О чём эта страница
О нобелевской премии 2021 года.
Как недавно сказал мне один студент, обратив внимание на мое давнее безумное пророчество о том, что органика никогда больше не получит нобелевских премий, – а какой смысл тогда заниматься органикой, если нет шансов съездить в Стокгольм к тамошнему королю. Это хороший настрой, одобряю, испытываю жгучий стыд, беру свои слова назад, да и вообще – давайте и правда посмотрим, как лауреаты этого года решили эту проблему. И вы тоже сможете, если постараетесь. Будьте как Б.Лист и Д.Макмиллан, и одна из нобелевок второй четверти 21 века – ваша.
На этой страничке мы уделим некоторое время общему трёпу, но главная цель страницы другая – действительно разобраться, за что дали нобелевку, причём довольно подробно, даже занудно, за что сразу извиняюсь, но этот сайт иногда читают не только наши студенты, изощрённые в органической химии и всё понимающие с полуслова, но и просто любители химии. Разберём, что такое органокатализ, какой он бывает, почему так важна энантиоселективность и как она достигается. Заодно разберемся в некоторых важных и тесно связанных с органокатализом вещах – механизме конденсации Кнёвенагеля, МБХ-реакции, как работает витамин В1 и при чём тут карбены, энантиоселективной альдольной конденсации. И ещё обязательно разберёмся, чем кормят свиней (и коров), и почему это очень важно для успеха химии.
Да здравствует Альдольная конденсация!
Органики долго ждали своей нобелевки. Такого долгого перерыва не было давно. В 20 веке органики получали премии регулярно, но к концу века скорость стала снижаться. В 21 веке органики получили три премии за одно и то же – достижения катализа комплексами переходных металлов, и это показало, что у чистой органики наступают тяжелые времена – она уже не тянет одна, ей нужны пособники в лице координационной химии и промышленного катализа. Как и хотели великие Коттон и Уилкинсон, органика стала сливаться с неорганикой в одну науку, а чистые идеи сами по себе теряют актуальность – химия должна перестать делиться на чистую и прикладную. Налогоплательщикам больше не хочется платить за то, что некие умники копаются в механизмах реакций, стратегиях синтеза, реакционноспособных интермедиатах и прочей оторванной от жизни чистой науке – люди хотят лекарств, вечной молодости, средств против морщин, комаров, ковида; ярких экранов и всяких доселе невиданных прибамбасов буквально везде – и для всего этого нужны тысячи готовых органических соединений, а в процессе разработки нужен быстрый доступ, и виртуальный и реальный, к миллионам новых молекул. Поэтому наука номер один в современной химии – синтез, и не такой как раньше, неспешное колдовстово над колбами, а быстрый, надежный, высокоселективный, желательно легко автоматизируемый. Думать больше некогда, нужно синтезировать. Три металлические премии (2001,2005, 2010) ровно про это, за каждой из них стоят лоббисты из огромных химических компаний, которые собственно и оплатили исследования, приведшие к тем великим открытиям и достижениям, которые радикально изменили органический синтез. В тех премиях было много конкурентов, выбор конечных имен для объявления был далеко не бесспорен. Премию получили достойные, но там за каждой было еще много других не менее достойных, поэтому пришлось кого-то просто отодвинуть в сторону, а с кем-то другим просто терпеливо дождаться естественной убыли претендентов, отчего каждую из тех премий вручали седым старцам, и некоторые из лауреатов уже с трудом вспоминали, что же такое они отчебучили десятилетия назад, что их вдруг вытаскивают с тёплого моря в холодный Стокгольм и заставляют произносить речи про то, что уже давно уехало в раздел “когда мы были молодыми…”. После последней металло-каталитической премии 2010 года повисла зловещая тишина. Премия за премией уходили в материалы, атмосферу, ну и в бесконечную химию жизни, которая и берет максимум из всех химических премий, и так повелось еще очень давно. Да это и правильно, потому что ничего более интересного и волнующего, чем раз за разом убеждаться, что жизнь устроена намного сложнее, чем можно было бы предположить, и никаких надежд на то, что еще пара усилий и отступят все болезни, не осталось даже у самых отъявленных оптимистов. Усилий потребуется ещё немало, и немало химических нобелевок ещё попадет в клетку. Это вообще парадокс – всем понятно, что молекулярная биология, биохимия и как там это еще называют под зонтичным брендом life sciences – это вроде всё про органическую химию, и кто скажет, что белки и нуклеиновые кислоты – не органические соединения, и ведь большинство процессов, стоящих за функционированием клеток и тканей – это просто самые обыкновенные органические реакции, даже не просто самые обыкновенные, а нарочито простейшие типа всевозможных вариантов ацилирования и замещения, в обычной органической химии мы их считаем рутиной и скукотищей и поскорее стараемся проехать, чтобы заняться чем-то более сложным и волнительным. Поэтому никто уже давно не рассматривает эту науку как что-то связанное с органической химией. Дети выросли, уехали и забыли дорогу в родительский дом, а самих родителей, не скрывая, откровенно стесняются, какие-то они простоватые и воняют пиридином. Да, чуть не забыл, в 2016 году была ещё прикольная премия за “молекулярные машины”, а это вроде все органика, там даже синтез вполне зачётный попадается, а среди лауреатов есть мощные органики с кучей других заслуг именно в синтезе. Но почему-то никто не вспоминает о той премии в контексте органики, да и сами “молекулярные машины” очень хороший пример, как некоторые малость переусердствовали с рекламой довольно спорных работ, сделанных сильно на вырост. С тех пор прошло уже 5 лет, но никто никуда на молекулярных машинах не уехал, и мы как-то нечасто сталкиваемся с этими штуками на страницах химических журналов. Возможно, надо заглянуть в журнал “За молекулярным рулём”, только никак не найду ни одного номера.
И вот наступил холодный октябрь 2021 года, второго года новой эпохи, эпохи ковида. А что – ещё присуждают нобелевки? Не перенесли на пару лет, как олимпиаду и чемпионат Европы по футболу? Да, там в Швеции особый путь, не боятся ни чёрта, нобелевский комитет продолжал, оказывается, заседать, наверное, в зуме. Посовещались, и присудили. Не ковид ли повлиял, что вспомнили про органику? Давайте разберёмся, это интересная история. И сразу скажу, на мой вкус выбор лауреатов в этом году почти безупречный – премия нашла тех, кто этого хотел и очень много для этого сделал. И дана наконец вполне молодым исследователям в самой кульминации научной карьеры – их новые работы последних 10 лет намного интереснее того, что они делали раньше и за что собственно и присуждена премия. Особый комизм делу придает то, что и тот и другой давно забросили органокатализ в оригинальном смысле этого слова, как бы собственным примером показывая реальный потенциал этой области. Но раскрутили область именно они, и именно сначала вдвоем, и смогли так ярко показать потенциал, что туда с рекордной скоростью набилась туча народа, включая нескольких легендарных патриархов органики вроде Барри Троста. Нобелевка отчасти именно за это – за демонстрацию возможностей новой химии в новом веке, когда никто не хочет долго ни в чем копаться. Вместо этого надо найти хорошую область, чутьём уловить возможности, придать даже самым очевидным идеям статус концепций, бросить большие силы и средства, заманить побольше последователей – и вот результат: проходит десять с небольшим лет и область готова – как хорошее поле засеяно, удобрено, полито, опрыскано, тучный урожай вырос и убран – и теперь те, кто не успел, могут грустно бродить по колючей стерне, пытаясь найти хоть какой завалящий колосок.
Те, кто не первый раз заходит на этот сайт или когда-либо встречались со мной как с преподом или лектором, скорее всего вспомнят, что я считаю альдольную конденсацию одной из важнейших даже не реакций, а типов реакций. Многие удивляются – подумаешь, реакция, ничего особенного. Но я не устаю доказывать, что это именно один из важнейших подходов к синтезу – реакция между органическим нуклеофилом типа “донорный олефин” и органическим электрофилом карбонильного типа, а уж под этими типами скрываются бесчисленные множества конкретных соединений. Множество частных вариантов альдольной конденсации являются именными реакциями (конденсации Кляйзена-Шмидта, Кнёвенагеля, Анри, Штоббе, Дарзана и т.п.), но сама альдольная конденсация как архетип всей этой химии никакого имени не носит и это понятно – у истоков этой реакции сразу несколько знаменитых имён – Кейн, Вюрц, Бородин, Фиттих и другие, – и выдать ей имя не получается, и даже трудно сказать, сколько ей лет, но никак не менее 150, а то и 170. Отчасти поэтому многие не обращают внимания на альдольную конденсацию, считая ее малоинтересным старьём. Между тем за эти 150 лет с ней много чего произошло, реакция превратилась в могучий метод селективного синтеза. На её пути даже встретился человек, наконец давший имя одной из самых гибких её разновидностей – конденсации Мукайямы. Введение этого метода сразу позволило научиться искусно управлять реакцией – она оказалась не только хемоселективна и региоселективна (получаете то, что хотите, из исходных, которые можно сочетать разными способами), но и стереоселективна, и даже энантиоселективна, позволяет получать конкретные энантиомеры альдолей с высокой оптической чистотой. После работ Мукайямы альдольная конденсация стала развиваться с бешеной скоростью, и если посмотреть книги и и обзоры после 1980, то не узнаете альдольной конденсации вашей, это не про кротоновый альдегид и окись мезитила – это изощренный стереоселективный синтез с почти идеальными возможностями стерео- и региоконтроля исхода реакции, хотя и с кучей уже новых проблем. Первооткрыватель этого метода, Тэруаки Мукайяма вполне мог бы получить нобелевку, но дают её органикам редко и этот вклад оценить не смогли или не успели, хотя он скончался всего 3 года назад, а с момента первой публикации прошло более 45 лет.
И вот – премия 2021 года за энантиоселективный органокатализ, Бенъямин Лист и Дэвид Макмиллан. Про что это? Сейчас разберёмся, поймём, что лауреаты сделали очень много для того, чтобы этот метод стал насколько возможно общим подходом к синтезу. И у них многое получилось. Но совершенно бесспорно одно – в начале был очередной и в принципе хорошо известный вариант альдольной конденсации, и многие реакции, разработанные лауреатами, это очередные варианты альдольной конденсации, а один из лауреатов начал с альдольной конденсации и через 20 лет снова пришел к ней, а что будет дальше мя пока не знаем, но не в последнюю очередь благодаря этому решению Нобелевского комитета, теперь постараемся не пропустить.
Да здравствует Альдольная конденсация!
О чём эта страница
О нобелевской премии 2021 года.
Как недавно сказал мне один студент, обратив внимание на мое давнее безумное пророчество о том, что органика никогда больше не получит нобелевских премий, – а какой смысл тогда заниматься органикой, если нет шансов съездить в Стокгольм к тамошнему королю. Это хороший настрой, одобряю, испытываю жгучий стыд, беру свои слова назад, да и вообще – давайте и правда посмотрим, как лауреаты этого года решили эту проблему. И вы тоже сможете, если постараетесь. Будьте как Б.Лист и Д.Макмиллан, и одна из нобелевок второй четверти 21 века – ваша.
На этой страничке мы уделим некоторое время общему трёпу, но главная цель страницы другая – действительно разобраться, за что дали нобелевку, причём довольно подробно, даже занудно, за что сразу извиняюсь, но этот сайт иногда читают не только наши студенты, изощрённые в органической химии и всё понимающие с полуслова, но и просто любители химии. Разберём, что такое органокатализ, какой он бывает, почему так важна энантиоселективность и как она достигается. Заодно разберемся в некоторых важных и тесно связанных с органокатализом вещах – механизме конденсации Кнёвенагеля, МБХ-реакции, как работает витамин В1 и при чём тут карбены, энантиоселективной альдольной конденсации. И ещё обязательно разберёмся, чем кормят свиней (и коров), и почему это очень важно для успеха химии.
Мы не будем сюсюкать и славословить, приписывая лауреатам несуществующие достижения. Попробуем разобраться серьёзно, за что дали, мне кажется, что это лучший способ убедиться в том, что выбор Нобелевского комитета в этом году, как и почти всегда, действительно хорош и почти безупречен.
Органики долго ждали своей нобелевки. Такого долгого перерыва не было давно. В 20 веке органики получали премии регулярно, но к концу века скорость стала снижаться. В 21 веке органики получили три премии за одно и то же – достижения катализа комплексами переходных металлов, и это показало, что у чистой органики наступают тяжелые времена – она уже не тянет одна, ей нужны пособники в лице координационной химии и промышленного катализа. Как и хотели великие Коттон и Уилкинсон, органика стала сливаться с неорганикой в одну науку, а чистые идеи сами по себе теряют актуальность – химия должна перестать делиться на чистую и прикладную. Налогоплательщикам больше не хочется платить за то, что некие умники копаются в механизмах реакций, стратегиях синтеза, реакционноспособных интермедиатах и прочей оторванной от жизни чистой науке – люди хотят лекарств, вечной молодости, средств против морщин, комаров, ковида; ярких экранов и всяких доселе невиданных прибамбасов буквально везде – и для всего этого нужны тысячи готовых органических соединений, а в процессе разработки нужен быстрый доступ, и виртуальный и реальный, к миллионам новых молекул. Поэтому наука номер один в современной химии – синтез, и не такой как раньше, неспешное колдовстово над колбами, а быстрый, надежный, высокоселективный, желательно легко автоматизируемый. Думать больше некогда, нужно синтезировать. Три металлические премии (2001,2005, 2010) ровно про это, за каждой из них стоят лоббисты из огромных химических компаний, которые собственно и оплатили исследования, приведшие к тем великим открытиям и достижениям, которые радикально изменили органический синтез. В тех премиях было много конкурентов, выбор конечных имен для объявления был далеко не бесспорен. Премию получили достойные, но там за каждой было еще много других не менее достойных, поэтому пришлось кого-то просто отодвинуть в сторону, а с кем-то другим просто терпеливо дождаться естественной убыли претендентов, отчего каждую из тех премий вручали седым старцам, и некоторые из лауреатов уже с трудом вспоминали, что же такое они отчебучили десятилетия назад, что их вдруг вытаскивают с тёплого моря в холодный Стокгольм и заставляют произносить речи про то, что уже давно уехало в раздел “когда мы были молодыми…”. После последней металло-каталитической премии 2010 года повисла зловещая тишина. Премия за премией уходили в материалы, атмосферу, ну и в бесконечную химию жизни, которая и берет максимум из всех химических премий, и так повелось еще очень давно. Да это и правильно, потому что ничего более интересного и волнующего, чем раз за разом убеждаться, что жизнь устроена намного сложнее, чем можно было бы предположить, и никаких надежд на то, что еще пара усилий и отступят все болезни, не осталось даже у самых отъявленных оптимистов. Усилий потребуется ещё немало, и немало химических нобелевок ещё попадет в клетку. Это вообще парадокс – всем понятно, что молекулярная биология, биохимия и как там это еще называют под зонтичным брендом life sciences – это вроде всё про органическую химию, и кто скажет, что белки и нуклеиновые кислоты – не органические соединения, и ведь большинство процессов, стоящих за функционированием клеток и тканей – это просто самые обыкновенные органические реакции, даже не просто самые обыкновенные, а нарочито простейшие типа всевозможных вариантов ацилирования и замещения, в обычной органической химии мы их считаем рутиной и скукотищей и поскорее стараемся проехать, чтобы заняться чем-то более сложным и волнительным. Поэтому никто уже давно не рассматривает эту науку как что-то связанное с органической химией. Дети выросли, уехали и забыли дорогу в родительский дом, а самих родителей, не скрывая, откровенно стесняются, какие-то они простоватые и воняют пиридином. Да, чуть не забыл, в 2016 году была ещё прикольная премия за “молекулярные машины”, а это вроде все органика, там даже синтез вполне зачётный попадается, а среди лауреатов есть мощные органики с кучей других заслуг именно в синтезе. Но почему-то никто не вспоминает о той премии в контексте органики, да и сами “молекулярные машины” очень хороший пример, как некоторые малость переусердствовали с рекламой довольно спорных работ, сделанных сильно на вырост. С тех пор прошло уже 5 лет, но никто никуда на молекулярных машинах не уехал, и мы как-то нечасто сталкиваемся с этими штуками на страницах химических журналов. Возможно, надо заглянуть в журнал “За молекулярным рулём”, только никак не найду ни одного номера.
И вот наступил холодный октябрь 2021 года, второго года новой эпохи, эпохи ковида. А что – ещё присуждают нобелевки? Не перенесли на пару лет, как олимпиаду и чемпионат Европы по футболу? Да, там в Швеции особый путь, не боятся ни чёрта, нобелевский комитет продолжал, оказывается, заседать, наверное, в зуме. Посовещались, и присудили. Не ковид ли повлиял, что вспомнили про органику? Давайте разберёмся, это интересная история. И сразу скажу, на мой вкус выбор лауреатов в этом году почти безупречный – премия нашла тех, кто этого хотел и очень много для этого сделал. И дана наконец вполне молодым исследователям в самой кульминации научной карьеры – их новые работы последних 10 лет намного интереснее того, что они делали раньше и за что собственно и присуждена премия. Особый комизм делу придает то, что и тот и другой давно забросили органокатализ в оригинальном смысле этого слова, как бы собственным примером показывая реальный потенциал этой области. Но раскрутили область именно они, и именно сначала вдвоем, и смогли так ярко показать потенциал, что туда с рекордной скоростью набилась туча народа, включая нескольких легендарных патриархов органики вроде Барри Троста. Нобелевка отчасти именно за это – за демонстрацию возможностей новой химии в новом веке, когда никто не хочет долго ни в чем копаться. Вместо этого надо найти хорошую область, чутьём уловить возможности, придать даже самым очевидным идеям статус концепций, бросить большие силы и средства, заманить побольше последователей – и вот результат: проходит десять с небольшим лет и область готова – как хорошее поле засеяно, удобрено, полито, опрыскано, тучный урожай вырос и убран – и теперь те, кто не успел, могут грустно бродить по колючей стерне, пытаясь найти хоть какой завалящий колосок.
Те, кто не первый раз заходит на этот сайт или когда-либо встречались со мной как с преподом или лектором, скорее всего вспомнят, что я считаю альдольную конденсацию одной из важнейших даже не реакций, а типов реакций. Многие удивляются – подумаешь, реакция, ничего особенного. Но я не устаю доказывать, что это именно один из важнейших подходов к синтезу – реакция между органическим нуклеофилом типа “донорный олефин” и органическим электрофилом карбонильного типа, а уж под этими типами скрываются бесчисленные множества конкретных соединений. Множество частных вариантов альдольной конденсации являются именными реакциями (конденсации Кляйзена-Шмидта, Кнёвенагеля, Анри, Штоббе, Дарзана и т.п.), но сама альдольная конденсация как архетип всей этой химии никакого имени не носит и это понятно – у истоков этой реакции сразу несколько знаменитых имён – Кейн, Вюрц, Бородин, Фиттих и другие, – и выдать ей имя не получается, и даже трудно сказать, сколько ей лет, но никак не менее 150, а то и 170. Отчасти поэтому многие не обращают внимания на альдольную конденсацию, считая ее малоинтересным старьём. Между тем за эти 150 лет с ней много чего произошло, реакция превратилась в могучий метод селективного синтеза. На её пути даже встретился человек, наконец давший имя одной из самых гибких её разновидностей – конденсации Мукайямы. Введение этого метода сразу позволило научиться искусно управлять реакцией – она оказалась не только хемоселективна и региоселективна (получаете то, что хотите, из исходных, которые можно сочетать разными способами), но и стереоселективна, и даже энантиоселективна, позволяет получать конкретные энантиомеры альдолей с высокой оптической чистотой. После работ Мукайямы альдольная конденсация стала развиваться с бешеной скоростью, и если посмотреть книги и и обзоры после 1980, то не узнаете альдольной конденсации вашей, это не про кротоновый альдегид и окись мезитила – это изощренный стереоселективный синтез с почти идеальными возможностями стерео- и региоконтроля исхода реакции, хотя и с кучей уже новых проблем. Первооткрыватель этого метода, Тэруаки Мукайяма вполне мог бы получить нобелевку, но дают её органикам редко и этот вклад оценить не смогли или не успели, хотя он скончался всего 3 года назад, а с момента первой публикации прошло более 45 лет.
И вот – премия 2021 года за энантиоселективный органокатализ, Бенъямин Лист и Дэвид Макмиллан. Про что это? Сейчас разберёмся, поймём, что лауреаты сделали очень много для того, чтобы этот метод стал насколько возможно общим подходом к синтезу. И у них многое получилось. Но совершенно бесспорно одно – в начале был очередной и в принципе хорошо известный вариант альдольной конденсации, и многие реакции, разработанные лауреатами, это очередные варианты альдольной конденсации, а один из лауреатов начал с альдольной конденсации и через 20 лет снова пришел к ней, а что будет дальше мя пока не знаем, но не в последнюю очередь благодаря этому решению Нобелевского комитета, теперь постараемся не пропустить.
Краткое оглавление
1. Общий трёп (сразу после)
2. Три источника и три составных части органокатализа
3. Начала органокатализа и достижения лауреатов
4. Зачем нужна энантиоселективность
5. Талидомид
Что собственно открыли лауреаты?
Лауреаты собственно не открыли ровным счетом ничего. Что самое интересное – и не должны были. И это – очень поучительная история, которая рассказывает, как устроена и как делается современная химия высокого уровня. Если мы этого не понимаем, то продолжаем пребывать в наивном убеждании, что большая наука (в данном случае химия, но во многом это относится и к другим серьёзным наукам, двигающим вперед материальную сторону жизни) – это блестящие идеи больших умников. Типа, сидит такой большой умник, сидит, сидит, вроде спит, глаза закрыты и уже носом клюёт – но вдруг как вскочит, да как заорёт и – хвать, кто пробирку, кто листок мятой бумаги – плюх, плюх, зоркий взгляд вперен в пробирку или вдаль – и готово великое открытие, можно дальше носом клевать, пока тысячи усердных исполнителей воплощают гениальную идею в новые лекарства и заводские корпуса, и всё прочее.
Бывало и так… приблизительно, но давно. Давно уже не было. Было недавно одно милое исключение – трогательный японский старичок, который всю жизнь в одиночку тискал какую-то противную медузу, пока не вытащил из неё Зелёный Флуоресцентный Протеин, вооруживший исследователей жизни совершенно потрясающим инструментом визуального наблюдения за тем, что происходит в клетках. Все остальное в химии давно уже и близко не подходит к такому творческому методу.
Самый рабочий инструмент для добычи нобелевских премий выглядит совсем не так. Идея берется уже известная, иной раз десятилетия, иной – бывает что и столетняя. Амбициозный ученый смотрит на идею и понимает, что нобелевку хочется, а своих идей всё равно нет, но есть эта, забытая, но вроде в ней есть какой-то смутный потенциал, если повезёт, и если этой идее приделать хорошие крылья. Но потенциал есть везде, в любой морковке можно найти потенциал, недаром детей при советской власти поили морковным соком. Потенциал превращается в нобелевскую премию, только если в него как следует вложиться – нужны будут хорошие деньги, потому что химия – это эксперимент, много эксперимента, очень много. Химия устроена так, что успех приходит к учёному не когда угодно ему или ей, а когда угодно богам. А боги – сущности капризные, и успехом жалуют только тех, кто не ленится, тех, кто умеет приносить жертвы. Поэтому нужно из первоначальной идеи сделать пару хороших статей и хорошо их опубликовать в одном из топ-журналов, из этого добыть много грантов, обосноваться в хорошем месте, где не будут мешать, и есть условия для развития. Набрать много молодых, которым пока еще интересно, поставить их к тягам, описать задачу в самом общем виде (а другого в этот момент нет, потому что ни один химик никогда не знает, что в конце концов получится из смутной и с виду весьма немудрёной идеи), а дальше молодые сами начнут придумывать, что бы это могло быть и грызть землю, и у некоторых что-то начнет получаться. А остальные? – да, у большинства ничего не получится, но мы про них ничего никогда не узнаем. Если вам кажется это жестоким и несправедливым, вы правы, это именно так и есть, но вывод из этого один – займитесь чем-нибудь попроще, не суйтесь в современную науку.
Для успеха еще желательно, а скорее совершенно необходимо, чтобы амбициозных ученых было минимум два, а лучше больше, – тогда они друг с другом начнут соревноваться, и это ускоряет дело в разы, потому что все знают правила – до финиша добегут максимум трое, но на самом деле не больше двух, потому что нобелевка с некоторых пор устроена так, что если ее дают троим, то третьего берут из смежной области, или иногда из кладовки с нафталином – бывают такие подзабытые основоположники, которые сами давно забыли, что они заложили какие-то основы, но спустя вечность из этих основ вырастает нечто величественное, и благодарные потомки вдруг с изумлением узнают, что основоположник еще жив, хотя никто не помнит как выглядит и нет ни одной путной фотографии – так, например, в премии по метатезису 2005 года возник Ив Шовен, в премии за энантиоселективное гидрирование 2001 года – Уильям Ноулз, а в премии за кросс-сочетание Ричард Хек (который один стоит всех остальных, но это было давно, и с тех пор великий ученый посвятил себя без остатка разведению попугаев). А значит – из конкурентов премию получат только двое, и надо биться по-серьёзному, а то останешься третьим, и это должно быть намного обиднее, чем бронзовая медаль на чемпионате мира по футболу. Становится совсем горячо, статьи сыпятся как из рога изобилия, хирши растут как на дрожжах, то один, то другой вырываются вперёд, на шум сбегаются другие ученые, которые толком не понимают, зачем вся эта давка, но раз тут такая движуха надо тоже что-то сделать в этой области, хоть хиршей своих подтянут. В таких живых областях всегда очень живенько растет цитирование, причём у всех, а это нынче вещь незаменимая. Это тоже здорово подхлёстывает гонку, а опасности, что кто-то из этих составит реальную конкуренцию нет – это такая научная пехота (многие обидятся, но зря, без пехоты еще не выиграно ни одно сражение в мире, так что не так уж это и обидно – быть хорошей, надежной пехотой, и вообще – хотите быть маршалом, будьте, никто не мешает). Хоть корсиканец Буонапарте и уверял, что плох тот солдат, у кого в ранце не лежит маршальский жезл, но врял ли он бы обрадовался, обнаружив на поле битвы вместо ровного строя готовых к бою верных солдат нестройную толпу пузатых маршалов.
