Методы и задачи в нуклеофильном замещении и элиминировании. Галогенпроизводные, спирты, простые эфиры, эпоксиды.
В этом разделе очень много стереохимии, но почти нет новых методов образования C-C связей. Наиболее интересный и богатый с точки зрения использования в синтезе объект в данном разделе – химия трехчленных простых эфиров – эпоксидов (или оксиранов – это полные синонимы). Впрочем не забывайте, что алкилирование ацетиленид-анионов из предыдущей темы – типичная SN2-реакция, и в этом смысле принадлежит и этой теме, причём мы теперь кроме галогенпроизводных можем использовать и тозилаты спиртов, только не забывайте проверять субстрат на соответствие критериям SN2-замещения. Все остальное носит в основном вспомогательный характер.
10.10.2020 – добавлена защита гидроксильной группы в Разные вспомогательные методы. Задачи по этому методу добавлю на днях.
Новые C-C связи
Раскрытие эпоксидов – основной метод синтеза в этом разделе. Он селективен и очень гибок. Обратите на него особое внимание. Многие задачи решаются с использованием этого метода.
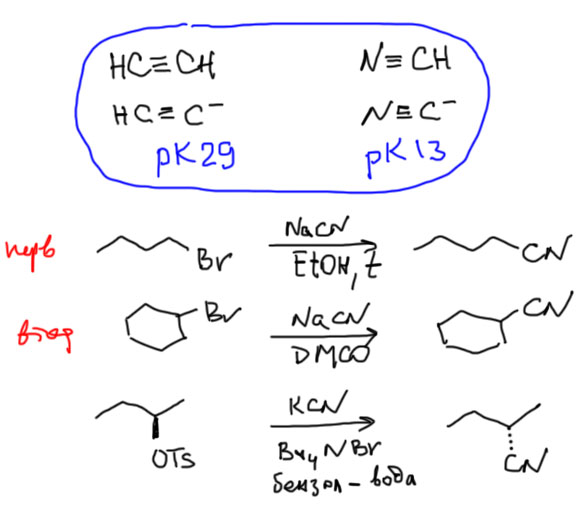 Цианиды используют в основном для гидролиза в карбоновые кислоты, и мы к этому еще вернемся.
Цианиды используют в основном для гидролиза в карбоновые кислоты, и мы к этому еще вернемся.
С этой реакцией связана еще одна интересная проблема. Цианид-ион – один из типичных амбидентных нуклеофилов – неподеленные пары есть и на углероде, и на азоте, и реакция может направляться на любой из этих центров. В случае атаки на азот получается изонитрил. Закономерности этого выбора мы как-нибудь обсудим отдельно, но пока будем считать, что в чистой SN2-реакции образуется обычный нитрил.
Эпоксидам не страшно элиминирование, поэтому ограничения на основность нуклеофила не работают. Эпоксиды отлично раскрываются литийорганическими соединениями (а еще лучше купратами R2CuI), реактивами Гриньяра (в реальной жизни хуже, но нам это не так важно), ацетиленид-анионами. Раскрытие идет строго по законам SN2 – атака на менее замещенный углерод и с обращением на этом атоме, хотя это обращение приходится почти всегда обозначать с помощью моно-дейтериевых производных, так как мы довольно сильно ограничены в возможности использовать дизамещенные эпоксиды из-за очень низкой реакционной способности.
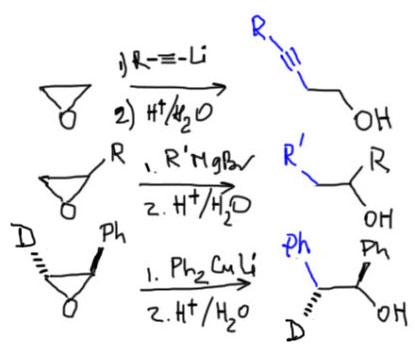
Раскрытие ди- и даже тризамещенных эпоксидов с выбором менее замещенного центра чаще можно встретить для циклических производных.
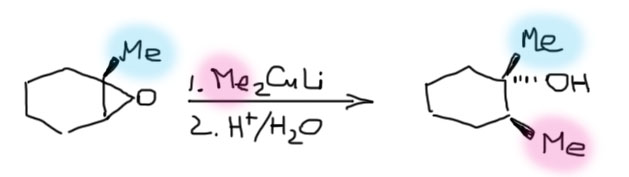
В этих реакциях есть одна проблема – раскрытие обычно проводят при низкой температуре, чтобы избежать побочных реакций, и в этих условиях требуется некоторая активация оксирана, поэтому часто еще добавляют кислоту Льюиса, обычно трифторид бора, который легкодоступен в виде комплекса с эфиром – так называемый эфират трехфтористого бора (BF3⋅Et2O). В нашей упрощенной химии это не обязательно.
Изменения реакционных центров
Нуклеофильное замещение
- Выбираем...
- Замена галогена на другой галоген
- Замена гидроксила на галоген
- Замена галогена на гидроксил
- Синтез простых эфиров
Не забывайте проверять структуру субстрата на соответствие механизму SN2. Использование третичных субстратов в нуклеофильном замещении в условиях SN2 – тяжкий непротительный грех!
Заменять можно не любой галоген на любой другой, но только а) хлор на иод; б) любой кроме фтора на фтор. Причина очевидна – кога речь идет о “тяжелых” галогенах, то есть хлоре, броме и иоде, то все они в виде анионов неплохие и сравнимые нуклеофилы (разница есть, нуклеофильность растет, как и положено, сверху вниз, но не настолько сильно, чтобы скорости отличались на порядки), и неплохие уходящие группы (опять таки, разница есть, но …). Поэтому любая такая реакция будет равновесием, и нам придется разделять смеси галогенпроизводных, что не очень просто. Только если найдется способ сместить равновесие по ЛеШателье, мы получим работающую реакцию. Такой способ есть в случае замены хлора на иод, и использует он простое свойство – растворимость. NaCl менее растворим в некоторых апротонных растворителях чем NaI – понятно, почему. Хлорид меньше иодида и кристаллы хлорида поэтому прочнее кристаллов иодида – гораздо больше должна быть энергия сольватации, чтобы разрушить кристалл NaCl. В апротонных растворителях, как мы уже усвоили, анионы сольватированы слабо, и почти вся сольватация уходит на катион. Если катион одинаков, получаем простое явление: иодид легче растворим, и в реакции обмена хлора на иод, хлорид натрия будет выкристаллизовываться, а равновесие – смещаться. Это называется реакция Финкельштейна, а растворителем для нее служит или ацетон, или ДМФА. В ДМФА реакция несколько быстрее, чем в ацетоне. Не забывайте, что это типичная SN2-реакция, и смотрите внимательно, чтобы не влипнуть в неправильный субстрат и не сделаться посмешищем.
