Обновления
Опубликована 10.01.2023
Енамины
Енолизуемые карбонильные соединения участвуют в реакциях не прямо, а через спонтанно образующиеся или специально полученные производные, являющиеся по своей природе электронодонорными нуклеофильными олефинами. Мы знаем основные производные такого типа: енолы, эфиры енолов, еноляты, енамины. Каждое из этих производных участвует в основных типах превращений енолизуемых карбонильных соединений, а это практически всегда начинается с присоединения к сильнодонорной двойной связи какого-то электрофила, и далее развивается по той или иной схеме в большинстве случаев приводя к продуктам α-замещения исходного карбонильного соединения. Поскольку этих производных так много (кроме перечисленных в современной химии используются и другие типы) органик получает в свои руки очень гибкий инструмент превращения карбонильных соединений в совершенно поразительное количество производных – и это очень важно, потому что если в том или ином случае не работает одно, мы не опускаем руки, а берем другое, и рано или поздно решаем задачу, причем хорошее владение химией позволит делать это не только селективно, но и мягко, и что немаловажно, дешево и просто. Много раз обращал ваше внимание на то, что разумный химик никогда не ставит своей задачей использовать самые дорогие, модные, сложные и т.д. реагенты, если есть возможность сделать то же самое дешевле, с помощью более старого, но проверенного метода, и чем проще, тем лучше. Хороший химик умеет работать, но не любит переутруждаться. Если что-то можно сделать без охлаждения, на воздухе, а не в инертной атмосфере, в необязательно тщательно высушенных растворителях (растворитель всегда должен быть отличного качества, перегнан, проверен, но абсолютным он быть не обязан, и если без этого можно обойтись, то это хорошо, а не плохо), с общедоступными реактивами – то это очень хорошие новости. Безусловно, все эти решения должны приниматься сознательно, исходя из точного знания химии, и делать что-то попроще там, где реально нужно посложнее, это уже не правильный выбор, а халтура.
Вот енамины – это отличный пример того, как химия нашла очень простое решение для многих важных реакций енолизуемых карбонильных соединений. Енамины – без вопросов самые простые стабильные производные этого типа, их легко получить в самой непритязательной лаборатории типа нашего практикума, из очень простых реактивов. Для этого не нужно ни сложной аппаратуры, ни тщательной очистки реактивов. Они легко выделяются и очищаются, без проблем хранятся практически не ограничено по времени, если в холодильнике и тара продута аргоном (как все амины они очень легко окисляются). Более того, хотя мы не будем рассматривать такие реакции в нашем курсе, в современной химии очень часто используют енамины, образующиеся прямо в реакционной смеси, без выделения, и это стало одним из основных приемов чрезвычайно модного органокатализа. Если немного поинтересоваться енаминами в органокатализе, вообще может сложиться впечатление, что это какая-то волшебная химия на все случаи жизни – у вас что-то не идёт? енамин пробовали? попробуйте, и дело в шляпе! Увы, это не совсем так, и это впечатление – самообман. Енамины могущественны, но при этом весьма коварны – если их использовать без точного понимания их структуры и реакционной способности, то во многих случаях не получится ничего, или получится не то.
Попробуем немного разобраться в химии енаминов и понять одну весьма странную вещь – не всё то енамин, что с виду похоже на енамин, и для того чтобы быть полезным обществу енамином, мало напялить на двойную связь аминогруппу. Химия енаминов весьма поучительна, она может научить внимательнее смотреть на структуру, точнее учитывать разные взаимодействия, не забывать, а активно применять то, что мы изучаем в органической химии.
Краткое содержание
Общие слова про преимущества и недостатки енаминов в ряду других нуклеофильных производных енолизуемых карбонильных соединений. Пирролидин или морфолин – вот в чём вопрос (спойлер – это туфта) Общие слова.
Как обычно, немного истории, кто и когда открыл енамины и почему вся слава досталась тому, кто этого не делал, и почему это справедливо, потому что мало что-то открыть, надо и крылья приделать – в данном случае крылья оказались журавлиными и полёт удался – на вкладке Откуда взялись енамины.
Как образуются енамины и почему для этого нужен кислотный катализ, хотя амины – основания. Взаимоотношения иминиевых катионов и енаминов. И немного про насадку Дина-Старка и как она работает – на вкладке Как образуются енамины.
Поскольку в химии енаминов очень важны эффекты сопряжения, я решил напомнить, что не всё то сопрягается, что находится рядом – на вкладке О сопряжении
И достигнув в этом деле некоторой ясности попробуем разобраться, какой енамин образуется в тех случаях, когда есть выбор в случае несимметричных кетонов, енолизуемых в обе стороны
И поняв, что при образовании енаминов никакой ясности нет – образуются оба возможных, – придется разобраться в том, почему же обычно считают, что образуется только менее замещённый енамин (спойлер – правильно считают, так и есть).
И разобравшись с этим, вновь не найдём успокоения, потому что придется разобраться, куда девается второй изомер
И наконец обретя ясность с то появляющимися, то исчезающими енаминами, обратим наш пытливый взор на самую популярную реакцию енаминов с акцепторами Михаэля, и разберёмся, как ей удаётся добраться до полезных продуктов, даже если в механизме этой реакции оказались заложены минимум две серьёзные проблемы, и почему в реакциях с некоторыми енонами образуются продукты аннелирования Робинсона
Ну и быстренько пробежимся по остальным полезным реакциям енаминов, отдельно помянув недобрым словом те, что являются плодом коллективной фантазии
И наконец я дошёл до покаяния – надо же разобраться в том, почему случился беспрецедентный массовый возврат баллов за енаминовую двухходовку (спойлер – это был новогодний сюрприз).
В конце, как чисто факультативный бонус, посмотрим на то, как можно использовать некоторые имины в реакциях, аналогичных реакциям енаминов, и таким образом обойти одно из навязчивых ограничений енаминовой химии.
Общие слова
Енамины – одни из важнейших нуклеофильных производных енолизуемых карбонильных соединений. Их достоинства очевидны:
- они стабильны, их можно получить и хранить, используя по необходимости. В этом они похожи на эфиры енолов.
- они очень легко получаются непосредственно из альдегида или кетона без необходимости использовать ни еноляты, ни какие-то катализаторы (кроме банальной толуолсульфоновой кислоты в количестве пары кристаллов). В этом они проще и доступнее большинства эфиров енолов.
- они обладают достаточной нуклеофильностью чтобы реагировать непосредственно с большинством углеродных нуклеофилов без необходимости прибегать к катализаторам и другим методам активации. В этом они превосходят эфиры енолов. И именно это принесло им новый невероятный успех уже в современной химии.
- реакции енаминов обычно селективны (в том числе региоселективны и стереоселективны) и дают желаемый продукт, если мы знаем, что желаем. В этом они похожи на эфиры енолов, но превосходят еноляты, многие реакции которых дают большие осложнения.
Но не бывает реагентов без недостатков. У енаминов их куча. И они ужасны. Каждое вбивает полноценный гвоздь в гроб енаминов. Кажется, нет у енаминов никаких шансов выползти из настолько хорошо заколоченного гроба – так много с ними проблем и недостатков. Это очень хорошо понял уже сам основоположник химии енаминов Джилберт Сторк, когда после первого громогласного заявления о том, что он нашел потрясающие реагенты, годные буквально для всего, ему потребовалось почти 10 лет чтобы попробовать реализовать это обещание, а в результате получилась статья, из которой ясно, что ничего универсального и решающего все проблемы не получилось, а многочисленные препятствия так и не удалось обойти.
Но, как мы любим, недостатки, даже смертельные, не умаляют достоинств, поэтому они и не думают ложиться в этот самый гроб, и химия енаминов живёт и побеждает. Более того, с началом нового столетия у енаминов обнаружилось новое дело – органокатализ. И на енаминах пара ловких чуваков в 2021 году доехала аж до нобелевской премии. Да, там полно еще всего кроме енаминов, но начинали оба именно с енаминов, и очень значительная часть их работ сделана с помощью химии енаминов. Енамины оказались просто бездонной бочкой всяких приятных сюрпризов. Но всего удалось достичь только когда в химии енаинов хорошенько разобрались, в том числе и основательно продумав их проблемы и научившись их обходить. Проблемы эти многочисленны.
- получаются они далеко не из всех енолизуемых карбонильных соединений. Можно даже сказать, что из большинства не получаются. Почти любые стерические препятствия рядом с карбонильной группой препятствуют образованию енаминов. Если вы будете читать про химию енаминов, или уже прикладывались к этому источнику знаний, не можете не удивиться или уже удивились, отчего там кругом один енамин циклогексанона. Ну и еще пары кетонов, или симметричных, или замещенных только с одной стороны и не самыми крупными заместителями. Это не случайно. мало из каких кетонов можно реально получить енамины обычным способом. От читателей, особенно малоискушенных, этот прискорбный факт тщательно скрывают. Некоторым утешением можно считать то, что в современной химии енамины получают еще дюжиной других способов, но все они довольно узки и проблему в целом не решают.
- енамины альдегидов образуются чрезвычайно легко, но имеют много особенностей, требующих особо тщательного отношения – иначе получите совсем н то, что хотели. Впрочем и с остальными производными (енолятами, эфирами енолов) у альдегидов всегда собственное мнение.
- высокая селективность енаминов в реакциях обычно имеет неприятную цену – низкие (иногда очень низкие) выходы продуктов при довольно жёстких условиях реакций. Если енамин не реагирует, ничего с этим сделать нельзя, приходится искать обходной путь или сдаваться. В современной химии их научились активировать, но мы не будем туда соваться, там все слишком сложно и много нюансов.
- не очень высокая нуклеофильность по сравнению с енолятами, поэтому енамины реагируют далеко не со всеми электрофилами, а только с самыми активными.
- любая реакция, которая за енаминами закреплена как важная и универсальная, на самом деле таковой не является ни в каком смысле – у всех этих реакций органичений больше, чем атомов углерода в молекулах енаминов. В этом они здорово уступают другим нуклеофильным производным енолизуемых карбонильных соединений – там, если вы научились, например, использовать еноляты или эфиры енолов, то решите действительно много самых разных задач, и получите инструменты, которые легко обобщаются на новые задачи. А вот с енаминами лучше не шалить, особенно если вы собираетесь работать с ними не на бумаге, а в стекле – сначала тщательно изучите литературу и найдите близкие аналогии, попробуйте прикинуть реальную структуру и так далее, – иначе только попусту потратите время.
- наличие атома азота создает еще один нуклеофильный центр, и с этим связаны некоторые трудности особенно в реакциях алкилирования, но мы не будем обращать на это внимания, потому что они не имеют систематического характера и проявляются довольно хаотично – в таких случаях в реальной химии просто говорят: “Не повезло, попробуем что-нибудь другое”
Хотя енамины можно получить множеством разных методов, мы будем использовать только один – реакцю енолизуемых карбонильных соединений с вторичными аминами в присутствии кислотного катализатора, обычно каталитического количества TsOH в условиях удаления образующейся в реакции воды. Это необходимо, потому что образование енаминов обратимо, константы равновесия, хоть и обычно на стороне енамина, но величины их невелики, поэтому енамины легко гидролизуются, чем, в частности, активно пользуется уже упомянутый модный органокатализ, а мы не будем. Мы всегда должны сначала получить енамин, а затем отдельно ввести его в реакцию, и тоже в условиях отсутствия воды в реакционной смеси. Но легкость гидролиза енаминов помогает выделять продукты после завершения реакции, потому что большиснтво реакций енаминов дают не сразу ожидаемые продукты (замещённые кетоны), а их енамины или соли иминия – и то, и другое немедленно гидролизуется просто при добавлении воды к реакционнй смеси, и только в случае использования наиболее нуклеофильных вторичных аминов типа пирролидина, нужно еще и добавить уксусную кислоту.
Хотя енамины можно получить практически с любым вторичным амином, очень быстро сложилась традиция использовать два циклических амина – пирролидин и морфолин (третий амин этого ряда, пиперидин, тоже используют, но реже – он во всем похож на морфолин, но дороже его, а запах пиперидина способен надолго привить отвращение к работе в лаборатории.
Пирролидин более реакционноспособен, быстрее образует енамины, реакции с ним обычно чище и дают более высокие выходы, но при этом он имеет некоторую избыточную реакционную способность, и поэтому он иногда не хочет уходить после реакции, легко образуя енамины или другие аддукты уже из продуктов реакции. Поэтому у реакций с пирролидиновыми енаминами часто бывают особые методики, более сложные, чем с морфолиновыми енаминами, и на это стоит обращать внимание.
Морфолин менее реакионноспособен, региоселективность образования енаминов из несимметричных кетонов ниже, выходы обычно ниже, зато амин сам по себе намного доступнее и дешевле, а недостатки по сравнению с пирролидином не так велики. Простой гидролиз реакционных смесей просто водой обычно сразу дает нужный продукт. Поэтому морфолиновые енамины использовали и будут использовать очень часто.
Откуда взялись енамины
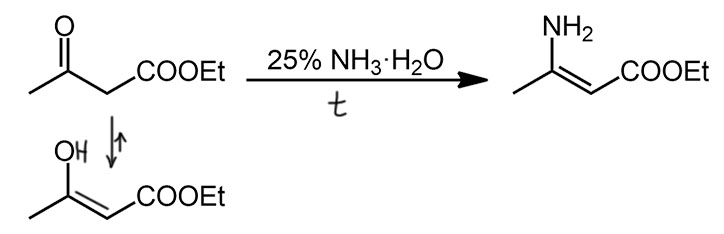
Енамины известны с 19 века, их получали многие знаменитые органики от Кляйзена до Робинсона и Виттига, особенно легко они получаются из 1,3-дикетонов и кетоэфиров, как правило прямо из исходных без каких-то дополнительных реагентов и усилий. Например, из ацетоуксусного эфира очень легко получается очень полезный в синтезе реагент, этил 3-аминокротонат, очень широко используемый в разных синтезах гетероциклических соединений. Это вещество известно с 19 века, с самого начала ацетоуксусного бума в химии, когда только самые ленивые химики не попробовали сделать новую реакцию из этого удивительного вещества, открывшего органикам глаза на то, что если в одной молекуле, недалеко друг от друга находится две функциональные группы, то свойства нового вещества это не простая сумма свойств каждого из классов, а нечто совершенно новое, восхитительно богатое возможностями. Роль ацетоуксусного эфира в развитии органической химии совершенно поразительна. Вот и в енаминах это соединение стало первым.
Енамин ацетоуксусного эфира просто с аммиаком, этил 3-аминокротонат совершенно устойчив, отлично хранится и никуда сам не превращается. Это может показаться странным, ведь енамины с незамещенной амино-группой очень неустойчивы иих невозможно выделить. А эти – пожалуйста. Такие же элементарно делаются из других кетоэфиров и дикетонов. Почему они такие стабильные разберем на отдельной вкладке. Впервые хорошенько изучил это соединение в 1884 году довольно известный шотландский химик Джон Норман Колли (одноименная порода собак тоже имеет шотландское происхождение, но с химиком никак не связана), ученик великого Вислиценуса старшего, который и дал нам всю основную химию ацетоуксусного эфира, и заодно и своих учеников заразил исследованиями этого бездонного соединения.
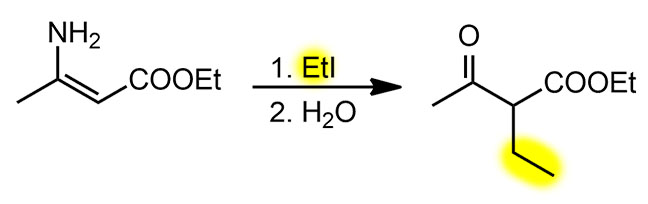
 Профессор Колли ещё известен тем, что был заядлым альпинистом и партнером во многих восхождениях совсем знаменитого шотландского альпиниста Мак-Кензи. Химиком Колли тоже был весьма дотошным, и сумел в то раннее время основательно разобраться в аминокротонате, в частности понять, как это соединение устроено (с поправкой на то, что под структурой в ранней химии понимали не совсем то, что мы понимаем сейчас), и попробовать его в разных реакциях, попробовав сравнить его поведение с работами своего учителя по самому ацетоуксусному эфиру. Колли обнаружил то, что в реакциях соединение неизменно теряет азот и возвращается к кетоэфирной форме, и среди прочего сделал алкилирование этилиодидом, получив этилацетоуксусный эфир – а это почти точный прототип того, что потом открыл Сторк, и что стало основой применения енаминов в синтезе. Возможно поэтому и реакцию Сторка стоило бы называть реакцией Колли-Сторка (невольно получился бы такой живой уголок, пёс и журавль, или очередная редакция бессмертной басни Эзопа-Лафонтена-Крылова). Кстати, такое алкилирование аминокротонового эфира иногда применяют и в более современной химии для того, чтобы чисто получить моноалкилирование ацетоуксусного эфира, когда обычная методика (алкилирование енолята ацетоуксусного эфира) дает частичное диалкилирование. Но это слишком небольшое преимущество, и за неимением более серьёзных достижений про енамины надолго забыли.
Профессор Колли ещё известен тем, что был заядлым альпинистом и партнером во многих восхождениях совсем знаменитого шотландского альпиниста Мак-Кензи. Химиком Колли тоже был весьма дотошным, и сумел в то раннее время основательно разобраться в аминокротонате, в частности понять, как это соединение устроено (с поправкой на то, что под структурой в ранней химии понимали не совсем то, что мы понимаем сейчас), и попробовать его в разных реакциях, попробовав сравнить его поведение с работами своего учителя по самому ацетоуксусному эфиру. Колли обнаружил то, что в реакциях соединение неизменно теряет азот и возвращается к кетоэфирной форме, и среди прочего сделал алкилирование этилиодидом, получив этилацетоуксусный эфир – а это почти точный прототип того, что потом открыл Сторк, и что стало основой применения енаминов в синтезе. Возможно поэтому и реакцию Сторка стоило бы называть реакцией Колли-Сторка (невольно получился бы такой живой уголок, пёс и журавль, или очередная редакция бессмертной басни Эзопа-Лафонтена-Крылова). Кстати, такое алкилирование аминокротонового эфира иногда применяют и в более современной химии для того, чтобы чисто получить моноалкилирование ацетоуксусного эфира, когда обычная методика (алкилирование енолята ацетоуксусного эфира) дает частичное диалкилирование. Но это слишком небольшое преимущество, и за неимением более серьёзных достижений про енамины надолго забыли.
Сам собирательный термин “енамины” предложил Георг Виттиг в 1927, рассуждая, какую структуру должны иметь продукты взаимодействия 1,3-дикетонов с аммиаком, и ему пришла в голову аналогия с енолами. Собственно, такую структуру аминокротоната рисовал ещё Колли, и из этого мы видим, как медленно складывались совершенно привычные для нас представления о взаимоотношениях классов органических соединений – от первых экспериментов до проблесков осознания чего-то типа системы прошло несколько десятилетий, то есть чья-то жизнь. Сейчас время движется побыстрее, и шевелиться надо немного шустрее, а то жизнь пройдёт, а осознание так и не наступит.