Итак, многое уже есть – живое направление, которое сообща долбят несколько крупных соединений под командованием честолюбивых командиров и туча народа попроще. Но из этого еще не следует, что в результате будет нобелевка. Живых направлений в химии сотни, химия – наука огромная и очень живая. А нобелевки дают редко и немногим. Вот в органике нобелевки не было 11 лет, и это не значит, что там царит тишина, поле усеяно костями, все умерли и только в одном углу гудит как улей какая-то жизнь, так что шведскому королю останется только обратить на это своё высочайшее внимание. Ничего подобного – органика в последние два десятилетия развивается невероятно быстро, так, что её и не узнать, если случайно пропустишь всего лет десять. В органике несколько раз были такие периоды, когда за пару десятилетий она изменялась до неузнаваемости. В начале второй половины 19 века, когда появились структуры и представление о том, как между собой связаны классы соединений. В послевоенные десятилетия, когда появились теории структуры и механизмов, и органика наконец стала настоящей наукой, а не аналогом поваренного искусства для избранных шефов. И в последние лет 20-25, когда воедино собрались достижения самых разных наук от теоретических расчетов до координационной химии, а новые методы эксперимента и анализа взвинтили темп исследований до головокружительного – всё это позволило проходить от первой идеи до тысяч статей и развитой области за менее чем десятилетие.
И вот – как выделиться в своей области, когда и другие не зевают. И кругом такая же жизнь, такое же гудение научных масс. Шанс один – идею надо хорошо продать. Хорошо – значит лучше всех. Продать – значит убедить видеть в этой идее и в том, что из неё получается (а в этот момент еще совсем не ясно, получается ли из неё хоть что-нибудь) нечто совершенно необходимое человечеству, да даже в общем такое, что можно сказать, что это последний шанс человечества – поддержать именно эти исследования и именно этих неутомимых, прозорливых, эх, да что уж там – гениальных организаторов научных побед. Давайте поднажмём, намекают они, а то хана, погибель, не спасётся никто. Убеждать надо не своих коллег учёных – те отлично понимают смысл таких речей и сами не прочь расхваливать свои собственные исследования точно так же; а политиков и инвесторов, а значит и вообще народ – люди же должны знать, что им повезло быть современниками священнодействия науки, а значит – не пропала еще надежда, будут, будут лекарства от всего сразу одной таблеткой, принимать три раза в день натощак после еды.
И ещё несколько ведёр дёгтя…
Сейчас мы приступим к серьёзному обсуждению науки, за которую совершенно справедливо присуждена в этом году нобелевская премия. Это очень хорошая наука, в ней стоит разобраться и найти и её сильные, и её слабые стороны. Но в начале я не могу не отреагировать на то, как подают нобелевские премии в общедоступном контексте. В науке вообще-то многие дюжины только крупных международных премий, а уж национальных вообще не счесть. И никто не обращает на них никакого внимания, кроме ближайших коллег лауреатов. Но нобелевка – особое дело. Так повелось, что эта премия имеет общечеловеческий характер, её все ждут каждый год, а значит её надо как-то объяснить широкой публике. Это просто несчастье, если подумать. Наука сложна и профанированному объяснению не поддаётся, ни сейчас, ни сто лет назад. Лучше всего было бы, раз уж это имеет статус важной новости для масс-медиа, как-то из этой ситуации выкручиваться покороче, минут на пять, обращая внимание в первую очередь, каким замечательным и ярким людям досталась заслуженная слава. Но нет, иной раз в самом обычном СМИ минут 10-20 терпеливо долбят что-то из стандартного набора – теперь будут лекарства, если речь про органику, ну или новые смартфоны, если про что-то другое. Люди спрашивают, почему до сих пор нет лекарства от… (длинный список). Или почему лекарства так дороги. Им намекают, что это потому что не было того, за что дали. И вот теперь дали – и будет всё, не волнуйтесь, платите налоги, прививайтесь и не переходите улицу на красный свет. Особое очарование этим речам придает то, что нобелевки всегда дают за прошлое, часто за очень далекое. А значит – и лекарства, и смартфоны на основе премии должны уже давно быть. И они и правда есть, и в них и правда много того, что следует из премий, но это же – про прошлое, а прошлое никому не интересно, интересно будущее. Новые лекарства и новые смартфоны – это будущие премии, мы ничего про них сейчас не знаем, и чествуем эту, сегодняшнюю, про то, что уже состоялось очень давно или просто давно, но лекарств всё равно пока мало, они дороги, а смартфоны тупят.
В общем, мы не будем здесь заниматься апологетикой, а попробуем разобраться в сути дела, тем более, что это не СМИ, а просто сайт для любопытных, и мы не обязаны попусту сюсюкать. Красота хорошей науки в сложности и заведомой неполноте, оставляющей поле деятельности для тех, кто только пришёл. Но прежде составим список того, что особенно раздражает (меня, но думаю, что не только меня) в речах, прославляющих органокатализ как новейшую прорывную методологию, без которой органика еще сто лет не могла бы ничего сделать.
- самая большая проблема органокатализа – он не открывает действительно новых методов синтеза. Все достижения органокатализа связаны с неким особым способом осуществления вполне классических реакций – в первую очередь всей этой могучей химии карбонильных соединений вокруг альдольной конденсации и ее многочисленных модификаций и обобщений. Много это или мало – очень много, невероятно много. Но если сравнить с тем, что было известно до 2000 года в этой химии, это в общем-то достаточно компактная надстройка. Красивая. Мезонинчик такой над огромным зданием.
- в рекламных целях органокатализ часто сравнивают с катализом металлами, причём почти неизменно уверяют, что это следующая эпоха в катализе, что органокатализ преодолевает недостатки металлокатализа и чуть ли не хоронит его, если уже не похоронил. По беспардонной наглости этому утверждению нет равных. Да, у металлокатализа действительно есть большой недостаток – и это сами металлы, даже не столько их цена, сколько проблемы с очисткой от следов металлов тех продуктов синтеза, которые применяются как лекарства. Но органокатализ никакой угрозы доминированию металлокомплексного катализа в синтезе не представляет. Ассортимент органокаталитических реакций до сих пор чрезвычайно узок, и это почти только реакции, относимые к химии карбонильных соединений, циклоприсоединения, ограниченной функционализации. Металлокомплексный катализ обслуживает намного более широкий, практически неограниченный круг типов органических соединений, начиная от еще нефункционализованных углеводородов. И что самое важное – среди органокаталитических процессов мы видим только хорошо нам знакомые органические реакции, в то время как металлокомплексный катализ ввел в органику и продолжает вводить сотни новых превращений, вообще не имеющих аналогов в классической органической химии. Металлокомплексный катализ радикально расширил органику, органокатализ просто дал некоторое количество остроумных и удобных альтернативных способов делать то, что и так все умели делать.
- ещё одно весьма беспардонное заявление, с которым носятся пропагандисты органокатализа – что это чуть ли не долгожданное окончательное решение проблемы энантиоселективности в синтезе. Это совсем не так. Да и нет такой проблемы. Энантиоселективный (или асимметрический синтез) достиг весьма высокого уровня до начала органокаталитической “революции”, и количество энантиоселективных реакций в органическом синтезе уже к началу нового века было огромно. Энантиоселективной может быть любая реакция, в которой образуется из ничего новый стереогенный элемент (центр, ось, плоскость, спираль). Если посмотреть наугад полные синтезы сложных молекул хоть в 2021 году, хоть десять лет назад, мы увидим огромное разнообразие энантиоселективных подходов. Органокатализ обязательно будет нам встречаться, но далеко не так часто как знаменитый динозавр на Тверской. Не каждая вторая энантиоселективная реакция будет органокатализом, хорошо если каждая десятая. И доля эта если и увеличивается, то весьма медленно. Может быть теперь, после нобелевки эта доля начнет слегка увеличиваться, но гегемонией тут не пахнет даже в воображении.
- часто говорят, что достоинство органокатализаторов – это простые и очень доступные молекулы. Но это справедливо только по отношению к первому поколению органокатализаторов Листа и Макмиллана. По мере развития области стало ясно, что у первого поколения возможности ограничены и пошли разработки все более сложных катализаторов, их опять стало бесконечно много – а это тоже одна из проблем катализа: химикам хочется иметь некий ограниченный набор, как это часто называют toolkit, ящик с инструментами, у каждого этикеточка, когда и где применять. В начале всегда так и бывает, но по мере развития области выясняется нехорошая вещь – универсальных, даже ограниченно универсальных катализаторов и реагентов нет, и у ящика скоро перестает закрываться крышка, а этикеточки првращаются в обзоры и списки сотен ссылок. Опять ничего не вышло из универсальности, и органокатализ просто добавил еще пару сотен методов к тысячам известных.
- даже поверхностный анализ сложных синтезов, каждый год сотнями публикующихся в топовых журналах, показывает, что органокаталитические стадии используются, мягко говоря, не часто. Хотел найти хоть один, в котором органокаталитическая стадия была бы хоть одной, но совершенно необходимой, ключевой, завершающей, решившей задачу. Не получилось. Надеюсь, всё же найду, тогда с удовольствием сделаю апдейт с рассказом про такую работу. Совсем недавно публиковали обзор методов, которые используются в современной высокопроизводительной разработке фармацевтики (Org. Process Res. Dev. 2019, 23, 1213−1242), составленный несколькими десятками синтетиков из университетской и промышленной науки, среди которых был и Макмиллан. Сводная табличка методов, отсортированных по частоте использования в разработках, получилась очень интересной. Органокатализ там ближе к концу, в доминируют в таблице разнообразные методы с использованием катализа комплексами переходными металлами, с безусловным лидерством реакции Судзуки-Мияуры. Органокатализ может утешиться только тем, что хоть немного, но обошёл метатезис (нобель 2005), но при этом безнадежно, с непристойным счетом что-то типа 50:1 проиграл кросс-сочетанию (нобель 2010) и сильно проиграл энантиоселективному гидрированию (нобель 2001). Ни о каком вытеснении металлов из синтеза говорить не приходится, они незаменимы, а проблему очистки продуктов от примесей приходится решать.
- у понятия органокатализ так и не появилось убедительного описания, которое помогло бы идентифицировать органокаталитические реакции среди других типов катализа. Что такое органокатализ? Коротко можно? Нет, увы, не получается, и очень много букв, которые ждут читателя в дальнейшем разборе, говорят не только о том, что здесь много интересного (это чистая правда), но и то, что у органокатализа есть немного свинская манера экспроприировать всё, что плохо лежит, например, обширнейшую и еще недавно вполне автономную область кислотно-основного катализа хиральными основаниями и кислотами Бренстеда-Лоури – “то моё, а то моё же”. Есть некоторая надежда, что эта экспансия в чужие огороды поутихнет после присуждения нобелевки, ибо вторую не дадут, а значит есть смысл перестать сгребать все в одно место, и не пытаться весьма искуственно объединять разнородные вещи под зонтиком одной методологии.
… и ложка мёда
Но если мы отделим слащавое славословие от действительных достижений, мы увидим очень хорошую науку. Мне однозначно понравилась эта премия. Даже больше можно сказать – мне сложно представить какую-то альтернативу, раз уж дело дошло до органики. В органике нового века невероятно много интересного. Вся органика фактически складывается заново, и это ни в коем случае не значит, что всё, что было до этого теряет смысл. Напротив – наконец приобретает более полный смысл. В органике 20 века очень много спешили и многое только поскребли с поверхности, и оставили – не хватало методов, информации, вычислительной мощи. Органика 20 века – лоскутки на огромной пока пустой доске, одни вопросы, ничего не ясно. Берёте любой метод, подход, механизм – на первый взгляд всё так ладненько, но только не вздумайте задавать лишние вопросы – почти любой вопрос остаётся без ответа, шаг в сторону – бездна. Один лишний заместитель, хоть метил – реакция не идет, катализатор не катализирует, оптический выход не виден без хорошей оптики. Всё это создало потрясающие перспективы для развития химии в новом веке, когда появлись огромные новые возможности исследования, было бы желание, средства и возможности. И да, химия стала развиваться очень быстро – все желающие брали те лоскутки с пустой доски и, делая многое заново с новыми возможностями, получали и получают намного больше. Еще недавно мы знали, что это невозможно, то недостижимо, такого не будет никогда и так далее – а теперь оказалось, что проблемы проясняются, барьеры оказываются преодолимы, а препятствия больше не препятствуют. Но плата за это велика – органика становится необозримой, найти в ней свой путь невероятно сложно, а выделить какую-то более-менее самодостаточную область – почти невозможно. Кстати, поэтому, скорее всего, предыдущие нобелевки даны за науку 20 века, 1960-х – 1980-х, даже в 90-е почти не заехали. И вот мы уже глубоко в 21 веке, сами не заметили, и уже нет возможности давать за прошлое, хотя бы потому что лауреатов уже не сыщещь. Пришлось искать что-то посвежее. А тут проблема – в любой стоящей области десятки претендентов, как их не выстраивай. Тупик однако. Но нашелся органокатализ, и это действительно как специально под нобелевку придумано. Во-первых, прямо ровесник века, первые и ключевые работы 2000-2001 года, символичнее некуда. Во-вторых, основоположников ровно два, и с этим трудно спорить. Да, если разобраться поподробнее, там найдётся ещё несколько очень важных персонажей с неменьшими заслугами. Но область как концепцию, а не просто поле для интересных исследований, раскрутили Лист и Макмиллан, и это видно прямо с самых первых работ, что уж совсем уникально – ну и чутьё у некоторых, дух захватывает!
Ну и, возможно, самое главное, для меня. Органокатализ по-хорошему парадоксален, потому что ищет решения проблем в самом очевидном месте. Под фонарем. Среди давно обрыдших трюизмов этот стоит на одном из первых мест – ищите желаемое где угодно, только не там, где светло. И вот все усердно ищут по самым темным и грязным углам, иногда что-то находят, но чаще нет, зато страшно устают, а не это ли признак усердного труда, и вообще, стыдно же искать где и так светло, там же уже и так всё обследовано. И толпы изможденных, покрытых пылью, тяжко дышаших людей продолжают усердно копаться в самых темных углах. И тут приходят элегантные господа, смотрят в самом светлом и чистом месте и говорят: Да вот же оно, искали? Измождённым искателям стыдно признаться, что там им не пришло в голову посмотреть, и говорят – не, это не наше, мы ищем большое и чистое – и опять шасть в темноту тяжко и усердно копошиться.
Статьи по органокатализу, особенно ранние, у многих вызывали одну и ту же реакцию. Сидит такой, потягивает кофе, лениво листает свежий джакс или ангевандте, скучно, господа, опять одно кросс-сочетание кругом да метатезис. Вдруг замирает, роняет пончик в кофе, бьёт себя по лбу, сильно бьёт и вопит: “Как же – я – такой умный и опытный – до этого – не – догадался!!!” Что же может быть проще!! Вон стоят все эти реактивы на самой нижней полке, но так же не может быть, что вот так просто, куда мы все глядели. Несчастного сотрясают рыдания. Да, так просто. Да, очевидно. Да, невероятно, что этого не сделали сто лет назад, десять лет назад, год назад.
Но вот так. Днём под фонарем некоторые нашли себе нобелевку. Но она одна, не ищите там больше, там больше нет. А где есть? Кто-то знает наверное. Ищите другие фонари. Пожелаем успеха.
Что такое органокатализ?
Премию дали за органокатализ. Не за химию, за слово. Химии там тоже полно, но для того, чтобы получить премию, очень важно придумать хорошее слово. В данном случае это было сделано в самом начале. И слово это было – органокатализ.
Лауреаты не открывали органокатализ в смысле собственно химии. Достижение состоит не просто в органокатализе, а в энантиоселективном органокатализе, хотя лауреаты даже это не открыли. Разберёмся сначала в том, что такое органокатализ и откуда он взялся. Слово ведь странное, и многих, меня например, долгое время сильно бесило. Мы ведь в органике хорошо знаем, что такое катализ, он у нас тут везде, особенно кислотно-основный – шагу без него не ступишь. Органический катализ, катализ в органической химии – а органокатализ это разве не сокращение? Раньше любили всё сокращать: партком, райпотребсоюз, ширнармассы и т.д. Может это из той же оперы?
Не совсем, хотя очень близко. Это слово, скорее всего, впервые появилось в статье одного из лауреатов, Дэвида МакМиллана (Ahrendt K. A., Borths C. J., MacMillan D. W. C. J. Am. Chem. Soc. 2000, 122, 4243). Почему “скорее всего”, ведь вроде бы это так и есть? Да слово настолько простое и очевидное, что трудно отрицать возможность того, что его кто-то уже употреблял раньше – но, тогда не зацепило и все пропустили – поди найди в терабайтах научных статей. Типичная история в науке – если термин не зашёл, то его нет. А зайдёт слово и станет термином только тогда когда в этом появится необходимость, и когда его подтолкнут заинтересованные амбициозные исследователи – ведь термин маркирует область, и бросить хороший новый термин всё равно что для колонизатора воткнуть флаг своей страны в новой земле или для золотодобытчика времён Золотой лихорадки вбить столбик на новом участке. Оттуда и словечко застолбить. МакМиллан вообще большой любитель изобретать понятия и вбивать столбики. Он делает это постоянно, в каждой второй работе столбит и столбит, и так наплодил уже не меньше дюжины понятий – такой неутомимый столбитель. Но МакМиллан – человек честолюбивый, но при этом добросовестный и честный, он не стал доказывать, что это он и придумал. Вместо этого он удивляется, почему идея такая явно есть, и приводит немаленький список ссылок, а слова нет. И как бы походя вворачивает – катализ небольшими органическими молекулами, пусть будет органокатализ, органокаталитическая реакция. Забавно то, что английский язык не очень любит словообразование сложными двухкорневыми словами, английский язык предпочитает более творческое словообразование за счет переносов смысла, метафоричности и тому подобной изящной игры короткими словами. Сложное слово бы лучше немец придумал – это типично немецкий способ словообразования. Б. Лист свою первую и ключевую работу опубликовал в том же году, и даже раньше, и Макмиллан на неё сослался как на яркий свежий пример принципа. Но немец Лист в этот момент работал в США и писал по-английски, но язык всё же чужой, и всегда у чужестранца есть робость предлагать новые понятия на неродном языке. Уже потом Лист попытался ввести свой термин – аминокатализ, но это была точно неудачная идея, потому что она сужает область (за аминокатализ нобелевку точно не получишь, хоть бы даже вся химия та же осталась), да и довольно двусмысленна. То, что большинство известных органокатализаторов так или иначе эксплуатирует соединения азота, это понятно, но, во-первых, это далеко не все амины, и во-вторых, зачем застолбив большую делянку добровольно отрезать от неё куски. С развитием органокатализа появились другие типы катализаторов, например, стабильные гетероциклические карбены, а в этих молекулах реакционный центр не азот, а углерод.
Так или иначе, слово неплохо зашло, и уже с середины нулевых стало общепринятым и очень популярным. Это уже большой успех сам по себе, половина нобелевской премии минимум. Дадут её или не дадут – это в будущем, но термин организует область, разрозненные работы вдруг выстраиваются в концепцию, и каждая следующая, кто бы ее ни сделал становится голосом в “хоре тысяч голосов”, слаженно выводящем Аллилуйю.
А почему это очень неоднозначное понятие, которое отлично могло и не зайти. Первые органокатализаторы (и исторически, и в работах лауреатов) – амины. Но амины – это основания Бренстеда-Лоури, и применение аминов для кислотно-основного катализа в органике – вещь настолько старая и привычная, что надо обладать огромной смелостью, чтобы попробовать сыграть на этом поле новую игру. Или фактически вбить столбик на участке, который до этого разрабатывали сто лет, перевернули всю землю на десять метров вглубь, просеяли через мельчайшие сита и обозначили как выработанный. И тут приходит весёлый шотландец, и говорит: Выкиньте ваши сита к чёрту, тут еще полно золота – сплошное золото, тут вообще больше ничего нет кроме золота.
Но самое интересное было дальше. Первые шаги органокатализа были вполне определённы – органокатализатор должен участвовать в реакции, образуя новые интермедиаты, в состав которых он входит, связываясь с субстратом ковалентной связью. И поскольку реакции энантиоселективны, то органокатализатор одновременно является и источником хиральности. В энантиоселкктивном органокатализе в первоначальном смысле оба явления были одинаково важны: органокатализатор связывается ковалентной связью и органокатализатор – источник хиральности. Если бы так и осталось, боюсь не получилось бы нобелевки, узковато, тема фактически исчерпала себя до 2010 года.
Но термин удачен, в начале производит много хайпа, цепляет, под него идет цитирование. И тогда происходит удивительное – под зонтик этого бренда ныряет вполне самостоятельная область – катализ хиральными кислотами и основаниями Бренстеда-Лоури, а это просто то, что давным давно существовало, но называлось общим кислотно-основным катализом. Никаких ковалентных связей, только водородные. Но здесь ключ в энатиоселективности – катализатор является источником хиральности. Ковалентных связей больше нет, но перенос хиральности есть. И на этой основе происходит слияние, точнее поглощение, и органокатализ в мгновение ока становится в несколько раз шире, потому что у кислотно-основного энантиоселективного катализа возможностей не в пример больше. Но не придумал бы Макмиллан удачный термин и не раскрутил его (вместе с Листом) до того, что куда более крупные соседи по катализу попросились под крышу.
А следующий, ещё более парадоксальный шаг сделал уже Лист. Он вообще придумал, как разделить две вещи – катализ отдельно, перенос хиральности отдельно. Катализ работает через ковалентную связь и образование интермедиата, а перенос хиральности через … ионную связь, просто в ионной паре. Дальше от первоначального смысла органокатализа некуда. Но – и это тоже ныряет под зонтик. И вот мы имеем уже огромную суперобласть, минимум с тремя очень разными областями внутри. Империя органокатализа. У империи должен быть один император? Не обязательно. У Римской после Диоклетиана было аж четыре, два главных, два поменьше. И у священной Римской после Карла Пятого было два. Добром ни то, ни то не кончилось, но прямых аналогий не бывает. Здесь такое слияние и поглощение закончилось нобелевкой, потому что вот в такой объединенной химии оказалось очень много впечатляющих результатов, методов, протоколов, типов катализаторов, всякого добра. Соединились и очень простые вещи, которые легко подавать, как исключительно дешевое и практичное решение. И очень сложные – для украшения и демонстрации мощи и изощренности мысли. Красиво задумано, чёрт возьми!
Откуда взялся органокатализ?
Повторю ещё раз: лауреаты не открывали органокатализ, и даже энантиоселективный органокатализ они тоже не открывали. Всё это было известно раньше. Но во-первых, так не называлось, и во-вторых, не расценивалось как отдельная область органического катализа, за которую можно огрести нобелевскую премию. Но заслуги лауреатов велики и неоспоримы. И чтобы лучше понять размеры этих заслуг, очень неплохо разобраться с тем, что было раньше, хотя бы потому что это как минимум несколько очень важных реакций и механизмов. Поэтому я остановлюсь на важнейших достижениях предшественников.
Дед органокатализа. Кнёвенагель.
Эмиль Кнёвенагель (18.06.1865 – 11.08.1921) впервые описал реакцию малонового эфира с альдегидами в 1894 (Knoevenagel E. Chem. Ber. 1894, 27, 2345). В общем, это типичная альдольная (поскольку почти всегда получается продукт дегидратации, то даже скорее кротоновая) конденсация. Необычное в ней сразу было то, что катализатором реакции являются амины, и у самого Кнёвенагеля это были вторичные амины. Это очень полезная реакция и в начале 20 века ей занимались многие, предложив кучу удобных вариантов, среди которых были и третичные амины, и соли аммония, и первичные амины и аминокислоты. Всё крутится вокруг азота и аминов, и никаких обычных катализаторов альдольной конденсации. Почему? Что в этой реакции особенного?
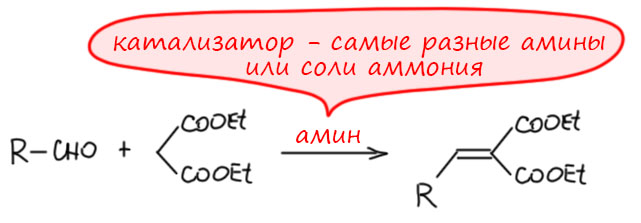
Механизм реакции Кнёвенагеля как правило представляют как обычную альдольную конденсацию. С одним нюансом – в роли метиленовой компоненты всегда используют соединения, обладающие высокой СН-кислотностью, с pK обычно в районе 10-12, иногда даже еще ниже. Это малоновый эфир, ацетоуксусный эфир и похожие соединения с двумя мезомерными акцепторами. Понятно, что это и объясняет, почему для этой реакции можно использовать более слабое основание, чем для обычной альдольной конденсации, где в роли метиленовых компонент берут енолизуемые альдегиды и кетоны, а это рК от 17-18 и выше. Вот поэтому в реакции Кнёвенагеля и используют амины, у них как раз pK отлично соответствует задаче. Вроде бы нет проблем. Обычный кислотно-основный катализ. Хотя если немного подумать, то по кислотности метиленовые компоненты Кнёвенагеля похожи на алифатические нитро-соединения, а сама реакция – на конденсацию Анри. Но в Анри охотно используют банальные основания, щелочи, даже обычную соду, иногда триэтиламин, но не вторичные амины и не соли аммония. А здесь – почти исключительно амины, и довольно часто в виде солей аммония со слабыми кислотами, тем самым лишний раз намекая, что дело, очень возможно, не в банальной основности. Амин в роли кислотно-основного катализатора. Почему кислотно-основного, а не просто основного? Потому что на разных стадиях механизма амин забирает протон (работает как основание) и дальше ион аммония отдает протон (работает как кислота), превращаясь обратно в амин. В этом и состоит катализ – катализатор возвращается в исходную форму через два переноса протона с участием амина и его сопряженной кислоты, иона аммония. Имеем обычный кислотно-основный катализ, и не так важно, что основанием или кислотой является органическая молекула. Здесь дело не в том, что она органическая. а в том, что она работает как кислотно-основная пара Бренстеда-Лоури.

Всё здорово. Только по-прежнему не покидает ощущение, что что-то тут может быть не так просто. Реакция Кнёвенагеля действительно в разных ситуациях идёт с самыми разными аминами и не только, но к вторичным аминам она особенно неравнодушна. Мы и в практикуме её делаем почти всегда по очень удобной методике Дёбнера, используя пиперидин в качестве катализатора и пиридин в качестве растворителя. И сам Кнёвенагель начинал с диэтиламина и пиперидина. В статьях тех времён никогда не поймёшь, почему исследователь взял то или иное соединение, но химия так устроена, что основоположники часто мистическим образом угадывают наилучший реагент, беззастенчиво пользуясь благорасположением и подсказками уполномоченных богов. Ну вот на полке стоял почему-то именно он. Потом ищут-ищут что-то другое, иногда находят, но часто нет. Ничего сверхъестественного, просто удача?
