Алканы. Электрофильные реакции.
Основными реакциями алканов (и, в общем, насыщенных фрагментов в более сложных молекулах) являются свободнорадикальные реакции. Так было раньше, и остается и поныне. Радости от этого никто не испытывает. Алканы (углеводороды нефти и газа) – важнейшее, главное сырье химической промышленности, и в этом отношении ничего за последние сто лет не изменилось. Попытки использовать в качестве сырья для промышленного синтеза углекислый газ, уголь, и так называемые восполнимые источники, то есть готовые органические молекулы из природы, до сих пор успехом не увенчались. Важные синтезы на основе этих типов исходных, конечно, есть, но по сравнению с нефте и газохимическим синтезом это жалкие крохи. Алканы – сырье незаменимое, и поэтому тем более обидно, что для основного типа их реакций, свободнорадикального замещения, характерная низкая селективность, трудность управления, энергоемкость и прочие такие же качества, заставляющие страдать любого современного промышленника – ведь все это плохо совместимо с современными требованиями к химическим процессам, приводит к росту издержек, снижению прибыли, и т.п. На поиск новой химии алканов выделяются колоссальные средства, любые новые идеи привлекают огромное внимание. Но пока успехов не очень много. В этом разделе мы посмотрим на основной альтернативный способ заставить алканы реагировать. Разработка этого способа принесла нобелевскую премию в 1995 году Джорджу Ола, который действительно вложил огромные усилия в то, чтобы заставить эту химию работать. В некотором смысле попытка удалась, и небольшой кусочек у свободнорадикальных реакций отвоевать удалось. Так как кусочек этот действительно небольшой, этот раздел можно пропустить всем, кроме тех, кому действительно интересно узнать, как это устроено. Формально такой вопрос есть в программе и билетах, но общий вес его так мал, что никакого невосполнимого вреда от того, что вы это пропустите не будет. И даже если эта химия вас заинтересует, никогда не применяйте ее для решения задач синтеза – в этой химии нет практически полезных методов лабораторного органического синтеза. Интерес к этой химии обусловлен решением задач промышленного синтеза, и научно-познавательными целями.
Основные идеи этого типа химии таковы.
- Связь C-H на насыщенном атоме углерода может быть местом атаки электрофильных реагентов, следовательно алканы являются нуклеофилами и основаниями, хотя и чрезвычайно слабыми;
- Чрезвычайная слабость алканов как нуклеофилов и оснований вынуждает использовать только самые мощные и реакционноспособные электрофилы, а обычные электрофилы, с которыми мы имеем дело, например, в химии алкенов, алкинов, ароматических соединений, с алканами не реагируют;
- Реакции этого типа относятся к реакциям замещения;
- Они очень часто осложняются перегруппировками, изомеризацией и разрывом скелета;
- Основной тип промежуточной частицы, участвующей в механизме электрофильных реакций алканов, – так называемый гиперкоординированный катион с трехцентровой связью, выглядит довольно необычно, но очень прост в обращении, так как всегда дальше вступает только в одну реакцию. Химии этой поэтому не стоит бояться, она гораздо проще свободнорадикальной.
Теперь, как всегда, пойдем по порядку. Жмите на заголовки.
Самый простой случай - реакция метана с протоном
Как мы уже выяснили, разбираясь с кислотами и основаниями, протон (несвязанный протон, протон как таковой, а что это такое поговорим отдельно) обладает невероятно высокой кислотностью, и по сравнению с протоном все остальные молекулы являются основаниями (ну, почти все, за инертные газы я не ручаюсь, но если мы ограничимся только органическими молекулами, то можно не сомневаться – все). Для большинства органических молекул написать, что получается при протонировании, очень легко: если есть атом с неподеленной парой – сажайте протон на него, если есть кратная связь в любом виде – на нее. А если нет ни того, ни другого, как в алканах и самом простом алкане, метане, – то куда сажать протон? Протон не имеет собственных электронов, и для взаимодействия с другими атомами ему понадобятся чужие электроны. Молекула метана так проста, что найти в ней электроны для образования связи можно только одним способом – взять их из связи C-H. Больше в молекуле метана ничего нет.
Но вопрос, что при этом получилось, не так прост. С точки зрения стехиометрии все просто – присоединение протона к метану дает катион CH5+ точно так же как протонирование аммиака дает катион аммония. Катион из метана по аналогии называют метонием (почему метонием, ведь исходная молекула метан? – формально потому что окончание –оний всегда означает результат протонирование, а –ан в слове метан не входит в корень слова). Но как это устроено? Самая простая мысль – по аналогии с аммонием на углероде висит пять атомов водорода, неверна, потому что у углерода нет возможностей обслужить пять независимых связей, ведь для этого потребовалось бы пять своих электронов, а Таблица Менделеева выделяет углероду всего четыре. Углерод не может образовать больше четырех связей. Тем не менее, просто исходя из формулы катиона можно сказать, что углерод в нем пятивалентен. И это верно, потому что понятие “валентность” приехало к нам из XIX века, когда химики ничего не знали о природе химической связи, и просто считали число ближайших соседей атома, связанных с ним связью. Атомов ближайших пять, и связью они точно связаны, иначе эта штука позорно развалилась бы, не успев образоваться. Что-то не то – ведь и валентность, несмотря на всю свою формальность, должна подчиняться Периодическому Закону, а в нем установлена верхняя граница валентности для атома, равная номеру группы. Это все верно, но отлично известно такое явление как гипервалентность, но оно характерно для элементов от 3 периода и ниже, уже познавших радость обладания доступными и пустыми d-оболочками. Для элементов 2 периода это явление нехарактерно, и если фактически наблюдается что-то подобное, как в катионе метония и его родственниках, то объяснение должно быть более сложным. Мы сейчас этим и займемся, но прежде замечу, что формально метоний все же является примером гипервалентности. Слово “валентность” в наше время стремительно выходит из моды и считается данью старине и традиционным ценностям, а вместо него предпочитают использовать термин “координационное число”, взятой из химии комплексных (координационных) соединений. В этом смысле любая сложная молекула есть комплекс, в котором есть центральный атом и лиганды. В данном случае лигандами придется счесть атомы водорода. И опять та же история – и в этих терминах углероду не положено иметь больше 4 лигандов, а поскольку их фактически пять, то такой атом углерода принято называть гиперкоординированным, или даже, с легкой руки нобелевского лауреата Джорджа Олы, просто гиперуглеродом.
