроксикомплекса палладия в случае Линдлара, затем добавляя восстановитель, формиат, формальдегид, гидразин или что-то типа этого – палладий восстанавливается и как-то связывается с поверхностью, даже трудно понять как, возможно просто физически, за счет электростатики или поляризуемости, но очень слабо. Кстати, когда говорят про всякие нанесенные катализаторы на подложках, часто воображают такую красивую поверхность (картинки Марса в этом момент вспоминаются из роликов NASA) и на ней такие блестящие шарики палладия или другого металла, ждут водород и алкин, как актиния на дне морском поджидает проплывающих рыбок. Увы, это не совсем точная картинка даже для более серьёзных катализаторов гидрирования. Палладий там не в виде отдельных атомов, связанных с поверхностью, а в виде довольно крупных частичек металла, буквально приклеившихся к поверхности обычно довольно слабыми физическими силами типа поляризаемости и кулоновского притяжения.
Когда поверхность такая слабая, как у сульфата бария или карбоната кальция, ни о каком равномерном распределении каталитических центров речи быть не может, это скорее похоже на почти механическую смесь крупных частиц палладия (крупных – это от нескольких нанометров до нескольких сот нанометров, то есть от сотен и тысяч атомов до сотен тысяч и миллионов атомов на частичку) и носителя. Носитель выполняет довольно примитивную роль – просто не дает частичкам палладия слипаться дальше, сохраняет проницаемость катализатора, ведь в реакции нужно, чтобы до поверхности свободно добирался водород и реагент (хлорангидрид или алкин).
Дальше, в случае Линдлара добавляют небольшое количество ацетата свинца. В условиях приготовления нет сомнения в том, что свинец в виде карбоната осаждается на поверхности. Зачем? Чёрт знает. Рассказы о том, что свинец-де тоже “каталитический яд” довольно смешны и явно идут откуда-то из проблем с автомобилями, где использование этилированного бензина с добавками тетраэтилсвинца убивает катализатор дожига. Больше никаких проблем от катионов свинца в катализе мы не знаем. Возможно, они даже помогают, например, стабилизируют поверхность и улучшают связывание каталитических частиц. В современной химии гетерогенного катализа очень много исследуют катализаторы с двумя или тремя металлами, там все очень сложно, металлы-помощники как-то правильно влияют на форму частиц катализатора, распределение, устойчивость, и так далее. Это не химия в нашем смысле, а исследование поверхности, поэтому не будем говорить глупости, а просто примем, что такая методика приготовления даёт лучшую активность. Если вас когда-нибудь занесёт в гетерогенный катализ и методы приготовления особенно промышленных катализаторов, увидите, что это поти священнодействие, куча всяких дополнительных компонентов создает оптимальный катализатор, и никто не знает почему, хотя работы, особенно современные, когда появилось куча приборов для исследования поверхности, будет вам впаривать красивые модели каталитического действия.
И это еще не всё. Когда ведут саму реакцию, а это и там, и там очень просто – обычная колбочка с магнитной мешалкой, соединенная с газовой бюреткой или просто резиновой камерой так как никакого давления для этих реакций не нужно, – в реакционную смесь добавляют немного реагента, который часто опять называют “каталитическим ядом”. Для катализатора Линдлара это обычно несколько процентов хинолина, для катализатора Розенмунда что-нибудь сернистое, например, тиомеочевина, или смесь хинолина с серой, но в отличие от Линдлара тот катализатор во многих случаях вообще этого не требует. Да и в реальных примерах использования катализатора Линдлара не всегда используют хинолин, потому что иногда это слишком сильно замедляет реакцию. Зачем эти добавки? Выражение “каталитический яд” это такая архаика, привет из прошлого. В современной химии мыслят не так, без ядов. Такие добавки это просто лиганды, довольно слабые (слабый лиганд – это лиганд с небольшой константой связывания), и роль их очень проста – уменьшать количество активных каталитических центров, то есть точно такая же, как использование “плохих” подложек типа карбоната кальция или сульфата бария. Поскольку каталитические частицы довольно большие и бесформенные, на поверхности есть разные атомы, с разной реакционной способностью, некоторые более селективны, некоторые менее – это зависит от положения атома на разных изломах, дефектах, вершинах, гранях и так далее, у разных атомов разное количество свободных координационных мест и разные возможности хватать за связи проплывающие мимо молекулы реагентов – некоторые хватают всех, а другие более разборчивы.
