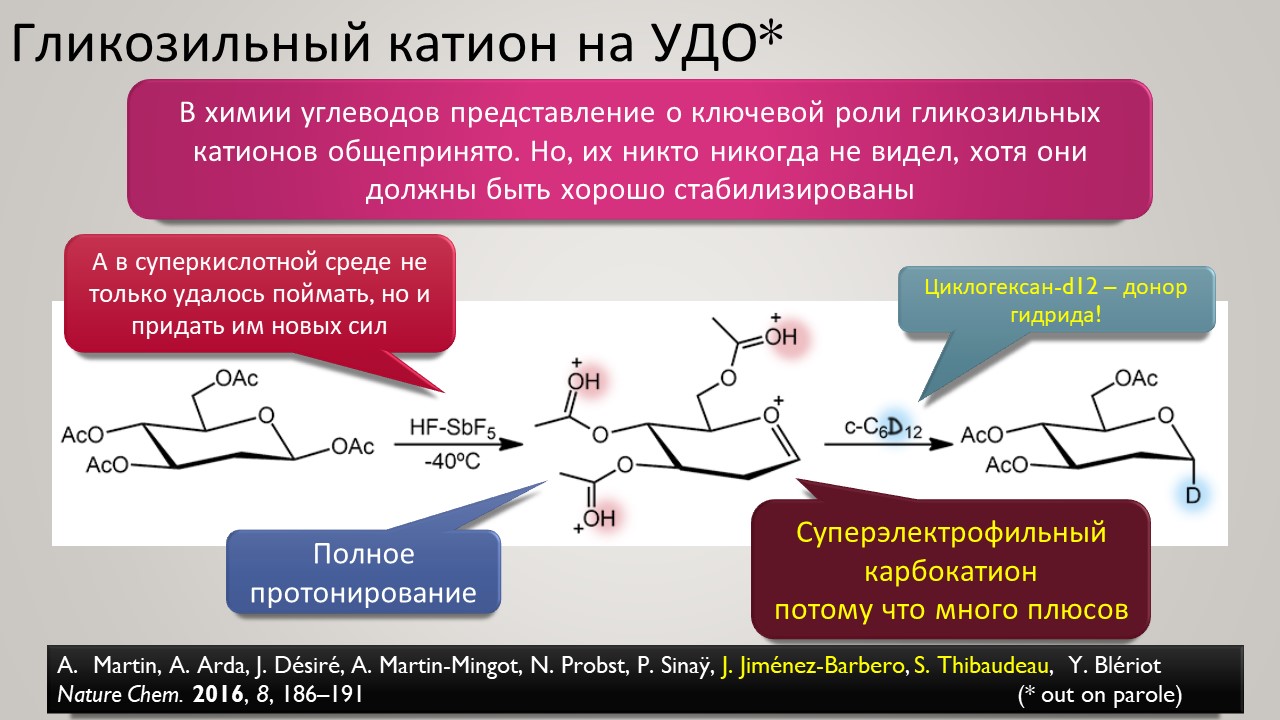
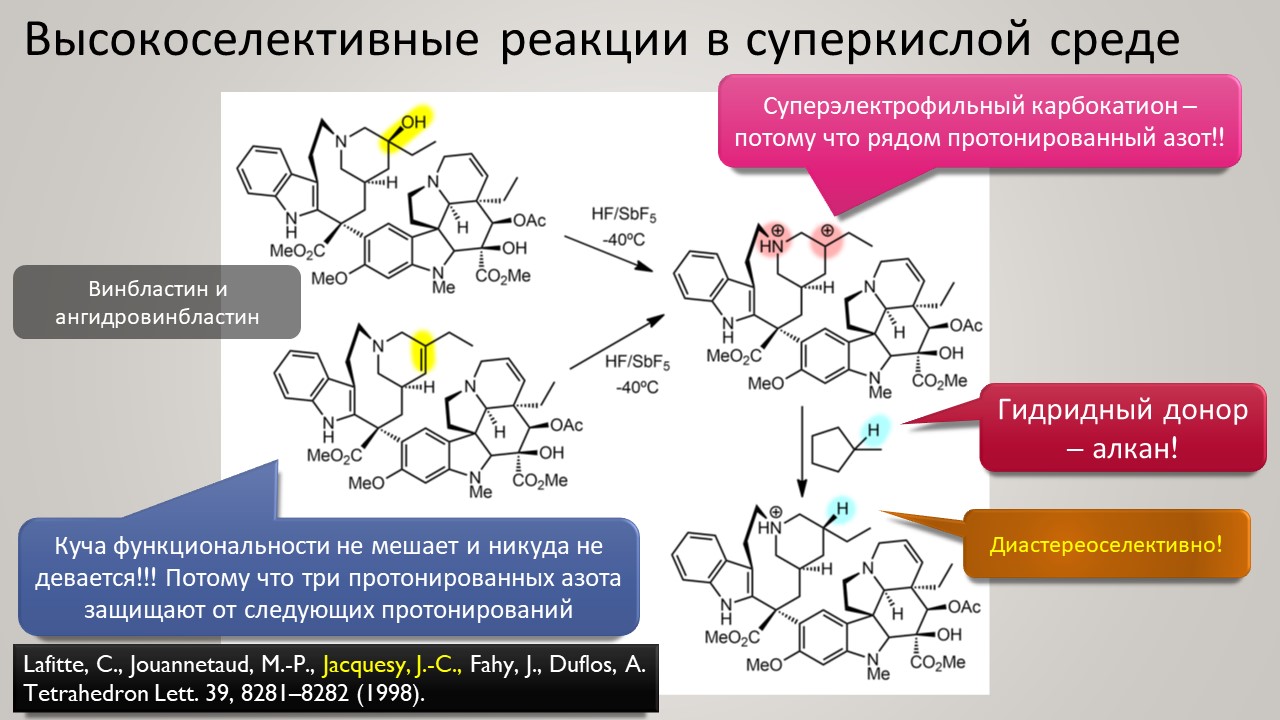
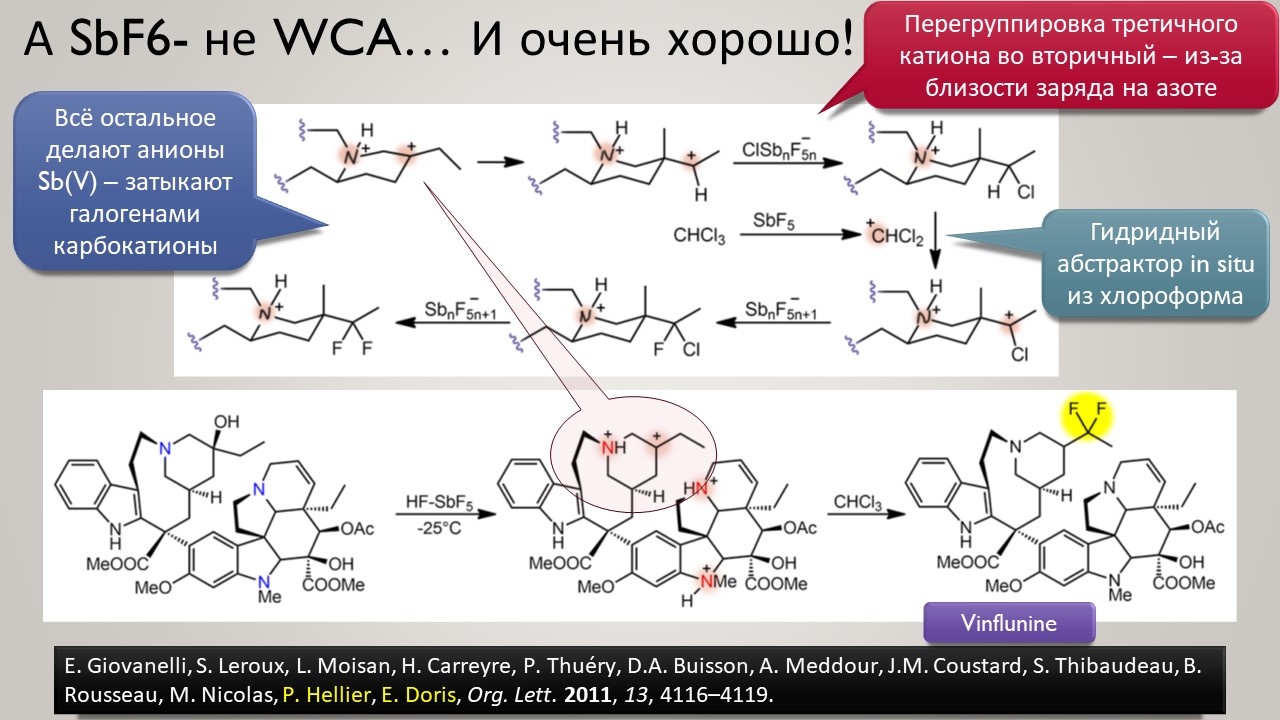
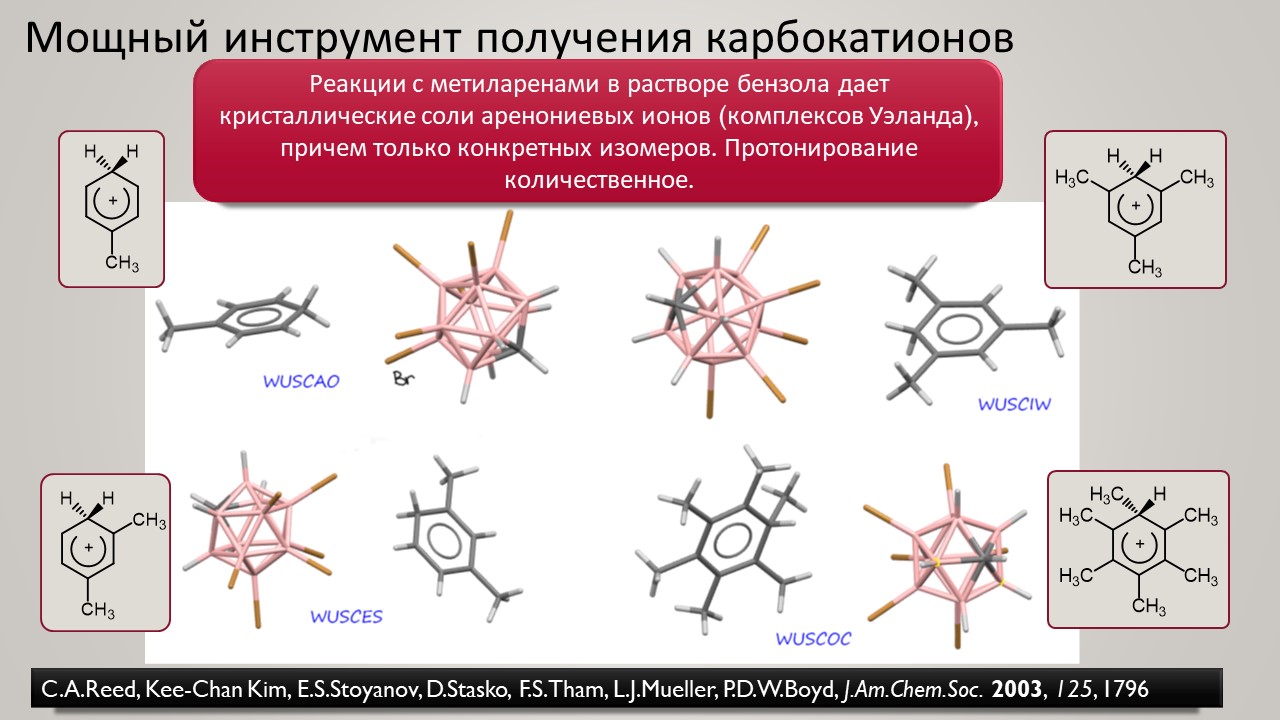
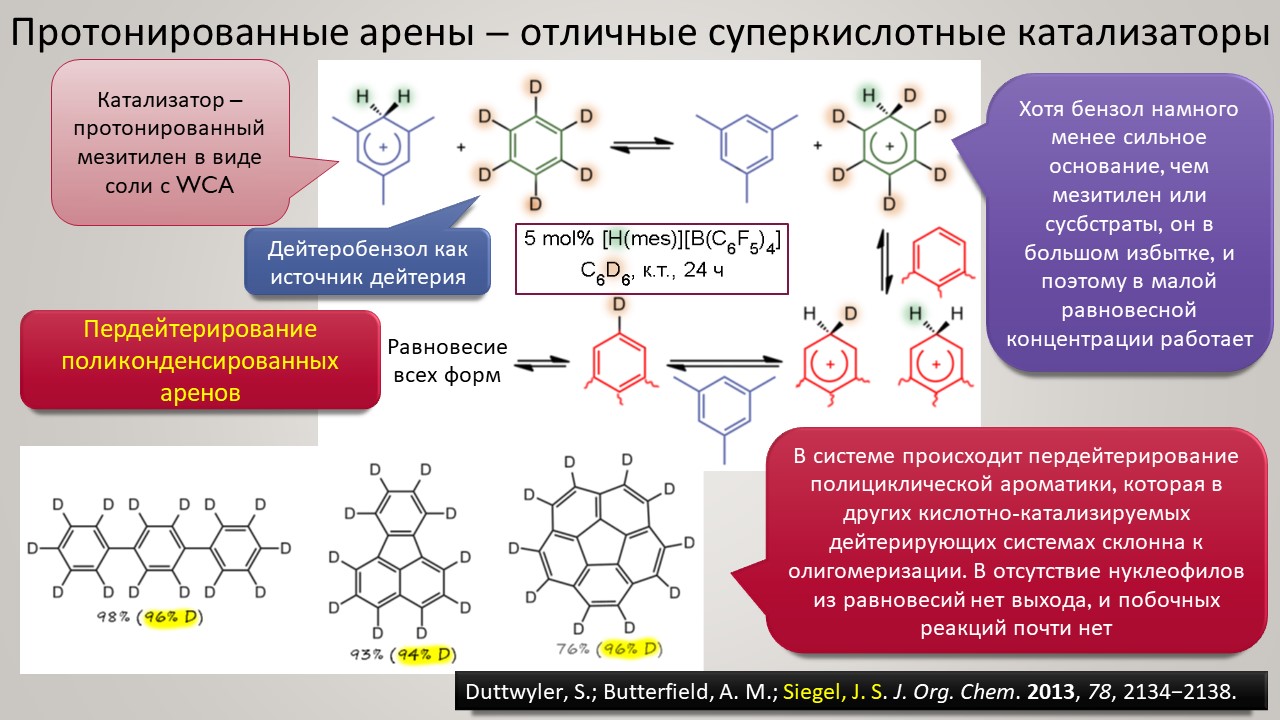
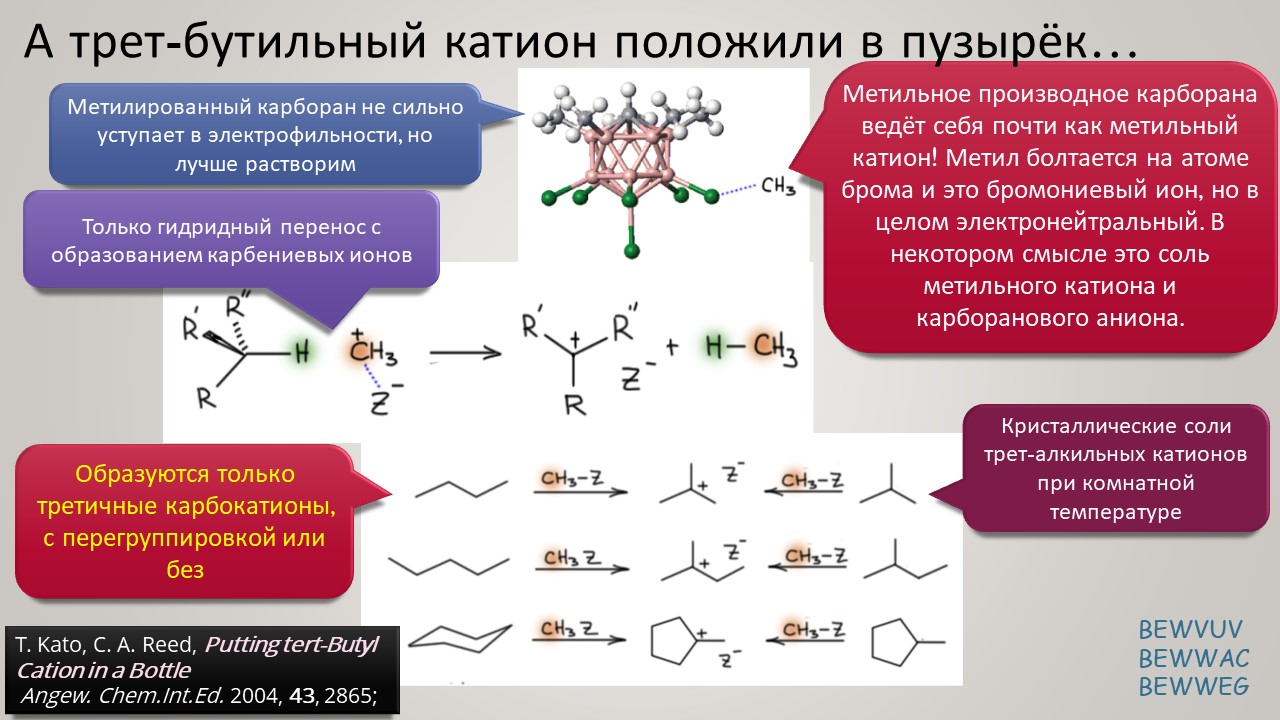
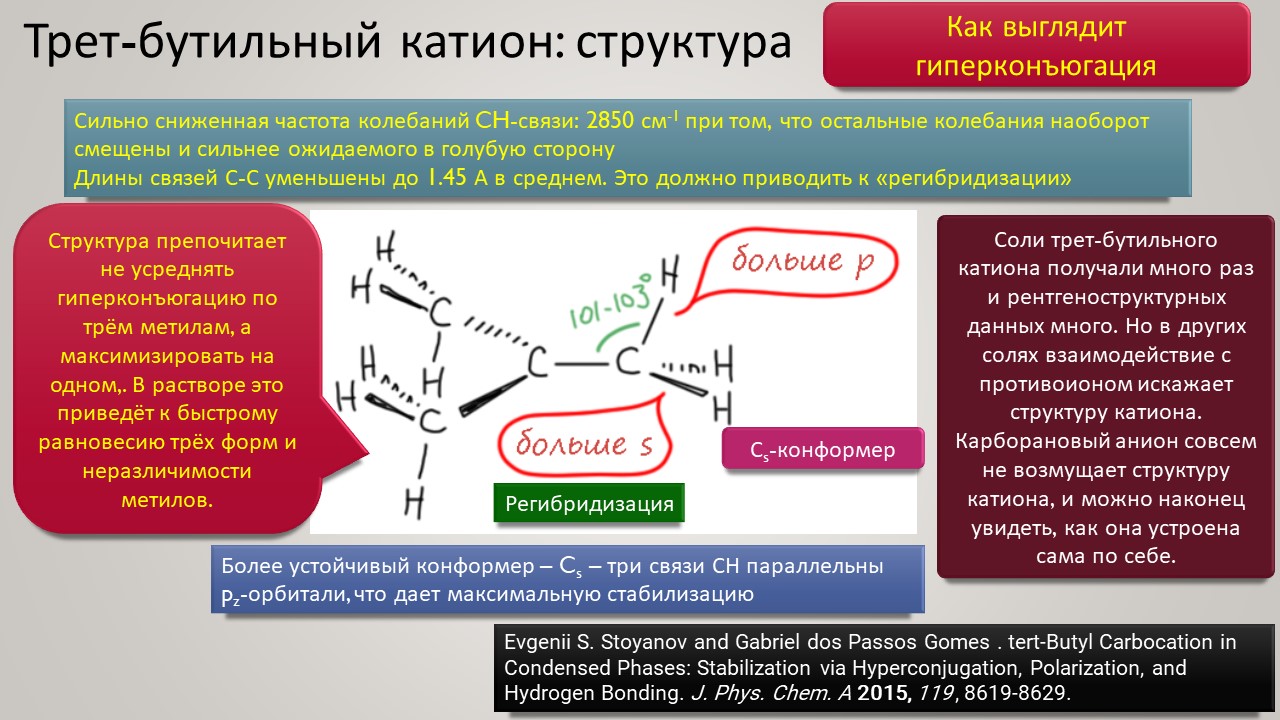
Есть ли предел кислотности и зачем он нужен
Среди основных понятий химии кислоты и основания занимают одно из первых мест – мы не можем понять химию ни на элементарном, ни на каком угодно высоком уровне, не понимая, что такое кислоты и основания, какие они бывают, и как кислотно-основные взаимодействия участвуют в химических процессах.
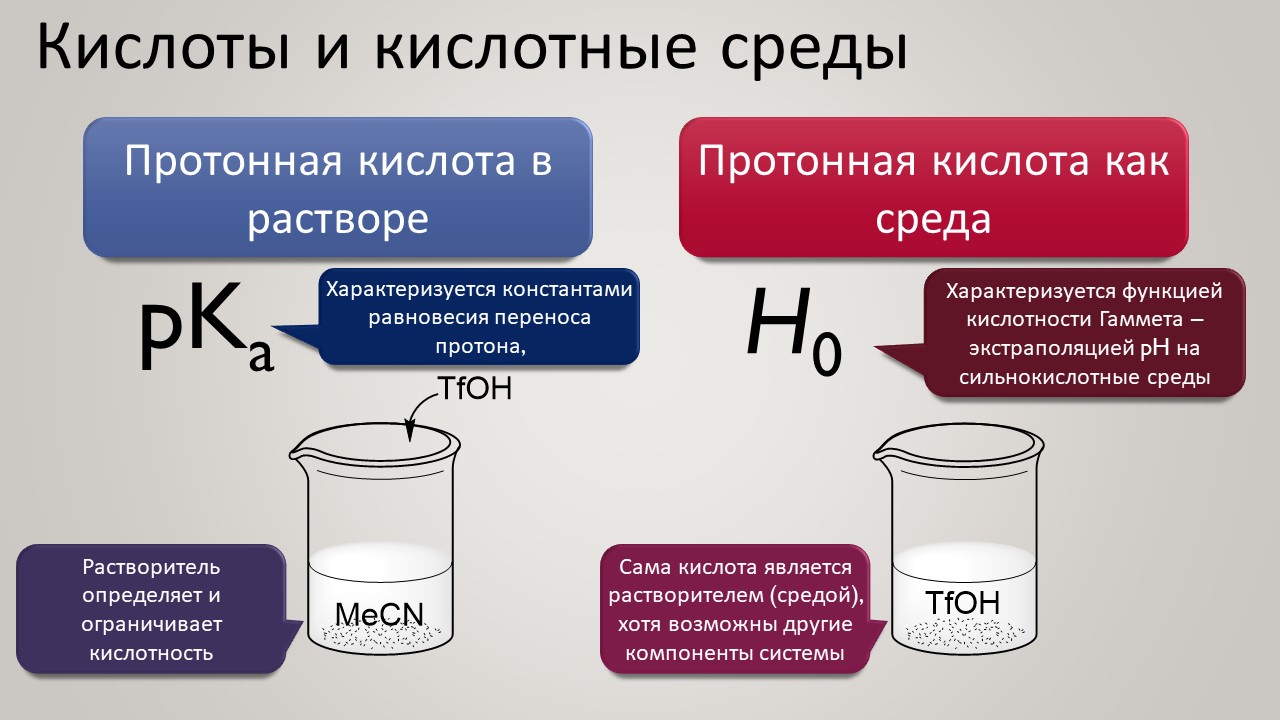
В 20-м веке возникло несколько теорий кислотности и основности, без сомнения самые важные – теории Бренстеда-Лоури или просто протонной кислотности (основность всегда