Теперь о фактической стороне дела. Как углерод не называй, но в отличие от слов, которые можно изобретать, ничем себя не стесняя, количество электронов, валентных оболочек и прочих инструментов для создания химических связей жестко ограничено структурой атома. И связей у углерода не может быть более 4. Далее возникает простой вопрос – пять атомов водорода в метонии равноценны и неразличимы или нет? Однозначно удалось установить, что это не так, и что в метонии пять делится на три плюс два, и эти два отдельных водорода держатся вместе друг с другом и с атомом углерода особым типом связи. Здесь обязательно нужно остановиться и весьма громко заявить общеизвестную истину, что химической связью в химии принято называть взаимодействие между двумя атомами, и не любое взаимодействие, а только такое, которое удерживает эти атомы рядом. Химическая связь поэтому по определению двухцентровая. И когда мы рассуждаем о всяких делокализованных системах, мы не имеем в виду образование одной многоцентровой связи, а говорим только о делокализации электронной плотности по системе связей, каждая их которых является двухцентровой. И вот впервые, когда мы думаем об устройстве катиона метония, нам не подходит определение химической связи как двухцентрового взаимодействия между двумя атомами. В катионе метония приходится представить себе трехцентровую связь между углеродом, водородом и водородом. Это одна связь. Это не вполне ковалентная связь – она обслуживается парой электронов, но эта пара пришла не поровну от каждого из участников, а взята из бывшей ковалентной связи C-H, а протон предоставил пустую орбиталь. Поэтому это скорее координационная связь донор (C-H) – акцептор (протон). В этом смысле это очень похоже на связь в аммонии, где донор (пара на азоте) и акцептор тот же протон. Но только не двухцентровая, а трехцентровая. Изображать такую связь приходится специфическим способом, рисуя такую пунктирную рогатину, направленную на все три связанных центра. Это очень дрянной способ, потому что в центре рогатины приходится рисовать точку, и начинает казаться, что в этой точке что-то есть – какой-то очень важный центр, какой-то пуп связи. Но это иллюзия, ничего там нет, кроме электронной плотности, но этого добра там везде полно, и в середине нет ничего особенного, нет даже максимума плотности. Других способов нарисовать трехцентровую связь изобрести не удалось даже нобелевскому лауреату, и приходится с этим смириться. А почему рисуют пунктир, ведь пунктир намекает на делокализацию, которой в этой связи нет, и не рисуют нормальные сплошные черточки? Просто чтобы не путать с обычными двухцентровыми связями. Смиримся. Привыкнем.
С точки зрения электронной структуры трехцентровая связь образуется взаимодействием связывающей орбитали связи C-H (состоит из гибридной орбитали углерода и s-орбитали водорода, которые перекрываются с одной фазой – обе на рисунке синенькие – и вдоль линии связи C-H, поэтому это σ-орбиталь) и пустой s-орбитали протона. Взаимодействие осуществляется с одной фазой – на рисунке все перекрывающиеся части синенькие, следовательно, новая орбиталь тоже связывающая. Более того, это тоже σ-связь, так как главный признак σ-связей – перекрывание осуществляется в области простанства между связывающими атомами. Вот и все. Напоминает, как указал один остроумный господин, пьющую мышь, вид сверху.
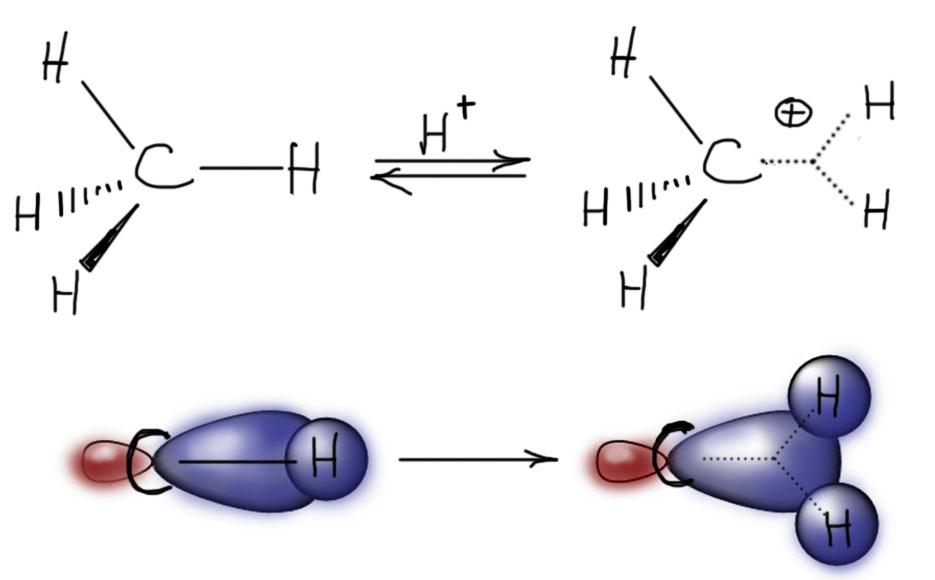
Таким образом, получается вполне нормальная химическая связь. Но, чудес не бывает, связать три атома одной парой электронов получается далеко не так хорошо, как два атома. Трехцентровые связи слабее обычных двухцентровых. Метониевый катион малоустойчив и с большим удовольствием распадается. Он легко образуется в газовой фазе и там молекулы метана охотно протонируются хорошими источниками протонов. Но если и там метоний встречается с органическими молекулами, обладающими хотя бы немного более значительной основностью чем метан, метоний легко их протонирует. На этом сонован один из методов масс-спектрометрии, называемый химической ионизацией.
В растворах, даже при очень низкой температуре, катион метония надежно поймать так и не удалось. Мы только предполагаем его образование, но как только он образуется, тут же распадается. Зачем же тогда вообще нужно об этом говорить?