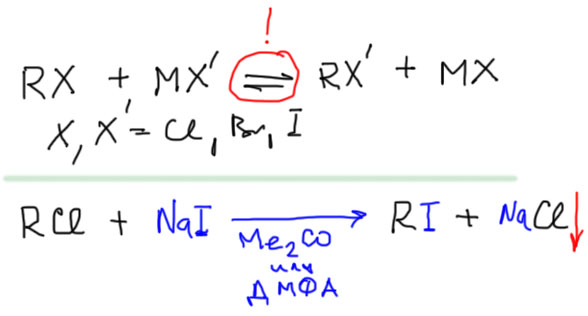
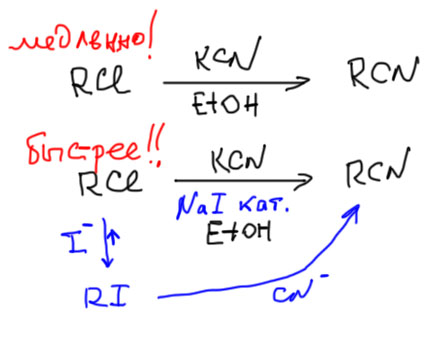
Во всех остальных парах галогенов такого эффекта достичь невозможно – у соседних галогенов маловата разница, и наоборот – иод на хлор – реакция тоже не идет по понятным причинам. Эту реакцию применяют прежде всего для “улучшения” уходящей группы – иодиды намного более реакционноспособны в реакциях SN2-замещения чем хлориды, и часто есть смысл потратиться на реакцию Финкельштейна, чтобы увеличить выход и селективность в последующей реакции. Иногда это делают даже каталитически – в реакцию алкилхлорида с не очень шустрым нуклеофилом добавляют немного иодида натрия, и прямо в реакционной смеси идет замена галогена, и затем значительно более быстрая реакция с нуклеофилом, например:
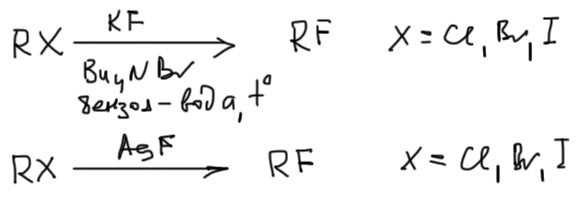
Замена “тяжелых” галогенов на фтор
Здесь работает другая идея: фтор вообще не является уходящей группой, и уж коли он куда встал, то бейсбольной битой не вышибешь: равновесия можно не бояться. Но есть проблема – фторид как нуклеофил это большая проблема. Проблема в том, что фторид – один из самых маленьких анионов (собственно, только гидрид еще меньше), поэтому он а) образует очень прочные кристаллические решетки с катионами мтеаллов, и растворимость фторидов в органических растворителях, даже апротонных биполярных, ничтожна. Реакции просто не идут. Б) В протонных растворителях, где растворимость неплохая (фторид очень ценит водородные связи), но и нуклеофильность ничтожная по той же причине. И там реакции не идут. Остается межфазный перенос -так мы надеемся затащить фторид в апротонную среду. И да, это работает. Годятся все способы – или готовый фторид тетрабутиламмония (есть в продаже, к этому реагенту много вопросов, но он есть и часто используется), или обычный фторид плюс соль четвертичного аммония, например, Aliquat 336 (см. про растворитель в Нуклеофильности), или краун-эфиры. Кроме этого, используют и еще один прием – фториды серебра или ртути. Эти два металла известны огромной любовью к тяжелым галогенам, и просто связывают уходящие группы, но в современной химии, ценящей экономное отношение к веществам и повышенное внимание к экологии и безопасности, такие реагенты (стехиометрические соли тяжелых металлов) стали признаком дурного вкуса.
SN2-замещение гидроксила на хлор, бром, иод
Ни в коем случае не применяйте “школьную” реакцию – действие галогеноводородных кислот или их солей в присутствии серной – бромистый этил так получить можно, но для хоть чуть более сложных субстратов получите кашу из продуктов перегруппировок, олигомеризации и прочих нечаянных радостей.
Для акуратной замены используют разные реагенты, но все они – галогенангидриды каких-нибудь простых неорганических кислот. Галогенангидридом кислоты, напомню, называются соединения, получающиеся из этой кислоты заменой гидроксильной группы на галоген, если гидроксильных групп несколько, то может быть несколько галогенангидридов. Механизм действия их всегда одинаков – сначала при взаимодействии со спиртом, обычно в присутствии какого-нибудь основания типа пиридина, образуются сложные эфиры неорганических кислот, то есть образуется хорошая уходящая группа и одновременно галогенид-ион, то есть нуклеофил. Этот нуклеофил SN2-замещает хорошую уходящую группу с образованием галогенпроизводного. Для получения хлорпроизводных используется чаще всего хлорангидрид сернистой кислоты – хлористый тионил. Вот как это происходит.
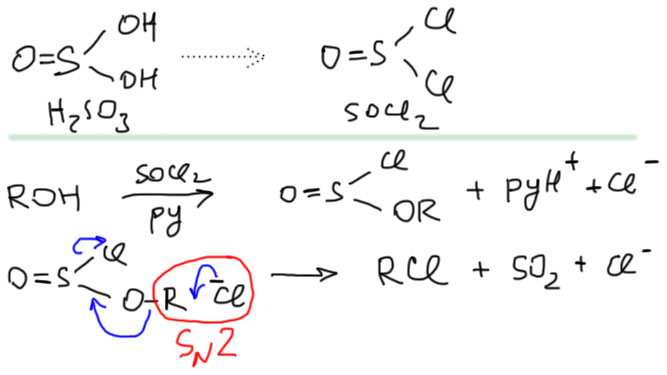
Видим, что нам нужен именно полуэфир-полухлорангидрид, потому что он легко распадается – уходящая группа превращается в газообразную двуокись серы и хлорид – и именно поэтому это хорошая уходящая группа (она буквально улетает в трубу). Если получится полный эфир (два моля спирта на моль тионила), то на этом все кончится – это вполне устойчивые вещества. В литературе можно найти и другие хлорангидриды, использующиеся для этой цели, в частности PCl5 и POCl3. Нам не нужно с ними связываться – это капризные вещества, про них нужно много знать, чтобы понять, как их реально можно использовать.
Для получения бром- и иодпроизводных чаще используют тригалогениды фосфора – то есть полные галогенангидриды фосфористой кислоты. А почему не бромистый тионил? В основном, потому что он дорог, и потому что из бромистого тионила на дело идет только один атом брома, а из трехбромистого фосфора – все три. С иодом то же самое, и впридачу реагент не берут готовый а делают прямо в реакционной смеси. Механизм можете попробовать написать сами по аналогии. Если не получится, не расстраивайтесь – это никому не нужная чисто умозрительная туфта. Это SN2 – и все.
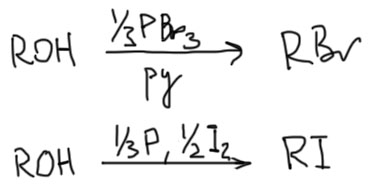
Новые реагенты для замены гидроксила на галоген
Смысл любого реагента для замены гидроксильной группы состоит в замене гидроксила на хорошую уходящую группу, желательно прямо в реакционной смеси без необходимости учинить отдельную стадию. Чем лучше получается уходящая группа, тем быстрее пойдет последующее SN2-замещение. Наибольшим потенциалом для создания таких групп обладают соединения фосфора. Одна из причин этого – чрезвычайно высокое сродство фосфора к кислороду, образование таких связей всегда очень выгодно. Именно это и нужно – ведь нам нужно забрать у спирта гидроксильную группу. Вот и отдадим ее кислород фосфору, а протон подарим кому-нибудь еще, и желательно так, чтобы не появлялась сильная кислотность за счет образования галогеноводорода. Эта кислотность мешает – она снижает нуклеофильность и подвергает угрозе всякие группы, которые могут оказаться в молекуле субстрата, например, двойные связи. Для замены гидроксила на хлор применяют смесь трифенилфосфина и CCl4. Механизм не очень прост, но результат очень просто понять – эти два реагента на пару растаскивают гидроксил – кислород идет фосфору (очень идет), а протон отходит углероду в виде хлороформа – совершенно устойчивого соединения, не проявляющего никакой кислотности.
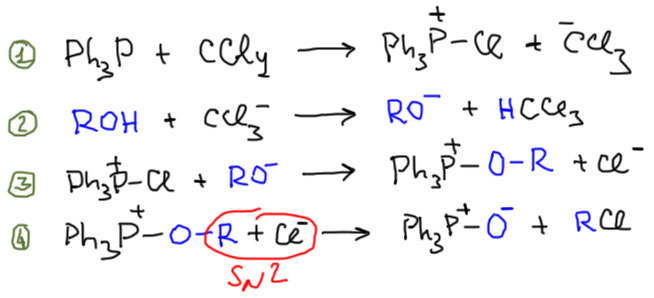
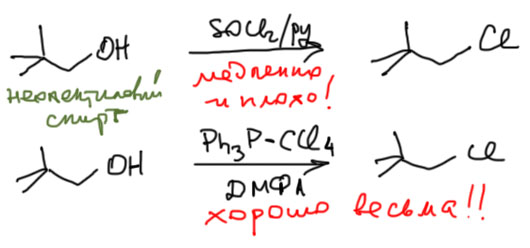
для замены на бром таким же образом используют или Ph3P-CBr4, или Ph3P-Br2. Для иода есть похожий реагент, но мы не будем его использовать. Эти модные (в середине прошлого века) реагенты применяют только в сложных случаях. Во-первых, к плохим субстратам, особенно вторичным и алкилам с разветвлением на β-атоме, в том числе пресловутому неопентилу. Во-вторых, когда в молекуле есть что-то, несовместимое с сильной кислотой, например, донорная двойная связь.