идёт в паре и её даже не обязательно явно упоминать) и теории Льюиса, возникшей одновременно и как составная часть теории об особой роли электронных пар в химии. В 20-м веке с этими теориями отлично разобрались, построили на них фундамент теории химии, как органической, так и неорганической и координационной, убедились, что нужны обе, и что невозможно их объединить в одну (попытки были, но большого следа не оставили), а приходится жить с обеими. Надо признать, что это немного неудобно, потому что эти теории оперируют совершенно разными методологиями. Если теория Бренстеда-Лоури оперирует термодинамическими характеристиками, константами равновесия кислотно-основных процессов, и таким образом хотя бы в принципе может претендовать на обладание количественными характеристиками, позволяющими объективно сравнивать силу кислот и оснований, то теория Льюиса позволить себе этого не могла из-за того, что в ней долго не было консенсуса о том, какое основание (или кислоту) можно было бы выбрать в качестве опорной для однообразной привязки шкал. Теория Бренстеда-Лоури хороша еще и тем, что она одинаково относится к кислотам и основаниям за счёт симметричности кислотно-основного равновесия: из кислоты и основания получается основание и кислота, поэтому вы можете очень удобно и кислоты и основания характеризовать одним набором констант (строго говоря – одним набором для каждого растворителя или среды), составляя такие лестницы кислотности (или основности), по которым можно взбираться из подвала самых слабых к сияющим высотам самых сильных. И вот это восхождение – одна из самых интригующих глав химии кислот (и оснований тоже, но это будет в отдельной лекции).