И довольно часто самым разным исследователям приходила в голову идея, что у реакции Кнёвенагеля, возможно, другой механизм, использующий именно вторичный амин, но не просто в качестве основания. Соображения эти возникли впервые тогда, гогда в органике появились енамины, открытые после войны Джилбертом Сторком. Енамины нам хорошо известны, и мы помним, что они довольно легко получаются из енолизуемых альдегидов и кетонов при действии вторичных аминов. В практикуме мы это тоже делаем. Но интересно то, что в реакции Кнёвенагеля чаще применяются неенолизуемые альдегиды, типа бензальдегида. Такие альдегиды не могут дать енамин, но с вторичным амином они тоже реагируют, и тоже отщепляется вода, но получаются катионы иминия. Пока не спрашивайте, откуда там взялся протон, это отдельная история, просто примите, что в смеси присутствует небольшое количество кислоты, годится даже карбоновая, ведь для отщепления воды из аддукта альдегида и амина большая кислотность не нужна.
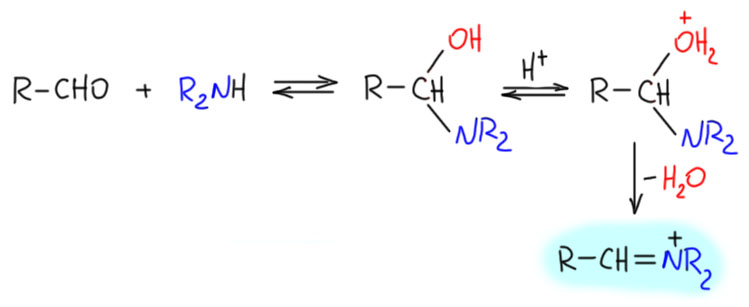
Ну и? Зачем портить альдегид? Нет, не портить, а наоборот даже активировать. Если мы сравним альдегид и катион иминия, несложно понять, что это соединения одного типа – углерод с двойной связью, на другом конце которой висит электроотрицательный атом. Электронная плотность смещена к этому атому, углерод становится электрофильным. Электрофильность углерода карбонильной группы – основа основ химии карбонильных соединений, но катион иминия не только не уступает в этом смысле, а даже и превосходит, просто потому что азот кватернизован. Хоть сам элемент и менее электроотрицателен, чем кислород, но в катионе иминия азот положителен и поэтому является более сильным акцептором. Это легко обнаружить, если нарисовать смещение плотности с помощью мезомерии.
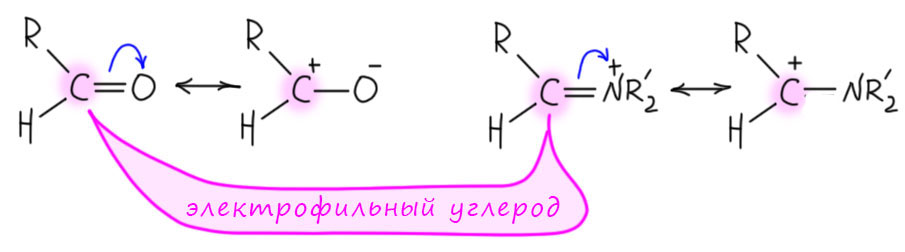
Итак, образование соли иминия увеличивает электрофильность. Соль иминия быстрее реагирует с нуклеофилами, чем сам альдегид. Но постойте, скажете вы, если уже хорошо пропитались химией альдегидов и кетонов – альдегиды ведь и сами по себе электрофилы хоть куда, зачем их еще активировать. Это верно, но в органике всё относительно, и всё познается в рядах. Альдегидов много, и не все они такие же удалые как простые алифатические альдегиды. Есть, например, бензальдегид и его замещенные и это совсем не такие хорошие электрофилы, ведь сопряжение с ароматическим кольцом снижает электрофильность. И вот тогда могут пригодиться катионы иминия – их появление в реакционной смеси будет равнозначно активации карбонильной группы. Механизм изменится, но в конце приведет к тому же продукту, что и кислотно-основный катализ. Здесь он, кстати, тоже нужен, потому что нужно создавать енолят.
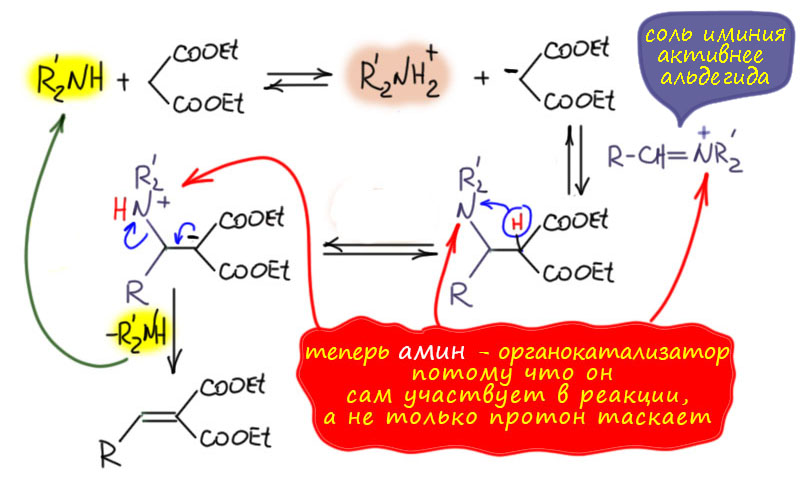
На последней стадии элиминирует амин. Это не злостное нарушение правила про то, что амины – очень плохие уходящие группы. В данном случае мы имеем дело с очень важной особой реакцией, присоединением по Михаэлю, которая известна своей обратимостью. И здесь это обратная к ней реакция, и это выгодно, потому что образуется двойная связь, сопряжённая с мезомерным акцептором. В теме про ненасыщенные альдегиды и кетоны мы это подробно разбираем.
Итак, мы получили особый механизм реакции Кнёвенагеля, в которой вторичный амин выполняет не только обычную роль основания и переносчика протона, но и прямо участвует в реакции, превращая альдегид в еще более активный электрофил. Скорость увеличивается, а это и есть катализ. Катализ небольшой органической молекулой. В 2000 году Макмиллан предложит это назвать органокатализом. Кнёвенагель, естественно, ничего этого не знал, в 1894 году ещё не заморачивались механизмами, да и средств не имели их изучать. Так или иначе, именно Кнёвенагель открыл реакцию, которая в будущем начнет заставлять думать об особом механизме, обращать внимание на вторичные амины в качестве катализаторов. Реальная конденсация Кнёвенагеля, кстати, в разных условиях и с разными субстратами, и с разными аминами, вероятно может происходить и по органокаталитическому, и по обычному механизму. У меня даже есть подозрение, что это до сих пор никто толком не исследовал, но это неважно.
Поэтому именно Кнёвенагеля можно считать нечаянным отцом, точнее дедом органокатализа. Лист в 2010 году даже посвятил этому отдельную статью, тем более, что он явно испытывал потребность из патриотических соображений ещё раз подчеркнуть, что старые немецкие классики открыли всю органическую химию, и выставить себя как наследника громкого имени – кто не знает про реакцию Кнёвенагеля. И как-то так получилось, что свою нобелевку он получил в 2021 году, ровно в столетие смерти Кнёвенагеля. Хорошо отметил!
Отцы органокатализа. I. Бреслоу.
Настоящий органокатализ начинается с витамина
Кнёвенагель Кнёвенагелем, исторический приоритет его реакции в истории органокатализа велик, но всё же сам Кнёвенагель не знал и не мог знать, какие тайны зарыты в реакции, которая получила его имя. Настоящая, оcознанная история органокатализа конечно, начинается много позже. На мой взгляд, совершенно бесспорным отцом органокатализа следует признать знаменитого химика второй половины 20 века Рона Бреслоу (он совсем недавно умер, в 2017 году). Это был по-настоящему великий исследователь, куда он только не совался, и везде делал нечто особенное, иногда даже немного курьёзное, но обязательно очень важное для развития мысли. Нобелевку он заслужил не один раз, но так и не получил её. Больше того, в самом конце своей карьеры, в 2012 году он стал жертвой довольно мерзкого наезда, из-за чего даже пришлось отозвать статью в джаксе. А в самом начале карьеры, когда ему было всего 27 лет, он сделал и опубликовал совершенно великую статью в том же джаксе (R. Breslow, J. Am. Chem. Soc. 1958, 80, 3719). В английском есть такое замечательное слово – seminal – что можно перевести, как “ставшее семенем”. Это слово почём зря, но всё же иногда не совсем зря применяют к статьям, которые стали важной вехой или могут стать таковым. Вообще, из семян может вырасти всё что угодно. Из каких-то вырастают чахлые былинки и сорняки, а из других – секвойи и баобабы. Вот та статья Бреслоу точно стала seminal, и из неё точно вырос впечатляющих размеров баобаб. И даже не один, минимум три баобаба. У некоторых растений есть такие семена – выглядят как одно семечко, а там, на самом деле целая гроздь, и вырастает сразу много. Эту статью с тех пор процитировали почти 1300 раз и продолжают это делать, и это невероятно много для экспериментальной статьи. а не обзора или мнения.
Эта работа изучила очень сложную проблему. Есть такой витамин, тиамин или В1. Это не очень сложное производное гетероцикла тиазола, как всегда в природных молекулах с дополнительным обвесом другими фрагментами, но центр молекулы – именно тиазол, точнее тиазолий. Этот витамин в клетке работает как коэнзим нескольких ферментов, в частности транскетолаз, которые умеют делать ацилирование альдегидами и пировиноградной кислотой. С точки зрения обычной органики это просто бензоиновая конденсация, которую, как мы знаем, в колбе делают с помощью цианидов. А в клетке – с помощью витамина В1. Попробуйте найти что-то общее между витамином В1 и цианидом.
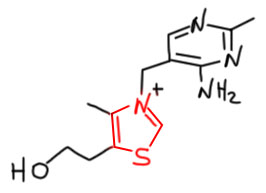
А вот Бреслоу нашёл. В принципе, мы тоже найдём. Во-первых, в молекуле витамина два гетероцикла, но основную функцию выполняет пятичленный тиазол в кватернизованной форме. Второй гетероцикл в молекуле играет вспомогательную роль, он помогает таскать протоны между реакционными центрами, что тоже, кстати, в будущей химии органокатализа всегда будет сопровождать реакции, когда дойдём, увидим. В обычной химии в колбе, а не в клетке, это не обязательно, в растворе найдётся кому таскать протоны. Поэтому Бреслоу попробовал несколько солей тиазолия попроще и нашёл, что они работают не хуже. Протон между двумя гетероатомами достаточно кислый, и легко снимается в водном растворе обычной щёлочью и даже более слабыми основаниями. Аналогия с HCN вполне прозрачная – углерод тремя связями связан с гетероатомом или гетероатомами, более электроотрицательными чем углерод. В тиазолии один из гетероатомов сера, менее электроотрицательная, чем азот, но зато азот кватернизован и имеет положительный заряд, что не может не увеличивать акцепторный эффект.
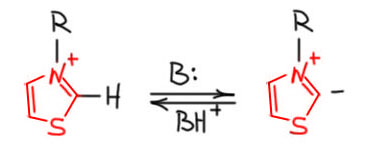
Анион тиазолия может быть нуклеофилом точно так же как цианид. И присоединяться с карбонильной группе альдегида. Такой аддукт, точно так же как в оригинальной бензоиновой конденсации имеет возможность переместить протон от бывшего альдегидного углерода, потому что такой карбанион стабилизирован, здесь – сопряжением с тиазолиевым кольцом. И в этом месте, нарисовав граничную структуру мы с замиранием сердца видим, что нет больше никакого аниона, так как плотность сместилась на четвертичный азот, закрыв его плюс. Интермедиат такого типа нейтрален. Это лишний раз говорит о том. что он должен быть достаточно устойчив. Более того, если помните настоящую бензоиновую конденсацию, то там важную роль в стабилизации карбаниона играл фенил, поэтому настоящая бензоиновая конденсация идет только с ароматическими альдегидами, и то не со всеми. А здесь основную роль в стабилизации играет тиазолий, и фенил больше не нужен, в реакцию могут вступать любые альдегиды. Бреслоу тут же это и продемонстрировал, успешно провернув реакцию с обычным ацетальдегидом. То есть получается, что это больше нет смысла называть бензоиновой конденсацией, коль скоро ограничение на альдегид не нужно. Надо бы это называть ацилоиновой конденсацией, потому что получаются ацилоины, но этот термин давно заиграли за другой реакцией. Вот этот нейтральный интермедиат стал настолько важен в механизме и связанных вещах, что впоследствии интермедиаты такого типа обозвали интермедиатами Бреслоу, и у них появилась своя, очень интересная химия. Несмотря на делокализацию, интермедиат имеет нуклеофильность и может вступить в реакцию с ещё одной молекулой альдегида, фактически это опять альдольная конденсация. Но аддукт может стабилизироваться отщеплением тиазолиевого аниона. Вот нам и продукт – ацилоин, теперь без ограничений. А тиазолиевый анион опять возвращается в реакцию – это катализатор. Типичный органокатализатор, вообще без вопросов – небольшая органичееская молекула, участвующая в реакции как катализатор, а не просто кислота или основание.
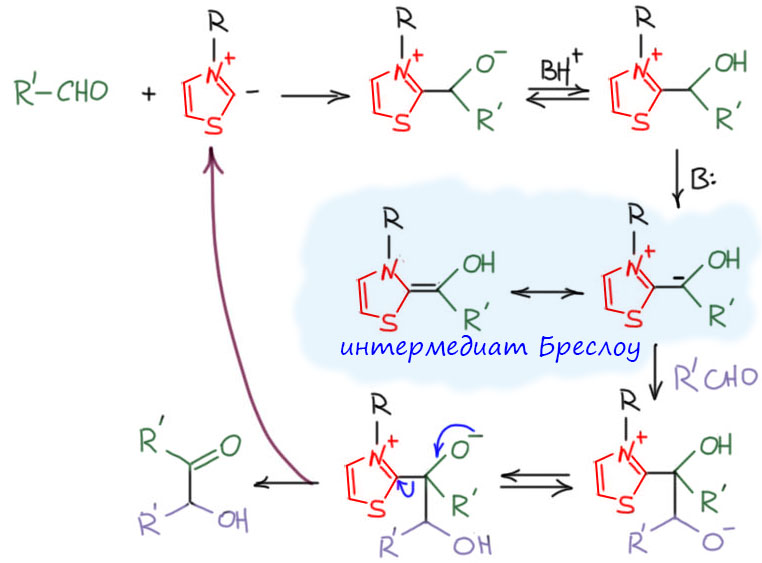
Это первая работа, где подобное не только предположено, но и доказано. И нет оснований сомневаться в том, что именно Бреслоу нужно считать изобретателем органокатализа, не слова, а сути. В данном случае нет стереоселективности. Нобелевку дали за энантиоселективный органокатализ, а не просто за органокатализ. И даже если бы Бреслоу дожил до 2021 (чуть-чуть ведь не дотянул), он вряд ли поучаствовал в этой нобелевке. Хотя, кто знает, Нобелевский комитет иногда бывает чрезвычайно щепетилен и к двум более современным и актуальным лауреатам добавляет третьего, из прошлого, но настоящего, величественного основоположника. А Бреслоу ох как заслужил нобелевки, и это понимали все, кто хоть сколько-нибудь помнит, кто делал современную органическую химию. Жаль, что не дотянул!
Чтобы немного точнее понять, какой уровень мысли был у Бреслоу, чуть-чуть еще покопаемся в этой работе. Бреслоу увидел еще одно интересное свойство депротонированного тиазолия – он похож на цианид, а у цианида есть вторая граничная структура – изоцианид, а это не что иное как карбен.
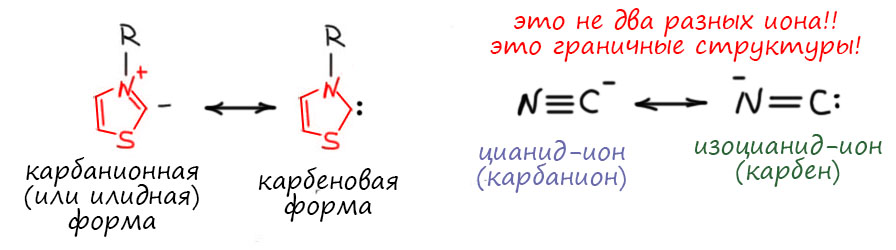
Это было сказано, хотя развивать эту мысль он не стал – в 1958 году это бы выглядело дико, и начинающего учёного быстро бы заклевали – всего несколькими годами ранее химия карбенов только-только возникла, и тогда все ассоциировали карбены с бешено реакционноспособной, короткоживущей частицей, которая присоединяется к двойным связям, влезает по связям С-Н и вообще страшно безобразничает, далеко за гранью допустимого в приличном обществе. Поэтому идея о том, что карбены могут иногда быть милыми и пушистыми, селективно участвовать в реакциях, да даже играть роль катализатора, совсем тогда никому бы не понравилась. Но намёк в работе настолько очевидный, что мимо пройти было трудно, и скоро по этому пути пошли другие. Это действительно оказалось карбеном, но особенным – гетероциклическим нуклеофильным стабильным карбеном. Через несколько десятков лет это будет окончательно доказано, тогда выясниться, что для таких карбенов лучше брать не тиазолий, а производные имидазола, два азота дают лучшую стабилизацию, чем азот и сера. И начался совершенно триумфальный успех этих карбенов (их принято называть сокращением NHC – нуклеофильные гетероциклические карбены, хотя чаще это читают как карбены из азотных гетероциклов) – они оказались и потрясающими лигандами в катализе комплексами переходных металлов, и сами по себе отличными органокатализаторами, и еще много чем. В принципе, эти карбены тоже стучатся в заветную дверь в Стокгольме, хотя я не уверен, что это можно назвать органической химией – это ближе к координационной химии, катализу, материалам и т.п.
Вот так работа! В ней одной открыт и объяснён будущий органокатализ, стабильные гетероциклические карбены, плюс это одна из первых работ, где показано, что у ферментативных реакций есть самые обыкновенные органические механизмы, и объяснена работа важных ферментов транскетолаз. все это спокойно и обстоятельно, без лишних воплей. Сейчас так не делают. Сейчас это бы разрезали на 20 работ, каждой дали бы по броскому заголовку и истерическому введению, запихали бы в цветные топ-журналы с дурацкими картинками на обложке и оглавлении. И хотя я понимаю почему наука стала такой – она стала сильно дороже и нужна куча денег, а деньги дают те, кто в науке не понимает ни черта, зато отлично знает законы маркетинга и PR – но есть у меня сильное подозрение, что этот новый гламурный стиль науке больше вредит, чем помогает, нередко надувая слона из мухи, отчего у нас в современных журналах что ни статья – великое открытие и ничего другого нет, а как из этого выбрать действительно важное, уже никто понять не может.
Отцы органокатализа. II. Морита, Бейлис, Хиллман.
Мы знаем, как в органической химии важно каждый день не менее десяти раз повторять мантры “нуклеофил с нуклеофилом не реагирует” и “электрофил с электрофилом не реагирует”. Никто не собирается в этом сомневаться, так как это всё равно что впасть в опасную ересь. Но есть одна реакция, в которой происходит ровно то, что запрещено. Это чрезвычайно популярная в новом веке реакция Бейлиса-Хиллмана, опубликованная в начале 1970-х двумя промышленными химиками из компании Целанез (подразделение Хёхста), причем только в виде патента. Нормальные химики патентов не читают, а если случайно попадется на глаза, то не верят, потому что патент служит не для сообщения результатов, а для защиты коммерческих прав, поэтому в патентах обычно содержатся заведомые выдумки, скорее скрывающие суть дела. Поэтому вплоть до конца прошлого века про эту реакцию никто не слышал. Но однажды её нашли и оказалось, что те ребята просто не умели писать патенты и совершенно наивно написали там сущую правду. Кто бы мог подумать, что в патенте написана правда! Это провал. К счастью, прошло так много времени, что авторов не могли уже привлечь к ответственности за раскрытие ноу-хау компании, а то ведь могла бы быть не реакция, а новое дело Бейлиса. Когда реакцию попробовали и убедились, что она очень даже рабочая, она всем очень понравилась и стала очень активно исследоваться и применяться. Можно сказать, что это одна из самых популярных реакций в последние 20 лет. И оказалось, что до Бейлиса и Хиллмана ту же реакцию описал японец Морита, тоже промышленный химик, из японской компании Тоё Раён (так вот оказывается что значит “на раёне” – Морита открыл новую реакцию на Раёне раньше Бейлиса и Хилмана на Целанезе), причем не в патенте, а в главном японском химическом журнале. И опять – это потом японцы стали одними из главных химиков человечества, а в 1968 к ним относились очень предвзято, так как никакой химии в Японии до середины прошлого века вообще не было. Японских журналов тогда почти не читали. Теперь реакцию принято называть реакцией Мориты-Бейлиса-Хиллмана, и это довольно часто сокращают в реакцию МБХ (MBH reaction). Понятно, что у нас с этим будут проблемы, и мы не будем ее так называть, чтобы не писать каждый раз про иностранного агента, а то ещё сайт заблокируют, а реакцию объявят нежелательной.
Реакция выглядит совершенно дико. Глаза хочется протереть, когда ее первый раз видишь, да заорать “вы что, не знаете, что электрофил с электрофилом не реагирует!!!!”. Реагирует альдегид и акцептор Михаэля. Акцептор Михаэля остается нетронутым, но к нему в α-положении пристыковывется альдегид, как карбонильная компонента в альдольной конденсации. Но когда первый шок проходит, и мы замечаем, что в реакции участвует еще третичный амин (у Бейлиса-Хиллмана) или фосфин (у Мориты), то механизм становится почти очевидным. Это органокатализ в чистом виде – органический нуклеофил присоединяется к акцептору Михаэля, образуется карбанион енолятного типа, и дальше происходит – опять она! – альдольная конденсация. Из аддукта нуклеофил отщепляется и может зайти на второй круг, он работает как типичный катализатор, органокатализатор.
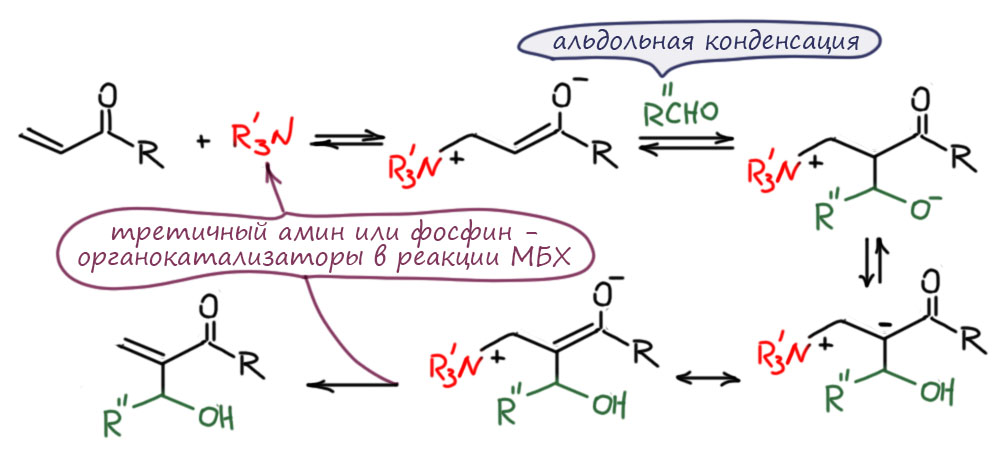
Когда реакцию исследовали, оказалось, что она мощна – в неё вступают самые различные акцепторные олефины – акриловые эфиры, нитрилы, винилсульфоны, и т.д. Самое приятное то, что в продукте (продукты реакции часто называют аддуктами Бейлиса-Хиллмана) сохраняется акцептор Михаэля и можно дальше развивать синтез на этой удобнейшей функции. Современное развитие реакции Мориты-Бейлиса-Хиллмана впечатляет множеством возможностей, в том числе есть и энантиоселективные варианты, и они плавно переехали в область энантиоселективного органокатализа.
Отцы органокатализа. III. Хайош, Вихерт.
Конденсация Кнёвенагеля, бензоиновая конденсация в присутствии стабильных нуклеофильных карбенов, реакция Мориты-Бейлиса-Хилмана безусловно являются важнейшими предшественниками химии органокатализа, но в своих основных версиях эти реакции не имеют стереоселективных вариантов (в современных версиях имеют все). А нобелевка дана именно за энантиоселективный органокатализ, и как мы увидим дальше, это действительно очень важно. Но и энантиоселективный катализ открыт задолго до начала работ лауреатов.
Непосредственным прототипом первых работ Листа послужила именная реакция Хайоша-Пэриша-Эдера-Зауэра-Вихерта, именно так её принято называть довольно необычно включив в название всех соавторов двух статей, а не только руководителей. Так иногда делают, когда работа приходит не из университетской, а из промышленной химии, где сам чёрт не разберёт, кто что делал, и кто кому начальник, потому что никто никогда не видел этих людей – промышленные химики не любят и не находят нужным светиться на открытых конференциях, так как всегда есть шанс проболтаться о чём-то важном конкурентам. Получилось поэтому такое трескучее название, впрочем, в новом веке эта реакция стала популярной и ее стали сокращать в реакцию Хайоша-Вихерта. Про реакцию эту до недавнего времни мало кто знал, хотя она есть в больших справочниках по именным реакциям. Но один из лауреатов, Лист, всегда на неё ссылался (вообще, оба лауреата показывают совершенно щепетильное отношение к этике, в каждой своей работе не забывают упомянуть всех предшественников и их достижения), и сейчас, после объявления нобелевки реакция всплыла и стала знаменитой, а в википедию срочно написали биографии руководителей тех работ Золтана Хайоша (венгерский химик, как многие другие бежал из Венгрии после жестокого подавления Хрущёвым антисоветского восстания 1956 года) и Рудольфа Вихерта (немецкий химик, который успел побывать в советском плену после разгрома Германии, впрочем он был совсем юным школьником, попавшим в самом конце войны в помощники наводчика зенитной пушки, и в плену не задержался, его не отправили в Сибирь, а быстро отпустили, и он продолжил учиться).
