Обновления
Опубликована 12.01.2024
Окисление спиртов
Окисление спиртов в карбонильные соединения, альдегиды или кетоны, это одна из самых востребованных реакций окисления в органическом синтезе. Методов синтеза спиртов великое множество, а для развития синтеза продожение через химию карбонильных соединений всегда приветствуется. Иногда даже делают так – собирают спирт, например, реакцией с гриньярами или другой металлоорганикой, затем окисляют в карбонил, пользуются карбонильной химией для достройки скелета и других украшений, и сделав максимум возможного, вновь восстанавливают до спирта, ведь возможно он и нужен в конечном продукте. Совсем современная химия стала косо смотреть на такие упражнения, но иногда по другому синтез и не построишь, поэтому хоть лопните от злости, но синтетики делали, делают, и будут делать синтезы, перемещаясь от химии спиртов в химию альдегидов и кетонов и обратно.
Поэтому и методов окисления спиртов тоже множество, даже если иметь в виду не все что опубликовано (это сотни), а только то, что реально используется в синтезах (это десяток с хвостом). Зачем так много? Синтез сложен, многообразие ситуаций, в которых может оказаться синтетик далеко по цепи синтеза, невообразимо – сочетание заместителей и функциональных групп, и других элементов структуры, все время изменяющаяся стерическая обстановка вокруг реакционных центров – все это делалет необходимым доступность разнообразных методов, каждый из которых может или сыграть или провалиться по причине несовместимости с тем, что уже есть в молекуле. Уместный вопрос – а разве синтетик, прежде чем пускается в длинный путь, не проигрывает все повороты синтеза? Конечно синтез планируется с учетом имеющейся информации, но учесть всё невозможно, и к тому же почти всегда с самых ранних стадий начинаются отклонения от первоначального плана. Поэтому так важно иметь разнообразие инструментов.
На этой странице я довольно подробно разберу основные способы окисления спиртов в карбонильные соединения, делая особенный акцент на разборе структуры реагентов и механизмов окисления, потому что именно это и позволяет понимать, почему одни реагенты более селективны, а другие менее, но при этом их можно применять, точно понимая, что нужно сделать, чтобы не получить переокисления или ещё чего-нибудь. Всё кроется в механизмах. Вообще в органике так повелось, что окисление и восстановление рассматривают довольно поверхностно, многие вещи вообще проскакивают, поэтому всегда так сложно понять, что происходит в реакциях, приводящих к переходу от одной функциональной группы к другой. С окислением при этом сложностей даже больше, чем с восстановлением. В реакциях востановления доминируют два механизма: перенос электрона с последующим превращением анионрадикальных интермедиатов, и перенос гидрида от доноров гидрида. Там, конечно, немало вариантов в продолжениях, но в целом всё одно и то же. А вот в окислении механизмов гораздо больше, и немаленькое количество реакций, котрые, на первый взгляд, не выглядят как окисления (напрмиер, всякие присоединения к кратным связям) сопровождаются увеличением степени окисления одного или нескольких атомов, а следовательно являются окислениями. Но, тем не менее, и в окислении есть ограниченное количество шаблонных механизмов, варьирование которых и дает разнообразие. Вот мы на этой странице и убедимся, что подавляющее большинство реакции вообще идут по одному механизму, хотя кажутся такими разными.
Предупреждение! Эта страница сильно выходит за рамки программы 3 курса и предназначена просто для тех, кому любопытно узнать больше. Пожалуйста, при решении задач на 3 курсе не используйте для окисления спиртов в карбонильные соединения методы, которых нет в программе. Хромовых реагентов (PCC или комплекса хромового ангидрида с пиридином), и диоксида марганца вполне достаточно для всех задач химии 3-го курса.
Краткое содержание
Превращение спиртов в карбонильные соединения это и почему самое простое не всегда самое лучшее. Любое обсуждение в органике лучше начинать с механизмов, и вот мы так и поступим – подумаем, могут быть в окислении спиртов в карбонильные соединения, и наметим себе самый перспективный.
Сначала займёмся реагентами на основе шестивалентного хрома, а это основные окислители спиртов в органике, и на 3-м курсе ими вообще можно и нужно обойтись. Самые про химию таких помогут нам двигаться дальше. А ещё лучше мы будем понимать реакции , если вспомним, что это переходный металл, все соединения хрома это комплексы, свойства которых зависят от лигандов. И немного отвлечёмся от спиртов и вспомним один из самых загадочных реагентов в органике – : мы теперь хорошо вооружены, чтобы разобраться в этой странной истории.
А теперь подробнее разберёмся в хромовых реагентах окисления спиртов, почему их так много и чем они отличаются. Начнём с самого старого, , но из-за простоты и удобства до сих пор вполне востребованного в простых случаях. Для сложного синтеза, где прежде всего нужна селективность один за другим делали более сложные реагенты. в методах Сарета и Коллинза работает достаточно аккуратно, но страдает от необходимости брать большие избытки. А самый удачный реагент, , получился, что неудивительно, у Кори, причём к делу остроумно пристроили и главный недостаток этого реагента – высокую кислотность. Там же помянем и другой полезный, но менее популярный реагент, дихромат пиридиния.
Хватит хрома, но возьмём ещё следующий элемент в ряду – марганец. Простейшее соединение, это почти незаменимый реагент для окисления спиртов бензильного, аллильного и пропаргильного типов, особенно если в молекуле есть другие спиртовые группы и их надо оставить в покое. Увы, реагент только с виду простой – активную форму двуокиси марганца нужно специально делать, и это даётся не всем.
Довольно металлов. Займёмся очень популярными методами окисления, использующими , но чтобы заставить это вещество кого-то окислять, его надо активировать и в этом и состоят секреты многочисленных методов этого типа, обычно связываемых с неким Сверном.
И наконец приступим к настоящему чемпиону современного синтеза – окислению . Придётся разобраться, как устроено это соединение и почему оно так странно называется, и если мы разберёмся в том, что ключом к пониманию структуры является гипервалентность, то и станет нам совершенно очевиден.
Ну и наконец, в 21 веке всё идёт лесом, если ваши методы недостаточно зелены, а по критериям зеленой химии все хромовое, марганцевое идет на свалку сразу, а за ними идут и Сверн с токсичными и агрессивными активаторами, и Десс-Мартин с опасным и дорогим реагентом. Но есть спасение – старый советский реагент TEMPO в изящной итальянской оправе – и эти методы считаются : за ними будущее, а всё остальное надо оставить в 20-м веке.
Заодно с некоторым удивлением поймём, что методов мы разобрали немало, почти ничего существенного не забыли, а механизм практически везде один, совсем один – E2-элиминирование, и вся разница только в уходящих группах.
Краткое сравнение методов окисления спиртов
-
методреагентспиртыдостоинстванедостатки
-
реагент Джонсараствор CrO3 в серной кислоте, реакцию ведут в ацетоневторичныеочень просто и эффективно, дёшево, минимальный избытокнизкая селективность, первичные спирты переокисляет
-
реагент Саретакомплекс CrO3 с пиридином в пиридиневторичныеболее селективно, нет сильно-кислой средыбольшой избыток реагента, мерзкий пиридин, первичные спирты переокисляет
-
реагент Коллинзакомплекс CrO3 с пиридином в дихлорметаневторичные, первичныеочень быстрая реакция, селективно, комплекс можно приготовить на местеочень большой (6-тикратный) избыток реагента
-
реагент Кори-Саггса, PCC, ПХХхлорохромат пиридина, реакцию ведут в дихлорметаневторичные, первичныебыстро, селективно, удобно, реагент можно купить и хранить, самый маленький избыток из всех хромовыхвсё равно избыток, высокая кислотность приводит к осложнениям (перегруппировкам и т.п.)
-
реагент Кори-Шмидта, PDC, ПБХбихрохромат или дихромат пиридиния, реакцию ведут в дихлорметаневторичные, первичныесамый дешевый и простой, негигроскопичный, обычно маленький избыток, низкая кислотность, неплохой окислитель для крупных синтезоввсё равно избыток, медленные реакции, переокисление первичных спиртов в присутствии воды
-
диоксид марганцаактивный диоксид марганцабензильные, аллильные, пропаргильные - первичные и вторичныедешево и надёжно, другие спиртовые группы не затрагиваютсячудовищный избыток, для больших загрузок неудобно выделять продукты, реагент нужно специально делать и может не получиться
-
окисление по Мофату, Сверну и др.ДМСО плюс электрофильные активаторы (оксалилхлорид, трифторукусусный ангидрид, DCC, и т.п.)первичные, вторичныеселективно, надёжно, быстро, никогда не бывает переокисления, большой выбор методик и активаторовв самом популярном варианте - окислению по Сверну - используются крайне агрессивные активаторы, и реакцию необходимо вести при низких температурах
-
окисление по Дессу-Мартинупериодинан Десса-Мартина, DMPпервичные, вторичныенепревзойдён по универсальности, селективности, мягкости. Незаменим для сложных многостадийных синтезов полифункциональных молекул на микро- и полумикроуровнедорого, большая молярная масса, не так легко получить хорошего качества, опасен при хранении и транспортировке. Почти непригоден для синтезов с большими загрузками
-
окисление по Анелли и по Пьянкателли-МаргаритеTEMPO (кат.), гипохлорит натрия (А.), PhI(OAc)2 (П.-М.)первичные, вторичныефаворит современного "зеленого" синтеза, просто, эффективно, в варианте Анелли предельно дешево, и довольно универсально (особенно вариант П.-М.)как всё зелёное довольно сырой и не очень надёжный метод, лучше не использовать как первый метод, а только на стадии масштабирования и "озеленения" синтеза
Дегидрирование или окисление?
Само превращение спирта в карбонильное соединение это реакция, обратная гидрированию карбонильного соединения. В этом месте сразу возникает важная особенность – эта реакция в направлении от спирта к карбонилу и молекулярному водороду (диводороду – так это в последние десятилетия стали называть, попробуем и мы привыкнуть) эндотермична, так как карбонил лежит выше по энергии чем спирт (например, для самой простой пары метанол-формальдегид эндотермичность дегидрирования составляет 90 кДж/моль). Последнее это очередное проявление общей закономерности – кратные связи дестабилизируют молекулы, хотя это тем менее выражено, чем более электроотрицательны соединяемые атомы. Впридачу ещё разрываются две вполне приличные связи C-H и O-H, что тоже затратно, но с другой стороны получается неплохая компенсация в вида одной из самых прочных связей в молекуле водорода (диводорода, привыкаем). Довольно мерзкий фокус общепринятой конвенции отсчёта энергетических термодинамических функций (энергии, энтальпии, свободных энергий) состоит в том, что их всегда отсчитывают от нулевых уровней, за которые принимают величины для обычных состояний простых веществ (элементов), соответственно водород представлен диводородом при стандартных условиях, и нам кажется, что это просто ноль и не о чем говорить. Но здесь такой же фокус, как тогда, когда в каком-то магазине вам затирают про бесплатную доставку – будьте уверены, что вы за доставку заплатили и скорее всего втридорога, только не прямо, а через вклад в цене купленных товаров. Так же и с циклами Гесса – если мы их составляем по стандартным энтальпиям образования, мы получаем правильную величину, но не понимаем, что в ней уже учтена энергетика простых веществ (их приняли за ноль, но это формальность выбора точки отсчета в равновесиях), а если бы мы могли тот же цикл расписать например, учитывая энергетику образования и разрыва связей, мы получили бы ту же величину и при этом более осмысленныю картину изменения энергии от исходных к продуктам. Пользуясь свойством равновесий – как бы вы ни посчитали разность энергий (энтальпий) между двумя участниками равновесия (какой бы цикл Гесса вы ни построили, а вариантов всегда много), если это сделано корректно, то величина будет одна и та же – мы имеем замечательную возможность прикинуть эти величины по стандартным энергиям (энтальпиям) образования, а смысл в эту разность привнести через мысленный расклад цикла по другим величинам (тем же энергиям связей). Такие несложные оценки очень много дают для понимания того, как и почему идут (или не идут) реакции. 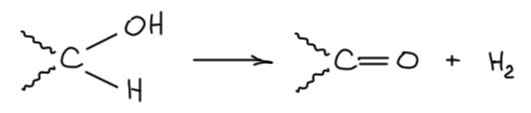
Дегидрирование спиртов до альдегида и кетона поэтому очень проблемная реакция. Мы хорошо знаем самый простой пример этой реакции – дегидрирование метанола до формальдегида, и это промышленный процесс, основной источник формальдегида, что очень понятно, так как метанол в промышленности одно из самых крупнотоннажных веществ, получаемое ежегодно в мире миллионами тонн по гетерогенно-катализируемой реакции оксида углерода с водородом. И всё что можно легко получить из метанола поэтому точно будут получать из метанола. Реакция дегидрирования гетерогенная эндотермическая, поэтом требующая постоянных затрат энергии на нагревание – процесс ведут пропуская пары метанола через нагретую до 650° серебряную сетку для того чтобы уменьшить время контакта, иначе будет разлагаться уже и формальдегид. В этом проблема дегидрирования спиртов – из-за эндотермичности процесс требует высокой температуры, чтобы скорость реакции была разумно высокой, но высокая температура плохо совместима с любой органикой, даже с самой простой, потомоу что всё органическое на свете при высокой температуре превращается в ацетилен и дальше в сажу. Этот процес тоже не исключение, и серебряные сетки тоже быстро покрываются сажей, процесс нужно прерывать, сажу выжигать, сетку реактивировать и так далее. И еще одна проблема, довольно странная – куда девать водород. Казалось бы это очень ценная вещь, все только и говорят про будущее ээнергетики с водородом в качестве основного топлива. Но сейчас-то с ним что делать, когда еще до будущего как до соседней галактики? Если на том же предприятии есть другой процесс, где нужен водород, то и прекрасно, можно туда его. А иначе плохо – водород в карман не положишь, в общем обычно это обуза, а не второй ценный продукт процесса.
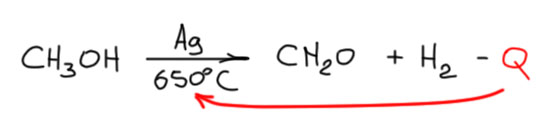
Вот по совокупности этих причин (энергозатратность, необходимость пристроить опасный водород, периодическая регенерация катализатора, снижающая производительность и т.п.) промышленность и бизнес давно пришли к вроде бы парадоксальному выводу – нужен другой процесс, более экономически выгодный. И в основном перешли (не знаю как сейчас, но в 1980-е в Москве был завод, производивший формальдегид именно дегидрированием метанола над серебряной сеткой – в реакторе было окошко, в которое была видна раскаленная сетка – завод был в Кускове прямо рядом со станцией электричек) на окисление формальдегида кислородом воздуха. Эта реакция катализируется гетерогенно разными оксидами металлов, но часто с низкой селективностью, и большая часть метанола переокисляется в оксиды углерода. Лучший катализатор нашёл в 1930-х знаменитый американский каталитик Гомер Симпсон Адкинс и это оказалась смесь оксидов железа и молибдена (у гетерогенных катализаторов обычно невозможно указать точный состав и даже степени окисления металлов, но это точно никакой не молибдат железа, как иногда пишут): реакцию окисления метанола на этом катализаторе иногда называют реакцией Адкинса-Петерсона (Adkins-Peterson reaction), немного жирно для единственной реакции с одним исходным, но такова традиция, к тому же на этой основе был разработан промышленный процесс, но не в США, а в Швеции компанией Персторп (входит в Perstorp Holdings AB) в 1959 был построен первый завод, где формальдегид стали делать этой технологией, получившей название Формокс (Formox). Катализатор ещё немного доработала, видимо, добавив ещё и ванадий, знаменитая английская компания Джонсон Мэтти (Johnson Matthey), которую знают все, кто хоть немного соприкасался с металлами платиновой группы и их производными – это такой основной источник всего связанного с благородными металлами, но компания делает еще и другие катализаторы из металлов попроще. Собственно так и практически только так – по процессу формокс от шведской компании Персторп и фирменным катализатором от JM и получают с тех пор формальдегид во всём мире – просто покупают готовую технологию и запускают её. Это удобно, потому что формальдегид нужен много где, но он не хранится и плохо транспортируется, поэтому там, где возникает потребность в этом соединении, проще поставить небольшую установку нужной производительности и гнать его прямо в следующий процесс.
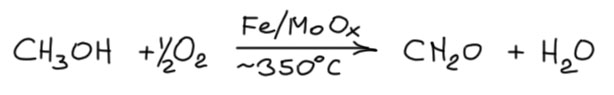
Я специально так подробно это рассказал, чтобы мы увидели, насколько важны в химии все детали. Казалось бы, реакция прямого дегидрирования даёт две ценные вещи – формальдегид и водород, а в формокс-процессе водород сгорает в бесполезную воду. Но если вы не можете использовать водород (а это требует отдельных вложений, и если потребности нет, то водород становится отдельной опасной морокой), то второй процесс выигрывает, потому что он экзотермический (вода сильно экзотермична и перевешивает всё), идёт при намного более низкой температуре, поддерживать которую помогает и использование тепла реакции, соответственно, энергоёмкость намного ниже; и еще прекрасная отлаженность технологии и очень устойчивый и производительный фирменный катализатор.
Для других спиртов прямое дегидрирование тем более не используют, потому что даже для превращения этанола в ацетальдегид нет хорошей технологии, и тем более для спиртов ещё сложнее. Исследования в этой области ведутся, и интерес есть и большой, потому что это было бы интересно с точки зрения современных требований к устойчивому развитию и безопасным технологиям. Но для этого нужно найти эффективные и селективные катализаторы, а это оказалось не так просто несмотря на огромные усилия. Для эндотермических реакций вообще найти хорошее решение всегда сложнее, чем для экзотермических, хотя эндотермичность сама по себе не приговор, а просто осложняющий фактор – вам придется доставить больше энергии в реагирующую систему, и соответственно, есть вероятность, что она пойдёт не туда, и будет истрачена на конкурирующий процесс, угробив селективность. Такая растрата по-химически, только уголовку завести не получится, потому что молекулы неподсудны – они просто используют возможности для того, чтобы вступить в реакцию, и это уже ваша проблема – постараться предсказать все альтернативные пути и найти методы избежать осложнений.
Поэтому пока приходится смириться и разбирать не способы дегидрирования, а способы окисления спиртов в карбонильные соединения.
Возможные механизмы окисления спиртов в альдегиды и кетоны
Попробуем просто представить, что нужно сделать, чтобы спирт превратить в карбонильное соединение. Это обычная предварительная работа, которую всегда делают, когда хотят представить возможные альтернативы – расписывают даже не механизмы в полном смысле слова, а общие черты возможных механизмов. И дальше уже пытаются известные реакции проанализирвоать с точки зрения этих общих схем, и найти уже более подробные разновидности механизмов. Так делают всегда, с этого начинали и основоположники механизмов Ингольд и Хьюз, когда намечали возможные механизмы нуклеофильного замещения и элиминирования.
Задача простая – убрать два атома водорода, причём даже интуитивно понятно, что тот, который на углероде, убрать сложнее, и механизмы должны различаться именно по тому, как убирается этот водород (обратите внимание что я не называю водород протоном, потому что в общем случае атом водорода вовсе не обязательно ведет себя как протон, то есть положительно заряженный ион или движение в сторону такого иона). И у нас получится три общих пути:
- отщепляем протон основанием и получаем карбанион. Это сразу видно, что путь плохой – такой карбанион будет сильно дестабилизирован соседством с атомом с неподеленной парой – альфа-эффектом, это всегда плохо, да и дальше непонятно, что с этим карбанионом делать, потму что надо теперь убирать гидрид, а какой к чёрту гидрид на атоме кислорода. Нет, это не кажется разумным путём. Но отложим пока, у этого пути есть модификация, которая его спасёт.
- отщепляем гидрид акцептором гидрида, таких немало. Вот тут всё видится просто блестяще – и образующийся карбокатион хорош, мезомерно стабилизирован, и дальше надо просто убрать протон с кислорода – да он сам улетит. Отмечаем путь как перспективный.
- отщепляем атом водорода каким-нибудь радикалом. Тоже выглядит неплохо – радикал сразу же потерят второй атом водорода – таких путей полно в свободнорадикальной химии. Отмечаем.
- ну и хоть и не нарисовано, но всегда имеем в виду согласованный путь, когда оба водорода улетят куда-нибудь одновременно, пока непонятно как, но это возможно, а примеры стоит искать в химии переходных металлов. Пока отложим, но забывать не будем.
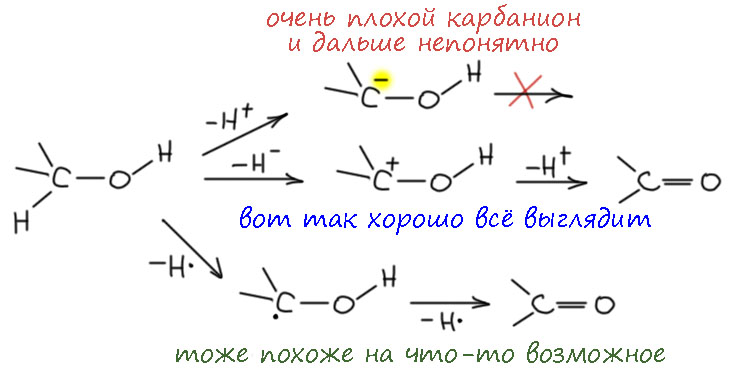
Вернёмся к первому пути, который мы забраковали по двум причинам. Там явно видится обычная проблема во многих механизмах – нет хорошей уходящей группы. Водород в виде гидрида – одна из худших уходящих групп на свете, и когда такие пути появляются на горизонте, они почти никогда не работают, потому что гидрид почти никогда не может уйти (всё же иногда может, и я когда-нибудь допилю страничку про гидрид, где расскажу про такие фокусы тоже). А что если… – если сначала превратить спирт в какое-то производное, которое создаст уходящую группу. Какую? Пока не знаем, но общее требование – она должна забирать пару электронов, то есть быть нуклеофугом. О, это мы знаем, полно таких, мы сто раз уже применяли. Стоп, не всё так просто – обычно мы создавали уходящую группу, чтобы она уносила из бывшего спирта и атом кислорода тоже, а здесь нам надо кислород оставить на месте, а забрать пару – формально у кислорода между прочим, что выглядит авантюрой, но и здесь не будем торопиться, кислород ведь не пострадает, потому что с другой стороны получит свою пару назад от бывшей связи C-H. Получится такой согласованный механизм, в котором отщепление протона и уход уходящей группы происходят более-менее одновременно в едином переходном состоянии.
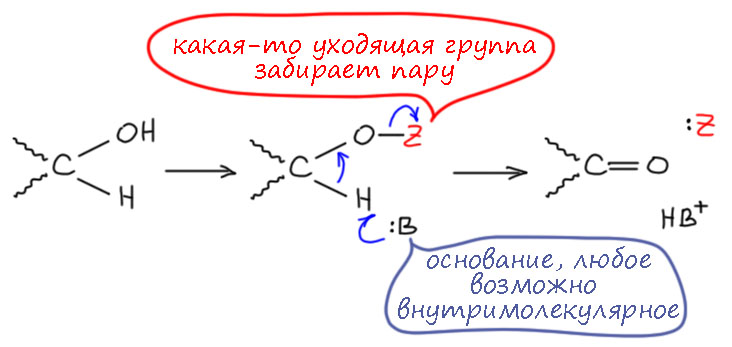
Разрисуем последнюю схему немного в другом ракурсе. Протон отщепляет основание, одновременно (более-менее, мы знаем этот фокус – в химии одновременно не значит синхронно, возможно легкое опережение или запаздывание) уходит уходящая группа и образуется π-связь. О, это я где-то видел… – ага, да это же как две капли соляной кислоты похоже на механизм E2-элиминирования. И мы недавно даже разбирались в том, что это механизм отлично приспособлен для образования не только C=C связей, но и связей углерода с килородом и азотом, и даже и других двойных связей. И дали себе задание поискать примеры. И вот – и специально не искали, так само в руки плывёт.
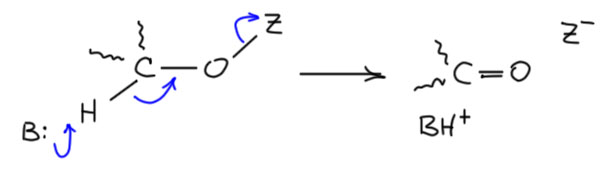
Да, но это пока не пример, а просто гипотеза, что такое может быть. Хорошо, ну вот нам примеры. Их чуть меньше чем большая куча. И среди них те, что мы особенно любим. И вообще, скоро мы убедимся что почти все методы окисления первичных спиртов в альдегиды и вторичных в кетоны соответствуют этой схеме, и скорее придётся мучительно искать примеры других механизмов.
Механизм с гидридным переносом мы уже знаем. Тут важно осознать, что превращение спиртов к карбонильные соединения имеет обратную реакцию – восстановление карбонильных соединений. И вот оно чрезвычайно часто идёт именно как присоединение гидрида из донора гидрида к карбонильному углероду. Если подумать, то это нечто обратное реакции отщепления гидрида, ведь точно так же как перемещение протона и перемещение атома водорода и перемещение гидрида всегда происходит как обмен между донором и акцептором.
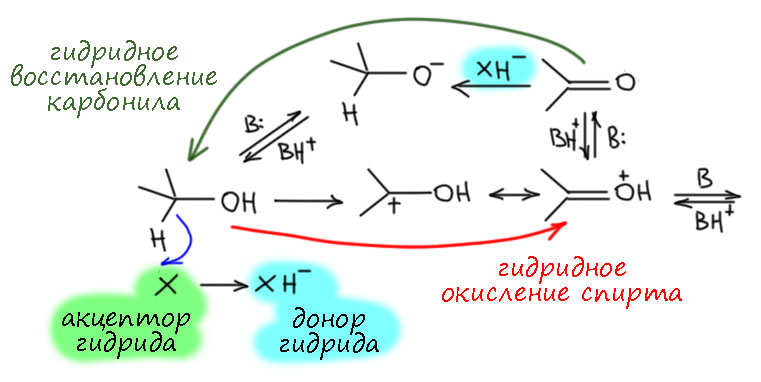
Есть целый класс соединений, называемых донорами гидрида (я уже давно ваяю страничку про это, но никак не доваяю, много там всего интересного, закопался). Чаще всего доноры гидрида необратимо сдают гидрид на карбонил – это, например, хорошо нам известные алюмогидрид лития и т.п. Это нам сейчас, когда мы хотим в обратную сторону, неинтересно. Но есть несколько очень интересных реакций, где это прямо обратимо – реакции Каниццаро, Эванса-Тищенко, Меервейна-Понндорфа-Оппенауэра-Верлея. Они разобраны на странице про химию карбонильной группы.
Общие вещи про шестивалентный хром
Самые популярные окислители спиртов в альдегиды, кетоны, кислоты это производные шестивалентного хрома. У них большая история, даже сейчас не смогу написать, когда и кто это применил первым. Окислительные свойства хромовой кислоты, хромового ангидрида известны c ранних времён химии. Собственно и сам элемент хром был открыт в самом конце 18-го века Луи-Николя Вокленом востсановлением именно хромового ангидрида углём, то есть обычной углеродной металлургией, а хромовый ангидрид он же и открыл, и способом, который кажется немного сомнительным. Интересно, что в Природе немало хромовых минералов, содержащих именно шестивалентный хром – хроматов и бихроматов. Воклен разбирался с образцом крокоита, который тогда называли сибирской красной, потому что минерал был открыт на Урале в окрестностях Екатеринбурга при Екатерине Великой, и использовался в качестве минерального пигмента. Химически это хромат свинца, и Воклен разложил его соляной кислотой, явно разбавленной, иначе получил бы он хлористый хромил, но он получил раствор хромовой кислоты, который упарил, и якобы получил хромовый ангидрид. Надо признать, что если это так, то работал этот старинный француз просто удивительно чисто, и умудрился хромовый ангидрид в этом процессе ничем не восстановить. Среди хромовых минералов, что совсем удивительно, встерчаютсяс и чистый бихромат калия (лопецит) и чистый хромат калия (тарапакаит), – их находят в месторождениях чилийской селитры. Про чилийскую селитру ходят мифы, что это как-то связано со знаменитым гуано, но это совершенно неверно, не связано никак, и природная сеитра образуется в тех местах, где из-за частых гроз дождевая вода содержит окислы азота, и такой дождь выпадает на места, где из осадочных пород выщелачивается карбонат натрия. Дальше понятно, и ясно, что это должны быть какие-то страшно засушливые места, где влаги ровно столько, чтобы принести немного азотной кислоты, но не столько, чтобы смыть всё это. И вот в таких местах получается сильно окислительный раствор, и если есть еще какие-то минералы, которые фонят хромом, то там и идёт окисление и диффузия и кристаллизация хромата и бихромата, причем именно калия, а не натрия – но это связано с условиями кристаллизации.
В общем, химики 19-го века имели и хромовую кислоту, и ее соли, и хромовый ангидрид, и органики рано стали ею окислять, и считали эти соединения сильными окислителями, которыми можно окислить спирт в кислоту, расщепить двойную связь в две кислоты, расщепить кетон и так далее. В те времена окислители делили на сильные и слабые; хромовую кислоту считали сильным, а сильным окислителям положено было сильно окислять. О селективности задумались много-много позже, и только тогда стали придумывать специальне реагенты на основе соединений хрома(6+).
Но мы займемся хромовыми окислителями поподробнее. И разберемся, как они окисляют спирты, какой там механизм. Если тот, в котором водород отщепляют в виде протона, то нам уже сказано искать уходящую группу. Поищем.
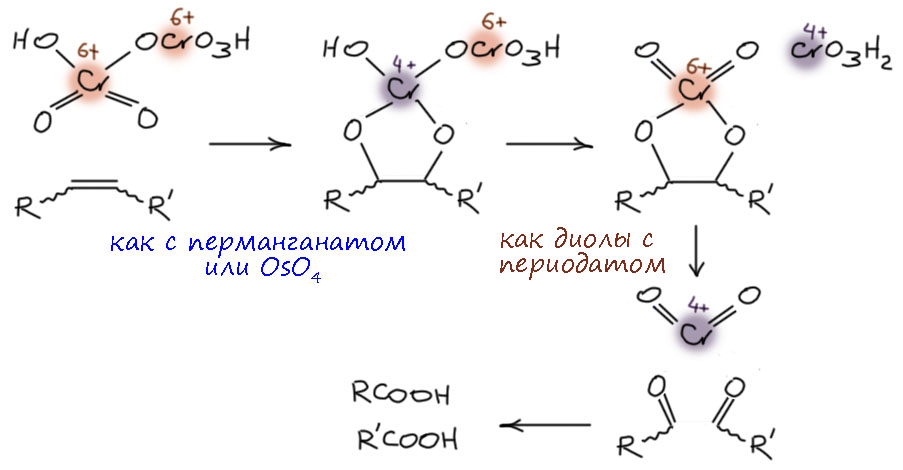
А это тут при чём? О какой уходящей группе может идти речь в реакциях с хромовым ангидридом? О самой настоящей, и именно в этом и состоит собственно окисление – уходящая группа уносит электроны, пару, два электрона, минус два электрона – двухэлектронное окисление.
Возьмём для начала сам хромовый ангидрид и посмотрим на него как на любой ангидрид. Ангидриды реагируют со спиртами с образованием сложных эфиров. Запишем:
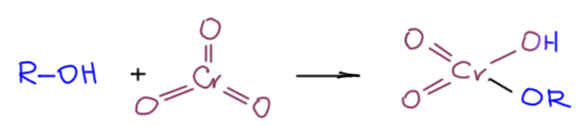
Ну и теперь расположим так, чтобы видеть атом водорода на углероде и примерим схему E2-элиминирования.