Распад метоний-катиона и других молекул с трехцетровыми связями.
В трехцентровой связи три центра, но она никогда не распадается на три части по очень простой причине – нельзя два электрона разделить на три. Электроны неделимы. Это элементарные частицы. Мощная мысль товарища Ленина про то, что “электрон так же неисчерпаем как атом” оказалась столь же полезной для человечества, как и все остальные идеи этого невероятно самоуверенного субъекта. Но хотя бы жертв и разрушений было меньше.
Остается только один вариант – распад на две части. Например, на те, из которых она и образовалась. Но это было бы совсем неинтересно. Но фокус состоит в том, что есть не один, а три способа, которыми три может распасться на два плюс один. Нарисуем это в общем виде:
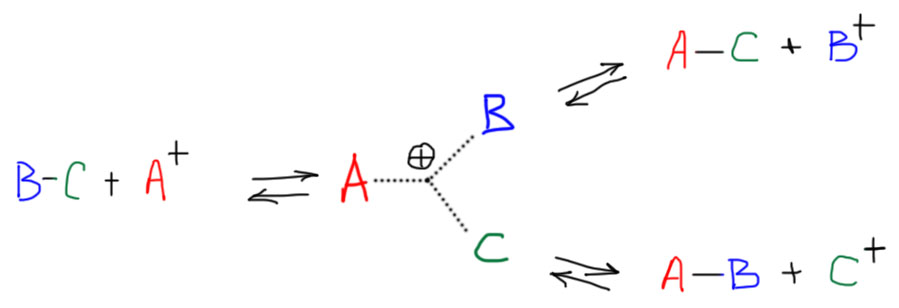
Получается прелюбопытнейшая ситуация – трехцентровой катион может как образоваться, так и распасться тремя разными способами. В этом смысле он симметричен относительно своих частей. При этом никто не говорит, что три пути распада (и образования) равноценны и равновероятны, но они возможны. И, как показали многочисленные эксперименты с большим количеством таких ионов, действительно реально осуществляются.
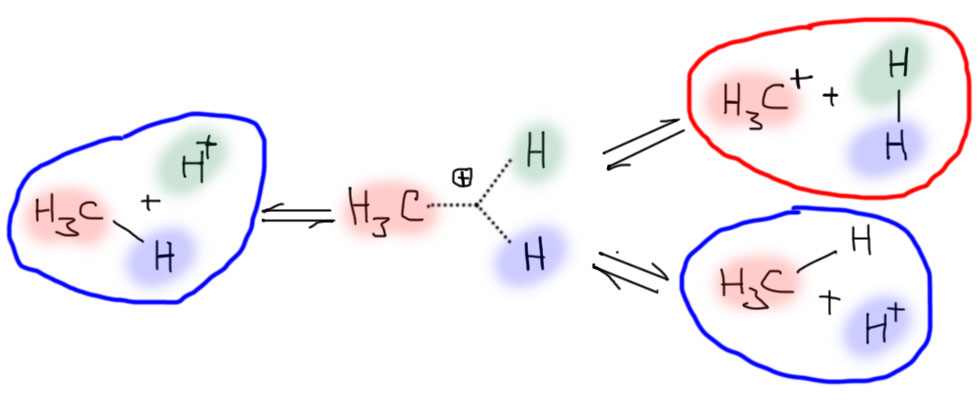
Попробуем применить эту схему к катиону метония. Получим три пути распада, которые одновременно являются и путями образования метония. Два из них (обведены синим) одинаковы и нам уже хорошо известны: метоний получается протонированием метана, и распадается на метан и протон. А вот третий (обведен красным) может удивить – метоний распадается на метильный катион и молекулярный водород. Но это прямо следует из общей схемы, и придется с этим смириться. Но это не фантазия, и не обычный в науках фантом, возникающей из-за избыточного доверия к абстрактным моделям – этот путь реализуется реально, и был неоднократно доказан экспериментально и в прямом, и в обратном направлении. Карбокатионы действительно обладают способностью реагировать как с молекулярным водородом, так и с разными веществами, содержащими атомы водорода в таких положениях, чтобы после отщепления водорода в виде протона образовывался бы более-менее стабильный другой катион.
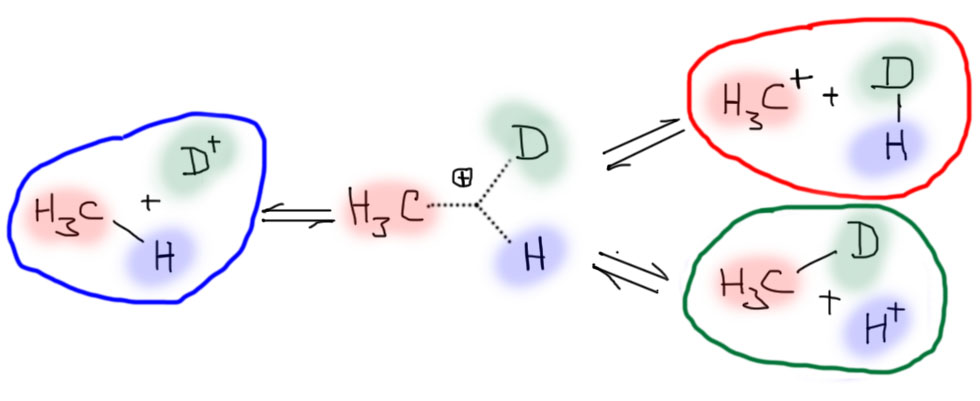
Два одинаковых пути распада метония тоже можно развести, если применить обычный прием – использование изотопов. Возьмем вместо обычного протона его дейтериевый аналог – дейтерон. Видим очень любопытный сценарий: в результате взаимодействия обычного метана с дейтероном должен происходить изотопный обмен, и частично получаться метан дейтерированный. Это можно было бы легко зафиксировать экспериментально, так как молекулы с дейтерием и без легко различимы, например, с помощью масс-спектрометрии – масса дейтерированного метана на единицу больше массы обычного. А дейтерообмен пойдет и дальше, появится дидейтеро-, тридейтеро- и в конце концов, полностью дейтерированный метан. Но кроме того в реакционной смеси должен получиться молекулярный водород, да не обычный, а “полутяжелый”, HD. И метильный катион, с которым дальше тоже что-то произойдет. Но пока что у нас одна проблема: метан – это понятно что такое, Газпром вам его нацедит кубическими километрами. А вот что такое протон, хоть обычный, хоть дейтерированный. Где дают протоны? Один ответ очевиден – на границе Франции и Швейцарии на большой глубине работает Большой Адронный Коллайдер, в котором по огромному кольцу летают настоящие протоны. Может можно сцедить немного в пузырек? Увы, нет. Они там все строгого учета, каждый посчитан и пронумерован, да и за многие годы работы БАК общее количество протонов не достигло и наномоля. Приличные химики таких количеств даже не замечают. В следующем блоке разберемся, откуда берут протоны для протонирования метана.