SN1-замещение гидроксила на хлор или бром, но не иод
Третичные спирты очень легко превращаются в хлор и бромпроизводные под действием концентрированных галогеноводородных кислот (если кто забыл, напоминаю, что первая называется соляной, и в концентрированном виде имеет концентрацию около 37%. А вторая – бромистоводородной, 48%). Это типичная SN1-реакция. Образование карбокатиона облегчается а) протонированием гидроксила и образованием хорошей уходящей группы – воды; б) подходящей средой – сильнокислой водной, для SN1 лучше не придумаешь – и высокополярная, и протонная. Образующийся карбокатион немедленно реагирует с имеющимися нуклеофилами, а это вода (очень много) и галогенид-ион (тоже немало, кислоту всегда берут в избытке, она дешевая, не жалко). И все это обратимо. И вот так бы это и гонялось по равновесиям туда-сюда, туда-сюда, если бы не то случайное обстоятельство, что алкилгалогениды не растворимы в воде и выделяются в виде отдельной несмешивающейся фазы. Налицо помощь от ЛеШателье. равновесие смещается в сторону продукта, выходящего из реакционной равновесной смеси.
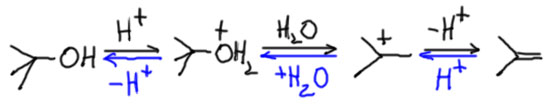
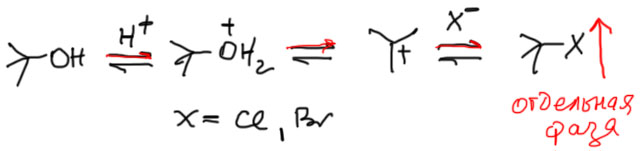 С иодистоводородной кислотой это не работает, так как образующиеся иодпроизводной той же кислотой и восстанавливается в алкан, который нам сейчас даром не нужен. А как же быть? А никак. Зачем вам третичные алкилиодиды? Все равно других применений SN1-химии нет, а там, где нужны третичные галогениды (например, в алкилировании ароматики по Фриделю-Крафтсу) хлорид лучше.
С иодистоводородной кислотой это не работает, так как образующиеся иодпроизводной той же кислотой и восстанавливается в алкан, который нам сейчас даром не нужен. А как же быть? А никак. Зачем вам третичные алкилиодиды? Все равно других применений SN1-химии нет, а там, где нужны третичные галогениды (например, в алкилировании ароматики по Фриделю-Крафтсу) хлорид лучше.
Если все же есть необходимость, то используют в качестве нуклеофила ацетат-ион (или бензоат, или другие карбоксилаты, иногда еще используют банальный карбонат или бикарбонат, но мы не будем, так как условия таких реакций сложноваты для наших целей). Это слабый нуклеофил и слабое основание, но с помощью межфазного переноса – самое то, что нужно – никакого элиминирвоания и скорость замещения вполне приемлема. Плуча.щиеся сложные эфиры гидролизуют спиртовой щелочью – это очень быстрая, почти мгновенная реакция, не затрагивающая атом углерода исходного субстрата. Поэтому, общий результат этого метода – чистое SN2-замещение галогена на гидроксид.
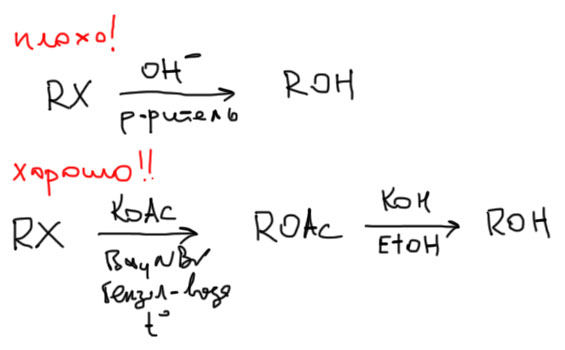
Иногда еще можно встретить довольно странный реагент для прямого превращения галогенпроизводных в спирты – супероксид калия KO2 в ДМСО. Супероксид калия – это довольно знаменитое вещество, его используют в изолирующих противогазах и системах регенерации воздуха на подводных лодках и тому подобных местах, где человек может оказаться длительно отрезанным от источников воздуха, так как при взаимодействии с двуокисью углерода образуется кислород. Анион этой соли – супероксид-анион, представляет собой молекулу кислорода с лишним электроном (анион-радикал кислорода), то есть это типичный нуклеофил с альфа-эффектом – хороший нуклеофил и плохое основание. SN2-замещение должно идти отлично. Но при этом образуется не спирт, а какая-то странная перекись, которая, видимо, восстанавливается в спирт диметилсульфоксидом. Все бы ничего, но супероксид калия нехило взрывается при контакте с влагой воздуха, поэтому реальных желающих получать так спирт в природе маловато, и найти применения этой методики в реальных синтезах непросто. Не будем это использовать, хотя метод поучительный (нуклеофил с альфа-эффектом).
Не нужно получать простые эфиры “школьной” реакцией – действием серной кислоты на спирты! Диэтиловый (и диметиловый) эфиры так получить можно, но все остальное будет давать в основном осмоление за счет кислотно-катализируемой олигомеризации побочно образующихся олефинов.
Основной метод – SN2 замещение с использованием алкоксидов в качестве нуклеофилов (реакция Вильямсона). Растворитель при этом – тот же спирт. Если спирт сложный и нежидкий, можно взять эфирный растворитель (ТГФ). Апротонный растворитель нельзя использовать, если есть возможность элиминирования. Если нет – тогда пожалуйста, ДМФА будет хорошим выбором. Межфазный катализ для этой реакции не используют. Если простой эфир несимметричный, то встает вопрос, что использовать в виде галогенпроизводного, а что в виде алкоксида. Выбор прост – минимизируйте возможность элиминирования. Например, вторично-первичный простой эфир лучше получать из вторичного алкоксида и первичного галоенпроизводного, а не наоборот. Алкоксид обычно получают прямо в реакционной смеси действием гидрида натрия.
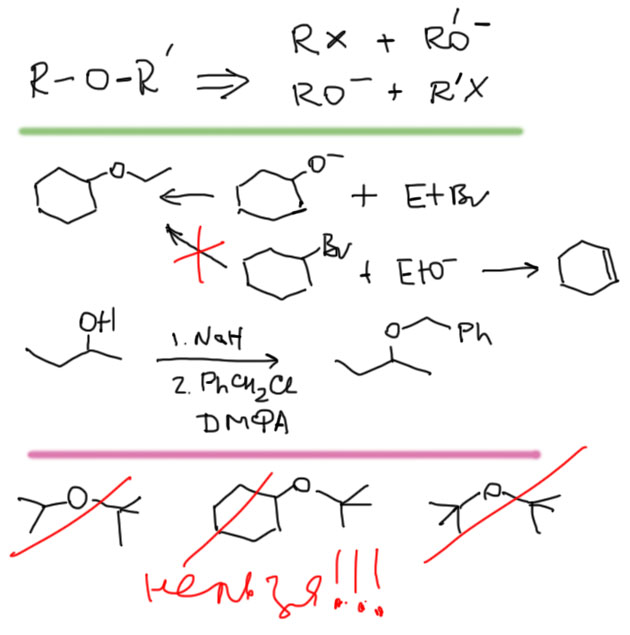
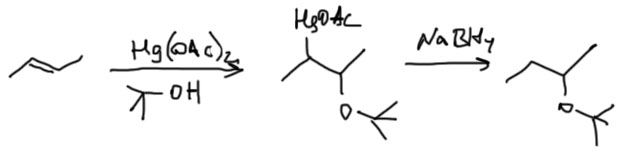
Ни в коем случае нельзя получать этой реакцией вторично-третичные и третично-третичные эфиры – 100% элиминирования при этом гарантировано. Если нужны такие эфиры придется вспомнить старую, но недобрую реакцию сольвомеркурирования – присоединения к олефину солей ртути(2+) и спиртов с последующим восстановительным демеркурированием. Дрянь реакция (в основном из-за необходимости использовать огромные количества ртути), а другой нет.
Элиминирование
Напоминаю, что используется только E2-элиминирование, то есть реакции, в которых явно показано основание, специально подобранное в соответствии с решаемой задачей. Даже для превращения спиртов в олефины лучше забыть про кислотно-катализируемую дегидратацию, а превратить OH в уходящую группу (галоген или тозилат) и элиминировать основанием.
- Выбираем...
- Выбор основания для элиминирования
- Чистое элиминирование по Гофману
- Смещение двойной связи в олефинах
Элиминирование очень часто зависит от стереохимии, и в задачах обычно совмещается с SN2-замещением. Будьте внимательны, и как следует отработайте стереохимические приёмы
Еще нам нужно основание. Выбор оснований для E2-элиминирования огромен и почти бесконечен. Но реальной разницы между ними нет, поэтому ограничимся простыми и надежными. Перед элиминированием нужно а) оценить субстрат; б) понять, куда мы хотим направить двойную связь; в) оценить стереохимию, но это разобрано отдельно.