В случае Линдлара это обычно объясняют так, что связывание хинолина блокирует поверхность от связывание алкенов – хинолин как бы встревает между алкинами и алкенами, первые продолжают активно взаимодействовать, вторые нет. Это обычная игра на константах равновесия между двумя равновесиями вставим третье. И большие константы связывания, скорее всего, у менее селективных центров. Конечно никто эти константы никогда не измерял, это просто гипотезы из общих соображений, но вполне разумные. Гетерогенный катализ увы до сих пор остается наукой, где результат достигается кропотливой оптимизацией и перебором условия и составов. А уж потом можно померить кучу всяких спектров и написать красивую модель.
Вполне очевидно только одно – все эти мероприятия по ограничению количества (или концентрации на поверхности) активных каталитических центрах приводят к тому, что преимущество получает основная реакция. Те же алкины, например, легче связываются с активным палладием (тройная связь – просто лучший лиганд), чем двойная, но когда активных центров много, это не играет роли, хватет и на тройные и на двойные связи. Когда мало, реагируют преимущественно тройные. Тем не менее, во всех методиках использования катализатора Линдлара всегда предупреждают, что нужно следить за поглощением водорода, и когда поглотится ожидаемое количество для восстановления в алкен, реакцию рекомендуется прекращать, иначе дальше пойдет восстановление дальше и селективность накроется. С катализатором Розенмунда то же самое, но там все еще сложнее, потому что на каждый эквивалент поглотившегося водорода выделяется эквивалент HCl и объём не изменяется, поэтому там поглощают HCl щелочью и титрованием следят за степенью превращения. Если передержать и там случается перевосстановление, но там даже сложнее, потому что альдегиды на палладии не гидрируются, но возможны другие побочные реакции типа декарбонилирования.
Для гидрирования алкинов обычно используют Линдлар, но можно найти многочисленные примеры и использования катализатора Розенмунда, палладия на сульфате бария, получается не хуже, а иногда лучше. Никакой специфики у выбора именно карбоната кальция вместо сульфата бария поэтому нет. А почему Линдлар все же победил, в том смысле, что катализатор применяется все же намного чаще? Скорее всего, потому что работа поновее, и катализатор готовится удобнее и лучше воспроизводится. Сульфат бария – совсем слабый носитель, палладий на нем практически не держится, и это не очень хорошо. Иногда даже бывает так, что магнитная мешалка в процессе реакции механически разрушает катализатор, из=за чего гидрирование часто продолжают по-старинке делать в таком специальном сосуде – утке, которую перемешивают не магнитной мешалкой, а снаружи качалкой, чтобы не тереть катализатор. Карбонат кальция по сравнению с сульфатом бария все же имеет некоторую химическую активность на поверхности – карбонат вполне зачетный лиганд для палладия, в начале возможно химическое связывание и на этих центрах уже растут наночастички. Поэтому при нанесении каталитические центры распределяются более равномерно и дисперсность выше, и стабильность.
А почему наоборот Линдлара не применяют для восстановления по Розенмунду? Потому что там выделяется HCl и катализатор бы с шипением накрылся.
Почему получается строго цис? Для этого надо знать механизм, но механизм для гетерогенных реакций установить очень трудно. По аналогии с гомогенными реакциями можно считать, что водород сначала окислительно присоединяется к палладию, затем гидрид палладия присоединяется к кратной связи (такие реакции в химии переходных металлов всегда имеют син-стереохимию), и последний водород садится за счет восстановительного элиминирования с сохранением конфигурации.