Суперкислоты.
Протон в химии – это синоним слова кислота. В разделе Кислоты и основания можно найти краткое введение в то, как устроена кислотность и основность в органической химии. Из этого раздела ясно, что никаких протонов в органической химии нет, а есть кислоты, то есть молекулы, способные отдать протон основаниям. Чем слабее основание, тем более сильная требуется кислота. Кислоты так и ранжируют, используя набор оснований-индикаторов от самых сильных до очень слабых. Каждую новую кислоту пробуют с этими основаниями-индикаторами. Но сегодня нас заинтересовало такое невероятно слабое основание как метан. Проблема в том, что известные наборы индикаторных оснований включают самые слабые основания типа 2,4,6-тринитроанилина (пикрамида), который остается незапротонированным в растворе концентрированной серной кислоты, но и этому основанию далеко до метана по слабости.
Получается интересная вещь. В водных растворах есть понятие “сильная кислота”, и там все сильные кислоты одинаковы, между ними нет никакой разницы кроме молекулярного веса. Как только мы в водном растворе прибываем на тот конец шкалы кислотности, где находятся сильные кислоты, то обнаруживаем, что дальше ехать некуда – это предел. А в неводных средах все только начинается. Сначала немного поупражняемся в словах и терминах.
Во-первых, что такое среда. Среда – это все, что находится в растворе, не только растворитель, но и все остальное. Из этого всего часто мысленно удаляют компоненты, которые оказывают слабое влияние на свойства среды. Например, если мы изучаем поведение метана в растворе в присутствии серной кислоты, то средой будет раствор серной кислоты во взятом растворителе, а метан можно не учитывать – он слабо влияет на свойства среды. В общем, органические вещества, которые мы берем для какой-то реакции в среду обычно не включают, а растворитель, кислоту, основание – включают.
Во-вторых, поскольку сейчас мы говорим о кислотах и кислотности, понятие кислотности относят не к самой кислоте, а к среде. Так и говорят – “кислотность среды”. Дело тут в том, что кислотность – это способность передавать протон основаниям. Если основание очень слабое, то протон от кислоты может перейти не к нему, а к растворителю, или зависнуть в самой исходной кислоте.
Растворитель активно ограничивает кислотность среды. Мы отлично видели это по воде, но то же самое, только в немного или много более широких рамках будет и в других растворителях. Скажем, если мы возьмем в качестве растворителя ацетон, то увидим, что это вещество отлично протонируется сильными кислотами, и в этом растворе (этой среде) протон не дойдет до более слабых оснований. Найти растворители, которые меньше ограничивали бы кислотность, очень непросто. Наиболее пригодны для этой цели очень слабоосновные производные 4- и 6-валентной серы – жидкий диоксид серы, или галогенангидриды серной кислоты типа SO2ClF. Эти жидкости почти не отвлекают протоны от решения задач по протонированию очень слабых оснований. Понятно, что растворители эти незавидные, вонючие, агрессивные, но как-то так вышло, что ничего лучше в смысле низкой основности создано не было, Творец не сильно увлекался химией и быстро переключился с растворителей на создание рыб, птиц, гадов, зверей, а также Адама и Евы. Так и живем. Гадов много, а растворителей мало.
Но даже если растворитель подобрать удалось, есть проблема с самой кислотой. Проблема эта имеет несколько граней. Возьмем серную кислоту, и это эталонная сильная кислота. Обычная концентрированная серная кислота имеет концентрацию 96%. Это значит, что в ней 4% воды. Вот, пустяк какой! Не пустяк. Это значит, что на каждый моль серной кислоты в концентрированной серной кислоте приходится почти 1/4 моля воды. И мы знаем, что протон очень хорошо взаимодействует с водой. Следовательно, концентрированная серная кислота, бесспорный эталон сильной кислоты в обыденном понимании этих слов, далеко не так сильна, так как содержащаяся в ней вода мешает протонам как следует разгуляться.
Хорошо, рецепт ясен, давайте уберем из серной кислоты эти остаточные 4% воды. К счастью, это довольно просто сделать, добавив расчетное количество олеума (раствора серного ангидрида в серной кислоте). Серный ангидрид жадно связывает воду до последней молекулы, и в результате мы можем получить жидкость, точно соответствующую формуле H2SO4. Эту жидкость часто называют моногидратом, имея в виду то, что это формально продукт взаимодействия одного моля серного ангидрида с одним молем воды. H2SO4 = SO3 + H2O. Но реально воды там нет, а есть чистейшая серная кислота. И действительно, кислотность моногидрата выше чем кислотность концентрированной серной кислоты, а это, напомню, значит, что моногидрат способен протонировать более слабые основания и в большей степени, чем концентрированная серная кислота. И именно моногидрат принято считать эталонной сильной кислотой. Моногидрат не протонирует метан в хоть сколько-нибудь заметной степени.
Отсюда следует еще один принцип:
нельзя добиться очень высокой кислотности, если в среде присутствует вода, даже в очень небольших количествах. Вода связывает протоны по рукам и ногам. Поскольку протон в отличие от электрона частица не элементарная, а очень даже составная, то найти в ней руки и ноги дело вполне реальное. Поэтому при работе с суперкислотами все очень тщательно обезвоживают, и строго защищают от влаги воздуха. Работать с суперкислотами в открытой посуде можно только днем посредине пустыни Сахара, когда влажность воздуха равна 0.