Оценка субстрата – это мы уже знаем. Основная идея – оценить, хорош ли субстрат для SN2. Если хорош, то нам нужно избежать конкурентного замещения, использовав боле сильное основание. Годится KOH в апротонном растворителе или с использованием межфазного переноса. Если субстрат плох для SN2 (вторичный, бета-разветвленный, третичный), то годится KOH в спирте. Если хотите немного повыпендриваться, то используйте сильное нейтральное основание – диазабициклоундецен (ДБУ) – оно отлично работает и никогда не создает проблем с конкурентным замещением. При элиминировании в основном получается более замещенный олефин – это называется элиминирование по Зайцеву. Чем больше степень замещения, тем больше селективность в сторону Зайцева.
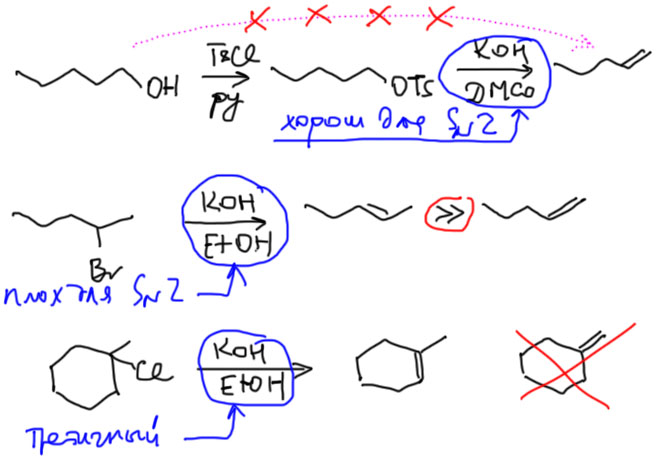
В другую сторону – менее замещенного олефина (элиминирование по Гофману) – элиминирование направляется в основном если а) основание сильно и стерически затруднено; б) уходящая группа похуже. Первое обстоятельство важнее. Самый простой выбор – трет-бутилат в трет-бутаноле или ДМСО. Если хотите еще круче, то можно взять еще более громоздкие третичные алкоголяты – трет-амилат, трет-гептилат, и даже трициклогексилкарбинолят. Все это уже точно в ДМСО, так как соотвествующие спирты твердые.
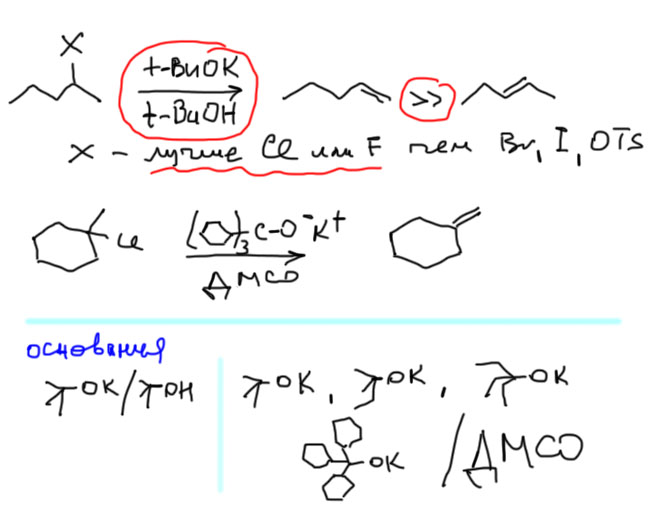
Даже самые сильные и самые громоздкие основания не дают чистого элиминирования по Гофману – второй олефин образуется в количествах до 20%. Нам для решения задач это не так важно, мы упрощаем и рассчитываем на основной продукт, пренебрегая побочным. Но для синтеза это очень скверно, так как делить изомерные олефины чрезвычайно трудно – очень часто они почти не отличаются ни по температурам кипения, ни по хроматографическим свойствам. Практически чистый путь элиминирования предложил еще в 19 веке как раз Гофман, ничего не зная ни про E2-механим, ни даже про основность. Но смысл его решения такой – нужно взять очень плохую уходящую группу, тогда и с основанием можно особенно не заморачиваться. В качестве уходящей группы используют триметиламин, а вся процедура выглядит как раз на 19 век: берем амин, обрабатываем его избытком иодистого метила – происходит три подряд SN2-замещения и образуется четвертичный аммоний с противоионом иодида. Обрабатываем это влажной окисью серебра, осаждается иодид серебра и получается гидроксид аммония. Дальше просто греем и происходит E2-элиминирование.
Этот старинный метод таит в себе несколько забавных открытий. Август Вильгельм фон Хоффманн, или по нашей традиции просто Гофман, открыл эту реакцию в 1851 году, когда еще даже и формул структурных писать не умели, и в результатах реакций разбирались, кропотливо сравнивая продукты с известными веществами. Тем не менее, Гофман фактически открыл два очень модных понятия современной органической химии – межфазный перенос и его влияние на основность, и ионные жидкости. Элиминирование в условиях Гофмана – реакция во многом удивительная, хотя бы потому что мы привыкли к тому, что обычное элиминирование требует более хороших уходящих групп чем триметиламин и более сильных оснований, чем банальный гидроксид-ион. По всем нашим идеям о механизме E2-элиминирования, реакция, подобная тому, что открыл Гофман, идти просто не может. Но идет ведь – и легко, и количественно и изумительно чисто! Секрет этой реакции, видимо, в том, что гидроксид-ион в этих условиях становится чрезвычайно сильным основанием. Во-первых, реакция идет фактически в условиях межфазного переноса, так как сам субстрат, гидроксид четвертичного аммония, является типичным межфазным переносчиком, а мы помним, как сильно влияет межфазный перенос на основность простых маленьких анионов. Во-вторых, реакция у Гофмана идет фактически в расплаве этого самого гидроксида четвертичного аммония, а это ни много ни мало, а то, что нынче называется ионной жидкостью и считается новым типом высокополярных растворителей. Основность гидроксид-иона в таких условиях может быть чрезвычайно высока и соответствовать pK намного больше 30. Вот он и действует именно так, как положено очень сильному основанию в реакции E2-элиминирования с E1cb-подобным переходным состоянием, то есть энергично отдирает протон, не дожидаясь, пока плохая уходящая группа решит все-таки попробовать уйти. Получается, что Гофман не только открыл чрезвычайно полезную реакцию, с помощью которой в до-ЯМРную эпоху успешно устанавливали структуры сложнейших природных азотсодержащих соединений, но и стал невольным первооткрывателем межфазного переноса и ионных жидкостей, естественно не заметив этого. Мы же заметим, и воздадим должные почести великому ученому.
Так как уходящая группа очень плоха, но и работает как индуктивный акцептор, отщепление протона опережает уход плохой уходящей группы, и тогда важно, какой из имеющихся протонов более кислый – а это именно протоны на менее замещенном атоме углерода.

Внимание: это теория, а реально мы этот способ применить не можем, так как не знаем, как получать амины. Это не так просто – во втором семестре разберемся. То, что сразу приходит в голову – реакция галогенпроизводного с диметил- или триметиламином – категорически не годится, потому что в большинстве случаев вам понадобится для элиминирования амин с вторичной алкильной группой, но вторичные алкилгалогениды, плохие SN2-субстраты, просто вообще не реагируют с относительно слабыми нуклеофилами аминами. И здесь вам не поможет ни ДМСО, ни межфазный перенос, так как эти средства работают только для анионных нуклеофилов. Как обойти эту неприятную засаду мы узнаем только во 2 семестре. Поэтому делайте эту реакцию, только если амин уже готов, подан в условии задачи.
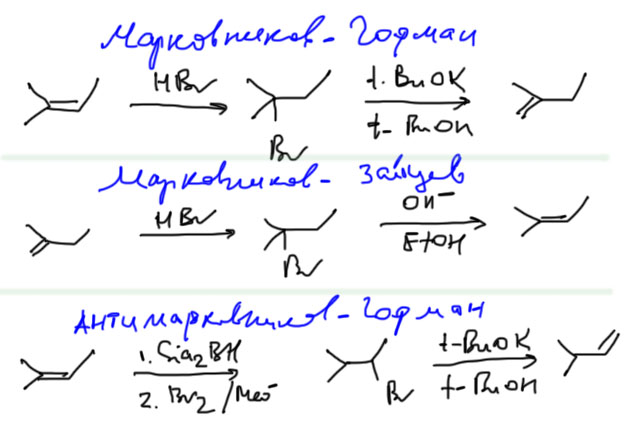
Разницу между элиминированием по Гофману и Зайцеву плюс присоединение HBr по Марковникову и против Марковникова удобно использовать для целенаправленного смещения двойной связи на одно положение либо изнутри наружу, либо в обратном направлении. Возможности метода ограничены, но он довольно полезен. Для вытаскивания двойной связи наружу есть еще и метод Брауна, связанный с обратимостью гидроборирования, но этот метод непросто применять и на 3 курсе его использование не рекомендуется. О методе можно почитать на странице про Алкены.