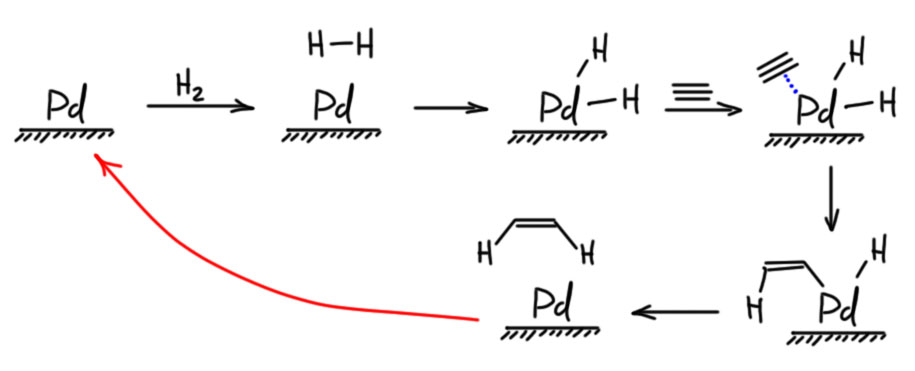
Восстановление по Линдлару годится для внутренних и терминальных алкинов, у которых можно обозначить конфигурацию дейтерием и он никуда не денется, потому что в реакции не используются сильные основания, а основности хинолина для отщепления терминального протона не хватает. Катализатор обозначают обычно просто как Pd/CaCO3, добавляют хинолин, и не забывают водород – катализатор сам собой не гидрирует.
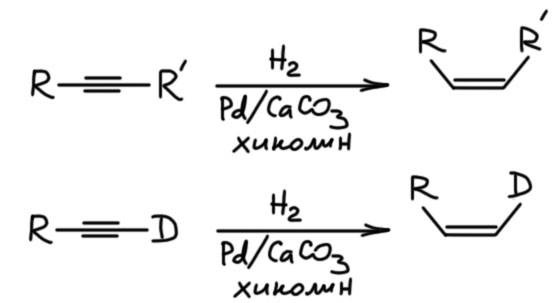
а также для соединений с несколькими тройными связями, в том числе сопряженных диинов.
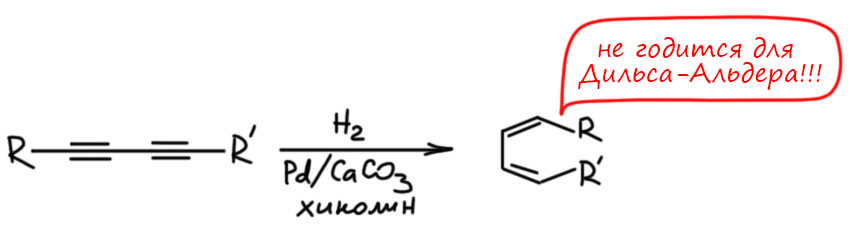
В конце замечу, что хотя катализаторы Линдлара и Розенмунда называют катализаторами, ни тот, ни другой не применяют повторно, их нельзя извлечь из реакционной смеси, помыть, посушить и положить в следующую. Результатом будет полная потеря селективности, гидрировать будет, но полностью. Поэтому их просто собирают, и перерабатывают на палладий, один из самых дорогих металлов, а из палладия уже сснова делаю новую порцию, если нужна.