Кислоты, более сильные чем чистая серная кислота, то есть моногидрат, принято называть суперкислотами. Это очень старое понятие, очень скоро ему исполнится сто лет. Но применить его к органической химии догадался только нобелевский лауреат Дж.Ола и сравнительно недавно. Прежде чем мы перейдем к его достижениям попробуем разобраться, что делает кислоту более сильной чем моногидрат. Сильно, но не беспардонно упрощая картину, можно сказать, что силу кислоты ограничивает взаимодействие протона с анионом собственной кислоты. Именно в этом отличие “физического протона”, наматывающего круги в адронном коллайдере в совершенном одиночестве, от “химического протона”, который неотделим от кислоты, которая является его источником. В химии к протону всегда полагается нагрузка в виде аниона кислоты. Протон взаимодействует с этим анионом хотя бы чисто электростатически, но и химически также. И чем слабее эта связь, тем свободнее протон, тем сильнее кислота и среда, в которой она содержится.
Создать кислоту, анион которой совершенно не взаимодействовал бы с протоном невозможно. Электростатику в любом случае не отменить. Это значит, что любая кислота заведомо слабее свободного протона. Впрочем, в растворах свободных протонов быть не может, поэтому это небольшое горе. Но можно последовательно идти по пути снижения взаимодействия аниона и протона. Во-первых, для этого нам понадобятся делокализованные анионы с отрицательным зарядом, который рассредоточен по атомам электроотрицательного элемента, кислорода. И чем меньше заряд, тем лучше. Поэтому, например, хлорная кислота сильнее серной: в обеих заряд рассредоточен по 4 атомам кислорода, но в серной два минуса, а в хлорной один (по другому можно сказать, что серная кислота сильна только по первой ступени, а в анионе HSO4– атомы кислорода неодинаковы и делокализация не так равномерна, как в анионе перхлората). Но по этой дорожке дальше перхлората не уедешь, а хлорная кислота еще очень далека до высот кислотности. Дальше можно добавлять сильные акцепторы, типа фтора или перфторалкильных групп. Так получаем две популярнейшие суперкислоты – фторсульфоновую HSO3F и трифторметансульфоновую HSO3CF3 (или чаще пишут наоборот CF3SO3H, по-английски она коротко называется triflic acid, откуда анион называется даже и по-русски трифлатом). Обе кислоты исключительно сильны. Они уже очень близко подобрались к метану, и отлично протонируют более податливые разветвленные алканы. Трифторметансульфоновая кислота еще и очень удобна, например, не травит стекло, ее легко очистить и использовать повторно, и, наконец, она в какой-то степени органическая, а значит родная для органических химиков, которые часто инстинктивно недолюбливают неорганические вещества.
Все, и здесь уперлись. Нет больше индивидуальных кислот с еще большей кислотностью. Теоретически, такими кислотами могли бы быть не кислородные, а полифторные кислоты за счет еще большей электроотрицательности фтора. Что-то типа HBF4 или даже HPF6. И эти кислоты реально существуют, а их анионы очень хорошо известны и как раз и используются, когда нужен анион, обладающий ничтожной, почти неизмеримо малой основностью. Увы, кислоты этого типа известны только в растворах, обычно водных, а в чистом виде неустойчивы. Тем не менее, мы на правильном пути. Попробуем обойти эту сложность, сделав кислоту такого типа в растворе, но не водном, а в растворителе, который не мешает высокой кислотностью – мы уже их нашли. Так как такие кислоты не бывают чистыми и их нельзя взять и растворить, просто сделаем их, взяв безводную чистую HF (это страшная вещь, но среди химиков есть крутейшие герои, которые умеют с ней работать) и эквивалент кислоты Льюиса, например, BF3 в растворе, мы и получим такую кислоту и срду с очень большой кислотностью. Когда перебрали все такие фториды, нашли, что наибольшая кислотность образуется с фторидами элементов 5 группы, особенно хорошо оказался SbF5, но от него почти не отстают AsF5, TaF5 и некоторые другие. Такие смеси – протонная суперкислота плюс кислота Льюиса – дают колоссальный рост кислотности, и мы почти вплотную приблизились к вершине. Метан уже наш – эти смеси отлично его протонируют и вызывают превращения гиперкоординированного катиона метония.
Но самой вершиной кислотности со слов нобелевского лауреата принято считать другую пару, составленную, в принципе, точно так же, только вместо HF берется HSO3F. Эквимольная смесь этой суперкислоты и кислоты Льюиса SbF5 в ненуклеофильном растворителе типа жидкого диоксида серы считается самой сильной из известных суперкислотных сред. Можно либо нарисовать комплексный анион по образцу SbF6–, хотя получится немного громоздковатая штуковина [SbF5(SO3F)]–, но для делокализации минуса чем более громоздкая, тем лучше. Или подумать о том, что в чистой фторсульфоновой кислоте кислотность ограничена тем, что анион фторсульфоната пристает к протону и не дает ему сорваться с цепи и укусить метан за связь C-H. Дадим фторсульфонат-аниону мощную кислоту Льюиса, она закроет минус своим телом, и протон уже ничто не будет сдерживать.
Дж.Ола никогда не получил бы нобелевки, если бы не умел великолепно подавать и рекламировать свои работы. Обнаружив эту суперкислотную среду и сделав с ней важнейшие исследования, Ола отправился на ёлку в Рождество, снял с ёлки свечку и под радостные крики детворы растворил свечу в принесенной жидкости, прикинувшись волшебником. Свеча якобы была не восковая, а парафиновая, то есть сделана из алканов, и ее растворение означало, что алканы протонируются. Пятилетние дети были приятно удивлены этим неслыханным явлением. Детали этого странного действа история не сохранила (селфи тогда еще делать не научились), но кислоту с тех пор прозвали Волшебной, и даже запатентовали это название (Magic Acid®) в патентном ведомстве США.