Разные вспомогательные методы
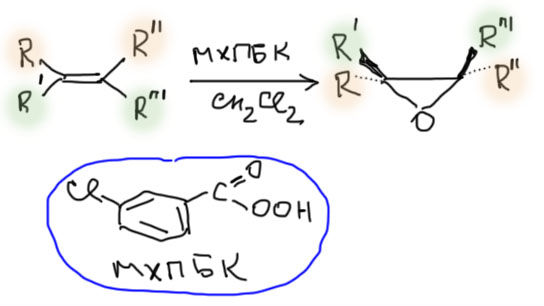
Все другие эпоксиды получаем реакцией эпоксидирования – приоединением электрофильного кислорода к двойной связи. Источников электрофильного кислорода море – это популярная область химии с чудовищной по объему литературой. Мы будем использовать для этой цели надкислоты, самая популярная из которых называется мета-хлорпербензойная кислота. Она очень недорога и удобна тем, что если реакцию вести в дихлорметане, получающаяся из нее мета-хлорбензойная кислота выпадает в осадок, и образующийся эпоксид выделяют просто фильстрованием и упариванием фильтрата на роторном испарителе досуха. Эпоксиды, несмотря на славу ну просто очень реакционноспособных соединений вполне устойчивы, и с ними работают точно так же как с любыми другими органическими веществами. Обратите внимание, что стереохимия в этой реакции сохраняется, то есть те заместители, которые в олефине были по одну сторону от двойной связи, и в эпоксиде останутся по одну сторону, то есть будут сидеть на связях одного типа (клин-клин, или зебра-зебра). Что из них вперед, что назад, не важно, так как это все равно рацемическая смесь, и мы просто, как уже однажды договорились, рисуем один энантиомер, зная, что там столько же и второго.
Тетрагидропиранильная защита
Здесь мы впервые сталкиваемся с тем, что в органической химии называется защитными группами или просто защитами. Зачем это нужно? Это очень просто. Даже в самом начале этой химии мы уже видим, что в молекулах органических соединений может быть много самых разных заместителей, групп. Органические молекулы могут быть очень большими, практически неограниченно большими, и в такие молекулы можно напихать вообще всё, что известно в органической химии, и каждой твари можно взять и по паре, и по паре дюжин. Ну, конечно, это не так просто, и некоторые группы не очень совместимы с некоторыми другими группами, поэтому наши фантазии придётся ограничить, но не очень сильно, только чуть-чуть. Органическая химия для того и существует, чтобы возбуждать самые смелые фантазии, даже, не побоюсь этого слова, разнузданные.
Но напихать-то мы сможем, а вот что делать, если мы захотим что-то делать с такой молекулой дальше – реакции какие-нибудь. В органической химии бывают такие реакции (точнее, последовательности реакций, их называют разными красивыми словами типа каскад, домино, тандем и т.п.), в которых участвуют сразу много заместителей и групп, и за один присест молекула изменяется очень сильно, просто до неузнаваемости – образуется множество новых связей, а множество других разрывается, и всё это обычно ещё и стереохимически чисто и красиво. Но таких процессов всё же пока не так много, и делать такие фокусы умеют только очень продвинутые синтетики. Гораздо чаще, органический химик проявляет разумную робость – и старается в каждой реакции работать только с маленькими частями молекулы, отдельными связями, группами, и завершив одну стадию синтеза спокойно переходить к другой, и так, шаг за шагом приходить к намеченной цели. Это правильная стратегия, и мы всячески продвигаем именно этот подход – тише едешь, дальше будешь. И чтобы так двигаться по многостадийному синтезу, мы должны быть уверены, что в каждой стадии, когда мы замышляем заняться какой-то конкретной группой, нам не будут мешать другие. Если мы об этом не позаботимся, вместо желаемого продукта очередной стадии можем получить мерзкую смесь всякой дряни, молекул, в которых желаемая группа даже не изменилась, зато в её окрестностях полный разгром и бардак. Напоминает, как современные вояки, похваляясь высокоточным оружием, целят особо точной ракетой прямо в глаз какому-нибудь злодею, но попадают в свадебную процессию километрах в 50-ти от намеченной цели.
С вояками сделать ничего невозможно, но в органической химии есть очень мощный набор инструментов для того, чтобы работа над превращениями отдельных групп была максимально точной и безошибочной. Это набор защитных групп и реакций, которые эти группы вводят, когда нужно, и снимают, когда в них отпадает надобность.
В органике особую роль играют функциональные (или, как их называет ИЮПАК, характеристические) группы. В этом разделе у нас первая такая – гидроксил. Представим себе, что у нас есть молекула, в которй есть гидроксил и еще много всякой всячины, и мы пытаемся производить какие-то работы в молекуле, но не на гидроксиле. Подумаем, чему может помешать гидроксил.
- во-первых, и это самое главное, гидрокси-группа – кислота Бренстеда-Лоури, довольно слабая, с кислотностью обычно в диапазоне pK 12-18, в зависимости от конкретных заместителей в окрестностях. Но это значит, что мы не сможем создать где-то в другом месте молекулы какой-нибудь оснóвный центр с высокой основностью (напомню, что основность мы характеризуем кислотностью сопряжённой кислоты). Например, мы не сможем создать ацетиленид-анион, или магнийорганику или литийорганику, ну вот, например:
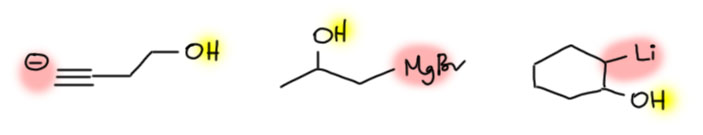
- во-вторых, спирты довольно легко окисляются, и могут мешать окислению каких-нибудь других групп. Надо сказать, что в органике очень много селективных окислителей, и для большинства задач есть свои реагенты. Поэтому задачи по селективному окислению чаще решают не защитой, а подбором селективного реагента. Но у нас пока не очень много реагентов, мы часто орудуем какими-нибудь допотопными реагентами на основе хрома(VI), да и марганцовкой не брезгуем, поэтому нам иногда не помешает защита и в этом случае. Представьте себе, например, что мы решили развалить алкен на две части марганцовкой, а в нём ещё спиртовая группа. Если мы её не защитим, то сожжём всё к чёрту – спирт окислится до кетона, а кетон окислится дальше.
- в-третьих, гидроксильная группа – нуклеофил, и в разных реакциях, например, в ацилировании тоже будет участвовать. И здесь нужно смотреть на конкретную проблему, во многих случаях можно подобрать более селективный реагент. Но иногда проще защитить гидроксил. Здесь та же проблема – у нас мало реагентов, и нам проще прибегнуть к защите, а в современном синтезе реагентов много, и есть возможность подобрать селективный реагент без защиты. Только не сделайте вывод, что вам надо срочно изучать современные реагенты по всяким модным книжкам и обзорам – подбор реагента для конкретных синтезов сделать очень непросто, просто выучить штук сто для этого недостаточно, в реальной работе приходится изучать литературу и искать примеры применения.
- в-четвёртых, и это очень важный момент – особенно опасайтесь конкуренции от гидроксильной группы в тех реакциях, где возможно внутримолекулярное взаимодействие, то есть образование циклов, особенно и в первую очередь 5- и 6-членных, если вам это не нужно. Это может быть где угодно. Участие гидроксила в таких реакциях часто называют реакциями с содействием. Мы как-то перестали их изучать в последние годы, и на сайте они пока тоже не обсуждаются. Например, в реакции электрофильного присоединения, результат с защитой и без может быть разным (защиту пока обозначим просто как R) – с защитой карбокатион (или мостиковый ион) связывает внешний нуклеофил, например, воду или спирт, а без защиты – внутренний нуклеофил с образованием циклического эфира.