Удивительным образом эта реакция была одновременно и независимо обнаружена двумя группами промышленных химиков в Штатах и Германии, и опубликована в патентах и статьях в 1971 году. Вообще-то это просто хорошо нам известное аннелирование по Робинсону, вторая его часть, циклизация уже готового аддукта Михаэля в бициклический кетон, который так и назвали кетоном Хайоша-Вихерта. Фокус был в том, что этот кетон предназначался для полного синтеза разных стероидов, и исследователи поставили задачу сразу сделать оптически чистый энантиомер, в промышленном синтезе дорого возиться с разделением энантиомеров, а если ключевой полупродукт в цепочке синтеза уже оптически чист, то всё очень сильно упрощается, потмоу что дальше чистить надо уже не энантиомеры, а диастереомеры. Поскольку стероиды в те годы были очень популярны, синтезом занимались многие и в компаниях и в университетах – синтез стероидов вообще долго служил мотором развития органического синтеза в целом. Вот и тут две разные компании: Офман Ля Рош в Штатах и Шеринг в Германии поручили свои ученым поработать над проблемой. Надо сказать, что ученые не просто поработали, а нашли эффективное решение, практически идентичное, и это, если немного подумать, было потрясающим достижением. Дело в том, что это мы сейчас привыкли к асимметрическому синтезу, и почти все реакции имеют асимметрические варианты. Разве что нитрование бензола обошлось без этого и то только потому, что нитробензол до сих пор не удалось разделить на энантиомеры. Но это не всегда было так. Более того, настоящие успехи асимметрического синтеза – это дело последних максимум 40 лет. До этого никак не удавалось понять, как же сделать так, чтобы в реакции, где образуется асимметрический центр, образовался бы ощутимый перевес одного энантиомера над другим. Было ясно, что для этого нужно использовать нечто хиральное, но как это выбрать, чтобы был большой эффект, долго было непонятно. А асимметрическом синтезе тех времен оптические выходы в 10-20% были свидетельством успеха.
И вот эти две группы исследователей делают реакцию, в которой сразу решают и эту проблему и фактически открывают асимметрический органокатализ. Мыслят они так – чтобы осущесвить внутримолекулярный альдоль нужно основание. Посмотрим, что тут. Исходное нехирально, потому что оба заместителя торчат из симметрично замещенного пятичленного кольца. Там есть метил, это очень важный метил, потому что именно ему предстоит стать заместителем на стереогенном центре в энантиомерах. Но пока он всего-навсего энантиотопный. Это слово означает, что пока он находится в нехиральной молекуле на атоме углерода, у которого есть два одинаковых заместителя с некоторой возможностью какой-то реакцией сделать их разными. Такой атом углерода называется прохиральным центром. И если такая реакция будет найдена, и в результате реакции симметрия будет разрушена, центр станет хиральным, и этот метил станет одним из заместителей на стереогенном центре. Проблема в том, что пока мы не знаем, как это сделать, и обычные основания, которые используются в альдольных конденсациях дадут без шансов рацемическую смесь.
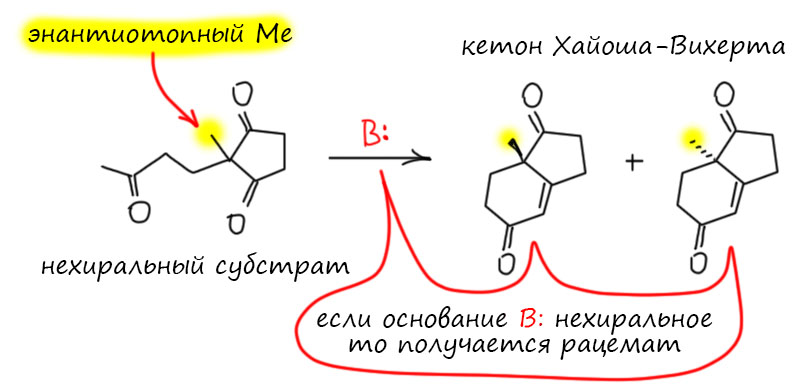
А почему бы нам не взять как раз основание хиральным? Но – такие фокусы уже много раз пробовали, и было ясно, что если хирально основание, переносящее протон, то серьезной эффективности асимметрической индукции (или переноса хиральности – процесс появления оптической активности в продуктах реакции от одного из хиральных компонентов реакционной смеси называют и так и так) ждать не стоит – протон переносится на приличном расстоянии, основанию не нужно сильно приближаться к реакционным центрам, более того, при переносе протона образуется нехиральный карбанион и куда там переносить хиральность не ясно. А стереоцентр образуется в стадии, которая не зависит от хирального основания – атаки карбаниона (или енолята) на карбонил. Тупик? Тупик. Но надо попробовать. В уме явно держат ракцию Кнёвенагеля, и ищут хиральный вторичный амин. Хиральные вещи лучше всего искать в Природе – они там готовые. Взгляд падает на аминокислоту пролин, единственную не с первичной, а вторичной аминогруппой, да еще и в составе циклического амина, похожего на пиперидин из реакции Кнёвенагеля. Кстати, не только. Обе группы пробуют каждая с полдюжины разных хиральных аминов, с каждым получается некоторое преобладание одного энантиомера над другим, но не очень большое, и толкьо с пролином получается великолепно. Для того времени, когда синтетики не были избалованы хорошими энантиоселективными реакциями, даже невероятно хорошо.
Вот у обеих групп получились очень высокие энантиомерные избытки около 85%, а это более 90% нужного энантиомера. Это триумф. Больше даже не нужно, потому что вещество с такой чистотой можно просто перекристаллизовать самым обычным способом, и это дает оптическую чистоту выше 90%, а при некоторой сноровке и умении перекристаллизовывать можно получить почти чистый энантиомер. Обратим внимание на то, что рацемическую смесь невозможно разделить простой перекристаллизацией на энантиомеры. И смесь с низкой оптической чистотой тоже нельзя. Возможность перекристаллизации повляется, когда оптическая чистота продукта уже высока. Это очень просто понять, потому что рацемат обычно кристаллизуется как кристаллы рацемата – в решетку входят оба энантиомера в соотношении 1:1. То есть рацемат ведет себя в отношении кристаллизации как отдельное вещество со свойствами, отличными от свойств чистых энантиомеров (только в кристаллическом состоянии! – в растворе рацемат – это не вещество, а смесь). И если оптическая чистота невелика, из раствора сначала будет выкристаллизовываться рацемат, и поди там пойми, когда можно дробной кристаллизацией в конце отделить немного кристаллов энантиомера с большим содержанием. Почти нереально, и даже если получится, выход будет очень мал. А вот когда один энантиомер преобладает и сильно, к 90% и выше, то сначала будет кристаллизоваться кристаллы чистого энантиомера, и только в конце вывалится немного рацемата. Умелый экспериментатор без труда получит отличный выход и мало что потеряет.
Ну и почему пролин дал такой эффект. Сами первооткрыватели поняли это только в самых общих чертах, но тем не менее предположили, что пролин участвует в реакции не только как основание, а скорее всего образуется енамин, и этот енамин вступает в альдольную конденсацию. Так это и есть. Образуется енамин, как и положено в химии енаминов (посмотрите в химии карбонильных соединений), менее замещённый, поэтому донорная двойная связь образуется в терминальном положении. Енамин может реагировать как одна из форм енолизуемого карбонильного соединения (другие формы: енолят, енол, эфир енола – смотрите в альдольной конденсации), способная реагировать с карбонильной группой, образуя альдоль в виде соли иминия. Фокус тут в том, что енамин просто так с карбонилом не реагирует, карбонил имеет недостаточную реакционную способность и должен быть активирован кислотным катализом – образованием водородной связи с какой-нибудь кислотой. А вот тут кислота и есть, в самой молекуле пролина есть карбоксильная группа, вполне достаточная для активации через водородную связь с кислородом карбонила.
Конденсация с енамином даёт соль иминия, которая легко гидролизуется, обрауя кетон и пролин, который может вступить в реакцию еще раз. Это третий гвоздь в этой программе – реакция оказалась еще и каталитической, пролина можно взять меньше чем эквивалент. Это настоящий катализ, и теперь мы знаем как это называется – энантиоселективный органокатализ. Альдоль легко теряет воду, образуя типичный продукт аннелирования по Робинсону – циклогексенон, но уже в виде одного из двух возможных энантиомеров. А почему не оптически чистый? Механизм же написан так, что должен получится один энантиомер. Но в реальности всегда есть возможности других путей реакции, например, активация может осуществиться межмолекулярно – это менее выгодно, но не совсем невозможно, достаточно для образования нескольких процентов другого энантиомера. Не бывает 100%-ной эффективности. Но та, что получилась, невероятно велика. Для 1971 года это был прорыв. Прорыв, который тогда никто не заметил. Работа сильно опередила своё время.
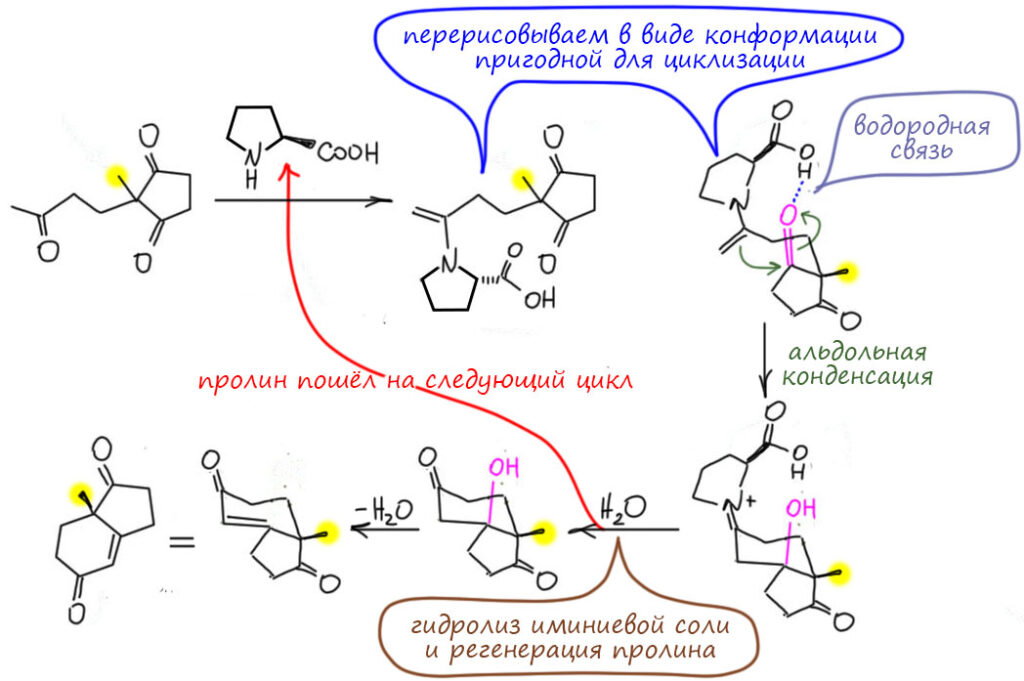
А мы не проходили, что енамины участвуют в альдольной конденсации! Еноляты, енолы, эфиры енолов – да, а енамины мы использовали только для алкилирования, ацилирования, и реакции Михаэля. Правильно, енамины и не используют просто так в альдольной конденсации, никаких противоречий. Но это принципиально можно сделать, потому что у них достаточна реакционная способность, но при условии активации карбонила протонной кислотой или кислотой Льюиса. В обычном варианте альдольной конденсации это достаточно трудно достичь, потому что в такой реакции возможны разные побочные реакции, прежде всего реакция Манниха, и образуется сложная смесь всякой дряни кроме желаемого продукта конденсации. А вот в таком варианте, который мы называем органокатализом, и где амин, используемый для енамина сам содержит активирующую кислотную группу, это оказалось возможным, как минимум в таком внутримолекулярном варианте. Получается, что в реакции Хайоша-Вихерта удалось сразу открыть несколько потрясающих вещей – а) органокатализ может запустить реакцию, которую без этого запустить трудно, она была бы неселективной; б) если органокатализатор хирален, органокатализ может достичь энантиоселективности, и очень высокой.
Нобелевку великим учёным Хайошу и Вихерту!!! Э, нет, чёрта с два. Они сделали великолепную реакцию, но только как частный пример, очень интересный, но частный. Как это применить для других реакций и как вообще это работает ни Хайош, ни Вихерт не выяснили. Да и не могли, это было давно, и методов исследования тогда было мало. Да и работали эти исследователи в коммерческих компаниях по строгим программам, связанным с целями производства, и просто так баловаться с чистой химией просто не могли – их бы повыгоняли с очень тёпленьких местечек с зарплатой, несравнимой с тем, что получают в университетах. Так что кому зарплата и страховка до конца жизни, а кому нобелевка.
Так что мы просто поздравим этих исследователей с великолепной работой, которая сыграла не сразу, а только через 30 лет, и еще через 50 лет из этого получилась немаленькая область химии и нобелевка, как вишенка на всём этом роскошном торте. И мы поняли, что в этой работе в неявном виде содержится весь энантиоселективный органокатализ. А что самое главное и явное в этой работе? Пролин. Это исключительно важная находка.
Вперёд – к нобелевке-2021!
И вот настал 2000 год. Всё уже известно. Реакций, в которых небольшие органические молекулы играют роль катализаторов, непосредственно участвуя в реакциях как нуклеофилы или электрофилы, а не просто как кислоты или основания, известно уже немало. Я разобрал несколько самых важных и без преувеличесния исторических, но реально их много больше – десятки. Но их никто не называет органокатализом и не считает, что это отдельная область исследований. И в это время в органике царят реакции катализируемые комплексами переходных металлов. Они творят чудеса, их открывают по десятку в год только совсем новых. Всем кажется, что именно это и есть ответ на все проблемы органического синтеза. И действительно – в первые 10 лет 21 века эта область даже ещё ускорится, и несколько раз подряд превзойдет саму себя. Вот где химия, вот где нобелевские премии (это так и получилось и все премии 21 века до нынешней, пошли на металлокомплексный катализ). С другой стороны, в органическом синтезе окончательно сложилось представление о том, что реакции должны быть селективны во всех типах селективности, которые заложены в самой реакции – хемо-, регио-, стерео- всех видов и т.п. (есть еще несколько видов более экзотических селективностей, но если они подразумеваются, то и их тоже подавайте). Если реакция предполагает стереоселективность, никому уже не нужен нестереоселективный вариант. Комплексы переходных металлов неплохо справляются с стереоселективностью, и первая же нобелевка 21 века отмечает именно это, правда на достижениях прошедшего века – за энантиоселективное гидрирование и эпоксидирование/дигидроксилирование. И та премия как будто закрыла тему – энантиоселективность уже отмечена, что вы еще хотите, нобелевская премия – это про самое важное, и те, кому судьбой довелось её получить фактически тянут на себе всех остальных.
И вот, в 2000 году появляется несколько работ двух молодых ученых, сверстников, Бенджамена Листа и Дэвида Макмиллана. Это очень простые и короткие работы, про очень несложную, почти рутинную химию – альдольную конденсацию, реакцию Манниха, реакцию Дильса-Альдера, и в них использованы очень простые реактивы. Но работы моментально цепляют, и именно этим – неправдоподобной простотой. Как будто химия за прошлый век ушла в дебри, заблудилась, не нашла хорошей дороги в светлое будущее, и от отчаяния решила всё начать сначала.
Первое слово дороже второго
Итак, мы видим, что то, что катализ с участием небольших органических молекул, в котором они не являются просто кислотами или основаниями, был хорошо известен в 20 веке, но никаких попыток как-то осознать эти реакции отдельно и обособить в новую область катализа никто не предпринимал. Кстати, остановимся ещё на секунду и спросим – а почему всё время говорят про маленькие или небольшие органические молекулы? Да просто потому, что большие органические молекулы – это, например, ферменты. А ферментативный катализ открыли ещё Либих с Вёлером в 1830-х, скоро 200 лет отмечать будем. И фокус в том, что во многих ферментах, не во всех, но во многих, катализ осуществляется не только за счет самого белка-фермента, но и за счет небольшой органической молекулы, коэнзима, большинство которых это хорошо нам знакомые витамины. Собственно, обсуждая великую работу Рона Бреслоу про тиазолиевый катализ бензоиновой конденсации, мы уже упоминали, что она выросла из анализа действия коэнзима транскетолаз, витамина В1, так что и эта тема тоже уже была вполне раскрыта, и к началу нового века все известные типы ферментов и их кофакторов были известны в мельчайших деталях. Ферементативный катализ область колоссальная, уже давно пожавшая урожай нобелевских премий и вполне нашедшая своё место не только в собственно реакциях в клетке, но и в обычном органическом синтезе.
И вот тут на сцену выходят Бен Лист и Дэвид Макмиллан. Оба молоды (по 32 года – тот самый возраст, когда в науке надо делать большие дела) и дерзки. Без последнего качества ничего бы не получилось, потому что въехать в область, где в общем-то почти все известно, и решить, что именно они смогут все это вдруг запустить заново – это настоящая дерзость, наглость, безрассудство. А что, спросит себя человек, лишенный этих качеств, до меня тут дураки паслись? Какие уж дураки, вон череда имён каких, ноги от благоговейного ужаса подгибаются! Что тут неизвестного могло остаться? И пойдёт восвояси искать что-то совсем-совсем новое, но не найдёт, естественно, что совсем нового могло остаться в органической химии после стольких лет такого бурного роста, и завянет непроснувшийся талант. Недаром же еще в 1990 один из крупнейших органических авторитетов Дитер Зеебах, который, например, изобрел наши любимые дитианы и вообще весь принцип умполунга, анализируя развитие органической химии, написал, что в ней уже все известно, и серьезные достижения могут быть только в применении переходных металлов. Своей нобелевки Зеебах тоже не дождался. Но с переходными металлами он поразительно сыграл в пророка – ровно пять лет спустя начался совершенно невероятный бум катализа комплексами переходных металлов, многократно расширивший возможности органического синтеза и в некоторой степени не завершившийся до сих пор. А со всем остальным Зеебах скорее проиграл – органика и не думает уходит на покой, и даже наоборот тоже стала развиваться бешеными темпами. И развиваться она стала в очень значительной степени за счет того, что ученые типа Листа и Макмиллана поняли, что уже достигнутое – не предел, а только прелюдия – берите любую известную реакцию и копайте дальше, благо современная техника эксперимента позволяет снова начать копать там, где кончили копать предшественники.
Копать начали с очень простых вещей, особенно Лист, который в этот момент работал в очень продвинутой биохимической лаборатории у проф. Карлоса Барбаса Третьего и исследовал возможность катализа альдольной конденсации некими специфическими белками. Возможность была показана, стала получаться красивая современная наука с пафосным названием “каталитические антитела”. При этом результаты не были выдающимися, особенно в смысле энантиоселективности – она оказалась довольно скромной. Но в ходе этой работы Лист обратил внимание, что альдольную конденсацию, возможно, получится сделать без всякого белка, а только с помощью одной аминокислоты. Перебрав их немало, выяснили, что ни одна не может сравниться с L-пролином, той самой аминокислотой, которая уже засветилась в реакции Хайоша-Вихерта. Получаются и выходы неплохие (больше 70%) и, что самое главное, вполне приличная оптическая чистота (ee 76%) – это не совсем фонтан для 2000 года, но очень неплохо, особенно если учесть издевательски простые условия – просто смешали ацетон, р-нитробензальдегид (взяли потому что он наиболее активен из ароматических альдегидов, а ароматический альдегид взяли, чтобы не связываться с побочными реакциями), одну треть L-пролина, растворили в ДМСО и поставили крутиться на сутки. Все, после обычная водная разделка, экстракция и анализ продуктов – не сложнее нашего практикума. Получилась короткая работа фактически с одним положительным результатом, и эта работа (List, B.; Lerner, R. A.; Barbas, C. F., III. J. Am. Chem. Soc. 2000, 122, 2395-2396) остается по сей день самой цитируемой во всём немаленьком наследии Листа.
Посмотрим, что же такого выдающегося в этой простой с виду (и по сути тоже, но простота не всегда хуже воровства, иногда она лучше цветущей сложности) работы. Пролин ведь уже известен в альдольной конденсации, даже энантиоселективной, это же и есть реакция Хайоша-Вихерта 30-летней давности. Да, но от той реакции с тех пор никто так и не продвинулся. Дело в том, что та реакция внутримолекулярная, это циклизация, аннелирование Робинсона по сути. А внутримолекулярные реакции очень часто идут намного легче межмолекулярных, когда дают 5-ти или 6-тичленные циклы. И если у вас есть хорошая циклизация, вы редко сможете в тех же условиях сделать межмолекулярную реакцию. Поэтому никто и не пробовал. Да и очень хорошо было известно, что ни альдегиды, ни кетоны с вторичными аминами никакой альдольной конденсации не дадут – там будет куча реакций, получатся енамины и иминии, пойдёт Манних (мы знаем что Манних – это вариант альдольной конденсации, но опять таки, хорошо он идет только с самыми простыми альдегидами, а с теми, что посложнее получаются такие смеси, что не расхлебаешь), другие реакции, в общем, это очень сложно и малопригодно. Но здесь фокус оказался именно в пролине, в том, что в нем кроме вторичного амина есть карбоксил, и этот карбоксил может помочь делу кислотным катализом. Итак, смотрим. Во-первых, при взаимодействии кетона (в первой работе это просто ацетон) образуется енамин, и карбоксил помогает и этому тоже – таскает протон. Вспомним, как мы получали енамины из обычных вторичных аминов, пирролидина или пиперидина – долго кипятили с азеотропной отгонкой воды в присутствии каталитического количества сильной кислоты. А здесь всё делает этот карбоксил – он внутри молекулы, сближен со всеми реакционными центрами и это колоссально облегчает реакцию, так что она идет просто при смешении.
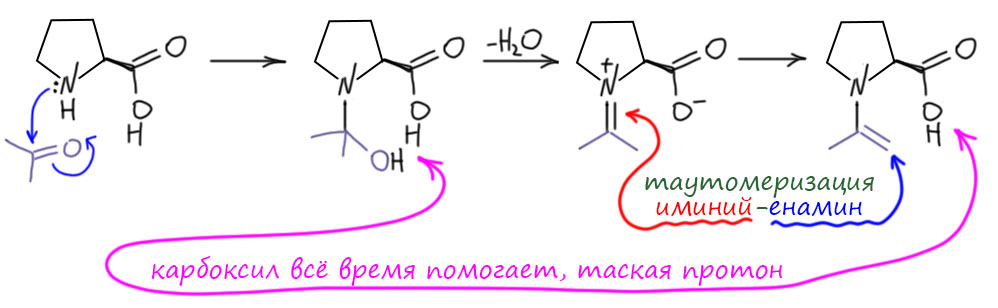
Отсюда первое поучительное – не думайте, что в новом веке в химии осталось всё только очень сложное, а всё простое съели противные и жадные предки. Ни фига подобного, у предков было так много всего неисследованного, что они, вытаращив глаза, метались между совершенно случайными находками, и почти никогда не пробовали копать посерьёзнее. Если сокровища валяются на земле, станете вы копать землю и долбить киркой – просто нагнётесь и подберете. Вот химики в 20 веке шли и подбирали, и не могли унести всего. Наступил 21 век, и да, на земле уже почти ничего не валяется, но стоит хоть чуть-чуть копнуть, даже не лопатой, а просто детским совочком – и вот оно, блестит, переливается. А в этом случае даже совочком не пришлось, валялось на поверхности, просто никто не заметил, хорошо что не затоптали. Так кстати, в химии бывает довольно часто – хорошая вещь, до ума доводится легко, получается отличная химия, но нет в ней блеска новизны, потому что оказывается, что она уже кусочками описана в дюжине мутных старых статей в Тьмутараканском бюллетене прикладной химии, который, как назло, все же попал в реферативные базы.
Хорошо, получился енамин, дальше уже более-менее понятно, так как мы уже разобрались в реакции Хайоша-Вихерта. Но та реакция внутримолекулярная, всё под рукой, а здесь надо еще изловить проплывающий мимо альдегид. Ну вот этим и займется опять карбоксил – водородная связь даёт то, что нужно, слабое взаимодействие, обеспечивающее обратимое образование реакционного комплекса. Здесь фокус опять в том, что енамин – не самый сильный нуклеофил, он не будет просто реагировать с альдегидом, альдегид надо активировать взаимодействием с кислотой Бренстеда-Лоури. Но вот она – карбоксил пролиновый. Без этого нет реакции. Поэтому прореагирует только тот альдегид, что попался на крючок водородной связи, и в этом еще один секрет этой реакции и вообще всех селективных реакций – реакция идет только в определенном реакционном комплексе, где все реагенты заботливо собраны всякими слабыми взаимодействиями до реакции – и уже в этом комплексе и только в нем и происходит реакция. 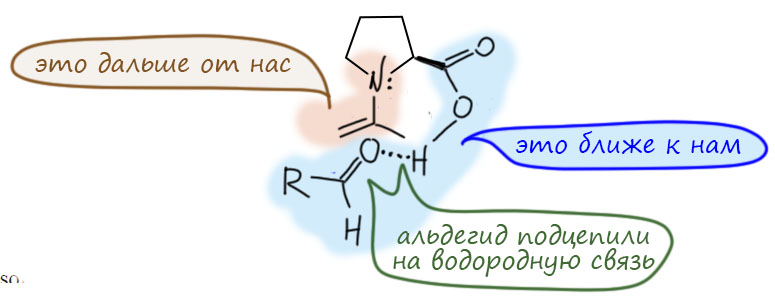 И ещё одна клёвая фишка. Пролин-то у нас не какой-нибудь, а настоящий, природный, энантиомерно чистый L-пролин (мог бы быть и D-, но он, как несложно догадаться, дороже, не сильно, но дороже, а зачем для первого эксперимента деньгами разбрасываться – всё должно быть максимально просто и дешево). А значит, и расположение групп в нем определенное, карбоксил идет вперёд, к нам, и там-то он и ловит альдегид. А остальная часть молекулы, включая енаминный кусок, немного сзади. Иными словами, хиральный пролин собирает на себе хиральный реакционный комплекс – и это-то и есть условие того, что реакция не только пойдёт, но и продукт тоже будет не рацематом, а хиральной молекулой. В этом месте мы, конечно, не должны думать, что тут всё так просто, что можно нарисовать что-то правдоподобное и все сразу станет понятно. Тут осталось еще много вопросов – например, а отчего альдегид расположился так, а не наоборот. Поэтому для более серьёзной работы нужно аккуратно считать модели таких реакционных комплексов, а еще лучше, то и переходных состояний, только тогда мы увидим по настоящему, какой путь и какое взаимное расположение выгоднее. И тогда мы узнаем, что другое расположение не сильно невыгоднее лучшего, поэтому реакция идет частично и по менее выгодному, поэтому и получается не 100%-ная оптическая чистота, а меньше. Но для того, чтобы понять принцип работы таких реакций, простого наброска реакционного комплекса вполне достаточно.