Реакция метана с суперкислотой
Теперь попробуем разобраться, что происходит в реальности, когда метан реагирует с очень сильной суперкислотой в растворе ненуклеофильного растворителя, соединив то, что мы разобрали в предыдущих блоках. Итак, при пониженной температуре булькаем сухой метан в раствор эквимольной смеси фторсульфоновой кислоты и пентафторида сурьмы в смеси жидкого диоксида серы с фторхлорсульфурилом. Мы уже знаем, что происходит протонирование и образование неустойчивого гиперкоординированного метониевого катиона, и он обратимо распадается на метан и протон (этого мы не увидим), но параллельно и на метильный катион и водород. Отлично – если у нас есть возможность анализировать состав газа над раствором (берем пробы и впрыскиваем в масс-спектрометр), то мы увидим там молекулярный водород. И это пока единственный признак того, что в растворе что-то происходит. А можем ли мы в растворе увидеть метильный катион. Можем попробовать, потому что растворитель вполне годится для ЯМР-спектроскопии, а раз уж делается такой непростой эксперимент, то и решить проблему, как регистрировать спектр прямо с реакционной смеси, можно. Но если мы это сделаем, то никакого метония или метильного катиона там обнаружить не получится – мы увидим только метан. Оба этих простых катиона чрезвычайно неустойчивы или реакционноспособны, и не накапливаются в растворе в измеримых количествах. На этом можно было бы остановиться, сделав вывод, что реакция, конечно, интересная, но бессмысленная. Но, если набраться терпения, и дать метану пореагировать с суперкислотой подольше, то в смеси вдруг появится небольшое количество совершенно неожиданного продукта – трет-бутильного катиона. Он в этой смеси вполне устойчив, медленно накапливается, а когда мы реакцию закончим, и как это водится, выльем в воду со льдом, и выделим продукты, то обнаружим небольшое, но вполне измеримое количество трет-бутанола. Откуда там трет-бутильный катион?
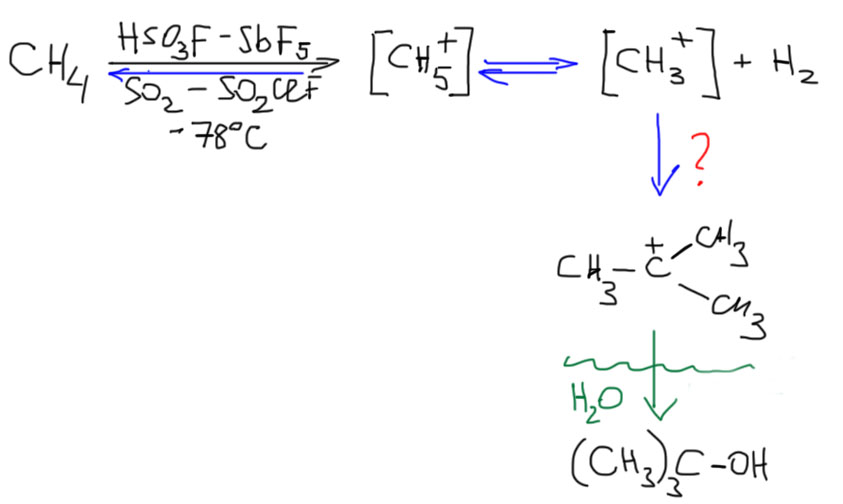
Образование трет-бутильного катиона, наверное, спмое интересное во всей этой истории. Оно показывает, что там происходит что-то более значительное, чем просто разрыв связей C-H. Если бы все сводилось только к этой реакции, проку от этой химии было бы немного. Но там как-то умудряются образовываться связи C-C и возникать разветвленный скелет, а это уже может быть перспективно для настоящего синтеза, если не лабораторного (для которого на первом месте всегда стоит селективность и образование трудноразделимых смесей не допускается), то хотя бы промышленного.
Чтобы понять, как там образовался трет-бутильный катион, нужно понять как может образоваться связь C-C. Так как в системе не так много органических молекул, то гадать долго не придется – связь C-C может образоваться только за счет реакции метильного катиона с метаном. Такую реакцию легко написать, если обобщить взаимодействие метана с протоном на другой очень мощный электрофил – метильный катион. Очевидно, что при этом промежуточно должен образоваться новый катион с гиперкоординированным углеродом и трехцентровой связью, только в этот раз связанными атомами будут два углерода и водород. Напишем этот ион и по уже принятой схеме распишем его возможные пути распада. 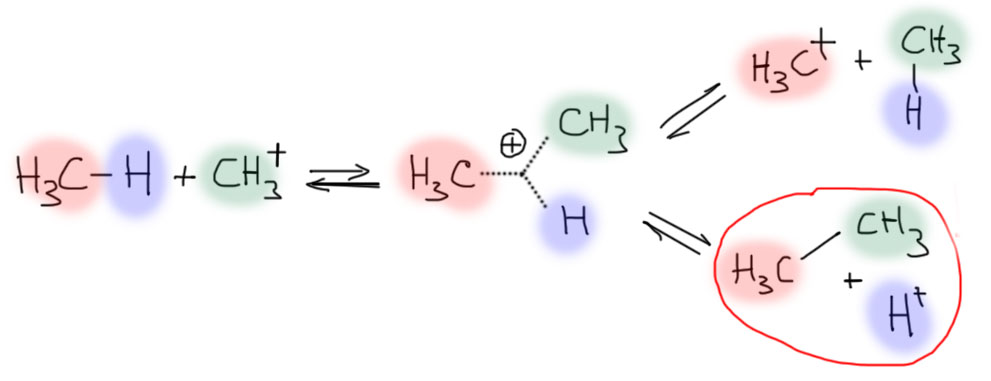
Получаем, как и в случае протонирования, два пути вхолостую, и один, приводящий к новому продукту – и это, ни много нимало, настоящий этан, то есть действительно произошло усложнение скелета и образование новой связи C-C. Если мы примем этот путь, то дальше дело пойдет уже вполне весело, и до трет-бутильного катиона рукой подать. Единственный вопрос, который останется, это почему система фиксируется на этом катионе. Но это достаточно очевидно. Карбокатионы (настоящие карбокатионы с секстетным углеродом, они же карбениевые ионы) имеют очень разную устойчивость – это мы знаем. Но почти никогда не задаем вопрос, а что это значит. Слово “устойчивость” в органической химии, как я уже не раз писал по разным поводам, используется достаточно беспорядочно, и под этим понятием подразумевают не только и даже не столько настоящую устойчивость в термодинамическом смысле, то есть свободную энергию образования – чем меньше, тем устойчивее. Но чаще имеют в виду реакционную способность: чем она выше, тем больше шансов, что молекула или ион успеют с чем-нибудь прореагировать раньше чем вы успеете эту молекулу или ион как-то обнаружить. Вот в случае карбокатионов дело именно в этом. По термодинамической устойчивости сравнивать метильный, этильный, изопропильный и трет-бутильный катион совершенно бессмысленно, ведь в них разные количества атомов и связей. И когда говорят, что метильный катион менее устойчив чем этильный (или любой первичный алкильный), а он менее устойчив чем изопропильный (или любой вторичный алкильный), а он менее устойчив чем трет-бутильный (или любой третичный алкильный) имеют в виду именно реакционную способность, и всю эту цепочку нужно читать как метильный катион более реакционноспособен чем этильный, этильный – чем изопропильный, изопропильный – чем трет-бутильный. А реакционная способность у карбокатионов одна – это электрофилы.