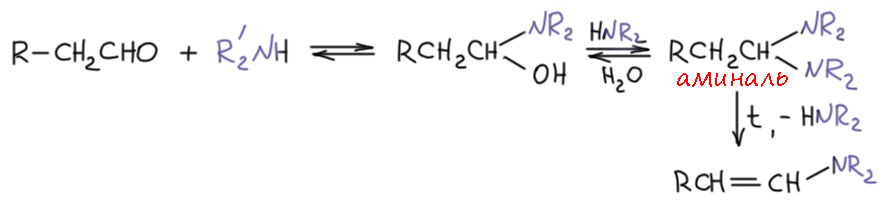
Но это все были енамины от кетоэфиров и дикетонов, а первые обычные енамины получил совершенно ожидаемый персонаж – Карл Манних. Мы отлично знаем реакцию Манниха, для которой формальдегид сначала реагирует с вторичным амином, и поскольку этот альдегид неенолизуем, получается иминиевая соль, которая работает как электрофил по отношению к енолам. Чувствуете, как близко мы подобрались к енаминам – всего-то и надо было взять енолизуемый альдегид. Манних и взял, и нашел (C. Mannich, H. Davidsen, Chem. Ber., 1936, 69, 2106), что реакция альдегидов с вторичными аминами, и у него тогда впервые активно материализовался пирролидин, в присутствии безводного поташа, который работает просто как водоотнимающее средство (этот наивный старинный термин нужно понимать не так, что некое средство буквально отнимает воду от каких-то интермедиатов реакции, а как то, что выделяющаяся вода связывается, например, гидратируя ионы соли, и так выводится из равновесия) и легко образуются такие аналоги ацеталей, их позже стали по аналогии и называть, аминали. Эти аминали, кроме самого маленького, из ацетальдегида и пирролидина, при перегонке в вакууме теряют одну молекулу амина и образуют енамины. Манних с этими веществами ничего делать не стал.
Потом случилась война. Новая жизнь енаминов с войной оказалась связана совершенно непосредственно. В новую жизнь енамины вытолкнул молодой химик Джилберт Сторк. Сторк – еще один легендарный химик, за которого можно поблагодарить катаклизмы 20 века. Бельгиец из Брюсселя по подданству родителей с еврейскими корнями, тогда еще Жильбер вместе с семьёй он переехал во Францию, там и учился чему-то непонятному, пока не началась Вторая мировая. Семья успела сесть на пароход и отправиться в Штаты в 1939, интуитивно поняв, что в Европе начинается нечто ужасное. Жильберу тогда было 18 – самое время отправиться на войну, а что такое война с Германией французы и бельгийцы еще очень хорошо помнили по битвам Первой мировой, когда сотни тысяч и миллионы молодых людей превратились в кровавую грязь окопных сражений. До сражений вообще у Жильбера и его семьи дело вряд ли бы дошло. В 1939 никому даже в страшном сне не могло приснится, что всего через год Франция будет молниеносно разгромлена, и станет наполовину оккупированной, наполовину марионеткой Гитлера. И оказался бы юный Сторк в концлагере с нулевыми шансами выжить. Вместо этого в Америке он выучился, случайно увлёкся химией, в 1953 году устроился в знаменитый университет Коламбиа в Нью Йорке, и уже на следующий год сделал знаменитую работу, описанную на одной страничке статьи (G Stork, R. Terrell, J. Szmuszkovicz J.Am.Chem.Soc., 1954, 76, 2029-2030) про получение 2-алкил и 2-ацилкетонов, с которой и пошла химия енаминов, енаминов Сторка, как их после назвали, в реакции Сторка.
Интересно, что Сторк не только не получил первым енамины, как уже понятно. Он даже не придумал хороший способ их получения. Всего за год до Сторка вышли две статьи неких Хейла и Херра из фармацевтической компании Апджон, которые попробовали использовать енамин в качестве защитной группы для кето-группы в первом кольце стероидов для селективной модификации карбонильных групп в других положениях. Енамины они получали с пирролидином и как раз и придумали использовать азеотропную отгонку воды с ксилолом с помощью знакомой всем насадки, которую они называют необычно – насадкой Бидвелла-Стерлинга (об этом ниже). Сторк этого и не скрывает, а ясно пишет, что именно это и использовал как наиболее удобный метод. Суть открытия Сторка, сделавшего енамины столь популярным инструментом в синтезе, в том, что он показал, что они реагируют с разными электрофилами по углероду, а не по азоту, как вполне мог и должен был полумать почти любой нормальный химик. В общем, это вполне очевидно – анамины это по природе своей типичные амины, да, с непредельным заместителем, но чем он хуже, например, ароматических аминов, анилинов. Анилины ведь тоже могут реагировать с электрофилами по кольцу, но, например, с галогенпроизводными анилины нормально дают четвертичные аммонийные соли, да и для реакций, скажем, с акцепторами Михаэля или ацилхлоридами нужен катализатор, это типичные реакции Фриделя-Крафтса со своими особенностями. Эта химия вполне хорошо была известна ещё в 19-м веке. Поэтому открытие Сторка, что с енаминами почти никогда не наблюдается реакция по азоту, и многие полезные электрофилы реагируют без всякой активации, просто путем нагревания смеси енамина и электрофила в безобидном растворителе, оказалось действительно неожиданным и открывающим глаза другим исследователям на возможности. Сам Сторк через 10 лет удивлялся тому, как естественно было воспринято это открытие – многим показалось, что так и должно быть, и пока Сторк копался со следующей статьёй, описывающей химию енаминов в деталях, появились довольно многочисленные работы, в которых реакциями енаминов с алкилирующими агентами пользовались как даром богов, не удосужившись сослаться на первооткрывателя метода. Впрочем, Сторк быстро стал чрезвычайно авторитетным исследователем, сделав много важных работ по синтезу, и никто уже не подвергал сомнению приоритет, и даже енамины стали называть енаминами Сторка (что несправедливо), а их реакции с алкилирующими и ацилирующими агентами – реакцией Сторка (что полностью справедливо).
Сторк был одним из первых органиков, кто предложил не какую-то одну реакцию или реагент, а сразу целую методологию. Енамины реашают задачи сложного синтеза – смотрите как здорово они это делают. Он просто показал на нескольких примерах, что они реагируют с электрофилами (слово это, кстати, тогда еще не было общеупотребительно, и он назвал это акцепторами). Все реакции шли просто при нагревании енамина с электрофилами в растворе метанола без каких-либо дополнительных реагентов. После окончания реакции в смесь добавляли воду и еще немного нагревали, чтобы гидролизовать промежуточную иминиевую соль. Как видим Сторк сделал очень точный выбор и одной работой закрыл три основных типа C-электрофилов: алкилгалогениды в SN2-замещении, галоидангидриды в ацилировании (кроме бензоилхлорида это еще и этиловый эфир хлоугольной кислоты, который является хлорангидридом полуэфира угольной кислоты)
, акцепторы Михаэля в сопряженном присоединении. Продукт с двумя циклами, который получается из метилвинилкетона – это обычный продукт циклизации (аннулирования) Робинсона.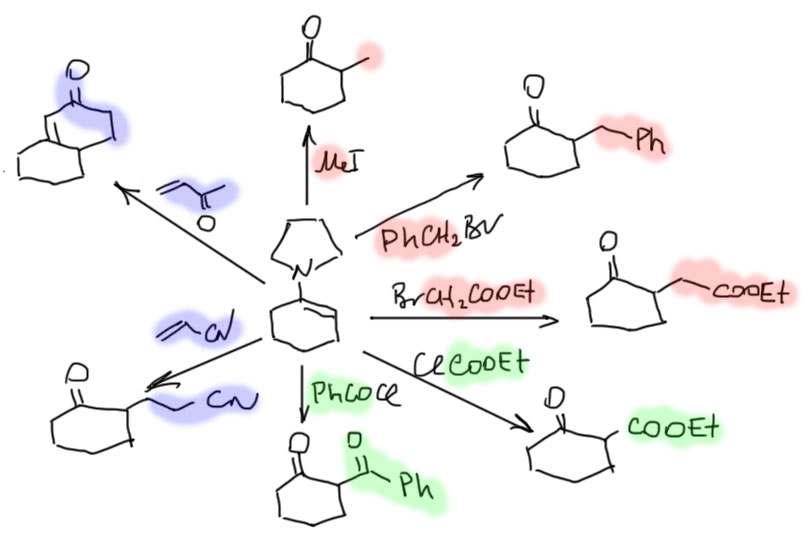
Как образуются енамины
Основной метод как был так и остался – реакция енолизуемых кетонов и альдегидов с вторичными аминами в присутствии кислотного катализатора.
В этом месте можно задуматься. Как? Зачем? Амин же основание, он будет кислотой протонирован и перестанет быть нуклеофилом. Посмотрим на механизм. Возьмём для простоты ацетон. Как обычно нарисуем при взаимодействии кетона и нуклеофила тетраэдрический интермедиат. Этот интермедиат может потерять воду, но для этого ему нужно перекинуть протон с первоначального места на азоте на кислород. Это очевидные стадии, но нужно понимать, что протоны сами собой не скачут, их должен кто-то переносить, этот кто-то – переносчик протона, пара кислота-основание Бренстеда. В растворе мы получим равновесие всех возможных форм в соответствии с относительными кислотностями и основностями каждого центра, но мы этих величин не знаем, и можем только нарисовать все возможности. Нас собственно, как обычно, цифры не интересуют, нас интересует только принципиальная возможность. Одна из таких форм потеряет воду. Все эти реакции – переноса протона и диссоциация воды – обратимы. Вот эту функцию – переносить протон между различными цетрами и поддерживать хорошие скорости реакций в равновесиях – и выполняет кислотный катализатор. В схеме мы обозначаем такую пару обобщённо, но в конкретной ситуации ясно, что кислоту бурут обычно сильную, а в растворе есть много вторичного амина, основания. В этом случае можно считать, что парой кислота-основание (HA/A–) служит протонированный и непротонированный амин (R2NH2+/R2NH). А поскольку сильная кислота берётся в каталитическом количестве, в растворе остаётся достаточно нуклеофила, свободного амина. 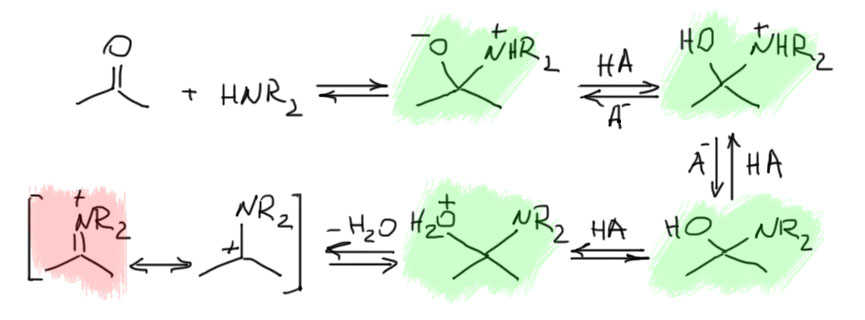
Но то, что непосредственно получилось при уходе воды от аддукта, это ещё не енамин, а иминиевый катион. Эта штука могла бы стать конечным продуктом взаимодействия карбонильной группы с вторичным амином, если бы карбонильное соединение было бы неенолизуемым. Ровно так и происходит, например, в реакции Манниха. В классической реакции Манниха карбонильное соединение всегда формальдегид, и в этом случае иминиевому катиону деваться некуда. Он остаётся, и при наличии рядом нуклеофила, например, енола, реагирует с ним.
Но если есть протон на α-углероде, легко происходит элиминирование и образуется енамин. Если рассмотреть эту реакцию внимательно, станет понятно, что это действительно типичное E1-элиминирование: иминиевый катион имеет вторую граничную структуру карбокатиона, и действие основания, а как мы выяснили, реакция всегда идёт в присутствии достаточного количества вторичного амина, вполне приличного основания. Катион иминия и енамин представляют собой пару сопряженная кислота и основание. Аналогия с обычными карбокатионами правильна, но не стоит забывать, что в отличие от обычных карбокатионов, стабилизированных индуктивными эффектами и гиперконъюгацией, здесь работает полноценное сопряжение. Поэтому если обычный карбокатион – очень сильная кислота (современная наука даже использует термин суперкислота), то катион иминия – кислота не очень сильная, а енамин, следовательно протонируется гораздо проще, чем обычный олефин. С этим связано, например, очень легкое превращение двух изомерных енаминов у несимметричных кетонов, с которым мы еще столкнёмся. Взаимопревращения катиона иминия и енамином происходит чрезвычайно легко, настолько что во многих реакциях вообще трудно понять, через какой интермедиат они идут, иминий или енамин.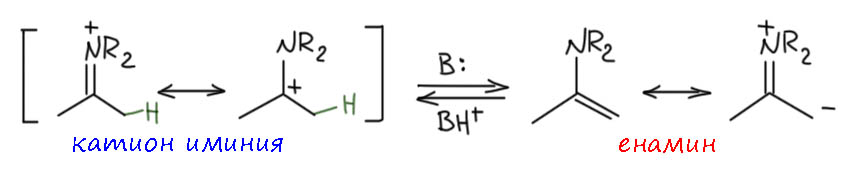 Итак, образование енамина из вторичного амина и карбонильного соединения катализирует кислота, достаточно сильная, но неважно какая конкретно, так как всё равно реальным кислотным катализатором является протонированный амин, а амин может запротонировать почти любая более-менее сильная и даже не очень сильная кислота. В таких случаях в совсем старой химии брали каплю серной кислоты, но ее сильные окислительные свойства часто приводят ко всяким осложнениям в момент добавления. Когда нашли тоуолсульфоновую кислоту TsOH, именно она стала стандартом сильной кислоты для катализа – это дёшево, доступно и удобно (она твёрдая, её легко взвесить, она не имеет окислительных свойств, а по силе мало отличается от серной). Ещё одна проблема – обратимость всей реакции взаимодействия амина и карбонильного соединения. В реакции выделяется вода, и пока она остаётся в реакционной смеси та же кислота катализирует гидролиз енамина. Воду обязательно надо убирать из реакционной смеси. Это можно делать многими способами, например, вести реакцию в присутствии веществ, связывающих воду, использующихся как осушители. Очень часто применяют, например, безводный сульфат магния, особенно если хотят получить енамины не с пирролидином или морфолином, а с небольшими и доольно летучими вторичными аминами, например. диметиламином. Также применяют активированные молекулярные сита, и еще много чего. Это удобно и когда вам не нужно делать много енамина, а как принято в современной химии, реакции ставятся на миллиграмовые количества и енамин делают непосредственно перед использованием.
Итак, образование енамина из вторичного амина и карбонильного соединения катализирует кислота, достаточно сильная, но неважно какая конкретно, так как всё равно реальным кислотным катализатором является протонированный амин, а амин может запротонировать почти любая более-менее сильная и даже не очень сильная кислота. В таких случаях в совсем старой химии брали каплю серной кислоты, но ее сильные окислительные свойства часто приводят ко всяким осложнениям в момент добавления. Когда нашли тоуолсульфоновую кислоту TsOH, именно она стала стандартом сильной кислоты для катализа – это дёшево, доступно и удобно (она твёрдая, её легко взвесить, она не имеет окислительных свойств, а по силе мало отличается от серной). Ещё одна проблема – обратимость всей реакции взаимодействия амина и карбонильного соединения. В реакции выделяется вода, и пока она остаётся в реакционной смеси та же кислота катализирует гидролиз енамина. Воду обязательно надо убирать из реакционной смеси. Это можно делать многими способами, например, вести реакцию в присутствии веществ, связывающих воду, использующихся как осушители. Очень часто применяют, например, безводный сульфат магния, особенно если хотят получить енамины не с пирролидином или морфолином, а с небольшими и доольно летучими вторичными аминами, например. диметиламином. Также применяют активированные молекулярные сита, и еще много чего. Это удобно и когда вам не нужно делать много енамина, а как принято в современной химии, реакции ставятся на миллиграмовые количества и енамин делают непосредственно перед использованием.
Но для обычной работы всё же первый по популярности метод – азеотропная отгонка воды. Хотя я в этом полностью не уверен, но кажется именно Сторк сделал этот метод очень популярным в органической химии, где его применяют во всех случаях, когда нужно непрерывно из реакции удалять образующуюся воду – синтез енаминов, иминов, сложных эфиров, другие случаи элиминирования воды, просто высушивание и обезвоживание – очень часто делают этим способом. Как мы уже выяснили, Сторк подцепил этот способ у своих предшественников Хейла и Херра, которые использовали некую насадку Бидуэлла-Стерлинга. Что за штука? И зачем?
Дело в том, что многие летучие органические жидкости при кипении захватывают в пары воду, если она им попадается в том сосуде, где происходит кипение. При этом, совершенно необязательно, чтобы эти жидкости с водой смешивались. Даже очень часто так бывает, что жидкости с водой не смешиваются, но при попытке перегнать, в пар уходит смесь с водой, – понятное дело, в пару разделения фаз нет – но когда пар конденсируется, конденсат снова расслаивается на воду и органику. В этом случае такую смесь называют гетероазеотропом, в отличие от гомоазеотропов, когда перегоняется при постоянной температуре смесь смешивающихся жидкостей (про этанол с водой знают все). Азеотропы очень удобно использовать для удаления воды и осушки, особенно гетероазеотропы. Гетероазеотропы кипят всегда ниже каждого из компонентов, поэтому если одна из жидкостей – вода, то ниже 100 градусов, даже если вторая жидкость кипит сильно выше. Это же явление, кстати, лежит в основе перегонки с водяным паром, только там все немного наоборот – не жидкость уносит воду, а вода – жидкость, поэтому перегоняются с водяным паром даже очень высококипящие вещества – перегонка заведомо идет не выше температуры водяного пара.
Причина образования гетероазеотропов (да и гомоазеотропов тоже) проста. В курсе физхимии обычно говорят про разные всякие законы фазовых переходов, газов, растворов и т.д. – и всегда не забывают напоминать, что они относятся к идеальным системам. Если речь идет о парообразовании, то пар должен быть максимально похож на идеальный газ, а в идеальном газе или нет межмолекулярных взаимодействий, или они совершенно одинаковы в любом составе. Если такими взаимодействиями действительно можно пренебречь парооразование из смеси жидкостей шло бы в строгом соответствии с законами идеальных систем – смесь кипела бы при температурах фазовых переходов, а состав паров соответствовал бы летучести компонентов. Увы или наоборот ура, особенно когда одно из веществ вода, об идеальности можно забыть – молекулы воды не взаимодействуют разве что с инертными газами, даже с алканами взаимодействуют, а уж с соединениями, образующими водородные связи – обязательно. И поведение испаряющейся системы становится неидеальным, сильно отклоняющимся и от закона Рауля, и от поведения чистых веществ, осуществляющих фазовые переходы. Можно сказать, что молекулы разных жидкостей вдруг буквально выпрыгивают из этой самой жидкости раньше времени только чтобы оказаться в объятиях молекул другого вещества, и те ведет себя точно так же. Ни те, ни другие даже не могут дождаться прописанных им температур кипения. Но парам все равно однажды придется сконденсироваться, и тогда молекулы опять разбредутся по своим фазам. Свидание было недолгим, результат – разделение. Очень хорошо и удобно, хотя грех так обманывать молекулы.