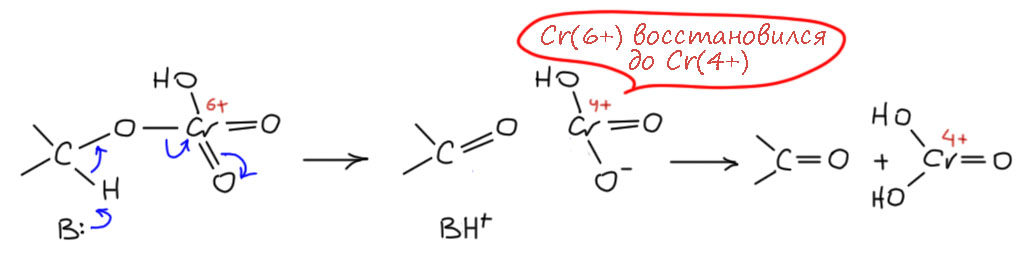
Видим, что всё рисуется довольно складно, и обращаем внимание на одно ранее не привлекавшее нашего внимания обстоятельство – уход уходящей группы с парой электронов равнозначен восстановлению. И если в начале у нас эфир хромовой кислоты с Cr(6+), то образуется нечто с степенью окисления 4+, что легко посчитать по обычным правилам. А что это? Оставим пока, разберёмся чуть позже. Сейчас нас больше заинтересует вопрос, а что там за основание, ведь вроде иногда такие реакции даже в кислой среде идут. Здесь мы в очередной раз сталкиваемся с особенностями согласованных механизмов, в которых не требуется высокая основность там, где требуется какая-то основность, потому что в таких механизмах депротонирование не происходит как отдельная обратимая стадия, а включено в общий согласованный путь перераспределения электронной плотности, и в таких случаях протон может отщепляться намного более слабыми основаниями и даже просто компонентами реакционной смеси, типа той же воды, других растворителей, иногда даже таких, которые мы рассматриваем как основания, типа пиридина.
Ещё одна проблема. Хромовые реагенты часто известны тем, что альдегиды частично переокисляются в карбоновые кислоты. Как это происходит? Скорее всего по очень похожему механизму через производные альдегидов, похожие на полуацетали, образующиеся обратимо в смеси. Проще всего это написать с участием хромовой кислоты.
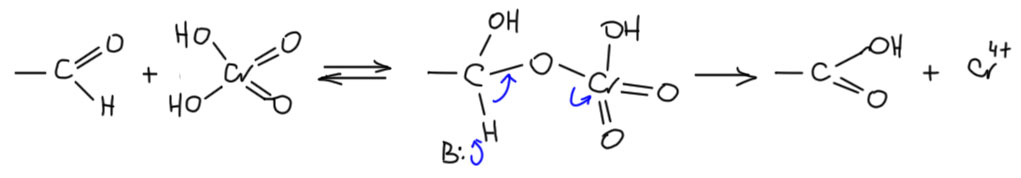
В этом месте мы видим, что для переокисления нужно обязательно иметь явную или неявную воду (например, в виде хромовой кислоты), а если мы придумаем реагент, в котором нет такой воды, скорее всего мы сможем достичь селективности окисления. И это весьма важно понять – более глубокое окисление присходит (может происходить) не потому что окислитель стал сильнее – строго говоря мы вообще не можем просто судить о том, какой окислитель сильнее, это отдельная проблема, и в общем виде практически нерешаемая. А потому, что есть или нет возможности для реализации механизма окисления.
Почему так много разных окислителей на основе Cr(6+)? Это связано с тем, что хотя механизм окисления общий и всё вроде нормально, но в реальной химии всегда возникают чисто практические проблемы – как совместить реагенты в одной колбе. Использовать чистый хромовый ангидрид нельзя. Можете заглянуть в любой хороший мануал по неорганике, и в статье про хромовый ангидрид почти неизменно увидите предостережение – воспламеняется при контакте с органическими соединениями. Иными словами, реакционная способность чистого хромового ангидрида такова, что окисление идёт быстро и сильно экзотеримично. Экзотермичность обусловлена двумя вещами – во первых два водорода из спирта идут в воду, а вода очень экзотермична, в любой реакции образование воды тянет реакцию в эту сторону. Во-вторых, само восстановление Cr(6+) также сильно экзотермично, высокая степень окисления у металла первого ряда переходных металлов очень неустойчива и переходит в нижние также с хорошей экзотермичностью. И как им тогда окислять реально, мы же не хотим бегать вокруг колбы с огнетушителем?
Поэтому в ранней химии окислительные свойства шестивалентного хрома были признаны, но применялось это довольно редко, пока новые поколения химиков не стали находить удобные способы переводить хромовый ангидрид в более мягкие и менее опасные реагенты. В ранней химии, то есть до Второй мировой войны, иногда окисляли хромовой кислотой, и это всегда были жесткие окисления, не спиртов в карбонильные соединения, а если уж спиртов, то в карбоновые кислоты, кетоны с расщеплением, олефинов в две кислоты, или алкилароматики в бензойные кислоты. Шестивалентный хром считался очень сильным окислителем, но реакции вели в сильнокислой среде. Учитывалась еще и взаимная растворимость – органика предпочитает реакции между растворами веществ, когда можно хорошо перемешивать, медленно прибавлять, охлаждать и тк далее. Есть и еще одна немного неожиданная проблема – во что превращается хром.
По механизму реакции хром превращается в некую кислоту 4-хвалентного хрома, но есть проблема – это валентное состояние хрома почти не имеет устойчивых соединений, если не лезть далеко в координационную химию и в сложные современные лиганды. Оксид хрома(4+) есть, и люди, много пожившие и видевшие Брежнева (тогда казалось, что ничего более смешного и убогого быть не может, увы, это оказалась ошибкой, может), помнят еще и эпоху кассетных магнитофонов, к которым полагалось правдами и неправдами добывать фирменные кассеты – лучшим магнитным покрытием для плёнки считался буро-рыжий диоксид хрома, и кассеты так и маркировались как хромдиоксидные. Но это кристаллический оксид, а в кристаллической решётке иногда стабилизируются необычные степени окисления металлов. И это тот случай, когда оксид есть, а гидроксида соответствующего нет. При попытке его получить, он окисляет сам себя, образуя Cr(3+) и Cr(5+), но и тут проблема – и пятивалентный хром неустойчив, и тоже окисляет себя сам – в общем часть хрома возвращается в 6+, а часть уходит в стабильную степень окисления 3+. Так можно весь хром заставить участвовать в окислении и стехиометрия будет 3 спирта на 2 хрома(6+)
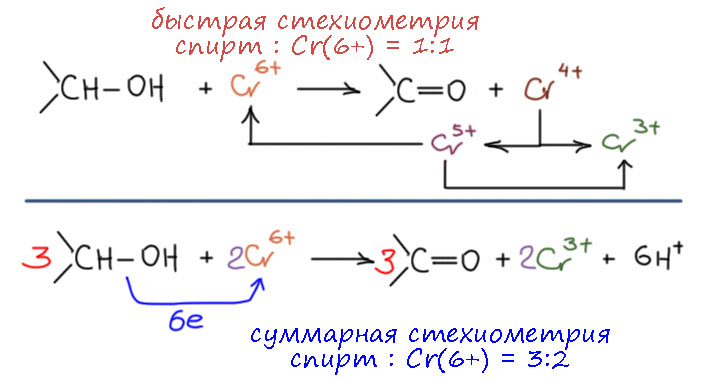
Но очень часто и со многими реагентами и реакциями, окисление сначала идёт быстро в стехиометрии моль на моль, а вся эта бадяга с диспропорционированием идут на этом фоне медленнее, и тогда предпочитают именно и брать соотношение 1:1, и через некоторое время просто гасить реакционную смесь или легкоокисляемым спиртов типа изопропанола, или просто водой, чтобы быстро высадить органику и экстрагировать ее, а что там будет дальше делать хром – его проблемы (в канистре для сливов).
А известно ли что-нибудь более конкретное про эти промежуточные соединения хрома 4+ и 5+? Насколько я знаю нет. Вообще в этом месте очень хочется попенять коллегам-неорганикам – что-то у вас белых пятен в вверенной вашим заботам химии до сих пор слишком много – вы там вообще работаете или только красивые речи про новые материалы толкаете? Но, отвечу за коллег, – вам органикам здорово повезло, у вас есть ЯМР как общий метод установления структуры, любую структуру, даже самую сложную можно быстро раздраконить даже имея пару миллиграмм вещества разными протоколами ЯМР. Да и вещества почти все стабильные, можно не торопиться. В неорганике нет такого общего метода установления структуры, к каждому элементу приходится подъезжать на особой телеге, да в придачу куча лабильных веществ, пока вы к ним пристраиваетесь, они уже во что-то другой превратились. И в хорошей современной неорганике без рентгеноструктурного анализа не проехать – а это на порядки более трудоёмкий и дорогой метод чем ЯМР. Так что не будем противными органическими снобами и войдём в положение других элементов, там всё очень сложно.
Ещё о структре соединений хрома(VI)
Это всегда вопрос к механизмам реакций и к свойствам окислителей, то есть мы должны очень много знать в том числе и из неорганической и координационной химии. Это сильно непростая задача, и в первую очередь стоит вспомнить, что хром – самый настоящий переходный металл, d-элемент, валентные возможности переходных металлов намного богаче чем то же самое у p-элементов. И нас не должны обманывать простые аналогии, типа хром и сера похожи. Это у нас от того, что мы привыкли к короткопериодной Таблице Менделеева, где эти элементы в одной группе, только в разных подгруппах. Как я уже тут замечал на страничке про валентность, короткая Таблица это плод химии 19-го века, она сделана по признаку валентностей, то есть рациональных соотношений элементов в соединениях, но не по электронной структуре, о которой во времена Менделеева слышали не больше, чем про новые модели айфонов.
Валентность это классный инструмент, когда вам надо просто упорядочить множество соединений элементов, но если мы хотим обсуждать реакционную способность и механизмы, нам нужно электронное строение и длиннопериодная Таблица, в которой между хромом (6-я группа) и серой (16-я группа) расстояние космическое. У хрома работают d-электроны, у серы в основном p-электроны, а в высшей степени окисления и s-электроны тоже – они глубже зарыты и до них не сразу доберёшься. Соединения хрома это координационные соединения, состоящие из металла и лигандов, а соеинения серы это обычные ковалентные соединения, а в степенях окисления 4 и 6 ещё и гипервалентные. У переходных металлов обычно есть все степени окисления от высшей до нулевой, через один; часть их может быть неустойчивыми, но это не мешает им образовываться в реакциях. У p-элементов степени окисления обычно идут через 2, а промежуточные хоть и могут иногда проявляться, но это почти всегда экзотика, и в реакциях и механизмах они участвуют гораздо реже (хотя нельзя сказать, что это совсем невозможно, но каждый раз требует очень серьезных усилий по доказательству). Поэтому сразу отметаем идею узнать что-то про хром по аналогии с известными соединениями и превращениями серы. Хромат ион вроде похож на сульфат-ион, и то и другое симметричные тетраэдры, но хромат это комплекс d-элемента с оксо-лигандами (в совсем современной номенклатуре оксидо-лигандами) и так его и надо рассматривать, а на сульфат придется смотреть совсем по другому. Сульфат корректнее рисовать с серой, с 4 ковалентными связями, чтобы соблюсти правило октета, и дополнительным гипервалентным связыванием за счёт ионности связей, но мы привыкли и ИЮПАК рекомендует рисовать с двумя двойными и двумя простыми, хотя это нарушает правило октета и требует для симметрии показать делокализацию каким-то способом. Ладно пока согласимся, это не наш предмет сейчас. Но хромат нельзя рисовать так же как сульфат ни тем, ни другим способом. Всё что остаётся, это нарисовать 4 оксолиганда на двойных координационных связях, и обозначить на уголке суммарный заряд всеё частицы 2-, а это просто сумма зарядов на атоме хрома и атомах кислорода, и как она конкретно распределена, можно узнать только из квантовохимических расчетов, причем это довольно бесполезная информация.
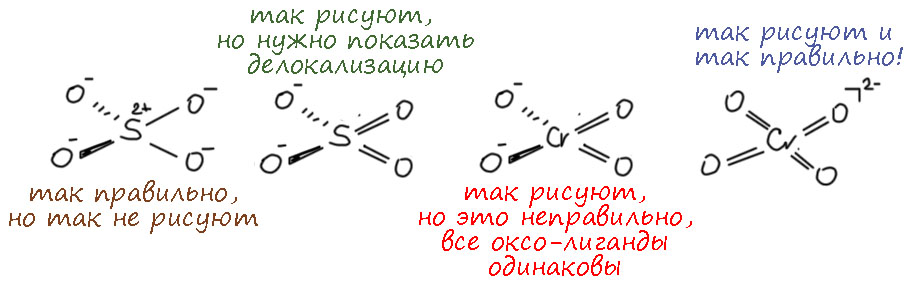
Оставим сульфат и посмотрим на соединения хрома(6+). Это максимальная степень окисления для 6-й группы, в ней у металла не остается валентных электронов, но остаются вакантные валентные орбитали. Максимальные степени окисления у переходных металлов последовательно дестабилизируются при движении к концу ряда: у первых элементов (в первом ряду это скандий и титан) это главные степени окисления, не проявляющие никаких свойств окислителя. Дальше они становятся все менее устойчивыми, но понемногу: ванадий(5+) это очень слабый окислитель, хром(6+) уже очень даже ничего окислитель, марганец(7+) это уже ого-го окислитель, а железо(8+) так до сих пор никто и не получил – никто не может окислить железо до этого состояния, а если кто-нибудь сможет, то немедленно пожалеет, потому что Fe(8+) немедленно окислит всё вокруг, включая незадачливого любителя дразнить поздний переходный металл. В второй и третий ряд соваться не будем, там высшие степени окисления становятся более устойчивыми, хотя тенденция сохраняется, только немного смягчается, и там в восьмой группе есть вполне устойчивые Ru(8+) и Os(8+), очень хорошие и полезные окислители, и даже есть в третьем ряду сведения о получении Ir(9+), ждём с нетерпением Pt(10+), хотя и немного опасаемся. Наверное всё же такую степень окисления в 10-й группе можно получить только в четвёртом ряду, но там как назло совсем короткоживущие элементы – распадется раньше, чем успеете отнять последний валентный электрон.
Теперь обратим внимание на два аниона: хромат и перманганат, а если по номенклатуре то это будут тетраоксохромат и тетраоксоманганат. Это изоэлектронные частицы, в середине металл с пустой d-оболочкой, на нём тетраэдром четыре оксо-лиганда (в координационной химии это четырёхэлектронные лиганды, висящие на двойных связях). Мы можем посчитать валентные электроны в обоих комплексах – их будет четыре по четыре итого 16.
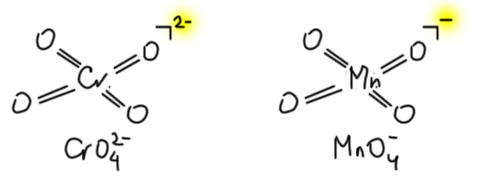
Это очень хорошо, почти полная валентная оболочка в 18 электронов – из этого следует, что в целом это устойчивые частицы (комплексы) и металл в середине практически близок к координационной насыщенности, но при этом марганцевый анион более сильный окислитель, но реагировать он будет с помощью кислородных лигандов.
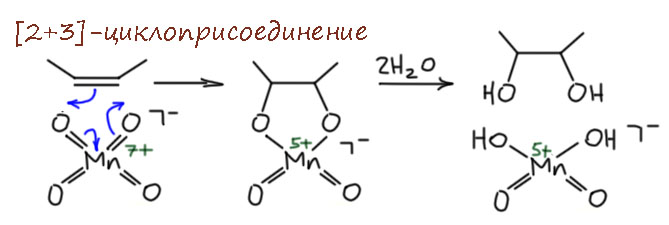
И мы это отлично знаем – ион перманганата – очень сильный окислитель, и нам лучше всего известно, как он реагирует с двойной связью – с образованием циклического комплекса, расщепление которого даёт диол. Механизм этой реакции – согласованное [2+3]-циклоприсоединение, которое идёт потому, что два оксо-лиганда превращаются в два алкокси-лиганда, и два электрона, раньше задействованные в оксо-лигандах отходят металлу, повышая его степень окисления до 5+ – мы можем по привычке как мы любим изобразить это кривыми стрелочками, хотя в химии переходных металлов у них нет такого же смысла как в обычной органике, но результат такой же, так и чёрт с ними, главное чтобы было удобно и всё сходилось. Дальше уже просто гидролиз, образование какого-то оксо-гидроксо комплекса Mn(5+), который неустойив и диспропорционирует, только дела до этого нет уже никому, потому что органическая реакция закончилось, а до неорганической у нас дела нет никому.
А вот хромат намного устойчивее, и мы, строго говоря, в органике вообще не знаем примеров использования хромат-иона (жёлтого). Можно было бы точно такой же механизм написать для реакции с двойной связью? Ну конечно можно, только реакционной способности хрома в хромате для этого не хватает, он просто намного стабильнее. Хромат в частности не окисляет и спирты, потому что с ними не может реагировать – нет механизма. Высокая устойчивость хромат-иона в частности делает возможным его образование в природных условиях – известно не менее 20 минералов с такими ионами, и когда хромат образовался в окислительных условиях, далее он уже вполне устойчив, ведь когда речь заходит о минералах, ясно, что их время жизни измеряется минимум сотнями тысяч, а то и миллионами лет. И мы ничего не знаем про минералы марганца, содержащие перманганат-ион: настолько велика разница в стабильности между соседними изоэлектронными частицами.
Чтобы шестивалентный хром стал окислителем, нужно как-то дестабилизировать это валентное состояние, слишком стабильное в нейтральнос хромате. Займёмся этим.
Теперь попробуем понять, что происходит, когда изменяется природа части лигандов у атома 6-валентного хрома. Здесь самая важная идея в химии переходных металлов – устойчивость высоких степеней окисления зависит от лигандов: донорные лиганды стабилизируют высшие степени окисления, а уменьшение донорности наоборот дестабилизирует. Следовательно, если мы хотим сделать производное высокой степени окисления более сильным окислителем, мы должны уменьшать донорность хотя бы одного лиганда (можно больше, но так можно дойти до таких окислителей, которые окислят и то, что мы не хотим). Теперь, внимание, вопрос: а оксо-лиганды, с которыми мы имеем дело, донорные? Да, и сильно. Потому что один из способов отобразить взаимодействие металла и оксо-лиганда очень нагляден – мы сначала связываем металл обычной координационной связью (σ-связью), а затем вторую пару на кислороде отправляем второй координационной связью (π-связью). Обратите внимание, что в химии металлов это показывают не совсем так, как мы привыкли – а ведь эта схема очень похожа на обычную мезомерию в органике, но для металлов вместо зарядов на атомах принято показывать общий суммарный заряд, а для металла следить за степенью окисления, которая не меняется от того, что на металл повесили какой-то лиганд. Зато меняется суммарный формальный заряд. И для того же хромата в целом мы увидим Cr(6+) – как будто 6+ в начале до связывания лигандов это и степень окисления и формальный заряд (не будем как в современной школе спорить где 6+ а где +6 – это собачий вздор и бессмыслица); и четыре оксо-лиганда внесли 8-, итого степень окисления осталась прежней 6+, а суммарный заряд стал 6+(-8)=-2. Замечательно удобно.
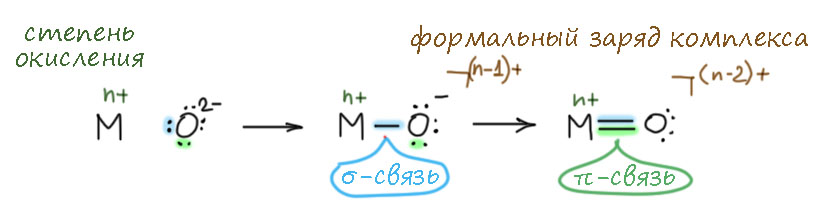
Следвоательно, оксо-лиганд – сильнодонорный лиганд, хорошо стабилизирующий высшую степень окисления, для хрома это 6+. И теперь перед нами открываются великолепные перспективы – если мы хотим увеличить силу окислителя, мы должны уменьшить донорность хотя бы одного оксо-лиганда. Что для этого нужно сделать, если мы хотим максимально не трогать ничего. А очень просто – хотя бы у одного оксо-лиганда отозвать π-связь.
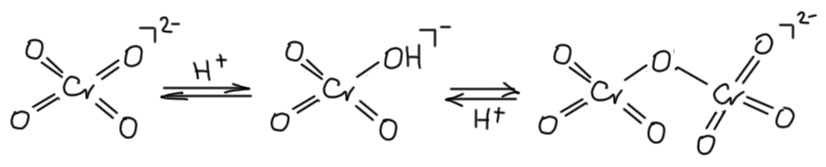
Самая простая реакция – подкисление, при этом по идее должен бы получиться просто ион гидрохромата (кислого хромата), но вместо этого происходит димеризация. Очевидно, что бихромат-ион выгоднее кислых хроматов. Почему? Можно предположить, что когда я написал про отзыв π-связи, это не совсем так, ведь пара на кислороде остается и может быть по прежнему использована, но не в такой степени- мы скорее не отозвали, а ослабили π-связь, снизили донорность лиганда. И тогда в бихромате легко можно увидеть, что мостиковый атом кислорода двумя разными парами может частично стабилизировать металл, и именно это может быть выгоднее. Но тут если это заинтересовало, нужно подробнее анализировать структуру и мы не будем этим заниматься, это детали.
Бихромат отличается от хромата тем, что он гораздо легче реагирует с нуклеофилами. И здесь – стоп – немного привыкнем к тому, что органика и неорганика немного по разному смотрят на очень похожие вещи. Вот две реакции – спирт реагирует с ангидридом карбоновой кислоты (не беру просто уксусный, потому что он запрещён в Российской федерации) и с бихроматом. В органике мы назовём это ацилированием – спирт как нуклеофил атакует электрофильный атом углерода, и происходит замещение, одна часть ангидрида уходит как уходящая группа, образуется сложный эфир кислоты. Бихромат действует очень похоже, хотя мы уже подчеркивали, что аналогия эта не полная, потму что хром это переходный металл, и действует в реакциях за счет d-электронов, но и здесь есть уходящая группа, и образуется сложный эфир хромовой кислоты (моноэфир, поэтому остаётся заряд) – но в этом случае это реакция лигандного обмена, и мы рисуем участников как комплексы поэтому такое немного странное обозначение заряда комплексов. И ещё – и там и там действует кислотный катализ, активирующий связи элемент-кислород к более лёгкому разрыву.
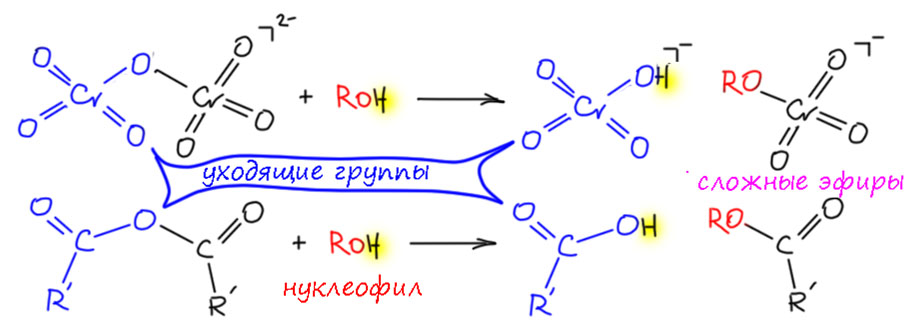
Образующиеся сложные эфиры хромовой кислоты это тоже комплексы хрома(6+), потерявшие часть стабилизации этого валентного состояния из-за замены одного оксо-лиганда на менее донорный алкокси-лиганд. Поэтому они не против как-то поучастовать в реакиях восстановления, и для этого мы уже нашли механизм при наличии хотя бы одного атома водорода на углероде остаток хромовой кислоты отваливает в E2-подобном элиминировании с уменьшением степени окисления. Похожим образом будут реагировать и альдегиды.
А какие ещё функциональные группы могут реагировать с такими хромовыми реагентами? На это легко ответить: другие нуклеофильные группы тоже будут замещать в отстатке хромовой кислоты один из лигандов со всеми последствиями. Поэтому не стоит например, испытвать на устойчивость к таким окислителям амино-групп, окиляться будут с гарантией, только там не очень просто понять, что получится, потому что промежуточные продукты окисления будут частично окисляться дальше. Поэтому у нас нет препаративных методов окисления аминов реагентами на основе хроматов (в ароматической химии есть такие, но пока оставим) не потому что реакции не идут, а потому что они неселективны. Но если амин защитить ацилированием (обычными защитными амидными группами), то нуклеофильность понизится настолько, что реакции с хроматами не будет и можно использовать. Что ещё? Тиолы вряд ли стоит брать по томй же причине. Остальное для нас экзотика. И мы видим, что здесь нет механизма для низконуклеофильных групп – двойных и тройных связей, CH-связей, ароматических колец, амидов, эфиров, производных карбоновых кислоти т.п.
Внимательный читатель в этом месте покопается в памяти и вскричит как герой Жюль Верна Паганель – а разве боковые цепи в алкилбензолах не окисляют горячей хромовой кислотой? А неужели олефины не сгорят в горячем хромпике? А реактив Этара?? Вот-вот, всё верно, не останавливаемся. То, что мы до сих пор разбирали это окисление образующимися на месте эфирами хромовой кислты в мягких условиях – такие реакции никогда н егагревают, а обычно даже охлаждают. А что будет если еще сильнее дестабилизировать Cr(6+) и ужесточить условия? Дестабилизировать как? Заменить не один оксо-лиганд, а два. А может три? Или все четыре? Нет, это уже лишнее, как мы сейчас увидим, двух уже достаточно для того, чтобы дестбилизировать Cr(6+) настолько, что мало не покажется никому. Посмотрим на типичный пример такого реагента, хлористый хромил, или реактив Этара.
Реактив Этара и аналоги
Про реакцию Этара почему-то знают почти все, видимо, потому что прямое окисление метильной группы в ароматике в альдегидную необычайно привлекательно – ароматические альдегиды нужны многим, а всякие замещённые толуолы доступны в количествах от килограммов до тонн по цене телеги с навозом. И если есть такая реакция, то это прямо гениально. Но вот мы смотрим разнообразные книги и статьи, где реально используются и получаются ароматические альдегиды и вообще не видим этой реакции, ни разу, ни одного примера. Что за чёрт! Хлористый хромил что ли какая-то труднодоступная экзотика? Ничего подобного, это очень простое вещество, делается легко из хромового ангидрида, хлорида натрия и серной кислоты. Тяжёлая жидкость, по виду практически неотличимая от брома – тёмно-бурая с бурыми тяжёлыми парами, и так и хочется сказать – с похожим запахом, но запах, что у брома, что у хлористого хромила лучше не пытаться почуствовать, потому что это обычно кончается необратимой порчей запахораспознавательного инструмента, коротко обозначаемого как “нос”. Тот момент, когда в нос попадают хоть следы паров одного или другого, становится поворотным в вашей жизни, и в вас зарождается сомнение, так ли нужен этот нос, в жизни, если проку от него больше никакого нет, а мучения он доставляет ужасающие – там всё горит и саднит. И вы начинаете завидовать майору Ковалёву и удивляетесь, зачем он носился по Невскому и пытался вернуть это орудие постоянной пытки вместо того чтобы благословлять судьбу, за то, что она его от этого избавила.
Хлористый хромил – жидкость крайне агрессивная, обращаться с ней надо очень уважительно, потому что она действительно часто самовоспламеняется, встретив какую-нибудь органику. Но сама реакция Этара делается очень просто, хотя точно не надо повторять оригинальную методику самого Этара – в те времена, а это 1881 год, химики обожали сероуглерод в качестве растворителя. Сероуглерод с хлористым хромилом просто так не реагирует, но – да не допустят этого боги! – что-нибудь саморазогреется и вы получите фонтан смеси одного и другого – ядерная война после этого покажется милой шалостью маленьких детей (чем собственно она и является, только в роли маленьких детей взрослые олигофрены). Патологоанатомы долго будут колдовать над вашими останками, но так и не смогут понять, что это за зловонная куча дымящихся ошмётков. Возьмите четырёххлористый углерод, он правда запрещён из-за высокой токсичности, но только в недружественных странах, а нам они не указ, хотя травиться назло этим самым странам тоже не нужно – работайте под хорошей тягой, и он хотя бы не горит. А ещё лучше десять раз подумайте, потому что реакция эта довольно паршивая, и реально получить ей почти ничего не получится.
Александр Леон Этар (Étard) это старый французский химик конца 19-го века, ученик великого Вюрца. Всю жизнь Этар исследовал этот свой реагент, видимо, решил, что занимается амбициозной задачей планетарного масштаба. Судя по тому, что жизнь его была не очень длинна (1852-1910), то, что он породил, его же в конце концов и убило – я уже много раз напоминал, что в ранней химии никаких тяг в лаборатория не было, и исследователи дышали всем, что исследовали. И после Этара его еще несколько десятилетий исследовали другие – минимум полсотни статей можно найти по этому поводу. В 1950-е некоторые предприняли последнюю попытку в этом реагенте разобраться, разобравшись в механизме реакции – это было как раз время, когда все хотели механизмов, и особенно постарарались румынские химике во главе главным румынским органиком Неницеску, любимцем Елены Чаушеску, той самой, которую в самом конце 1989 года вместе с супругом по имени Николау хорошенько нашпиговали свинцом. Из серии крайне мутных работ Неницеску и сотрудников так ничего и не получилось по причине их крайней наивности и слабого понимания химии. Этар хотя бы вошел в историю и в справочник именных реакций. Все остальные со своими работами растворились, потому что ничего путного сделать не получилось. Реактиву Этара можно выдать премию по номинации “самый бессмысленный реагент в орагнической химии, унёсший с собой на свалку истории множество человеко-лет труда первооткрывателя и других исследователей”. Я не знаю более достойного конкурента в этой номинации.
Реакция Этара фактически хорошо удалась только один раз – с толуолом, из которого получается бензальдегид с хорошим выходом (заявлено 70%). проблема только в том, что по стехиометрии хлористого хромила нужно 2 эквивалента на эквивалент метиларена. Как делают реакцию Этара? Одним способом, как собственно и нашёл сам Этар. К раствору толуола (или другого субстрата) в CCl4 или CS2 при интенсивном перемешивании и охлаждении прикапывают раствор хлористого хромила в том же растворителе – реакцию лучше вести около ноля, чтобы не получить саморазогревание и смертельный фонтан. Смесь перемешивают при комнатной температуре до исчезновения бурой окраски CrO2Cl2, иногда для этого требуются несколько дней, при этом всегда выпадает мелкий бурый осадок, который называют комплексом Этара. Этот комплекс просто разлагают водным раствором бисульфита, чтобы восстановить весь хром в трёхвалентный. Ну и дальше как обычно выделяют продукт, хотя каша получается малоприятная, но это довольно типично для всех хромовых окислений – в конце приходится фильтровать раствор от осадка с Cr(3+) в какой-то форме типа гидратированной окиси, долго мыть осадок растворителем, ну и дальше по обычной схеме мокрого разделывания.
Что же такое “комплекс Этара”. Судя по цвету, это что-то с четырёхвалентным хромом, который неустойив в растворах, но в твёрдом состоянии может оказаться более живучим. В литературе полно исследований комплексов Этара для реакций CrO2Cl2 с разными соединениями – реакция Этара всегда визуально идёт одинаково, что бы ни брали для окисления, а за годы интереса к ней брали почти всё, включая довольно сложные природные соединения типа терпенов, и даже алканы – и с ними выпадает “комплекс Этара”. Беда в том, что исследовать эти коплексы серьёзно невозможно, и для них делали элементный анализ и на его основе гадали, что это. Элементные анализы выходили худо, с огромными рассхождениями, но это не мешало гадать и делать глубокомысленные выводы. Мы не будем, а просто подумаем, как могла бы идти реакция Этара.