Зачем нужно переться всё выше и выше в поисках самой-пресамой сильной кислоты? Для того, чтобы ей присвоить звание кислотиссимусы (сохраним женский род, и немного порадуемся тому, что в русском языке кислота женского рода, а основание – среднего, что порождает нехорошие предположения о нетрадиционности кислотно-основных отношений), а себе славу человека, первым взобравшегося на Эверест? В прошлом веке такой чемпион чемпионов нашёлся, и место Самой Сильной Кислоты было занято, и дальнейшие попытки штурмовать вершину прекратились, правда фантазии первооткрывателя хватило только на довольно банальное определение Magic Acid, для убедительности оформленное как торговая марка. Что в этой кислоте волшебного сказать не мог никто и никогда, ведь рядом с ней расположилось ещё несколько составов практически такой же кислотности, но победителей не судят.
Победителей побеждают следующие победители, тогда предыдущие становятся проигравшими, а проигравших очень даже хорошо судят. Путь, предложенный создателем Волшебной Кислоты (вам больше нравится Магическая? – но вот магического в ней точно нет ни грана, в волшебным, как известно, может быть даже идиот, так что однозначная позитивность этого эпитета не очевидна) оказался в прямом смысле тупиковым – он и привёл в тупик (волшебный?), выхода из которого не было: ведь все возможные комбинации протонных кислот и кислот Льюиса были быстро исчерпаны, и всё упёрлось в одно действительно уникальное соединение – пятифтористую сурьму, обладающую какой-то совершенно нечеловеческой потребностью расширять координационное число сурьмы до шести и становиться октаэдром, подбирая буквально любые нуклеофилы, даже самые слабые. Ничего похожего больше нет. Поэтому гонка за кислотностью в 20-м века просто заглохла, а сами кислотно-основные отношения приобрели неприятный запах протухшей рутины, которую надо просто знать, как таблицу умножения, но ничего нового там не достанешь, а нобелевка и так уже уплыла, и второй не дадут.