Для удаления воды гетероазеотропной отгонкой годятся самые разные растворители, но удобнее всего простые ароматические углеводороды – бензол, толуол, ксилол (смесь изомеров). Хлороформ, например, точно так же летит с водой, но после разделения фаз хлороформ оказывается снизу, вода сверху. А углеводороды легче воды. Вообще этот метод использовали еще в 19-м веке, просто перегоняли смесь, отгон делили в делительной воронке и воду выбрасывали. Сейчас можно сделать точно так же, если у вас нет модного устройства, о котором речь ниже. Можно даже снабдить перегонную колбу капельной воронкой, и органический растворитель возвращать туда, если отогналось уже много, а не вся вода вышла. Вышла вода или нет определить легко – отгон будет мутным пока есть хоть малейшие остатки воды. Как только станет прозрачным, дело сделано. Не пренебрегайте этим простым способом, потому что на самом деле это довольно эффективно, ведь вода улетает довольно быстро, и возможно даже перезаправлять не придется. Так делали все старые химики и не жаловались. И, что самое противное, сами ничего не придумали. А вот всякие промышленные, прикладные химики, скорее даже лаборанты, которые тоже уже очень давно приспособили гетероазеотропную отгонку воды для определения влажности всяких продуктов или субстанций, и которым надо было эту процедуру повторять сотни раз на дню, додумались до очень простого устройства. Впервые его описали для нужд определения содержания воды в эмульсиях нефтепродуктов в 1920-м году американские химики Эрнест Дин и Дэйвид Старк (E W Dean, D D Stark, Ind. Eng. Chem., 1920, 12, 486). Как раз в те времена начался нефтяной бум – появилось уже много автомобилей – и стала быстро расти нефтепереработка, а наука, с этим связанная, стала быстро развиваться. Через несколько лет насадку немного усовершенствовали некие Бидуэлл и Стерлинг, просто сделав внизу тонкий отвод с грудуировкой, что облегчило определение уже небольших количеств влаги в обычных продуктах. Именно этот вариант насадки через 30 лет попал в руки органиков, и через статью Сторка устройство, уже в оригинальном виде Дина и Старка стало популярным. Сторк, что забавно, избегает имен изобретателей, видимо, не хочет, чтобы Сторка путали со Старком. Спите спокойно, Сторк и Старк, мы не путаем, вы оба классные ребята, давшие нам отличные способы делать химию. Кстати, в наших краях вариант Бидуэлла-Стерлинга довольно часто раньше попадался, причем это было именно советское стекло, подозреваю, что это такое штатное оборудование для лабораторий в пищевой промышленности. Но нам это неудобно, и вообще это не работает потому что сначала в эту узкую трубочку попадает эмульсия, капиллярные силы мешают ей нормально разделиться, и там застревает пузырек толуола, и вся точность определения объема идет к чёрту. Перемудрили, поэтому и имена почти пропали, и если бы не первая статья, с которой начинаются енамины, и знать бы их никто не знал. У нас обычно поступали просто – отрезали этот отросток и на резинке туда насаживали краник.
Итак, реакция кипит в колбе, пары поднимаются в обратный холодильник и скапывают в эту штуковину, там довольно быстро эмульсия разделяется и более тяжёлая вода собирается снизу. А вот органика скоро наполняет колено полностью и начинает скапывать обратно в колбу за новой порцией воды. Так, небольшим объёмом органики можно вытащить всю воду. Когда пользуетесь этой удобной фиговиной, не забывайте про несколько простых вещей. Первая – жидкость должна интенсивно кипеть, чтобы через насадку все время шла циркуляция. Это оычная ошибка, – прибор ставят на слабый нагреватель (какое-нибудь китайское дерьмо, которого у нас в практикумах немеряно) – жидкость в колбе вроде кипит, но тепла недостаточно и пары как будто застревают где-то в отводной трубке, и почти ничего не конденсируется. Так можно кипятить часами и днями без толка. Поменяйте нагреватель, или хотя бы сначала попробуйте закутать прибор в обычную алюминиевую фольгу. Следите за скоростью кипения и конденсации. Если скорость хорошая, процесс обычно занимает не больше часа. Если плохая, можете греть сутками, пока все в колбе не осмолится. Вторая важная вещь – растворителя должно быть достаточно, чтобы после полного заполнения насадки, в колбе еще оставалось приличное количество – бывают довольно крупные насадки Дина-Старка, куда влезает кубов по 50, и так можно незаметно испарить в колбе все досуха и тогда содержимое пригорит и будет разлагаться.
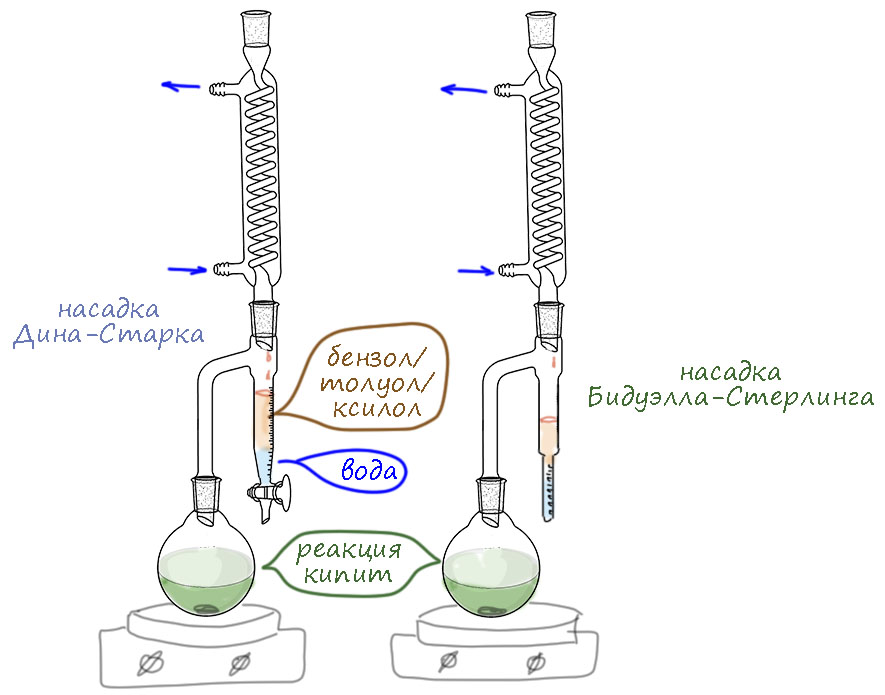
Для такого удаления воды можно использовать бензол, толуол и ксилол (это смесь изомеров случайного состава, хотя вы можете найти и чистые изомеры, но это не нужно). Что лучше? На самом деле, почти одинаково. Гетероазеотропы кипят ниже кипения воды, даже с ксилолом – чистый ксилол кипит при 135-140 градусах, а азеотроп приблизительно при 90°. Азеотроап с бензолом кипит около 70°, с толуолом при 84°. Способность вытаскивать воду у всех трех сравнимая; бензольный азеотроп вроде бы дает самое тщательное обезвоживание, но здесь это не нужно. Бензольным азеотропом очень тщательно высушивают самые гигроскопичные вещи. Нам же просто нужно убрать по возможности большую часть воды, ведь мы всё равно сразу после будем дальше чистить получившийся енамин, и промоем смесь водой, чтобы отделить кислотный катализатор, высушим и перегоним. Азеотропная отгонка нужна нам, чтобы максимально сместить равновесие в реакции, а не для того, чобы очистить продукт. Поэтому после завершения реакции в какой-то момент придется отгонять избыточный растворитель на роторном испарителе. И если возьмёте ксилол, процесс этот может оказаться непростым – ксилол нормально отгоняется, если у вас хороший ротор, фирменные шлифы держат вакуум вообще без смазки, хорошо работает насос – тогда ксилол улетит ласточкой при совсем небольшом нагревании бани. Но ротор может быть старым, шлифы на ловушке перепаянными, в насосе застрял дохлый таракан, а шланг от насоса испещрен вековыми трещинами и помнит Марковникова. Тогда ксилол отогнать или вообще не получится, или придется баню кипятить ключом, и в клубах пара еще и подогревать ловушку феном. При этом ксилол не дает никакого преимущества – он не вытаскивает больше воды, чем его более бедные метилами товарищи. В общем, я бы остановился на толуоле, хотя ненавижу его запах, от которого плавятся остатки мозгов. Бензол, к сожалению, хоть пахнет намного приятнее, но это доказанный канцероген, контакты с которым надо по возможности минимизировать.
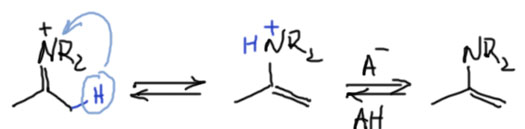
О сопряжении
Сопряжение – отличный способ стабилизации в органических молекулах. Это очень важная идея, которая определяет в органике очень много всего – от относительной устойчивости изомеров до многих особенностей, с которыми мы сталкиваемся, пытаясь понять механизм той или иной реакции. И мы так привыкаем к сопряжению, что начинаем относиться к этому способу взаимодействия частей молекул как к чему-то обязательному, неизбежному, лищь бы рядом оказались фрагменты или атомы с π-орбиталями, то есть либо кратными связями или ароматическими кольцами, либо атомами с неподеленной парой или вакантной орбиталью (радикалы сразу вынесем за скобки). И мы стараемся всегда сделать так, чтобы сопряжение возникло, такие фрагменты оказались рядом, и считаем, что здесь и думать нечего – ничего выгоднее сопряжения в органической химии нет, и отказаться от сопряжения могут только безумцы, а среди молекул таковых нет.
Но, увы, это заблуждение. Сопряжение действительно штука неплохая, но вот во-первых, не всегда, и во-вторых, соседство подходящих фрагментов “на бумаге” это самое сопряжение не гарантирует. Есть еще третий фактор – сопряжение безусловно стабилизирует только в том случае. если мы имеем дело с цепью сопряжения, на концах которой можем выделить донорный и акцепторный фрагменты. Но бывают еще цепи сопряжения, на обоих концах которых либо только доноры, либо только акцепторы – и такие сопряжения дестабилизируют, а значит невыгодны. А бывают еще случаи, когда сопряжение образует не цепь, а разветвление – то, что называется кросс-сопряжением, и такой тип сопряжения отлично бывает как стабилизирующим, так и дестабилизирующим. Я разбирал это на страничке про мезомерию и граничные структуры. В случаях кросс-сопряжения часто возникает конкуренция фрагментов за возможность сопряжения, и ее кто-то может выиграть, в случае чего, выигрывший образует нормальную цепь сопряжения, а проигравший будет выведен из сопряжения. Из этого следует, что иногда нужно даже уметь оценивать фрагменты по эффективности сопряжения, чтобы понимать, у кого больше шансов в борьбе за сопряжение.
Начнем с первого – соседство фрагментов на бумаге не гарантирует сопряжения. Это просто. Для споряжения нужно, чтобы сопрягающиеся орбитали были коллинеарны, а значит соответствующие фрагменты могли быть в одной плоскости. Увы, есть такая вещь как стерика, и она отлично может этому помешать. А поскольку стерика – штука чрезвычайно суровая, потому что имеет дело не с какими-то деликатными стабилизациями-дестабилизациями, а с очень грубым и безусловным эффектом отталкивания электронных оболочек из-за принципа Паули – невозможности нахождения в одной области простанства электронов с одинаковыми значениями всех квантовых чисел, а такие электроны как пить дать найдутся у почти любых атомов, то сближение становится невозможно. Радиус сближения, на котором этот эффект становится невыносимо силен обычно принимают равным ван-дер-ваальсовыми радиусам атомов. В общем, если у нас в молекуле есть сближения каких-то групп на такие рассояния, то группы найдут способ вывернуться – повернуться, отогнуться, и т.п., но этот эффект точно перевесит все выгоды от того же сопряжения. Но даже и с несколько большими расстояниями отталкивание все равно остается немаленьким – электронная плотность не заканчивается на ван-дер-ваальсовой границе, и сохраняются другие компоненты отталкивания, прежде всего обычная электростатика.
И не нужно больших групп для того, чтобы сопряжение сильно ослабить или почти совсем разрушить. Например, стирол – плоская молекула с сильным сопряжением, и это хорошо видно по многим свойствам и даже по длинам связей. Транс-метилстирол такой же плоский и сопряженный. А вот цис-метильной группы вполне хватает, чтобы фенил не смог находиться в одной плоскости с метилом. 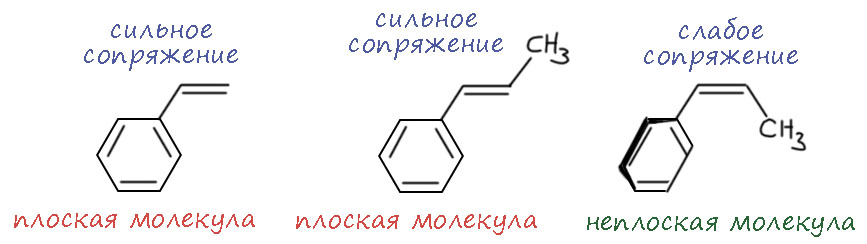
Немного допилим эту картину. Стирол и метилстиролы – нежесткие молекулы. Фенильное кольцо может вращаться вокруг связи между фенилом и винилом. Как положено, это заторможенное вращение, и мы должны рассматривать, как положено для нежестких молекул, конформации и искать устойчивые конформеры и барьеры вращения. В транс-метилстироле нам становится очевидно, что конформером – конформацией, соответствующей минимуму энергии (если забыли, перечитайте страничку про нежесткие молекулы) – является как раз плоская система с углом между ароматическим кольцом и винильным фрагментом (он тоже плоский) равным нулю. Нарисуем зависимость энергии от угла, пометив важные конформации проекциями Ньюмена как раз вдоль связи вращения (плоскость фенильного кольца обозначим жирной магентовой линией). Все понятно – есть единственный конформер, плоский, но вращение возможно, оно происходит с некоторым барьером как в любой нежесткой молекуле. Конформер соответствует минимуму энергии и этот минимум – а любой минимум, опять напоминаю, это признак стабилизации данной конфигурации атомов – ровно и обусловлен энергией стабилизации за счет сопряжения двойной связи и ароматической системы (это основной вклад в энергию, но там есть и другие, но нам это неважно). Теперь цис-метилстирол. Вращение тоже есть, но конформер уже не плоский, а плоская конформация теперь соответствует не минимуму, а максимуму энергии – это как раз барьер, который надо преодолеть, чтобы кольцо перекрутилось с одной стороны плоскости на другую. Барьер невелик – опять не будем обсуждать величину, просто примем, что она позволяет молекуле крутится точно так же как это делают, например, кольца в дифениле. Теперь внимание – в плоской конформации есть сопряжение. Как оно проявляется? Очень просто. Эта конформация дестабилизирована стерическим отталкиванием фенил-метил и стабилизирована сопряжением. Если бы стабилизация сопряжением была сильнее, чем стерическое отталкивание, мы бы тоже имели плоский конформер. Но здесь стерика сильнее сопряжения. Тем не менее, сопряжение уменьшает величину барьера вращения – он меньше, чем в случае транс-изомера, крутится легче, но конформеры – их два, они одинаковые, и соответствуют фенилу, повернутому на большой угол относительно плоскости винильного фрагмента. А почему не один и не под прямым углом? А потому что сопряжение продолжает стабилизировать любую конформацию, в которой угол не 90 градусов, чем больше угол, тем в меньшей степени (орбитали перекрываются, но с поправкой на косинус угла). Конформер соответствует балансу между стерическим отталкиванием и сопряжением. Как видим, поскольку угол большой, отталкивание доминирует почти везде.
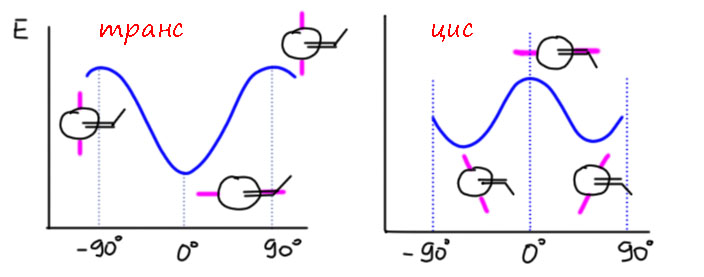
Напомню главный принцип стереохимии нежестких молекул. Для всех практических целей нежесткая молекула полностью представлена своими конформерами. Если мы их опознали и поняли, как они устроены, нам больше не нужно рассуждать про вращение. И даже барьеры нам не понадобятся, потому что они ни на что не влияют, пока не становятся достаточно большими, чтобы вообще заблокировать перекручивание из конформера в другой конформер – тогда они становятся изомерами (ротамерами) и появляется атропо-изомерия. Здесь такого не будет.
Я так подробно это проговорил, чтобы мы хорошо понимали, как устроены взаимодействия стабилизации (сопряжением) и дестабилизации (стерикой). А теперь возьмёмся за енамины.
Пункт первый. Амино-группа сопряжена с двойной связью и стабилизирует ее. Насколько сильно? Сильнее, чем тот же фенил или слабее? Пока не знаем, попробуем разобраться. Но начнём с начала.
Начнем с самого простого енамина – диметиламиноэтилена (это енамин ацетальдегида с диметиламином). Азот с неподеленной парой отлично сопрягается с двойной связью, которая работает как акцептор (напомню, что кратные связи и бензольные кольца в сопряжении могут играть роли и донора, и акцептора в цепях сопряжения, если они на конце, и роль проводника сопряжения, если они в середине; здесь двойная связь на конце и берёт роль акцептора, так как роль гетероатома с парой жёстко задана – это всегда донор). Самое лучшее сопряжение требует коллинеарности взаимодействующих орбиталей и максимального перекрывания. Поэтому выгодна регибридизация азота из обычного для аминов sp3-гибридного состояния (тетраэдра, в котором одна вершина это пара) в плоское sp2-гибридное состояние. Это даёт а) более короткую связь C(sp2)-N(sp2); б) полноценную p-орбиталь на азоте (гибридная орбиталь в тетраэдрическом азоте тоже можно сделать коллинеарной, но она будет отогнута наружу большей долей, и перекрывание будет заведомо слабее). Именно это и есть самая лучшая структура для енамина, обусловливающая его реакционную способность, за которой мы и охотимся. Но электронную структуру обсудим отдельно.