В этм месте придется пережить еще одну удивительную встречу с фантазёрами. Если заглянуть в википедию, английскую или немецкую или французскую, там приведён механизм этой реакции, выглядящий крайне внушительно. Вот так, я добросовестно перерисовал этот тупой вздор вместе со всеми игривыми стрелочкми.
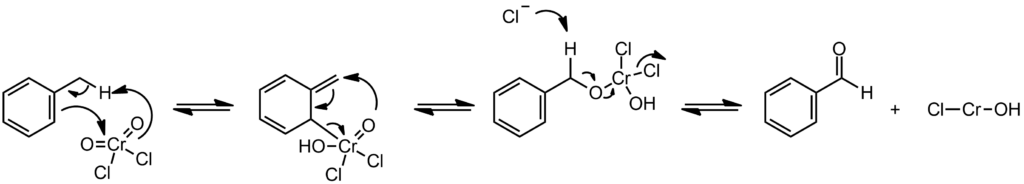
Одного этого механизма достаточно, чтобы присоединиться к недавнему предложению Илона Маска переименовать википедию в дикипедию, и даже без английской скабрезности, а просто по-русски в дикопедию. Впрочем наша версия ещё много раз хуже. Википедия может быть полезна, но при одном условии – нужно проверять каждое сведение из неё. В западных википедиях для этого есть ссылки, что сильно облегчает проверку, в нашей обычно и того нет, просто выдумка на выдумке. Но с этим механизмом вышел прокол и там. Ссылок на него нет, а пара ссылок в английской и немецкой, обещающие что-то про механизм, совсем не про этот. Получается, что этот бред выдумал анонимный автор статьи. Автор, который вообще ни черта не смыслит ни в координационной, ни в неорганической химии, и единственно, что умеет, это гонять кривые стрелочки – нехитрое, но опасное занятие. Стрелочками можно иллюстрировать уже хорошо продуманную схему, но просто рисовать стрелочки и думать, что они что-то объясняют – это одно из тупейших занятий, дискредитирующих профессию. Здесь же фантазёр вообще в голову не берёт ничего, кроме стрелочек. Первую стадию собезьянил из механизма окисления диоксидом селена, типа еновая реакция, но она приводит здесь к совершенно несуразному металлоорганическому производному 6-тивалентного хрома, шансов на жизнь у которого не больше, чем у комара в доменной печи, дальше идет еще одна несуразная перегруппировка, явно попытка выйти из неудобного положения, потому что точно такой же вполне разумный интермедиат можно было бы получить намного проще. Ну и в завершение выясняется, что автор механизма не в курсе как реально делают ту реакцию, механизм которой он отчаянно придумывает – где в этой схеме комплекс Этара? А реакцию Этара всегда – слышишь, неизвестный фантазёр – всегда делают через комплекс Этара, альдегид образуется не в самой реакции, а при разложении комплекса Этара. Реакция должна была бы кончится где-то здесь, но у фантазёра еще и совсем дикая стадия второго восстановления хрома до двухвалентного – то есть у него один хлористый хромил забирает 4 электрона, превращаясь в сильнейший восстановитель. Забудем этот несуразный вздор, высосанный из пальца неведомым шарлатаном.
А сами поробуем придумать что-то более разумное. Придумывать придётся, потому что просто взять это из какой-то вызывающей доверие работы не получится. Есть серия румынских статей Неницеску и соавторов, так и озаглавленных “механизм реакции Этара”, но там всю дорогу исследователи занимаются совершенно бессмысленной работой, пытаясь понять как устроены комплексы Этара для разных субстратов, не имея в общем никаких других данных кроме состава продуктов и элементного анализа – в социалистической Румынии, разорённой в пух и прах бездарным, но спесивым вождём Чаушеску, “гением Карпат”, ничего другого и не было. Вместо нормальной науки была его жена, у которой вроде бы даже со школьным образованием было скверно, но Елена служила президентом румынской академии наук, выдавала себя за великого химика, и покровительствовала Неницеску.
С другой стороны есть немало работ по окислению алкилароматики и множества других соединений раствором хромового ангидрида в уксусном ангидриде (запрещён в Российской федерации) – в этой системе, как считают, образуется непосредственный аналог хлористого хромила, диацетат хромила CrO2(OAc)2, и это тоже, как мы теперь понимаем, производное шестивалентного хрома, в котором это валентное состояние сильно дестабилизировано – ацетат, как и хлорид, лиганды скорее акцепторные, что оставляет на стабилизацию высшей степени окисления только два оксо-лиганда (большинство этих работ перечислены как ссылки в этой, одной из самых последних: M. Sowifiska, A. Bartecki Transition Met. Chem. 1985, 10, 63-66). Из них мы видим более-менее устоявшийся консенсус по составу комплексов Этара, так как в 1970-е к проблеме были подключены более серьёзные инструментальные методы, с помощью которых можно считать доказанным, что в состав комплекса входят два атома хрома в степени окисления 4+. И тогда, скорее всего, комплекс Этара это производное гидратной формы альдегида, такой ацеталь, только вместо остатков спирта висят простые комплексы хрома(4+). А как это образуется? Здесь тоже есть хорошая идея, отчасти подкрепленная спектроскопическими и кинетическими исследованиями, но по ацетату хромила (F. Freeman, C. R. Armstead, M. G. Essig, E. M. Karchefski, C. J. Kojima, V. C. Manopili, A.H. Wickman, J. Chem. Soc., Chem. Commun., 1980, 65). Ацетат хромила это раствор хромового ангидрида в уксусном ангидриде (запрещён в Росссийской федерации). Ключевая идея здесь – это свободнорадикальная реакция. И хлористый хромил и раствор хромового ангидрида в запрещённой субстанции, как показывает обобщение огромного количества работ за полвека до этих, окисляет много что, и даже алканы, но в основном это алкилароматика, непредельные соединения, имеющие аллильные положения и т.п. – всё это проще взять именно свободнорадикальными реакциями, которые всегда начинаются с отщепления атома водорода (по современной терминологии HAT – hydrogen atom transfer) – даже не с отщепления, а с переноса атома водорода между двумя молекулами или радикалами. Селективность у таких реакций минимальная, – просто выбираются атомы водорода, отщепление которых даёт радикалы постабильнее. Соединения хромила (сейчас мы назовём их диоксокомплексами хрома(6+)) отлично могут это делать по простым причинам: а) хром в этой степени окисления в диоксокомплексах дестабилизирован и готов снижать степень окисления, то есть забирать электроны; б) снижать степень окисления проще всего по одному электрону, забирая их от лигандов, хотя есть пути и сразу по два, как мы видим на окислении спиртов более стабильными триоксокомплексами хрома(6+), но здесь такой путь не виден; в) после изменения степени окисления, хром становится атомом, имеющим неспаренные электроны – у нечетных степеней окисления атомов из четных групп это очевидно, и может принять участие в переносе атома водорода. Нарисуем:
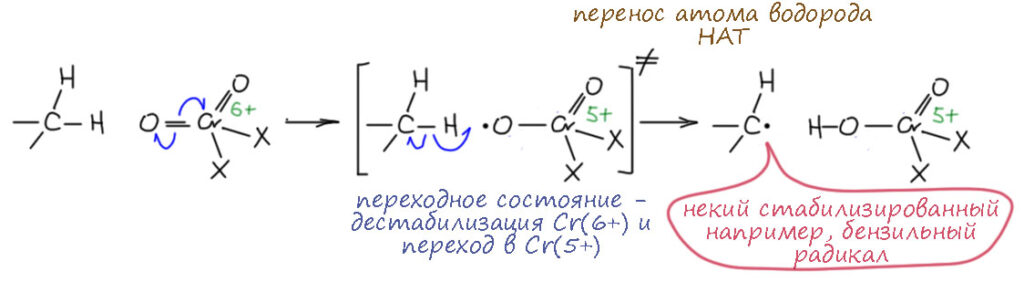
Видим, что за счет перехода Cr(6+) в Cr(5+) оксо-комплекс приобретает способность отрывать атом водорода от C-H связи – именно атом с одним электроном, а не протон, и не гидрид. Я специально изобразил это как переходное состояние процесса переноса атома водорода. Так образуется радикальная пара. А где второй радикал? Это комплекс хрома(5+) – у этого валентного состояния нечётное число электронов, и есть хотя бы один неспаренный электрон в валентной оболочке (а почему только один? – а потому что комплекс может быть высокоспиновым, если вспомните что-то из химии соединений переходных металлов, и тогда неспаренными может быть больше электронов, но это нам до лампочки, нам достаточно одного). Большим испытанием для органика является то, что в химии металлов не принято изображать неспаренные электроны на металле точками, это просто подразумевается. Что дальше? А дальше, скорее всего (напоминаю, что механизм гипотетический, а доказанного нет) неустойчивое валентное состояние Cr(5+) опустится дальше до Cr(4+), что можно сделать точно так же – смещением еще одного электрона с оставшегося оксо-лиганда, точно так же, но вместо переноса атома происходит рекомбинация радикалов и образуется такой комплекс хрома(4+), а это валентное состояние само по себе устойчиво, пока не попадёт в воду, а воды пока нет. Что дальше? Если бы это соединение осталось, то после гидролиза реакционной смеси, мы получили бы спирт, а Cr(4+) диспропорционировал бы на Cr(3+) и Cr(6+), который мог бы успеть уже во время обработки реакционной смесиокислить этот спирт. Но, во-первых, реакция на этом не остановилась, что известно, а во-вторых, реакции такого типа обычно сразу обрабытывают не просто водой, а раствором восстановителя типа бисульфита натрия, что сразу гасит все степени окисления хрома выше 3+. Но, как минимум тогда, когда окисляют метилароматику, очень легко происходит второй раунд точно такого же превращения и второй атом водорода уходит, и на углерод бывшей метильной группы сядут два комплекса хрома(4+). Это и есть комплекс Этара, если X = Cl, или что-то похожее, если X = OAc, что много раз выделяли, определяли что на органический остаток приходится два атома хрома и в виде Cr(4+). Гидролиз таких комплексов и даст карбонильную группу. А почему третий водород не уходит так же? Хлористый хромил и другие подобные вещи (хорошо известно, например, что некоторые замещенные толуолы тоже окисляются в альдегиды раствором хромового ангидрида в запрещённом в Российской федерации уксусном ангидриде, что и проверить нельзя, так как нет никакой возможности использовать запрещённое соединение, поэтому ну и чёрт с ними, пусть в недружественных странах разбираются) почти всегда используют даже не в двукратном количестве, а в избытке, но реакция останавливается на комплексе Этара 2:1. Дело либо опять в стерике – не подберёшься уже к этому третьему водороду. Либо в более тонких материях, связанных с тем, что у радикальных реакций есть свои предпочтения – я касаюсь этого в новом курсе лекций про химию 21 века, а здесь не буду.
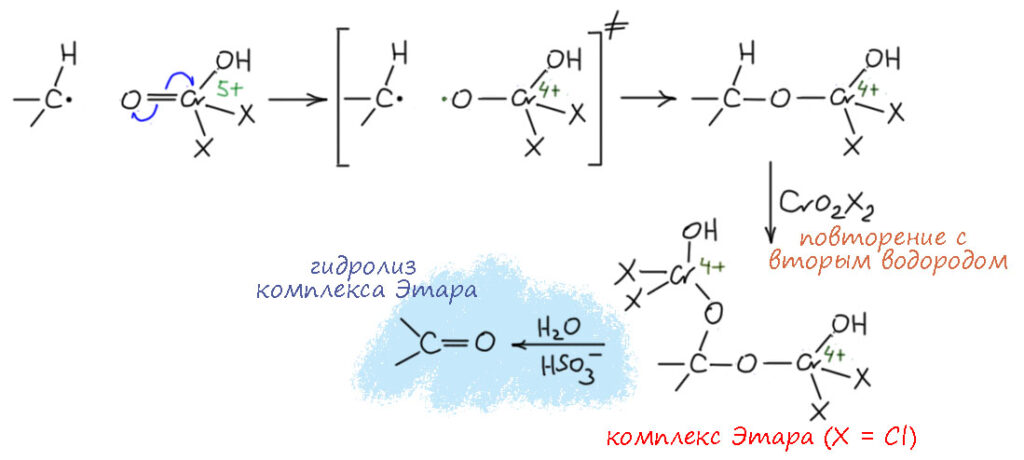
Поскольку механизм по природе радикальный, не удивительно, что реактив Этара окисляет даже алканы, быстрее те, у которых есть третичный атом водорода, но и вторичные тоже – но там получаетя страшная каша из спиртов, хлорпроизводных и чёрти чего ещё, описания этих реакций старые, воспроизвести их сложно и вряд ли кому-то нужно, ведь никакой практической ценности у них нет. Были времена, когда за любую реакцию алканов учёных хвалили, давали конфеты и гладили по голове, но с тех пор как-то понемногу стало понятно, что никакой доблести в том чтобы превратить алкан в трудноразделимую смесь малополезных продуктов нет, и нужно искать только высокоселективные и обязательно каталитические процессы, чтобы заслужить конфеты и ласковое отношение тех, кто финансирует науку.
По такому же механизму, скорее всего, окисляет и метилароматику и вообще много что, почти совершенно неселективно и просто раствор хромового ангидрида в серной кислоте, то есть двуххромовая кислота и что-то еще более сложное – обратите внимание, что и в таких комплексах хрома остается только два стабилизирующих Cr(6+) оксо-лиганда, и в общем, это почти то же самое что хлористый хромил или ацетат хромила. На альдегтде только остановиться нельзя, потмоу что в этих условиях все комплексы сразу гидролизуются, а альдегид будет окислен в кислоту. Реакции ведут при нагревании. Если захотите так окислять боковые цепи в алкилароматике, обязательно ищите готовую методику, потому что там все страшно зависит от конкретных условий (кислотности, температуры, длительности, соотношения), а если делать из общих соображений, либо недоокислите, либо переокислите.
Реактив Джонса
Приступим наконец к обсуждению собственно методов окисления спиртов именно в карбонильные соединения. Известные в старой химии окисления подкиcленным бихроматом для этого не годятся – они приводят к переокислению, даже кетонов, если окисляется вторичный спирт. А ещё они крайне неудобны для серьёзной препаративной работы, потому что работают фактически в водных средах, или гетерогенно, а это крайне неудобно и не позволяет контролировать ход реакции.
Тем мболее удивительно, что первый исторически хромовый реагент прост как средство для мытья посуды – это хромовая кислота, получаемая растворением хромового ангидрида в крепкой серной кислоте, ну по составу чисто хромпик (в современной химии пользоваться хромпиком для мытья посуды крайне нежелательно, соединения хрома(6+) канцерогенны, а над ёмкостью с хромпиком всегда есть аэрозоль, содержащая хромовый ангидрид и хромовую кислоту). А что в этом нового? А вот сейчас увидим.
Реагент называют в честь английского химика валлийского происхождения сэра Иварта Джонса (Sir Ewart Ray Herbert Jones, 1911-2002), чрезвычайно влиятельного деятеля британской науки, основателя и первого президента Королевского Химического Общества (RSC). Вы как и я будете удивлены, но это знаменитое общество, издающее наши любимые журналы типа Chemical Communications, образовалось в год Московской олимпиады, в 1980-м. Как, не может быть, а до этого что было?? В Англии же химией занимались с самого начала, в 18-го века, кем были, спрашивается, Джон Дальтон, Майкл Фарадей, Хемфри Деви. Неужели у них общества не было, англичане же так обожают всякие общества и клубы, где можно заседать, потягивая добрый виски и пыхтя сигарой, и не спеша перетирать общие проблемы или хотя бы погоду. Было, конечно, в Англии химическое общество, но не королевское. Вот удивительно, наконец нашлось что-то в Англии не королевское, видимо, монархи не торопились связывать себя с какой-то вонючей и подозрительной наукой. Видимо, так оно и было: я тут где-то на сайте уже рассказывал историю, как в середине 19-го века его королевское высочество князь-консорт Альберт решил всё же учредить в Лондоне колледж для обучения наукам, и даже выписали из Германии одного из величайших химиков той эпохи Гоффмана для руководства учебным заведением, но к идее быстро охладели, финансирование кончилось, Гоффман вернулся на родину, и единственное, что из этой истории родилось стало крупнейшим английским органиком Перкином, что тоже неплохо, и вообще Англия как-то всегда обходилась без назойливого участия королевского двора в своей жизни. Но для порядка учредить королевское общество всё же надо было. Но очень нескоро до этого руки дошли и не у кого-нибудь, а у автора первого хромового реагента.
Оказывается, было просто Химическое общество (the Chemical Society, в старой доброй Англии всегда считали, что никаких дополнительных эпитетов их организации не требуют, всем и так понятно, что стоит за артиклем “the”, и пусть другие, американцы там всякие, французы, немцы, русские, японцы и т.д. добавляют к своим аналогичным обществам дополнительные национальные эпитеты), президентом которого был сэр Иварт Джонс, и еще Королевский Институт Химии, как высокое собрание химических промышленников, и сэр Иварт Джонс в свою очередь служил президентом и там. Наконец сэру Иварту Джонсу показалось, что быть президентом очень славно, а во всех этих обществах как назло совсем долго сидеть было нельзя, в Британии химиков немало и им тоже хочется. Тогда сэр Иварт Джонс собрал всё своё влияние при дворе и добился объединения просто Химического общества и Института Химии в Королевское Химическое Общество – и, не поверите, тут же и возглавил его. И если бы не уже преклонный возраст, так бы и правил сэр Иварт Джонс британской химией, но пришлось уйти в отставку в 1982-м. А когда будущий величественный научный сановник был молод и ещё не был сэром Ивартом (титул выдали в 1963-м, не иначе как за заслуги по окислению спиртов в кетоны), он много занимался модной химией стероидов, а там очень часто надо окислять вторичные спиртовые группы в кетонные, и так однажды, в 1946-м он с коллегами опубликовал очередную статью в Journal of Chemical Society, в которой между другими реакциями нашли, что вторичные спирты отлично и чисто окисляются в кетоны хромовой кислотой в ацетоне, и этот метод не затрагивает, например, тройные связи рядом. Потос оказалось, что и двойные не затрагивает.
Так и появился реагент Джонса, который очень понравился особенно другим стероидным химикам. Я сам его пробовал для вещества ряда стероидов и нашёл совершенно превосходным – реакции идут быстро и чисто, а выделение продукта при некоторой сноровке очень удобно. Если всё делать правильно, и после непродолжительной реакции погасить оставшийся хром изопропанолом, то выпадает зеленый тяжелый порошок, видимо, гидратированного оксида хрома(3+) (или какой-то основной соли, чёрт знает, что это такое, но оно тёмно-зелёное и на фильтре выглядит как тяжёлый порошок), совершенно не мешающий отделению продукта (но порошок на фильтре надо тщательно промыть горячим ацетоном, потому что он иначе унесёт с собой немалую долю продукта).
Когда неискушенные люди видят, что из себя представляет реактив Джонса, а это просто раствор хромового ангидрида в крепкой, но не концентрированной серной кислоте, то есть не что иное как раствор двуххромовой кислоты, часто можно услышать брюзжание – и вот за этот всем известный примитив, мы этим раньше посуду мыли, дают имя реагенту! Вот же проходимцы наглые эти англичане, везде пролезут! Не торопитесь брюзжать. Самые ценные вещи всегда просты, и это надо было найти и описать. И секрет реактива Джонса еще и в том, что его всегда применяют в ацетоне: спирт растворяют в ацетоне, а ацетон – отличный растворитель, растворяющий почти любую органику, недаром им споласкивают посуду после мытья (ха-ха, хромпиком) – и к раствору довольно быстро и при адском перемешивании прикапывают раствор Джонса. Всегда в хромовых окислениях применяйте лучшие мешалки, потому что там бывают проблемы с выпадением каких-то мерзких клейких соплей, видимо это и есть что-то промежуточное, и если это быстро не промешать, вся реакция превратится в мучение, мешалка приклеится к стенке, в одной части колбы повалится непрореагировавший спирт, а в другой заварится ведьмино зелье коричневого цвета – и это тоже видимо те самые хром-четыре или хром-пять, решающие что делать дальше. Но если мешалка будет хороша, то все быстро и окончательно позеленеет, вывалится порошок гидрата окиси хрома(3+), а выходы быдут после выделения продукта высоки. Отличный реагент. Но только для вторичных спиртов. Первичные он стабильно переокисляет в кислоты, которые частично связываются с хромом(3+) и образуются мерзейшие зелёные тягучки, из которых добывать эту самую кислоту придется долго и не без шансов потерять минимум половину. Карбоксилаты хрома – это невероятно устойчивые комплексы очень сложного строения, и разложить их непросто. И в соединении не должно быть и вторичных, и первичных спиртовых групп – первичные окисляются быстрее и просто угробят вещество. Но когда в соединении есть только вторичные группы, особенно в циклах, то реактив Джонса хорош и удобен и поэтому его очень активно применяли и применяют особенно в стероидных синтезах.
Реактив Джонса отлично окисляет и двойные связи, если в соединении нет спиртовых групп – окисляет исчерпывающе, и это вполне можно встретить в реальных синтезах, например, в одном из самых знаменитых синтезов 20-го века – витамина B12: для того чтобы в начале 1970-х справиться с этой головоломной молекулой понадобилось два великих химика – Роберт Вудвад и швейцарец Альберт Эшенмозер (Эшенмозер на 8 лет младше Вудварда, но пережил того почти на полвека и скончался, немного не добив до сотни, летом прошлого 2023 года). Как раз в части Эшенмозера в самом начале окислением двойной связи реагентом Джонса сразу получают важную заготовку, причем сразу за окислением следуют две лактонизации с гидратной формой кетона – это похоже на внутримолекулярный ацеталь, но это не ацеталь, а ацилаль – когда вместо спирта карбоновая кислота. Исходное для этого расщепления получено реакцией Дильса-Альдера с катализом кислотой Льюиса – такой катализ работает не всегда, а только когда диенофил содержит карбонильную группу, сопряжённую с двойной связью, в этом случае координация карбонильного кислорода по кислоте Льюиса увеличивает акцепторность диенофила, а следовательно и реакционную способность:
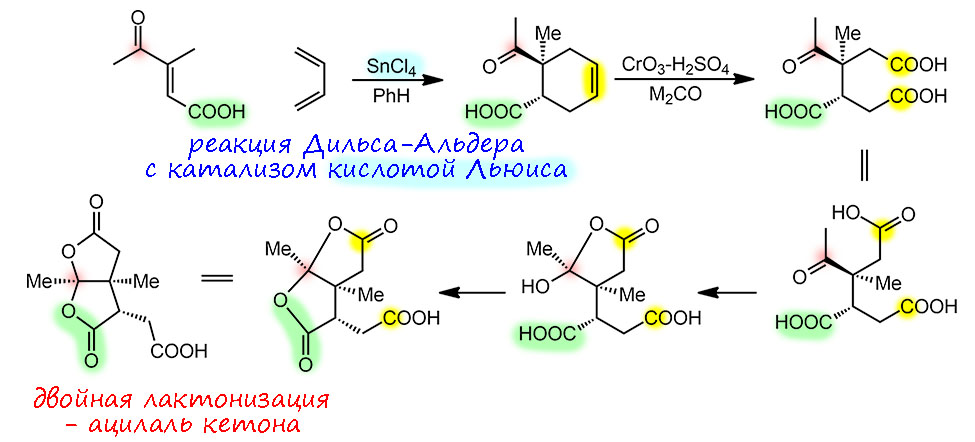
Так, и почему это происходит, ведь мы условились, что для любого окисления нужен механизм: нет механизма, нет окисления (восстановления это тоже касается, как и любой другой реакции), и что шестивалентный хром работает через образование эфиров хромовой (или двуххромовой) кислоты, которые дальше распадаются по общему механизму элиминирования. Но дальше мы убедились, что в более жестких условиях или в случае более сильной дестабилизации этого валентного состояния Cr(6+) может включать и другие, скорее всего, свободнорадикальные механизмы, и окислять уже всякие CH-связи, хоть немного активированные к отщеплению атома водорода. А двойная связь? А здесь стоит вспомнить то, с чего мы начинали – хромат изоэлектронен перманганату, и не окисляет как перманганат только потому что в хромате Cr(6+) отлично стабилизирован четырьмя оксидо-лигандами. Так, а если мы его чуток дестабилизнём? Ну, мы знаем как, в кислой среде хотя бы, превратим хромат в бихромат, а в реактиве Джонса ровно оно и есть – двуххромовая кислота. Ну и тогда нам кто-то из уполномоченных богов велел попробовать приспособить этот механизм, но – придётся ещё подумать, почему окисление олефина перманганатом или четырёхокисью осмия идёт до диола, а здесь – разрывается и связь C-C. Но то же самое происходит при окислении диолов периодатом или, сразу, олефинов четырёхокисью рутения (этого мы не знаем, но поверьте на слово). Это просто значит, что когда образовалось производное диола, металл (или элемент) сохряняет способность окислять – у периодата она есть сразу, а у рутения дестабилизирована и степень окисления 6+, в отличие от осмия, у которого она уже никого неспособна окислить. Хорошо, ну так и представим, что когда с двуххромовой кислотой прошла первая стадия, и образовался неокисляющий Cr(4+), второй атом хрома доокислит его обратно – это частая история с комплексами, у которых образовалось две разные степени окисления. Два альдегида доокислятся до кислот, это мы знаем. Вот такой получится механизм:
 Сразу скажу, что в современной химии так олефины окислять сильно не любят, потому что требуется очень много эквивалентов Cr(6+) на олефин (сами прикиньте сколько), и как обычно ещё и избыток нужен. Но в исторических синтезах вы это встретите и не только у Эшенмозера. В современной химии для таких расщеплений стали особенно популярны соединения рутения в каталитических системах со стехиометрическим реокислителем типа периодата. Об этом как-нибудь в другом месте.
Сразу скажу, что в современной химии так олефины окислять сильно не любят, потому что требуется очень много эквивалентов Cr(6+) на олефин (сами прикиньте сколько), и как обычно ещё и избыток нужен. Но в исторических синтезах вы это встретите и не только у Эшенмозера. В современной химии для таких расщеплений стали особенно популярны соединения рутения в каталитических системах со стехиометрическим реокислителем типа периодата. Об этом как-нибудь в другом месте.
Комплекс хромового ангидрида с пиридином
В любой книжке по неорганике сказано, что хромовый ангидрид воспламеняется в контакте с органическими растворителями, в первую очередь с пиридином. Это истинная правда – реакция очень экзотермична, и если просто налить пиридин на хромовый агнидрид, получается быстрый разогрев, и в таких условиях хромовый ангидрид становится сильнейшим окислителем, начинается окисление пиридина, дополнительный разогрев и воспламенение. Тем не менее, еще в 1948 году известный американский неорганик Гарри Сисслер с сотрудниками нашли (H.H. Sisler, J.D. Bush, and O.E. Accountius. J. Am. Chem. Soc. 1948, 70, 3827), что если смешивать реагенты аккуратно, обязательно именно ангидрид прибавлять мелкими порциями к пиридину, и при хорошем перемешивании и охлаждении получается желтый комплекс состава два пиридина на один хром-о-три, но ещё очень важно, чтобы всё было очень сухим, свежеперегнанный пиридин, и хромовый ангидрид, высушенный в вакууме при нагревании. В более современныъ методиках это делают без такой паранойи, хромовый ангидрид берут из хорошей фирменной банки, пиридин перегоняют, но уже за следами воды не бегают – достаточно чтобы все было просто сухо, а не сухо-сухо-сухо.
Что такое хром-о-три с точки зрения химии переходного металла хрома? Это комплекс хрома(6+) с тремя оксо-лигандами. У такого комплекса легко посчитать число валентных электронов – ноль от хрома и три по четыре от оксо-лигандов, итого 12. Это очень мало, из чего следует, что это очень сильно координационно-ненасыщенный комплекс, и хром в середине обзадает высокой льюисовой кислотностью и просто жаждет найти себе ещё донорные лиганды. Можно сравнить структуру CrO3 и серного ангидрида. И то, и другое плоские молекулы, очень похожие по форме, но структура у них разная, потмоу что хром это d-элемент, а сера – p-элемент, у серы надо соблюдать октет, а у хрома не надо, и для переходного металла рубежом числа валентных электронов является число 18, причем это не такой упор, как в химии p-элементов, это число можно преодолеть, хотя это сильно нежелательно. Поэтому мы не только вправе, но и хорошо делаем, когда рисуем CrO3 с тремя двойными связями, а вот для серы так рисуют, но это неверно, хотя и общепринято. Правильно рисовать граничные структуры. Именно потому что не хочется рисовать мезомерию и заряды, мы продолжаем пользоваться старинной структурной формулой SO3 где изображены не связи, а валентности, как в 19-м веке (я разбирал эту странную традиционную ценность на страничке про валентность). Чёртов ИЮПАК даже однажды издал рекомендацию, в которой признал, что формулы, нарушающие октет, неверны, но зато красивы и не требуют мезомерии, поэтому ими нужно пользоваться, а про себя помнить, что это неправильно. Плевать я хотел на ИЮПАК, буду писать и рисовать правильно, чего бы мне это не стоило. Я ещё добавил четвертую граничную структуру, в которой все связи одинарные с дополнительной ионностью, а на сере 6 электронов, секстет. Видимо, эта структура действительно вносит немалый вклад, иначе было бы непросто объяснить чрезвычайно высокую электрофильность серного ангидрида, просто ужасающую – он реагирует буквально с любыми нуклеофилами, даже с очень слабыми, чем можно пользоваться для электрофильной активации. У нас на кафедре этим долго и плодотворно занимался Н.С.Зефиров с сотрудниками – с помощью серного ангидрида раскачивали множество ленивых молекул и получали суперэлектрофильные реагенты для присоединения к кратным связям.
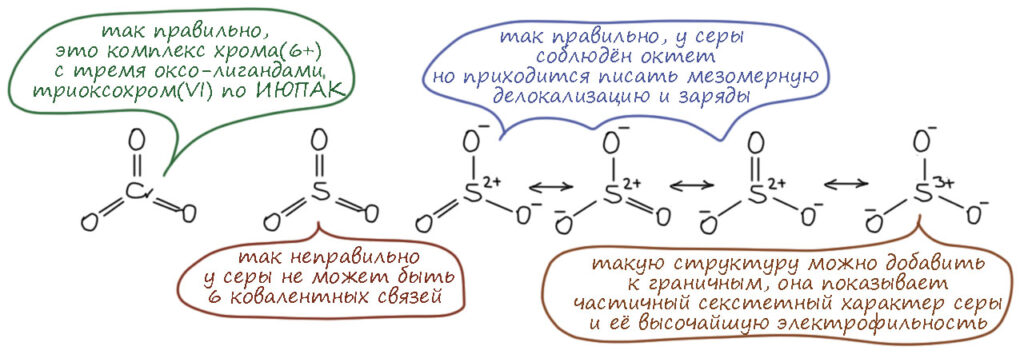
Зачем я вспомнил про SO3 хотя речь про хромовый ангидрид? Сравнивать похожие молекулы, но произведенные от разных типов элементов очень полезно – мы видим и сходство и различие. Оба ангидрида чрезвычайно высоко электрофильны (для переходных металлов чаще используют не электрофильность, а кислотность Льюиса – в той химии это одно и то же буквально, а вот в химии p-элементов некоторое различие есть, хотя и небольшое: будем пользоваться этими терминами взаимозаменяемо), серный просто чудовищно, но и хромовый весьма изрядно. И то, и другое бурно реагирует с пиридином. Реакцию пиридина с серным ангидридом нормально можно сделать только при очень аккуратной работе, бешеном перемешивании и охлаждении, так что проукт такой реакции обычно делаю немного иначе. А вот структуры продуктов разные. И там и там, электрофильный атом в середине реагирует с азотом пиридина, забирает его пару. Но хром может взять две молекулы – он координационно ненасыщен, от двух молекул пиридина получит ещё 4 электрона и валентных электронов станет 16 – ровно как в хромат-ионе. Это очень близко к координационной насыщенности, но еще одна молекула пиридина просто не лезет – места нет. Обратите внимание, что на азоте пиридина мы не рисуем плюс, потмоу что пиридин это лиганд, и вкоординационной химии не принято обозначать донорно-акцепторную связь между лигандом и металлом с разделением зарядов. И что мы оставляем двойные связи на оксо-лиганды, ведь они прямо не участвуют в связывании нового лиганда, вакантные d-орбитали для этого даёт хром. А вот с серным ангидридом может реагировать только одна молекула пиридина, больше туда не влезет, потмоу что на сере должен быть октет. И структура другая – поскольку у серы теперь не три, а четыре связанных атома, это уже тетраэдр. Правильную структуру проще всего нарисовать, если приделать пиридин к последней секстетной граничной структуре SO3 и обозначить заряд на азоте – это вам не химия металлов, с привычкой все рисовать как комплексы, здесь честная донорно-акцепторная связь с разделением зарядов. И опять, принято рисовать другую структуру, явно нарушающую октет. Попробуйте понять с этой структурой, почему сера не может подцепить еще один пиридин.