И ещё одна клёвая фишка. Пролин-то у нас не какой-нибудь, а настоящий, природный, энантиомерно чистый L-пролин (мог бы быть и D-, но он, как несложно догадаться, дороже, не сильно, но дороже, а зачем для первого эксперимента деньгами разбрасываться – всё должно быть максимально просто и дешево). А значит, и расположение групп в нем определенное, карбоксил идет вперёд, к нам, и там-то он и ловит альдегид. А остальная часть молекулы, включая енаминный кусок, немного сзади. Иными словами, хиральный пролин собирает на себе хиральный реакционный комплекс – и это-то и есть условие того, что реакция не только пойдёт, но и продукт тоже будет не рацематом, а хиральной молекулой. В этом месте мы, конечно, не должны думать, что тут всё так просто, что можно нарисовать что-то правдоподобное и все сразу станет понятно. Тут осталось еще много вопросов – например, а отчего альдегид расположился так, а не наоборот. Поэтому для более серьёзной работы нужно аккуратно считать модели таких реакционных комплексов, а еще лучше, то и переходных состояний, только тогда мы увидим по настоящему, какой путь и какое взаимное расположение выгоднее. И тогда мы узнаем, что другое расположение не сильно невыгоднее лучшего, поэтому реакция идет частично и по менее выгодному, поэтому и получается не 100%-ная оптическая чистота, а меньше. Но для того, чтобы понять принцип работы таких реакций, простого наброска реакционного комплекса вполне достаточно.
Ну и нарисуем саму реакцию. Нуклеофильный енамин в реакционном комплексе атакует активированный альдегид, образуется, как обычно, тетраэдрический аддукт, и после гидролиза это и есть альдоль. Гидролиз тоже идет мягко и это тоже следствие того, что карбоксил пролина помогает всем кислотно-катализируемым реакциям, в том числе и гидролизу. И если мы не потеряем стреохимию, и не забудем, что тройка связей альдегида ближе к нам, а енамин подходит сзади, то у нас получится и стереохимия продукта тоже. В стереохимии энантиоселективных реакций такой подход к прохиральной молекуле (прохиральная – значит сама нехиральная, но после реакции станеет хиральной) обозначают символом re, что означает, что атакующий атом видит перед собой тройку заместителей на прохиральном центре в конфигурации R.
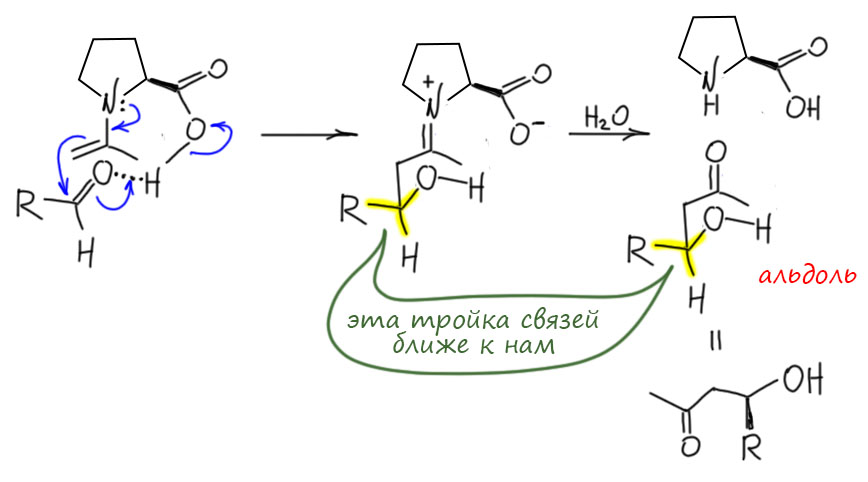
В этой маленькой работе Лист показал сразу весь потенциал этого подхода – простая и доступная хиральная молекула (здесь – банальная природная аминокислота) может успешно катализировать отличную органическую реакцию, причем при этом происходит весьма эффективный перенос хиральности с пролина на продукт, и пролин при этом возвращается, то есть может работать как катализатор. Лист в этом момент не сам себе хозяин, а работает на профессора Барбаса. Листу повезло, что его начальник не интересуется такой примитивной химией, он биохимик, ему интересны антитела, белки, клетки, и Лист ставит звездочку на себе, а не на профессоре. Лист наверняка сильно обрадовался такому интересному результату, но вряд ли мог знать, к какой богатой химии это быстро его выведет, уже как самостоятельного исследователя. Но быстро получил свой грант, и отделился от Барбаса. Самое забавное, что и профессор Барбас немного подумал, послал к чёрту свои антитела и клетки, и тоже занялся органокатализом. В начале нулевых он даже обогнал Листа в количестве публикаций на эту тему, как будто пытался нагнать упущенное, сделать что-то в этой области очень крутое и забрать её обратно себе, на что он имел совершенно полное право. Но бедняга умер в 2014 от редкой и скоротечной формы рака и не дожил до триумфа своего постдока, которому он подарил самую первую звёздочку, за 20 лет выросшую в нобелевку.
И тут на сцене появляется Макмиллан.
...входит Макмиллан
В том же 2000 году вышла первая органокаталитическая статья Макмиллана (Ahrendt K. A., Borths C. J., MacMillan D. W. C. J. Am. Chem. Soc.2000, 122, 4243), всего на несколько месяцев позже статьи Листа. Но статья Листа это коротенькое письмо в редакцию с одной реакцией и с некоторой, впрочем довольно туманной заявкой на общность найденного метода. Статья Макмиллана намного серьёзнее – хотя это тоже письмо в редакцию на две страницы, но в нём довольно много данных по реакциям с разными субстратами. И, что самое главное, с явной заявкой на то, что описываемая реакция открывает общий подход, который тут же впервые и называется органокатализом. Так прямо и написал в первом же абзаце:
Herein, we introduce a new strategy for organocatalysis that we expect will be amenable to a range of asymmetric transformations.
– здесь мы представляем новую стратегию для органокатализа, которая может стать пригодной для разных асимметрических превращений. Обратите внимание на это for – для. Макмиллан сделал потрясающий ход, настоящий блеф, bluff – здесь ведь нет никакого нового термина, и это явно просто сокращение словосочетание “органический катализ” – новая стратегия для органического катализа, всего-то навсего. Наверняка решил, что надо красивое словечко придумать, но статья горит, ничего путного в голову не приходит, а какой-то Лист уже танцует твист, откуда он только взялся – потом придумаю. Статья вышла, дело закрутилось, и как-то само собой случилось так, что невинное сокращение стало желанным новым термином, новой концепцией. Bluff удался. Ведь если бы это сразу был термин, надо было бы написать …we introduce a new strategy – the organocatalysis – that we expect… Вот так, совершенно случайно, в химии закрепился этот странный, ничего не значащий, максимально расплывчатый термин органокатализ – но это оказалось очень хорошо, потому что потом станет очень удобно под это чёрти-что подгребать всё, что плохо лежит. Был бы термин поконкретней, фокус бы мог не пройти, новый катализ остался бы немного куцым, и кто знает, может именно это и определило блестящую судьбу и нагадало встречу с шведским королём.
Отсюда и пошло это слово, хотя формальный приоритет остался за статьёй Листа, и Макмиллан успел её процитировать. Макмиллан вряд ли ожидал появления конкурента, и наверняка потом укорял себя за то, что не опубликовал совсем короткое сообщение из одной реакции – это наверняка получилось бы сделать раньше, – а набирал материал на более существенное сообщение. В отличие от Листа, который был сотрудником другого профессора, Макмиллан к этому времени уже был самостоятельным со своей группой, и конечно, мог работать более серьёзно в отличие от Листа, котрый, скорее всего, еле уговорил своего профессора Барбаса ненадолго отвлечься от пафосной работы на каталитическими антителами, и опубликовать куцую заметку из одной простенькой реакции. Вот так бывает – в современной науке зевать не стоит, особенно если вы чувствуете, что сделали нечто очень привлекательное, из чего может вырасти нечто очень значительное.
Работа Макмиллана к тому же намного интереснее с химической точки зрения, чем работа Листа. У Листа это было альдольной конденсацией, и при всей невероятной и фундаментальной важности этой реакции, возможность катализа органическим нуклеофилом, а по совместительству основанием и кислотой в одном флаконе, для альдольной конденсации вполне очевидна – проблема только найти подходящее соединение. Это как если мы в июле-августе идём в хороший смешанный лес, можно предположить, что мы найдём там белый гриб, может быть даже не один, а если не найдём, это просто значит, что плохо искали и вообще грибник – это не профессия, а призвание. Лист вот был грибник – пошёл и нашёл. Но Макмиллан не пошёл в лес за грибами. Он пошел в лес за рыбой. Но в лесу нет рыб. Отчего же, может там речка в середине течёт или пруд с карпами, а у грибника оказалась раздвижная удочка, а личинка проволочника, добытая из ножки подосиновика, пойдёт на наживку.
Макмиллан тоже искал органокатализатор среди вторичных аминов – в этом смысле он вроде бы такой же грибник как Лист. И в субстрате реакции, который должен взаимодействовать с амином он увидел альдегид. Всё вроде то же. Но на эту наживку, вполне похожую на листовскую, он стал ловить … реакцию Дильса-Альдера. В этом месте хочется протереть глаза и спросить, не запутались ли мы в именных реакциях, их так много, что немудрено не туда заехать. Ведь реакция Дильса-Альдера, как учим мы на 3 курсе – реакция между диеном и диенофилом – имеет согласованный механизм и вообще ни в чём не нуждается, это, например, одна из немногих реакций, вообще не зависящих от растворителя, отчего ее вообще часто делают без растворителя – разбавлять, только скорость снижать. И какой к чёрту там может быть органокатализ? Что там могут делать нуклеофилы или электрофилы?
Но оказалось, что может быть в реакции Дильса-Альдера и просто катализ, и органокатализ. И, надо сказать, эту мысль Макмиллана трудно не признать и оригинальной, и невероятно изящной. Я даже не могу сказать, были ли у этой мысли какие-то прототипы. Мы ведь знаем, что в химии все идеи уже давно высказаны, и дело современных химиков – только раздувать хорошо известное. А здесь я не уверен, что Макмиллан просто взял известную идею, и как-то по своему её применил. Похоже, что идея была весьма свежей.
Но пойдём по порядку. Я позволю себе разобрать этот кейс поподробнее, потому что мы на 3 курсе как-то слабо разбираемся в Дильсе-Альдере, кстати, одной из считанных реакций, которая удостоилась отдельной нобелевской премии. Выбор Макмиллана для первой работы поэтому можно назвать дважды удачным. Реакция Дильса-Альдера, как нам известно, всё же имеет свои капризы. Например, в ней реагирует не любой олефин, а обычно только такой, у которого есть акцепторные заместители. Такие олефины называются диенофилами. Из этого следует несколько вещей. Одна состоит в том, что в этой реакции диен играет роль донора, а диенофил – акцептора. Это вовсе не значит, что в реакции электронная плотность переедет от диена к диенофилу – мы знаем, что там плотность перемещается симметрично – шесть электронов как по команде в циклическом переходном состоянии перемещаются, разрывая одни и образуя другие связи. Когда мы говорим “электроны перемещаются” это не значит, что они по каким-то причинам вдруг приходят в движение и покинув старые свои молекулы, в которых уютно жили не тужили, вдруг загораются идеей сменить квартиру и устремляются в новые молекулы, а иногда и в одну. Вряд ли электронам свойственно это “весьма мучительное свойство, немногих беспокойный крест” – на самом деле, конечно, речь идет о том, что перестройка структуры в процессе реакции сопровождается перераспределением электронной плотности. Но нарисовать это удобно именно стрелками, как научил нас великий Боб Робинсон, сражаясь с не менее великим Кристофером Ингольдом за первенство в создании теории механизмов реакций, мы так привыкли, нам так удобно, пусть будет так, тем более что во многих реакциях стрелки действительно показывают направление смещения плотности – от донора к акцептору, от нуклеофила к электрофилу. Абстрагируясь от заместителей получим обычную картинку
Но проблема в том, что реакция Дильса-Альдера, как и многие другие согласованные реакции с циклическими переходными состояниями, не имеют направления смещения плотности. Поэтому, кстати, мы и можем стрелки нарисовать в другом направлении и ничего не изменится. И при этом – парадокс – олефин должен быть акцепторным, а диен донорным (есть другой вариант, но он очень странный и им мало кто пользуется). Такое сочетание реакционной способности обеспечивает начало взаимодействия – когда молекулы сближаются и сталкиваются, а делают они это в жидкости непрерывно, им надо понять, стоит им реагировать, или лучше разойтись, почёсывая ушибленные бока. И самое лучшее, что придумала химия, для того, чтобы молекулы разобрались в своих пристрастиях и состоит в том, что мы называем словами донор – акцептор или нуклеофил – электрофил. Реакция Дильса-Альдера начинается точно так же как любые другие электрофильно-нуклеофильные реакции, но дальнейшее развитие в циклическом переходном состоянии и приведет к тому, что плотность распределится симметричным способом в новой молекуле.
С точки зрения орбитальных взаимодействий это всегда значит, что самое главное взаимодействие обеспечивается двумя орбиталями. Совершенно очевидно, что у молекулы донора это заполненная орбиталь, обычно самая высшая заполненная, ВЗМО (по-английски highest occupied – HOMO). А у акцептора – пустая, незаполненная, свободная орбиталь, НСМО (lowest unoccupied, LUMO). Поскольку мы уже распределили роли в Дильсе-Альдере, то понимаем, что от диена нам нужна ВЗМО, а от диенофила НСМО.
Из этого следуют многие важные вещи, как например, орбитальный контроль, региоселективность, экзо-эндо-стереоселективность и мы здесь все это трогать не будем. Нам здесь понадобится другое, реакционная способность. Все, кто делал реакцию Дильса-Альдера хорошо знают, что не такая уж она лёгкая, как её иногда малюют. В большинстве случаев, даже если у нас и диен неплох, и диенофил не последний нужно и концентрации взять побольше (вот почему ее делают часто без растворителя), и либо поднагреть от души, если диен позволяет, либо просто оставить надолго, на много дней в тёплом месте у батареи. Но оказывается, есть способ сильно ускорить реакцию, но это получается далеко не всегда. Уже в 1960 Питер Йейтс и Филип Итон обнаружили, что реакцию Дильса-Альдера очень сильно ускоряют кислоты Льюиса, но только в том случае, если двойная связь сопряжена с акцепторной группой (почти всегда это карбонил в кетонах, производных непредельных кислот, хинонах и т.п.), в которой есть донорный атом, за который и может зацепиться кислота Льюиса. И зацепившись, сделает акцептор ещё акцепторнее (кислотное взаимодействие забирает плотность от донорного атома, а он в своб очередь ищет компенсацию потерям в той молекуле, на которой висит). И диенофил – ещё диенофилистее. Например, добавка безводного хлорида алюминия ускоряет реакцию антрацена и малеинового ангидрида в невероятное количество раз – строго говоря, без катализатора она практически вообще не идет в растворе при комнатной температуре, и 4800 часов – это просто грубая оценка, скорее всего сильно заниженная.
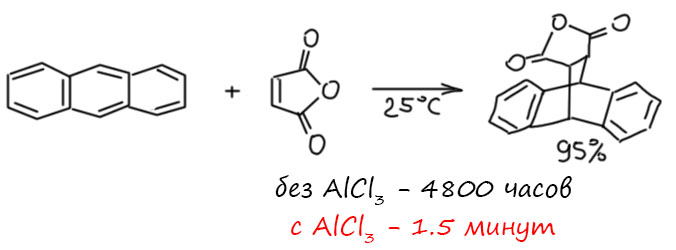
А в терминах орбиталей это означает понижение энергии орбитали. Это всегда так – акцептор понижает энергию, это довольно легко понять причем кучей способов: например, представив, что акцептор создает положительный заряд на атомах, а это усиливает взаимодействие с ними электронов. Или представив себе, что акцептор откачивает электронную плотность, тем самым усиливая связь остающихся на фрагменте, которого так обижают, электронов. Ну или ещё как-нибудь. Донор, в той же логике, всегда повышает энергию орбиталей, создает избыточную плотность, ослабляет связь электронов – они же не любят друг друга, и если бы не притяжение к ядрам, так и бросились бы врассыпную.
И вот, в реакции Дильса-Альдера диенофил должен быть акцепторным, тогда его НСМО опускается и становится доступной для взаимодействия с ВЗМО диена. И если акцепторность диенофила недостаточна и реакция не идет или идет, но в более жестких условиях, значит НСМО высоковато, и надо бы её малость подопустить. Хорошо бы у молекул был бы такой винтик сбоку под шлиц маленькой отвертки, чтобы можно было подкрутить – в одну сторону крутишь – НСМО опускается, в другую ВЗМО поднимается. Но винтика нет, вместо винтика иногда может быть неподеленная пара на сопряженном атоме, на ней можно повесить кислоту Льюиса и НСМО понижается, насколько, мы не знаем и не можем это контролировать и настраивать, но даже в таком, слепом варианте это исключительно хорошо работает. Это весьма эффективно, и такие диенофилы можно здорово раскачать.
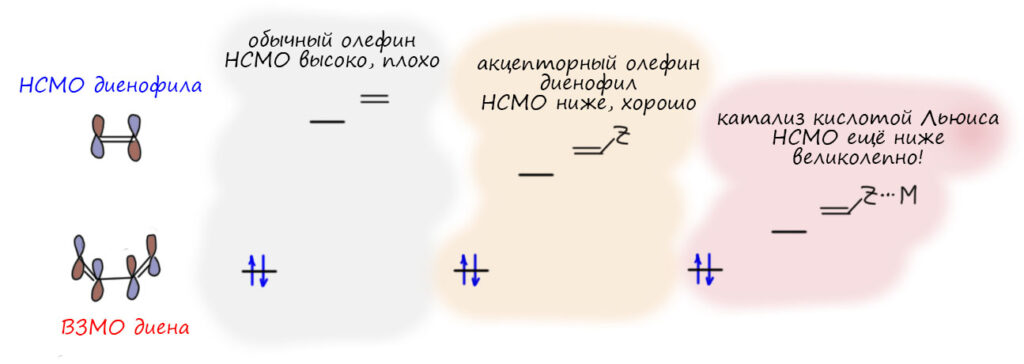
Диенофилы такого типа почти всегда содержат сопряженную карбонильную группу – это еноны, хиноны, акриловые кислоты и т.п. И тогда бывает, что просто с диенофилом реакцию вообще иной раз не запустишь – надо долго греть, а диенофил при этом может и осмолиться. Но если добавить кислоту Льюиса типа хлористого алюминия, трехфтористого бора или четырёххлористого олова, реакция идет почти моментально и даже при сильном охлаждении. Это очень хороший метод, и его давно развили. Один из главных основоположников энантиоселективного синтеза, француз Анри Каган в конце 1980-х развил на этой основе энантиоселективный вариант реакции Дильса-Альдера, использовав в качестве кислот Льюиса комплексы металлов с хиральными лигандами.
Прежде чем посмотреть на идею Макмиллана, осмотрим, откуда берется энантиоселективность в реакции Дильса-Альдера. Даже в самой простой реакции циклоприсоединения простого диена с монозамещенным олефином образуется один стереогенный центр. Но поскольку оба реагента нехиральны, реакция всегда дает рацемические смеси.
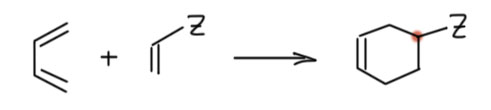
Тем не менее, с открытием катализа кислотами Льюиса появилась возможность переноса хиральности с катализатора, если использовать комплексы металлов с хиральными лигандами. Таких реакций с конца 1980-х было сделано очень много, и оптические выходы иногда достигали очень высоких величин выше 90%. В большинстве случаев энантиоселективность запускали с более сложными диеном и диенофилом, самый популярный диен в этой химии – циклопентадиен, а диенофилы, в осоновном дизамещенные. Более сложная и более жесткая форма молекул облегчают более эффективный перенос хиральности, так как у таких молекул меньше вариантов взамиго расположения. Обратим внимание, что в такой реакции возникает сразу 4 стереогенных центра, а кроме того возникает еще и проблема диастереоселективности, так как возможно образование и диастереомеров (эндо и экзо). Когда возникает сразу 4 стереоцентра, может появиться впечатление, что это более сложный случай, чем простая энантиоселективность – мы же знаем, что когда стереоцентров два и более, возникают диастереомеры и число стереоизомеров становится большим. Но в данном случае (и во всех аналогичных) четыре стереоцентра не независимы – их конфигурация фиксирована циклом. Поэтому здесь речь всё же идет об энантиомерах, образование которых очень легко понять, если увидеть, что диенофил к диену может подойти двумя способами (если не рассматривать вторую диастереомерную пару) – и эти два пути являются зеркальными отражениями.
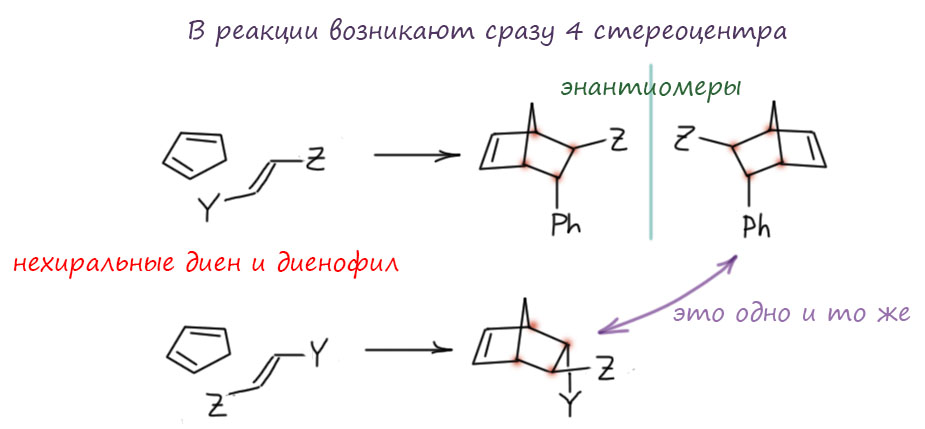
Оба пути по энергии одинаковы, так как оба реагента имеют плоскости симметрии. Но если мы уберем плоскость симметрии с одного из реагентов, например, с диенофила, тогда два пути перестанут быть одинаковыми, перестанут быть отражениями друг друга, и появится разница в энергиях активации, а значит и в скоростях. В варианте катализа кислотами Льюиса это достигается тем, что некая хиральная кислота Льюиса даёт комплекс с диенофилом, и такой комплекс уже хирален.
Мысль Макмиллана была в этом направлении, но металл хотелось заменить чем-то более аккуратным. Металлы – вещь хорошая, но в тех случаях, когда синтез делается в практических целях, особенно когда цель синтеза – что-то для медицины, возникает проблема необходимости тщательной очистки продукта от следов металлов, и там настолько жесткие требования, что очистка может стать дороже самого синтеза, а может и вообще ни к чему не привести по очень простой причине – сложные органические молекулы типа тех, что применяются в лекарствах, часто содержат пропасть всяких функциональных групп, которые как лиганды удерживают металлы так, что достать металл оттуда становится вовсе невозможным – молекула таскает металл с собой. Макмиллан всю дорогу работал на гранты от больших фармацевтических компаний и делал науку, нацеленную на решение практических задач. И если можно придумать, как активировать, например, диенофил без металла, то это будет неплохо. А дальше тот же способ пригодится для других реакций, химия в этом смысле проста как гвоздь – у вас или электрофил, или нуклеофил. Способ активации электрофила сработает везде, где нужен электрофил. То же можно сказать про нуклеофилы. Кто вообще придумал эти два гениальных понятия? Ингольд, Ингольд, да благословят его уполномоченные боги и приветствуют в царствие своём. Ингольду, кстати, нобелевки так и не дали, противный был, со всеми ссорился, вот и не дали. А вся органика стоит на его плечах и не падает скоро уже сто лет как.
Идея Макмиллана состояла в том, чтобы карбонил обратимо заместить на более акцепторную группу. И в качестве такой взять иминий – ведь хоть азот и менее электроотрицателен, чем кислород, но положительный, кватернизовынных, аммонийный азот более акцепторен. Откуда Макмиллан это знал? Вообще-то и мы это знаем, только не догадываемся. Знать, но не догадываться – удел многих. И так мимо проходит большая, красивая химия – и другие едут в Стокгольм к тамошнему королю. Знаем хотя бы потому, что знаем реакцию Манниха – в ней иминиевая соль формальдегида оказывается более сильным электрофилом, чем сам формальдегид. Если бы это было не так, не было бы никакой реакции Манниха, и была бы только альдольная конденсация с самим формальдегидом. И вот, мы знаем реакцию Манниха, но не догадываемся, что она хочет нам сообщить нечто общее – что иминий более электрофилен, более акцепторен чем карбонил, и это может пригодиться не только для Манниха. А Макмиллан догадался.
Вот его замысел. Взять диенофилом непредельный альдегид, еналь. Такие диенофилы реагируют с диенами только при нагревании. А что если найти такой вторичный амин, который даст иминиевую соль, и она станет лучшим диенофилом, чем исходный альдегид, и будет реагировать прямо при комнатной температуре. Эх, а ещё бы этот амин был хиральным, и чтоб реакция Дильса-Альдера стала энантиоселективной! Для этого, кстати, мягкость условий особенно важна, потому что при повышении температуры растет хаос и разупорядоченность в том числе и реакционных комплексов, и стереоселективность падает драматически. Размечтался! Безнадёжная затея, ведь мы же знаем, что с сопряженными карбонильными соединениями амины реагируют не по карбонилу, а по Михаэлю. Но урождённого шотландца Михаэлем не напугаешь, и вот он уже пробует. Прямо надо сказать, что с начала от Михаэля всё-таки решили немного подстаховаться, и в β-положение на всякий случай поставили фенил, чтобы снизить константу равновесия 1,4-присоединения. И поиск хорошего амина привёл Макмиллана к не самой тривиальной вещи, особенно с виду – гетероцикл, производное имидазолидина, хиральный. Но если вглядеться, это оказывается довольно простое производное самой банальной аминокислоты, L-фенилаланина.