Цис-гидрирование, другие методы
Гидроборирование
Второй способ цис гидрирования алкинов мы и так знаем, это гидроборирование большими боранами, с разложением продукта гидроборирования уксусной кислотой (ни в коем случае не сильными кислотами, сильные кислоты бораны разлагают очень неохотно, а побочных реакций могут натворить немало). Этот метод не очень хорош для внутренних алкинов, если с двух сторон есть разветвления, так как гидроборирование очень чувствительно к стерическим препятствиям. А вот где это вещь незаменимая, это с териминальными алкинами и тогда, когда нужно вводить дейтерий точно в нужные положения с нужной стереохимией. Мы это уже разбирали, просто напомню:
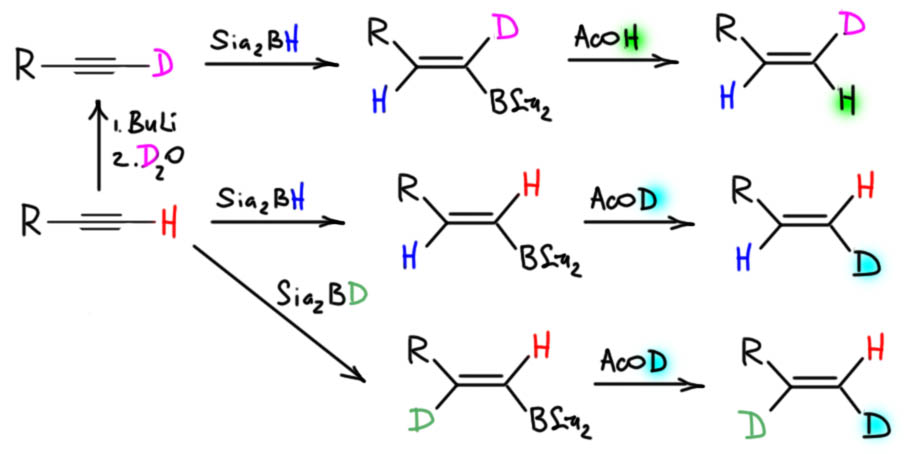
Все остальное про гидроборирование уже разобрали.
Гидрирование на активном никеле
Еще один очень популярный метод, уступающий только Линдлару, гидрирование с использование особых форм никелевого катализатора. Никель как катализатор гидрирования кратных связей появился очень рано, с самом конце 19 века, это вообще первый гетерогенный катализатор гидрировавния, за который была дана нобелевская премия 1912 года Полю Сабатье. Никель как катализатор гидрирования чрезвычайно активен и неселективен, и долго с ним не могли ничего сделать для более тонкой, селективной работы. Если надо прогидрировать всё, что можно прогидрировать – никель идёт к вам. Если нужно хоть немного выбирать, никель – худший выбор, и надо искать катализаторы у более дорогих его родственников, палладия или платины, а вообще катализаторов гидрирования очень много. Но никель так привлекателен своей дешевизной, что попыток сделать из него что-то поаккуратнее в химии с начала прошлого века было несметное количество, и многие дали очень интересные вещи. С точки зрения гидрирования мы хорошо знаем никель Ренея (по имени американского инженера Маррея Рейни, придумавшего этот совершенно ослепительно гениальный трюк – растворять в щелочи сплавы металлов с алюминием, алюминий уходит и остается высокодисперсный нерастворимый в щелочи металл – в 1926 году), но для гидрирования алкинов в алкены он не подходит – гидрирует до алканов, да еще и вызывает перегруппировки.