Впервые применил этот комплекс для окисления вторичного спирта в кетон весьма известный американский химик Льюис Сарет, один из основоположников синтеза стероидов (Сарет – очень близкий родственник, кажется двоюродный брат или типа этого, не менее знаменитого, многие скажут – печально, министра обороны США Дональда Рамсфельда при президенте Джордже Буше младшем, одного из главных руководителей вторжений США в Афганистан и Ирак после терактов 11 сентября 2001 года). Вот ровно для очередного стероидного окисления он и искал окислитель, не имеющий кислотной реакции, а большинство известных тогда окислителей, включая двухромовую кислоту в ацетоне или уксусной кислоте были кислыми. Он и заметил работу Сисслера про комплекс хром-о-три с пиридином и приставил его к делу с успехом, именно как суспензию в пиридине, без выделения и связанных с этим опасностей. Метод остался, хотя его путают с методом Коллинза, но окисление по Сарету идет в пиридине и гетерогенно, с суспензией комплекса, полученного непосредственно в той же колбе прямо перед добавлением окисляемого субстрата. Реагент всегда берут в избытке, обычно в немаленьком типа 4-6-кратного, и стехиометрия не имеет значения, после реакции смесь просто выливают в воду или разбавляют эфиром, и не обращая внимания на приключения остатков хрома, экстрагируют продукт как обычно органикой, иногда перед этим отфильтровав осадок соединений хрома, обычно коричневый. Метод вполне рабочий именно для вторичных спиртов, а первичные спирты этим методом частично переокиляются, и это очень неудобно, потмоу что образующаяся кислота даёт соль с пиридином и её просто из такой смеси очень трудно выделить. Метод по прежнему очень популярен в химии стероидов, где окисления спирта в кето-группу встречаются очень часто. Сам делал, неприятно, много пиридина, комплекс надо заваривать очень осторожно, но результат отличный. Двойные связи метод не затрагивает, можно не волноваться. Конечно, здорово напрягает огромный избыток хромового ангидрида, но это так получается потому что реакция фактически идёт на поверхности частичек осадка комплекса, частички быстро покрываются продуктом восстановления, и всё, что внутри, просто перестаёт работать.
Нерастворимость комплекса в пиридине (на самом деле, скорее малая растворимость, чем нерастворимость) подвигла следующих искателей улучшений искать замену пиридину. Так появился метод Коллинза по статье Джозефа Коллинза и сотрудников в 1968 году (J.C. Collins, W.W. Hess, F.J. Frank. Tetrahedron Lett., 1968, 3363). Скверно, что метод назвали по имени не химика, а топ-менеджера большой фармацевтической компании Стерлинг Драгз, в конце 1980-х растворившейся в слияниях и поглощениях, а этот Коллинз до сих пор жив и занимается какой-то околонаучной фигнёй. Написался большой начальник в авторы первым, вот и реагент Коллинза остался в химии. Даже так – Коллинз подрядил на эту работу двух химиков из провинциального университета в Иллинойсе, понятно, что именно они работу и сделали, но реагент всё равно носит имя мистера директора. Эти забытые парни, Хесс и Франк, были умелыми химиками и неплохо разобрались в реакции. Они сделали две вещи – во-первых, нашли, что после выпадения желтого мелкого осадка из пиридина не надо торопиться его оттуда доставать, он в этот момент плохо фильтруется, очень чувствителен к влаге воздуха и способен даже самовоспламеняться, но если смесь покрутить дальше, осадок самопроизвольно перекристаллизовывается, укрупняется, становится кристалличным и приобретает красный оттенок, и такой уже легко отфильтровать, высушить и даже хранить. И такой комплекс легко растворяется в дихлорметане. Что собственно и стало методом Коллинза. Состав комплекса при этом не изменяется.
Реакцию со спиртами ведут при комнатной температуре, прикапывая раствор спирта к раствору комплекса – буквально через 10-15 минут окисление заканчивается, из раствора вываливается коричневое дерьмо, а в растворе не остается даже следов хрома(6+) ни в какой форме. Чтобы достичь высоких выходов приходится использовать большой избыток реагента, до 6-тикратного (не по стехиометрии, а по эквимольному соотношению), просто потому что окисляется только то, что успевает до выпадения осадка, а осадок это какие-то полимерные частицы, содержащие и хром(6+) и хром(4+), бесполезные для реакции. Поэтому бесполезно пытаться уменьшить количество окислителя – просто вырастет количество непрореагировавшего и уменьшится выход. Более аккуратные исследования показали, что на выпадение осадка очень сильно влияют даже следы воды, и чем суще реагенты, тем лучше идет реакция, и даже можно немного уменьшить избыток. Самое главное отличие метода Коллинза от метода Сарета – возможность окислять первичные спирты в альдегиды. Если всё сделано правильно, реагент свеж и растворитель сух, переокисления в кислоты практически можно не бояться. Это непостредственное следствие того, что окисление альдегида в кислоту хромовыми реагентами идёт через аддукт альдегида и какой-то формы хромата, а здесь этого просто нет. К тому же реакция идёт очень быстро. Это говорит о том, что механизм собственно окисления видимо внутримолекулярный. Попробуем представить. Первое, что происходит, это замещение лиганда пиридина на алкоксид. Как-то так:
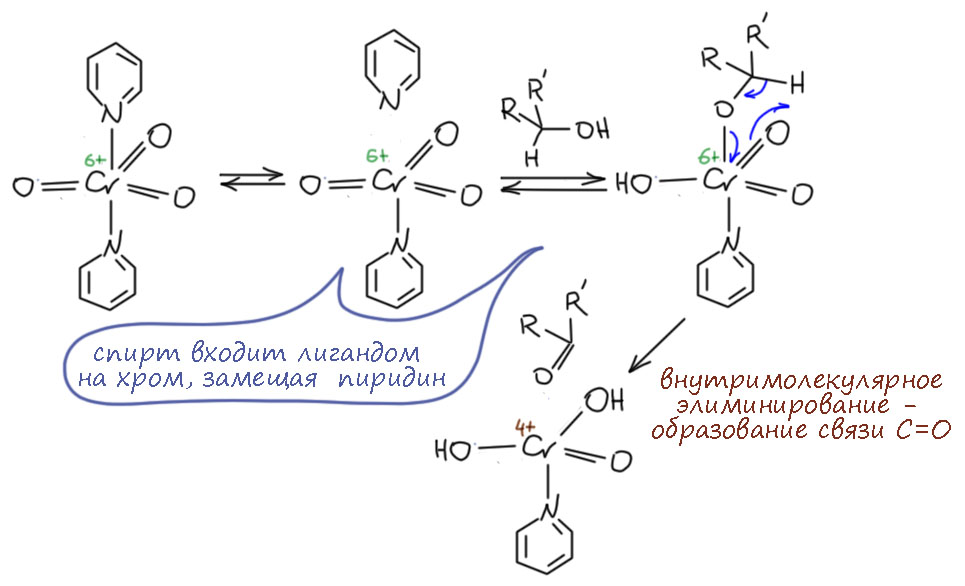
Дальше очень похоже на другие окисления хромом(6+), но рисуем всё как комплексы, как положено, но для органиков слегка непривычно. Поскольку реакция идёт в малополярном растворителе, дихлорметане, в котором оказался растворим исходный комлекс, просто потому что он симетричный, а значит в целом неполярный, и с двух сторон закрыт органической молекулой, пиридином, – а продукт реакции, некий комплекс хрома(4+) уже вполне чужой для малополярной среды – он несимметричный, полярный, из него торчат кислотные группы. Поэтому этот продукт восстановления начинает вываливаться из малополярной среды, скорее всего из-за координационной ненасыщенности (нарисованный комплекс имеет всег 12 валентных электронов) становится зародышем образования олигомерных комплексов, подхватывая и исходный комплекс тоже – поэтому все это хозяйство вываливается из реакционной смеси и выносит непрореагировавший окислитель.
Тем не менее, метод оказался очень удобным и в отличие от окисления в пиридине, неплохо и селективно окисляет спирты в альдегиды, и точно так же не трогает другие друппы кроме других высоконуклеофильных типа аминов. Двойные связи не трогает совсем, не только не окисляет, но и не двигает (такое бывает, если окисление идет в сильнокислотной среде, как в случае реактива Джонса и не только) и не изомеризует. Вот пример из оригинальной статьи Коллинза, Хесса и Франка
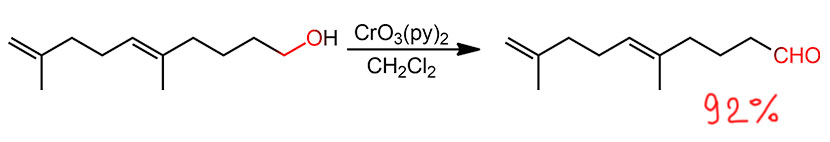
Хлорохромат пиридиния
Называемый также реагентом Кори, или для того чтобы хоть немного не повторяться в десятке других реагентов Кори, реагентом Кори-Сагса (Corey-Suggs reagent). Хромовые реагенты окисления спиртов забавны тем, что каждый очередной объявлялся окончательным решением, свободным от недостатков предыдущих (переокисления, опасности, сложностей хнатения, необходимости использовать избыток и т.д.), по проходило несколько лет и в реальной практике синтеза оказывалось, что недостатков всё ещё выше крыши и надо опять что-то искать. Все реагенты от Джонса до Коллинза предлагали химики, занятые исследовательским или практическим синтезом стероидов. Стероиды и правда были первой целью первых десятилетий органического синтеза – из них делали много лекарств и потребность в хороших синтезах и методах была огромной. К 1970-м синтез уже настолько развился, что появились гораздо более сложные цели, и потребность в высокоселективных реагентах стала совсем острой. За дело берутся уже настоящие мастера синтеза, которым нужно иметь под руками много инструментов синтеза, чтобы решать самые головоломные задачи. И вот на сцену выходит Кори, вице-бог синтеза, который в это время как раз решил поспорить с верховным богом синтеза Вудвардом, кто же первый придумал идею правил Вудварда-Гофмана: Кори уверял, что первая идея была его. Между этими спорами Кори в очередном синтезе сталкивается с окислением спирта в карбонил, пробует реагент Коллинза и удовольствия не получает. Сложно, выход далек от желаемого, да еще и избыток совершенно беспардонный. Кори поручает своему аспиранту Уильяму Сагсу поискать что-то получше, тот берется за дело и находит этот реагент, и заодно диссер доделывает. Все довольны. Реагентом оказывается вполне известное соединение, хлорохромат пиридиния, желтый кристаллический осадок, легко вываливающийся при добавлении пиридина к раствору хромового ангидрида в солянке. Соединение очень простое: с точки зрения обычной химии это полухлорангидрид хромовой кислоты в виде соли. С точки зрения координационной химии это комплекс хрома(6+). Напишем сначала как это обычно делают, не принимая во внимание законы координационной химии. а по аналогии с химией р-элементов: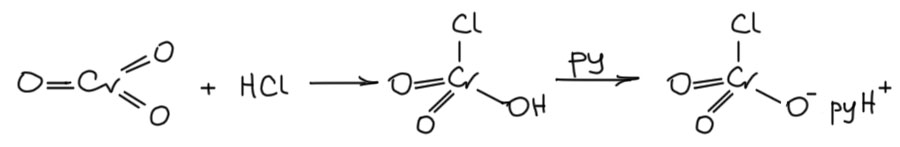
Но поскольку мы уже немного приобщились к координационной химии d-элементов, то видим проблемы в такой структуре. Она очень координационно ненасыщена – здесь те же 12 электронов как и в исходном триоксохроме(6+) – и зачем, спрашивается, было огород городить и хлорид присоединять? Обычно в химии металлов лиганд присоединяют, чтобы получить более насыщенную координационную сферу и пополнить счет валентных электронов. Лигандов стало 4, а электронов столько же? Конечно нет, ведь кислород с минусом это не что иное как ионная форма оксо-лиганда, и эту пару электронов легко тоже пустить на вторую координационную связь – это тоже оксо-лиганд. Тогда мы понимаем, что случилось: сильно ненасыщенный атом хрома в триоксохроме(6+) присоединил еще один лиганд, хлорид с образование комплекса хлоротриоксохромата(6+) – окончание -ат говорит нам о том, что на всём комплексе заряд -1, формально мы считаем его на хроме, в реальности он делокализован по лигандам, но в координационной химии это никак не обозначают кроме общего заряда (хотите за квадратными скобками, хотите от скобок оставьте маленький уголок в углу). Вот – это уже хром(6+) с 14 валентными электронами, как в хлористом хромиле (дихлородиоксохроме), как в хромовой кислоте (дигидроксидиоксохроме) – это в скобках названия вробе бы как по номенклатуре координационных соединений, просто чтобы почувствовать разницу, но увы. ИЮПАК в очередной раз пересмотрело эту номенклатуру (это называется Красной книгой – не поверите! – но ровно так и называется!! вот она в сети), и теперь хлористый хромил называется дихлоридодиоксидохром(VI), а хромовая кислота дигидроксидодиоксидохром(VI), хромат это тетраоксидохромат(VI)(2-). Вам, надеюсь, уже плохо? Это ИЮПАК, детки! Кто тут детки? Мы все детки перед этой могущественной организацией, мировым химическим правительством. Но номенклатура получилась настолько неуклюжая, что ей почти никто не пользуется. Чаще видим то, что было номенклатурой раньше. Раньше был оксо-лиганд, теперб оксидо. Раньше гидрокси, теперь гидроксидо. Вместо хлоро – хлоридо. Более-менее понятно. Важно понимать, как устроены комплексы, а название приложится. Итак, вернемя к хлорохромату и нарисуем его правильную, координационную формулу, и назовём по Красной книге: хлоридотриоксидохромат(VI)(-):
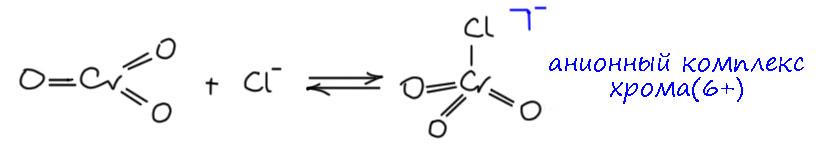
Эта соль с пиридинием получила название хлорохромат пиридиния (pyridinium chlorochromate, PCC) и оказалась весьма удачной формой хромового окислителя. Она растворима с том же дихлорметане, что и пиридиновый комплекс хромового ангидрида (по Красной книге: бис(пиридино)триоксидохром(VI)). Окисляет она немного медленнее, реакции идут от часа до нескольких чатов, и заканчиваются выпадением темно-коричневого липкого дерьмища. Более медленная реакция оказалась удачнее тем, что комплекс хрома быстро не выводится из реакции, как в случае метода Коллинза, поэтому шестикратный избыток не нужен, хотяя небольшой избыток (50% к соотношению 1:1, то есть хром воссанавливается в реакции до Cr(4+), а диспропорционирование и частичный возврат Cr(6+) как в Джонсе, не происходят и не учитываются. Полуторакратный избыток по сравнению с шестикратным это очень хорошо. Вы можете спросить – что им, хрома жалко? Во-первых, и жалко тоже, химия, особенно современная, не должна разбрасываться избытками почём зря – всё это отходы, а реакции окисления нередко используют в промышленно синтезе лекарственных субстанций с загрузкаи на десятки и сотни килограмм, и тогда задача куда-то деть полтонны вонючей хром-содержащей, а значит токсичной и канцерогенной дряни, может стать непреодолимой проблемой для производителя. Реагент Кори-Сагса, он же PCC точно так же, как реагент Коллинза окисляет вторичные спирты до кетонов, и первичные до альдегидов без переокисления до кислот. Механизм реакции будет очень похож – замещение более лабильного хлоридного лиганда на алкоксид и элиминирование, скорее всего тоже внутримолекулярное.
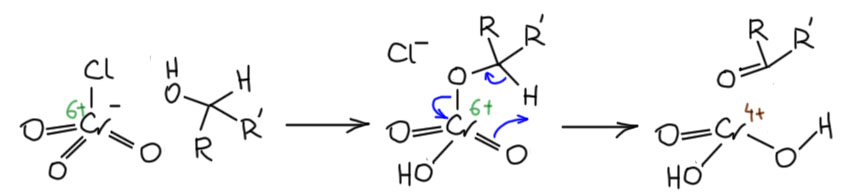
Образующийся комплекс хрома(4+) координационно ненасыщен и сильно – всего 10 валентных электронов, и это говорит о том, что с ним обязательно что-то произойдёт, чтобы хром хоть немного, но пополнил оболочку. Это что-то это обычно полимеризация – комплексы хватают друг друга за лиганды, которые становятся мостиковыми; а при такой ненасыщенности просто цепочкой дело вряд ли кончится, и мостиков образуется не один а два, то есть получится такой полимерный нерастворимый комплекс хрома(4+) с включением частично непрореагировавшего комплекса хрома(6+) – это и есть тот самый липкий тёмно-коричневый осадок, который после реакции положено тщательно отмывать растворителем типа эфира, чтобы дость оттуда частично залипший продукт, осадок при этом становится рыхлым и теряет липкость, что удобно – можно просто декантировать весь раствор, осадок тяжёлый и почти не взмучивается.
Получилась отличная методика, и селективная, и довольно экономная (Corey E. J., Suggs, J. W. Tetrahedron Letters. 1975, 16, 2647–2650). Стоит не забывать, что из хромных реагентов только Джонс используется даже иногда вообще стехиометрически в соотношении 2 к 3 (в расчете на полное восстановление хрома до трёхвалентного), но чаще эквимольно, а небольшой избыток по этой стехиометрии гасят в конце изопропанолом, что хорошо видно – полностью уходит желтый оттенок, и все становится чисто зеленым. Все остальные реагенты используют в кратных избытках (двух-трех-кратных Сарет, шестикратных Коллинз), так что полуторакратный избыток Кори-Сагса это, как бы сказал один президент США, хорошая сделка. Ещё один бонус – реагент очень легко получается, почти не гигроскопичен и поэтому отлично хранится, он не опасен. Поэтому он просто есть готовый в продаже для особо ленивых. Можно купить готовый.
Но у хлорохромата пиридиния все же есть одна проблема, которую иногда пытаются обратить на пользу, но чаще она доставляет хлопоты. В реакционной смеси возникает довольно большая кислотность из-за того, что остается ион пиридиния, а это сопряжённая кислота пиридина, приблизительно равная уксусной по силе. И ещё освобождается хлорид-ион, увеличивающий кислотность пиридиния за счет малополярной среды и поэтому хлорид очень хорошо вцепляется в протон на азоте пиридиния и существенно увеличивает его кислотность. Это такие фокусы малополярных сред, в которых нельзя пренебрегать взаимодействиями ионов, потому что в таких средах нет ионов, а есть ионные агрегаты. Результатом является то, что в процессе окисления возникает сущесвтенная кислотность, и если в субстрате есть что-то, чувствительное к кислоте, могут быть осложнения. Одно из таких – слетают некоторые слабые защитные группы типа тетрагидропиранильной, которую как назло, мы на 3-м курсе и используем как основную. Эту проблему предлагают решать, добавляя сухой ацетат натрия в реакцию окисления, чтобы снижать кислотность среды.
Но Кори не был бы самим собой, если бы вместо того, чтобы расстроиться из-за того, что и новый реагент иногда дает что-то не то, не задумал бы новый поворот темы. Главное отличие выдающегося синтетика от обычных химиков и состоит в том, что фантазия работает как у хорошего шахматиста – сделал ход (попробовал окислить какой-то спирт новым реагентом) – увидел неожиданный ответ противника (продукт окисления оказался не тем) – сразу задумал продолжение, использующее это осложнение к своей выгоде и новым синтезам. Раз этот реагент вызывает осложнения, связанные с кислотно-катализируемыми побочными реакциями, значит сделаем эти побочные реакции основными и выкатим новый изящный синтез. Что там катализируют кислоты? Например, внутримолекулярные циклизации карбокатионов на кратные связи. Это происходит весьма легко, если в этом процессе может образоваться стабилизированный карбокатион, третичный. И Кори выкатывает синтез терпена пулегона. Пулегон содержится в эфирных маслах разных видов мяты: такие минорные компоненты природных ароматов придают им более сложный букет запахов, чем можно получить, заменив природное масло чистым главным компонентом этого запаха, ментолом. Даже интереснее, синтез Кори исходил из чистого энантиомера другого хорошо известного терпена, левовращающего цитронеллола, содержащегося в эфирных маслах розы, герани и множества других ароматных растений, и легкодоступного буквально тоннами. Из такого энантиомера получился (S)-пулегон, которого нет в Природе: вот и здорово – можно было понюхать и убедиться, что у энантиомеров разные запахи. Пулегон вообще штука не очень приятная. Его довольно долго производили синтетический и использовали как компонент ароматических смесей. Но если мы посмотрим на эту молекулу, увидим, что это по структуре сопряженный ненасыщенный кетон, то есть по свойствам – акцептор Михаэля, а все акцепторы Михаэля веществас сильной биологической активностью, часто канцерогенной, потмоу что в клетках вступают в присоединение по Михаэлю с нуклеофильными группами нуклеиновых кислот и белков, изменяя их функции. Пулегон в этом смысле по структуре очень похож на такие заведомо неприятные соединения как окись мезитила и форон. И когда однажды изучили его токсикологию, обнаружили то, что и ожидалось – это плохое вещество, не стоит им дышать. В США синтетический пулегон был запрещён к использованию в качестве ароматизатора для всего, что непосредсвено попадает в организм, например, в вейпах. Но мятные масла, где пулегон есть, но в очень малых количествах, не запрещены, потому что уровни концентраций почти безопасны. Впрочем, если у вас есть дурацкая привычка целыми днями парить мятным вейпом, знайте, что риск есть, и не так мал.
И вот синтез Кори и коллег (E. J. Corey, H. E. Ensley, J. W. Suggs, J. Org. Chem., 1976, 41, 380). Он фактически одностадийный – спирт окисляется в альдегид, дальше обратимое протонирование, и атака электрофильного оксокарбениевого иона на двойную связь так, что замыкается шестичленный цикл и образуется третичный карбениевый ион. Дальше E1-элиминирование, которое даёт не более замещённый олефин, а менее стерически затруднённый, который оказывается устойчивее более замещённого – что нарушает букву, но не дух правила Зайцева. Такое направление элиминирвоания почти всегда наблюдается в системах, где есть выбор между дву- и тризамещённым олефином. Поэтому получился не пулегон, а изопулегон, правда неизветстно, какой диастереомер. Транс-изопулегон применяют вместо пулегона как ароматизатор – запах похож, но поскольку это не акцептор Михаэля, то и токсичности нет. Но Кори всё же хотел именно пулегон, и просто перегруппировал двойную связь действием основания в сопряжённое положение – можете сами потренироваться в механизме через енолят.
Очень быстро этой особенности хлорохромата пиридиния нашли весьма умное применение. Мы хорошо знаем, что есть такая интересная, но неприятная вещь – несимметричные аллильные системы, мы видими их в самых разных разделах – и в электрофильных, и в нуклеофильных, и в свободнорадикальных реакциях – и везде у нас проблема с тем, что могут образоваться два разных продукта с разными положениями двойных связей (аллильный сдвиг), а закономерности запутаны и непонятны – где-то стерика, где-то растпределение зарядов или электрона, где-то вообще какая-то смесь. Поэтому на 3 курсе мы всячески избегаем таких примеров, да и в настоящей исследовательской химии и в синтезе, к таким реакциям относятся настороженно: есть много частных случаев, когда можно сделать разумные предсказания, но в общем случае нарваться на ненужный продукт или вообще смесь. Тем не менее, всегда, когда аллильные сдвиги можно сделать управляемыми и предсказуемыми, это всегда отличные новости и превосходный метод синтеза. С окислением хлорохроматом получилась такая история. Если взять третичный спирт аллильного типа, то в присутствии кислот возникает равновесие перегруппировки, в котором присутствуют оба спирта, исходный и перегруппированный. Но поскольку в смеси есть хлорохромат, который не может окислить исходный третичный спирт, но зато может окислить второй, если он оказывается первичным или вторичным. Это окисление выводит конкретный продукт из равновесия, и смещает весь процесс в сторону одного конкретного продукта. Замечу, что этот процесс чаще рисуют иначе – сначала получается хромат третичного спирта, который перегруппировывается в перегруппированный хромат, который уже и даёт продукт окисления. Я не видел доказательств именно такого механизма и не считаю, что это само собой разумеется, а в таких случаях всегда стоит предпочесть более постой и более общий механизм, что я и сделал. Будут доказательства образвоания третичного хромата и сигматропной перегруппировки со сдвигом именно хромата, тогда посмотрим, а пока обойдёмся достаточно очевидной схемой. Здесь только один вопрос – достаточно ли кислотности в растворе хлорохромата пиридиния, чтобы генерировать карбокатион из исходного спирта. Скорее всего да, это третичный, стабилизированный карбокатион, и есть множество примеров карбокатионных реакций, инициируемых протонированным пиридином в разных растворителях, в том числе малополярных, так что противопоказаний нет.
Эту возможность практически одновременно заметитили и реализовали два американских химика, и ранее проявлявших интерес к аллильным перегруппировкам – Бейблер и Даубен, второй немного позже, но с гораздо лучше проработанной статьёй в отличном журнале и с большим количеством примеров (Dauben W. G., Michno D. M. J. Org. Chem. 1977, 42, 682–685). А первый, наоборот, с очень короткой и сырой статьёй да ещё и в журнале с неоднозначной репутацией места, куда сливают недоделанные работы (Babler J. H., Coghlan M. J. Synth. Commun. 1976, 6, 469–474). Метод поэтому называют и окислением по Бейблеру и по Бейблеру-Даубену. Метод получился отличным, – кому не понравится аккуратная перестановка двойной связи с получением еналя или енона, с которыми дальше можно делать всё, что угодно, мы умеем, а уж серьёзные синтетики и подавно. Даубен это так и прозвал, цветасто и немного завирально, внутримолекулярной транспозицией карбонила (Intramolecular Alkylative-1,3-Carbonyl Transposition), причем развил это в даже еще более интересную методологию, когда исходный третичный аллиловый спирт получают реакцией виниллитиев с карбонильными соединениями. В общем виде получается так:
![]()
Изящно, не правда ли? Ещё симпатичнее получилось, когда к обычному кетону присоединяли винилмагнийбромид (реакцтив Нормана, напомню, что из винилгалогенидов магнийорганику делают не в эфире, а в ТГФ), и при действии хлорохромата пиридиния на получающийся третичный аллиловый спирт образуются чрезвычайно ценные в синтезе непредельные альдегиды (сопряжённые енали). Единственная проблема – если возможно образование диастереомеров по двойной связи, то и получаются их смеси, поэтому лучше всего дело идёт с симметричными, например, циклическими кетонами.
![]()
С этим реагентом случаются и другие фокусы, связанные с его кислотным характером, поэтому не надо удивляться иногда необычными продуктам, ну и если сами будете применять, имейте это в виду, и не рискуйте неожиданными перегруппировками и циклизациями, ну, или наоборот, пытайтесь это как-то применить. Реагент этот оказался последним крупным развитием темы хромовых окислителей спиртов; после было немало модификаций типа нанесённых на всё, что под руки попалось, но ничего столь же общего и полезного. Реагент до сих пор довольно популярен в синтезе.
В конце упомянем ещё один хромовый реагент – дихромат пиридиния, PDC. Эту штуку тоже выкатил Кори, поэтому метод иногда называют окислением по Кори-Шмидту (Corey, E. J.; Schmidt, G. Tetrahedron Lett., 1979, 20, 399-402). Не исключено, что однажды наигравшись с хлорохроматом пиридиния, кто-то из постдоков Кори неплотно закрыл баночку с реагентом, тот нахватал влаги и превратидся аккурат в дихромат пиридиния. Однажды баночку открыли чтобы сделать новый синтез, с удивлением обнаружили, что вместо жёлтого порошка там оранжевые кристаллы, вполне симпатичные. И использовали их, а потом разобрались. В то же самое превращается и комплекс хромового ангидрида с пиридином во влажном воздухе. И если не заниматься утилизацией прокисших реактивов, то PDC можно очень легко получить – он прямо вываливается из раствора двуххромовой кислоты при нейтрализации пиридином. Из этого следует одно великолепное качество этого реагента – он не гигроскопичен, удобно хранится (но никогда не делайте такие вещи впрок сотнями грамм или больше – случайный перегрев банки, а всякое бывает, может вызвать бурное разложение). Используют реагент в виде суспензии в дихлорметане, и в этом виде он удивительно неплохо работает и для первичных и для вторичных спиртов, никого не переокисляет, и поскольку он намного менее кислотен, чем PCC, он обычно не вызывает побочных реакций. А разве и там, и там кислотность не связана с ионом пиридиния? Это же что-то типа уксусной кислоты по кислотности? В PDC именно так, а вот в PCC кислотность связана с образующимся прямо на месте HCl, а это намного более сильная кислота, чем пиридиний. Окисления PDC проводят очень похоже на реакции с PCC, но они обычно намного медленнее. Первичные спирты окисляются до альдегидов, но если раствоитель не очень сухой, или реагент старый и нахватался немного влаги, часто бывают переокисления. В целом, PDC далеко до популярности PCC, его чаще применяют в простых случаях с вторичными спиртами, когда нужно много продукта и экономия на реагента становится существенным фактором.
Диоксид марганца
Перманганаты калия, натрия, и т.п. слишком сильные и неселективные окислители, имеющие сразу несколько механизмов окисления спиртов, альдегидов, некоторых C-H связей в алкилах, двойных и тройных связей, аминов, сернистых соединений и т.п. – проще сказать, что перманганат не может окислить. В общем, он может окислить всё, даже алканы, вот только ничего конкретного в этих реакциях не получится, потому что все промежуточные точно так же окисляются, и окисление закончится только когда исчерпается окислитель, и в совершенно произвольном месте. Поэтому с перманганатом шутки плохи, хотя есть немало специальных методик, которые позволяют в разных специяических случаях добиться от перманганата вполне предсказуемого поведения и хороших результатов. Но там так много чисто препаративных нюансов, что разбираться в этом можно только если очень нужно.
Перманганат может окислять точно так же как хромат – через образование эфира кислоты HMnO4 (мы предпочитаем смотреть на все производные переходных металлов как на комплексы, и тогда это по старой версии номенклатуры гидрокситриоксомарганец(VII), а по новой Красной книге гидроксидотриоксидомарганец(VII)) и элиминирование, через свободнорадикальный механизм, тоже похожий на то, что мы видели с реактивом Этара и действие двуххромовой кислоты в жестких условиях. Чрезвычайная легкость включения двух типов механизма делает перманганаты совсем трудноуправляемыми окислителями. Поэтому смысла в использовании перманганата почти нет, кроме самых простых случаев окисления первичных спиртов в кислоты. Вторичные спирты тоже лучше не окислять, потому что кетоны также легко окисляются дальше в том случае, если они енолизуемы хотя бы в одну сторону. Причина такой сильной окислительной способности нам тоже хорошо понятна – марганец это последний элемент первого ряда переходных металлов, имеющий высшую степень окисления, и это валентное состояние уже настолько сильно дестабилизировано, что ищет все способы дать металлу хоть немного своих электронов (оксо-стабилизация и здесь работает, ион перманганата, а по номенклатуре тетраоксоманганата(VII)(-) или тетраоксидоманганата(VII)(-), это более стабильный комплекс, чем сама марганцевая кислота или её эфиры, но и он в растворах разлагается, медленно окисляя воду).