Макмиллан придумал другое – образование иминия с хиральным вторичным амином, молекулой, сделанной из природной аминокислоты L-фенилаланина. Почему такая странная молекула? Это экспериментальный результат, Макмиллан с сотрудниками перебрал довольно много разных, в том числе и некоторые производные пролина, и именно эта оказалась наилучшей. В дальнейшем, этот, как любят это называть, структурный мотив, станет визитной карточкой Макмиллана, так же как пролин – Листа. Надо сказать, что в этом отношении Лист немного выиграл, у него органокатализатор проще, его вообще не надо делать, все природные аминокислоты доступны тоннами, ими, извините, свиней кормят. Катализаторы Макмиллана всё же надо делать, хотя это и простая химия, и как только они стали популярны, химические компании тотчас наладили их производство. Но свиней этим всё же не кормили. Вообще это забавно – еще недавно в химии большой доблестью было сделать что-нибудь позаковыристей, чем сложнее тем больше были гордость и спесь синтетика. В новой химии свиней всё чаще приглашают экспертами качества результатов: химия должна стать не сложней хрюшкиной еды – помните это гениальное “я свинья и ты свинья, все мы вместе свиньи, нынче дали нам, друзья, целый чан ботвиньи”. Реакции надо теперь вести в ржавом ведре в воде из сточной канавы, вместо аргона хорошо идут миазмы из канализации, а катализатором надо взять вот что-то не сложней ботвиньи. Вот тогда статья в Nature обеспечена. Это называется sustainable chemistry и отлично идёт – хирши от таких результатов растут как хрюшки на добрых жирных помоях.
Итак, при добавлении катализатора Макмиллана в реакционной смеси из альдегида образуется соль иминия, а кислотный катализ для этого обеспечивает то, что амин берут в виде соли аммония. Этого достаточно.
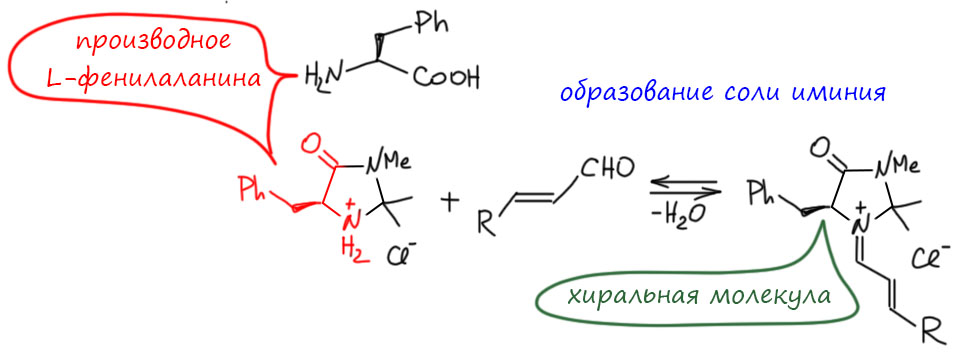
Вы спросите (надеюсь) – а почему это мы сами так реакции не делаем, что просто оба реагента положить в колбу и они сами прореагируют – а всё продолжаем что-то дополнительно добавлять, нагревать, перемешивать, удалять воду и так далее. Это очень просто – здесь не нужно, чтобы иминий образовывался количественно, чтобы его можно было выделить и получить высокий выход. Соль иминия образуется в равновесии в некотором количестве, но дальше тут же вступает в реакцию циклоприсоединения, и выходит из равновесия. А равновесие само сделает так. чтобы на место выбывшего образовался ещё иминий. Исходный альдегид в таких условиях (в разбавленном растворе и при комнатной температуре) просто не реагирует. Второй очевидный вопрос – а почему не идёт Михаэль, 1,4-присоединение. Ответ можете поискать на страничке, где я разбираю закономерности 1,2 и 1,4-присоединения и не оставляю камня на камне от популярной, но жалкой и бесполезной “теории” ЖМКО. Где-то тут есть такая страничка. Если коротко, то реакция по карбонилу всегда быстрее 1,4-присоединения, но 1,2-присоединение обратимо, и более медленное 1,4-присоединение имеет большую константу равновесия, потому что продукт более устойчив. В данном же случае, продукт 1,2-присоединения реагирует с диеном, и не успевает распасться обратно и дойти до 1,4-продукта. Кстати, даже если часть и дойдёт, не страшно, ведь 1,4-присоединение тоже обратимо, и в условиях когда из равновесия удаляется именно и только продукт 1,2-присоединения, весь непредельный альдегид рано или поздно перекочует в продукт через 1,2-присоединение, то есть иминий.
Итак, иминий образовался, и этот диенофил более реакционноспособен, чем исходный альдегид. По той же причине, что и комплекс диенофила с кислотой Льюиса – заместитель на олефине становится более акцепторным, и НСМО двойной связи понижается, а диены со своей ВЗМО это любят. Раньше ВЗМО диена смотрела куда-то вверх, где маячила НСМО диенофила, и трудно ей было дотянуться, высоковато. А теперь ей эту НСМО, можно сказать, под нос подсунули, взаимодействуй – не хочу. Хочет ведь, хочет. И взаимодействует гораздо веселее, реакция сильно ускоряется, идёт в разбавленном растворе при комнатной температуре. Но это еще не всё. Поскольку диенофил теперь хирален, два возможных пути подхода к диену, которые раньше давали рацемическую смесь, теперь не одинаковы. Схематически мы это нарисуем так, что нам покажется, что в одном из путей, заместитель на стереоцентре в иминиевой части в одном из подходов оказывается ближе к диену, и за счет стерики просто не дает нормально расположиться для образваония циклического переходного состояния – оно поэтому оказывается выше по энергии, чем в другом подходе. Поэтому получается не рацемическая смесь, а серьёзное преобладание одного энантиомера – энантиоселективность.
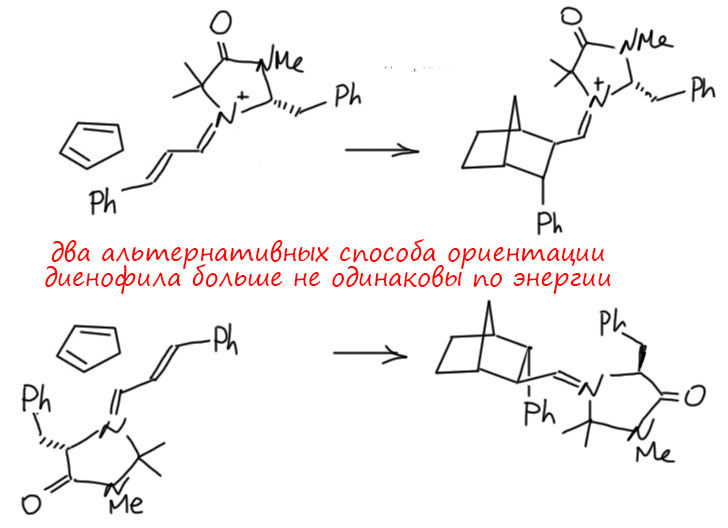
Это такой принцип – разрушение равноценности правого и левого. В этом же и состоит секрет распознавания энантиомеров – в обычном, изотропном мире, где есть зеркало, в котором право отражается в лево, разницы нет. Но стоит нам построить мир, в котором больше нет зеркала и право отличается от лево, возникает разница, и чем сильнее искажение, тем сильнее разница. Хиральный вторичный амин в катионе иминия разрушает симметрию взаимодействия диен-диенофил, и возникает энантиоселективность. В некотором смысле это можно проиллюстрировать простой аналогией. Вот, старинный офицер идет с дамой в оперу, и берет даму под руку с той стороны, где платью дамы угрожают проезжающие по Невскому кареты – может встать и слева, и справа. Офицер нехирален, и лево и право одинаковы. Но на самом деле офицер прохирален, и теперь представим, что он напялил парадный мундир с огромной шпагой или саблей в ножнах на одном боку. Всё, офицер с саблей теперь хирален, лево и право больше не одинаковы, и даму можно подхватить только с одной стороны, там где нет шпаги. Вон та бензильная группа в катализаторе Макмиллана в некотором смысле и играет роль этой дурацкой сабли.
В мире молекул такая картинка, конечно, только претендует на убедительность, там всё сложнее, и взаимодействия могут быть как отталкиваниями, так и притяжениями (водородные связи, электростатика и прочее), но общий принцип именно этот – несимметричная молекула разрушает симметрию, создавая энантиоселективность. Если немного вдуматься, то мы увидим, что пока вот эта штука в одном из реагентов, которая и разрушила симметрию, находится в деле, два возможных продукта вовсе не энантиомерны, а диастереомерны, собственно именно поэтому у них разная энергия. Подробнее не будем, ведь мы разбираем нобелевку, а не энантиоселективность в общем. Но это полезно, потому что все энантиоселективные реакции эксплуатируют один и тот же принцип – разрушение симметрии хиральным участником реакции, а это может быть один из реагентов, катализатор, или даже что-то еще. Важно только добиться, чтобы это разрушение симметрии было серьёзным и создавало значительную разницу в направлениях реакции, а для этого нужно добиться того, чтобы источник разрушения симметрии был близко к реакционному центру, и за счет суммы взаимодействий ориентировался бы максимально более определенным способом. Этого непросто добиться, приходится перебирать много вариантов и в эксперименте и в теоретическом моделировании, пока не найдётся хороший. Этого добились далеко не сразу, асимметрический синтез, как это называли раньше, или энантиоселективный синтез, как предпочитают сейчас, очень долго давал жалкие результаты – оптическая чистота не превышала 20%. Но в 1970-80-х понемногу разобрались, что лучше делать, одно из ключевых достижений принадлежит уже упомянутому Анри Кагану, который так и не получил нобелевской премии, и с тех пор реакций с высокой энантиоселективностью стало огромное количество – практически любая реакция, в которой возможно образование стереогенных центров (или даже осей, плоскостей, спиралей) в новой химии имеет множество энантиоселективных вариантов высокой эффективности.
Ну и в конце убедимся, что у будущего нобелевского лауреата всё должно быть прекрасно – и замысел, и реализация, и оптическая чистота, и выходы, и катализ. Кстати, где тут катализ? А вот он – как только циклоаддукт образовался, уже ненужный вторичный амин отщепляется гидролизом, и возвращается в реакцию, а это и есть катализ.
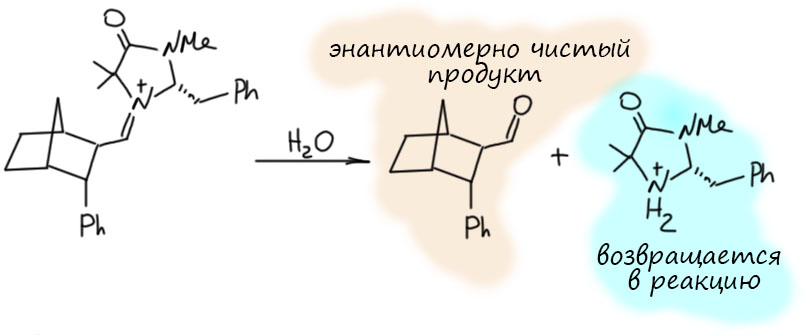
Всегда, когда глядишь на такие реакции, думаешь, как же классно у них всё получается – когда нужно садится, когда не нужно отщепляется, красота! Не врут часом? Больно гладко всё, как в армии, всё по линеечке, покрашено, надраено, все мероприятия по расписанию. Нет, не так. В мире молекул все подчиняется теории вероятностей, там все случайно, полный хаос, а то что мы видим – это среднее, такая равнодействующая хаоса. Руководят всем равновесия, их оптимальное соотношение в данном сложном процессе, состоящем из множества равновесий. Так устроены в химии равновесия, что возникает ощущение потрясающей согласованности связанных друг с другом реакций. Работает только тогда, когда такая система будет найдена в исследовании, выбрана из множества менее удачных вариантов. Из тысяч подобных процессов заработает как следует только один, и любое изменение, даже незначительное, обычно всю идиллию безжалостно разрушает. Хорошая реакция подобна хорошей поэзии – бегут друг за другом строчки, ни одной не выкинуть, не переставить, не заменить ни одного слова.
Что было дальше
Оба исследователя вполне поняли потенциал того, что они обнаружили. И свой собственный тоже, хотя они в нём явно и раньше не сомневались. И бросились сначала копать именно в том месте, где обнаружилась первая находка. Лист решил выбить из пролина всё, что в это небольшое соединение заложил Творец. Вообще, это удивительно, как много всего заложено в любое соединение, каким бы простым оно не казалось. С молекулой водорода носятся уже второй столетие, и никак не исчерпают её возможностей. Про воду вообще говорить неудобно – мало там обычной науки, тысяч статей ежегодно, так до сих пор находится немалое количество шарлатанов, которые видят в этом соединении мистические тайны, не видимые в обычных экспериментах. Так что невероятные новые возможности пролина, открытые в 21 веке, совсем не могут вызывать удивления. Это вообще удивительная аминокислота, единственная без свобоной амино-группы, и в Природе у неё особые функции и в сворачивании третичной структуры белков, и в формировании активных центров ферментов и много чего ещё. То, что пролин легко дает с енолизуемыми карбонильными соединениями, открывает возможности пересмотреть всё, что мы знаем про енамины, и приспособить всё это для энантиоселективного катализа. Заодно, на правах первооткрывателя, Лист долго избегает термина “органокатализ”, использует слова енаминокатализ и аминокатализ, и в упор не замечает параллельно существующего Макмиллана, который в общем-то изучает очень похожие реакции и фактически ту же самую реакционную способность. Когда в 2007 Лист уже набрал достаточно материала на большой 100-страничный обзор в Chemical Reviews (Chem. Rev. 2007, 107, 5471-5569), он уже смирился со словом органокатализ, употребляет его часто и с удовольствием, но откуда его взял не пишет, а Макмиллан удостоился первого упоминания в обзоре на 28-ой странице и с 201-ой ссылкой.
Пролиновый метод органокатализа реакций енолизуемых карбонильных соединений через образование енамина за 6 лет развился необычайно. Его применили в множестве энантиоселективных альдольных конденсаций, как межмолекулярных, так и внутримолекулярных, заодно выяснили, что метод оказался очень гибким, но не универсальным. Но универсальных методов в химии и не бывает, любой имеет границы применения. Главное состояло в том, что метод понравился, его стали применять разные исследователи, начался поиски других органокатализаторов, в основном на основе разнообразных производных пролина, но не только. Более сотни работ были сделаны за 6 лет. И это очень важно. В органике ежегожно публикуются тысячи работ, и огромное количество новых методов и модификаций, но подавляющее большинство никого не привлекает. Вообще, когда встречаете работу, которая кажется вам интересной, посмотрите, привлёк ли этот метод кого-нибудь ещё именно с практической точки зрения. Если работе 5 лет или больше, но цитируют ее только в обзорах и предисловиях к статьям, это значит, что метод или не понравился, или, что еще хуже, плохо воспроизводится, а то и вовсе является лажей – никто про это не напишет, это не принято, да и любой, кто попробовал, но получил плохой результат, сначала подумает, что может виноват не метод, а просто не получилось. А вот если метод и правда хорош и полезен, его попробуют сначала единицы, получат свои результаты, опубликуют, это привлечет новых исследователей, и метод полетит. Вот с органокатализом произошло именно это – метод полетел, многим стало любопытно, а что и правда так просто можно делать альдольную конденсацию, да ещё и энантиоселективную. Это точно проще классической Мукайямы – ведь не надо получать никаких производных, типа силиловых эфиров, не надо покупать довольно дорогие хиральные хелатные комплексы – реакция, описанная Листом, обещала, что досточно взять карбонильную и метиленовую компоненты, добавить пролин или что-то типа того, покрутить раствор денёк-другой при комнатной температуре, и можно выгружать продукт. В общем, надо признать, что ничего проще с времен открытия альдольной конденсации не предлагалось.
Среди сотен работ по органокаталитической альдольной конденсации особо выделяется работа… Макмиллана – он смог сделать кросс-альдоли из двух разных енолизуемых альдегидов. Надо сказать, что это та ещё задача в любом варианте направленной альдольной конденсации, еноляты от альдегидов получить непросто, силиловые эфиры тоже выкидывают всякие фокусы, так что в реакциях часто получаются непригодные смеси продуктов. Мы от вас на 3 курсе это скрываем, уверяем, что направленно можно сделать любую кросс-конденсацию, или через еноляты, или Мукайямой. Но это упрощение, на самом деле есть множество комбинаций компонент, когда проблемы почти неразрешимы в любом подходе, и далеко не любой кросс-альдоль можно получить с хорошим выходом. И вот как раз кросс-конденсация двух енолизуемых альдегидов – один из главных затыков альдольной химии всех времён и народов. И тут приходит блистательный Макмиллан…
Макмиллан и Нортроп в 2002 году показали, что пролин позволяет сделать и эту конденсацию, причем и диастерео и энантиоселективно (Northrup A. B., MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 6798). В этой статье Макмиллан с удовольствием посчитался со свои будущим коллегой по нобелевке, при перечислении источников для своей работы указав на первом месте не Листа, а Барбаса, а саму реакцию пролин-катализируемой альдольной конденсации обозвал реакцией Хайоша-Вихерта-Барбаса-Листа, намекнув на то, что ничего особенно нового во вкладе Листа по сравнению со старой реакцией нет, да и вообще, знаем мы кто в той статье был руководителем. Но результат получился отличный и такой, которому сначала даже не веришь. Я поэтому разберу этот случай подробнее, это, на мой взгляд, довольно поучительно.
Обычная проблема такой конденсации в том, что у альдегидов близкая и очень большая реакционная способность, и почти невозможно избежать обеих самоконденсаций и кросс-конденсации наоборот. Чтобы избежать побочных реакций самокондесации и перекрестной конденсации в другом порядке исследователи применили редкий в наше время приём – вместо того, чтобы, как это нынче принято, закинуть все компоненты реакционной смеси в пузырёк и оставить перемешиваться, даже устроили медленное прибавление одного альдегида к смеси другого с пролином. Правда, выбор был немного странным, в смеси с пролином была карбонильная компонета, а не метиленовая, но об этом чуть ниже. И всё! Если учесть, что пролин был взят в каталитическом количестве (10 мольных процентов), это могло сработать только в одном случае – если самоконденсация карбонильной компоненты по каим-то причинам не может конкурировать с перекрестной конденсацией. И даже в этом случае не вполне понятно, почему первый альдегид почти не дает самоконденсацию. В общем, это непростой трюк, но он получился и впоследствии был не раз использован для синтезов. Реакцию еще и ведут при охлаждении, что тоже способствует селективности. Вот один такой пример из статьи (там на один углерод меньше в первом альдегиде, но это не важно) – видим, как гладко всё получается, энантиоселективность практически количественная. Более того, в реакции образуется не один стерегенный центр, а два, поэтому к такой реакции два вопроса – энантиоселективность и диастереоселективность.
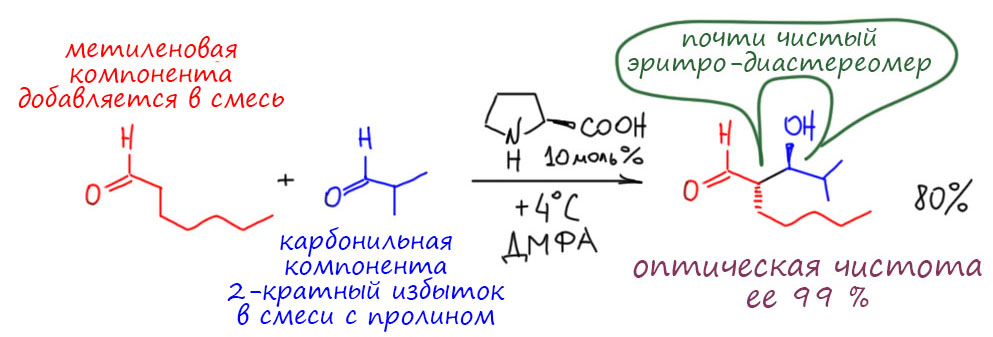
Диастереоселективность оказалась высока, образуется практически исключительно эритро-изомер (точнее смесь эритро:трео 24:1), а у этого диастереомера исключительно один энантиомер (S,S). Круто! Вот что такое современная химия – старая классическая органика не могла толком сделать такой альдоль хотя бы рацемическим и смесью диастереомеров. Органокатализ – хлоп! – и решил сразу все проблемы, да еще и предельно простым способом, какой только можно себе представить. Поэтому возникаёт чёртов соблазн сказать – вот она, настоящая химия, а что мы тут возимся на 3 курсе с этой альдольной конденсацией, разбираем все закономерности, как и что реагирует, как выбрать карбонильную компоненту, как подготовить метиленовую и в каком виде, какая у кого реакционная способность и так далее. Ух, тоска какая (автор тихо воет от обиды за бесцельно прожитую жизнь)!
Спокойно! Берём себя в руки, внимательно вчитываемся в строки статьи, обязательно лезем в supplementary information, куда давно уже принято сваливать от греха подальше все экспериментальные подробности исследования, и начинаем кое-что понимать. Нет, не то, что вы возможно подумали. Всё тут правильно, и сделано всё по-настоящему, никакого вранья тут нет – если вы хотите долгой жизни в науке и признания заслуг вплоть до нобелевки, будьте готовы, что и через 20-30 лет вас ткнут носом в старую ошибку, когда кто-то ее обнаружит, поробовав воспроизвести ваши эксперименты. Просто дело в том, что в химии очень многое определяется никогда заранее не известными константами скоростей отдельных стадий, из которых состоит сложный процесс – основной и побочных реакций. И умением химика играть на таких различиях, понимая их иногда умом и опытом, иногда ощущая чутьём, иногда просто делая сотни экспериментов, из которых будет в конце отобрано несколько наиболее удачных. Виртуозная игра на тонких различиях в реакционной способности и отличает серьёзного синтетика от ремесленников, воспроизводящих готовые методики, точно так же как виртуозная игра на музыкальном инструменте отличает знаменитого солиста от рядового музывканта, играющего на 25-м пульте в огромном оркестре.
Посмотрим на работу внимательно. Во-первых, восхитимся техникой эксперимента. Объём реакционной смеси не превышает 1 миллилитра, и из этого полмиллилитра прибавляются медленно и равномерно в течение 10-15 часов. Попробуйте как-нибудь полмиллилитра добавлять медленно и равномерно в течение 10 часов. Полмиллилитра это приблизительно 20 капель. Две капли в час, и нет, капать не пойдёт – нужно именно равномерно. Для этого у Макмиллана есть автоматический шприцевой микронасос, который и дает вот такую тончайшую струйку через тонкую иголку. У меня вопрос – а почему бы не сделать этот эсперимент нормально, на 10 мл хотя бы, ведь все реактивы в этом исследовании – дешёвка, доступная не то что в Принстоне, а и на краю Земли килограммами. Что экономим? Ответ простой – так сейчас принято делать химию, в микроколичествах. Одна из причин этого состоит в том, что для хорошей статьи реакции делаются сотнями и тысячами, а то, что публикуется, это наилучшие эксперименты, доли процента. Тогда проще купить специальное оборудование для таких микрореакций, которое позволяет делать их сразу десяткаи и сотнями. Денег у Макмиллана с самого начала было много, стоит только почитать раздел благодарностей в его статьях, и оборудование было самое современное. Вторая причина связана с первой – анализ смесей максимально автоматизируется, а для этого тоже лучше использовать микроколичества. Ну а побочный результат такой химии – если другой химик захочет использовать описанную методику на более солидных количествах, придется поработать над масштабированием, и это далеко не всегда просто и очень часто является источником ошибок.
Попытаемся понять, как у Макмиллана сделана конденсация. Во-первых, обратим внимание на то, что с пролином смешивается карбонильная компонента, а не метиленовая, и карбонильная компонента берется в 2-кратном избытке. Вроде бы с пролином надо смешивать метиленовую компоненту, ведь именно она дает нуклеофильный енамин. Почему же смешивают карбонильную – ведь это должно приводить к её самоконденсации. Загадка? Скорее всего нет – и если внимательно посмотреть на список использованных альдегидов, то мы видим, что карбонильной компонентой всегда во всех парах используется альдегид покрупнее, часто разветвлённый. Фокус, видимо (видимо, потому что это наша интерпретация, а с статье об это прямо не говорится, это такие милые фигуры умолчания, которых много в современной химии – вранья нет, но секретиков полно) в том, что такие альдегиды намного медленнее образуют енамины: енамины, в отличие от енолов предпочитают быть менее разветвленными, так как иначе возникают стерические взаимодействия с вторичным амином, снижающие сопряжение между парой азота и двойной связью. Поэтому в смеси карбонильной компоненты, выбранной такими способом, и пролином самоконденсация идёт достсточно медленно, чтобы при прибавлении альдегида поменьше, даже при том, что прибавляемый альдегид находится в заведомо намного меньшей концентрации, именно он будет перехватывать пролин, образуя енамин, и тогда карбонильная компонента в избытке обеспечит именно перекрестную конденсацию, потому что самоконденсация метиленовой компоненты будет невыгодна из-за намного меньшей концентрации. Вот такая хитрость. Эта хитрость говорит нам – нет, это не общий метод перекрёстной конденсации, все наши старые знания никуда не делись и даже напротив, помогли нам разобраться. И если нам захочется по методу Макмиллана сделть конденсацию двух альдегидов, придется все предусмотреть, продумать сочетание и только тогда реакция может получиться.
А то, что эта реакция получается, ясно потому что её стали применять в синтезах. Потрясающим применением этого метода стал стереоселективный синтез углеводов несколькими последовательными альдольными конденсациями, причем Макмиллан сначала сделал синтез, в котором сначала собирается половинка углевода пролиновым методом, а затем углевод окончательно собирается с помощью конденсации Мукайямы. Но последователи доработали подход и нашли условия полной сборки только пролиновым методом. Подробнее не будем, если заинтересуетесь без труда найдёте статьи.
Пролиновый метод активации енолизуемого карбонильного соединения стали применять дальше для других реакций, где может использоваться енамин. В первую очередь это конденсация Манниха. Для нас это такая частная реакция, когда енолизуемый кетон реагирует с формальдегидом и вторичным амином. Это классическая реакция Манниха, с помощью которой вводят диалкиламинометильную группу в α-положение кетонов. Раньше только так и делали. Но возникает вопрос, а почему нельзя вместо формальдегида взять другие альдегиды или даже кетоны. А вместо вторичного амина первичные. Ответ такой – можно, но в обычных условиях Манниха реакция очень неселективна и дает кучу разных продуктов, ведь там полно возможностей реакций между компонентами в самых разных комбинациях. Обобщённую реакцию Манниха тем не менее уже давно исследуют, используют в качестве катализаторов комплексы металлов, давно появилась и энантиоселективная реакция Манниха. Но пролиновый катализ дал отличные возможности делать эту реакцию проще и с высокой селективностью. Лист сделал первую работу про это в том же 2000 году, уже отделившись от своего профессора Барбаса. Так и хочется тупо сострить про Листа-буратино и Карабаса-Барабаса, но здесь ситуация ровно противоположная – профессор благородно дает возможность талантливому ученику немедленно обособиться и начать свои собственные исследования, фактически отдав тему, но при этом оставив за собой возможность заняться параллельными исследованиями наперегонки. Барбас и начал копать того же Манниха и нашел много своего, причем он страшно торопился и на одну статью Листа отвечал пятью своими. Такая работа наперегонки дала интересный результат – энантиоселективного органокаталитического Манниха стали называть реакцией Листа-Барбаса-Манниха.