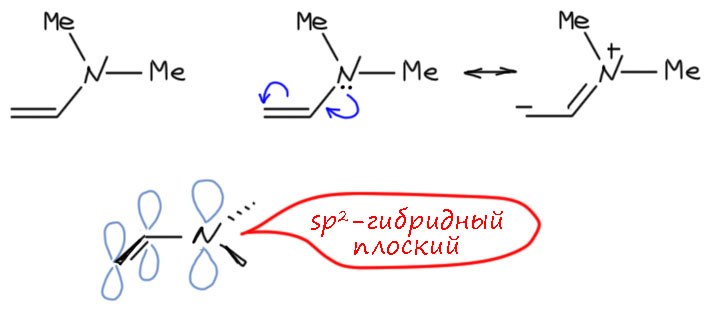
Сейчас же посмотрим, что будет, если мы начнем крутить амино-группу вокруг связи C-N, то есть тоже изучим возможности конформаций и конформеров. Что будет, если аминный фрагмент уходит от коллинеарности? Довольно понятно, что это приведет к ослаблению сопряжения, а следовательно и стабилизации за счет сопряжения. Но это еще не вся правда. Во-первых, с ослаблением сопряжения, уменьшается выгодность sp2-гибридного состояния, и азот регибридизуется в sp3. Почему? Потому что для элементов 2-го периода в ситуации когда все связи одинарные, выгоднее тетраэдр, потому что в нем достигается самое большое удаление валентных электронов друг от друга, и соотвественно и отталкивание тоже (в данном случае чисто электростатическое). Только сопряжение перевешивает этот фактор и способствует регибридизации из тетраэдра в плоский треугольник. Далее, регибридизация в sp3 удлиняет связь C(sp2)-N(sp3), и это дополнительно ослабляет остаточное сопряжение. И ещё – мы ведь не забыли, что у азота есть и индуктивный эффект, и вполне себе акцепторный – азот, между прочим и между нами, один из самых электроотрицательных элементов Периодической системы. И индуктивный эффект есть всегда. В плоской, полностью сопряженной форме он тоже есть, но эффект сопряжения его перевешивает. Как только мы начинаем вращение, индуктивный эффект быстро отвоевывает своё и выходит вперёд. Парадоксальным образом этот эффект тоже является стабилизирующим – вообще, как мы уже видели, двойная связь сама по себе, это дестабилизирующий элемент структуры органических соединений из-за избытка электронной плотности в очень небольшой области пространства, поэтому всё, что помогает как-то смягчить нехороший эффект собственно двойной связи, стабилизирует молекулу (или конформации нежесткой молекулы). Эффект сопряжения создает стабилизирующие многоцентровые взаимодействия, разводит электронную плотность по большему пространству. Акцепторный индуктивный эффект и акцеторный эффект гиперконъюгации делают в общем то же самое, только другими способами. Однозначно дестабилизирующим является только донорный индуктивный эффект, никак не компенсированный гиперконъюгацией – поэтому, в частности, так нестабильны простые еноляты, но мы сейчас не про них, а про енамины. Итак, при вращении аминогруппы происходит а) быстрое ослабление сопряжения; б) регибридизация азота в sp3; в) перехват инициативы акцепторным индуктивным эффектом. Еще раз повторю – все эти факторы являются стабилизирующими, и минимум энергии плоской формы просто объясняется тем, что сопряжение стабилизирует несколько сильнее, и потеря этого эффекта приводит к увеличению энергии. Но небольшому. Поэтому енамины так легко вывести из плоской формы – они мало что теряют при этом. Теряем мы (и приобретаем одновременно), но об этом позже.
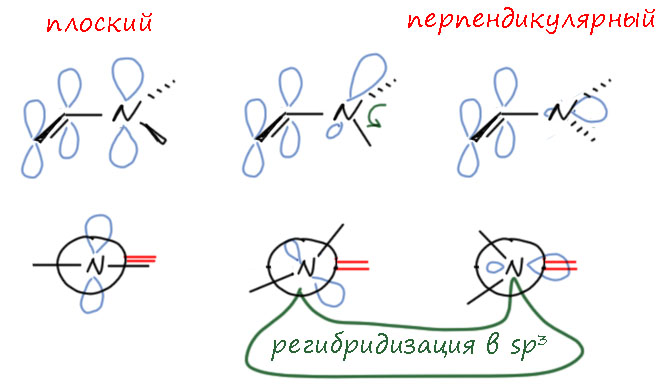
Когда енамин плоский, полностью соответствующий ожиданиям от эффекта сопряжения пары азота и двойной связи? И когда мы можем ожидать существенного разворота амино-группы с регибридизацией? Это довольно просто представить, и самый главный эффект оказывает стерика. Это довольно нетрудно представить, если посмотреть на енамин с точки зрения того, как расположена относительно двойной связи амино-группа. Поскольку мы хотим плоского енамина, ничто не должно мешать такому расположению. И тогда мы видим, что главная проблема, от которой никуда не денешься, это органические остатки на азоте. Даже если это самый маленький метил, он обязан расположиться в плоскости. И тогда мы видим, что если на двойной связи на него смотрит водород, проблем нет – енамин сможет быть плоским. А значит будет, потому что мы уже согласились с тем, что это выгодно, если ничего не мешает. Где может быть такое? У циклических кетонов прежде всего – циклопентанона, циклогексанона, и более крупных тоже. Ага, смекнули советские пионеры, так вот почему во всей этой енаминовой химии так популярны циклические кетоны! Такое впечатление, что енамины вообще больше ни для чего не годятся. В стероидной химии енамины просто бешено популярны, там как раз пропасть циклических кетонов. Да, ровно так и есть, в том смысле, что с циклоалканонами енамины работают особенно хорошо, и в этой химии в прошлом веке все были без ума от енаминов. Но второе неверно, енамины полезны не только для циклоалканонов. Метилкетоны тоже отлично дают плоские енамины. Пока не будем обращать внимания на второй возможный енамин, если с другой стороны кетона тоже енолизуемая группа. И третий тип кетонов, который может дать плоские енамины, это кетоны, из которых получается нециклический, но монозамещенный енамин.
А вот енамины, которые хотят, но не могут стать вполне плоскими, гораздо более многочисленны. Даже одна метильная группа, попавшая на дальний атом двойной связи, не позволит фрагменту стать плоским, причем и на азоте ничего крупного не нужно. Понятно, что если это не метил, а что-то покрупнее, проблемы становятся совсем серьезными и там уже недалеко и до перпендикулярного расположения, а это, как мы выяснили ведет к регибридизации и превращению амино-группы в чистый индуктивный акцептор.
Ну вот мы и увидели основные варианты структуры енаминов. А теперь попробуем ответить еще на два вопроса:
1. Какой енамин образуется из несимметричного, енолизуемого в обе стороны кетона
2. А как структура енамина отражается на его реакционной способности
Какой енамин образуется
Селективное превращение несимметричных кетонов – одна из основных задач в химии кабонильных соединений. Мы уже довольн многое здесь умеем, работая с енолятами. Обычный вопрос – более замещённый или менее замещенный, обычно в несколько упрощенном виде, когда менее замещённый – это либо метил, либо незамещенная сторона циклоалканона. Решение, которое мы применяем опирается на различие между кинетическим и термодинамическим контролем – то, что ыстрее образуется против того что устойчивее в равновесной смеси.
А енамин – что это? Ответ очевидный – енамины почти всегда получают в равновесных условиях – долго нагревают смесь карбонильного соединения и амина в присутствии кислотного катализатора, часто удаляя воду азеотропной отгонкой или еще как-то. В таких условиях не может быть сомнений – это именно равновесные условия. И какой же енамин образуется?
В химии енаминов по разным причинам укоренился неправильный ответ: образуется менее замещенный енамин, потому что он более устойчив, а более устойчив он по причине, которую мы уже рассмотрели – он плоский и поэтому сопряжённый. Вернее, это ответ не совсем неправильный, а полуправильный.
Правильный ответ такой – образуется равновесная смесь енаминов. И в этой смеси очень часто нет существенного преобладания одного из изомеров. Мы и это уже разобрали – плоский енамин действительно более устойчив чем неплоский, но ненамного, потому что оба енамина стабилизированы: один сопряжением, второй остатками сопряжения плюс индуктивным акцепторным эффектом. Разница мала, константы равновесия не сильно отличаются от единицы, и преобладание плоского енамина невелико или вообще пренебрежимо мало.
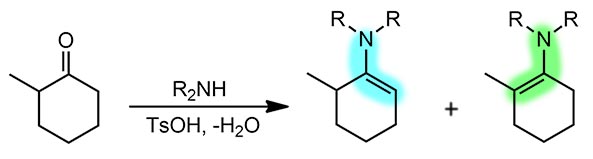
Когда-то из этого получилась небольшая сенсация, потому что в начале химии енаминов представление о том, что образуется селективно менее замещенный енамин, преобладало. Но в 1967 Гуровиц и Джозеф сделали работу, которая внесла изрядную сумятицу в стройные ряды енаминщиков и енаминщиц (W. D. Gurowitz, M. A. Joseph, J. Org. Chem. 1967, 32, 3289). Использовав только недавно ставший доступным ЯМР, они определили состав енаминов для нескольких популярных несимметричных кетонов. И выяснили, что в смеси всегда присутствуют оба, и более замещённый, и менее замещенный часто никак не преобладает, и еще – что это здорово зависит от амина.
 Почему это так мы уже в общих чертах поняли и без Гуровица – оба енамина стабилизированы, но по раному. Менее замещённый имеет плоский енаминовый фрагмент и стабилизирован сопряжением амин – двойная связь. Более замещённый енамин имеет скрученный относительно плоскости двойной связи амин и нарушенное сопряжение, но он все равно стабилизирован и потому что двойная связь более замещённая. а это никто не отменял, и потому что у амино-группы остается акцепторный индуктивный эффект, и он тоже стабилизирующий. Поскольку величины стабилизирующих эффектов крайне трудно оценить, мы можем предположить, что они находятся в некотором паритете, и оба енамина довольно близки по энергии.
Почему это так мы уже в общих чертах поняли и без Гуровица – оба енамина стабилизированы, но по раному. Менее замещённый имеет плоский енаминовый фрагмент и стабилизирован сопряжением амин – двойная связь. Более замещённый енамин имеет скрученный относительно плоскости двойной связи амин и нарушенное сопряжение, но он все равно стабилизирован и потому что двойная связь более замещённая. а это никто не отменял, и потому что у амино-группы остается акцепторный индуктивный эффект, и он тоже стабилизирующий. Поскольку величины стабилизирующих эффектов крайне трудно оценить, мы можем предположить, что они находятся в некотором паритете, и оба енамина довольно близки по энергии.
Собственно, именно это и получено в статье Гуровица и Джозеф. Вот распределения енаминов для четырех разных вторичных аминов. Три из них циклические, наиболее популярные в химии енаминов – пирролидин, морфолин, пиперидин. И обычный диметиламин. С диметиламином енамины делать не любят, но только потому что невозможно использовать удобную методику с азеотропной отгонкой воды – летучий диметиламин улетит первым. Поэтому приходится использовать молекулярные сита, в больших количествах, так как надо убрать много воды, а ёмкость сит не очень велика. Тем не менее, енамин получить можно, и он мало чем отличается. Итак, мы видим, что наибольшую разницу менее и более замещенного енамина дает пиперидин (позже эту величину измеряли другие исследователи, и у них обычно получалось похуже, что-то около 70:30). Остальные амины дают почти равнове соотношение.
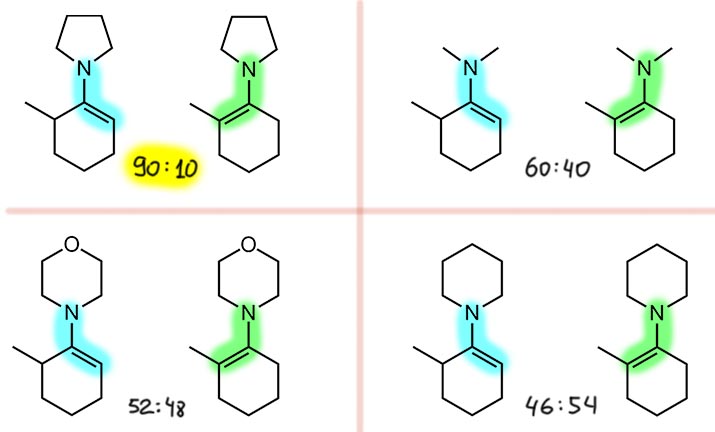
Почему такая разница? Это следствие немного разного баланса стерических взаимодействий. Замечу, что для других кетонов часто получается другая картина, и пирролидин ничем особенным не отличается от морфолина и пиперидина, поэтому все три амина были и остаются главными для енаминовой химии, причем более доступный и дешёвый морфолин всё же по статистике большого количества работ применяется в среднем чаще.
И что из этого следует? Что химия енаминов становится очень сложной, потому что надо учитывать распределение енаминов? Что она неселективна? А почему тогда и Сторк и многие его последователи считали, что наоборот она великолепна и селективна. Вряд ли они заблуждались.
Посмотрим на реакционную способность енаминов.
Почему обычно получается продукт из менее замещённого енамина, если присутствуют оба
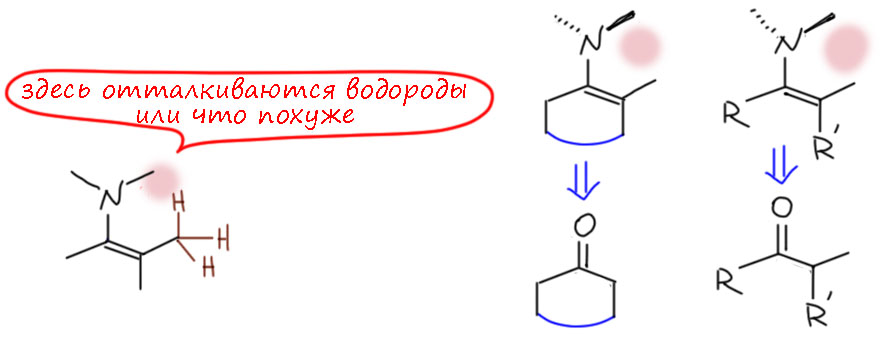
Соотношение изомерных енаминов в равновесной смеси практически никак не влияет на основной продукт, образующийся в результате реакций с типичными электрофилами – акцепторами Михаэля, и это установил очень быстро сам Сторк. Он был уверен, что это связано с тем, что именно такой енамин образуется (раздел про несимметричные кетоны в его главной статье так и начинается: The less substituted enamine is formed from unsymmetrical ketones such as 2-alkylcyclohexanones), и это попало во многие учебники. Интересно, что Сторк это не написал прямо с потолка, а привел данные ЯМР – но этот метод в 1963 году был еще настолько груб и несовершенен, разрешение спектров было ужасным, а Сторк, скорее всего, взял для этого опыта пирролидиновый енамин 2-метилциклогексанона – так что неудивительно, что была совершена такая ошибка. Стерическое объяснение этого явления тоже было сформулировано сразу, и мы с ним почти соглашаемся, действительно основной причиной является стерическое взаимодействие протонов метильной (или алкильных) групп с группами на атоме азота – то, что получило название аллильного напряжения (allylic strain). Почти соглашаемся – это значит, что мы тоже видим эту причину, но понимаем, что дело там вовсе не в относительной устойчивости енаминов. А в чём – сейчас разберёмся. Сторк там же особенно удивляется тому, что даже 2-фенилциклогексаноновый енамин ведет себя точно так же несмотря на вроде бы очевидное сопряжение фенильного кольца с двойной связью. Но мы уже поняли, что там нет и не может быть никакого сопряжения – фенильное кольцо повернуто относительно плоскости двойной связи из-за того же стерического взаимодействия. Более того, фенильное кольцо мешает и амину нормально сопрягаться с двойной связью, в общем – такое соперничество, в котором нет выигрывших.
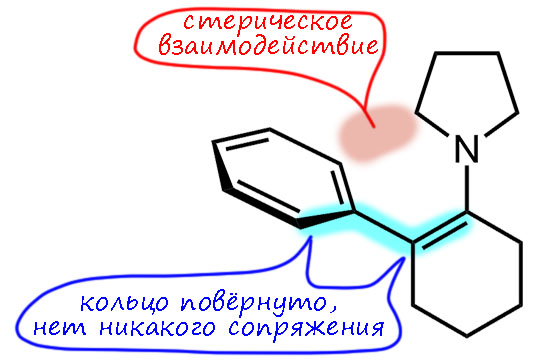
То, что это не совсем так, как мы уже видели, установлено было довольно быстро (самая главная статья Сторка по енаминам вышла в 1963, а статья Гуровица и Джозеф – в 1967), но мало повилияло на уверенность многих химиков в том, что прав был именно Сторк, и дело именно в этом. Химикам важнее продукты реакции, а объяснение – дело пятое, вроде убедительно, а как там на самом деле пусть разбираются всякие теоретики, а практикам до этого нет ни дела, ни времени – синтез требует реагентов и методов, а не болтовни.
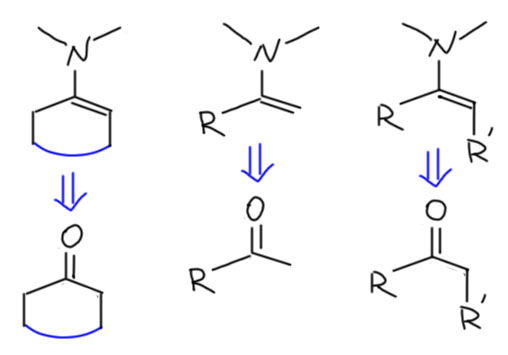
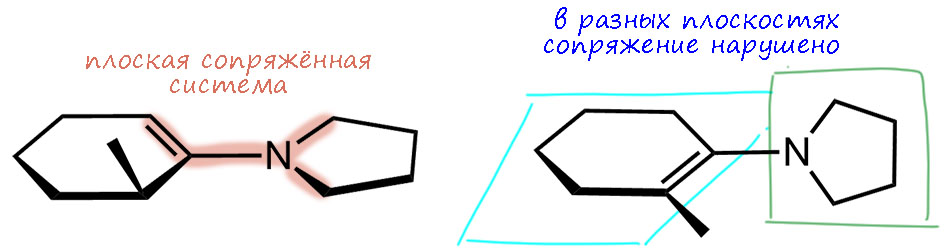
Посмотрим ещё раз, чем отличается менее замещённый енамин от более замещённого. С точки зрения структуры мы уже разобрались: в менее замещённом енамине двойная связь и амин копланарны, сопряжение выражено полностью. В более замещенном стерическое отталктвание протонов метильной группы (или другого заместителя, тем более) и протонов на углероде рядом с азотом выворачивает амин относительно двойной связи – сопряжение нарушается, возможно не полностью, но сильно.
Вот как это выглядит на расчетной структуре. Обращаю внимание, что связь азота с бывшим карбонильным углеродом не жестко закрепляет два эти фрагмента друг относительно друга, и мы вновь имеем дело с конформерами, и то, что дает расчет это и есть самый выгодный конформер (собственно, в этих молекулах он единственный). Как мы помним их химии сопряжённых диенов, сопряжение не блокирует вращение вокруг простой связи (связь между сопргающимися фрагментами остается простой, сопряжение приводит к небольшому укорочению связи и небольшому увеличению электронной плотности на ней, что можно интерпретировать как небольшой увеличение порядка сверх единицы). И вот эти структуры (для информации DFT B3LYP 6-311G(d,p)) сверху с водородами, снизу без, слева менее замещённый, справа более. Картинки красноречиво подтверждают ожидания даже с некоторым запасом. Одна структура плоская, вторая перпендикулярная. Единственное. что может вызвать некоторые сомнения – обещали, что в менее замещенной все будет плоским и азот sp2-гибридным, а здесь все же видно, что первые углероды от плоскости немного, но заметно отходят. Да, это потому что такую геометрию требует пятичленный цикл – он не хочет становиться совсем плоским (в теме про циклоалканы мы узнаем про торсионное напряжение и про то, почему оно немного гнет циклопентановый цикл – вот это оно и здесь слегка безобразничает), но это очень слабое отклонение, которое практически не влияет на сопряжение, или по-другому говорят, что у азота очень небольшой барьер инверсии, и поэтому затраты энергии на отклонение от идеальной гибридизации в любую сторону обходится недорого. В более тонкой химии енаминов, впрочем, эта неуступчивость пирролидина иногда портит некоторые проявления химии енаминов с этим амином, но мы с таким вряд ли встретимся. Но вот что важно – мы видим, что более замещенный енамин совсем сильно скручен, у него разрушено сопряжение полностью (потеряно около 2.5 ккал/моль энергии стабилизации – это оценки), поэтому это очень невыгодно, и поэтому с этим амином в равновесной смеси так мало более замещённого.