Для окисления определенного типа спиртов оказалось очень полезно другое соединение марганца – диоксид марганца, но не любой, а специально приготовленный, свежий. То, что двуокись марганца, самая обычная, из банки окисляет некоторые спирты было открыто в одной исторической работе ливерпульским химиком Ричардом Мортоном, одним из первых исследователей, кто стал применять спектроскопию в органической химии, а первой практической спектроскопией стала спектроскопия в видимом и ультрафиолетовом свете (ИК была открыта даже раньше, и даже неплохо исследована, но практически она сложнее и применять ее стали немного позже), и он же сделал очень важное открытие, связав витамин A1 (он же ретинол) и вещество, которое участвует в том, как у нас и не только у нас устроено зрение, которое после открытия назвали ретиненом потому что в нём есть ненасыщенность. Мортон и его сотрудники обнаружили, как раз наблюдая изменения в спектре поглощения, что ретинол в растворе над осадком диоксида марганца медлено превращается в ретинен, и несложные умозаключения на основании элементного анализа (это 1946 год, Англия после войны тоже была изрядно истерзана, и исследования еще были очень экономны) показали, что ретинен это альдегид от ретинола, поэтому его переименовали в ретиналь, и этим названием мы пользуемся с тех пор. Реакция эта сама себя выдаёт. У исходного ретинола всего пять двойных связей в сопряжении, а мы знаем, что сопряжение смещает полосу поглощения в сторону видимой области света, но понемногу, и пяти сопряженных связей хватает только для того, чтобы максимум поглощения (325 нм) приблизился к границе видимой области (считается, что это около 340 нм). Поскольку полосы поглощения почти всегда очень широкие, то она немного заезжает спадающим участком в видимую область – и это всегда воспринимается, как жёлтый цвет, но только в достаточно концентрированном расторе или в кристаллах – в этм несложно убедиться, посмотрев на разные формы витамина А, которых полно в продаже в аптеках. Окисление создаёт альдегидную группу, которая активно встраивается в сопряжение, тем лучше, что это откровенный акцептор, а сопряжение всегда сильнее, когда хотя бы на одном конце цепи сопряжения мезомерный донор или акцептор. Результат – сильное смещени полосы поглощения в видимую область, максимум (380 нм) уже хорошо заехал в синюю область спектра, а поскольку мы видим результат поглощения, то есть то, что осталось, мы видим белый свет минус синий, а это нечто, в зависимости от конкретной формы полосы и концентрации, оранжевое или даже красное: в растворе инертных растворителей ретиналь оранжевый, – то есть Мортон и его сотрудники прямо и увидели, как исходный жёлтый раствор становится оранжевым. Выход их тогда не интересовал, поэтому они не стали исследовать как сделать двуокись марганца более активной и получить высокую степень окисления исходного спирта. Мы же без труда увидим в ретиноле аллиловый тип структуры спирта.
Для больших любителей именных реакций, окислени диоксидом марганца иногда (крайне редко) называют окислением по Боллу-Гудвину-Мортону в порядке аторов в статье, где это было описано (Ball, S.; Goodwin, T. V., Morton, R. A., Biochem. J., 1948, 42, 516). Это не совсем справедливо, потому что никакого метода окисления в статье описано не было, она не про это.
Полезным реагентом это вещество стало только после аккуратного исследования Аттенборо и сотрудников, установивших удобную воспроизводимую процедуру приготовления заведомо активной формы (Attenburrow, J.; Cameron, A. F. B.; Chapman, J. H.; Evans, R. M.; Hems, B. A.; Jansen, A. B. A.; Walker, T.; J. Chem. Soc. 1952, 1094). Это простая реакция сульфата марганца(II) и перманганата в стехиометрическом соотношении в щелочной среде, но делать ее нужно строго по методике этих химиков, а это, как говорят на только что скончавшемся английском языке, a tricky busyness – растворы сульфата марганца и крепкой щёлочи одновременно прикапывают к раствору перманганата из двух капельных воронок. После реакции коричневый осадок фильтруют, хорошо моют водой до бесцветного смыва, и сушат в сушильном шкафу при нагревании, растирают и порошком это можно хранить, но недолго – активность всё время падает, даже в холодильнике. Недели точно можно, но через год это уже просто обычная коричневая бесполезная дрянь. Вроде бы такой порошок можно активировать, покипятив с разбавленной азоткой, и опять высушив, но проще сделать новый. Все, кто хоть раз с этим окислителем связывался в реакциях с не мизерной загрузкой, обыно потом согласны руками отдирать водороды от каждой молекулы спирта, лишь бы больше не делать ничего с активной двуокисью марганца. Фильтровать ее после реакции мучение, она мелкая, забивает фильтры (здесь здорово помогает порошок неактивного кремнезёма, Целит, Celite – если в вашей лаборатории нет этой замечательной вещи, обязательно заведите, сэкономит много времени на трудных фильтрованиях), всё коричневое, колбы, фильтры, стол, стены, руки, лица. Правда этот коричневый цвет довольно легко отмывается влажной пастой щавелевой кислоты. И некоторыми усилиями можно сделать так, что коричневая дрянь останется в пределах посуды и наружу не полезет – раза с десятого это начинает удаваться. В общем, противная вещь. Но совершенно незаменимая в синтезе, потмоу чо обладает исключительной избирательностью – окисляет только спирты аллильного, пропаргильного и бензильного типов до карбонильных соединений (альдегиды дальше не окисляет), и больше не трогает практически ничего за очень-очень редкими исключениями. Ничего альтернативного в органике так и не придумади, а задачи такие в сложных синтезах встречаются нередко. Вот пример из оригинального синтеза природного соединения каллистатина, и это уже одна из последних стадий, где рисковать нельзя (Lautens M., Stammers T. A. Synthesis 2002, 14, 1993.). Окисляется только спирт аллильного типа, а вторая спиртовая группа сохраняется – там двойная связь на один атом углерода дальше (это называется гомоаллильный спирт). На следующей стадии вторую спиртовую группу окислят реагентом Десса-Мартина (разберём дальше), снимут защиту с третьей и искомый продукт готов.
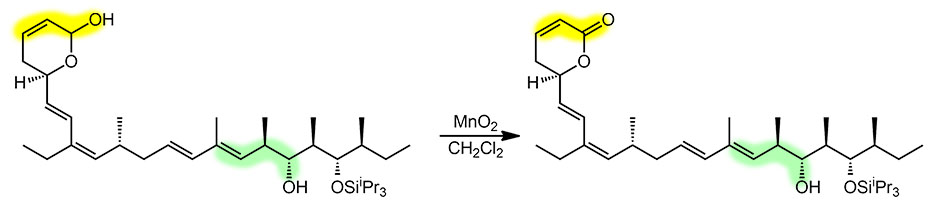
Итак, окисляются только аллильные, пропаргильные и бензильные спирты, но при этом допустимы самые разные заместители и близко и далеко от спиртовой группы. Под “бензильными” спиртами и здесь и вообще в органике часто понимают любые спирты с гидроксильной группой на первом углероде боковой цепи ароматического соединения, не обязательно бензольного ряда, но и, например, любого гетероциклического (фурана, пиррола, индола, пиридина и т.п.) – такие спирты часто отлично окисляются диоксидом марганца, и из-за проблем с теми же гетероциклическими группами этот окислитель вообще может оказаться единственно возможным.
Немало примеров довольно сильно замещённых спиртов, то есть кажется, что этот окислитель не сильно зависит от стерических препятствий (такие выводы всегда относительны, потому что стерика – вещь страшная, и если что-то не блокирует метил или этил, то вставьте на их место изопропил или трет-бутил, и узнаете, что такое стерика). И никого поэтому не останавливает ещё одна совсем неприятная особенность этого окислителя – его применяют даже не в больших (как реагент Коллинза, например, напомню, что там избытки 6-тикратные), а в очень больших, огромных, и неприлично колоссальных избытках – от 15-20-кратных (это редко) к 40-60-ти кратным (это обычно), но можно увидеть и ста-стапятидесятикратные избытки. Дело спасает то, что чаще всего этот реагент используют в очень сложных синтезах с миллимолярными и микромолярными количествами, так чо даже стократный избыток это всё равно граммы. Но если вам когда-нибудь придется окислять двуокисью марганца что-то в количествах хотя бы десятков миллимолей, что типично для препаративной работы в органике, то диоксида марганда уже пойдут десятки грамм, а это лёгкий и объёмистый порошок, и вот тут уже мало не покажется, и проклянёте тот день, когда узнали про этот реагент, и если есть хотя бы малейшая возможность обойтись без него (а если у вас в молекуле нет разных гидроксильных групп, а есть только одна аллильная или бензильная, то с задачей справится пости любой хромовый реагент, впрочем, не забывайте, что хлорохромат пиридиния иногда устраивает фокусы с некоторыми – третичными – аллильными спиртами), то к чёрту диоксид марганца. И никогда не начинайте работу с диоксидом марганца, если у вас нет под руками щавелевой кислоты.
Как окисляет диоксид марганца в общем непонятно, потому то изучать реакции, которые идут на поверхности очень трудно, но стократ труднее делать это с твердым материалом с неизвестной структурой. А что за проблема со структурой – наверняка структура диоксида марганца известна во всех деталях? Это верно, но есть очень серьезный нюанс – под структурой твердого тела имеют в виду то, что внутри кристалла, где есть решетка, и там и правда всё ясно. Здесь мы задаем первый вопрос – а диоксид марганца случайно не аморфный – с аморфными веществами всё совсем плохо, у них нет определённой структруры даже внутри. Нет, слава всем подходящим богам, диоксид марганца внутри кристаллический. Но работает поверхность. С поверхностью всё тоже могло бы быть нормально – у хороших кристаллов вполне определенная поверхность, с которой можно работать – там вполне определенные грани кристалла, на которых понятно что происходит. Увы, таких аккуратных кристаллов в реальной химии почти нет, а гораздо чаще мы имеем дело с сильно дефектными материалами, и если внутри кристаллическое вещество находит способы создать порядок даже при не самых идеальных условиях получения, то на поверхности царит страшный бардак. Большинство решеток оксидов металлов, кроме самых простых щелочных и щелочноземельных это бесконечные трехмерные структуры – фактически это просто полимерные комплексы, где каждый атом металла имеет свою координационную оболочку, состоящую из оксо (теперь оксидо) лигандов, часть из которых работает как мостики к следующему такому атому металлу, и так и строится трехмерная упорядоченная решетка. На поверхность все это хозяйство выныривает и тут выясняется, что для тех оксо-лигандов, котрым надо было бы стать очередными мостиками, не хватило следующего атома металла, мостик повисает в воздухе – и, скорее всего, подхватывает атом водорода, из оксо- (оксидо-) лиганда превращаясь в гидроксо (гидроксидо) лиганд. Фактически это означает, что на поверхности диоксид марганца гидратирован (протоны-то откуда?), и дальше мы еще понимаем, что поверхность таких кристалликов, которые растут быстро, как придется, это не такие гладкие плоскости, время от времени красиво пересекающиеся с другими красивыми плоскостями, образуя роскошные грани правильного кристалл – глаз не отвести – а полный хаос сдвигов, трещин, разрывов, даже дырок (пор), и там везде поверхность, увы, сильно напоминающее то, что мы нынче часто видим из мест, где много месяцев рвутся бомбы, ракеты и прочие мерзкие штуки, которые положено называть стыдливым эвфемизмом “различные средства вооружённой борьбы”. А когда речь идёт об особой, активной форме того же диоксида марганца, мы понимаем, что сами условия осаждения подобраны так, чтобы сделать поверхность максимально такой нерегулярной, сильно гидратированной, причем поскольку это вода связанная на поверхности (по другому мы можем сказать, что на поверхности оксид частично переходит в гидроксид). Более того, мы даже точно не знаем, нет ли на поверхности центров с другими степенями окисления, например, Mn(5+) – ведь реакцию ведут в сильно-окислительных условиях из перманганата, а иногда рекомендуют дополнительно активировать препарат диоксида марганца кипячением с разбавленной азоткой. Иногда бывали даже идеи, что ровно такие центры на поверхности, с пяти или даже шестивалентным марганцем, и отвечают за особые окислительные свойства активной двуокиси марганца, которую мы и применяем для окисления спиртов. Возможно, так и есть, но доказано это не было, хотя ничего необычного в этом нет, ведь сам окислитель применяется в огромных избытках, так что даже один процент таких центров (они на поверхности, а процент берется по общему содержанию марганца в реагенте) вполне мог бы справиться с окислением.
Что можно более-менее разумно сказать о механизме, что позволило бы принять некоторую правдоподобную гипотезу? Во-первых, это практически наверняка свободнорадикальная реакция, что и определяет избирательность к аллильным, пропаргильным и бензильным спиртам – и это значит, что первым отщепляется атом водорода под гидроксильной группой, чтобы как раз и образовать радикалы такого типа. Было бы неплохо, если бы кто-то озадачился определением изотопного эффекта по дейтерию в этом положении. О, есть такой! Вполне известный химик из фармацевтической компании Пфайзер, Ирвинг Голдман (1931-2021), очередной потомок эмигрантов из Украины и Российской империи, сделал очень обстоятельную работу по мезанизму окисления двуокисью марганца, которую можно вполне взять за основу, немного поправив выводы под более современное представление о свойствах соединений металлов (Goldman, I. M.; J. Org. Chem. 1969, 34, 3289). Никто с тех пор ничего лучше не сделал. Вот он как раз и измерил изотопный эффект. Эффект оказался не просто большим, а совсем неприлично большим – такое иногда бывает, хотя мы не можем исклчать, что в 1969 году исследователя немного подвела точность измерений. Но может и нет, статья очень серьёзная, результаты много раз перепроверены и доведены до статистически достоверной точности. Аномально большие изотопные эффекты иногда действительно бывают, это тема для отдельного обсуждения. Величина изотопного эффекта у Голдмана достигает 14 (напомню, что это отношение скорости реакций по протию к скорости реакции по дейтерию), так что, например, можно считать вполне препаративным окисление монодейтерированного бензилового спирта в дейтерированный бензальдегид, чистота будет более 90%. Степень обогащения продукта дейтерием настолько высока, что можно вполне использовать эту двухстадийную методику для препаративного получения дейтерированных альдегидов.
Дальше мы немного домыслим рассуждения Голдмана в нашем стиле. Диоксид марганца это комплекс Mn(IV), но, увы, мы не можем использовать мономерный комплекс, так как имеем дело с поверхностью, и должны смотреть на отдельные марганцевые узлы в кристаллической решетке, рассматривая ее как трехмерный полимерный комплекс. Внутри решётки, это известно, посмотрите любую большую книжку по структурной химии, марганец имеет координационное число шесть, и все шесть атомов кислорода мостиковые, соединяют с соседними марганцевыми узлами. Степень окисления 4+, своих электронов у металла 7-й группы остается 7-4 = 3, нечётное число, значит у металла есть неспаренные электроны в любой конфигурации (когда число четное, может бть и так, и так, но с нечетным вопросов нет). Внутри решетки на металле 6 кислородных лигандов, итого число валентных электронов 3+6*2 = 15, мало, металл координационно ненасыщен, но внутри это никого не волнует, туда все равно не попадешь, да и и с металлами первого ряда, имеющими небольшие ковалентные радиусы, очень часто бывает так, что просто места нет в координационной сфере для дополнительных лигандов. Но на поверхность мы вылезаем с заведомо еще менее полной координационной сферой, хотя она может быть и гидратирована. Мы точно этого не знаем, но гипотезу разумную построить можем. Октаэдру на поверхность проще всего вылезти так, что три лиганда останутся в глубине и будут держать атом. В этом месте ещё немного подумаем. От Mn(4+) часто в учебниках производят гидроксид, который могут нарисовать и с четырьмя гидроксилами Mn(OH)4, и в частично дегидратированной форме как будто как нечто изоэлектронное сернистой кислоте H2MnO3 или Mn(O)(OH)2. Мы понимаем, что это нереалистичные формулы и таких молекул мы в реальной жизни не увидим. Повторю почему. Марганец – переходный металл, а соединения переходных металлов это комплексы, для которых очень важны закономерности d-элементов. В частности, не должно быть высокой степени координационной ненасыщенности в комплексах с простыми маленькими лигандами. Если лиганды большие и сложные, они могут заставить металл сохранять высокую координационную ненасыщенность, просто создавая стерические препятствия другим возможным лигандам. Но если простые, то металл будет иметь возможность связать больше лигандов, и они всегда найдутся, как минимум в виде второго такого же комплекса, с которым можно поделить лиганд, который станет мостиком. Вот, у гидроксидов марганца число валентных электронов: своих три и восемь от лигандов (по паре от гидроксида, по 4 от оксо) – итого 11. Это очень мало, и это значит, что металл будет стараться подцепить ещё, до октаэдра, то есть всего щести. Почему не больше? Потому что во-первых, координаационная сфера не резиновая, и шесть шариков хорошо заполняют пространство вокруг центрального шарика, особено если он не очень большой: внизу Таблицы у элементов может юыть координационное число и семь, и восемь, и с особо мелкими лигандами даже 9. Во-вторых, связывание лигандов идет через орбитали валентных электронов, которые имеют направление в пространстве, и как раз очень хорошо соответствуют октаэдру. Итак, когда гидроксиды марганца(4+) образуются, а они обязаны это делать в обычных реакциях, то будучи координационно ненасыщенными, они просто начинают собираться во все более крупные кластеры – олигомеризоваться – и так в конце концом и получается частичка диоксида марганца. Поскольку мостикам проще быть оксо (оксидо) чем гидроксидо, то внутри частички все связи будут через такие оксидо-мостики, а гидрокси-группы останутся только на поверхности. Там же могут быть и оксо-группы, и вообще одни в другие очень легко превращаются просто как кислота и сопряжённое основание.
Теперь подберемся к окислению спиртов. Спирт как нуклеофил является донорным лигандом ничем не хуже воды или гидроксида, а на самом деле и лучше, потому что более нуклеофилен, а металлы точно так же это чувствуют, как и субстраты в SN2 замещении. Логично заместить спиртовой лиганд, то есть алкоксид, на то же место где был гидроксид у металла. В комплексах металлов обычно легко проходят реакции замещения лигандов, особенно если новый лиганд чем-то лучше старого. Давайте сократим схему, только символически показав не участвующие в деле лиганды, обеспечивающие удержание марганцевого комплекса в частичке.
Теперь осталось совсем немного – изобразить отрыв атома водорода от углерода под спиртовой группой. Мы уже делали нечто подобное, когда разбирались с реакцией Этара и вообще жёстким окислением хромовыми реагентами, когда включается свободнорадикальный путь. Работает оксо-лиганд и сам металл, получающий один электрон с лиганда и восстанавливающийся до Mn(3+). Образующийся радикал стабилизирован аллильным, пропаргилным или бензильным сопряжением. Очевидно, что такие марганцевые комплексы на поверхности оксида могут оторвать атомы водорода только от самых слабых положение, а это, как мы знаем из свободнорадикальной химии, прямо связано со стабилизацией образующегося радикала. Поэтому реакция очень селективна.
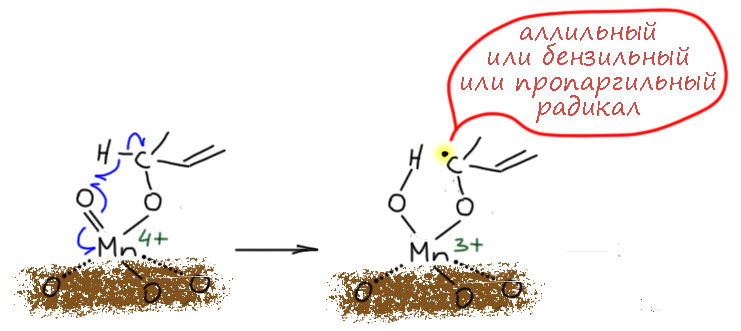
Образовавшийся радикал остается в координационной сфере, а марганец в состоянии Mn(3+) продолжает оставаться окислителем и не против забрать еще один электрон, чтобы достичь устойчивой степени окисления Mn(2+). Это очень просто делается очень распространёной в радикальной химии реакцией
элиминирования – со второго атома рядом с радикальным центром отваливает другой радикал, а из оставшихся электронов сооружается двойная связь.
А где тут радикал? А вот марганец в состоянии 2+ имеет семь минус два – пять своих электронов, нечётное число, один точно неспаренный (могут быть и все пять в высокоспиновом состоянии, но нам нужен один, а с остальными пусть разбираются неорганики).
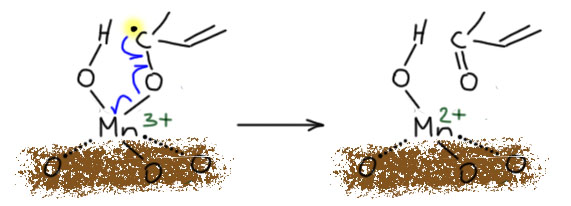
Всё, окисление состоялось, слезаем с поверхности. На поверхности остается уже никого не способный окислить комплекс Mn(2+). Когда поверхность частичек в основном будет состоять из таких центров, окисление замедлится и остановится, потому что Mn(4+) под поверхностью в реакции не участвует – это и объясняет необходимость огромных избытков. Способ приготовления активной двуокиси марганца дает совершенно случайные размеры частичек и совершенно разную площадь поверхности, поэтому так сильно различаются эти избытки в разных работах от 10-15-кратного до 100-150-кратного: понятно, что берут сколько-то, крутят, делают ТСХ и если реакция сильно неполная и пятно исходного велико, добавляют ещё окислителя.
Такой вот окислитель. Очень неудобный, поэтому если нужно просто окислить аллильный, пропаргильный или бензильный спирт, и других спиртовых групп нет, предпочтут другие окислители – хлорохромат пиридия, методы Моффата-Сверна, периодинан Десса-Мартина. Но если есть проблема хемоселективности, диоксид марганца был и остается незаменимым окислителем.
Окисление активированным ДМСО
Хромовых реагентов много, они решают много задач в синтезе и пользуются популярностью как проверенные, чуть не написал, веками, но даже самому старому именному реагенту Джонса всего 70 лет. Тем не менее, хромовые реагенты вызывают очень много вопросов и у тех, кому нужен синтез в больших количествах какой-нибудь субстанции для лекарств; и у тех, кому нужно разматывать длиннющие цепочки стадий, приближающих к цели, какому-нибудь природному соединению, где каждый неверный шаг перечеркивает уже затраченные усилия. Хромовые реагенты довольно селективны, и есть выбор между теми, кто использует основные условия, и теми, кто использует кислотность, но увы, ни то, ни другое иногда бывают неприемлемы даже в мягком варианте. Самое противное в хромовых реагентах – почти всегда избытки, и образование большого количества часто очень неприятных, густых и липких осадков, увлекающих в себя часть продукта – их нужно тщательно промывать после реакции, иначе потери могут быть огромными. А в промышленности, где работают с сотнями килограмм и тоннами, это крайне неприятные отходы, которые воообще невозможно нормально утилизовать и приходится нанимать мафию для того, чтобы избавиться от ржавых бочек теми же методами, которыми избавляются от трупов жертв преступлений.
Поэтому поиск других окислителей всегда приветствуется. Иногда мы видим неожиданные реагенты и спрашиваем, как вообще в голову могло прийти и что в этом может быть хорошего? Вам что хромового ангидрида мало? Ну да, не просто мало, а глаза бы его не видели, проклятого. Что у вас там? Диметилсульфоксид, превращающийся в конце в невероятно вонючий диметилсульфид? Ничего, тяги хорошие, давайте посмотрим, как сами реакции устроены, нет ли там преимуществ, но уже даже одна перспектива больше не перетирать с горячим ацетоном коричнево-зелёную тянучку, в которой к концу реакции уже намертво залипло пять мешалок одна за другой, наполняет сердце химика почти экстатическим вожделением.
На уходящем диметилсульфиде основано даже больше методов, чем на шестивалентном хроме. Часто их всех объединяют под зонтичным брендом “окисление по Сверну”, но это очень несправедливо и совершенно неверно; я могу это объяснить только странной привлекательнотью необычной фамилии Сверн, почему-то на ум приходят всякие сочинения мировой литературы или что-то около этого – гибрид Свифта с Жюль-Верном, а может у Марселя Пруста кто-то такой был, а нет, там был Сван, ну ещё был английский художник Северн, который почему-то затесался в память как поэт, скорее всего потому, что лежит на некатолическом кладбище в Риме рядом с Китсом.
А все такие методы используют одну и ту же реакцию, и просто варьируют прелюдию – как затащить спирт в виде алкоксидной группы на диметилфульфоний, и как только это получилось, такой интермедиат очень легко вступает в точно такое же элиминирование, как то, что мы видели у хромовых окислений – отщепляется протон любым, самым слабым основанием, и уходит хорошая уходящая группа, превращающаяся в диметилсульфид.
То, что такое элиминирование возможно и легко идёт обнаружил хорошо нам знакомый по истории с амбидентными нуклеофилами Нейтан Корнблам, придумавший очень полезный способ превращение первичнх алкилгалогенидов или тозилатов в альдегиды. Эта история тоже имеет отношение к амбидентным нуклеофилам, потому что ДМСО отлично можно считать таким – у него два нуклеофильных центра, кислородный, по всем правилам жёсткий, и сера – явно мягкий. Многие очень хорошо знают, что MeI реагирует с ДМСО по углероду, образуя кватеринизованный ДМСО, который при действии оснований даёт илид, используемый в реакции Кори-Чайковски. О, вот, все правильно – наверное в реакциях с жёссткими субстратами можно получить и реакцию по кислороду, подумает любитель ЖМКО и вульгарного толкования правила Корнблама (об это желающие могут прочитать на страничке про Амбидентные нуклеофилы). Мы сейчас увидим, что да, так и есть, но не всё так просто, и в очередной раз с ЖМКО ничего не выйдет. Нуклеофильные свойства ДМСО обнаружил другой великий основоположник механизмов, Сол Уинстейн, который и показал, что самые активные именно в SN2 субстраты довольно легко сольволизуются в чистом ДМСО с образованием продуктов по кислороду (S. G. Smith and S. Winstein Tetrahedron, 1958, 3, 317-319). Нам вдвойне поучительно узнать, что сольволиз это не монополия SN1 реакций, и не монополия протонных растворителей – вполне может быть SN2-сольволиз. Поскольку ДМСО всё же нуклеофил не самый сильный, приличные скоорости реакций можно получить только с активными SN2-субстратами типа MeI или бензилгалогенидов. С вторыми всё еще сложнее, о чем ниже, а с MeI образуется O-метильное производное, но – при повышении температуры и увеличении времни реакции получается продукт кватернизации серы. ВОт собственно и вся история, очень похожая на все остальное того же типа, и не имеюшая ни малейшего отношения к ЖМКО. У амбидентного нуклеофила есть два центра, один из них более реакционноспособный с алкилирующими агентами, понятно, что здесь это кислород, так как на сере хоть и есть пара, но и значительный положительный заряд, не очень располагающий к реакциям с электрофилами. Но – реакция по кислороду обратима. А реакция по сере, намного более медленная – нет! Весь ужас в том, что это даже нельзя однозначно классифицировать как кинетический и термодинамический контроль, раз вторая реакция необратима. Но нехитрая уловка – объявить необратимую реакцию обратимой с очень большой константой равновесия, делает дело -мы можем объявить О-продукт кинетическим, а S-продукт термодинамическим. И неудивительно, что именно продукт по сере, гипервалентный сульфуроний, становится основным при нагревании – медленная реакция становится побыстрее, а из-за необратимости (или большой константы равновесия0 продукт накапливается в смеси и становится основным.
Почти одновременно ту же ракцию делает Корнблам с рядом очень активных SN2-субстратов типа α-бромзамещенных кетонов (если кетон это ацетофенон, то такие производные традиционно называются фенацил бромидами) и бензилбромидов, и находит, что реакция идёт по О, но даже ещё интереснее – если её оставить на несколько часов просто стоять – раствор галогенпроизводного в ДМСО и больше ничего! – то образуются альдегиды (N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand, W. M. Weaver J. Am. Chem. Soc. 1957, 79, 6562). Учтите это, когда будете работать с ДМСО как растворителем – это не совсем инертный растворитель, и можно получить неожиданную реакцию вместо ожидаемой. Получается, что из такого продукта так легко идёт элиминирование, что с этим справляется своя собственная основность реакционной смеси – хотите бромид берите как основание, хотите сам диметилсульфоксид, разницы нет, в растворе все равно будет кислотно-основное равновесие. Здесь наверняка играет роль и повышенная кислотность протонов рядом с карбонильной группой.
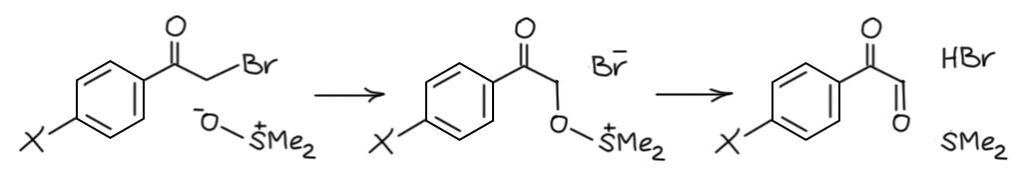
Но это занятный особый случай, а в нормальный метод синтеза – а иметь прямое превращение галогенпроизводного в альдегид неплохо, хотя мы и ограничесны активными в SN2-замещении субстратами – удалось очень просто, добавив явное основание, причем несильное, напрмер, триэтиламин или даже бикарбонат натрия. Неплохо реагируют первичные алкилиодиды или тозилаты, бензильные галогениды. Чтобы использовать первичные хлор или бромпроизводные чуть позже предложили вести реакцию в присутствии иодида, получается такая in situ реакция Финкельштейна, заодно видим, как работает настройка реакционной способности через замену уходящей группы, если не хвататет нуклеофильности (Dave, Paritosh, Byun H.-S., Engel R. (1986). “An Improved Direct Oxidation of Alkyl Halides to Aldehydes”. Synthetic Communications. 16 (11): 1343–1346). И наглядно видим разницу между иод-производными, и даже бромпроизводными в реакции SN2. Добавки иодида, кстати, используют нередко в реакциях SN2-замещения, когда нуклеофил не самый сильный и не тянет бром и хлорпроизводные с разумной скоростью.
Но с еще менее реакционноспособными галогенпроизводными это не работает, слишком медленно. Но придумали еще одно усовершенствование. Есть такая фишка, как содейтсвие замещению галогена солями серебра. Этим особенно любили пользоваться в 19-м веке, когда еще ничего не смыслили в механизмах и закономерностях замещения. Очевидно, что иодны серебра координируются по атомам галогенов и облегчают уход не в виде галогенид-иона, а в виде галогенида серебра, а это очень прочные комплексы. В применении к реакции Корнблама Ганем и Бекман предложили сначала перемешивать или даже нагревать галогенпроизводное с тетрафторборатом серебра в ДМСО, и затем добавлять триэтиламин (Ganem B., Boeckman, R. K. Jr. Tetrahedron Lett. 1974, 15, 917–920). На первой стадии выпадает обильный осадок галогенида серебра, и это признак того, что реакция идёт, образуется О-замещённый ДМСО. Триэтиламин вызывает элиминирование. 
Такое улучшение уходящей группы позволяет довольно сильно расширить возможности метода и даже применить его к окислению вторичнх галогенпроизводных, хотя условия придется сильно ужесточить.