Вместо того, чтобы разбирать, что там сделали Лист или Барбас приведу одну поразительную работу на эту тему, принадлоежащую японской лаборатории под руководством Юдзиро Хаяси (Hayashi, Y.; Tsuboi, W.; Shoji, M.; Suzuki, N. J. Am. Chem. Soc.
2003, 125, 11208.). Проблема первых методик Листа и Барбаса состояла в довольно низких выходах и обилию побочных пробуктов. Хаяси заметил, что трехкомпонентная реакция должна ускоряться давлением. Но как устроить давление в жидкости? Безусловно, можно нагнетать в автоклав под давлением инертный газ, но это опасно и в обычной лаборатории не получится. Хаяси придумал нечто такое, что мы бы обязательно назвали “голь на выдумки хитра”, но в Японии голь не водится уже давно, с тех самых времён, когда её как следует разгромили, отобрали все военные игрушки и заставили взяться за ум, что хоть и не сразу, но получилось на славу. Голь в Японии не водится, а хитрость на выдумки японцам всегда была свойственна, не из-за нужды, а по эстетическим причинам. Что бывает, если в закрытом сосуде заморозить воду? Его разорвет, даже если сосуд металлический. Вода при кристаллизации довольно сильно увеличивается в объеме, а если объем замкнут, то нехилое давление его размыкает. Хаяси прикинул это давление, нашел что это что-то типа 200 атмосфер, и это значит, что можно взять хороший автоклав, атмосфер на 500, бросить в него завинченную тефлоновую пробирку (тефлон не теряет гибкости при пониженной температуре) с реакционной смесью, залить под завязку водой и положить в морозилку. Хаяси не преминул кокетливо заметить, что морозилка – обычная бытовая. И там, в компании с мороженой курицей автоклав с водой замерзнет и в пробирке поднимется давление до 200 атмосфер. Дополнительный бонус в том, что если давление делать обычным способом, нагнетая газ или жидкость насосом, то если что-то пойдет не так и автоклав разорвет, будет много жертв и разрушений. А если автоклав с водой все же разорвет, этого не заметит никто, и даже мороженая курица не пострадает. Вот так Хаяси с сотрудниками и сделали реакцию Листа-Барбаса-Манниха, и действительно, выходы оказались почти количественными. При -20 градусах! Органокатализ работает при отрицательной температуре! Правда держать приходится долго, но это просто лежит в морозилке, и если мороженая курица не возражает против такого соседства, то вообще проблем нет. Обратите внимание на амин – п-анизидин, кажется, почему такая частность, но это очень популярный приём, потому что п-анизильную группу очень легко убрать мягким окислением, и тогда у нас будет первичная аминогруппа на стереоцентре – и делайте с ней дальше всё, что в голову придёт.
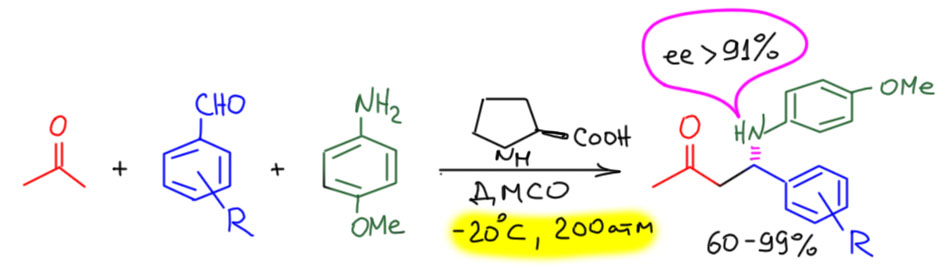 По органокаталитическому Манниху за 20 лет вышли сотни статей и много задач было решено, много разных кетонов, альдегидов, аминов соединено. Это очень полезная и важная реакция в синтезе, так как дает разнообразные аминокетоны, очень гибкие полупродукты разных сложных синтезов – две мощнейшие функции, да еще и с чистой стереохимией, мечта. Мечты сбываются.
По органокаталитическому Манниху за 20 лет вышли сотни статей и много задач было решено, много разных кетонов, альдегидов, аминов соединено. Это очень полезная и важная реакция в синтезе, так как дает разнообразные аминокетоны, очень гибкие полупродукты разных сложных синтезов – две мощнейшие функции, да еще и с чистой стереохимией, мечта. Мечты сбываются.
Следующая реакция, на которую оказался направлен пролиновый метод, как нетрудно догадаться это реакция Михаэля. Енамины отлично реагируют в присоединении по Михаэлю, а если к этому приделать энантиоселективность, совсем получится отлично. Как и в случае Манниха, энантиоселективные Михаэли были известны во множестве, в основном тоже с катализом хиральными кислотами Льюиса. Лист ринулся в Михаэля сразу после Манниха, но этот орешек оказался покрепче (List B., Pojarliev P., Martin, H. J. Org. Lett. 2001, 3, 2423.). Сам по себе катализ пролином вполне работал, например, на реакции енолизуемых кетонов с нитроолефинами, но энантиоселективность получалась довольно бледная, редко за 20%, а из такого энантиомерного избытка много не сделаешь. Причина такой невыдающейся асимметрической индукции проста – в этой реакции реакционный центр (электрофильный углерод акцептора Михаэля) довольно сильно удален от то группы в акцепторе, за которую пролин в енамине может дотянуться водородной связью – реакционный комплекс получается рыхлым и плохо ориентирует реагенты друг относительно друга.. Зато диастереоселективность была неплоха, в основном получался трео-продукт. В некоторых случаях, впрочем энантиоселективность была неплоха, как например:
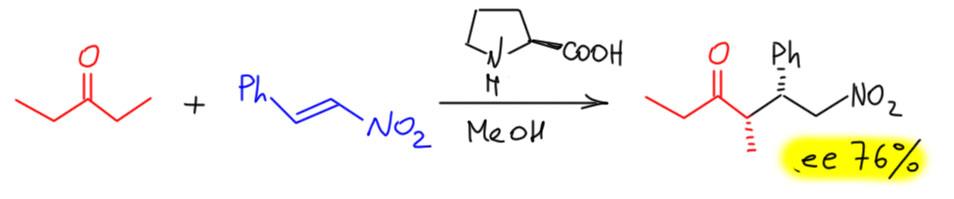
Но несмотря на невыдающиеся первые результаты (у Барбаса получилось еще хуже – просто рацемат), за 20 лет здесь многое получилось, но уже с более сложными органокатализаторами, не с пролином.
Дальнейшее развитие енаминового катализа пошло и в сторону приспособления этого подхода к различным частным случаям. Это обычный удел всех методов органического синтеза – сначала появляется общий подход, простой и красивый. После быстро выясняется, что не такой уж он общий и к конкретным случаям начинают разрабатываться модификации. При разработке модификаций первоначальный посыл метода часто оказывается забыт. Так и здесь начали с природного дешевого пролина, но быстро вяснили, что во многих случаях он дает довольно жалкие результаты, и катализаторы стали понемногу усложняться и становиться довольно громоздкими. Ну и с другой сторону, метод стал расширяться на другие реакции, в которых могут помочь енамины. Зная химию енаминов, нам будет не очень сложно догадаться какие. Поэтому оставим это, тут уже море химии.
Посмотрим лучше куда в то же время понесло куда более изобретательного Макмиллана.
А Фридель и Крафтс-то и не знают...
Макмиллан, как мы уже видели, в начале своего пути практически строго дополняет Листа – у обоих карбонильные соединения и вторичные амины, но у одного нуклеофильная активация и енамин, а у другого иминиевая соль и электрофильная активация (или акцепторная, или, как он сам любит – НСМО-понижательная). Начав с Дильса-Альдера, дальше он попробовал реакцию, которую стали называть реакцией Фриделя-Крафтса. Как это стали называть? Это же классическая старая реакция, 19 век, хлористый алюминий и все такое! Да, но если вдуматься в то, что это такое, то это электрофильное алкилирование ароматических соединений. Не ацилирование – нам на 3 курсе вдолбили, что из двух реакций Фриделя-Крафтса ацилирование – это шедевр, а алкилирование – ни к чему непригодная дрянь. А вот в современной химии всё наоборот, ацилирование – скучная рутина, из которой уже ничего не выдоишь, не то что нобелевку, а и статейку какую завалящую в приличный журнал. А алкилирование теперь – кладезь всяких фокусов. Ведь только алкилирование может быть стереоселективным (энантиоселективным, диастереоселективным). Итак, возвращаемся к первой работе Макмиллан, делаем иминий из сопряженного непредельного альдегида, и вспоминаем, что на примере Дильса-Альдера мы увидели, что замена карбонила на иминий увеличивает акцепторность, а значит и электрофильность. Тогда мы можем получить альтернативный способ активации, ведь обычно в Фриделе-Крафтсе активируют кислотами Льюиса, но здесь роль карбонила, координированного по иону металла, может играть как раз иминий. Активация без металла.
И что, хорошо активирует? Не очень, поэтому бензол мы этим алкилировать не сможем, и даже анизол не сможем, а вот что-то по-настоящему сильнодонорное – что там у нас в курсе самое активное в электрофильном ароматическом замещении – фенолы (пока отложим, они по кислороду скорее реагируют), анилины (пока отложим, они не кажутся чемпионами донорности, а мы для начала хотим, чтобы наверняка, впрочем чуть позже их так алкилировать тоже стали) – а, вот, пятичленные гетероциклы, особенно азотистые: пиррол, индол – вот где огромная реакционная способность, превосходящая возможности человеческого воображения. Забавно, что Макмиллан мог быть первым в органокатализе, но немного опоздал. Мог быть первым в энантиоселективной реакции Фриделя-Крафтса – но и тут немного опоздал. В тот же 2000 год это опубликовал мощный датский химик Карл Анкер Йоргенсен (Jensen K. M., Thorhauge J., Hazell R. G., Jørgensen K. A. Angew. Chem., Int. Ed. 2001, 40, 160), и он и дальше будет активно мешаться под руками у Листа и Макмиллана, но на соучастие в нобелевке так и не набрал, хотя сделал очень много интересного и часто бывал первым. В этом случае Йоргенсен сделал первым энантиоселективное алкилирование индола, но у него был катализ кислотой Льюиса, комплексами меди или цинка с хиральными лигандами, из очень модной серии лигандов, производных оксазолидина, собираемых по две штуки на каком-нибудь фрагменте и так, чтобы получалась хиральная молекула, диастереомер с одной осью симметрии второго порядка (поэтому такие лиганды называют C2-симметричными) – такие лиганды называются BOX-лигандами (от Bis-OXazolidine) и это один из основных типов в энантиоселективном катализе. Несмотря на довольно хищный вид, это весьма несложные соединения, легко получаемые из оптически чистых аминокислот, не обязательно природных, и легкодоступных нитрилов.
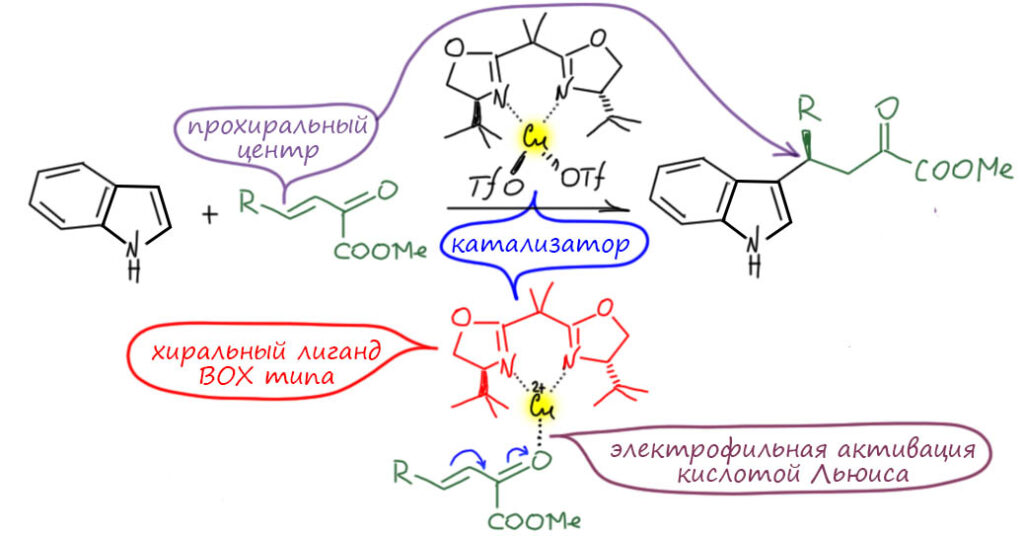
А Макмиллан сделал именно органокаталитическую версию, и с этим стал первым (Paras N.A., MacMillan D.W.C. J. Am. Chem. Soc. 2001, 123, 4370). И по сравнению этих почти одновременных работ довольно легко оценить преимущество нового подхода. Здесь всё точно так же, как в уже разобранной реакции Дильса-Альдера. Видим, что этот способ активации выглядит гораздо проще того, что предложил Йоргенсен с кислотным катализом – вместо весьма громоздкого комплекса металла используется довольно небольшая и несложная органическая молекула. И внимательные заметят, что даже субстрат немного проще – Йоргенсену понадобился кетоэфир, а Макмиллану просто альдегид. Там это было связано с проблемами взаимодействия с кислотой Льюиса, а здесь все проще и в этом смысле. В общем, получилось неплохо и это был отличный старт для еще одного важного применения органокатализа в синтезе. Так что даже неплохо, что Йоргенсен немного опередил, зато было с чем сравнить, и показать явное преимущество.
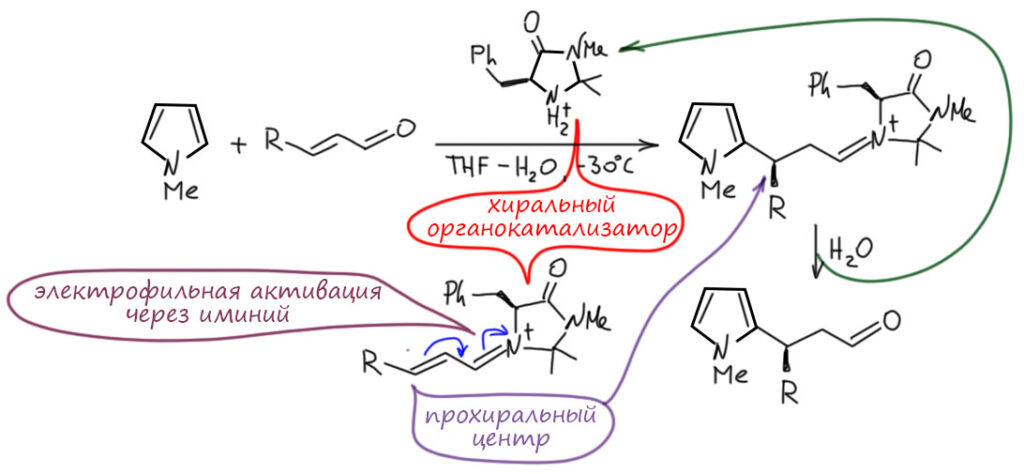
С тех пор прошло 20 лет. Макмиллан получил нобелевку, а Йоргенсен нет и уже никогда не получит, хотя это один из виднейших исследователей энантиоселективного катализа с множеством важнейших работ. Те две работы столкнулись во времени, и там вроде бы нет вопроса, чей катализ круче. Но вот спустя 20 лет можно спросить – и что, органокатализ окончательно победил кислотный катализ хиральными комплексами металлов? Ничего подобного, продолжают развиваться оба и применяются оба, никто никого не победил. А нобелевка одна.
А зачем вообще нужна энантиоселективность?
Это уже вторая нобелевская премия, в которой подчеркивается важность энантиоселективности. Так что в некотором смысле железное правило нобелевских премий – два раза за одно и то же не дают, немного просело. И это действительно так – в обоих премиях энантиоселективность – ключевое понятие, важнейший маркер достижений. Способов достижения оптической чистоты продуктов реакции очень много, но так или иначе в основе подход один – уничтожение симметрии (рассимметризация). Не любой симметрии – у многих хиральных объектов есть отличные элементы симметрии, оси симметрии второго и даже больших порядков – и мы особенно любим такие объекты, часто находя среди них особенно эффективные катализаторы или части катализаторов энантиоселективных реакций. Бескомпромиссному уничтожению должны подвергнуться все зеркала, где бы они ни находились.
И зачем нам эта энантиоселективность. Почти всегда на этот вопрос отвечают одинаково – чтобы получать чистые энантиомеры. Далее нам рассказывают про отражение в зеркале, и все остальное, что мы и так хорошо знаем. Но еще дальше начинаются рассказы про то, что энантиомеры (оптические изомеры) очень важно получать чистыми, потому что у них разные свойства. Один сладкий, другой горький; одно лекарство, другое яд; одно пахнет лимоном, другое апельсином; на одно клюют самцы, на другое самки – и так далее. В общем, да, это так и есть. Живые организмы различают энантиомеры разных молекул, потому что живое распознает молекулы с помощью взаимодействий со своими молекулами или комплексами молекул в рецепторах, и все такие штуки заведомо асимметричны, и по определению должны по-разному взаимодействовать с энантиомерами.
Все это так, но это далеко не вся правда, и даже точнее, это полуправда. Полуправда это потому что далеко не все молекулы, попадающие в организмы, действительно так надежно распознаются по энантиомерам. Это в общем точно та же проблема, как и сам энантиоселективный синтез – не любой хиральный реагент или хиральный катализатор дает высокую оптическую чистоту продуктов. И даже наоборот – редкие такие реагенты и катализаторы дают. Собственно ровно поэтому это такая сложная наука, так много времени ей понадобилось, чтобы добиться реальных успехов, и поэтому у нас уже вторая нобелевка за это.
Надежное распознавание энантиомеров происходит только тогда, когда взаимодействие между молекулами надежно фиксирует их взаимное расположение, то есть требуются несколько разных точек взаимодействия (самых разных, и водородных связей, и электростатики, и ароматического стекинга, и даже просто обычной стерики за счет сложной формы взаимодействующих молекул, подходящих друг к другу как ключ к замку). В реальности это случается не часто. И для организмов хорошее распознавание достигается заведомо для тех молекул, которые организму известны и для чего-то нужны, отбор таких молекул произошел в процессе эволюции и с этим проблем нет. Но мы хотим синтезировать и использовать молекулы, с которыми организмы раньше не встречались, и с хорошей вероятностью у них нет возможности точно распознать энатиомеры, потому что взаимодействия будут далеки от оптимальных.
Из этого следует одна довольно простая вещь – в практических прменениях за чистыми энантиомерами гоняются нечасто. Примеров того, что фармакология требует чистый энатиомер какого-то действующего вещества, а не рацемат, не так много. Очень часто для иллюстрации этого принципа приводят историю про скандал с талидомидом, но эта история не вполне корректно воспроизводится, потому что на ранних стадиях расследования того инцидента были сделаны неверные гипотезы, они разошлись по литературе, а настоящая история далеко не так однозначна – она разобрана на отдельной вкладке.
Так что, нам не нужны оптически чистые соединения? Нужны, конечно, еще как нужны. Но в большинстве случаев проблема не в энантиомерах, а в диастереомерах, то есть соединениях, у которых не один, а два или больше стереогенных центров. Вот диастереомеры реально различаются очень сильно, это совсем разные молекулы, и разница там не в лево-право, а буквально во всем, в том числе в форме молекулы, взаимном расположении частей и функций. В отличие от энантиомеров, различать которые непросто и не очень надежно даже средствами биополимеров клетки, в диастереомерах разбираются все, и ошибиться в диастереомере – то же самое, что подсунуть просто другое соединение. Так не бывает.
Отсюда следует жесточайшее требование к любым практически применяемым соединениям с несколькими стереоцентрами – диастереомер должен быть один и только тот, который требуется. Синтез таких молекул требует того, чтобы по мере построения структуры стереоцентры появлялись в нужной конфигурации, потому что потом их исправить не получится. В этом одна из особенностей синтеза диастереомеров. Если мы делаем энантиомер с одним стереоцентром, и получили плохой оптический выход, мы можем исправить дело, добавив допольнительно расщепление и дополнительную очистку, и проблем не будет, кроме дополнительных расходов. А если мы по дороге построения диастереомера плохо сделаем любой из стереоцентров, исправить уже не получится, только вылить в помойное ведро. Только самый последний по ходу построения стереоцентр можно подправить, потому что в этом случае образуется смесь диастереомеров, нужного и ненужного, и эту смесь придется поделить, выделив нужный, любым способом разделения (перекристаллизацией или хроматографией), прежде чем вводить следующий.
Теперь самый главный вопрос. Вот мы заходим на очередную стадию, чтобы сделать очередной стереоцентр, и у нас в уже готовой части есть несколько стереоцентров с правильной конфигурацией. Вопрос – какой тип селективности нам нужен, чтбы получить новый стереоцентр правильной конфигурации? Ответ очевиден – диастереоселективность. Так, а откуда она берется? Какого типа реагент или катализатор нам нужен? Хиральный или нехиральный? Одного ответа нет.
Здесь дело в том, что для того, чтобы новый центр стал хиральным или, точнее, стереогенным (это тонкости терминологии, не очень важные, поэтому примем здесь для простоты, что это одно и то же, хотя это не одно и то же), он может получить это качество из уже существующей части молекулы, от уже существующих стереоцентров. Это диастереоселективность в чистом виде, и для этого не нужен хиральный реагент или катализатор, но нужно, чтобы реагент или катализатор направлялся на прохиральный центр с одной или другой стороны за счет взаимодействия с уже существующими стереоцентрами. Такое бывает и нередко, но эти случаи требуют, чтобы реагент как-то цеплялся на существующую часть молекулы прежде чем он осуществит атаку на прохиральный центр – водородными связями, кординацией, еще чем-то.
Вариант второй. Реагент или катализатор не может зацепиться за существующую стереогенную часть молекулы. Возможно, она далеко. Или стереогенные центры там слишком простые (например, просто третичные алкилы какие-нибудь, как, например, в аминокислоте изолейцин) , или стерически прикрытые, или еще какая-то фигня. Тогда для реагента или катализатора молекула фактически нехиральна, и перед ними стоит точно такая же задача, как если бы тех стереоцентров вообще бы не было – различить лево и право, и обеспечить перенос хиральности от себя, раз больше неоткуда. Ну и что это за реагент или катализатор? Ответ очевиден – энантиоселективный. И если все получится и новый стереоцентр окажется таким как нужно, мы вспомним про то, что там уже есть другие стереоцентры, и общая конфигурация будет определяться всеми – а это значит, что результатом будет диастереоселективность. В частности, это будет означать то, что мы не можем характеризовать результат оптическим выходом или энантиомерным избытком. По той простой причине, что второго энантиомера нет и быть не может, ведь все остальные стереоцентры уже заданы.
Проиллюстрируем простым примером. Допустим, у нас была молекула RRSR и нам хочется еще стереоцентр S. Мы делаем реакцию и получаем искомый диастереомер RRSRS, но увы, селективность не 100% и у нас есть примесь RRSRR. Это другой диастереомер. Энантиомер имел бы конфигурацию SSRSR, но откуда ему взяться? А раз неоткуда, то откуда может взяться энантиомерный избыток. Неоткуда, и использовать эту характеристику нельзя. Вместо ней мы используем характеристику диастереоселективности, а это просто отношение диастереомеров, dr (diastereomeric ratio). Замечу только, что энантиомерный избыток (ee) в таких случаях все же довольно часто используют, но под этим понимают разность выходов диастереомеров, если в них отличается только один центр, то есть принимая, что мы имеем дело с “энантиомерами” по одному центру. Осторожнее с этим, здесь очень легко запутаться, и нужно очень аккуратно изучать публикуемые в таких случаях характеристики, чтобы понять, о каких энантиомерах и диастереомерах идет речь.
Итак, мы пришли к очень важному выводу. Энантиоселективные реагенты или катализаторы используются не только для энантиоселективного синтеза, но и для диастереоселективного синтеза, и даже намного чаще, потому что это намного более важная задача. И естественно, это предполагает, что диастереомер образуется в оптически чистом виде, а так будет всегда, если хоть один стереоцентр по дороге был определенным, а не рацематом. Бывает по-другому? Да, можно везде брать рацематы, но так давно уже перестали делать, потому что это сложнее, а не проще – можете сами убедиться, если проследите по цепочке, что будет, если на отдельных стадиях использовать энантиоселективные реагенты или катализаторы – конфигурации начнут расходиться, и в конце концов приедете в дикую смесь. Но если такой стереоцентр только один и в конце, а так бывает, и в этом случае все будет как в обычном энантиоселективном синтезе с одним стереоцентром, и можно будет использовать оптический выход или энантиомерный избыток.
В конце разберем один такой синтез из Макмиллана (Chem. Sci., 2011, 2, 308), не целиком, естественно, а по стадии, в которой вводится стереоцентр энатиоселективным органокатализом. Вот какое чудище надо синтезировать. Это метаболит из одной морской губки, не губки, но типа того, не важно. Вообще эти странные существа из самых простых многоклеточных, с самого низа эволюционных цепей, сидящие на дне морском и просто фильтрующие всю жизнь воду в поисках съедобной мелочи, один из самых крутых источников головоломно сложных молекул, многие из которых интересны как потенциально очень активные вещества – среди них, например, ищут агенты для химиотерапии. У всех этих губок и тому подобных существ жизнь однообразная, тоскливая, вот они и развлекаются органическим синтезом высшей сложности на радость крупнейшим синтетикам, которые обожают играть в игру “иль я хуже губки, или не сумею”. А синтезировать это надо, потому что из губок много не получишь, и исследовать на активность нормально не получается. Эта штука называется диазонамид А (это не химическое название, хоть и дико похоже, а производное от имени этой губки Diazona). В этой забавной раскоряке всего 4 стереоцентра, но три из них, отмеченные красным, можно быстро ввести на ранних стадиях синтеза из уже готовых оптически чистых исходных, аминокислот и их производных. В этом тоже один из сектретов длинных синтезов – если что-то можно взять готовое, в том числе стереоцентры, никто не будет это синтезировать или вводить энантиоселективным синтезом. Но четвертый центр, четвертичный, зелененький, очень хитро устроен и вводить его надо синтезом.