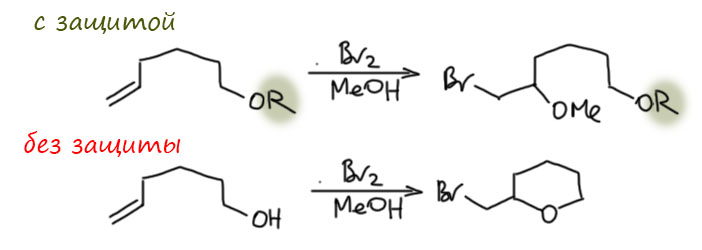
Защитами нужно уметь пользоваться. Без толку их тоже не стоит использовать. Можно ведь и так – чем разбирать, нужна защита или нет, просто повесить её – пусть себе болтается, не помешает. Чаще всего это просто признак непрофессионализма. А в реальной химии это просто не работает, потому что нужно думать не только о самих реакциях, но и о том, как производится обработка реакционных смесей, выделяются и очищаются продукты – защитные группы очень часто или не выдерживают этих манипуляций, или затрудняют их. Поэтому защитные группы нужно вводить тогда, когда они нужны и снимать, когда они больше не нужны.
Ну а теперь наконец обсудим типичную и очень популярную защиту для гидроксила – тетрагидропиранильную. Это странное называние происходит из номенклатуры гетероциклов. Максимально ненасыщенный гетероцикл с 6-членным циклом и одним кислородом называется пиран. Но пирана есть два, и чтобы их различить помечают, какой углерод цикла насыщен: поэтому есть 2Н-пиран и 4Н-пиран. Гетероциклы всегда нумеруются от гетероатома. Реагент полностью поэтому называется 3,4-дигидро-2H-пиран. Вы, конечно, можете спросить, а почему не 4Н, ведь после гидрирования одной связи уже трудно понять, что было в начале. Именно поэтому – если нет разницы, то номенклатура всегда предпочитает цифры поменьше.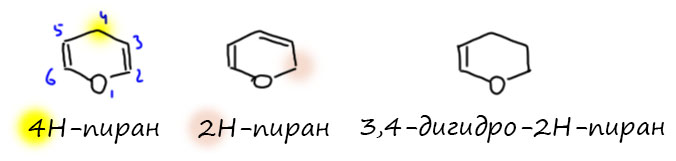
Вот этот реагент, сокращённо называемый ДГП или DHP, очень недорог и легкодоступен. По природе он представляет собой донорный олефин, он же простой эфир енола. Про енолы мы поговорим в теме Карбонильные соединения, но про донорные олефины уже знаем. Такие олефины чрезвычайно легко реагируют с электрофилами, в частности, протонируются, а образующийся катион жадно хватает любой нуклеофил, например, спирт. 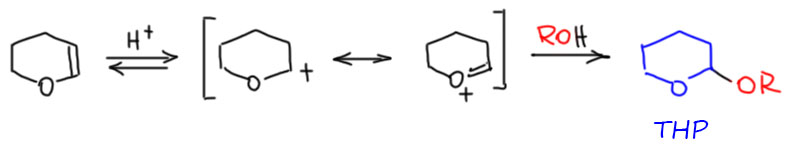
Образующиеся производные содержат уже полностью гидрированный остаток пирана, поэтому их называют тетрагидропиранильными эфирами (остаток сокращённо называют ТГП или THP), но это не просто эфиры – эфиры такого типа, когда на углероде висит два атома кислорода эфирного типа, называют ацеталями. Это производные альдегидов, очень специальный вид эфиров, с одним очень важным отличием от обычных эфиров – ацетали очень легко образуются и очень легко гидролизуются, и то, и другое требует кислотного катализа. Как это работает при образовании мы уже увидели, а гидролиз происходит столь же легко. Только при гидролизе получается не ДГП, а продукт его гидролиза – 5-гидроксипентаналь, а защищённый спирт возвращается. Кстати, если вам когда-нибудь придётся использовать ТГП-защиту, при выделении продукта не перепутайте то, что вам нужно, с этим гидроксиальдегидом. 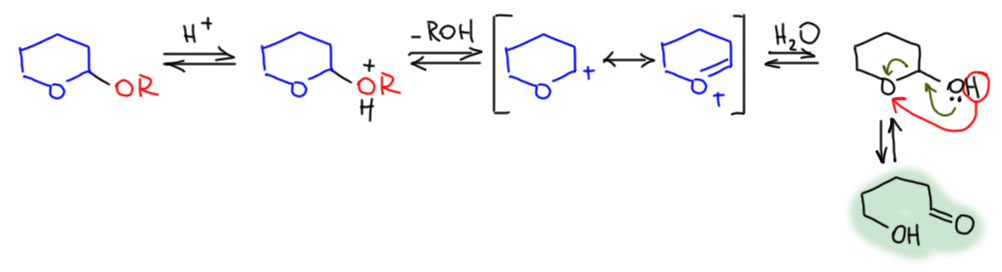
Ну и перейдём к практическим вещам. Как повесить ТГП-защиту на гидроксил. Для этого годится почти любая кислота, протонная или Льюиса, в каталитических количествах. Важно, чтобы в реакционной смеси не было воды, которая смещает равновесие в обратную сторону. Самый распространённый метод – толуолсульфоновая кислота в растворе дихлорметана или хлороформа. Эти растворители почти не содержат воды и не являются гигроскопичными, поэтому достаточно взять свежеперегнанный растворитель над щепоткой гидрида кальция, а в хорошей лаборатории реакции всегда ставят в свежеперегнанных сухих растворителях, даже если никто этого не просит. В литературе вы найдёте без преувеличения десятки других методик, в которых всегда будет какая-нибудь кислота, иногда твёрдая или полимерная, например, монтмориллонит (это алюмосиликатный минерал, глина, твёрдая кислота Льюиса) или нафион (перфторированный полимер с сульфо-группами, сильная протонная кислота). Реакция хороша тем, что в ней из двух компонентов образуется одно вещество, поэтому для выделения надо просто профильтровать смесь через небольшой слой силикагеля и упарить растворитель на роторе.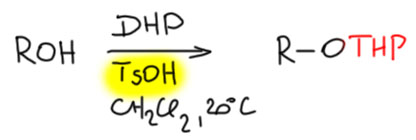
Учтите, что если у вас не один, а несколько гидроксилов, защищены будут все, поэтому нужно рассчитать достаточное количество ДГП. Невозможно защитить одну группу из двух (или из нескольких), взяв один эквивалент ДГП – получите просто смесь недозащищённых, на фиг никому не нужную. Единственное, что может защитить какую-нибудь гидроксильную группу от защиты – какая-нибудь тяжёлая стерика, когда до группы просто не доберёшься. Но такие случаи очень непросты для планирования синтеза, и нам на 3 курсе точно неподвластны, – пришлось бы делать молекулярное моделирование, чтобы понять, как устроена молекула – будем таких историй избегать.
Для снятия ТГП-защиты равновесие нужно обратить назад. Это тоже происходит очень легко, нужен кислотный катализатор и вода – именно вода смещает равновесие в сторону свободного гидроксила. Кислоты достаточно слабой. Самая распространённая смесь для снятия ТГП-защиты – водная уксусная кислота в растворе ТГФ, который растворяет все это – и ТГП-производное, и воду и уксусную кислоту. Смесь просто перемешивают при небольшом нагревании для ускорения. Но в литературе вы найдёте ещё десятки рецептов для снятия ТГП-защиты. 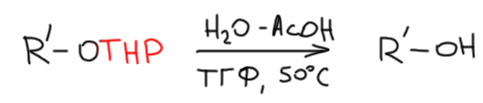
В качестве примера приведём простое превращение с ацетиленидным анионом. Попробуем проалкилировать пропаргиловый спирт. Без защиты получим смесь продуктов алкилирования по углероду и кислороду, даже если сначала получим дианион. Моноанион мы получить чисто не сможем, потому что кислотность гидроксила больше кислотности ацетиленового протона. Но если получим дианион, мы могли бы попробовать предсказать, что алкилирование по углероду будет преобладать, именно потому что кислотность больше, а значит основность больше, а отсюда мы можем предположить, что и нукелеофильность будет больше. Это правильно, но есть проблема – во-первых, правило больше основность – больше нуклеофильность качественное, а не количественное, оно показывает общую тенденцию, но не способно предсказать количественные соотношения скоростей даже очень приблизительно. Во-вторых, в апротонных растворителях основность (а значит и нуклеофильность) увеличивается, но никто не знает насколько, точнее, на сколько порядков или единиц pK. Поэтому в ТГФ основность (и нуклеофильность) ацетиленид-аниона и алкоксид-аниона различаются намного меньше, и конкуренция между этими нуклеофилами неизбежна. Придётся защитить гидроксильную шруппу, чтобы не путалась под ногами. 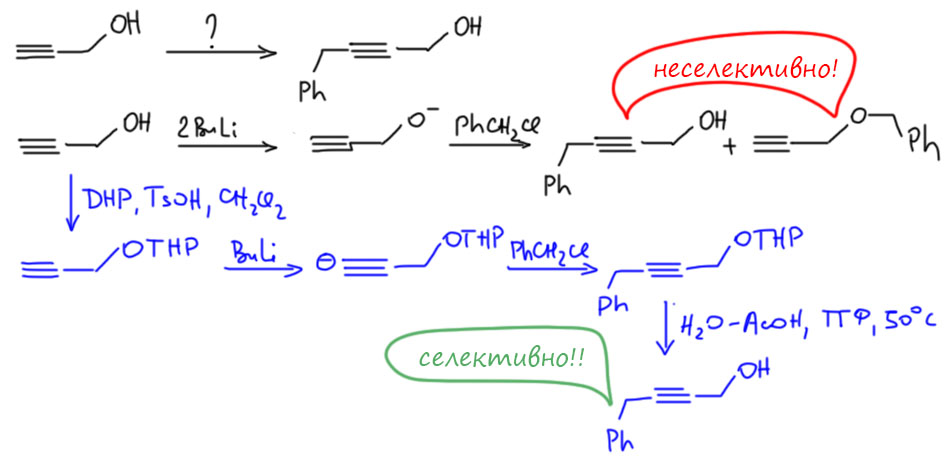
Стереоселективные реакции
- Выбираем...