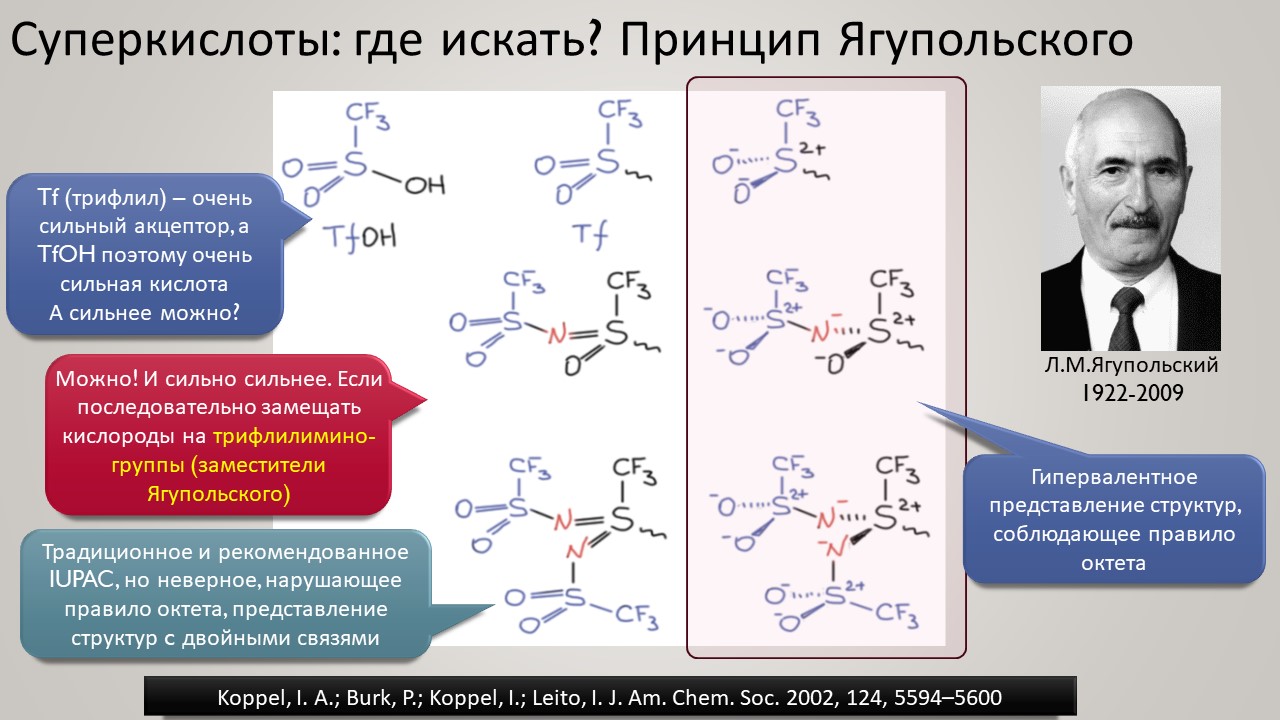
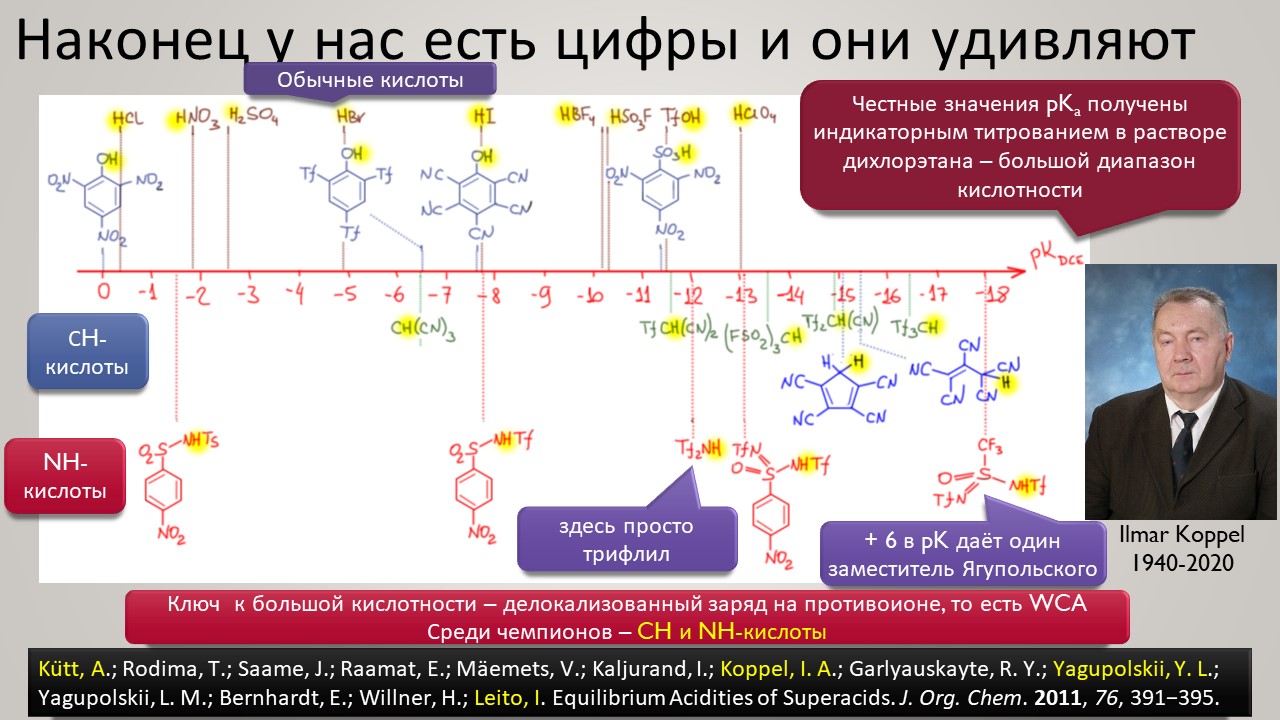
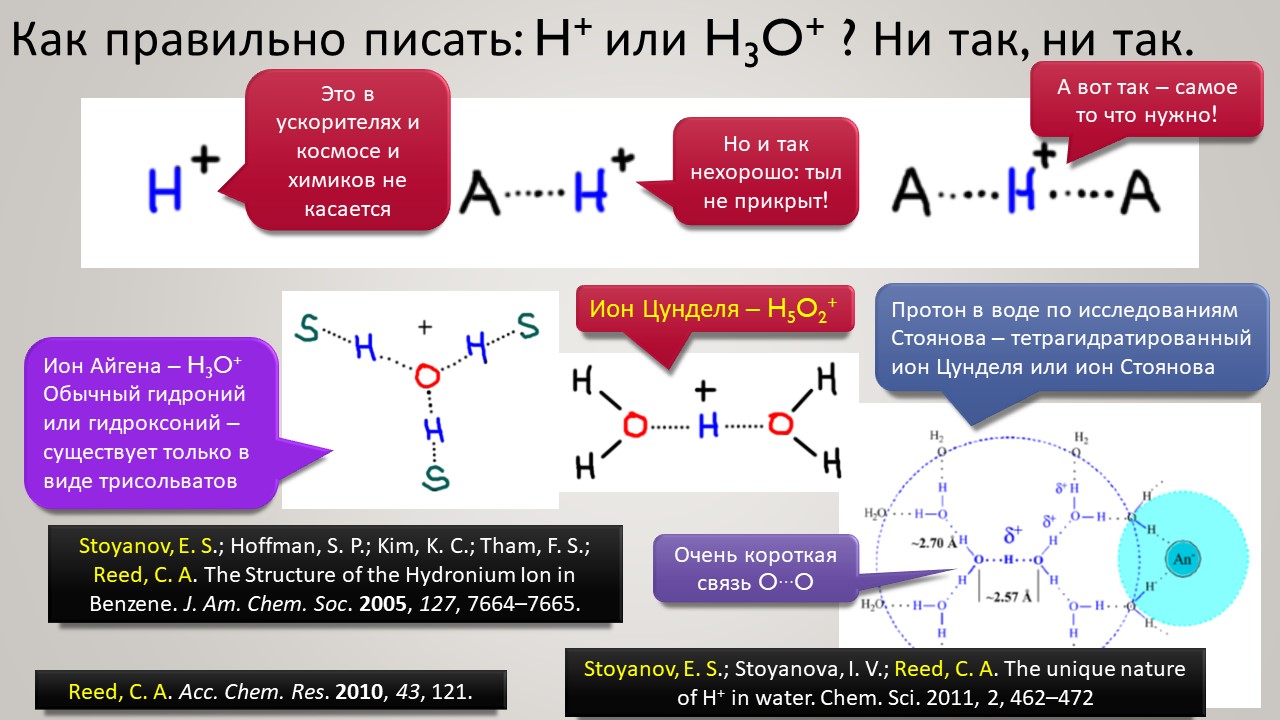
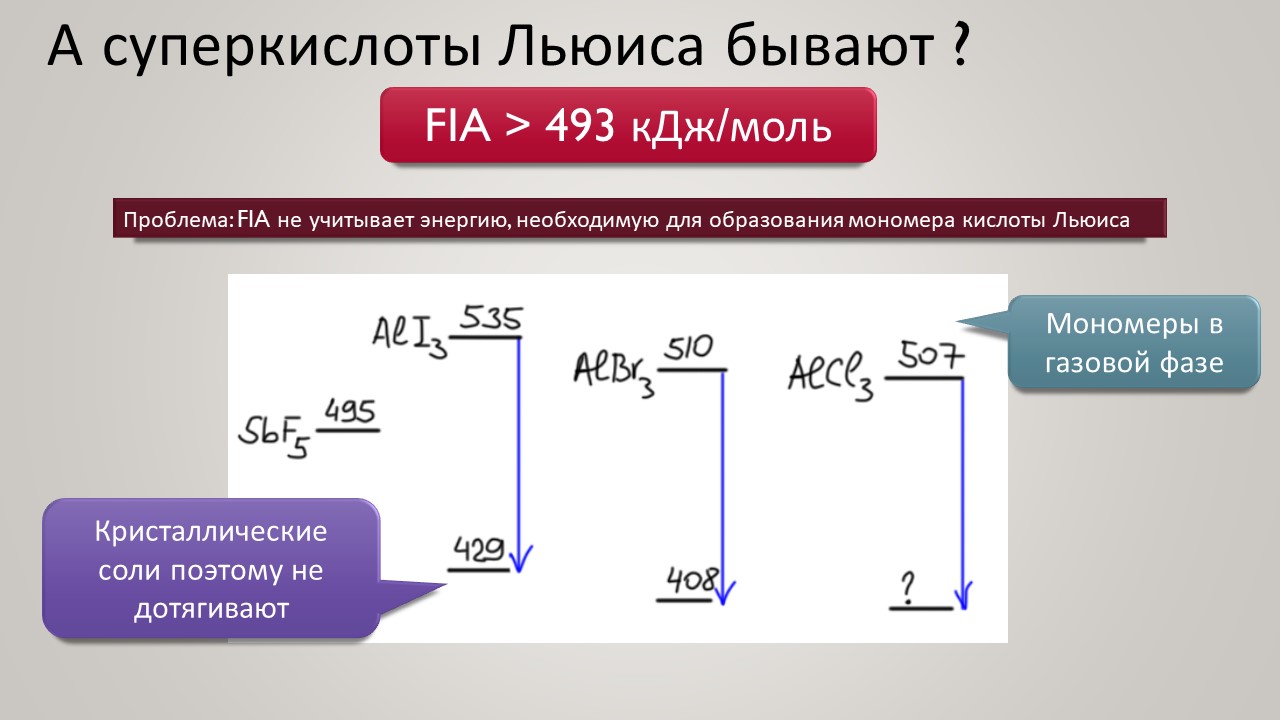
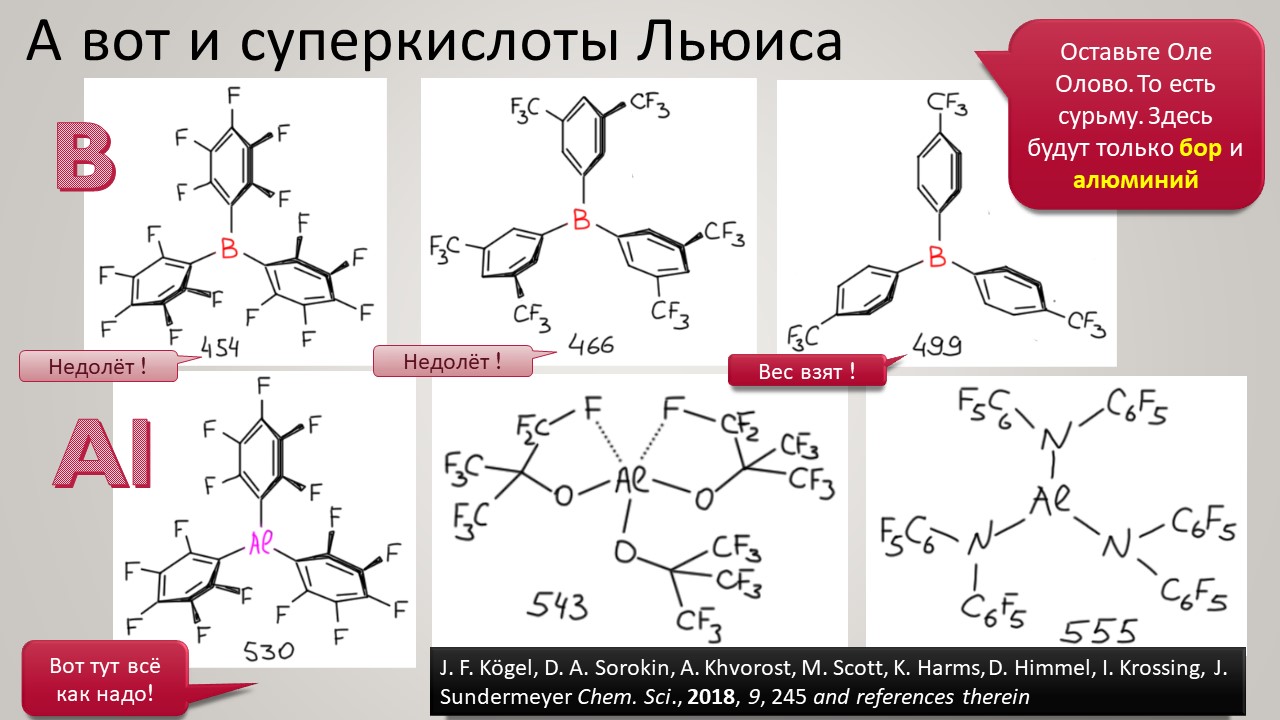
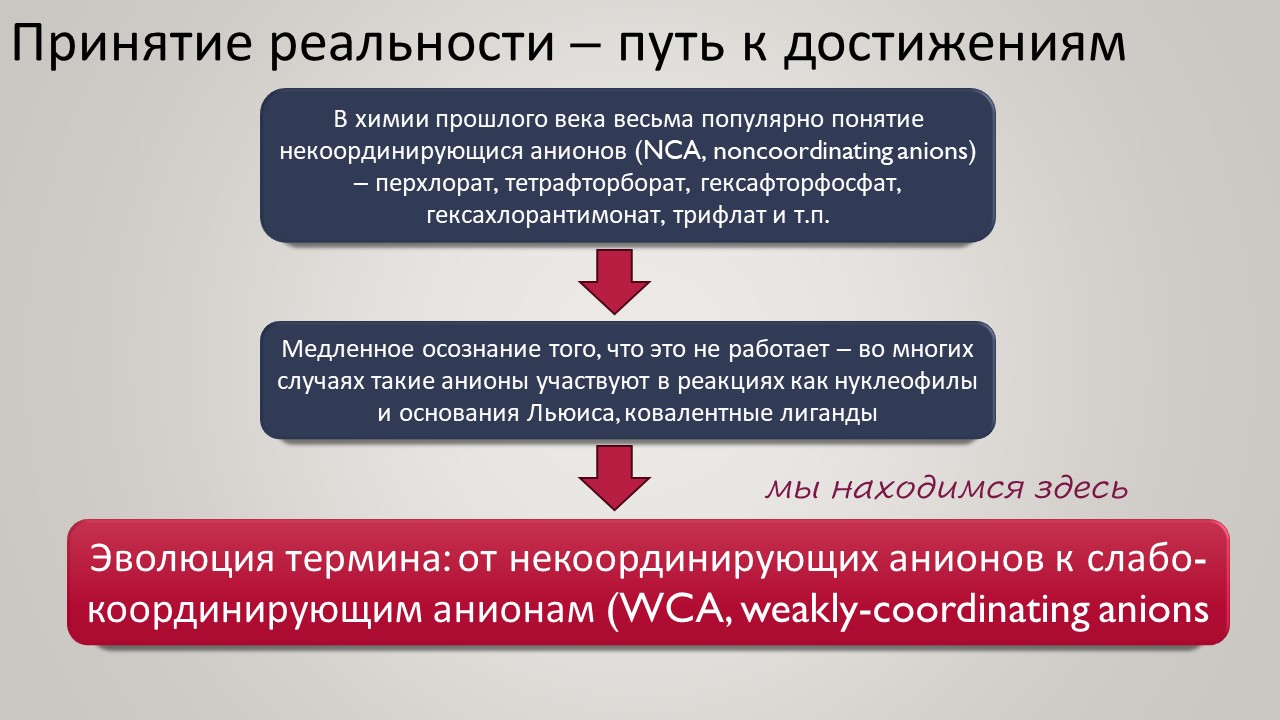
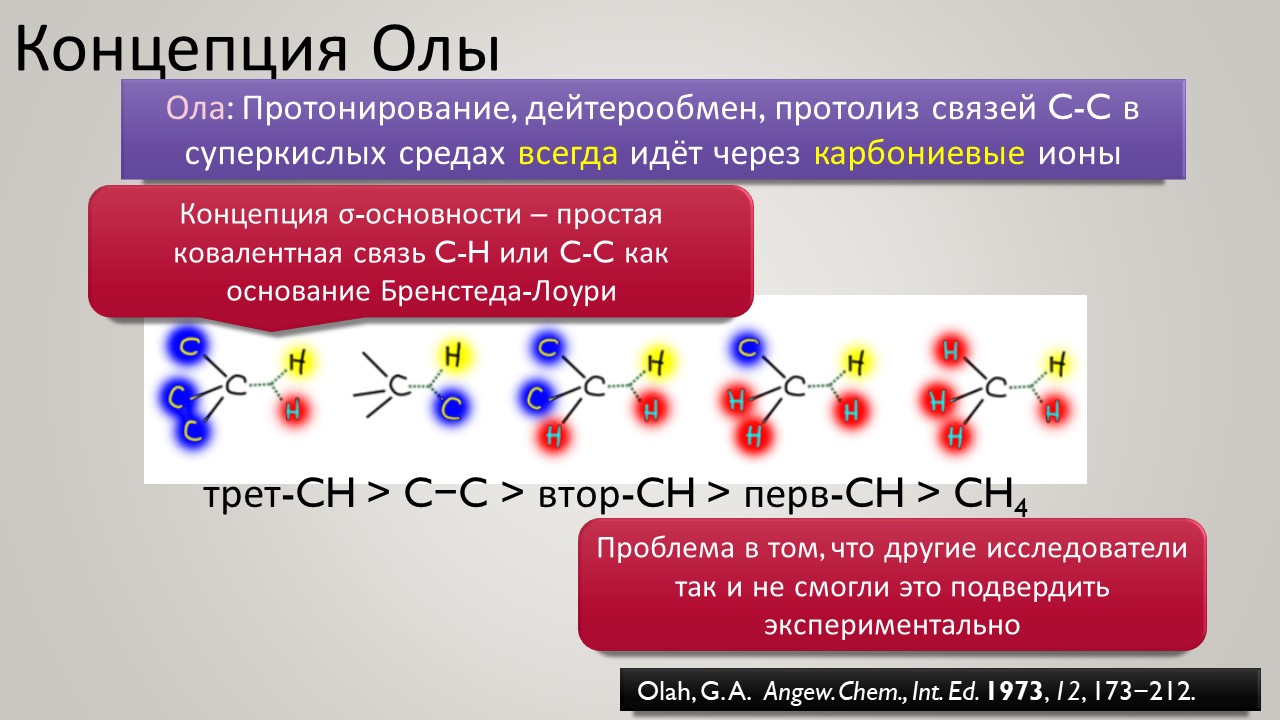
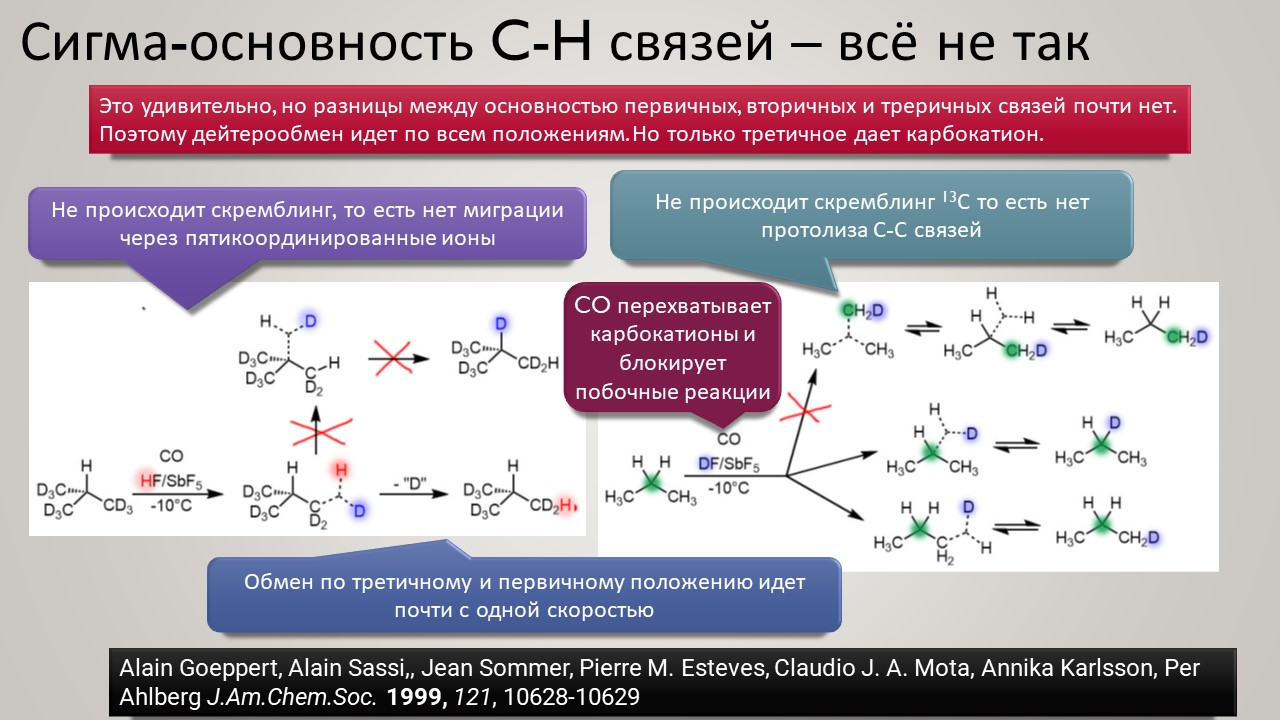
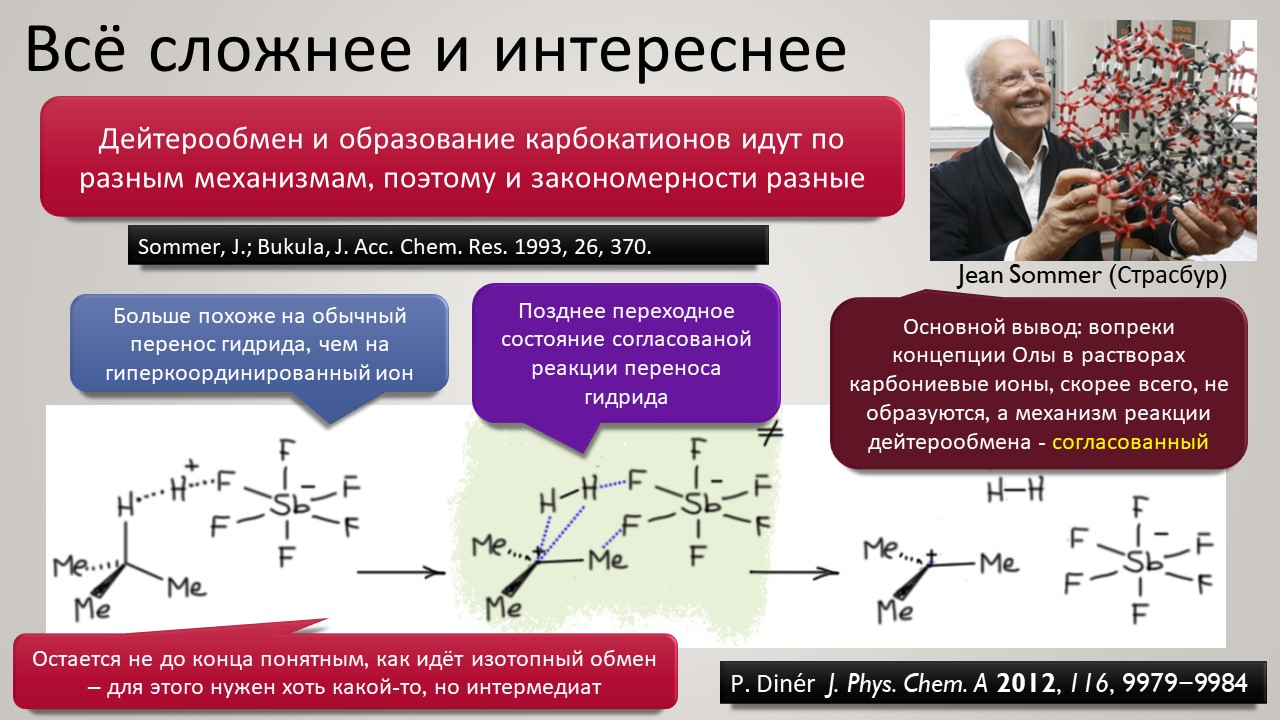
Но новый век всё же и здесь принёс оживление. Оказалось, что чтобы двигаться дальше, нужно получше разобраться в том, как ведёт себя протон в конденсированных средах, где делается химия. Протон – штука удивительная, это единственная химическая частица (то есть, частица, обозначаемая символом элемента из Таблицы Менделеева), у которой вообще нет собственных электронов, и это делает её поведение в химических системах совершенно особенным – она цепляется вообще за всё просто потому что это её единственный способ приобщиться к электронной плотности и стать чем-то похожим на химию, а не на субатомную частицу для космоса или ускорителя-коллайдера. Но фишка в том, что если мы хотим иметь кислотность побольше, нам надо придумать что-то, к чему протон хорошо цепляться не сможет – не совсем не сможет, что невозможно – но будет цепляться слабо, чем слабее тем лучше. И в этом месте сразу рухнуло всё обаяние фтористых производных сурьмы, потому что за фторные лиганды на сурьме протон как раз цепляется очень хорошо, и это и определяет потолок кислотности. Свободу протону! Ищите такие анионы, которые взаимодействуют с протоном намного слабее гипервалентных фторидных комплексов Sb(5+). И дело пошло – таких противоионов нашлись целые серии, поскольку были найдены принципы построения таких анионов сразу нескольких типов. И этот поворот оказался чрезвычайно плодотворным не только в спортивном смысле погони за суперкислотность. Впервые нашлись очень хорошие точки соприкосновения двух кислотностей – протонной и Льюиса – оказалось, что штурмовать одновременно две вершины проще, чем одну за другой.