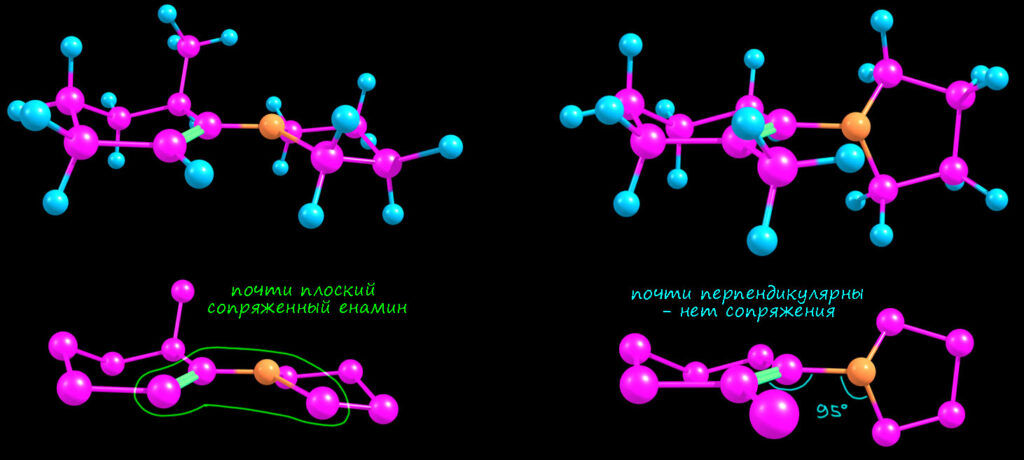
Но если взять морфолин, или пиперидин – шестичленные и более гибкие циклы – в них и стерика будет немного полегче, и затраты энергии на регибридизацию поменьше, поэтому в них меньше разница между менее и более замещенным енамином, и поэтому равновесная смесь содержит их почти поровну.
А теперь посмотрим как это отражается на реакции. Плоский енамин полностью пользуется сопряжением, а значит смещением электронной плотности от азота на крайний углерод. Мы выражаем это граничной структурой, в которой этот углерод выглядит как карбанион, это нуклеофил, и он отлично реагирует с электрофилами. И это то, что мы любим в енаминах Сторка.
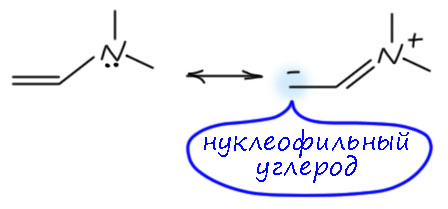
Скрученный енамин так не может. Азот у него полностью или частично выведен из сопряжения, нет больше смещения электронной плотности, и нет особой нуклеофильности. Азот даже немного работает как индуктивный акцептор, но это довольно слабый акцептор, и дополнительной деактивацией можно пренебречь – мы все равно не можем установить ее вклад, а в настоящий акцепторный олефин типа акцептора Михаэля он не превратится. Надо понимать, что любой олефин (даже акцептор Михаэля) обладает способностью реагировать с электрофилами, но только с достаточно сильными. А плоский енамин отлично реагирует с углеродными электрофилами, с которыми обычные олефины не реагируют. Теперь понятно, что у несимметричных кетонов реагирует только менее замещенный енамин. И вот почему набюдается это явление, а вовсе не потому что такой енамин, как Господь Бог наш, един. Можно ли считать такой контекст упоминанием Господа всуе, что является нарушением заповеди? На мой взгляд нет – мы же рассматриваем не какую-то суету, а поведение молекул, которых многие сочтут вполне божественными объектами. Мы разгадали одну из загадок, кто скажет Природы, кто скажет Творения, в любом случае это хорошо и никакой суетой точно не является. Заповедь не нарушена.
Повторим этот важный вывод еще раз. Если в смеси присутствуют оба енамина из несимметричного кетона, а это, видимо, всегда так и есть, потому что разница в энтальпиях образования невелика, и константы равновесия несильно отличаются от единицы, то в реакциях с электрофилами реагирует практически исключительно менее замещённый енамин. Удивительно, но это правило работает просто поразительно хорошо. Это означает одно – из двух путей, из менее и более замещенного енамина, второй путь имеет существенно большую энергию активации. Но и этого мало, потому что важно ещё одно – путь из менее замещенного енамина еще и быстрый (естественно, относительно, потому что мы знаем, что реакции енаминов не очень-то и быстрые). Это дает ситуацию, когда один путь быстрый, а другой настолько медленнее, что мы так и не можем узнать насколько. Обратите на это внимание, потому что это довольно редкий случай, когда кинетический контроль не просто контролирует (в большинстве случаев кинетического контроля, вспомните хоть дурацкое 1,2-присоединение к диенам, образуется не один продукт, а оба продукта, но один преобладает) – а прямо чисто выбирает одно направление. На следующей вкладке мы покажем, что этому способствует и взамопревращение енаминов. И это чертовски важно, потому что в химии енаминов бывают и довольно странные истории, когда мы отходим от стандартных аминов типа пирролидина и морфолина, а берём всякие замещенные, стерически затруднённые амины – в этом случае капризы конформаций делают именно более замещенный енамин более стабильным и преимущественно образующимся – но и они не могут сломать эту разницу в реакционной способности. Нам это не нужно, но в современном органокатализе такие эффекты с успехом эксплуатируют. Вот ситуация с двумя изомерами и двумя путями на картинке профиля энергии вдоль координат реакции. Система выбирает путь через меньший барьер, и не хочет двигаться в сторону высокоэнергетического переходного состояния. Если к этому добавить еще и быстрое взаимопревращение изомеров, получим работающую систему выбора пути. Это хорошо известная система, которую я уже однажды поминал, как встречающуюся и там и сям, где не ждёшь, – принцип (или кинетика) Кёртина-Гаммета, один из ее крайних случаев. Если есть два пути, смотрите скорее на барьеры, а не то, сколько каждого из альтернативных реагентов в смеси. Может быть и один процент того, кто реагирует существенно быстрее второго (которого в этом случае 99%), но продукт будет получаться больше (или даже почти только) из этого малого.
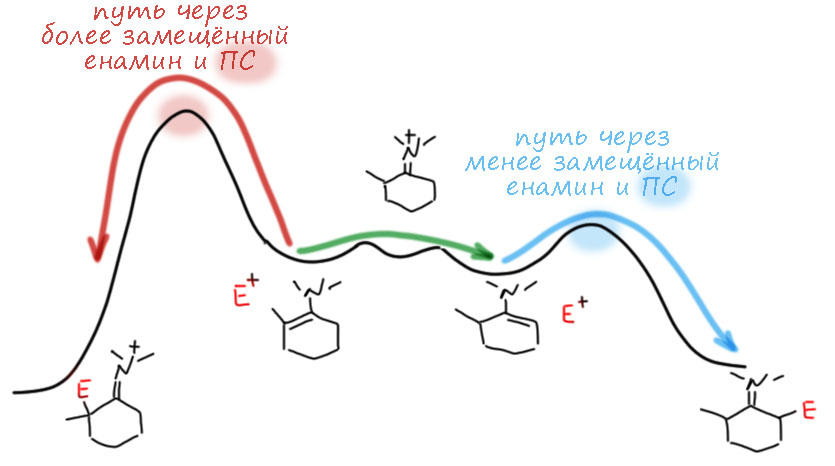
Отлично, передохнули, а теперь возьмёмся за следующую загадку – куда девается более замещённый енамин, ведь во многих случаях его около половины, а выходы продуктов реакции со многими электрофилами от 70% и даже выше. Если бы это было не так, мы бы всегда видели требование использовать пирролидин, с которым менее замещенного енамина больше всех (это далеко не всегда так, кстати). Но нет, в реальных работах очень часто используют морфолиновые енамины, с которыми соотношение менее:более далеко от такого преобладания нужного. Что-то там, видимо, ещё происходит, что позволяет не так сильно зависеть от равновесного соотношения, не только на уровне селективности реакции, но и на уровне выхода.
Займёмся этой проблемой на отдельной вкладке.
А куда девается более замещённый енамин?
Прежде чем заняться этой проблемой на несимметричном кетоне, просто аккуратно разберем, что происходит в реакции енамина более простого кетона с типичным электрофилом. Для начала посмотрим алкилирование галогенпроизводным. В химии енаминов есть традиция под самым простым кетоном всегда подразумевать циклогексанон, а не ацетон, потому что с енаминами маленьких нециклических кетонов химия енаминов это мучение, почти никогда не дающее нужных продуктов. Увы, химия енаминов не всесильна.
Реакции с активными галогенпроизводными ведут просто при нагревании смеси енамина и галогенпроизводного в растворе какого-нибудь несложного органического растворителя, например, бензола или ацетонитрила. После нескольких часов нагревания просто добавляют воду, недолго перемешивают и приступают к обычному выделению продуктов реакции (экстракция, промывка водой, осушка, отгонка растворителя, перегонка, перекристаллизация или хроматография). Получается, что в самой реакционной смеси ничего лишнего нет. Первым делом при взаимодействии с галогенпроизводным получается замещённая соль иминия, большая часть которой доживает до конца реакции и после гидролиза даёт ожидаемый продукт алкилирования. Но, частично соль иминия может превратиться в енамин, точно так же как это происходит в самом синтезе енаминов, путем депротонирования, что есть не что иное как E1-элиминирование (соль иминия это с другой стороны просто сильно стабилизированный карбокатион). Основанием может служить как галогенид-ион (слабое основание, но здесь вполне годится, мы это уже много раз обсуждали). В результате образуется новый енамин, а точнее, как мы уже знаем, смесь более и менее замещенного, и небольшое количество протонной кислоты. Образовавшийся енамин может подцепить вторую молекулу галогенпроизводного, которое почти всегда в этой реакции берется в небольшом избытке, с образованием продукта диалкилирования (после гидролиза), и действительно, продукты 2,6-диалкилирования практически всегда образуется как побочный продукт в алкилировании, а с самыми активными галогенпроизводными типа аллилбромида и избытком побольше может дыже стать и основным продуктом. Обратите внимание, что правило – образуется менее замещённый продукт – работает и здесь.
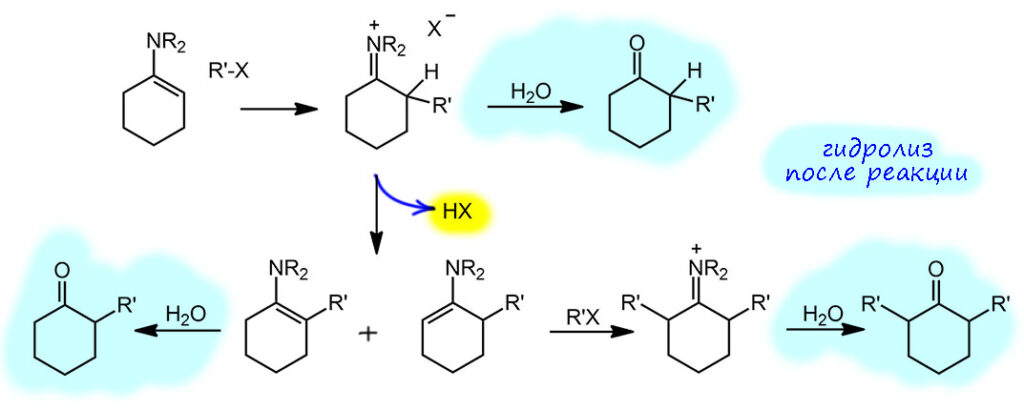
Это понятно, но пока это не то, что нас интересовало. Но обратим внимание на это образование протонной кислоты, в малых количествах, каталитических, но не может ли эта кислота учинить какой-нибудь кислотный катализ. Давайте попробуем разыграть тот же сценарий для несимметричного кетона. Итак, у нас смесь более и менее замещенных енаминов, в которой менее замещенный активно расходуется в реакции с алкилирующим агентом, а более замещенный просто не реагирует и накапливается (относительно менее замещённого, конечно). Вот, в смеси образовалось небольшое количество протонной кислоты. По уже рассмотренному сценарию в смеси появляется протонная кислота, которая отлично вызывает равновесную перегруппировку енаминов, потому что протон обратимо присоединяется-отщепляется к/от двойной связи – менее замещенный енамин снова появляется и реагирует дальше. На вопрос, а отчего это не протонируется аминный азот, отвечаем, как у нас повелось – протонируется конечно, но это один из кислотно-основных путей в системе, просто дополняющий еще одним равновесием (есть данные, показывающие, что енамины сначала протонируются по азоту, а затем медленнее перегуппировываются в иминиевые ионы – это говорит о том, что протонирование по азоту быстрее, это еще один случай кинетического контроля, а в химии переноса протона иногда говорят о кинетической основности или полнее, о кинетически-контролируемой основности). И таким способом в реакцию вступает большая часть исходного кетона, и менее замещённый енамин не пропадает.
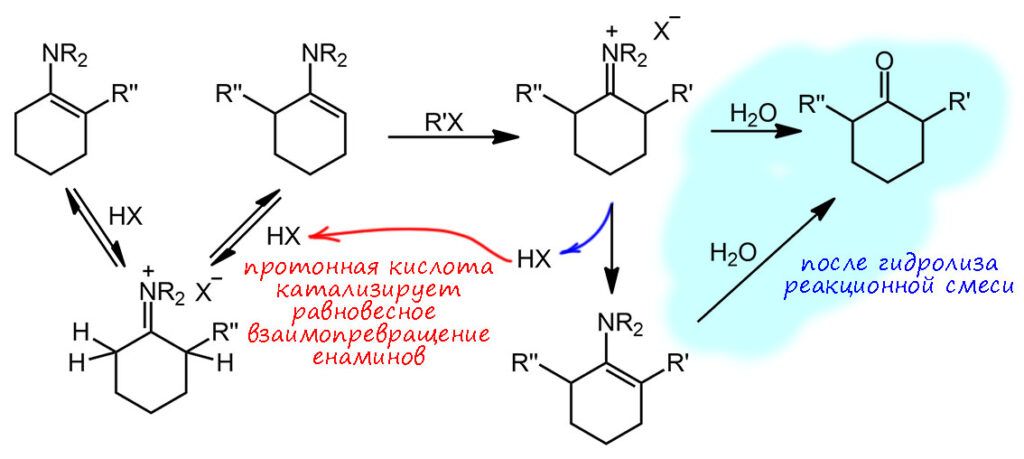
При этом, что касается протонирования енаминов, то даже в такой простой реакции разница в реакционной способности более и менее замещенного енамина значительно, что однажды позволило Джонсону и сотрудникам сделать забавный эксперимент (Johnson, F., Duquette, L. G., Whitehead, A., Dorman, L. C. Tetrahedron, 1974, 30, 3241–3251). Они взяли не просто метилцикогексанон, а его трет-бутильное производное, что в данном случае неважно, а им понадобилось потому что они одновременно еще и изучали конформационные фокусы. Так вот, если быстро обработать смесь енаминов сильной кислотой, а затем быстро эту кислоту нейтрализовать, то можно получить чистый более замещённый енамин, а менее замещенный уходит в виде гидролизованного кетона. Вот как это выглядит. Из кетона получили енамины, в этом случае тоже почти равную по соотношению смесь. Быстро встряхнули ее с разбавленной водной солянкой, проэкстрагировали гексаном. Из гексанового слоя достали исходный кетон. А водный слой быстро подщелочили разбавленным едким натром и экстрагировали оттуда чистый более замещенный енамин, который оказался совершенно стабилен хоть в чистом виде, хоть в растворе.
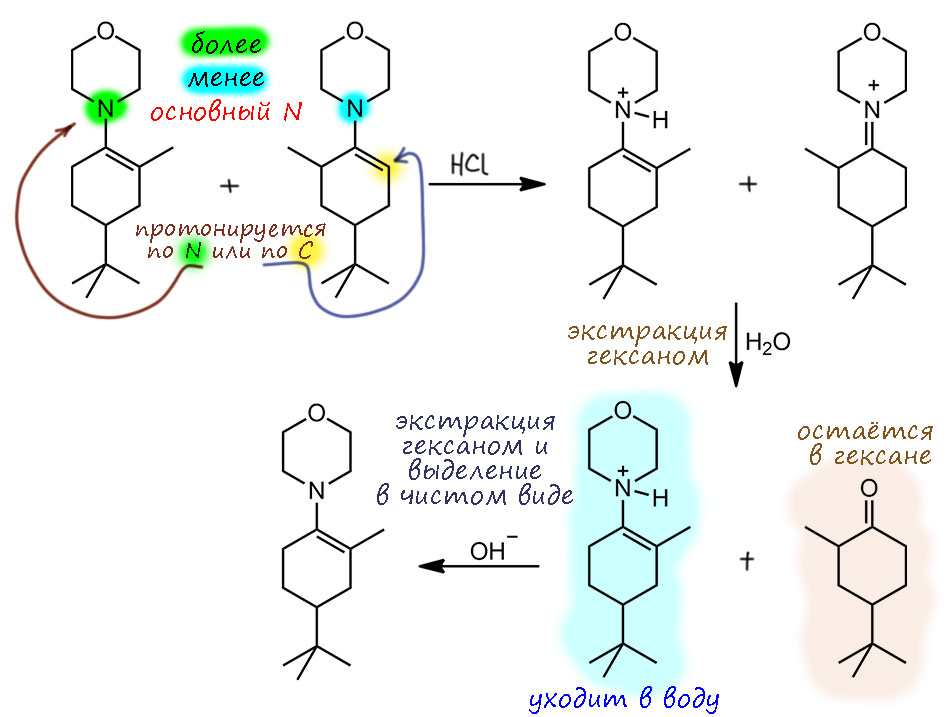
Из этого остроумного эксперимента мы видим несколько вещей. Первая – у менее замещённого енамина азот менее основен. Это понятно, так как он включен в сопряженную систему и регибридизован в sp2. Этот енамин быстро присоединяет протон по двойной связи, и иминиевый катион быстро гидролизуется водой. Вторая – у более замещённого енамина азот не включен в сопряжение и поэтому более основен. Он протонируется, протонированная форма не изменяется дальше и может быть с успехом экстрагирована в воду, как любая соль аммония. И дальше депротонирована и сохранена как более устойчивый енамин для других экспериментов. Здорово, молодцы, а не противоречит ли это идее о том, что енамины в присутствии кислоты быстро превращаются друг в друга, снова давая равновесную смесь? Нет, тот же Джонсон показал, что даже в присутствии следов кислоты это происходит весьма быстро, особенно в органическом растворителе, а в описанном эксперименте просто пришлось поторопиться со всеми манипуляциями, чтобы обогнать более медленное превращение более замещённого енамина. Позже другие исследователи проделывали похожие опыты с бромированием – менее замещенный енамин давал бромкетон, а более – оставался неизменным (в опыте использовалось точное количество брома, соответствующее менее замещенному енамину, потому что иначе более замещенный тоже присоединил бы бром – здесь игра была на скоростях реакции).