В 1962 году интересное решение нашел Герберт Браун. После того как он в 1940-м открыл борогидрид натрия, стал буквально везде его совать и искать ему применения. В 1956 из этого родилась химия гидроборирования. А еще раньше он же исследовал гидролиз борогидрида в водном растворе и обнаружил, что эту реакцию катализируют соли никеля, водород выделяется, а из раствора выпадает черный осадок. Чуть позже другие обнаружили, что этот осадок имеет стехиометрию борида никеля Ni2B (R. Paul, P. Buisson, N. Joseph, Ind. Eng. Chem., 1952, 44, 1006). И тогда Браун решил вернуться к этому материалу, и в 1962-63 обнаружил, что у него есть две очень сильно различающиеся формы. Первая получается восстановлением водного раствора соли никеля, вторая – раствора ацетата никеля в этаноле. Первую назвали P1-Ni, вторую – P2-Ni. Буква P – это просто сокращение фамилии Paul, хотя есть и второе толкование – это первая буква университета Purdue, в котором работал Браун. Получились очень интересные и разные восстановители, особенно второй. Интересно, что эти работы руками делал другой Браун, молодой Чарльз Аллен Браун, просто однофамилец, и это тот самый Браун, который через несколько лет откроет выдающуюся реакцию ацетиленового зиппера, а следовательно тоже нехилый ученый. Увы, соседство с великим Гербертом Брауном сыграло с ним жестокую шутку – младший Браун полностью растворился в нобелевском лауреате, тем более что у того второе имя тоже Чарльз, и все поиски того Брауна тонут в миллионах ссылок на этого. Интересно, что этот катализатор в литературе очень часто называют катализатором Брауна, не догадываясь, что Браун там не тот, который везде и всюду, потому что настоящую популярность это реагент получил после статьи именно младшего Брауна 1973 года (Brown C. A., Ahuja V. K. J. Chem. Soc., Chem. Commun. 1973, 553), в котором тот показал, что особенно здорово этот катализатор работает при добавлении этилендиамина, и именно этот вариант с тех пор все и используют.
Надо сказать, что P2-Ni был любимым восстановителем автора нашего главного учебника, профессора Курца. И он заразил им всю кафедру. Лет 30 назад нельзя было пройти по коридору, чтобы не стать свидетелем диалога: У меня не восстанавливается то-то и то-то – А ты пробовал(а) пэдваникель? – Ах, как же я сразу не догадался, пэдваникель, конечно, пэдваникель. И второй собеседник убегал в свою комнату немедленно пробовать пэдваникель. Я сам однажды пробовал пэдваникель. Ничего не получилось, и я решил, что просто не умею его готовить. И больше никогда не пробовал.
Так вот, если пэодинникель был заявлен как аналог никеля Ренея, только более селективный и активный, то пэдваникель оказался весьма необычным катализатором гидрирования. Во-первых, он гидрирует не все двойные связи, а только наименее стерически затрудненные, в первую очередь терминальные, а другие не трогает. Во-вторых, это очень чистый катализатор цис-гидрирования ацетиленов, и за поледующие полвека гладко вышел на второе место после Линдлара. Он часто работает быстрее и чище, не нужен хинолин, от которого в продуктах непросто избавиться, да и это банальный никель, а не палладий. Использовал – выбросил, а палладий надо сохранять и время от времени, накопивши, регенерировать. Да и делается он элементарно, с ним легко работать, он не пирофорен, вполне выдерживает недолгое пребывание на несвежем воздухе (и на свежем, наверное, тоже, да где ж его взять).
Восстанавливать можно буквально любые тройные связи в любых соединениях с любыми заместителями и функциями. И при наличии нескольких связей при дозировании водорода и контроля времени можно добиться еще и селективного гидрирования по принципу меньшей стерики. И диины, и триины, и тетраины, и енины, и ендиины, и триентриины. И можно использовать дейтерий, чтобы вводить два D в цис-конфигурации. И почти не надо трястись, замеряя поглотившийся водород, потому что просто так он не перевосстанавливает.
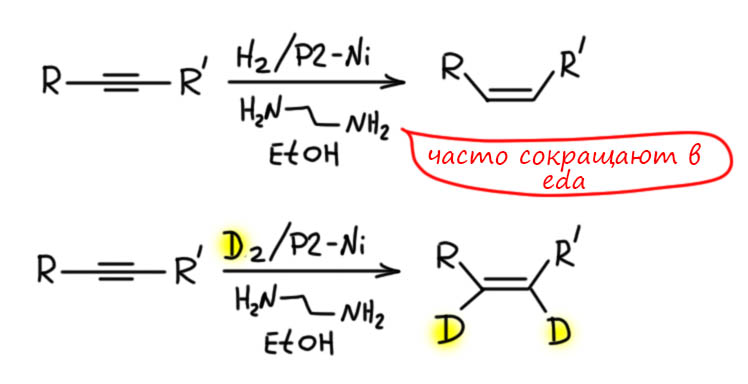
В общем – мечта. Но, видимо, не один я не умею его готовить, и время от времени кто-нибудь ещё обязательно пожалуется, что ни черта не вышло.