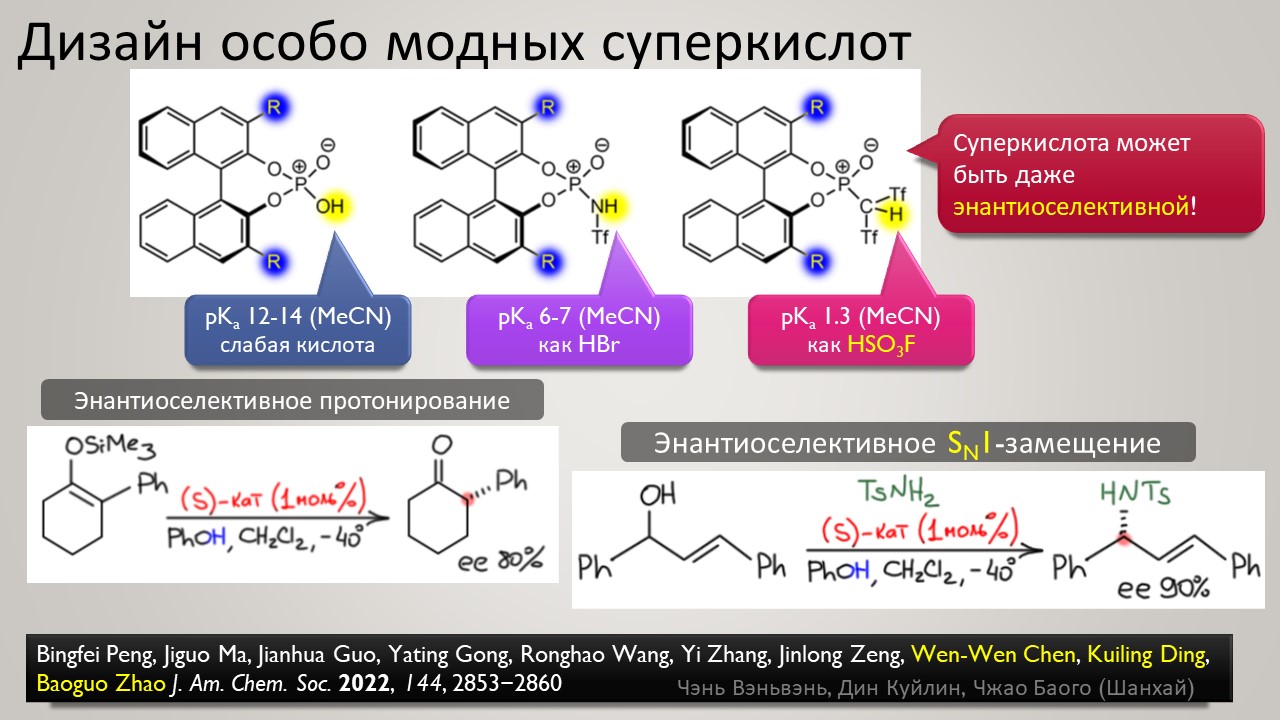
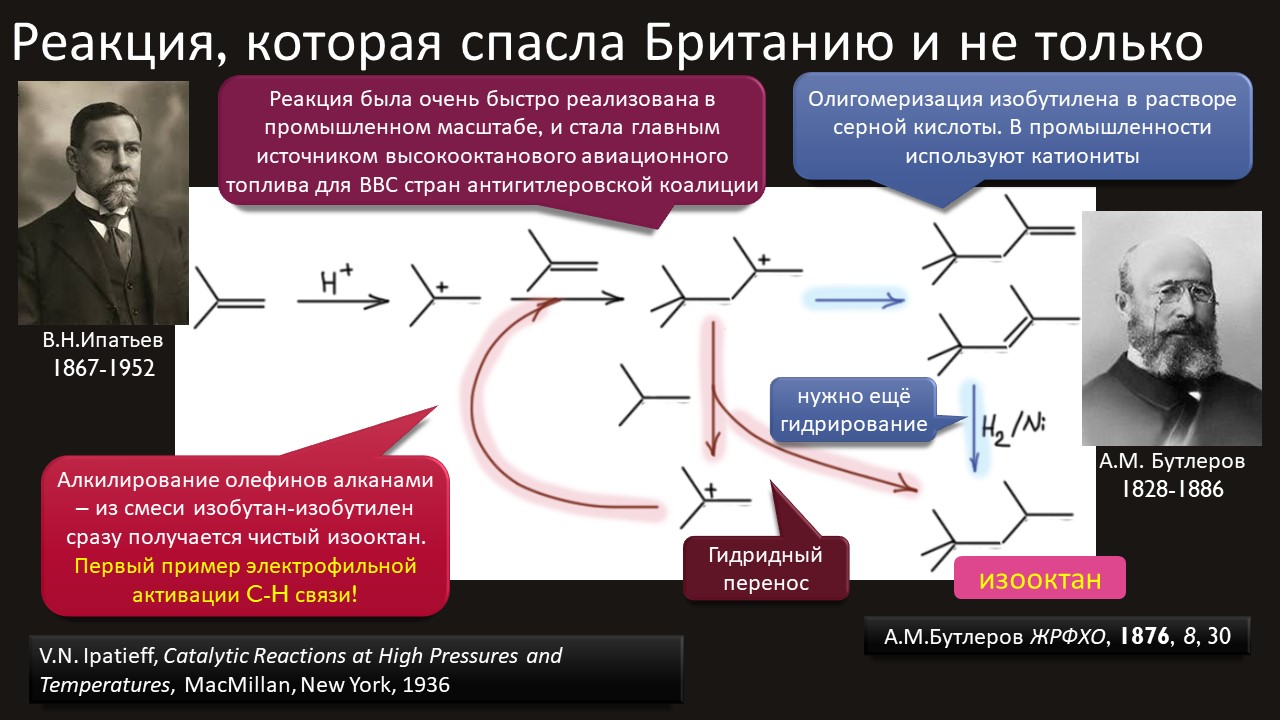
Высокая кислотность – дело, безусловно, общехимическое, и неорганики, и координационные химики тоже очень ценяят сильные протонирующие среды и суперкислоты обоих типов -протонные и льюисовы. Но органики особенно заинтересованы в том, чтобы под рукой всегда были суперкислотные среды и реагенты – высокая кислотность даёт ключ к электрофильной активации самых разных групп и реакционных центров, как показал ещё Владимир Ипатьев, открывший в 1930-х совершенно беспрецедентный для современной ему химии процесс алкилирования олефинов и ароматических соединений алканами, то есть первый пример электрофильной активации C-H связей в парафинах, до этого считавшимися почти безнадёжными с точки зрения применения как исходных в органическом синтезе. Мощное развитие этот подход получил только через полвека в работах Джорджа Олы, получившего за исследования сверхкислотных сред и активации алканов нобелевскую премию в 1995 году. Одно из важнейших применений этой химии – быстрый прогресс в исследовании карбениевых ионов (карбокатионов), а в новом веке введение в исследования новых классов сверхкислот и слабокоординирующих противоионов позволили расчистить множество проблем этой химии и отбросить немало заблуждений. Прогуляемся по этой химии и найдё там много интересного и даже сенсационного, а главное, немного приведём в порядок наши представления о кислотности и кислотах, а там с прошлого века накопилось немало странного мусора, с которым стоит расстаться без сожаления.