Получаем в итоге очень простую картину – если по дороге образуются первичные или вторичные катионы, они сохраняют способность образовывать трехцентровую связь со связями C-H. А трет-бутильный катион уже совсем ленив и такой фокус провернуть не может. На нем Природа решила отдохнуть и перестать суетиться, все время усложняя скелет. Попробуем это расписать в схему. Начнем с этана, до которого мы уже добрались.
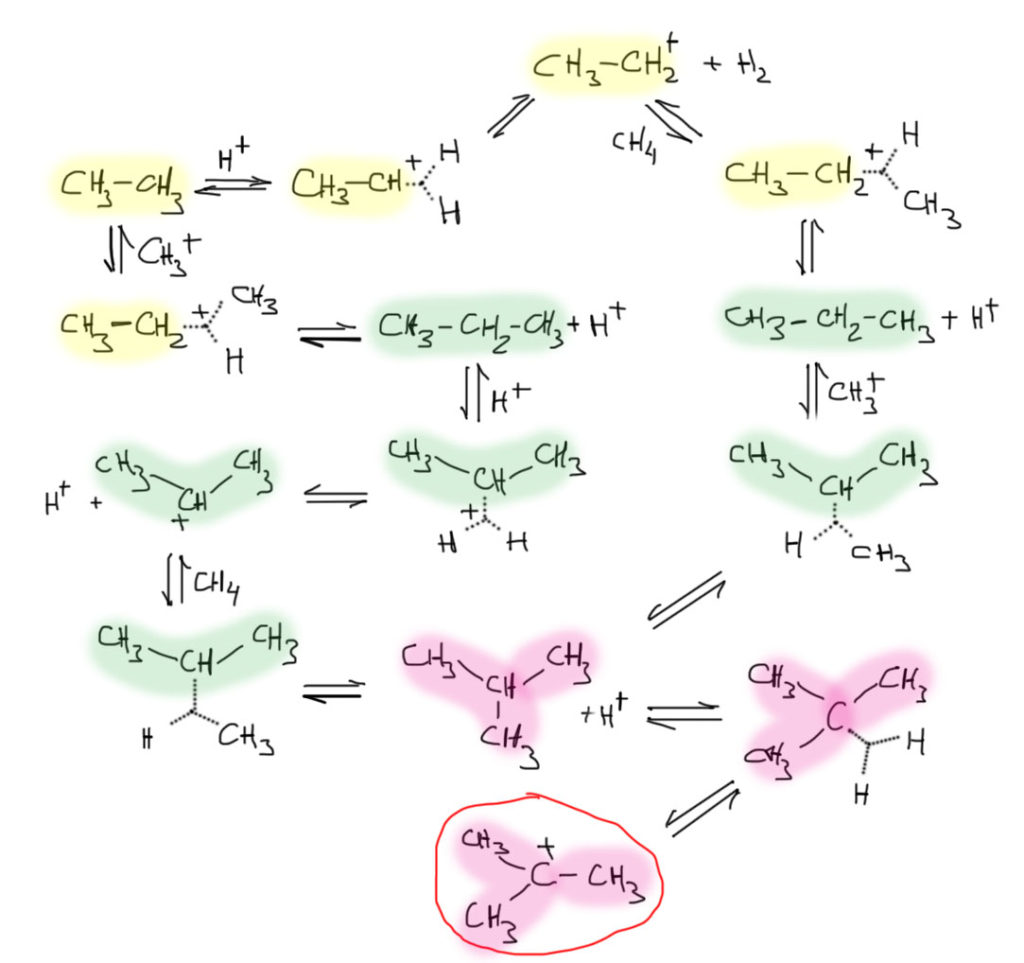
Это только часть возможных превращений. Если вы поняли принцип, то сами нарисуете еще множество таких реакций образования гиперкоординированных ионов и их распада. Берем сильные электрофилы, а в этой системе это протон и все врзможные карбокатионы кроме ленивого трет-бутильного и по очереди пристраиваем их ко всем связям C-H, рисуем трехцентровые связи и их возможные распады, часть из которых будет просто путем в обратную сторону, а часть приведет к новым молекулам и ионам, скелет понемногу будет усложняться – сначала два углерода, потом три, потом четыре. Дальше дело не пойдет, потому что образуется ленивый трет-бутильный катион, и к нему ведут все дороги в этой системе. Он терпеливо дождется, когда вам надоест булькать метан в суперкислоту, или кончится сухой лед для охлаждения, и превратится в трет-бутанол или изобутилен. По-английски такие ленивые частицы, завершающие все возможные пути в таких многостадийных процессах кратко и точно называют catch-all.