А кстати, так что же это такое? В литературе кроме катализатора Брауна и #пэдваникеля эту штуку часто называют боридом никеля. Но последующие исследования показали, что точной стехиометрии у этого вещества нет, в той статье Пола и сотрудников немного упростили реальность, а реальная стехиометрия там ближе к Ni5B2, но и это ничего не значит, потому что это некий почти аморфный материал, не имеющий определенной ни структуры, ни состава, бор там действительно есть, но это не какая-то конкретная бинарная фаза, а просто смесь, и там много еще и всякого мусора типа соды. В общем, это типичный конгломерат наночастиц, слиплись они просто в процессе образования. Именно поэтому этот катализатор может не получиться – материалы такой неопределенной природы всегда зависят от совокупности факторов, иногда мистических. Возможно нужет этанол определенной марки, а может быть надо готовить только в определенные дни, как садоводам рекомендуется сеять рассаду по лунному календарю.
А можно без водорода обойтись?
Катализаторы Линдлара и Брауна (пэдваникель) – это катализаторы гидрирования молекулярным водородом. Водород – это несложно, если он у вас есть, а это либо баллон, либо генератор водорода. Баллон с водородом – это опасно, их запрещают держать в лаборатории, нужно ставить снаружи и делать подводку по фасаду здания, а это вам никто не разрешит делать, если ваша лаборатория выходит окнами не во двор. Если нет, то нужно искать у кого есть, наполнять камеру, а камера не любая держит водород, он умеет утекать через резину. Очень хорошая штука – генератор водорода, но это дорого и мало у кого есть. А по старому, из цинка и соляной кислоты в аппарате Хиппа делать водород нельзя – в нем бывают примеси фосфина и арсина, и накроется ваш катализатор. В общем, если вы часто делаете гидрирование, то все эти проблемы давно преодолели, и можно гидрировать дальше. А если нет, и это – эпизод в синтезах совсем другого рода, то призадумаешься, а нельзя ли без водорода. Да, есть гидроборирование, аккуратная чистая реакция, но весьма дорогая, нужен источник борана, надо заваривать дисиамилборан (его не хранят и не покупают). Тоже есть проблемы.
В более-менее современной химии стал очень популярен еще один способ, он стал встречаться в статьях даже чаще Линдлара, но только в сложных случаях. Гидрирование одиночной тройной связи по-прежнему делают чаще всего Линдларом или пэдваникелем.
Альтернативный метод основан на весьма давнем наблюдении, что тройную связь иногда удается восстановить до цис-алкена просто цинком. Цинк нужен активированный. Есть очень много способов активировать цинк, но самый лучший способ предложил немецкий химик Вильгельм Боланд, много занимавшийся синтезом и изучением сигнальных молекул (феромонов) всяких насекомых. Среди таких соединений очень много длинных цепочек в которых в разных положениях встречаются двойные связи, цис и транс. И это надо синтезировать, на чем очень здорово набили руку исследователи типа Боланда. Одна из задач, кторую он никак не мог решить с Линдлром и другими известными методами было селективной цис-восстановление тройных связей, сопряженных с транс-двойными. И нашел он, что цинковую пыль можно очень хорошо активировать очень простым способом. Цинковая пыль это порошок металлического цинка, получаемый прямой перегонкой металла – его плавят в больших ретортах и нагревают выше 1000 градуcов, и цинк просто перегоняется, застывая в охлаждаемом приемнике сразу в виде порошка. Такой порошок, цинковая пыль (zinc dust) – дешевый и легкодоступный материал, но поверхность цинка у такой пыли из банки покрыта оксидом, карбонатом цинка и всякой дрянью. Чтобы получить поверхность именно цинка этот материал активируют десятком разных способов. Иногда не просто очищают, но и вносят электроположительные металлы типа меди, образующие гальваническую пару – тогда цинк принудительно становится анодом и готов отдавать электроны. Боланд придумал делать двойную пару, сначала цинковую пыль обрабатывают раствором соли меди, затем раствором соли серебра. Образующийся материал (его обозначают Zn(Cu/Ag)) восстанавливает тройные связи при нагревании в метаноле, который поставляет протоны. Механизм реакции вполне очевиден, но с непривычки может ошарашить. Цинк передает два электрона π-связи, связывая дианион в цинк-органическое соединение, трёхчленный цикл, цинкациклопропен (буква –а– правильная, она обозначает то, что в молекуле циклопропена один атом углерода заменен на гетероатом, здесь, цинк).