Итак, мы убедились в том, что в присутствии малых количеств протонной кислоты изомерные енамины превращаются друг в друга, образуя равновесную смесь, поэтому в условиях реакций с электрофилами, а это обычно многочасовое нагревание, такое равновесие успевает эффективно перекачать почти весь енамин в более реакционноспособную менее замещенную форму, выводящуюся из системы реакцией с элекрофилом. Отсюда и иллюзия, многократно воспроизведенная в разных статьях, книжках и учебниках, о том, что при получении енаминов несимметричных кетонов образуется только менее замещенная форма, потому что она более устойчива.
В конце зададим вопрос – но второй (более замещённый) енамин ведь тоже олефин. Ну что-то он должен же присоединять. Неужели никогда не образуется продукт из этого енамина? Ответ будет довольно уклончивым: в литературе есть примеры таких реакций, но а) это довольно мутные старые работы, не очень хорошо написанные, настолько, что возникает больше вопросов, чем ответов; б) по крайней мере, в некоторых случаях ясно, что или механизм может быть более сложным, чем прямое электрофильное присоединение, или даже возможна перегруппировка продукта. Не буду этого приводить, потому что все это странные случаи. Но есть и еще одна возможность – взят настолько реакционноспособный электрофил, что ему будет все равно, какой олефин атаковать. Такой электрофил надо еще поискать. Может быть галоген? Но нет, данные по реакциям галогенов с енаминами очень мутные, но вроде бы и там преимущественно атакуется менее замещённый. Может быть дейтерирование сильной дейтерированной кислотой? Есть такая работа, и в ней дейтерий в основном встает на менее замещённое место, а примесь более замещенного, скорее всего, объясняется уже дейтерообменом в продукте. Но вот один пример найти удалось, и это очень странный элекрофил, сульфонилированная с двух сторон перекись водорода (R. V. Hoffman, C. S. Carr Tetrahedron Lett., 1986, 27, 5811-5812). Это такая группа, аналогичная тозилу, называется нозил (Ns) и это ещё более лучшая уходящая группа, чем тозил. А почему это не свободнорадикальная реакция, ведь перекись? Потому что идет при -78 градусах, а при такой температуре представить гомолиз даже очень слабой связи трудно. А вот электрофилом это может быть совершенно, извиняюсь, отмороженным, ведь отличная уходящая группа создаёт возможность для поляризации молекулы с развитием электрофильного, почти секстетного кислорода, а это очень сильный электрофил, что-то в одной лиге с синглетным карбеном. И вот, этот реагент с пресловутой равновесной смесью морфолиновых енаминов 2-метиоциклогексанона дает ровно такую же смесь продуктов – то есть ему совершенно все равно, к какому енамину присоединяться. Попробуйте найти что-то похожее. Нам, конечно, это не нужно, но я специально привел пример, показывающий, как трудно бывает преодолеть разницу в реакционных способностях с виду очень похожих соединений, разница в которых объясняется только тонкостями взаимодействия заместителей.
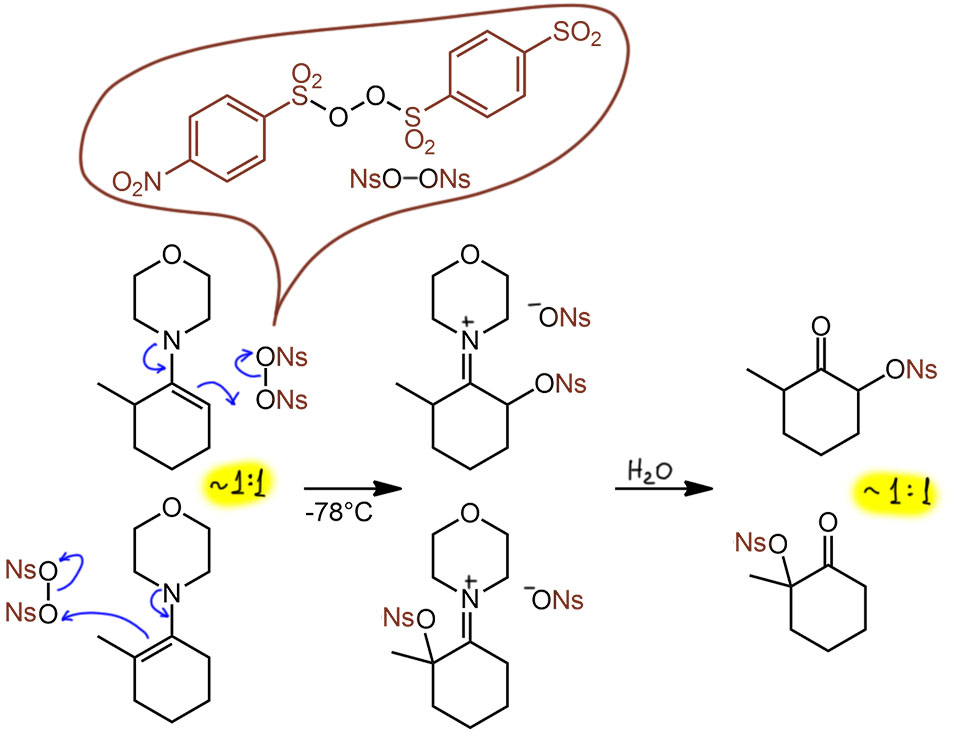
Не всё так просто
Без сомнения, самая популярная реакция енаминов прямо с самого начала и до сих пор – реакция с акцепторами Михаэля, электрофильными олефинами – непредельными сопряженными альдегидами, кетонами, сложными эфирами, нитроолефинами, винилсульфонами и т.п. – таких соединений в органике море, и это одно из самых любимых решений в синтезе, потмоу что позволяет сразу очень сильно усложнить структуру, снабдив ее еще и возможными продолжениями, а в современном синтезе все это еще и стереоселективно. Енамины очень хорошо реагируют с такими соединениями, причем вариантов оргомное количество, и на этом сделано огромное количество и исследований, и сложных синтезов, и львиная доля популярности енаминов связана именно с этим типом взаимодействия. Когда говорят об именной реакции Сторка, обычно имеют в виду именно это.
Мы так далеко не полезем, но всё же чуть-чуть копнём, чтобы понять, как непросто работают с виду вполне ясные реакции. Эта непростота, с одной стороны, осложняет понимание, особенно в начале, но с другой стороны, она же предоставляет возможности умного использования реакции.
Особую ценность реакции придает то, что для неё ничего не нужно кроме енамина и акцептора Михаэля. Реакции акцепторов Михаэля с нуклеофилами обычно требуют или кислотного или основного катализа для активации или нуклеофила, или самого акцептора, и это часто приводит к побочным реакциям. А здесь не нужно ни того, ни другого – реакции ведут почти всегда протсо при нагревании двух этих реагентов в растворах разных простых растворителей типа спирта, ацетонитрила, бензола, хлороформа. Потом добавляют воду, недолго перемешивают и приступают к выделению продукта. Но если мы просто напишем реакцию, сразу увидим одну проблему, которой нет, например, в реакции енаминов с галогенпроизводными, где сразу образуется соль иминия, только ожидающая гидролиза. Здесь сначала образуется доаольно страшная штука – карбанион (стабилизированный акцептором Z) с одной стороны, и соль иминия – с другой, то есть нечто, имеющее и нуклеофильный, и электрофильный центры в одной молекуле. По всем законам химии эти центры должны взаимодействовать. Спасает то, что между ними нехорошее число атомов, и замкнуться они могут только в 4-хчленный сильно напряжённый циклобутан. Действительно, когда реакцию проводят с особо активными енаминами и акцепторами Михаэля не при нагревании, а при охлаждении, такие циклобутаны даже иногда выделяются, но они неустойчивы из-за сильного напряжения (подождём до темы циклоалканы, чтобы узнать, что это такое) и потому, что на соседних атомах находятся сильно-донорный (амин) и сильно акцепторный (Z) заместители – такие соединения, у которых на соседних атомах могут возникнуть стабилизированные катион и анион, часто называют пуш-пульными (push-pull, тяни-толкай), такими бывают олефины, циклопропаны, циклобутаны. Хорошо, если такой циклобутан не образуется, особенно при повышенной температуре, то надо что-то сделать хотя бы с карбанионом, иначе эта штука начнёт накапливаться и тогда просто будет реагировать одна молекула с другой с орбразованием ненужных олигомеров, которые для простоты называют смолами, а процесс их образования – осмолением. Есть ещё один процесс, который может “успокоить” карбанион – мы знаем, что отщепление протона от иминиевого катиона может давать енамин, и к счастью, здесь это очень легко организовать, если исходный енамин получен из кетона, енолизуемого на обе стороны – тогда такой перенос протона может легко произойти внутримолекулярно, так как карбанионный центр довольно легко достает до другой стороны. Так образуется енамин уже продукта, и в таком виде он обычно доживает до конца реакции, и при гидролизе дает конечный продукт. Иногда, когда акцептор Михаэля очень активен, этот енамин может подцепить вторую молекулы олефина и дать двойной продукт, и даже в обычных случаях это образуется как побочное в количествах до 10%. Вот так это можно разрисовать, если взять очень популярные енамины циклогексанона, но с другими енаминами получится то же самое – мне просто надоело рисовать общие случаи.
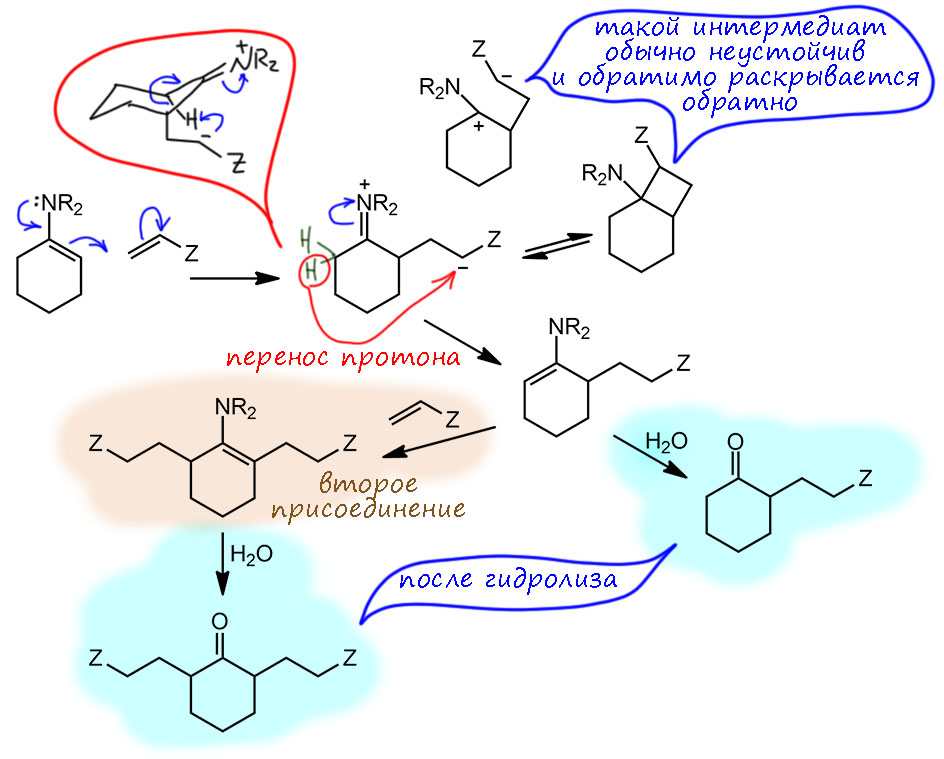
Если реакция идет в спирте, а так делают часто, причем спирт берут абсолютный, чтобы избежать преждевременного гидролиза енаминов, проще представить себе перенос протона с помощью растворителя. Кстати, это может помочь и если у нас енамин сделан из кетона, енолизуемого только наодну сторону, или из альдегида. С альдегидными енаминами, впрочем, я не рекомендую связываться, потому что с ними редко реакции идут чисто – слишком много там альтернативных путей.
![]()
Теперь посмотрим на еще одну проблему, которую, впрочем, проблемой не считают и наоборот очень часто применяют в синтезе. Если акцептор Михаэля это производное акриловых кислот, нитроолефин, винилсульфон и т.п., то получается ожидаемый продукт обычно с хорошим выходом и без больших осложнений. А вот с непредельными альдегидами и кетонами хлопот намного больше. С альдегидами вообще лучше не связываться, они слишком реакционноспособны, и имеют возможности для того, чтобы вступить в миллион побочных реакций. С кетонами дела обстоят намного лучше, и даже совсем хорошо, только продукт часто получается не тот, что мы ожидаем по аналогии. Вот что обычно происходит. Сначала реакция, как с другими акцепторами Михаэля даёт цвиттер-ионный интермедиат, только в этом случае это енолят. Я опять возьму для простоты циклогексаноновый енамин, хотя в этой реакции охотно применяют самые разные енамины, даже нециклических кетонов.
Этот енолят, в принципе, тоже должен был бы снять протон с противоположной стороны и превратить иминиевый катион в енамин, и ждать конца реакции и гидролиза. Но вместо этого происходит то, что обычно просходит с енолятами несимметричных кетонов, если в реакционной смеси кроме енолята есть еше какой-нибудь переносчик протона. Здесь таких до чёрта. Проще всего заметить, что эти реакции чаще всего делают в спиртовых растворителях – вот и переносчик готовый. Сам Сторк начинал с простых апротонных растворителей типа бензола, но тогда таким переносчиком может быть даже соль иминия, частично превратившись в енамин. Это нас не должно интересовать, ведь мы слишком хорошо уже знаем, что удержать чистый енолят с одной стороны кетона можно только при полном отсутствии в реакционной смеси и еще непрореагировавшего кетона, и всего остального подозрительного (поэтому так аккуратно делают кинетические еноляты – любое отклонение от методики чревато потерей селективности). Итак, тем или иным способом первоначально образовавшийся енолят изомеризовался в другой енолят. Обращаю внимание еще раз на то, что мы в очередной раз встречаем ситуацию, когда в равновесии есть два (или больше) реакционноспособных частиц, и нас не волнует, какая из них более устойчива, если превращаться во что-то еще имеет возможность только одна из них – именно через неё и пойдёт дальше реакция.
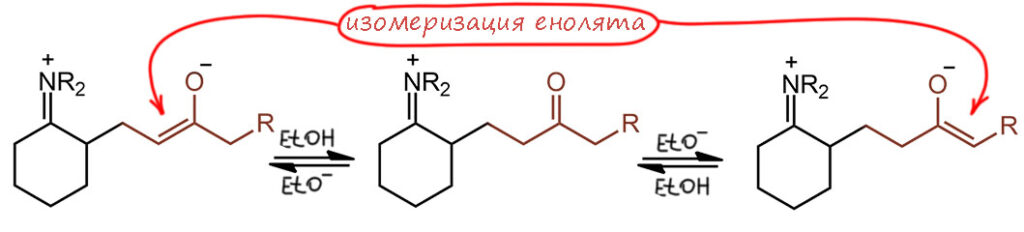 Теперь расстояние между нуклеофильным углеродом енолята и электрофильным углеродом иминиевого катиона стало равно шести, мы это любим и немедленно начинаем пристраивать одну часть к другой, что легко сделать перерисовав енолят в виде подходящего для циклизации конформера (можно не париться и просто выгнуть цепь приблизительно, но так симпатичнее и отражает тот совершенно бесспорный факт, что куски углеродных цепей любят складываться в подобие кресла). Следует замыкание цикла и элимирирование вторичного амина – и это нас уже не удивляет, потому что мы отлично знаем про обратимость присоединения по Михаэлю, очень ярко выраженному для азотных нуклеофилов. Вот он, продукт реакции, хорошенький. Сторк получил такой впервые, использовав самый простой енон, метилвинилкетон, и это стало одним из самых популярных применений реакции Сторка.
Теперь расстояние между нуклеофильным углеродом енолята и электрофильным углеродом иминиевого катиона стало равно шести, мы это любим и немедленно начинаем пристраивать одну часть к другой, что легко сделать перерисовав енолят в виде подходящего для циклизации конформера (можно не париться и просто выгнуть цепь приблизительно, но так симпатичнее и отражает тот совершенно бесспорный факт, что куски углеродных цепей любят складываться в подобие кресла). Следует замыкание цикла и элимирирование вторичного амина – и это нас уже не удивляет, потому что мы отлично знаем про обратимость присоединения по Михаэлю, очень ярко выраженному для азотных нуклеофилов. Вот он, продукт реакции, хорошенький. Сторк получил такой впервые, использовав самый простой енон, метилвинилкетон, и это стало одним из самых популярных применений реакции Сторка.
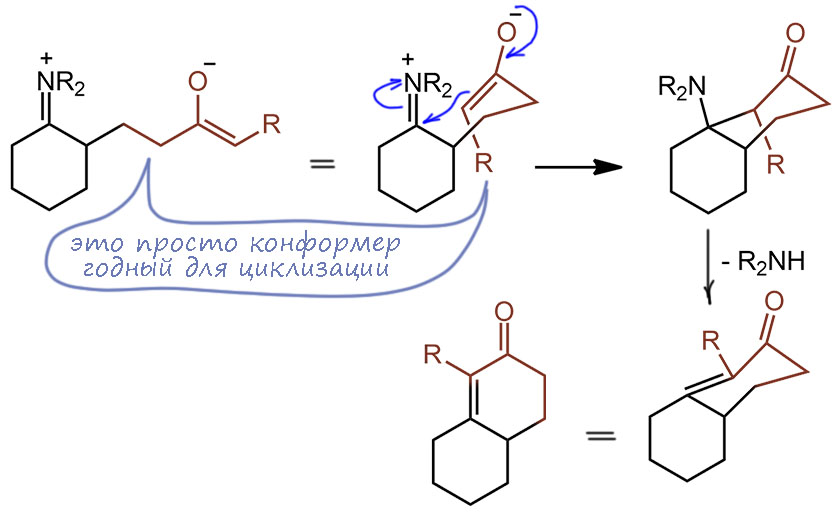
Ещё раз повторю, что реакция идет просто при нагревании раствора енамина и енона в подходящем растворителе, часто абсолютном спирте, без добавления оснований. Это очень удобно. Если мы внимательно на всё это посмотрим, узнаем, что получается то же самое, что в знаменитом аннелировании по Робинсону (этим мы займемся в теме про алициклы). Фактически это альтернативная методика выполнения этой реакции, не требующая никаких катализаторов, ни сильных оснований. В синтезе она не менее, а даже более популярна, чем первоначальный вариант аннелирования Робинсона. Но здесь есть еще одна возможность. Реакция Робинсона идет через равновесный енолят, а значит с несимметричными кетонами идет в основном по более замещенному положению. А енамин реагирует по менее замещенному. Получаем замечательный пример, как подбирая метод можно направлять реакцию по разным путям. Вот как в этом самом простом примере. Обратите внимание на еще один замечательный факт – ни енолят, ни енамин в таких реакциях не образуются в виде одного изомера, но только всегда смеси более и меенее замещенного. Но особенности течения каждой из этих реакций приводят к преимещуственному образованию только одного продукта.