Вот даже пример из реального синтеза
сложного природного дитерпенового метаболита из какого-то коралла (R. A. Craig II, R. C. Smith, J. L. Roizen, A. C. Jones, S. C. Virgil, B. M. Stoltz J. Org. Chem. 2019, 84, 7722−7746)
Как видим, это вторичное бромпроизводное с кучей других заместителей и функций, и поскольку вторичное греть пришлось долго, но всё прошло отлично, выход почти количественный и ничего не сломалось.
С Корнбламом понятно, кстати, поскольку реакция идёт и с тозилатами, её можно приспособить и для окисления спиртов через тозилаты. Кислород при этом приходит от ДМСО. Реакция Корнблама полезна, но из-за бешеных ограничений по реакционой способности из-за механизма SN2 это, как нынче говорят, нишевый продукт. Куда популярнее методы окисления с помощью ДМСО прямо из спиртов с сохранением спиртового кислорода.
Первый метод этого ряда обнаружили случайно. Сделал это Джон Мофат из весьма знаменитой мексиканско-американской фармацевтической компании Синтекс. Настолько знаменитой, что заслуживает пару слов. Это компания, которая изобрела и впервые вывела на рынок противозачаточную таблетку (эту таблетку, быстро ставшую необычайно популярной, стали называть просто the pill, с определенным артиклем), и на этом страшно разбогатела, производя их сама и продавая патенты другим. А еще она знаменита тем, что это такой, по современному говоря, стартап, основанный учёными, занимавшимися синтезом стероидов, и понявших, что на этом можно отлично заработать. После войны синтез стероидных соединений стал едва ли не главным занятием синтетиков. Стали быстро развиваться методы и синтеза и исследования, и дело пошло, в него стали вкладывать огромные деньги крупные фармацевтические компании (крупные по тем меркам, бизнес на лекарствах только начинался, и еще не достиг того масштаба, до которого дошли в конце века). И вот, несколько химиков решили не работать на уже известные компании, и основали новую, причем не в Штатах, а в Мексике, которая в 1950-х подавала большие надежды в развитии бизнеса. И руководить компанией кроме профессиональных бизнесменов стал настоящий учёный химик Карл Джерасси, один из пионеров применения спектроскопии в органической химии – по его собственным воспоминаниям, он согласился на эту роль и переезд в Мексику потому что компания владела одним из первых УФ спектрометров, созданных компанией Бекман. Этот знаменитый прибор уже однажды сыграл историческую роль, когда помог Вудварду сделать первое в его жизни исследование, ставшее правилами Вудварда (не путайте с правилами Вудварда-Хоффмана, до которых ещё далеко). Идея с Мексикой быстро провалилась – проблем там оказалось намного больше чем преимуществ – компанию перевели в Калифорнию, в Пало Альто, бизнес пошёл отлично, Джерасси стал миллионером – а это едва ли не единственный в истории пример ученого, который совмещал исследования с бизнесом, – но исследования не только не бросил, а именно и только раззадорился, став одним из главных пионеров применения разных видов спектроскопии в органической химии, а заодно еще и писателем-фантастом сделался вполне популярным.
И вот на той же компании Syntex работает и Джон Мофат, и делает какую-то химию с неким нуклеозидом, пробуя в те годы ещё только недавно появившиеся растворитель, ДМСО, и реагент, дициклогексилкарбодиимид. Тогда ещё никто толком не знал, что ждать от этих веществ, а специальные свойства апротонных биполярных растворителей еще не были открыты. И вместо ожидавшейся реакции, неважно какой, вдруг ощущает необычайную вонь диметилсульфида – в колбе произошло нечто странное, ведь никто не собирался ничего восстанавливать или окислять, и почему вдруг сульфоксид превратился в сульфид. В нуклеозиде была первичная спиртовая группа, но представить себе, что она окислилась – чем? – судя по вони, диметилсульфоксидом – ну нашли тоже окислитель, уж скорее восстановитель, до сульфонов сульфоксиды окисляются. Но запах не оставлял других вариантов – ДМСО восстановилось, а значит, что-то окислилось. Что?
Мофат оказался отличным химиком (неудивительно, что его взяли работать в такой амбициозной компании, скорее всего сам Джерасси и нанимал) и быстро понял не только что окислилось, но и предложил механизм реакции, разобрался, что там делает карбодиимид, и вообще оказался родоначальником, наверное, самой длинной серии именных реагентов, действующих по единому принципу, использующих один окислитель, ДМСО, и целую кучу разных активирующих агентов почти на любой вкус. И мы отдадим дань таланту этого химика, но не будем воспроизводить его схему, а сначала разберем ситуацию в общем виде, потому что вслед за Мофатом в последующие пару десятилетий появилось не менее полдюжины методов, использующих с общих чертах тот же приём и в общем тот же механизм.
В этих методах всегда сначала активируют ДМСО, создавая на сере хорошую уходящую группу. Затем добавляют спирт, который вступает с таким производным ДМСО в реакцию нуклеофильного замещения на атоме серы, и это самое обычное SN2-замещение, только не на атоме углерода, как мы привыкли, а на атоме серы. В химии разных элементов много типичных механизмов, которые в общих чертах воспроизводятся, но в деталях могут отличаться, потому что у элементов разная валентность и стереохимия. В SN2 важно то, что это нуклеофильное замещение (спирт это нуклеофил), что оно бимолекулярное – здесь гарантией этому невыгодность просто диссоциации производного серы, так как на сере уже положительный заряд, а при диссоциации он должен был бы еще увеличиться, что явно невыгодно. На третьей стадии происходит уже хорошо известное нам элиминирование с образованием двойной связи C=O как в большинстве методов окисления спиртов.
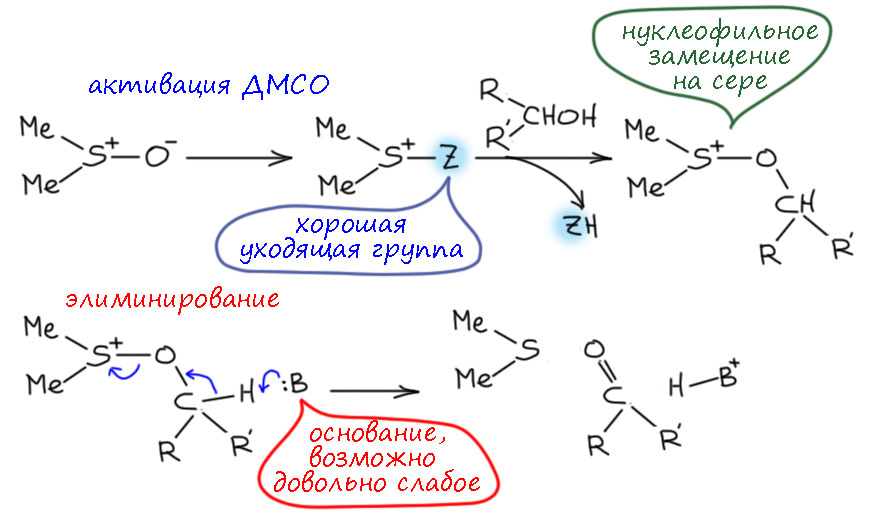
Теперь более конкретно посмотрим по стадиям и уже реальным вариантам.
Стадия 1 – активация ДМСО
ДМСО активируют самыми разными электрофильными реагентами, создавая на месте кислорода хорошую уходящую группу. Зачем много вариантов? Чтобы создать многообразие условий: некоторые позволяют замесить все вместе при комнатной температуре, некоторые требуют отдельной стадии при низкой температуре, третьи дают что-то посредине. А от реакционной способности зависит и, например, селективность и толерантность к другим функциональным группам.
Самая первая система, обнаруженная и описанная Мофатом и Пфицнером (Pfitzner K. E., Moffatt J. G.; J. Am. Chem. Soc. 1963, 85, 3027) с виду довольно неуклюжая, но надо помнить, что обнаружили её случайно. Дициклогексилкарбодиимид (DCC) это хорошо известный и самый популярный сшивающий реагент для прямого получения сложных эфиров и амидов из кислот и спиртов или аминов, требует для собственной активации кислотный катализ. В методе Моффатта-Пфицнера использовали малоприятную вещь, просто фосфорную кислоту, но немного после нашли, что можно брать другие протонные кислоты средней силы. Слабые не работают, а сильные дают не то. Поэтому используют кроме фосфорной, трифторацетат пиридиния, дихлоруксусную, и еще несколько подобных кислот. Карбодиимид активируется протонированием, ДМСО присоединяется к среднему углероду, и так создается хорошая уходящая группа – дициклогексилмочевина, реакция со спиртом даёт активированное производное, немедленно вступающее в элиминирование. Все месят вместе при комнатной температуре. DCC берут избыток, в конце гасят уксусной кислотой. Все, кто когда-либо работал с DCC, знают какая это гадость – всё происходит вовсе не так быстро и количественно, и потом из продукта приходится выковыривать дициклогексилмочевину хроматографией и перекристаллизацией, а она всё лезет и лезет в ЯМР своими могучими циклогексановыми мультиплетами. А запах! Вроде слабый, но мерзкий и приставучий, я называю его запах протухшей свечки. Бррр… Оно еще и токсично весьма.
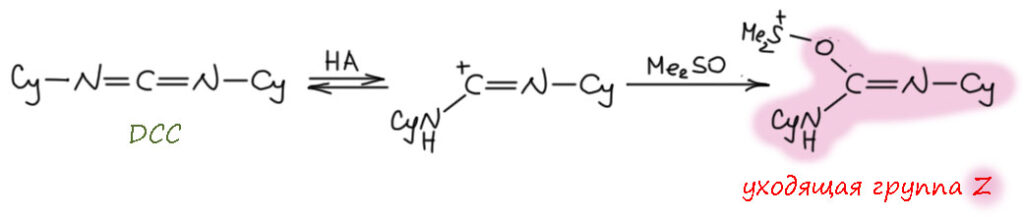
Потенциал метода Мофата был оценен органиками, и понемногу началась работа по доведению этой славной химии до ума. Наиболее преуспел в этом деле Дениэл Сверн, известный полимерный химик, известный своим вкладом в технологию винильных полимеров, но не избегавший и препаративной органической химии. Вот именно там он и оставил своё имя в истории химии, потому что наиболее далеко продвинулся в исследовании методов окисления с использованием ДМСО и сделал самый популярный медод, который долгое время удерживал лидерство в препаративной химии (это место смог отнять только периодинан Десса-Мартина и только в новом веке). При этом самый главный метод Сверна выглядит довольно жестоко – ДМСО в нём активируют оксалилхлоридом, а это чрезвычайно агрессивное и мерзкое вещество, уступающее фосгену только меньшей летучестью (Mancuso A. J., Huang S.-L., Swern D. J. Org. Chem. 1978, 43, 2480). Но секрет популярности медода Сверна именно в мягкости, а мягкость достигается тем, что реагент настолько активен, что работать с ним надо при значительной отрицательной температуре, причем сначала делается активация ДМСО, так что орагническое вещество само с оксалилхлоридом не контактирует. Активация происходит за счет сначала ацилирования, и тут же расщепления очень нестойкого и очень активного производного хлоридом. Активированный реагент это хлордиметилсульфоний, противоионом хлорид. Этот реагент устойчив только при низкой температуре, и до прибавления спирта нельзя упускать температуру выше -20º.
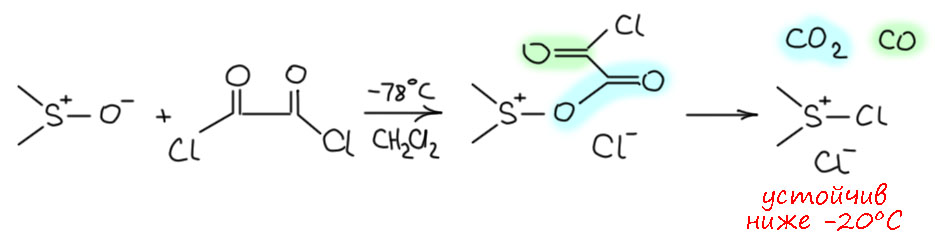
На третьем месте по популярности стоит тоже разработка Сверна, но использующая вместо оксалилхлорида вещество, которое могло бы с ним поспорить по мерзости, но оно всё же намного спокойнее – трифторуксусный ангидрид: сильно летучая и страшно агрессивная дрянь. На меня в свой время чрезвычайно большое впечатление произвело зрелище капли этого ангидрида, падающего на поверхность воды: в момент касания слой контакта бурно вскипает, и выделяющаяся энергия отправляет каплю в почти неизменном виде обратно, откуда прилетела, и кажется с ровно такой же скоростью. А ведь просто уксусный ангидрид (запрещён в Российской федерации) с водой реагирует настолько лениво, что нужно перемешивать смесь несколько часов, чтобы слой ангидрида разошёлся – к счастью, мы не сможем этого сделать, так как вещество запрещено. Но трифторуксусный ангидрид (TFAA) не запрещён, и его вполне можно применять как реагент. Если соберетечь это делать, будьте осторожны – это очень тяжёлая и очень летучая жидкость: невидимые пары от неё при переливании невидимо польются и будут стекать вниз, но любое препятствие их упруго отбросит, так что ваш нос может совершенно неожиданно попасть в невидимый фонтан паров ангидрида и подвергнуться активации: для обычной эксплуатации в виде органа обоняния он станет непригоден. Работайте под реально хорошей тягой, держите нос подальше от места переливания, лучше за створкой тяги, и ни в коем случае не экспериментируйте на открытом столе или под плохой тягой с неопущенной створкой.
Метод активации трифторуксусным ангидридом был разработан Сверном даже чуть раньше, чем метод с хлористым оксалилом (Omura K., Sharma A. K., Swern D.; J. Org. Chem. 1976, 41, 957), применяется он нередко, но реже основного метода. Обычно его тоже называют методом Сверна, но особые педанты, чтобы не путать, называют полностью – метод Омуры-Шармы-Сверна (довольно несправедливо основной метод всегда называют просто Сверном, хотя там тоже были соавторы-исполнители). В этом метода тоже сначала выполняют активацию, добавляя по каплям TFAA к раствору ДМСО в дихлорметане при низкой температуре: активированный реагент обычно выпадает в осадок, хотя это обычно непросто разобрать в реакциях, которые делают при хороших минусах – всё дымится, наружные стенки покрываются инеем, и вообще хочется поскорее это закончить, потмоу что любая оплошность в низкотемпературных реакциях обычно заканчивается испорченным синтезом.
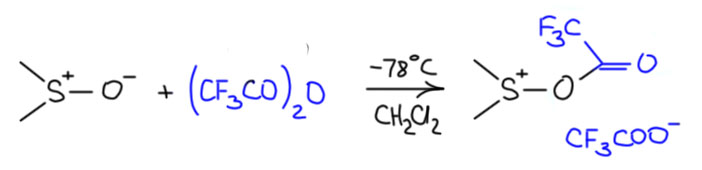
Все упомянутые методы активации требуют пониженной температуры и очень аккуратной работы. Как уже не раз замечал, даже самый великий синтетик, не боящийся вообще ничего, с большим удовольствием будет хоть иногда делать реакции в более простых условиях. Если можно без охлаждения (которое экспериментально гораздо сложнее и ответственнее нагревания), то хотелось бы. Вот и в методах активации ДМСО условия попроще искали многие. По популярности в синтезе впереди метод, разработанный одним из самых мощных химиков 20-го века Уильямом Дёрингом (мы с ним уже много раз встречались), использующий хорошо известный реагент (он нам пригодится как минимум в химии гетероциклических соединений) пиридинсульфотриоксид – это такой продукт сульфирования пиридина по азоту, довольно легко получающийся при взаимодействии пиридина и раствора серного ангидрида при хорошем охлаждении (часто используют хлорсульфоновую кислоту, это не намного более деликатная вещь, но условия чуть проще). Вообще этот реагент есть в продаже (там где есть, конечно), он вполне спокойный, не дымит, не кусается, не летит, и работать с ним можно очень просто – единственное условие, что всё должно быть сухо, иначе он начнёт гидролизоваться, появится сильная кислота и всё пойдёт наперекосяк. Зато реакция с ДМСО идет при комнатной температуре. Метод называют методом Париха-Дёринга (Parikh J. R., Doering W. von E. J. Am. Chem. Soc. 1967, 89, 5505), и он довольно популярен в синтезе, хотя реагент существенно слабее классического Сверна и может подвести в сложных случаях. Активация заключается с сульфировании (переносе сульфата на ДМСО). В очередной раз напоминаю про идиотскую традицию продолжать рисовать структурные формулы гипервалентных соединений с помощью старинных валентностей, а не связей, и только для того, чтобы избегать формальных зарядов на атомах. Но мы будем настаивать на том, что соблюдать правило октета для p-элементов важнее, чем бояться запутаться в зарядах, и что так получается не только правильнее, но и намного удобнее, потмоу что мы видим суть дела и то, как происходят реакции: вот и в этом случае правильная запись показывает, что это не что иное как нуклеофильное замещение (SN2(S)) на атоме серы, и происходит оно так же как в химии углерода – с инверсией, хоть мы здесь и не видим последствий инверсии, так как нет стереогенного центра на сере с тремя одинаковыми кислородами. Приятным бонусом являеся ещё и образование пиридина, который ещё пригодится в последней стадии.
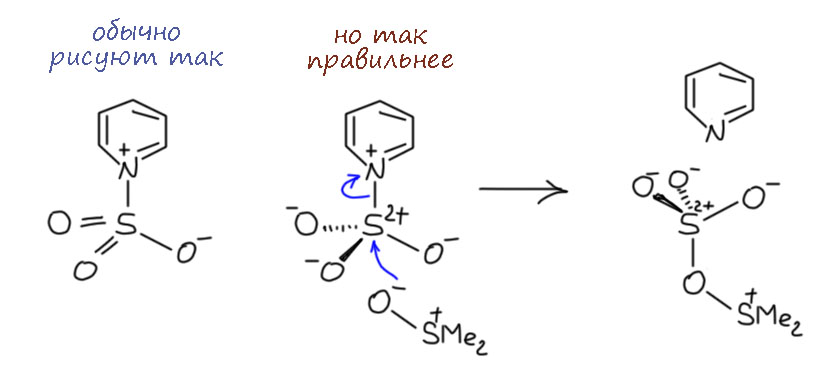
Это основные способы активации. Но есть еще несколько намного менее популярных. Был еще такой американский химик Дональд Олбрайт, всю жизнь проработавший на фармацевтической компании Ледерле, и он очень рано включился в совершенствование метода Мофата, ещё до Сверна сделав несколько полезных улучшений, причём он старался сделать как можно проще и без агрессивных и опасных реактивов, явно с прицелом на применение в масштабах промышленного тонкого синтеза. Не учёл он только того, что в Российской федерации запретят уксусный ангидрид, потому что именно этот реагент использован для активации ДМСО в методе Олбрайта-Гольдмана. Активацию рисовать не буду, она совершенно аналогична активации трифторуксусным ангидридом, да и незачем нам. Метод Олбрайта-Гольдмана хорош тем, что все делают прикомнатной температуре, смешивают все реагенты сразу и просто перемешивают, проще не придумать. Недостаток – просто колоссальный избыток уксусного ангидрида, побочные реакции ацетилирования спирта и других функций, довольно медленная реакция (перемешивают часами), низкая реакционная способность – окисляется далеко не всё. Олбрайт на этом не успокоился (возможно, гениально предвидев сложности росссийских химиков через полвека с этой запрещёнкой) и предложил довольно курьёзную замену – фосфорный ангидрид. Этот метод стали называть методом Олбрайта-Онодеры, но не использует его, кажется, никто, потому что хотя фосфорный ангидрид и дёшев, и не запрещён, но работать с ним – удовольствие крайне сомнительное: из-за убийственной гигроскопичности он успеет покрыться коркой раньше, чем вы его куда-то добавите. Но если получится – можете попробовать. Реакцию делаю также очень просто, прикомнатной температуре смешивают тоже почти всё разом и долго перемешивают. Механизм активации рисовать не буду, потому что нарисовать что-то определенное не получится – можете почитать страничку про полифосфорную и метафосфоную кислоту, чтобы понять, что это химия олигомеров неопределннного состава. Кратко процесс можно назвать фосфорилированием, и увидеть в нём много общего с тем, что происходит в живой клетке с ролью АТФ и так далее.
Итак, с активацией мы более-менее разобрались (в литературе можно найти еще немало заходов на ту же тему, но больше ничего особенно интересного), но здесь всплывает ещё один метод, который исходит не из ДМСО, но в остальном просто повторяет ту же химию – это метод Кори-Кима, одно из великого множества улучшений, введённых в химию вторым после бога синтетиком, Элайей Кори (напомню, что он родом из того самого Ливана, где гремят взрывы и десятилетиями нет житья от междоусобиц и беспокойных соседей; Кори это немного американизированная форма очень популярной среди ливанских арабов-.христиан фамилии аль Кхури, что-то типа Попова по-нашему). Вместо сульфоксида берут диметилсульфид – очень летучее вещество (температура кипения как у эфира, и пары немного легче) с ужасающим запахом, но во всех сульфоксидных окислениях он же получается в конце, а тут его берут в начале – разницы мало, разве что есть вероятность грохнуть баночку, тогда от невыносимой вони всё живое в радиусе пяти километров в ужасе бежит и вы окажетесь хозяином своего факультета, университета или даже города. Если конечно, позаботителсь заранее о хорошем противогазе. Впрочем, он не поможет. Итак, берём диметилсульфид и аккуратно окисляем его при пониженной температуре N-хлорсукцинимидом, как всегда в арстворе дихлорметана. Сначала на серу садится электрофильный хлор, то есть получается то, что в основном методе Сверна, но его тут же вышибает более нуклеофильный сукцинимидат – такое двойное нуклеофильное замещение – сначала на хлоре (да, именно так, нуклеофильная сера атакует атом хлора, а имидат это уходящая группа, это SN2(Cl) – вот до чего универсальный механизм!), а потом на сере SN2(S) – прямо молекулярный бильярд такой, с отскоком. Реагент выпадает в осадок и ждёт спирта, а имидатный остаток будет уходящей группой.
Стадия 2 – реакция со спиртом
Активированный сульфоксид, сульфониевая соль с одной хорошей уходящей группой, легко вступает в реакцию со спиртом. В зависимости от качества уходящей группы эта реакция может очень быстро идти уже при хороших минусах, и это важно, потому что многие такие активированные производные просто не выживают при температурах выше -20º и разлагаются. Поэтому спирт обычно тоже добавляют при охлаждении. Сама реакция со спиртом это типичное нуклеофильное замещение на атоме серы. Сера в сульфониевых солях имеет тетраэдрическую геометрию, если учесть неподелённую пару, и тогда замещение развивается прямо как наше любимое SN2 на углероде. Но есть важный нюанс, касающийся атомов 3-го периода и ниже: если во втором периоде мы должны строго придерживаться согласованности механизма и рисовать в середине переходной состояние, то для атомов из периодов ниже можно представить и реальный интермедиат, тригональную бипирамиду, в которой всё, что было на сере становится плоскостью, а входящая и уходящая группы повисают на гипервалентной трёхцентровой связи (четырёхэлектронной, если учитывать несвязывающую пару, и двухэлектронной, если учитывать только связываюшие электроны). Для этого нужно, чтобы входящая и уходящая группы имели высокую электроотрицательность – лучше всего фториды, но годятс яи кислородные группы. Точно здесь ничего не известно до сих пор, потому что о таких вещах стали вспоминать совсем недавно, и самое главное – такие интермедиаты, если и есть, очень слабые (минимум на вершине маленький), поэтому мы это вспоминаем только для того, чтобы понемногу привыкать во-первых, к обобщениям механизмов на другие элементы, и во-вторых, получше понимать в чём специфика других элементов по сравнению с углеродом.
С практической точки зрения, почти всегда спирт добавляют после стадии активации, выдержав некоторое время, обычно тоже при низкой температуре, и сама реакция со спиртом почти всегда очень быстрая. Методы, использующие более мягкую активацию, типа уксусного ангидрида, обычно не разделяют стадии активации и прибавления спирта, но они так редко применяются, что можно про это забыть.
Стадия 3 – элиминирование
Как только алкоксисульфониевый интермедиат образовался, он готов к элиминированию диметилсульфида и превращению в карбонильное соединение. В этом смысле завершение всех реакций этого типа совершенно одинаково: уходящая группа всегда одинакова – диметилсульфид. Поэтому по идее все должно быть одинаково в смысле завершения. И это довольно интересно, потому что разница всё же есть. В завершении, как и вообще в реакции элиминирования основание забирает протон с одновременным (одновременным в смысле согласованного механизма, а не в смысле синхронности – эта проблема всякий раз будет всплывать в согласованных механизмах: согласованный не значит синхронный).
Связь кислород-сера, тем более кватернизованная, довольно слабая, поэтому диметилсульфид можно считать отличной уходящей группой, и поэтому не нужно никаких сильных оснований. В исходном методе Пфитцнера-Мофата, где берут карбодиимид, вообще в явном виде нет основания, но берут небольшое количество не очень сильной кислоты, в оригинале вообще просто фосфорной, но дальше стали брать трифторацетат пиридиния. Очевидно, что именно анион этой кислоты (трифторацетат, дигидрофосфат, иногда что-то похожее) и работает как основание, причём реакция идёт при комнатной температуре. То, что это именно основание и что оно нужно выдаёт тот факт, что реакция не идёт в присутсвии настоящих сильных кислот типа сульфоновых или хлорной. Никаких дополнительных оснований не нужно и в методах Олбрайта, где используют активацию уксусным ангидридом (запрещённым в Российской федерации) или фосфорным ангидридом – очевидно, что основанием там служат образующиеся на месте ацетат-ион или какие-то полифосфаты. Эти реакции тоже идут долго и при комнатной температуре.
Но в остальных разновидностях окисления активированным ДМСО (а также в методе Кори-Кима) берут основание в явном виде, и это почти всегда триэтиламин, причём добавляют его обычно последним, после активации и добавления спирта. Самые популярные методы Сверна (активация оксалилхлоридом или TFAA) проводят при низкой температуре стадии активации и добавления спирта, только потом добавляют триэтиламин и уже дают нагреться до комнатной температуры. В методе Париха-Дёринга, триэтиламин добавляют со спиртом и реакцию ведут при комнатной температуре или слабом охлаждении выше нуля.
До раскрутки окисления периодинаном Десса-Мартина, методы Мофата-Сверна, особенно именно Сверна были хитами синтетических методов и встречались чуть реже чем в каждом синтезе. Наличие нескольких вариантов позвояло довольно легко подбирать метод под конкретную молекулу, чтобы избегать побочных реакций. В целом методы очень селективны, совсем избегать нужно только нуклеофильных первичных или вторичных аминов.
Все методы этой группы окисляют и первичные и вторичные спирты, первичные обычно быстрее просто по стерическим причинам, и это касается практически всех методов окисления, в механизме которых есть последняя стадия элиминирования, а это, как мы уже поняли, совершенно львигая доля методов. Вторичные спирты получают преимущество перед первичными только в методах, где водород уходит в результате гидридного переноса (окисление активированным ацетоном в реакции Меервейна-Понндорфа-Вердея-Оппенауэра). Если водород уходит протоном через согласованное элиминирование, то значение имеет только стерика, она играет против стерически затруднённых вторичных спиртов, и в некоторых случаях получается окислить первичный спирт в молекуле, где есть ещё и вторичные спиртовые группы, но это всегда выясняется путем эксперимента. Проблема еще в том, что есть манера использовать активированный ДМСО в избытке относительно окисляемого спирта, так что манипулировать просто соотношениями непросто.
Периодинан Десса-Мартина
Это последний из крупных методов окисления спиртов в карбонильные соединения, ставший чрезвычайно популярным в сложном синтезе, где это без сомнения метод номер один. Особую ценность методу придают мягкость условий (комнатная температура и никаких дополнительных реагентов), стехиометрический характер, и исключительная селективность, позволяющая применять метод соединениям с большим количеством других реакционноспособных групп и фрагментов. Надо понимать, что сложность органических соединений такова, что никогда нельзя предусмотреть всех сюрпризов структуры, и абсолютной селективности не существует – найдётся и на самый селективный реагент нечто, что его обманет. Но в сравнении со всем другим, этот реагент великолепен именно своей надежностью и селективностью, при том, что в остальном это очень плохо – дорого, неэкономично, и даже опасно – в больших количествах реагент ни делать, ни хранить не рекомендуется – соединения гипервалентного иода в чём-то очень похожи на соли диазония – они нестойки на свету, и иногда довольно неприятно хлопают. Я сам однажды на это нарвался, когда получал трис(трифторацетокси)иод и все было нормально, пока дело не дошло до сушки кристаллов в вакууме. Хлопнуло без предупреждения и без очевидных причин, хотя и без особенных последствий кроме разлетевшейся склянки Шленка – в такие минуты всегда начинаешь сильно сомневаться в правильности выбора профессии.
Итак, что за зверь и почему он так странно называется. О, это прямо клубок недоразумений, памятник тому, что большинство химиков пренебрегает понятиями гипервалентности и ищет объяснений из обычной валентности, а тут ничего кроме проблем не найдёшь. Реагент был открыт в начале 1980-х, когда возник очень большой интерес к органическим производным иода с валентностью более единицы – мы смело назовём их гипервалентыми, но тогда этим словом пользовались немногие, а большинство находилось в убеждении, что так просто проявляют себя d-оболочки, и проблем нет, просто соединения такие. Из-за этого практически везде, включая специализированные монографии, структуру соединения представляют неправильно, а механизм окисления остаётся запутанным. И это жаль, потому что в этом соединении нет вообще ничего особенного – это соершенно типичное производное гипервалентного иода, строго соблюдающее все закономерности структуры таких соединений, да и механизм окисления ничего особенного из себя не представляет.
Жаль, что я так до сих пор и не допилил страницу про гипервалентность – можно было бы здесь не тратить время на напоминание простых вещей. Допилю, но не скоро. Придётся напомнить. Проблема в том, что p-элементы, к которым относятся все неметаллы и металлоиды главных подгрупп от 3-й до 8-й групп короткопериодной таблицы или 13-й – 18-й групп длиннопериодной системы не могут образовать более четырёх ковалентных связей, потому что вынуждены следовать правилу октета, которое не блажь, а банальное отражение того неоспоримого факта, что валентная оболочка этих элементов состоит из одной s- и трёх p-орбиталей и больше взять им валентные возможности неоткуда – никаких d-орбиталей в их распоряжении нет и это доказано-передоказано всеми известными методами квантовохимических расчетов. Все вроде просто, но есть проблема – у элементов от 15-й до 18-й (дальше только длиннопериодная) есть соединения и их много, которые вроде бы очевидно нарушают это правило. У этих элементов есть нормальные валентности равные 8-(старый номер группы) или 18-(новый номер группы): у азота это 4 и 5, у серы 4 и 6, у хлора 2,3,4,5,6,7. У элементов из периодов ниже 3-го то же самое, с дополнением инертных газов снизу у которых есть 2,4,6,8. Откуда эти валентности? Если разрисовать структуры молекул с такими валентностями привычными нам структурными формулами и посчитать каждую черту связи за пару, правило октета будет явно нарушено.