- Вальденовское обращение - один центр
- Сохранение конфигурации
- Раскрытие эпоксидов
- Анти-элиминирование
- Стереоселективный синтез олефинов - важное напоминание
В этом разделе почти вся стереохимия нашего курса. Помучайтесь и хорошо разберитесь с тем, как всё это делается. Зато потом про стереохимию услышите нескоро.
Основная идея в стереохимии нуклеофильного замещения – строгая обязательность обращения конфигурации при SN2. Это называется Вальденовское обращение и считается законом природы почти столь же строгим как закон сохранения энергии. Это настолько распространенная вещь, что говорят просто “обращение”, не имея в виду – в ислам, иудаизм, или православие, – а только и всегда стереохимический результат одного механизма – SN2. Так и говорят, типа, “в этой реакции наблюдается обращение”, “что-то мы не видим обращения – механизм-то не SN2…” и т.п., и все сразу понимают о чём речь.
Другое дело, что для того, чтобы заметить это явление, нам нужно иметь стереохимию (выражение “иметь стереохимию” это игривый жаргон, но мы тут не учебник пишем, можем себе и не такое позволить и обязательно ещё позволим), то есть четыре разных группы на центральном атоме углерода. Одна из них уходящая, одна может быть водородом, ну а две должны быть разными. То есть либо мы имеем дело с вторичным субстратом, либо с первичным с одним атомом дейтерия.
Далее все просто – если есть SN2, выкручивайте стереоцентр, проще всего показав входящую группу противоположно уходящей. Удобно водород при этом отправлять назад в исходном и больше не показывать. Второй вариант – если уходящая группа показана “в плоскости”, а стереохимия маркирована одним из заместителей, тогда крутите его. Осторожно: если требуется R/S-разметка, не превращайте автоматически эр в эс и наоборот – правила старшинства формальны и могут не сработать – не ленитесь определить конфигурацию заново. Впрочем, можно сначала проверить – в большинстве случаев уходящая группа – старший заместитель, и если входящая остается старшим, то “правило обращения буквы” работает.
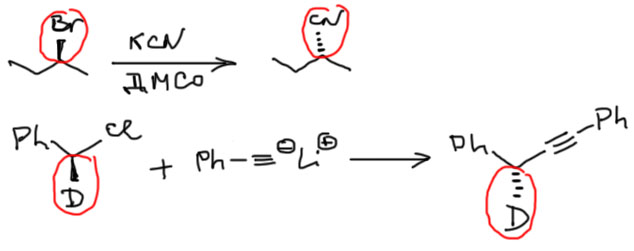
Если в субстрате больше одного стереоцентра, то крутим только тот, который подвергается замещению. Тогда эритро превращается в трео и наоборот 9если центра рядом). Все остальные стереоцентры должны сохранить конфигурацию! Это совершенно банально, но нет таких ошибок, которые не смогли бы совершить большевики. Не уподобляйтесь большевикам! Обратите внимание на третий пример – там вроде бы все это нарушено, что надо не вывернуто, что не надо вывернуто. Но продукт – диастереомер, мезо-форма, для нее не важно абсолютное направление связей на стереоцентре, важно относительное – было трео, стало эритро. Будьте бдительны! Не рисуйте стереохимию машинально, каждый раз думайте и анализируйте результат.
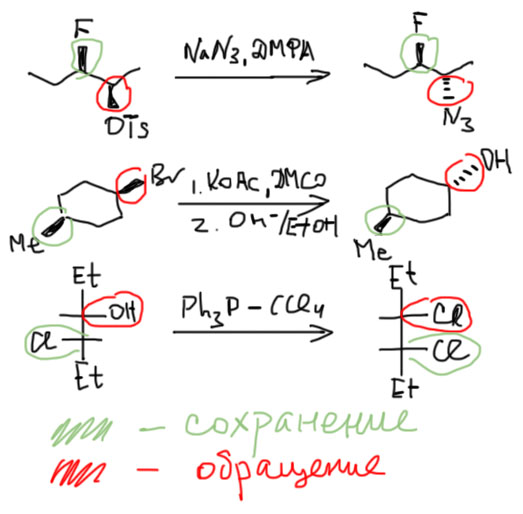
По учебникам с 50-х годов прошлого века кочует миф о том, что если превращение гидрокси-группы в хлор проводить хлористым тионилом в растворе диоксана, то в результате будет сохранение конфигурации. Статья такая (не в уголовном, а в научном смысле) и правда была, но описывала она теоретическое исследование влияния растворителя на стереохимический исход замещения гидроксильной группы на хлор, и было действительно на паре моделей показано, что эфирные растворители диоксан и ТГФ способствуют сохранению конфигурации. Был даже сформулирован как будто третий механизм замещения SNi, совершенно, как показали последующие исследования несостоятельный, но страшно живучий – эту бессмертную гадину можно найти даже в текстах 21 века. Встретите – давите без сожаления. Более разумное и, видимо, правильное объяснение этому явлению – именно две последовательные SN2 реакции, первая с растворителем, который является слабым нуклеофилом, но в огромном избытке. Второй – уже с хлоридом. Довольно занятно. Такое явление называется межмолекулярным нуклеофильным содействием. Но с практической точки зрения оно малополезно, потому что в препаративных условиях плохо воспроизводится и ненадежно. Это – реликт времен увлечения механизмами реакций, когда каждый хотел изобрести свой механизм и войти в историю. У некоторых почти получилось. Но нам не нужно это подбирать, и всегда будем брать хлористый тионил в присутствии пиридина, когда результат – чистое обращение. А когда нам нужно сохранение конфигурации, используем двойное обращение, как показано в начале этой вкладки.