Практическое применение гиперкоординированных ионов. Изомеризация алканов.
Трет-бутильный катион – это хорошо и интересно, но на нобелевку не тянет. Если вы когда-нибудь читали вердикт Нобелевского комитета по поводу любой из премий, не могли не заметить, что после короткого описания собственно достижения идет многословное рассуждение о том, как это достижение облагодетельствует человечество: излечит больных, накормит голодных, напоит жаждущих, согреет замерзших, умножит счастливых, и так далее. Лабораторными опытами с суперкислотами, как бы неожиданны и интересны они ни были, человечество осчастливить не получится. У химии гиперуглеродных катионов и электрофильных реакций алканов должно быть какое-то важное применение. И оно есть. Нефть состоит в основном из углеводородов нормального строения потому что получается в недрах из остатков живых организмов. А самое важное применение нефти до сих пор – производство бензина. Качество бензина (октановое число) прямо зависит от содержания в нем разветвленных углеводородов. Поэтому изобретение эффективного процесса изомеризации алканов очень важная задача. По крайней мере, была такой в прошлом веке. Оле повезло, задержись он со своими работами лет на 20, остался бы без нобелевки, потому что в нашем веке идея “больше бензина” уже не кажется такой актуальной.
Попробуем применить химию гиперуглерода. Возьмем для простоты нормальный бутан и подвергнем его действию суперкислоты. Это неизбежно приведет к образованию карбокатионов нормального строения, один из них будет первичным, другой вторичным. Когда карбокатионы изомерны, слово “стабильность” можно понимать в классическом смысле свободной энергии образования, а это значит, что равновесие превращения изомерных катионов друг в друга будет смещено в сторону более устойчивого, первичный изомеризуется в вторичный, а вторичный, если сможет, в третичный. Карбокатионы изомеризуются за счет сдвига соседнего атома водорода или алкильной группы. Дальше все очень просто: рисуем все возможные сдвиги в виде равновесий, и если сможем такими шагами прийти в третичный катион, значит точно придем, и все равновесие будет смещено именно в эту сторону. Рисуем.
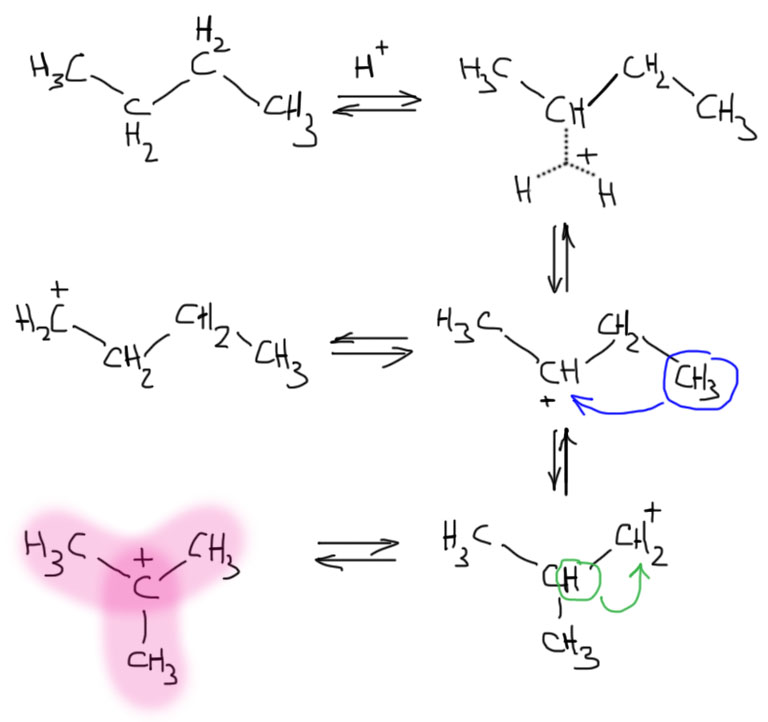
Как видим, проблем нет. Вторичный бутильный катион сдвигом метила превращается в первичный, но уже с изомеризованным скелетом. А он сдвигом водорода (правильнее говорить гидрида) превращается в самый стабильный трет-бутильный. Катион еще нужно превратить в алкан, изобутан, но это произойдет, если в системе кроме суперкислоты есть водород, находящийся в равновесии с участием гиперкоординированного катиона.
Если у нас не бутан, а длинный алкан типа октана, то равновесных сдвигов будет гораздо больше, но некоторые из них обязательно дадут третичные катионы. Вполне можете это сами проверить, расписав часть схемы равновесий по аналогии с бутаном. Результатом всегда будет изомеризация. Это ровно то, что нужно для улучшения качеств нефтяных углеводородов при производстве высокооктанового бензина.
Одна вещь только не дает покоя. Для изомеризации нужна суперкислота, а это, как мы видели, явно не те вещества, которые можно применить в крупнотоннажной промышленности. Но как только стал понятен принцип, началась охота за практичными суперкислотами. Промышленность терпеть не может реакций в растворах даже если реактивы дешевые, потому что продукты реакций нужно из растворов доставать, а это удовольствие совершенно запредельно сложное и дорогое, когда речь идет о тысячах тонн. В промышленной химии предпочитают гетерогенные реакции, в которых реагенты или катализаторы – твердые тела, а реакцию ведут пропуская исходные вещества над поверхностью. Для промышленности нужны твердые суперкислоты. Их быстро нашли, сначала устроив некоторое подобие трифторметансульфоновой кислоте, навесив сульфо-группы на перфторированный полимер (это называется Nafion). Но лучше всего оказались синтетические пористые неорганические материалы на основе алюмосиликатов, цеолиты, которые обладают свойствами ионообменников и в форме, заряженной ионами водорода. При нагревании поверхность таких материалов обладает выраженными свойствами суперкислоты и отлично катализирует изомеризацию алканов, и это очень дешево и практично. Ну а еще дальше начинается уже химия переходных металлов, которые обладают способностью взаимодействовать с C-H связями с образованием очень похожих на гиперкоординированные ионы комплексов. Но это уже совсем выходит за рамки курса.