На самом деле нет, и это очень давно известно. К сожалению, в химии так и не появилось конвенции, которая бы позволила явно и недвусмысленно обозначать такие ситуации, и мы пользуемся в химии непереходных элементов теми структурными формулами, которые возникли в 19-м веке до появления электронных представлений. Ну, так получилось. Они очень удобны, те структуры. Но в реальности нам надо что-то сделать с ситуацией, когда нужно использовать для образования связей не одиночный электрон валентной оболочки (как мы это делаем у углерода – четыре электрона, четыре орбитали – четыре ковалентные связи), а пару, которую невозможно разбить – места нет в валентной оболочке самого элемента. Выход простой – нет места у самого элемента, попросите тот элемент, который собирается с вами связаться, помочь – отдайте ему второй электрон пары. Тогда можно смело завязывать связь – эта связь будет обслуживаться парой, и правило октета не нарушит (была пара и осталась пара). А что второй элемент? Во-первых, он должен быть достаточно электроотрицателен, и даже точнее – важна разность электроотрицательностей – второй элемент должен быть более электроотрицателен, и чем больше, тем лучше. Поэтому лучше всего работает фтор, кислород тож, но хуже, и ещё хуже азот. На самой-самой грани возможностей углерод, там это редко, но соединения даёт чрезвычайно важные в синтезе, илиды. Можно найти примеры соединений с хлором, иногда серой, совсем редко бромом – но вся эта троица очень редко работает на такие связи. Следующий момент очень важен: таких гипервалентных связей всего два типа. Одна – то, что мы в стандартных структурных формулах рисуем двойной связью – но это не двойная связь, а одна ковалентная с парой и вторая ионная – лишний электрон отъехал на более электроотрицательный элемент, оставив с одной стороны плюс, а с другой минус. С этим более-менее понятно, хотя требует рисовать заряды (это формальные заряды, в реальной структуре такие связи за счет поляризации деформируют плотность в сторону плюса, и разница зарядов не такая явная, но это нельзя изобразить совсем). Второй тип связи сложнее – в ней пара связывает не два атома, а три – это трёхцентровая двухэлектронная связь. Формально и очень удобно делать такую связь из “двойной” – посадите третий атом в виде аниона на центральный атом в виде катиона – получите симметричную незаряженную структуру, в которой вы тут же должны забыть, кто был первым, а кто вторым – получается, что как будто черточка связи на оба атома одна и мы только для красоты прерываем её на центральном атоме.
Чтобы не уходить в общие рассуждения далеко, потренируемся сразу на соединениях иода. У иода одна нормальная валентность, и мы легко обрауем иодпроизводные. На иоде остаются три пары – проще всего считать, что это перпендикулярные p-орбитали. Тут не все так просто, потому что с одной стороны, привычная нам гибридизация это особенность только элементов 2-го периода, и ниже валентные орбитали стараются участвовать в связывании по отдельности. Но некоторая степень смешивания все же есть. Мы можем условно считать, что в тех же соединениях R-I единственная обычная ковалентная связь со стороны иода подразумевает смешение s и px-орбиталей. А это значит, что пара с другой стороны атома иода тоже смешанная, и это делает ее отличной от двух других пар – чисто py– и pz-орбиталей, и мы особенность этой пары хорошо увидим на структурах – она ниже по энергии (за счет примеси s) и будет до последнего сопротивляться использованию.
Получается такая картинка, немного школьная, но вполне разумная.
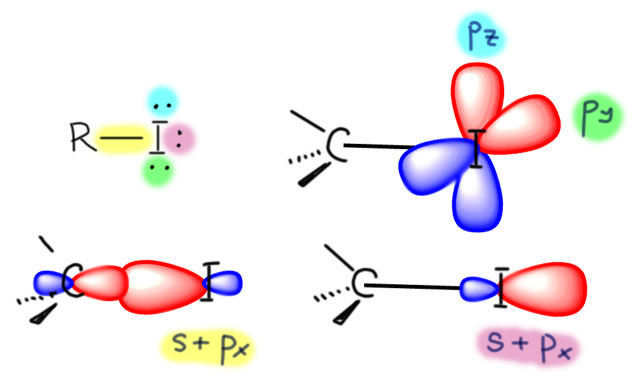
Использование этих пар возможно с помощью одного из двух типов гипервалентных связей. Сначала попробуем одну. Получим производное трёхвалентного иода. Заметим, что именно трёхвалентного, а не иода(3+) – степень окисления нам не поможет, а только собьёт с толку. Со степенью окисления у иода вообще всё плохо, он очень сильно зависит от используемой шкалы электроотрицательностей, в частности соотношения иода и углерода – они меняются местами в разных шкалах. Мы можем по рекомендации ИЮПАК пользоваться только Алленом, но тогда мы получим малоприятную историю с тем, что похожие соединения иода и брома окажутся разведены в классификациях на основе электроотрицательностей. Вот почему так хороша старая добрая классическая валентность – она не формальна, как электроотрицательность, а неразрывно связана с самыми простыми вещами в химии – стехиометрии и рядами аналогичных соединений. Здесь мы получим очевидные ряды для трёхваленого иода два типа соединений IX3 и XI=O (например, трифторид иода и оксифторид иода). Здесь мы видим одну из фишек гипервалентности – ограниченность вариантов. В том типе связи, которая обозначается традиционно как двойная, второй элемент практически только кислород. Азот и углерод тоже возможны, но это большая экзотика и мы с ней не встретимся, если только специально не будем искать такие соединения. В роли X может быть и другой галоген, и группа, присоединённая через атом кислорода. Группы, присоединённые через другие более-менее серьёзные неметаллы (азот, углерод, сера) могут быть только поодиночке и в двумя другими галогенными ли кислородными, но и это экзотика и интермедиаты в реакциях. Гипервалентность очень сильно ограничивает нас в вариации структуры. Сейчас нам это и не нужно. Итак, берём R-I и одну из перпендикулярных пар на p-орбитали. Связь с атомом кислорода (и если приспичит всё-таки, то и с азотом или углеродом) устраивается просто – берём атом с шестью электронами (секстетный, просто сингдетный атом кислорода, или если таки приспичило, нитрен или карбен) и вешаем его на эту пару донорно-акцепторным взаимодействием. Поскольку пара от азота и он ее отдает, то на нем возникает формальный плюс (один свой электрон ушёл), а на кислороде (или азоте или углероде, если приспичило) – минус: два электрона пришло, но один законный свой, а второй лишний – формальный минус. Итого – связь, обслуживаемая парой, то есть обычная ковалентная (мы много раз уже обращали на это внимание: донорно-акцепторная связь это не тип связи, а способ образования связи, обычной ковалентной связи, и после того, как связь образовалась, вы не можете отличить ковалентную связь от донорно акцепторной – именно поэтому не надо злоупотреблять особыми обозначениями типа стрелочки или пунктира). Поскольку на соседних атомах противоположные заряды, это дает дополнительное связывание фактически ионного типа, оно весьма нехилое, потому что атомы рядом, уже притянуты друг к другу ковалентной связью. Более точное рассмотрение этой второй связи показывает, что в ней есть некоторая доля ковалентности – это выражается в деформации электронной плотности на атоме-доноре (кислороде, азоте, углероде) в сторону атома-акцептора (здесь это иод), но это видно только в квантово-химических расчетах очень высокого уровня с использованием сложных базисов, учитывающих возможность таких деформаций (условно говоря, обычную восьмёрку p-орбитали немного “ведёт” в сторону второго атома, как быдто она немного приняла на грудь и стоять прямо больше не может) – но нам это совершенно до лампочки, эти детали ничего не прибавят к картине связывания, а только голову заморочат. Итак, приняли: делаем из RI соединение с кислородом (в химии других элементов это бы назвали оксидом, как у азота есть N-оксиды, а здесь были бы I-оксиды, но в химии иода закрепилось старое название иодозо или иодозил, весьма неудачное, потому что просто использует формальное сходство с нитрозил-производными, а это плохая аналогия, потому что нитрозильные производные не гипервалентные, а нормально-валентные, это стандартная валентность азота три). Обращаю внимание на то, что иодозильная группа образует прямой угол со связью C-I – уголковую структуру, потому что ее обслуживает p-орбиталь, по картинке выше примем что это py-орбиталь, (или pz– – сейчас это совершенно одинаково)
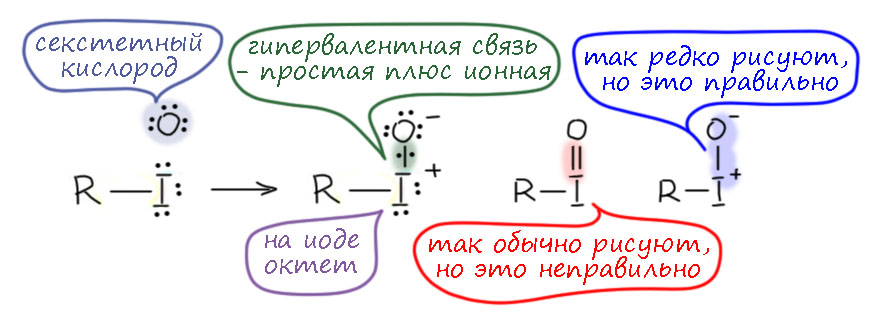
Кроме такой, есть еще и второй тип гипервалентной связи. Я его уже рассматривал на страничке про химию фосфора. У всех элементов, проявляющих гипервалентность, то есть валентность выше, чем восемь минус номер группы в короткопериодной системе (или 18 минус номер группы в длиннопериодной), или же в других словах – выше, чем нормальная валентность (которая как раз и равна упомянутым разностям) – всё совершенно одинаково: два основных типа гипервалентных связей. Первая – как бы двойная, а на самом деле простая плюс ионная. И вторая – два связанных атома на одной p-орбитали центрального атомоа – трёхцентровая связь. И здесь – внимание – тоже есть путаница: в литературе такую связь обычно называют трёхцентрoвой четырёхэлетронной (3c4e), но в валентную оболочку центрального атома она даёт два электрона, а ещё два остаются на присоединяющемся атоме. Вот как она образуется на примере того же иодозобензола – попробуем присоединить к нему воду. Один протон посадим на отрицательный кислород, и воспользовавшись формальным положительным зарядом на иоде, подгоним к нему с другой стороны гидроксид. Смотрите, что получается: гидроксид садится на иод, оставив при себе свою пару, так что счёт валентных электронов на иоде не изменился – был октет и остался октет. Связь трехцентровая – три атома пользуются одной валентной орбиталью иода, поэтому они сидят на прямой, кислороды друг напротив друга. А почему связь называют четырёхэлектронной? А потому что гидроксид пришел со своей парой, и эта пара осталась фактически на несязывающей молекулярной орбитали (посмотрите текст про фосфор – там это разрисовано), а связывающая одна, она трёхцентроая, на ней пара. И ещё один повод для недоразумений: когда мы пары на иоде рисуем, как нас научил Льюис, то из-за двухмерности рисунка получается так, как будто две пары друг напротив друга. Но атом трехмерен и три р-орбитали перпендикулярны, так что вторая пара не стоит на пути присоединяющейся группы.
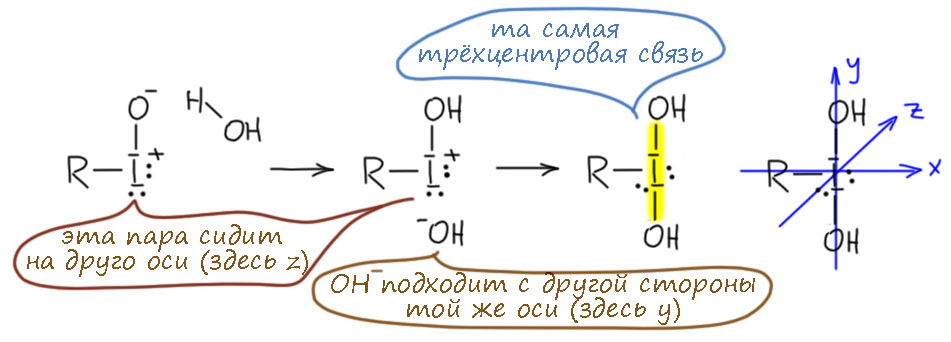
ОК, поверим, а правда у иодозобензола есть такой гидрат? Нет, гидрат неустойчив, но иод очень хочет образовывать такие трехцентровые связи. В отличие от фосфора и вообще элементов 3-го периода, которые предпочитают “двойные” гипервалентные связи, элементы 5-го периода предпочитают трехцентровые, потому что атомы больше, и могут принять больше связанных атомов вокруг себя. В химии металлов это называют координационным числом, иногда этот термин применяют и в химии непереходных элементов, но не всегда, и здесь есть повод для того, чтобы не путать терминологию, потому что связывание принципиально разное – то, что d-металлы делают с помощью d-орбиталей, p-элементы вынуждены делать с помощью гипервалентности. Такой гидрат неустойчив просто потому, что является протонной кислотой, и из-за обратимого протонирования-депротонирования будет вступать в реакции. Но сам иодозобензол все равно предпочтитает перейти в форму с такими связями, что и успешно делает просто за счет полимеризации – твердый иодозилбензол это полимер. Поэтому он не кристаллизуется, и представляет собой мелкий гигроскопичный порошок – поэтому он довольно неудобен в работе – плохо растворим в простых растворителях (из-за полимерности). Получаются такие длинные гибкие цепочки (на атомах кислорода получается угол, поэтому это не прямая – фрагменты O-I-O линейные, но соединяются они под углом – кислорода, скорее всего sp2-гибридный. Интеерсно, что полимеризация, кажется, начинается как раз через гидратацию, и на концах звенья протонированы (Richter, H. W.; Cherry, B. R.; Zook, T. Z.; Koser, G. F. J. Am. Chem. Soc. 1997, 119, 9614-9623). Цепочки в твердом иодозобензоле еще и склеены модными галогенными связями (об этом можно узнать в моей последней лекции из нового курса Органика 21 века), а в некоторых полиморфах этого соединения вместо линейных цепочек образуются большие циклы, тоже сшитые чем-то типа галогенных связей.
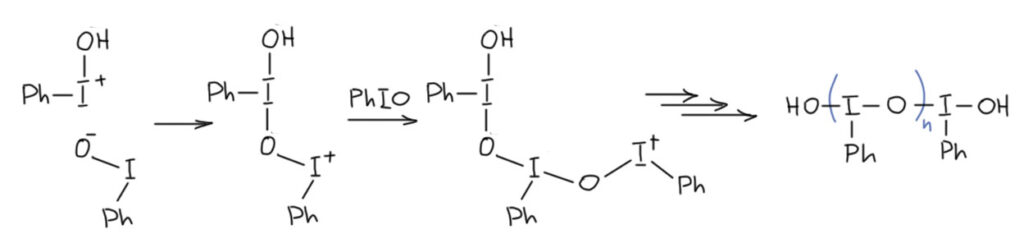
Но, например, взаимодействие с сильными кислотами деполимеризует иодозобензол, и тогда получаются вполне мономерные (или димерные) реагенты, очень популярные в органической химии, например, реагент Козера или реагент Зефирова, очень важные и популярные в химии гипервалентного иода.
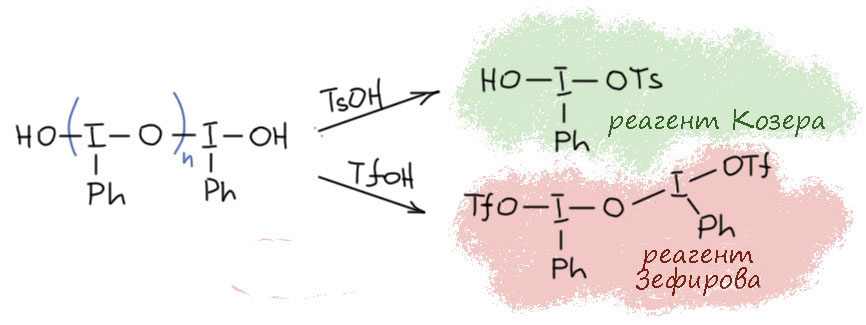 И ещё много интересного. Однажды мы подробнее про это поговорим, а пока мы просто пытаемся разобраться как устроен реагент Десса-Мартина и немного подбираем основы гипервалентных соединений иода. И вот мы к этому идём понемногу. Следующий шаг – многочисленные производные иодозобензола, в которых на гипервалентной трёхцентровой связи висят другие группы. Какие? Гипервалентность требует, чтоб они были электроотрицательными. Это могут быть галогены и не все – только фтор и хлор, электроотрицательности брома и тем более иода недостаточно. Или кислородные группы, вот их может быть много разных – карбоксилаты, даже алкоксиды. Среди таких соединений самые знаменитые и простые фенилиодозодиацетат и бис-трифторцетат.
И ещё много интересного. Однажды мы подробнее про это поговорим, а пока мы просто пытаемся разобраться как устроен реагент Десса-Мартина и немного подбираем основы гипервалентных соединений иода. И вот мы к этому идём понемногу. Следующий шаг – многочисленные производные иодозобензола, в которых на гипервалентной трёхцентровой связи висят другие группы. Какие? Гипервалентность требует, чтоб они были электроотрицательными. Это могут быть галогены и не все – только фтор и хлор, электроотрицательности брома и тем более иода недостаточно. Или кислородные группы, вот их может быть много разных – карбоксилаты, даже алкоксиды. Среди таких соединений самые знаменитые и простые фенилиодозодиацетат и бис-трифторцетат.
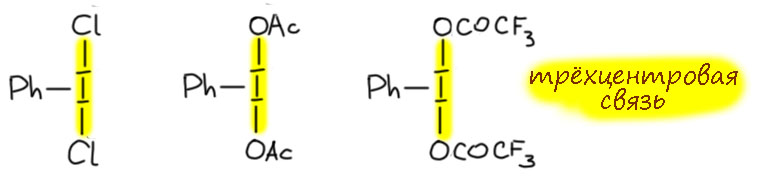
И ещё очень удобно использовать в качестве одной из групп карбоксильную группу прямо из того же фенила, из орто-положения. Тогда второй группой может быть и гидрокси, и ацетокси, и другой карбокси, и трифлат и много чего ещё. Из-за стереохимических требований пятичленного кольца (хотя оно сильно искажено, потому что связи с иодом намного длиннее) две группы на иоде несколько отклоняются от прямой, но не сильно, и это никак не меняет природу трехцентровой связи – это обычное отклонение от идеальной геометрии, которое встречается в химии чаще, чем не встречается.
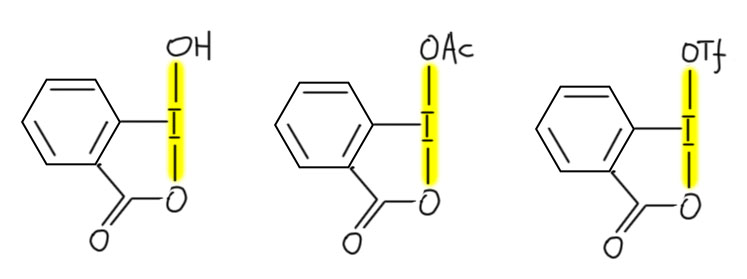
А вместо фенила может быть что-то? Да, замещённые фенилы в первую очередь. А вот алкилов быть не может, и это очень похоже на соли диазония – такие соединения просто имели бы огромныую реакционную способность в нуклеофильном замещении и немедленно развалились бы. Так, возвращаемся к к группам на трехцентовой связи: а группы с другими элементами? Даже с азотом уже проблемы, тем более с серой или углеродом. Гипервалентность требует, чтобы группы эффективно забирали пару, чтобы из четырёх валентных электронов на этой связи, два находились на несвязывающей орбитали, а это требует большой электроотрицательности от связанных атомов (групп). Поэтому лучше всего, чтобы они были на одинаковом элементе (кислород лучше всего, хлор и фтор тоже, но это только два соединения, а кслородных – пропасть). Почему? Потому что эта пара должна быть делокализована на двух группах. Ой, а как это? Да, это хорошо видно по молекулярным орбиталям (опять отсылаю на страничку про фосфор). А так, без орбиталей, можно нарисовать граничные структуры, представив трехцентровую связь как мезомерию двух двухцентровых. Нам это непривычно, но ничего странного в этом нет.
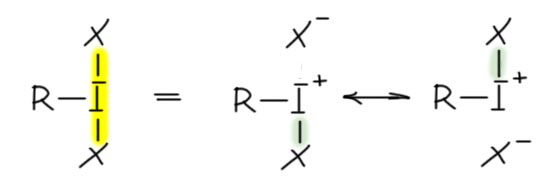
Такое представление ещё помогает понимать, что трехцентровая связь весьма полярна, причём тут есть парадокс – если взять её целиком, все три атома, то если она симметрична, то естественно неполярна. Но каждая её половинка полярна, и весьма, и нам ещё придется как-то примириться с тем, что эта самая половинка это просто фигура речи, и что трёхцентровая связь не состоит из двух обычных, как мы показываем чёрточками. Впрочем, и это тоже хорошо видно, если по какой-то причине одна из связанных групп решит отвалить, то отвалит она в виде аниона, а остаток превратиться в обычный катион иодония. И наоборот, катион иодония может связать анион и образовать гипервалентную трёхцентровую связь – и здесь нам приходится напрячься, чтобы не перепутать граничные структуры (которые существуют только у нас в голове) и реальный процесс обратимой диссоциации производных трёхвалентного иода.
Но и это, чёрт возьми, не всё. Такие производные иодония кроме просто диссоциации могут работать и как электрофильные реагенты и тогда смещение электроной плотности произойдёт в обратную сторону. Например, арилиодозодихлориды довольно часто применяют как мягкие электрофильные хлорирующие агенты. В этом случае, электроны остаются на оставшемся куске трёхцентровой связи, и она закономерно развалится, причём произойдёт восстановление иода до обычного одновалентного состояния.
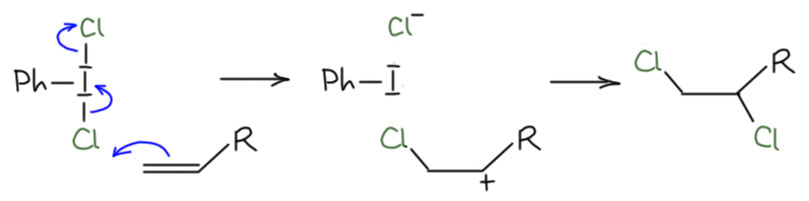
Немного познакомившись с особеностями строения производных трёхвалентного иода, возьмёмся за следующую пару электронов и пустим её в следующую гипервалентную связь. Взяв, например, иодозобензол, сделаем (мысленно) реакцию с вторым атомом кислорода. Образуется вторая такая же связь, и она будет перпендикулярна первой. Должна быть перпендикулярна. В отличие от иодозобензола, который всегда является полимером, такое производное, иодилбензол кристаллизуется без полимеризации. В старину, из немецкого использовалось в русской литературе название иодобензол, очевидно по аналогии с нитро. Именно это название для группы IO2 было у нас главным, никто особенно не парился потому что такие соединения были экзотикой, и с ними почти никто никогда не сталкивался, и не путал иодбензол и иодобензол. Но в английском такое название невозможно, так как PhI и так уже назывался iodobenzene, в английском соединительная гласная o обязательна между согласными в частях слов, но раньше у них было в ходу название iodoxybenzene. Сейчас и по-русски это иодилбензол, так по ИЮПАКу. Структура иодилбензола известна из рентгеноструктурных данных, и там даже не одна структура, так как у этого соединения несколько полиморфных форм – отдельные молекулы ассоциированы друг с другом в трёхмерную структуру, но это не такой полимер как у иодозобензола – у иодилбензола расстояния иод-кислород очень сильно отличаются для своих и чужих кислородов, поэтому это межмолекулярные взаимодействия, а не полноценные гипервалентные трехцентровые связи. Так или иначе, структура близка к ожидаемой, хотя угол между кислородами не 90 градусов а сильно больше, но это понятно – отрицательно заряженные атому отталкиваются. Тем не менее, в общих чертах мы видим то, что и можем предсказать из способа образования – на две пары на двух перпендикулярных р-орбиталях садятся кислороды.
Сам иодилбензол (и замещённые) соединения очень неудобные и им долго не могли найти никаких применений и считали экзотикой. Химия у них должна быть очень похожа на химию иодозобензолов, только с учетом двух перпендикулярных гипервалетных связей, но их общих соображений они должны быть более сильными окислителями, но это долго оставалось неиспользованным, хотя самое перспективной производное иодилбензола было получено уже в конце 19-го века. Эта штука обычно называется реагентом IBX, по устаревшему названию орто-иодоксибензойная кислота. Как видим, её легко можно представить, если в уже известной нам орто-иодозобензойной кислоте, представляющей собой гетероцикл с трёхцентровой связью (бензиодоксол), ещё одну пару отправить на секстетный кислород, как собставенно и делают, используя более сильные окислители типа бромата или надсерной кислоты, а это и есть переносчики секстетного кислорода (эту тему пока раскрывать не буду, и просто схематически обозначу такой окислитель-переносчик). Как видим, в реагенте одна связь трёхцентрвая, а вторая – ковалентно-ионная, которую принято рисовать двойной, но мы нарисуем правильнее. Эти связи перпендикулярны. Спереди у иода остаётся еще одна пара, но мы ее не трогаем, ее окислить намного сложнее, и строго говоря, в органических производных иода окисление до иода семивалентного вообще неизвестно, органическая часть сгорает раньше.
Вот этот реагент IBX долго пытались пристроить в органическую химию как окислитель, и с некоторым успехом, но он тоже не очень удобен и довольно опасен в обращении – может долбануть хлопнуть при нагревании.
Однажды этой химией очень серьёзно занялся американский химик Джеймс Мартин, большой любитель гипервалентных соединений, и как раз стал искать что-то более удобное и менее опасное, перебрал всякие производные и в 1983 году опубликовал очень простой вариант этого реагента, образующийся при реакции с избытком уксусного ангидрида (запрещён в Российской федерации) – и мы, уже съев собаку на гипервалетных структурах легко понимаем, что получилось – этот кислород на “двойной” связи перешёл в структуру с трехцентровой связью, и все гидроксилы заместились на ацетокси-группы. Соавтором в статье у Мартина был его студент (или постдок) Дениэл Десс (Dess D. B., Martin J. C. J. Org. Chem. 1983, 48, 4155–4156). Соединение сразу предложили как селективнй окислитель для спиртов, что неудивительно, ведь исходный реагент IBX тоже это умел, но был не очень удобен.
Итак, это соединение пятивалентного иода, содержащее одну обычную ковалентную связь с фенилом и две трёхцентровые, на которых висят три ацетокси-группы и карбоксилат от бензойной кислоты попарно. Именно так и надо его рассматривать и рисовать – фенил вбок, и две трехцентровые связи под прямым углом – мы без труда увидим тут октаэдр без одной боковой вершины (в сторону этой вершины тычет последняя пара, и можете её нарисовать для полноты многогранника). Вместо этого структуру часто представляют так, что четыре кислорода в экваториальной плоскости образуют квадрат, а фенил растёт вверх как апикальная группа, и называют структуру тетрагональной пирамидой. С точки зрения геометрии что в лоб, что по лбу. Но с точки зрения химии я очень советую обычную ковалентную связь никогда не отправлять в апикальное положение. Когда мы однажды разберемся с гипервалентностью, как феноменом, свойствнным множеству элементов, то увидим, что есть очень большой резон апикальными всегда держать именно одну из трёхцентровых связей, а чисто ковалентную часть структур держать в экваториальной плоскости – получится очень красиво и будет видна общность множества структур разных элементов.
Соединение оказалось удобно в получении, устойчиво в хранении и поначалу даже показалось, что оно преодолевает известную проблему органических производных иода(V) – склонность иногда неожиданно бурно разлагаться, то есть производить громкий хлопок с разлетанием во все стороны сосуда в котором они хранились, а также неизбирательным поражением живой силы научных исследований осколками и продуктами разложения. Особенно это не нравилось компаниям, производящим и торгующим реактивами. Хороший реагент, но опасный, с кучей ограничений по транспортировке и хранению – так часто бывает. Так вот реагент Десса-Мартина обещал быть не только полезным в синтезе, но и безопасным. Дальше произошло курьёзное расхождение – реагент все равно оказался склонен к самовоспламенению и хлопкам, поэтому его не стоит накапливать в больших количествах. При этом, он настолько понравился синтетикам, что спрос резко вырос, и пришлось как-то обходить сложности за счёт маленьких фасовок и больших предосторожностей.
Прежде чем мы перейдём к обсуждению механизма действия реагента, разберёмся, почему он так странно называется – периодинан. Это вообще-то недоразумение, странный результат того, что ИЮПАК слишком долго возится с номенклатурами. Органическую они ещё как-то слепили довольно быстро, просто собрав кучу уже сложившихся традиций, но и то потом пришлось десятилетиями доделывать. А вот с номенклатурой других элементов возня получилась бесконечная, и номенклатуру родили корявую донельзя. Мало кто ее знает и ей пользуется. Мартину пришлось сочинять что-то на тему.
В чем загвоздка? Мы знаем, что номенклатуру соединений углерода делают замещением из углеводородов. А номенклатуру других элементов, возьмем только p-элементы, потому что номенклатура d- и f-элементов строится как названия комплексных соединений: металл-лиганды. Номенклатуру p-элементов тоже пытались сделать так же (серная кислота стала тетраоксосерной(VI) кислотой), но получилось слишком громоздкая система. Тогда пошли по другому пути – недолго думая слизали главный принцип у органики – всё от гидридов, и даже суффикс исходного гидрида – ан. То, что у множества элементов гидридов нет вообще, никого не смутило – возьмём те, что просто получаются по законам валентности, а есть они или нет, не важно. Дальше вышло еще смешнее. Вот иод. Какой у иода гидрид? Ежу понятно, HI. Как назовём? Ежу понятно, иод-ан, иодан (читается ёдан). Но пока возились, заинтересованные химики стали стряпать свои названия. Тот же Мартин по аналогии с химией фосфора и серы (там есть фосфораны и сульфураны) придумал для трёхвалентного иода навзание иодинан (iodine – iodinane), иодинан (читается ёдинан). Дальше ещё интереснее – а как называть соединения элемента с другми валентностями, отличающимися от нормальной – здесь разработчики точно понимают, что такое нормальная валентность, и что такое гипервалентность (есть ещё низковалентность, но там одна экзотика и система названий своя). Так вот, пока ИЮПАК копался, тот же Мартин решил просто добавлять приставку пер к следующей валентности. Производное иода трёх – иодинаны, пяти – периодинаны. Вот и вся разгадка названия. Что делать с семивалентным иодом Мартин вроде бы не разъяснил, но и органических производных у такого тогда точно не было. Наверное это стало бы гипериодинанами.
Но тут наконец вылез ИЮПАК. И говорит – пусть всё будет иоданами. А валентность мы обозначать буквой лямбда с индесом. Вот так производные трёхвалентного иода буде считать производными гипотетического иодоводорода IH3 и обозначать будем λ3-иоданами. А то, что Мартин навал периодинанами назовём от гипотетического иодоводорода IH5 и обозначим как λ5-иоданами. А семивалентного, понятно, например, семифтористый иод это λ7-гептафториодан. Иными словами закрепившееся за реагентом Десса-Мартина название периодинан не соответствует ИЮПАКу. Правильное название будет такое 3-оксо-1λ5,2-бензиодоксол-1,1,1(3H)-триил триацетат или 1,1,1-триацетокси-3-оксо-1λ5,2-(3Н)-бензиодоксол. Иод здесь в гетероцикле, который мы уже видели, поэтому здесь нет иодана, но есть лямбда-пять, указывающая на валентность иода. И тогда, поскольку лямбда-пять, а две валентности встроены в цикл, то остаются три, и по ним замещение на ацетат (или ацетокси) обозначено локантами 1,1,1 – от иода идёт нумерация в цикле. Иными словами здесь родоначальный гетероцикл гипотетический 1λ5,2-бензиодоксол таков, и вот так нужно по ИЮПАК это называть. Понятно, что проще оставить авторское Dess-Martin periodinane (DMP).