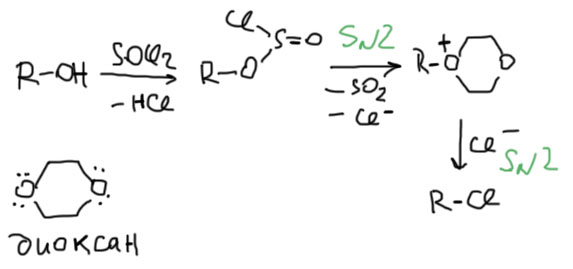
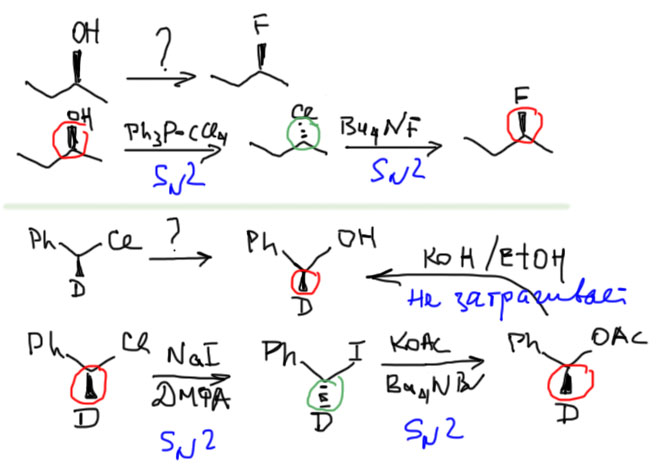
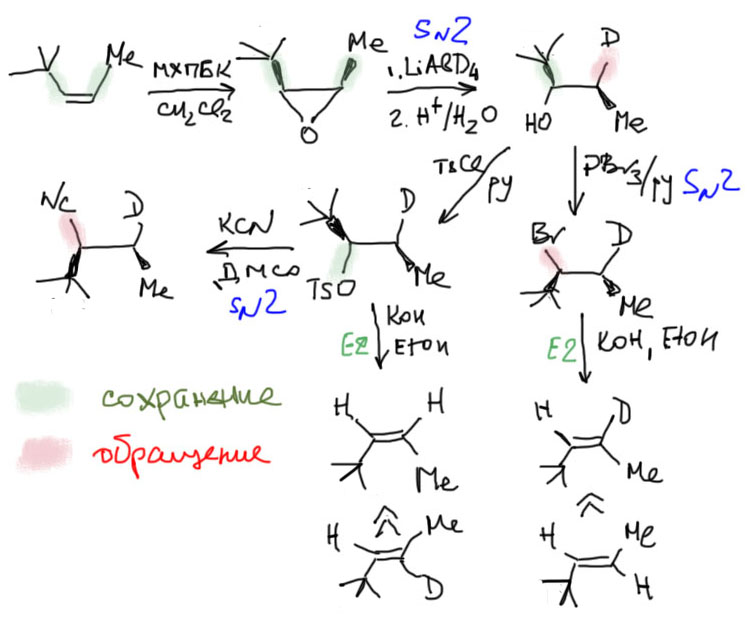
Раскрытие эпоксидов – не только мощнейший метод органического синтеза, используемый в сотнях и тысячах реальных синтезов сложнейших молекул, но и неисчерпаемый источник стереохимических задач, идеально приспособленный для дрессировки студентов с целью выработки условных рефлексов манипулирования стереохимическими формулами и отвлечения от вредных мыслей. В таких задачах сшиваются стереохимические закономерности химии олефинов, SN2-замещения и E2-элиминирования. Незатейливый, но неумолимый ход мысли в таких задачах чем-то напоминает рецепт приготовления пиццы из заплесневелых сухарей:
- эпоксид получают из олефина – это задает расположение заместителей;
- эпоксид раскрывают SN2-замещением – нуклеофил выбирает менее замещенный атом углерода и происходит обращение; на втором углероде при этом образуется спирт с сохранением конфигурации;
- спиртовая группа превращается в уходящую с сохранением (тозилат) или обращением конфигурации;
- выполняется еще одно SN2-замещение уже на этом центре;
- или E2-элиминирование.
Кушать подано. Угощайтесь.
Потренируемся в задачах. А пока пример…
Мы разобрали стреохимию анти-элиминирования и увидели, что так часто можно получать конкретные стереоизомеры (E или Z, цис или транс-диастереомеры) олефинов. Но эта реакция почти никогда не дает чистые стереоизомеры. Её используют только для того, чтобы поиграться со стереохимией. Поэтому, если в задаче потребовался чистый цис- или транс-изомер какого-то олефина, первое и последнее, о чем нужно думать, это не элиминирование, а стереоселективное гидрирование дизамещенных ацетиленов. Это очень надежный и довольно универсальный метод, который не нужно забывать никогда. Никогда!
SN2-замещение, стереохимия
- Превратите (R)-2-бутанол в (R)-2-метилбутанонитрил, (S)-2-фторбутан
- Превратите (R)-энантиомер монодейтеробензилового спирта PhCHDOH в (R,S) и (S,S)-диастереомеры дидейтеродибензилового эфира (PhCHD)2O. Что можно сказать об оптической активности полученных соединений?
SN2-замещение, алкилирование ацетиленида
- Из (R)-1,2-дифенилэтанола и метилацетилена получите (R)-1,2-дифенилпентан
- Из фенилацетилена, окиси этилена и иодистого метила получите 1-метокси-4-фенилбутан
Смещение двойной связи, синтез простых эфиров
- Из 2-метил-2-бутена (триметилэтилена), и изопропилового спирта получите 1-метокси-2-метилбутан и изопропил-трет-амиловый эфир (не брезгуйте гуглом, если забыли, что такое трет-амил или любое другое несистематическое название)
- Из 1-метилциклогексан-1-ола получите вот такой простой эфир. Решая задачу, будьте внимательнее и не поддавайтесь на провокации.
Эпоксиды - просто реакции с участием
- Из изопропилбромида и окиси этилена получите 4-метилпентанонитрил
- Из пропилена и бромбензола получите аллилбензол (осторожно: дешевая разводка – не попадайтесь!)
- Из метилацетилена, иодистого метила, диэтиламина получите 3-диэтиламинобутан-2-ол
Эпоксиды - стереохимия раскрытия цикла
- Из цис-β-дейтеростирола, иодистого метила получите 1-фенил-1-метокси-2-дейтеропропан с трео-конфигурацией метокси-группы и дейтерия. Внимание: если вам показалось (как мне самому через 2 года после того, как она была написана), что в этой задаче есть ошибка в условии, это не так, ошибки нет, задача решается.
- Из цис-стильбена получите 2,3-дифенил-3-дейтериопропанонитрил с трео-конфигурацией дейтерия и циано-группы.
Раскрытие эпоксидов, SN2, E2-элиминирование - стереохимия
- Стильбен (дифенилэтилен) подвергли следующим превращениям: а) эпоксидированию; б) раскрытию эпоксида метилмагнийиодидом с последующим гидролизом; в) превращению спирта в тозилат; г) элиминированию под действием спиртовой щелочи. При этом получился 1,2-дифенилпропен. Напишите схемы реакций с учетом стереохимии для двух разных изомеров стильбена – цис и транс. Объясните, почему элиминирование в одном случае (каком?) идет легко, а во втором существенно труднее. Что изменится, если гидроксил вместо тозилата превратить в другую уходящую группу – хлор?
Определяем изомер олефина по данным по стереохимии элиминирования
- β-Дейтеростирол (PhCH=CHD) подвергли следующим превращениям: а) эпоксидированию; б) получающийся эпоксид обработали фениллитем с последующим гидролизом реакционной смеси; в) получившийся спирт перевели в тозилат действием тозилхлорида в присутствии основания; г) элиминирование из образовавшегося тозилата под действием этилата натрия. В результате образовался транс-стильбен, не содержащий дейтерия. Какой изомер (E или Z) исходного дейтеростирола был взят.
Отдельно были проделаны почти те же превращения, но вместо тозилата было получено хлорпроизводное действием трифенилфосфина и тетрахлоруглерода, и далее опять элиминирование. Опять получился транс-стильбен без дейтерия. Тот же самый или другой изомер дейтеростильбена был использован во втором эксперименте?
Все умозаключения проиллюстрируйте схемами реакций с использованием стереохимических формул (для тренировки проделайте стереохимические манипуляции с разными стереохимическими проекциями – Фишера, Ньюмена и естественными).
Определяем диастереомер по результатам SN2 и элиминирования.
- (R)-втор-бутилфенилкетон (он же (R)-1-фенил-2-метилбутанон-1) восстановили боргидридом натрия, при этом получили оптически активный спирт. Спирт превратили в тозилат, тозилат обработали иодидом натрия в ДМФА, а иодид подвергли элиминированию действием гидроксида калия в этаноле. Основной продукт – (Z)-1-фенил-2-метилбутен-1. Определите, какой диастереомер (трео или эритро) спирта получился при восстановлении кетона. Разрисуйте всю стереохимию.
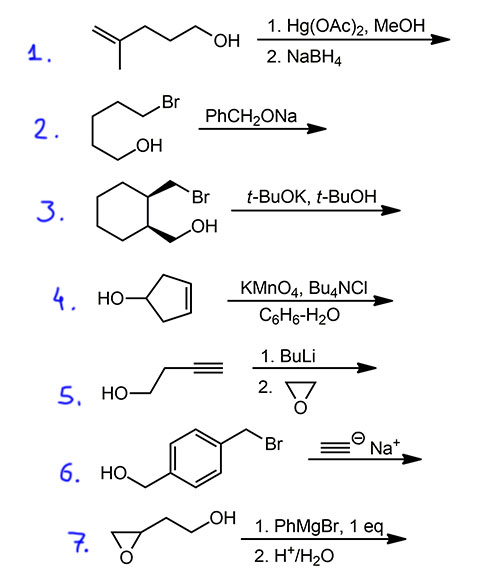
Защита гидроксила
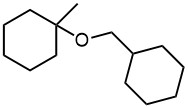
Вот семь реакций. Напишите продукты, сначала оставив гидроксил свободным, а затем сначала защитив гидроксил ТГП-защитой и после реакции сняв её. Сравните результаты и порадуйтесь различиям продуктов. Различия должны быть везде!