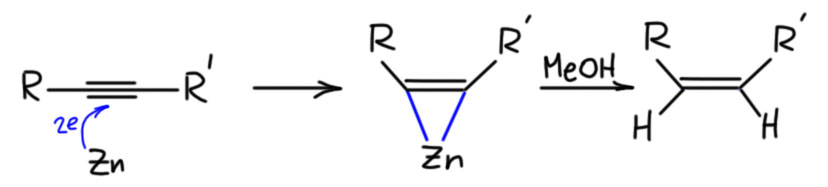
А так бывает? Да, металлоорганика вообще штука удивительная, привыкайте. Таких металлоорганических соединений много, желающие могут побольше об этом узнать в курсе про переходные металлы. Цинк – не переходный металл, но примыкает к ним; он отличается от нормальных переходных металлов тем. что в его металлорганических производных связи цинк-углерод больше похожи на ионные, это ковалентные связи с высокой степенью ионности; поэтому они легко расщепляются протонными кислотами. Это делает цинк очень важным металлом в органическом синтезе – он очень здорово сочетает в себе свойства, близкие к металлам переходным, в том, что образует более прочные связи с углеродом с высокой долей ковалентности, поэтому соединения цинка выгоднее. они легче образуются в самых разных реакциях, даже там, где мы этого не ждём. Цинковая металлоорганика стала невероятно популярна в современной химии, когда получше разобрались в закономерностях и перестали пугаться непривычно выглядящих молекул. Фокус цинка еще в том, что цинкорганика может образоваться в протонных растворителях, в спиртах, воде, даже в растворах кислот, и подвергается протолизу только после образования. Близость цинка с другой стороны к непереходным металлам второй группы выражается в высокой реакционной способности; соединения цинка вступают в реакции того же типа, что магнийорганика, но намного спокойнее, удобнее.
Метод Боланда особенно часто применяют в сложных синтезах. Приведу просто один из примеров из первой статьи самого Боланда (Boland W., Sieler C., Feigel M. Helv. Chim. Acta 1987, 70, 1025). Все связи в этих молекулах сопряжены.
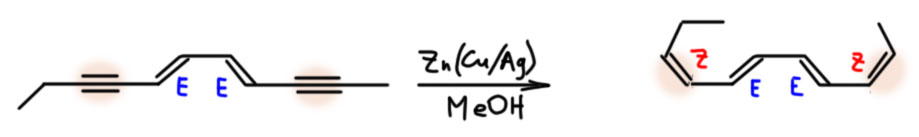
Задачи и вопросы к коллоквиуму по теме алкины
Гидроборирование алкинов
Гидроборирование алкинов - стереохимия
Из 1-бутина и других необходимых реагентов получите вот такой стереоизомер монодейтерированного бутандиола, естественно, в виде рацемической смеси с энантиомером двумя разными способами. Это эритро- или трео-диастереомер? Перерисуйте в проекции Фишера и разметьте центры по R/S-номенклатуре.