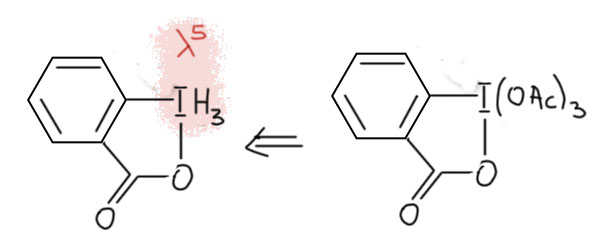
Окисление периодинаном Десса-Мартина
Периодинан Десса-Мартина (дальше DMP) сразу пристроили к окислению спиртов совершенно закономерно, потому что его предшественник, реагент IBX уже с этим отлично справлялся, но у него была масса недостатков – очень низкая растворимость в обычных органических расворителях, необходимость избытка, очень неприятный нрав и так далее. DMP преодолевал большинство этих недостатков, хотя при применении дальше выявились и у него проблемы. Но высокая растворимость в самых популярных растворителях типа дихлорметана, кристаллический характер (с кристаллическими реагентами всегда приятнее работать, чем с чем-то бесструктурным, аморфным), и возможность использовать практически ровную стехиометрию (в исходной статье избытки минимальные, 5%) создали реагенту очень хорошую основу для завоевания умов синтетиков, намаявшихся с хромовыми реагентами и методами из серии Моффатта-Сверна.
Механизм окисления с одной стороны довольно очевиден, но с другой – это тот самый случай, когда знать и уметь обращаться с гипервалентными соединениями очень важно. Если в реагенте есть трёхцетровая связь, работать он будет в общих цертах так:
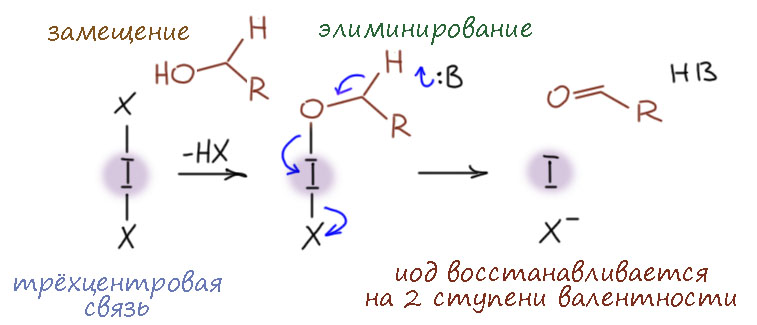
Спирт как нуклеофил замещает один из концов трехценровой связи. Почему? Из-за большей нуклеофильности, чем обычные группы на иоде типа ацетокси, трифторацетокси и т.п. Само замещение скорее всго идет как обратимая диссоциация-ассоциация. Это работает уже для производных трехвалентного иода, но там нужна серьёзная активация кислотами Льюиса, и это неудобно. В пятивалетных иоданах типа DMP замещение идёт намного легче, что отчасти даже мешает, но не сильно. Не забудем принцип гипервалентности – в таких связях легче участвуют более электроотрицательные элементы, поэтому такая высокая склонность к кислородным остаткам карбоновых кислот, спиртов, воды. Это очень хорошо сказывается на селективности, что кажется удивительным, но ни азотные, ни серные группы значимо не мешают работе этого реагента в сложных молекулах. С другой стороны, он оказывается очень гигроскопичным, и это мешает при хранении, но как мы увидим дальше, сильно помогает в реакции.
Берём уже сам DMP. У него три ацетокси-группы. Какая из них замещается легче? Ответ на это знали уже основоположники – та, что напротив карбоксилата из цикла. Это легко объяснить. Если в трехцентровой свяи две группы разные, то она будет поляризована в сторону лучшей уходящей. Поскольку разрыв цикла невыгоден, то именно ацетокси напротив кислорода цикла и становится более лабильной.
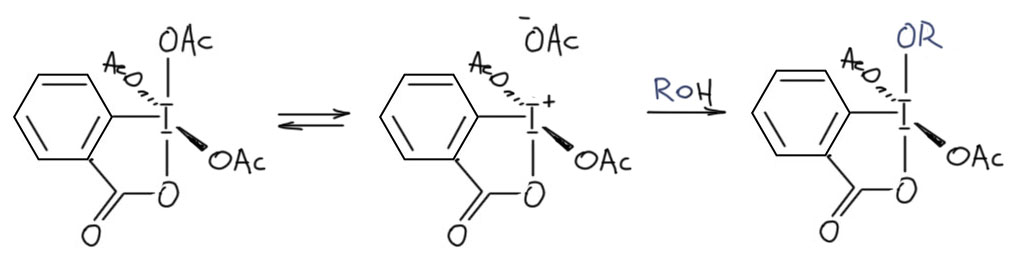
В этом месте получается небольшая проблема. Что дальше? Дальше нужно элиминировать из спиртового остатка действие основания, это легкая реакция, и годится и ацетат ион и что-то типа этого из реакции, но – поскольку это происходит на трехцентровой связи, то одновременно должна уйти с другой стороны группа с минусом. Но там цикл, а разрыв цикла невыгоден. Обращу внимание – не невозможен, а просто невыгоден. Всё же нарисуем.
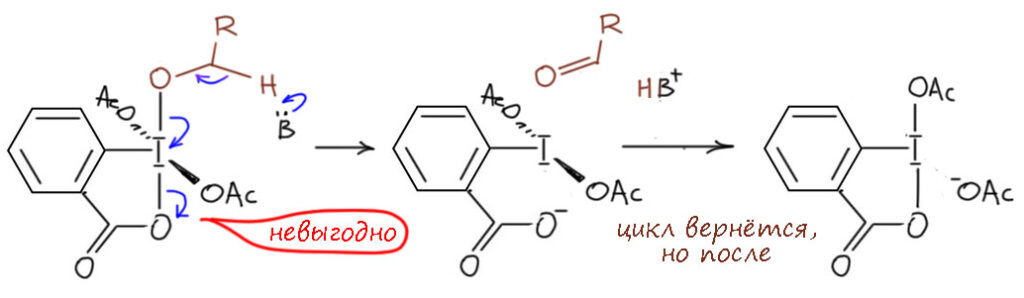
Скорее всего, именно это является причиной того, что окисление просто реагентом в сухом инертном растворителе типа дихлорметана идёт медленно, что подметили авторы реагента сразу. Они же и тоже сразу заметили, что если брать не один эквивалент спирта, а два, реакция идёт еамного быстрее, но это плохая идея, если спирт сложен, а этот метод почти всегда применяют к очень сложным и дорогим молекулам. Пробовали просто добавить эквивалент этанола или трет-бутунола, и это давало ускорение, но работало ненадёжно. Огромное и принципиальное улучшение метода сделали в 1994 году знаменитый американский всё-в-одном (синтетик, биохимик, биолог, медхимик, бизнесмен) Стюарт Шрайбер и Стефани Мейер, занимавшиеся тогда синтезом и исследованиями рапамицина. Статья про влияние воды на окисление периодинаном едва ли не перекрыла по цитированию исходные статьи Десса и Мартина, что неудивительно, так как именно она дала настоящиую путёвку в большой синтез этому реагенту (Meyer, S. D.; Schreiber, S. L. J. Org. Chem. 1994, 59, 7549-7552). Статья собственно проста как гвоздь и именно этим хороша – авторы задались вопросом, почему многие жалуются на плохую воспроизводимость окисления новым реагентом – а к этому моменту уже сотни статей так или иначе пробовали, и сами Десс и Мартин дополнительно его исследовали и пытались разобраться в проблемах. Появилось несколько улучшенных методов синтеза, но всё это окончательно проблем не решало. Мейер и Шрайбер нашли, что дело в… воде. Все, кто увлекается особо аккуратным экспериментом – все растворители свежеабсолютированные, посуда прожарена в вакууме, реактивы высушены и так далее – имели проблемы с тем, что окисление бывало медленным, а выходы далеки от желаемых. Но стоило просто дать дихлорметану “подышать” – постоять несколько минут в открытом сосуде, как реакция в таком растворителе ускорялась. Ну и если просто добавить эквивалент воды, то ускорение становилось значительным, а результаты воспроизводимыми, причем становилось не так важно, каким методом получали DMP. Очевидно, что дело в том, что одна ацетокси-группа замещалась на гидроксид (точно так же как в присутствии лишнего спирта – на алкоксид)ю Дальше я чуть-чуть додумаю за авторов той работы, потому что они всё свалили на более донорный гидроксид или алкоксид вместо ацетата – он, типа, облегчает реакцию. Я уверен, что дело именно в гипервалентных связях. Эти группы блокируют трехцентровую связь, включающую карбоксил кольца и начинает работать ацетат с другой связи – там чуть-чуть медленнее само замещение на остаток окисляемого спирта, но зато никаких проблем в элиминировании – второй ацетат отваливается синхронно.
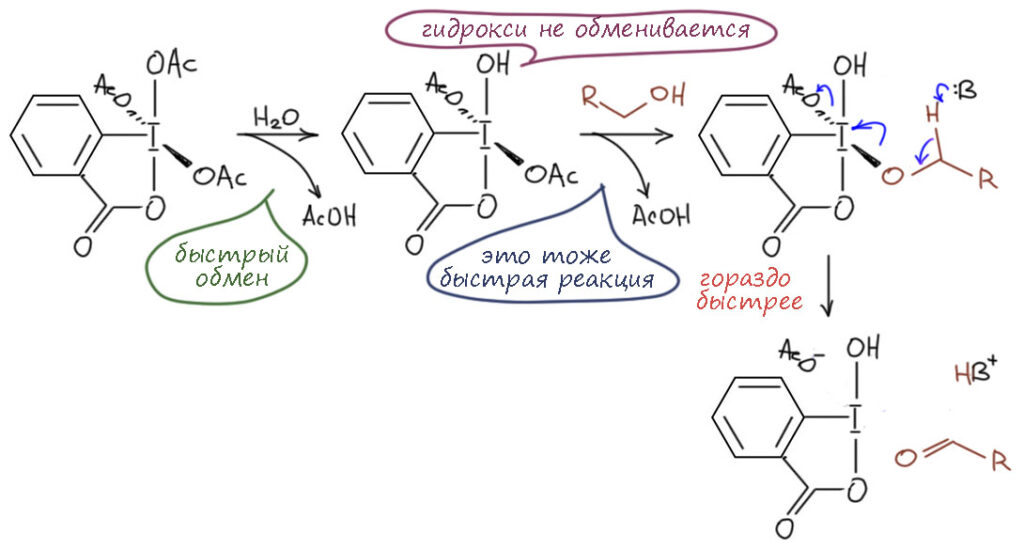
Метод Мейер-Шрайбера был отлично принят. Дальше все, кто применял DMP имели выбор. Если по каким-то причинам вода неприемлема, реагент в принципе работает и так, но часто требуются избытки, иногда кратные. Если нет проблем в воде, добавляют расчетное количество микрошприцем. Или используют ещё один остроумный приём – добавляют кристаллический бикарбонат натрия. Бикарбонат всегда содержит на поверхности немного воды для затравки, а дальше кадая высвобождающаяся молекула AcOH добывает из пищевой соды молекулу воды – получается просто идеальная стехиометрия, а избыток бикарбоната никому не вредит – реакции окисления DMP в конце обычно водным бикарбонатом и гасят. Вот эта методика – эквивалент DMP с маленькой горкой (10-25% избытка) в сухом дихлорметане и NaHCO3 на дне, крутим при комнатной температуре обычно несколько часов и гасим или водным бикарбонатом или водным тиосульфатом.
Ещё один вопрос – а что за основание снимает протон? Здесь такая же история, как и в других окислениях по тому же механизму элиминирования (все хромовые реагенты, системы Моффатта-Сверна) – любая частица, имеющая атом с неподеленной парой. Легкость этих реакций обеспыивают уходящие группы (производное 4-хвалентного хрома, диметилсульфид, трехвалентный иодан), это настолько выгодно, что даже в кислой среде, как у некоторых хромовых реагентов, это происходит легко. Уксусная кислота – замечательно, годится. Но в окислениях DMP особенно в присутствии бикарбоната это уже вполне полноценный ацетат-ион. Нередко эти окисления проводят в присутствии пиридина или лутидина – всё это обеспечивает дополнительную мягкость. Не исключен и вариант, когда работают какие-то атомы кислорода или азота прямо из субстрата, в этих окислениях очень часто берут сложные многофункциональные молекулы. Ещё один вариант, который рассматривают, но без доказательств – внутримолекулярное отщепление за счет другой ацетокси-группы в DMP. Всё это неважно, и в конкретных случаях немного варьируют состав системы. Так или иначе, DMP это точно самый универсальный реагент окисления первичных и вторичных спиртов в альдегиды и кетоны.
Зелёное окисление
Мы рассмотрели практически все важные и полезные методы окисления спиртов до карбонильных соединений, и нужно было бы остановиться. Но, у нас тут 21 век, и новые проблемы. Химия должна стать зелёной и отвечать задачам устойчивого самоподдерживающегося развития (об этом лекция в курсе про 21 век, возможно, ещё не выложенная). И все методы химии приходится пересматривать. Если бы химия оставалась в лаборатории для исследований, то может быть и проблем бы не было, но современная химия обязательно имеет в виду возможность применения для практического синтеза, в первую очередь, лекарственных субстанций. И там новые требования уже жестко определяют перспективы: хотите, чтобы ваш синтез мог быть реализован в серьёзных масштабах и приносил пользу – извольте доводить его до современных требований. А окисление спиртов чрезвычайно часто применяется в синтезах, хотя сейчас есть тенденция редокс-экономии, то есть ограничения окислительно-восстановительных стадий в синтезах, но это пока ещё редко удается последовательно реализовать. И вот, ревизия методов окисления спиртов показала неутешительный результат – все они никуда не годятся с зелёной точки зрения. Хромовые методы используют избытки реагента, шестивалентный хром очень токсичен, много отходов. Методы Моффатта-Сверна используют агрессивные токсичные реагенты один другого хуже. Периодинан при масштабировании опасен, взрывчат, испльзует стехиометрические количества производных редкого и рассеянного элемента иода. Для лаборатории все это нормально, но в современном промышленном синтезе чревато большими издержками. Среди хорошо исследованных методов окисления спиртов оказался только один более-менее проходящий по требованиям зелёной химии. И мы им сейчас займёмся. В принципе, метод и сам по себе неплох, хотя довольно сильно уступает особенно периодинану в универсальности и селективности, но при этом имеет возможности дополнительной доводки и усовершенствования. И нам этот метод тем более интересен, потому что в своей основе открыт в России, в далёкие советские времена, в начале 1960-х, хотя как обычно до практичной и удобной методики был доведён в других странах.
Метод использует очень интересные реагенты, с которыми и просто так неплохо познакомиться, потому что у них всякого интересного воз. Прежде всего это аминоксильные радикалы, самым знаменитым представителем которых является TEMPO. TEMPO был открыт профессором Семёном Николаевичем Казарновским и его аспирантом Олегом Львовичем Лебедевым в Нижнем Новгороде (в СССР город Горький) (Лебедев O.Л., Казарновский С.Н. Каталитическое окисление алифатических аминов перекисью водорода. II ЖОХ, 1960, 30, 1631-1635). Вещество это чрезвычайно удачное по совершенно потрясающему сочетанию свойств и реакционной способности, и по лёгкости получения из простых исходных. Исходное – продукт самоконденсации ацетона форон, который даёт с аммиаком двойной аддукт Михаэля, его восстанавливают модификацией Хуана реакции Кижнера-Вольфа и получают очень полезный стерически затруднённый амин, тетраметилпиперидин (TMP), а его уже Лебедев и Казарновский окислили перекисью водорода в присутствии вольфрамата. Доказательство того, что полученное вещество не что иное как стабильный свободный радикал было получено сильно не сразу, и, судя по всему, случилось тогда, когда другой исследователь, А.В. Ильясов применил метод электронного парамагнитного резонанса, построив для этого самодельный спектрометр (серийных тогда в СССР просто не было).
Название TEMPO появилось сильно позже. С одной стороны это вроде бы актроним, ну почти – от тетраметилпиперидина и кислорода. С другой, а зачем там буква Е? Я не знаю и пока не нашёл, кто первым запустил это эффектное название, но учитывая что дальше в химии этого радикала будет много итальянцев, я подозреваю, что это их мозгов дело: tempo в итальянском слово многозначное, это и время во всех смыслах, и темп, и даже погода. Многозначительный как бы акроним это большое дело для популярности вещества. Много было в химии 20-го века всяких стабильных радикалов и ловушек, но пришло время TEMPO, и все остальные отстали, потеряли темп.
К окислению спиртов это соединение тоже приставили советские учёные, из Института Химфизики, понятно почему – это школа Николая Семёнова, нобелевского лауреата за исследование свободнорадикальных цепных реакций, и свободными радикалами в этом институте занимались и занимаются многие, совершив много впечатляющих открытий. Весьма широкие исследования стабилизированных нитроксильных радикалов проводили в лаборатории Моисея Неймана, который, увы, скончался в 1967 году, и дальше исследования вёл Эдуард Розанцев и сотрудники. Кроме прочего, кажется, они переоткрыли метод получения нитроксилиных радикалов из аддукта форона с аммиаком и разных производных, так что можно подозревать небольшой спор о приоритете между Горьковской и Московской группами, во всяком случае на это похоже, если посмотреть как они друг друга цитируют. Ничего страшного, в науке такие тёрки дело обычное и только говорит, что тема хорошая, копать не перекопать, и каждому хочется быть первым во всём. В группе Розанцева тогда сделали работу, которая стала сенсацией – молекула со свободнорадикальным центром может реагировать по другому реакционному центру с сохраненим радикала – то есть можно прямо как-то модифицировать свободнорадикальный реагент. Реакций было множество – карбонил наверху (это предшественник TMP c карбонилом, он точно так же даёт нитроксильный радикал) реагировал со всем, чем обычно реагирует карбонил, а нитроксильный радикал на всё это чихал, и сидел между своими четырьмя метилами как у Христа за пазухой, и не обращал внимания на возню с карбонилом (Э Г. Розанцев, В. Д. Шолле Успехи Химии, 1970, 40, 417; Rozantsev E. G., Sholle V. D. Synthesis 1971, 401). Я хорошо помню, как заметки про это открытие тогда публиковали популярные научно-популярные журналы Наука и Жизнь и Химия и Жизнь. Дело выглядело удивительным – все же были в убеждении, что свободнорадикальные частицы настолько активны именно за счет неспаренного электрона, а тут получалось, что его можно обойти. На мой взгляд, как раз это немного преувеличено: поскольку свободный радикал сволне стабилен, что мещает реакции по другой функциональной группе, например, какой-нибудь обычной карбонильной химии. Ну, ничего, мы и сейчас видим, как в науке раздувают из мух слонов, этим с утра до вечера занимаются все химические гламурные журналы, а журналы типа Science и Nature только этим и занимаются. А тогда это была большая редкость, что про работу в химии можно написать привлекательную заметку в научпоп – в СССР тиражи НиЖ и ХиЖ были миллионными. Попутно исследовали и другие свойства и реакции нитроксильных радикалов, в частности обнаружили, что нитроксильные радикалы окисляются например солями меди(2+) или хлором и продукт окисления сам окисляет спирты в альдегиды (реакцию вели в метаноле, который окислялся в формальдегид, также пробовали этанол и изопропанол). Этми исследованиями занимался молодой химик Валерий Голубев и именно с этой работы (В. А. Голубев , Э. Г. Розанцев , М. Б. Ηейман, Изв. АН СССР, сер. хим., 1965, 1927) и надо начинать приоритет открытия реакции окисления спиртов в карбонильные соединения с участием TEMPO и других нитроксильных радикалов и стехиометрического реокислителя, назовём это реакцией Голубева-Розанцева-Неймана, но так не называют потому что эти ученые не были органиками, и никак не могли ставить перед собой задачу придумать новый метод органической химии, да и вообще, были настолько увлечены реакциями с сохранением радикального центра, что на всё остальное внимания не обратили. Но то так и осталось забавным курьёзом, а эта реакция стала прототипом метода, который грозится в новом веке отправить на свалку истории химии и весь хром, Джонсона, Коллинза, Кори, и Сверна с Моффаттом, и даже Десса и Мартина. Кто ж знал, что нам придётся быстро-быстро зеленеть.
В настоящий метод органической химии эту реакцию стал превращать крупный американский химик Мартин Земмельхак. Он полностью и исчерпывающе признал приоритет Голубева и сотр. в открытии самой этой химии (первая ссылка в его основной работе по теме), и принялся делать именно метод для использования в синтезе. Сначала было использовано электрохимическое окисление с каталитическим количеством TEMPO, а затем и системы с химическими окислителями типа хлорной меди, если стехиометрически, или каталитического количества хлористой меди в атмосфере кислорода. Земмельхак подтвердил, что TEMPO одноэлектронно окисляется до катиона, который сначала можно нарисовать с плюсом на кислороде, но поскольку это секстетный и крайне невыгодный кислород, то очевидно, что гораздо лучше аммонийная форма (её поэтому часто называют оксоаммониевым катионом)
Этот катион окисляет первичные спирты намного быстрее вторичных. В стехиометрическом соотношении первичный алканол количественно окисляется за 5 минут при -60ºC. В каталитических количествах TEMPO служит эффективным катализатором окисления, и вполне хорошо работает при комнатной темппературе. Земмельхак же предложил механизм окисления (Semmelhack, M. F.; Schmid, C. R.; Cortes, D. A. Tetrahedron Lett. 1986, 27, 1119). Я думаю, что мы сами сможем сделать то же самое, если немного подумаем, и получим то же самое. Посмотрим на катион и подумаем, на что он похож. Азот с плюсом изоэлектронен углероду.

Значит эта группа изоэлектронна карбонильной, а вся молекула изоэлектронна циклогексанону, естественно, с 4 метилами. Карбонильный углерод – электрофил. Значит, тем более будет электрофилом положительный азот. Стоп! Нас же учили, что аммонии неэлектрофильны, так как имеют полный октет и им ничего не нужно. Верно, но здесь двойная связь на кислород с хорошим потенциалом смещения электронной плотности в сторону кислорода и освобождением места для входящего нуклеофила. Следовательно, такая группа точно так же как карбонильная может присоединить нуклеофил, например, спирт. Скорее всего, даже лучше, так как электрофильность азота с положительным зарядом должна быть больше. Изобразим, заодно перерисовав шестичленный цикл, как положено, креслом.
Осталось нарисовать уже надоевшее элиминирование. Уходящей группой будет нейтральная молекула, циклический гидроксиламин, вполне выгодно, и очень похоже особенно на то, что происходит в методах, где уходит диметилсульфид.
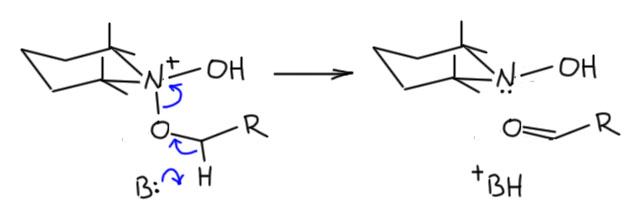
Реакция быстро идёт в основной среде, и сильно зависит от стерики, и это очевидно, учитывая, что спирт должен присоединиться к электрофильному азоту, что сопровождается регибридизацией из sp2 в sp3, и страдает от стерических препятствий от четырёх метильных групп, особенно тех, которые в аксиальных положениях. Все эти работы были интресны, но в органической химии никакого особенного резонанса не вызвали – это была какая-то физхимия, электрохимия, и прочая, с точки зрения органика-синтетика, неинтересная дребедень.
Но в 1987 итальянские химики Пьер-Лючо Анелли и сотрудники из Милана сделали очень удачное улучшение метода (Anelli P. L., Biffi C., Montanari F., Quici S. J. Org. Chem. 1987, 52, 2559), создав ему совершенно блестящие перспективы на будущее. В тот момент мало кто думал о том, что скоро начнётся жестокая борьба за зелёную химию, и ревизии подвергнутся все существующие методы, а преимущество получат те, которые создают меньше отходов, используют более простые реагенты, желательно в каталитических количествах и так далее. Анелли – химик весьма знаменитый, один из тех бедняг в химии, которые были в шаге от нобелевской премии, можно сказать, что следующие в очереди: он был соавтором в крайне амбициозном исследовании вместе с Фрейзером Стоддартом под названием Молекулярный челнок (Pier Lucio Anelli, Neil Spencer, J. Fraser Stoddart A molecular shuttle J. Am. Chem. Soc. 1991, 113, 5131–5133), причём итальянец, видимо, и был основным автором этой идеи, из которой скоро выросло чрезвычайно модная химия молекулярных машин, за которую дали в 2016 году нобелевку Стоддарту и ещё двум учёным, но не Анелли, которому зато от Стоддарта досталось почётное прозвище Мистер Молекулярный Челнок, чтобы не сильно расстраивался. В общем, с нобелевкой у Анелли дело не выгорело, но вот казавшаяся куда менее амбициозной и почти проходной работа по разработке протокола окисления спиртов прославила его намного надёжнее, уж больно хорош оказался протокол. Подчеркну это забавное обстоятельство: Анелли не открывал реакцию, это сделали советские химики Голубев и сотрудники. Анелли не исследовал реакцию с точки зрения механизма и условий перевода в каталитический режим – это сделал Земмельхак. Он всего навсего придумал, как сделать ее просто, дешево и надежно – и именно это сыграло главную роль в том, что реакция стала крайне перспективной и в новом веке практически основной с точки зрения практического окисления спиртов в альдегиды и кетоны.
Анелли с сотрудниками придумал использовать в качестве реокислителя для каталитического количества TEMPO обычный гипохлорит натрия, один из самых дешёвых окислителей в химии. Это то, что обычно просто называют отбеливателем, и продают в виде раствора, который перед использованием надо оттитровать, чтобы понять, какой там окислительный эквивалент. Реакция идёт в двухфазной систем, состоящей из органической фазы, в которой и идёт реакция окисления спирта оксоаммонийным катионом из TEMPO, и водной фазы, в которой находится стехиометрический окислитель, то есть гипохлорит, а также бикарбонат натрия, обеспечивающий слабощелочную реакцию. Желательно ещё добавление каталитического количества бромид-ионов, скорее всего, окисляющихся гипохлоритом до гипобромита, который и реокисляет гидроксиламин обратно в оксоаммониевый ион. Этот метод или по-модному протокол стали называть протоколом Анелли (не реакцией Анелли – это было бы сильно несправедливо, и в начале каждой статьи итальянцы описывают историю реакции и приоритеты Голубева и сотрудников, а также Земмельхака).
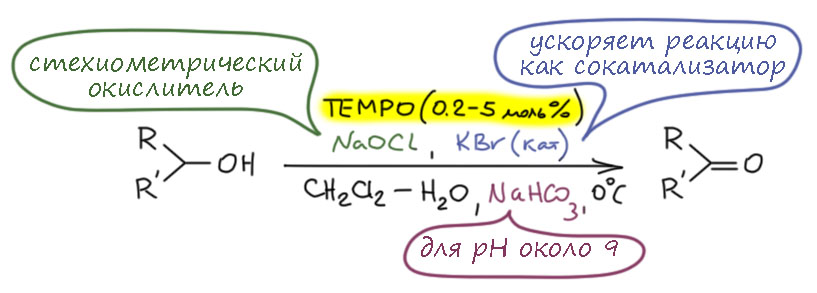
Система, как видим, весьма занятная. Это один из первых примеров того, что станет модно в современной химии – двухфазная система с разделением продуктов и реагентов по фазам: реакция идёт в органической фазе, где хорошо растворимы органические вещества, исходные и продукты. Вспомогательный процесс- реокисление каталитического окислителя (здесь, оксоаммониевого иона) идёт в водной фазе, где находится стехиометрический окислитель и прочие соли. Каталитический окислитель (TEMPO и его окисленная/восстановленная пара) обладает растворимостью в обоих фазах и так челночит между ними. Обратим внимание, что здесь нет в явном виде межфазного переносчика, и вся межфазная жизнь обслуживается растворимостью реагента (TEMPO и его пары) в обеих фазах. Это дополнительно обеспечивает очень высокую селективность процесса – окисляется сначала первиный спирт, затем вторичный, и обе эти реакции легко разводятся просто за счет количества реокислителя и увеличенного времени реакции, как в этом примере (Anelli P. L., Biffi C., Montanari F., Quici S. J. Org. Chem. 1989, 54, 2970):
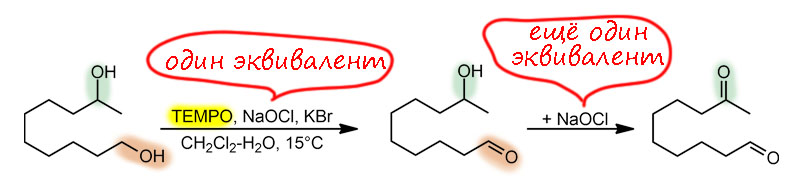
Больше того, если в смесь добавить межфазный переносчик, например, весьма популярный аликват-336, то, вероятно, за счет затаскивания гипохлорита в органическую фазу, увеличивается скорость реакции, и альдегид дальше окисляется до карбоновой кислоты. Надо только не прозевать при выделении, потому что значительная часть ее уйдёт в водную фазу в виде соли. И количество гипохлорита должно соответствовать по эквивалентам. Такое свойство окислителя весьма привлекательно, потому что иначе часто приходится делать окисление спирта в карбоновую кислоту в два приёма двумя разными реагентами.
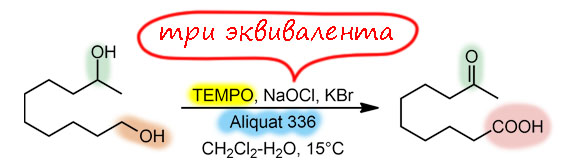
У протокола Анелли есть один действиельно серьёзный недостаток – если в молекуле спирта есть двойные связи, возмлжны реакции присоединения галогена и образование галогенгидринов как побочных. Несмотря на то, что гипохлорит и субстрат разведены по разным фазам, есть и частичная растворимость, и реакция на границе раздела фаз. Но система на основе TEMPO тем и хороша, что можно подбирать разные стехиометрические реокислители. Самый удачный выбор сделали другие итальянцы, но из Рима, из старого римского университета Ля Сапьенца, и так появился ругой популярный протокол Пьянкателли-Маргариты, в честь Джованни Пьянкателли, маститого итальянского химика, и его сотрудника Роберто Маргариты (Маргарита в итальянском это не женское имя, а в данном случае фамилия, а имя женское чуть другое – Маргерита, вот прямо пиццу Маргерита знают все , а она названа в честь королевы Италии Маргериты, одной из всего двух итальянских королев). Пьянкателли и Маргарита (вот уж прямо Мастер и Маргарита по-итальянски) заменили гипохлорит на другое производное галогена, только иода, фенилиодозодиацетат (это несчастное вещество как только не сокращают, и PIB, и PIDA, и BAIB и ещё как-то, но смысла нет, его проще целиком написать PhI(OAc)2, а о том, как это устроено, мы обсуждали на вкладке про Десса-Мартина.
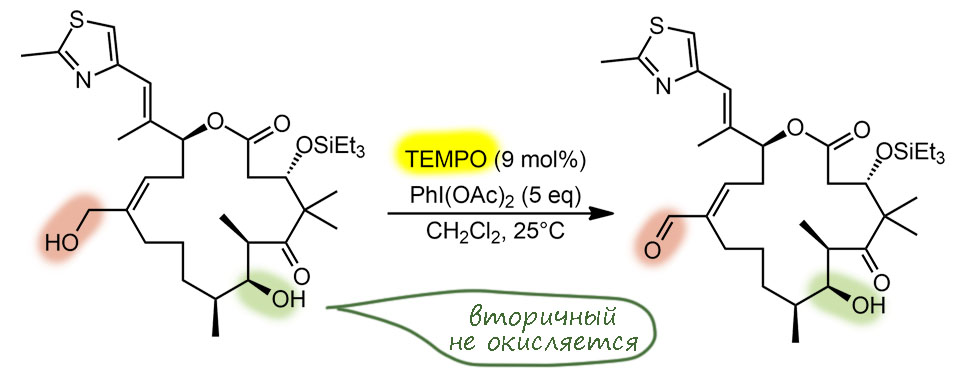
Chappell, M. D.; Harris, C. R.; Kuduk, S. D.; Balog, A.; Wu, Z.; Zhang, F.; Bom Lee, C.; Stachel, S. J.; Danishefsky, S. J.; Chou, T.-C.; Guan, Y.; J. Org.
Chem. 2002, 67, 7730