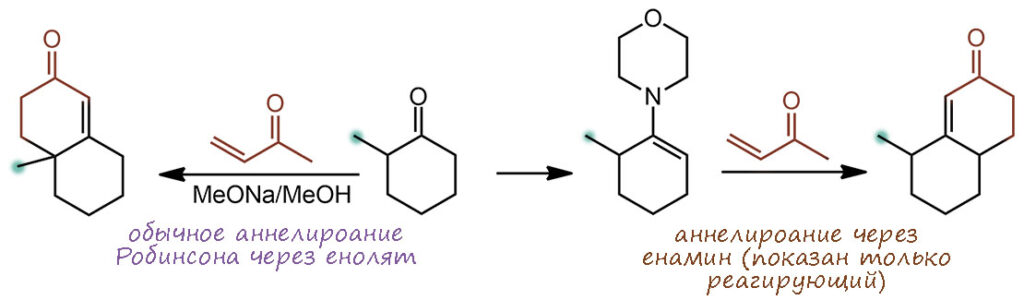
Основные реакции енаминов
Самая главная реакция енаминов, мы её уже разобрали, это реакция с акцепторами Михаэля. Большиснтво примеров использования енаминов в реальных синтезах это именно это, или в простом варианте, или в виде альтернативного аннелирования Робинсона.
Алкилирование
Схему реакции уже рисовали. Важно понимать, что это SN2-замещение с точки зрения алкилирующего агента. Никогда не забывайте законы SN2 и не ошибётесь – они работают в сотнях реакций (в тысячах, в десятках тысяч). Енамины реагируют только с самыми реакционноспособными субстратами, а это галогенпроизводные (или тозилаты) из обычного ряда: метил, аллил, бензил, пропаргил, галогенпроизводные карбонильных соединений. Первичный алкил, если неразветвлённый, взять можно, но лучше тогда иодпроизводное, и даже с ним выходы будут меньше 50% даже при очень долгой реакции. Ряд вполне приличный, так что можно наполучать много разных α-замещённых кетонов, если несимметричных, то с менее замещённой стороны. Енамины альдегидов в этой реакции ведут себя плохо, но для 3-го курса сойдёт, не будем совсем в мелочах копаться. Для кетонов енамины – это лучший способ алкилирования, потому что альтернативные еноляты ведут себя плохо, дают много побочных продуктов диалкилирования, самоконденсации, а с галогенпроизводными карбонильных соединений вообще дадут конденсацию, а не замещение. Кроме того, еноляты несравненно сложнее делать. В серьёзном синтезе алкилирование енолятов применяют тем не менее довольно часто, но почти всегда с более сложными субстратами, когда диалкилирование не идет по стерическим причинам, а у енолятов всё же есть одно преимущество – намного более высокая нуклеофильность, а значит и диапазон реакции шире в сторону более разнообразных алкилов.
Ацилирование
Очень полезная альтернатива сложноэфирной конденсации, и обычно вполне рабочая. Ацилируют хлорангидридами или иногда ангидридами кислот. Реакции делают просто, точно так же как алкилирование – просто перемешивают смесь при нагревании. После охлаждения обычно выпадает осадок хлорангидрида енамина дикетона – это те самые енамины дикетонов, с которых все начиналось, они очень устойчивы из-за хорошего сопряжения донор-акцептор, хотя здесь соль, но она тоже стабилизирована водородной связью (не показана). Соль фильруют, моют, и быстренько гидролизуют разбавленной солянкой, чтобы оттащить в виде соли аммония амин. Почти всегда для этой реакции берут морфолиновые енамины. Получают дикетон без всякого натрия или гидрида натрия, из легкодоступных исходных (напоминаю, что енамин циклогексанона в этой химии рисуют просто как типичный представитель класса).
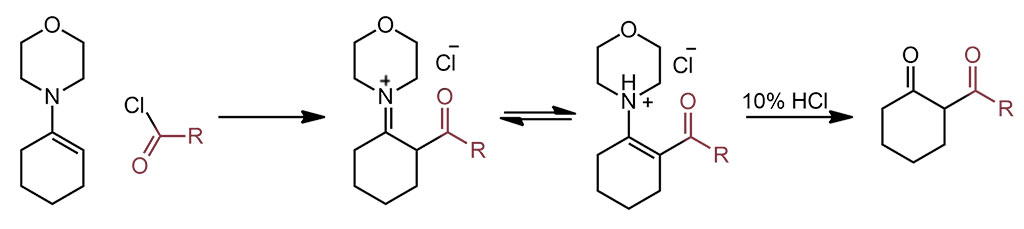
Есть еще такая штука как хлоугольный эфир – тогда получают кетоэфиры. Тоже такой вариант сложноэфирной конденсации без натрия или гидрида натрия. Реакцию ведут точно так же, и в конце получают кетоэфир, а это обычно енольная форма.
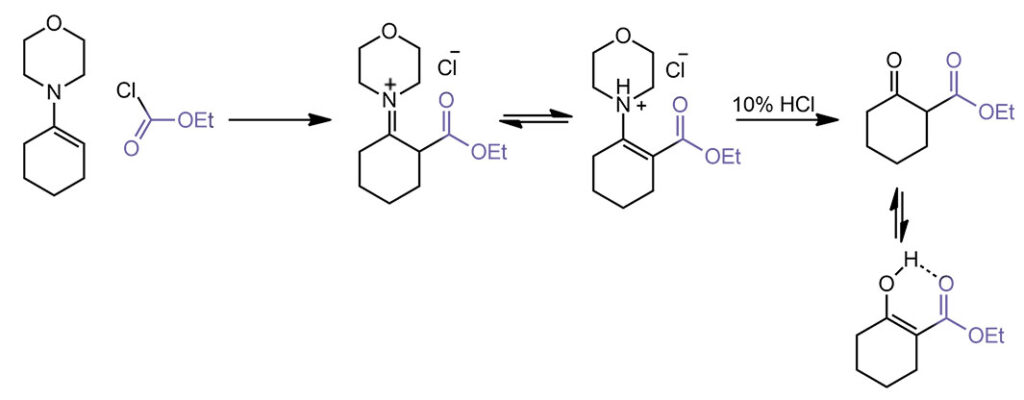
Есть ещё чёртова пропасть других реакций енаминов, но мы ими пользоваться не будем. Ничего особенного в них нет, и делают их по аналогии.
Что не работает
Альдольная конденсация. Она вполне могла бы идти с енаминами, и даже идёт с ними, потому что с этого как раз и начинается органокатализ, о чем можете прочитать на страничке про нобелевскую премию-2021. Но, как ни странно, в обычном варианте, а не в режиме органокатализа, просто конденсация не идёт, а вместо неё начинаются всякие фокусы типа реакции Манниха. Там все очень непросто и нет общих рецептов, поэтому реакцию просто не применяют. А когда появился органокатализ, о простой реакции забыли. Мы не будем даже пытаться.
Реакция с оксиранами. Это самая досадная история, потому что вроде должна, и это из общих соображений попало в некоторые учебники и задачники. Нельзя делать химию из общих соображений – вот мораль, вынесенная из этого случая. Эту реакцию делать пробовали, и даже есть статьи, где ее изучают, но она дает не то, что мы ожидаем, а некие продукты циклизации, в очень жёстких условиях, никаких преимуществ не просматривается. Енамины слабоваты как нуклеофилы для раскрытия оксиранов – мы ведь часто преувеличиваем их реакционную способность и желание во что бы то ни стало раскрыться. Ничего подобного – для этого нужны или сильные нуклеофилы, или активация кислотами Льюиса, которая для енаминов работает плохо, так как кислота сначала садится на азот и енамин убивает как нуклеофил. В общем, если встретите эту реакцию в задачах – используйте, вы же не обязаны отвечать за чужие грехи. А если никто не просит, не используйте, так как использовать в общем нечего.
Разбор спорной задачи
В последней контрольной 2022 года был вопрос про образование енамина и реакцию с акцептором Михаэля, акрилатом. Мне показалось, что с этим не должно быть проблем, так как, по моему мнению, мы должны про енамины знать их особенность давать в реакциях с электрофилами менее замещенное производное. Данный пример вполне в это правило вписывается, в нём с более замещенного положения создана достаточно убедительная стерика, которая не должна позволить обойти это правило химии енаминов. Пример не взят из реальной литературы, а сильно адаптирован по ряду аналогий, как всегда приходится делать, потому что реальная химия обычно сложнее, и для 3-го курса жестковата. Скорее, я руководствуюсь другим принципом: если бы мн пришлось планировать такой синтез в реальности, счел бы я возможным и разумным попробовать такую стадию. Да, счёл бы, да, здесь все вполне вписывается в химию енаминов. Тем не менее, я принял решение отозвать этот вопрос и оценить положительно все возможные ответы по двум причинам. Первая состоит в том, что хотя конечный продукт реакции должен быть именно таким, который и был задуман, но в схеме надо было написать и сам промежуточный енамин, а здесь никакой определенности нет, и почти наверняка образовалась равновесная смесь обоих енаминов, и что в ней было основным компонентом сказать очень трудно, и уж точно этого нельзя требовать от студента 3-го курса. Это очевидный косяк. Вторая проблема – могу ли я требовать корректного анализа сопряжения? Если сильно призадуматься и не лезть на баррикаду сразу, придется признать, что нет, не могу, это сложновато, уж точно для контрольной общего потока, хотя лучшие студенты с амбициями после окончания университета работать в науке, это вполне должны способны сделать. Итак, задача отозвана, баллы скомпенсированы, но давайте разберемся, что тут можно было бы ожидать, и действительно ли фенильная группа может нарушить правило менее замещённого продукта.
Итак, предполагался вот такой ответ. Менее замещённый енамин, и менее замещённый продукт.
Насколько можно было бы действительно ожидать образования единственного и менее замещённого енамина? Или могла, как в большинстве реальных случаев образования енаминов образоваться равновесная смесь:
Или вообще в данном случае более замещенный енамин образовался бы преимущественно или только, а менее замещенный не играл бы большой роли. В этом, и только этом случае, и конечный продукт реакции соответствовал бы более замещённому енамину. Какие аргументы можно привести в пользу преимущественного образования именно второго енамина? Он более замещен, но этого мало, мы это уже разобрали. Кто-то может увидеть здесь сопряжение с фенильной группой, и это был бы действительно весомый аргумент, хотя и здесь пришлось бы доказать, что сопряжение в плоской енаминной системе хуже, чем сопряжение с фенилом – а это, видимо, не так, потому что есть интересное свидетельство уже упомянутогоПитера Хикмота – в равновесии аллильной перегруппировки преобладает енамин, а не аллиламин (P.W. Hickmott, Tetrahedron, 1982, 38, 1975-2050, на странице 1977), собственно это взято из статьи еще более знаменитого химика Джека Хайна (это тот исследователь, который догадался, что при действии щелочи на хлороформ образуется дихлоркарбен, став одним из основоположников новой химии карбенов). Константа равновесия изомеризации оказалась немаленькой, около 40, на стороне именно енамина (J. Hine, S.-M. Linden, A. Wang, and V. Thiagarajan, J. Org. Chem. 1980, 45, 2821). В данном случае можно говорить о чистом сравнении – оба изомера плоские, с возможностью полного сопряжения двойной связи с каждым из фрагментов, поэтому вывод о том, что в плоском енамине стабилизация сильнее, чем в плоском фенилалкене. Из этого вовсе не следует, что преимущество одного над другим достаточно велико, чтобы вторым изомером вообще можно было бы пренебречь, но это важный довод.
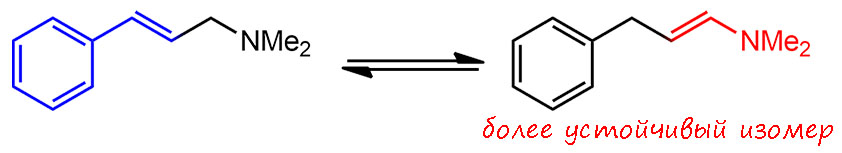
Теперь посмотрим на более замещенный енамин, и подумаем, а есть ли в нём вообще существенное сопряжение двойной связи с фенилом. Вообще-то ответ очевиден – нет и быть не может, так как это типичная система, в которой на двойной связи в цис расположении фенил и метил, который не дает фенилу стать копланарным по отношению к двойной связи. На этом месте вообще-то обсуждение можно было бы закончить, ведь у нас есть менее замещенный енамин, нормально стабилизированный сопряжением между двойной связью и амино-группой, и более замещенный енамин, где сопряжение нарушено и по фенилу (видимо, почти полностью, в цис-метилстироле угол поворота фенила около 70°), но и амино-группе (обычное для более замещённых енаминов стерическое взаимодействие, часто называемое аллильным напряжением).
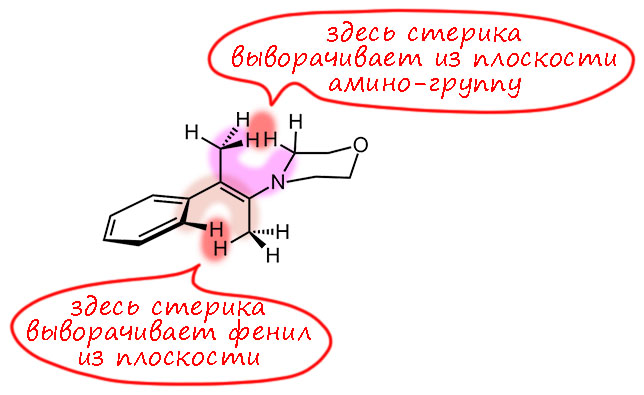
В общем, получается, что этот енамин не только ничем не стабилизирован, но существенно дестабилизирован. Есть все основания считать, что в равновесной смеси будет преобладать менее замещённый. Чтобы уже совсем однозначно разобраться с этой историей (а вдруг нарисованные картинки врут), я даже посчитал молекулярную геометрию этого енамина, больше для развлечения, но и для того, чтобы на таком примере показать, что дают квантово-химические расчеты. Чисто для информации тех, кому может это показаться важным – DFT B3LYP, полная оптимизация геометрии в двух базисах, 6-31+G(d,p) и 6-311G(d,p), минимумы проверены, результаты почти одинаковые. Разглядываем, и… приходим к выводу, что из общих соображений получилось ровно это. Фенил повернут, угол 67° относительно двойной связи, это очень большой угол, можно считать, что от сопряжения там даже рожек да ножек не осталось, а на виде сверху вообще можно поять, что это почти перпендикуляр. И амин скручен очень сильно, а азот заметно уходит от плоскости из-за регибридизации в sp3. И очень хорошо видны метилы, которые и мешают и фенилу, и амину быть копланарными с двойной связьб и эффективно сопрягаться.
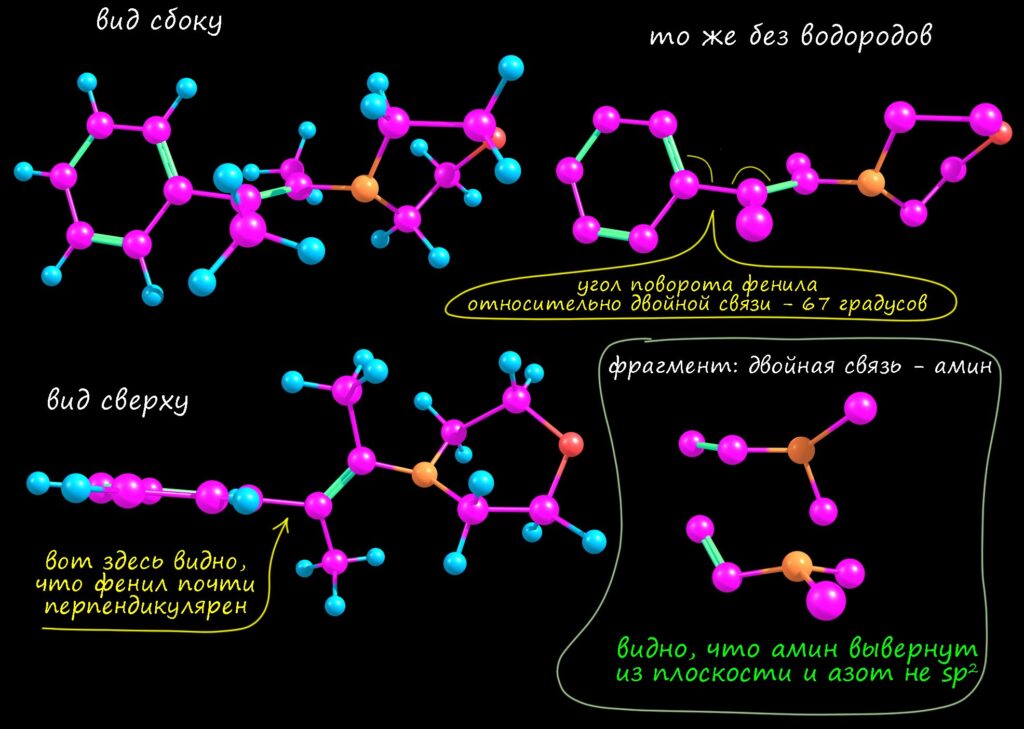
Любопытства ради посмотрим на менее замещенный енамин. Ну вот, здесь всё гораздо приятнее. Енаминовый фрагмент практически плоский, что говорит о хорошей степени сотряжения. Ни фенил, ни метилы этому не мешают, они отогнуты в другие стороны. То, что сопряжение в нужном нам енамине лучше еще и следует из длин связей: двойная заметно длиннее (1.353 Å против 1.349 Å в более замещённом енамине), связь C(sp2)-N короче (1.396 Å против 1.431 Å), а это следует из взаимодействия, обеспечиваемого мезомерией.
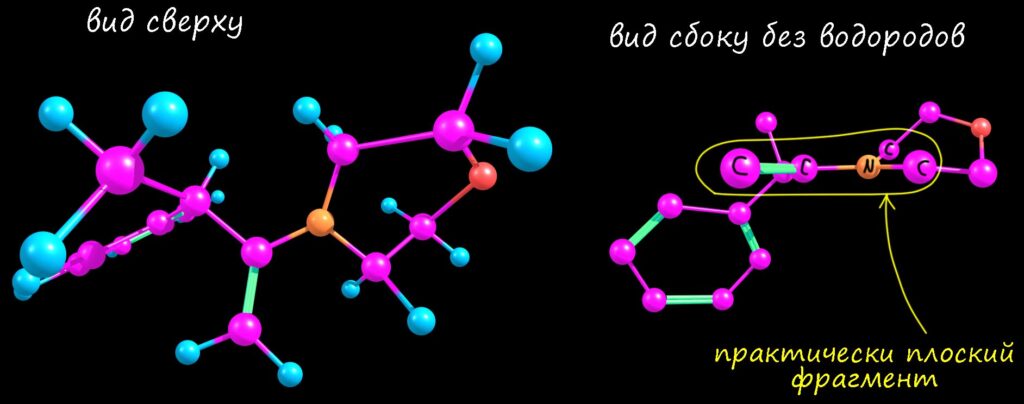
Резюмируем. Есть все основания считать, что поведение енаминов кетона из задачи вполне типично для енаминов кетонов, у которых с одной стороны заместители, а с другой или метил или незамещенный углерод алицикла. Возможно образование двух енаминов в равновесии, но преобладать будет менее замещенный, потому что он лучше стабилизирован. В этом случае преобладание может быть очень солидным, так как стерика явно дестабилизирует структуру с обеих сторон двойной связи. И в реакции можно без опасения ждать менее замещенного продукта, даже если более замещённый енамин присутствовал бы в равновесной смеси, птому что более замещенный енамин не имеет хорошего сопряжения двойная связь-амин, а только это сопряжение увеличивает нуклеофильность енамина для ракции с акцептором Михаэля.
Но корректный анализ ситуации (даже без расчета, который, как мы видим, просто подтвердил анализ структуры) явно выходит за рамки возможного для 3-го кура, поэтому я и снял эту задачу.
А если амин первичный?
строго факультативно! пожалуйста, не используйте эту химию для решения задач на 3-м курсе!
А что будет, если вместо вторичного амина в реакции с карбонильным соединением взять первичный? Речь идет про обычные альдегиды и кетоны, не те, что являются СН-кислотами с повышенной кислотностью, типа ацетоуксусного эфира и т.п. – эти дают с первичными аминами и даже аммиаком только енамины, и мы с этого и начинали историю енаминов. А что делают простые альдегиды и кетоны? Это давно и хорошо известно – они дают имины, или как их ещё называют, основания Шиффа, по имени итальянского химика Уго Шиффа из Пизанского университета, где очень увлекались ранней химией альдегидов (подробнее можно прочитать на вкладке про бисульфитные производные в разледе Ответы). Интересный вопрос – почему просто карбонильные соединения дают имины, а 1,3-дикарбонильные – енамины.
Это очень просто – дело в балансе стабилизирующих и дестабилизирующих взаимодействий. И очень важно понимать, что в химии мы никогда не имеем дело со стабильностью или нестабильностью как таковыми. Эти вещи а) всегда относительны; б) относительны по отношению к тому, во что можно легко превратиться.
Поэтому мы сравниваем каждый раз взаимопревращающиеся молекулы. Например, когда мы обсуждали енамины из дизамещённых аминов, мы сравнивали конформации, и выяснили, что в зависимости от вариантов стерического взаимодествия более стабильна или плоская форма (конформер), или форма, в которой амино-группа скручена на значительный угол, но не 90°. А что будет с енамином из монозамещенного амина? О, здесь появляется более интересная альтернатива просто вращению – таутомерия. Перемещение протона от азота на углерод. Одно из соединений это енамин, второе – имин, то самое основание Шиффа, как хотите так и называйте, Уго Шифф был неплохой парень, да еще и итальянец, да и работал в Пизе, а это, поверьте, очень хорошее место. А что стабильнее, енамин или имин? Это практически та же проблема, что сравнить кето-форму и енол. Только здесь мы вообще не можем оценить кислотности – нет у нас даже приблизительной оценки NH-кислотности имина. Попробуем с другой стороны. И в имине, и в енамине есть двойные связи. Двойные связи – дестабилизирующий фактор. Но чем более электроотрицательны атомы на концах двойной связи, тем лучше. Это довольно очевидно, раз мы считаем, что проблема двойной связи в избыточной электронной плотности в малой области пространства – но более электроотрицательные элементы лучше справляются с избыточной плотностью, собственно в этом и состоит электроотрицательность. Поэтому в этой паре стабильнее имин, и ровно это и наблюдается. Если получить производное с первичным амином почти любого моноальдегида или кетона, то по всем свойствам это будет именно имин – это будет очевидно по всем спектрам. В общем, мы здесь ничего нового не видим: для тех же карбонильных соединений основной будет карбонильная форма, и с этим мы уже разобрались очень хорошо.
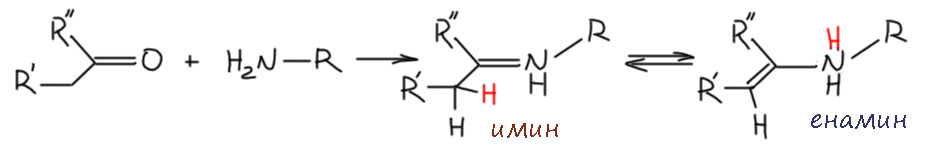
Хорошо, но в кетонах и альдегидах мы знаем, что малейшая примесь енольной формы, которую не видно ни в каких спектрах, работает во множестве реакций. А в имине есть такая же примесь енамина, и если есть, можно ли ее применить в реакциях? Ответ на это положительный, и мы сейчас посмотрим на одну такую реакцию, но сначала разберемся, почему у 1,3-дикарбонильных соединений енамины стабильнее иминов. Безусловно, можно просто сослаться на аналогию – енамин это как енол, а в таких дикарбонильных соединениях енольная форма всегда преобладает. Значит и енамин тоже должен. Но здесь объяснение легко увидеть. Имин в этом случае ничем особенным от имина монокарбонильного соединения не отличается. А енамин – отличается, и очень сильно. Енамин в этом случае это полноценная цепь сопряжения донор-акцептор с двойной связью в роли связующего звена. А такие цепи сопряжения это лучшее, что есть в сопряжении, это наиболее стабилизированная форма, и мы даже можем не обращать внимания на дополнительные факторы стабилизации – электростатическое притяжение минус-плюс, или водородную связь. Это очень серьёзный фактор, и он не только делает енамин преобладающей формой, но способствует его очень легкому образованию без всяких дополнительных ухищрений типа азеотропной отгонки, молекулярных сит и т.п. И даже не только с первичными аминами, но и с самим аммиаком, как мы уже видели, потому что первым в истории енамином был как раз такой енамин из ацетоуксусного эфира, аминокротонат.
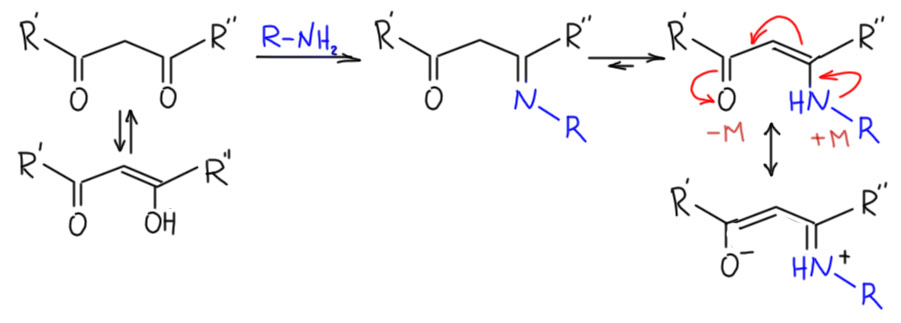
Возвращаемся к иминам простых альдегидов и кетонов, и будум искать реакцию или реакции, выдающие наличие в нем таутомерной формы енамина. Реакция такая нашлась (и не одна, но нам достаточно одной), и оказалась очень интересной. Это обнаружил, немного случайно, швейцарский химик, всю жизнь работавший во Франции, Мишель Пфау в 1970 году (Pfau M., Ribière, C. J. Chem. Soc., Chem. Commun. 1970, 6647), и дальше он, и его французские ученики и коллеги лет тридцать изучали эти реакции и сделали из них неплохие методы синтеза. Пфау заподозрил наличие таутомерии с енамином, и стал его ловить. Для начала он взял и снял (нет ничего плохого в этом слове вместо в 4 раза более длинного “зарегистрировал”, спектр это разновидность показаний прибора, в данном случае прибором является спектрометр, а показания приборов именно снимают, поэтому употребление этого короткого глагола вполне корректно) протонный ЯМР-спектр изопропилимина ацетона в дейтерометаноле. И с удивлением обнаружил, что прямо в ампуле очень быстро происходит дейтерообмен в метильных группах. В этом месте надо удивиться первый раз, потому что в самом ацетоне дейтерообмен так быстро не происходит, да и требует или кислотного или основного катализа. Дейтеорообмен явно идёт через енамин, и это значит, что таутомерия действительно есть, и равновесие таутомерии устанавливается намного быстрее, чем в енолизуемых кетонах. Тогда даже неважно, сколько енамина находится в равновесии, потому что если найти реакцию, в которой енамин расходуется, равновесие будет быстро восполнять убыль. Такое ускорение дейтерообмена в иминах по сравнению с кетонами, а значит и ускорение таутомерного превращения, связано или с тем, что константа равновесия имин-енамин все же больше, чем константа равновесия кетон-енол (это можно объяснить, можете сами потренироваться), или с тем, что сам же имин реакцию и катализирует, ведь это вполне себе приличное основание в отличие от кетона (константу основности мы не знаем, но это соединение азота).
Но дейтерообмен это важно, но интереснее более сложные реакции. Было обнаружено, что имины енолизуемых кетонов реагируют с акцепторами Михаэля, и эта реакция явно идет через таутомерный енамин. В самой первой статье сделали реакцию того же имина с диэтилмалеатом. И получили ожидаемый продукт реакции таутомерного енамина с этим олефином, причем в отличие от реакций более привычных нам енаминов, где гидролиз промежуточной соли иминия сразу возвращает кето-группу, здесь имин сохраняется в продукте и при желании достать кетон, придётся делать гидролиз. Получилась довольно интересная реакция. Дальше ее много изучали, там оказалось немало сложностей, поэтому она так и не получила такую же популярность в синтезе, как превращения енаминов Сторка.
У этой реакции есть одна интересная особенность, которую обнаружил в 1980-е очень активный в химии енаминов английский ученый Питер Хикмот. Как раз в это время он сделал странную вещь – переехал работать в Южно-Африканскую республику. Это сейчас эта страна вполне уважаемое и даже довольно высокоразвитое государство Африки, а тогда это было просто настоящий изгой, о которого отвернулось практически всё человечество, включая и запад и восток, – там практиковалась ужасная расистская система апартеида, полного разделения белых и черных граждан страны. ЮАР находилась под строжайшими санкциями ООН, которые вынудили ее правительство в 1990-х отказаться от апартеида и начать демократические реформы, но в 1980-х санкции и презрение человечества были в самой высшей точке. И вот этот Хикмот не побрезговал туда переехать работать, что весьма странно, но придется признать, что он там сделал отличное исследование. Что его туда понесло мы не знаем, может был он закорнелым расистом, а может просто что-то связывало сентиментальное. В ЮАР при всем апартеиде и санкциях была очень неплохая наука, в том числе химия, и медицина – именно в этой стране была впервые в мире осуществлена пересадка сердца и создана целая школа трансплантологии. В общем, ещё раз убедимся, что жить и работать можно везде, и если сделать что-то однозначно позитивное и полезное человечеству, то отрицательная аура места не испортит будущего признания заслуг конкретных людей.
Итак, это Хикмот посмотрел на уже многочисленные статьи Пфау и сотрудников, которые пока им большой славы не снискали, и понял, что нужно сделать одну простую вещь – попробовать реакцию на несимметричном кетоне, в первую очередь на самом популярном для этой химии 2-метилциклогексаноне (Hickmott P. W., Rae B. Tetrahedron Lett. 1985, 26, 2577–2580). Он подумал, что таутомеризация в основном будет давать более замещённый енамин, так как в этом случае выгода более замещенного олефина не будет омрачаться плохой стерикой, ведь амин первичный (он пробовал несколько разных, и анилин, и циклогексиламин, и бензиламин, и в конце остановился на этом последнем), и в сторону метильной группы будет смотреть водород на азоте.
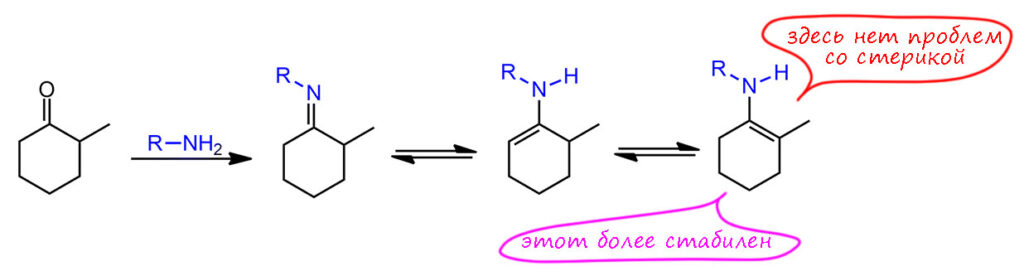
Идея полностью оправдалась. При реакции с типичными акцепторами Михаэля типа акрилата, акрилонитрила, винилсульфона получался только продукт по более замещённому положению енолизации. Реакции были довольно медленны, что неудивительно, ведь в реакцию вступает малая равновесная концентрация енамина, но при стоянии несколько дней всё получалось нормально. Хикмотт пробовал всякий очевидный катализ, и вполне успешно, но без блеска, и в дальнейшем эту реакцию обычно продолжали делать, просто оставив на несколько дней раствор имина и акцептора в ацетонитриле.
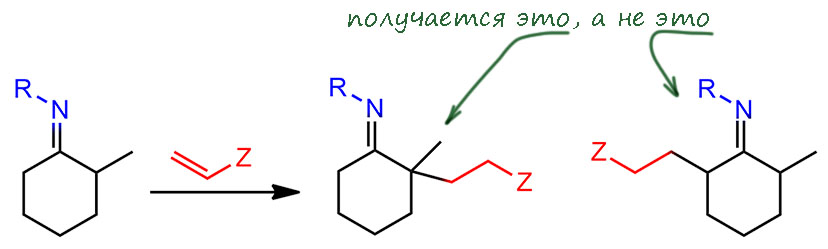
Этот подход позволяет сделать очень привлекательную вещь – селективно получать продукты реакий с акцепторами Михаэля и по более, и по мене замещённому атому. Обычный енамин дает менее замещенный, а таутомеризуемый имин – более замещенный продукт. Органики очень ценят такие методы, которые позволяют целенаправленно направлять реакцию в одно из возможных направлений. Кетоны получаются после гидролиза первоначально образующихся иминиевых солей и и иминов.
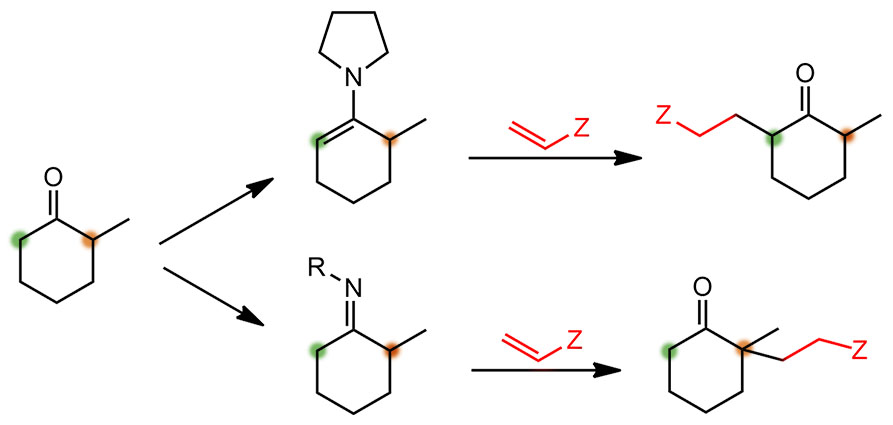
Пфау и его французские коллеги практически одновременно сделали еще одно интересное дополнение (Pfau M., Revial G., Guingant A., d’Angelo J. J. Am. Chem. Soc. 1985, 107, 273–274). Тоже взяв 2-метилциклогексанон, они использовали оптически чистый первичный амин, весьма легкодоступный (S)-β-фенилэтиламин. Получив имин (точно так же, как енамины – азеотропной отгонкой воды, катализ TsOH), оставили на несколько дней реагировать с метилвинилкетоном. Продукт мягко прогидролизовали разбавленной уксусной кислотой, и получили продукт по более замещённому положению, причем оптически активный амин был регенерирован – хоть он и недорог, но и недёшев, и возможность использовать повторно очень привлекательна. Продукт оказался оптически активен – стереогенный центр в енамине привел к тому, что реакция спереди (относительно плоского фрагмента двойной связи) оказалась более вероятна, чем реакция сзади (если бы хирального амина не было, оба направления были бы равновероятны и получился бы рацемат). В первом приближении это просто следствие того, что сзади оказалась более объёмистая фенильная группа. Такое явление называют переносом хиральности (или асимметрической индукцией), и в данном случае она оказалась довольно эффективна. Продукт получился в виде преобладающего (R)-энантиомера с энантиомерным избытком в 91%, а это соответствует смеси энантиомеров R:S = 95.5% : 4.5%. Надо только понимать, что сначала получается не энантиомер дикетона, а диастереомер имина (в продукте до гидролиза два стереогенных центра), поэтому это не совсем энантиоселективная реакция, а типично диастереоселективная, а этим нас не удивишь. Подробнее не будем, я уже в разных местах и этого и второго сайта много описывал разницу между энантиоселективностью и диастереоселективностью. Но отмечу еще одно выдающееся достоинство этой системы в сравнении с реакцией обычного енамина – здесь отлично можно применять непредельный кетон и остановиться на продукте замещения, в то время как обычные енамины в такой реакции обычно пролетают дальше и дают в значительной степени продукт циклизации, даже тогда когда он даром никому не нужен. Почему так происходит можете попробовать понять сами, если аккуратно разрисуете оба механизма, не пропуская никаких стадий.
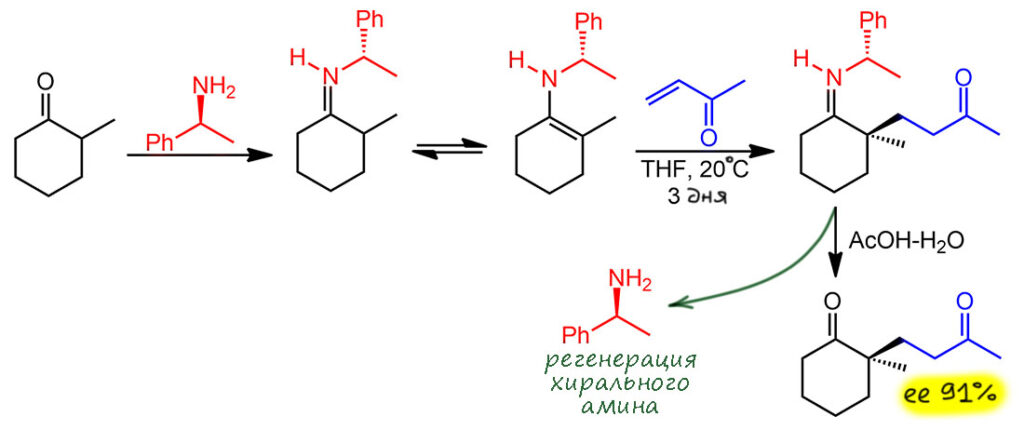
В общем, из химии иминов енолизуемых с первичными аминами получилась очень симпатичная химия и несколько полезных методов, дополняющих химию обычных енаминов. Особенно хорошо эта химия заработала в новом веке, когда ее запустили в режиме модного органокатализа. Но это мы пока оставим.