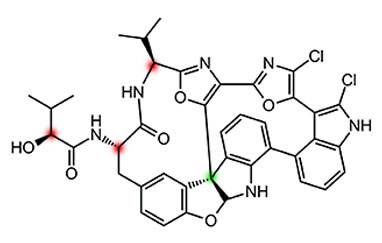
Макмиллан увидел, что есть хороший заход через реакцию, которую мы уже разобрали – энантиоселективный органокаталитический Фридель-Крафтс. Узнаёте? Чёрта с два. Так как крупный синтетик “читает” сложную молекулу, нам и в голову не придёт, но нас и к королю не зовут. В этом месте (и многих других) мы видим еще одну грань Макмиллана – это настоящий мастер синтеза, первого ряда, из гроссмейстеров – головоломно сложная молекула строится сильными, острыми, неожиданными ходами. Понятно, что за каждым стоит лошадиный труд его аспирантов и постдоков, но усилия, приводящие к цели только увеличивают удовольствие. Нобелевский комитет его ценит не за это, а нам незазорно восхититься.
Итак, кусок молекулы уже есть, три первых стереоцентра успешно установлены и никто их больше трогать не будет, хотя это тоже иллюзия, ведь до конца синтеза надо строго следить, чтобы не случилась рацемизация, а все три центра в этом смысле вызывают опасения, так как это енолизуемые положения. А там дальше будет и альдольная конценсация, условия которой придется подобрать так, чтобы не было мучительно больно за бездарно утраченную конфигурацию. Новый стереоцентр ставится органокаталитической реакцией Фриделя-Крафтса, мы ее тут уже разобрали и здесь она применена, причем в качестве акцептора Михаэля выбрали ацетиленовый альдегид, что позволило получить двойную связь в продукте, а ее дальше безжалостно порвут озонолизом, чтобы избавиться от двух лишних углеродов. В молекуле уже есть три стереоцентра (все три имеют S-конфигурацию), и они создают асимметрию, но слишком далеко от реакционного центра. Поэтому когда взяли рацемическую форму органокатализатора получили два продукта в соотношении 1:1. И это два диастереомера, SSSS и SSSR. Как видим, никакого переноса хиральности с хиральной части молекулы вообще не происходит. Вообще это довольно редкий случай, обычно все же существующие центры наводят некоторую хиральность на новый центр, но недостаточную, и тогда применяют энантиоселективный катализатор или реагент, чтобы увеличить селективность. Бывает даже так, что сущесвующие центры мешают, наводя хиральность в другую сторону, и тогда их приходится пересиливать – это особенно сложная ситуация. Здесь ситуация оказалась попроще, никто не мешает и не помогает. Когда применили хиральный органокатализатор и поработали над условиями реакциями (там все зависит от всего, от растворителя, противоиона, температуры, дня недели, расположения светил и т.д. потому что фиксация реакционного центра зависит от слабых взаимодействий, на которые влияет всё) добились впечатляющих 20:1, что соответствует оптическому выходу в 90%.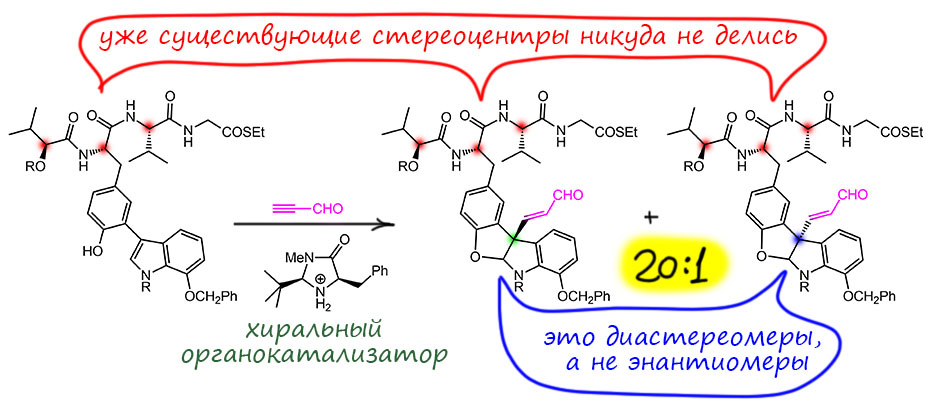
Энантиоселективнгый катализатор успешно создал новый стереогенный центр и завершил стереоселективную часть работы. Дальше молекула уже спокойно достроена до конца, и все стереоцентры остаются в заданной конфигурации. Если будет интересно, посмотрите по ссылке, там очень интересный синтез, в самом конце даже использующий уж совсем нашу химию типа электрофильного бромирования и защиты пара-положения в ароматическом кольце, правда совсем не так, как это принято делать у нас. Результатом будет большая, оптически активная молекула. Это безусловно чистый энантиомер одного из диастереомеров (SSSS – хотя надо бы проверить, вокруг последнего стереоцентра много чего происходит и формальное старшинство могло измениться, но это не важно). Второго энантиомера здесь нет и быть не может.
Во многих других сложных синтезах стереоцентров может быть еще больше, они создаются друг за другом в синтезе, и так получается искомый диастереомер. Многие шаги в современных синтезах будут делаться с помощью энантиоселективного катализа, не обязательно органокатализа. Энантиооселективные стадии создают искомый, и оптически чистый диастереомер. В этом и есть главная роль энантиоселективных реакций в органическом синтезе.
Трагическая утка. Талидомид.
Необходимость обязательно делать оптически чистые действующие вещества для использования в медицине обычно иллюстрируют жуткой историей ошибки с лекарством талидомид (Thalidomide). Но эта популярная история – типичный пример wishful thinking, как у них говорят. То есть обман во благо. Но не бывает обмана во благо. Реальность всегда сложнее.
Эту немудрёную штуку разработала одна небольшая немецкая фармкомпания и предложила на рынок как простое и безвредное успокоительное, мягкое снотворное на замену барбитуратам с приятным эффектом снятия тревожности. Таблетки стали популярны особенно в Германии, где у многих после 1945-го развилась бессонница и тревожность, и расходились как горячие пирожки. И через несколько лет с ужасом выяснили, что стало рождаться много детей с страшными дефектами, в основном с деформированными или вовсе неразвитыми руками и ногами. Таких детей родилось несколько тысяч, пока не поняли в чём дело и не убрали препарат с рынка. Это ведь не так просто – понять в чём причина. Скандал был страшный, эта история стала просто хрестоматийным примером того, как ужасны фармацевтические компании, в погоне за прибылью готовые убить и изуродовать миллионы людей.
Немного сразу снизим градус гнева – булки не растут на деревьях, а новые лекарства не появляются без фармацевтических компаний. А за прибылью обязана гнаться любая уважающая себя коммерческая компания, иначе она просто улетит с рынка и ничего не выпустит, потому что не найдёт средств на разработку и тестирование новых препаратов. А сделать фармкомпании некоммерческими, оплачивая их расходы из госбюджета, можно, такие эксперименты ставили в одной большой стране и ее сателлитах, только почему-то новых лекарств там получилось так мало, что даже и вспомнить особенно нечего, а представители правящего класса той большой страны правдами и неправдами добывали себе лекарства от ужасных заграничных коммерческих фармкомпаний.
Проблема с талидомидом была в том, что в 1950-х, когда это лекарство разрабатывали и выводили на рынок, не было еще жёстких требований к тому, как нужно лекарства проверять. А откуда, как вы думаете, эти требования вообще появились? А вот из таких историй как с талидомидом они и появились. Когда фарминдустрия только появлялась – а это произошло именно в Германии в конце 19 века, никто вообще ничего не проверял – помогает, и хорошо, всё остальное – ваши проблемы. Но случались отравления, и понемногу стали вводить требования к проверке токсичности. Сначала на общую токсикологию – и по этой части талидомид проверяли вполне тщательно и он оказался в небольших дозировках не вреднее шоколадной конфеты. Поэтому производитель и пустил его на широкий рынок, фактически безрецептурный. Кстати, в Штатах уже тогда требования были намного выше, чем в Европе – не стоит забывать, что это было послевоенное десятилетие, когда Европа откапывала себя из руин и ей было не до тонкостей, откуда и развивалась у людей тревожность, – а вдруг откопать не получится. В Штаты этот препарат официально не пустили. Но, конечно, нашлись те, кто привозил классные таблеточки от тревожности из Европы и туда, поэтому несколько десятков случаев есть и в США. После талидомида все страны ввели жесткое требование проверки любого вещества, которое может попасть в организм, на тератогенность, мутагенность, онкогенность. Да и маленьких фармкомпаний больше нет, потому что им не по силам затраты на полные испытания новых препаратов. С некоторых пор вообще повелось так, что новые лекарства разрабатывают небольшие стартапы, но когда по какому-то препарату становится понятна коммерческая песпектива, стартап выкупает одна из больших фармкомпаний и уже доволит дело до конца. Ужесточить требования заставил в том числе и талидомид. Дорогой, увы, ценой. А по-другому не бывает. Человечество всему учится весьма дорогой ценой. Вот, например, человечество научилось не устраивать мировых войн и не заигрывать с воинственными тираниями только когда убедилось дорогой ценой, чем это заканчивается. А другая часть человечества научилась не пытаться построить коммунизм, потому что по дороге случается такая убыль народа, что даже в случае успеха наслаждаться обществом всеобщей справедливости будет некому. И так везде. Сейчас вот с потеплением боремся, а еще недавно надеялись, что нефти хватит ещё на пару сотен лет.
Так что там с энантиомерами? Вот тут, увы, повсеместно повторяется история довольно далекая от реальности, зато красиво подтверждающая потребность в энантиомерно чистых веществах. Я всегда к этому отношусь скептически – может ли неправда что-то красиво подверждать? Мой ответ – нет. Неправда создаёт мифы и уводит от истины. Истина в каждом случае своя. Где-то нужны энантиомерно чистые вещества, где-то нет. Химия должна уметь обслуживать и ту, и другую потребность.
Повсеместно тиражируемая картинка такова: талидомид опасен, потому что содержал неправильный энантиомер. Правильный энантиомер безвреден и эффективен как лекарство. Неправильный – неэффективен и стал причиной трагедии. Поэтому надо было делать синтез энантиомерно чистого вещества. Вы наверняка это или слышали или читали, и не раз. И вывод – органический синтез должен всегда быть энантиоселективным.
Правда то, что из двух энантиомеров талидомида действительно только один имеет активность, обусловливающую его фармакологические свойства. Неправда то, что на плод действует другой, и что стоило бы талидомид выпустить в виде чистого активного энантиомера, всё было бы в порядке. Эту проблему долго исследовали и нашли, что во-первых, тератогенны оба энантиомера, а во-вторых, даже если бы один, это бы не помогло, потому что в организме талидомид быстро рацемизуется, что неудивительно, так как фталоильная группа висит на енолизуемом положении. Скорость рацемизации так велика, что какую форму не вводи, в течение нескольких минут получаются обе в виде рацемической смеси. Собственно, про это знали и производители – невелик секрет, любой химик посмотрит на молекулы и это скажет – поэтому его сразу производили в виде рацемата, и это тоже вполне распространенная стратегия – это намного дешевле, а если организм забирает один энантиомер (действие лекарства предполагает его взаимодействие с молекулами клетки и метаболизм), то второй будет непрерывно превращаться в рацемическую смесь, пополняя запас активного.
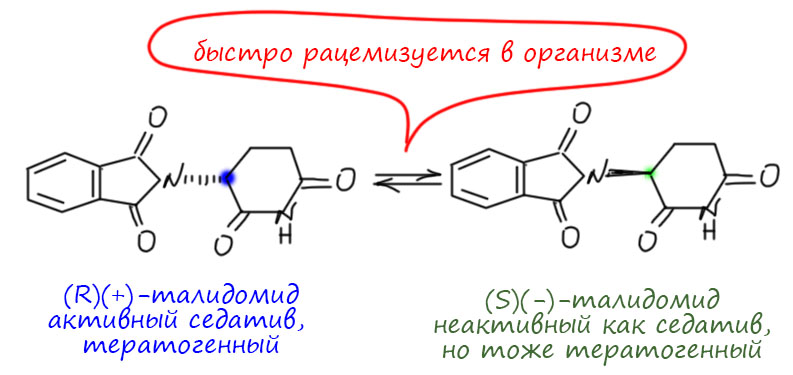
Интересно, что талидомид (да, по-прежнему рацемический) до сих пор используют в медицине, с другими дозировками и показаниями. Когда разобрались с механизмом действия на плод, выяснилось что это ровно те функции (ингибирование ангиогенеза и т.п.) что требуются для химиотерапии в онкологии и других тяжёлых патологий. И в рецептуре строго указывают на применение под контролем врача и строгий запрет назначать беременным и кормящим. Для всех остальных состояний талидомид не представляет опасности, и конечно, он не может быть безрецептурным успокоительным в виде любого энантиомера.
Только на своей химии до нобелевки не добраться
Первоначального замысла органокатализа надолго не хватило. Не только потому что сама стратегия ограничена, но и потому что набег химиков во всем мире на новую область был невероятно мощным. К концу нулевых было уже издано несколько монографий и больших обзоров, и идея начала выдыхаться. И если бы все осталось в тех рамках, то к 2021 году про эту историю бы уже подзабыли, и осталась бы она как интересный эпизод в истории органической химии, давший исследователям несколько полезных методов и протоколов. Но органокатализ в целом спас обычный рецепт империй в стадии упадка – не знаешь что дальше делать и как выполнять щедро выданные обещания, захватывай соседние страны. Будет шум, но рано или поздно все смирятся, и хотя бы получится нечто одно, безбрежное и бесформенное, сшитое даже не белыми нитками, а грубой проволокой, но по определению большое и прекрасное*. В химии эта стратегия иногда неплохо работает, и органокатализ, наверное, самая яркая история такого типа из тех, что посвежее. Интересно, что в химии такой процесс происходит спонтанно, им никто не управляет, но сами исследователи с охотой уходят под красивый термин как под зонтик или пресловутую эгиду, потому что это тренд, а хороший тренд здорово повышает цитирование, вспучивает хиршей и увеличивает вероятность получения хороших грантов, так что не важно, эгида так эгида, мы тут все с этой стороны под ней поместимся, места много. Только много лет спустя выясняется, что по другую сторону эгиды тоже кто-то стоит, и это не Афина Паллада, а довольно улыбающиеся господа, покупающие билеты в Стокгольм.
* Прим.автора – этот текст написан в октябре 2021 года и я не буду его редактировать, хотя последующие события придали этой банальной сентенции новые и неожиданные смыслы
Шаг назад, десять шагов вперёд
Ещё раз: что такое органокатализ?
Когда это понятие возникло в работах Макмиллана и Листа, и затем множества последователей в начале 2000-х, возник и консенсус – это такие каталитические реакции, в которых небольшая органическая молекула непосредственно играет роль катализатора, участвует в механизме каталитического процесса, в образовании промежуточных соединений. Мы в самом начале уже это обсуждали и пришли к выводу, что иначе было бы невозможно отделиться от такой банальной вещи, как кислотно-основный катализ, известный испокон веков химии как науки. Маленькое органическое соединение, уксусная кислота катализирует реакцию, например, бромирования ацетона – это органокатализ? Конечно нет, это просто кислотный катализ енолизации. Отлично. Чуть позже с некоторым осмыслением пришло такое определение – органокатализатор должен образовывать ковалентные связи, ну хоть одну, с участниками процесса. Интермедиаты каталитической реакции должны включать молекулу органокатализатора, связанную хоть одной ковалентной связью.
Отлично. Все первые работы Листа и Макмиллана – енаминовый, иминиевый и т.п. катализы идеально подходят под это определение.
Следующий шаг тоже очевиден – нас не интересует органокатализ как таковой, нас интересует только стереоселективный, и желательно энантиоселективный органокатализ. Это довольно странно, если вдуматься – почему бы нам просто не катализировать ту же альдольную конденсацию, циклоприсоединение, Фриделя-Крафтса, и т.п. каким-нибудь обычным вторичным амином, чтобы просто получить продукт, возможно он по природе нехиральный, а может нам и рацемическая смесь для чего-нибудь сгодится. Да, но с смого начала про это никто даже не думает. Возьмем любую работу – там только энантиоселективные реакции, даже никто не удосуживается проверить, не работает ли это для обычных реакций. Вот так повернулась органика на рубеже веков – если в какой-то реакции возможно образование стереогенных центров, разрабатываются только реакции, обеспечивающие стереоселективный исход (отдельно немного подробнее обсудим, в чем разница между стереоселективностью и энантиоселективностью). Это так вообще у людей устроено. Сначала они довольствуются малым и даже не задумываются о том, что где-то есть другая жизнь. Почти 150 лет органика была рацемической, а энатиомеры разделяли после синтеза. Синтез отдельно, расщепление отдельно – так это было устроено. Во второй половине прошлого века начались робкие попытки встроить одно в другое, возник асимметрический синтез, но очень долго был таким прикольным придатком к обычному, прежде всего потому что оптическая чистота получалась мизерной и все равно приходилось расщеплять энантиомеры – разницы мало начинать с полного рацемата или с энантиомерного избытка в 10%. Но строили-строили, и наконец зацепились за сначала отдельные реакции, затем многие реакции, и в конце доехали до правила – если в реакции образуется стереогенный центр, значит должен быть хотя бы один (а значит и бесконечное множество, как гласит моя любимая теорема Больцано-Коши) способ сразу сделать реакцию энантиоселективной с оптической чистотой не менее хотя бы процентов 75-80, а это уже уровень с которого не надо ничего расщеплять, досточно разок-другой аккуратно перекристаллизовать.
Всё, с этого момента пути назад не было. Если вам нужно что-то простое и не можете купить, берете старую методику из проверенных руководств. А все остальное уже делается по-новому, без рацематов.
Но тут-то и возникла одна загвоздка, которая позволила посмотреть на органокатализ пошире. Почему мы не хотели считать кислотно-основный катализ органокатализом и требовали ковалентного? Потому что кислотно-основное взаимодействие неспецифично – это в каком-то смысле обезличенный обмен протонами. Уже нужно уточнить – в рамках теории Бренстеда-Лоури, где кислоты и основания трактуются просто как переносчики протона. Протон пришел или ушел, начинается реакция; по ходу реакции протон еще не раз переместится, всякий раз не сам, а с помощью кислот или оснований, переносчиков протона. Это закон химии – протон сам по себе не гуляет, не умеет – его таскают кислоты и основания, переносчики протона.
И есть еще одна важнейшая вещь – водородная связь. Довольно часто в химии протон даже далеко не уходит – он остается в водородной связи между реагентами и по ходу реакции меняет положение, потому что изменяются роли донора и акцептора водородной связи. Этот фокус называется общим кислотным катализом. Протон движется очень быстро, да и вообще это квантовая частица, так что ему часто двигаться и не нужно, он и так находится там, где он нужен, в любой момент времени. Такой идеальный воин.
В этом месте возникает смутное подозрение – а не могут ли водородные связи способствовать энантиоселективности, например, если донор или акцептор водородной связи хиральны – не может ли произойти перенос хиральности за счет разности водородных связей при взаимодействии с энантиомерно-чистым донором или акцептором? А почему нет, ведь именно так работают многие процессы в живой клетке. Но это ложный след. В живом работают ферменты, белки, макромолекулы, там все очень хитро устроено и зависит от пространственной структуры комплекса макромолекула-субстрат, где тех же водородных связей видимо-невидимо и вместе они могут всё. А мы тут все же договорились ограничиться небольшими органическими молекулами. И тут уж скорее наc ждёт скепсис – всё же водородные связи штука слабая, и вряд ли они смогут так хорошо удержать реагирующие молекулы, чтобы они смогли почувствовать разницу между энантиомерами, ведь у маленьких молекул и водородных связей мало, не смогут, наверное несколько слабых связей делать то эе, что делают ковалентные или координационные.
И такая идея долго динамила развитие этого направления – время от времени кто-то пробовал перенести хиральность с помощью хиральной кислоты или основания в какую-нибудь важную реакцию, катализируемую кислотами или основаниями, но оптические чистоты были такими, что впору было назвать их оптическими нечистотами, забыть этот срам и больше не позориться. Но время шло, опыт накапливался, а с ним и понимание того, где надо искать.
Первая реакция на которой получился этот фокус нам отлично известна – это синтез Штрекера, осовремененный его вариант, в котором синильная кислота присоединяется не к альдегиду в присутствии амина, а сразу к имину. Эта реакция катализируется основаниями, но сам имин может выполнять роль такого основания, основности достаточно для обратимого депротонирования синильной кислоты. Она весьма популярна, так как позволяет получать разнообразные α-аминокислоты (через нитрилы, но они очень легко гидролизуются). В оригинальном варианте аминокислоты естественно, получаются рацемическими, но соврешенно понятно, что соблазн сделать так, чтобы они сразу были энантиомерно чистыми, невероятно велик. Естественно, никому не придёт в голову так получать природные аминокислоты – их и так полно, и они дешевы, как мы тут уже выяснили, ими кормят свиней (и коров) – но разнообразные синтетические аналоги аминокислот очень востребованы в синтезе и они вовсе не так доступны как природные, и долгое время для синтеза таких аминокислот требовалось расщепление энантиомеров кристаллизацией с огромными хиральными основаниями типа алкалоида бруцина. Это довольно дорогое удовольствие, особенно если аминокислота нужна не для баловства, а для синтеза реальных действующих веществ для лекарств.
И поэтому добиться энантиоселективного синтеза сразу энантиомерно чистых аминокислот было очень важной задачей, и многие этим занимались, медленно, но верно приближаясь к хорошим реакциям. Первые методы естественно использовали катализ хиральными комплексами металлов, но в этом случае это особенно неудачное решение, ведь сами аминокислоты тоже лиганды отличные, и отдавать металл после реакции категорически не хотят, что создаёт просто неразрешимые проблемы, если продукт вам нужен для медицины.
В таких синтезах обычно используют первичные амины, а если нужна в продукте незамещенная амино-группа, заместитель на азоте берут такой, который можно удалить (бензил, аллил и т.п.).
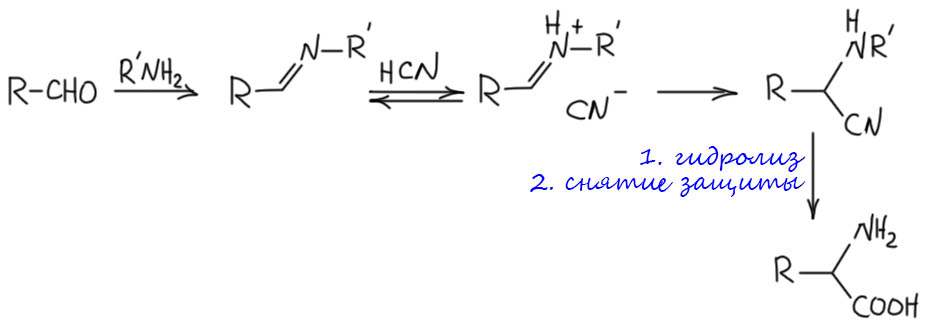
И вот, наконец, появляется первое серьёзное безметальное решение этой задачи, и сразу три группы исследователей предложили свои варианты. Первая работа Марка Липтона и сотрудников вышла в 1996 (Iyer M. S., Gigstad K. M., Namdev, N. D., Lipton, M. J. Am. Chem. Soc. 1996, 118, 4910), там использовался в качестве катализатора циклический дипептид, и результаты были очень хороши, но работа немного опередила время и сразу не зацепила. Оцените скорость развития химии в “проклятые девяностые” – тогда было столько всего, что органики немного, кажется, даже растерялись и не знали за что хвататься. Но через два года началось настоящее наступление на эту проблему, даже практически штурм, настолько быстрым был прогресс после этого.
Наиболее проработанное и осмысленное решение предложил в 1998 американский химик Эрик Джейкобсен (так и хочется назвать его Якобсеном, явно тоже предки с севера Европы, но это неправильно)(Sigman M. S., Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 4901). Работа получилась немного случайно – искали хорошие лиганды для металлов, чтобы опять катализировать Штрекера кислотой Льюиса, причем редкоземельной, но обратили внимание, что сами лиганды работают не хуже, а лучше. В 1998 Джейкобсен с сотрудниками опубликовали работу, в которой показали, что хиральные тиомочевины отлично наводят хиральность в синтезе Штрекера, ровно с помощью водородных связей и больше ничего. Вот какая молекула попалась исследователям.
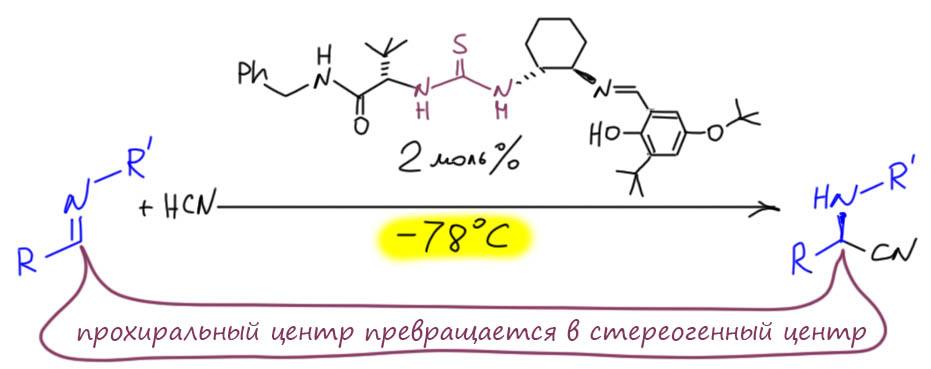
В этом месте, как следует рассмотрев это чудовище, уместно спросить, а как вообще в голову приходит делать что-то подобное. А в том-то и дело, что в голову в таких исследованиях никому ничего не приходит. Прошли те времена, когда новые молекулы придумывали, долго синтезировали, потом изучали, убеждались, что ничего хорошего кроме очередной статьи не получилось, дальше думали, опять синтезирвоали, и т.д., и так проходила жизнь, а шведский король так и не узнавал о вашем существовании. Иногда некоторым везло и придуманное неожиданно действительно оказывалось чем-то выдающимся. Одно из тысяч. С некоторых пор решили, что так дальше жить нельзя, надо ускоряться. И те, кто может себе это позволить перешли на технологию высокопроизводительного скрининга библиотек, не тех библиотек, где пыльные тома, а так стали игриво называть большие коллекции близких по структуре молекул, которые массово и быстро делают за еду в макдональдсе или отчаявшиеся, оголодавшие химики в бедных странах, или с некоторых пор автоматические системы в богатых странах. Берется реакция, которая с высокой надежностью работает для большого множества исходных – аминов, спиртов, тиолов и тому подобных функциональных соединений. Таких реакций в химии немало. Одна из них – синтез мочевин или тиомочевин из аминов и изоцианатов или изотиоцианатов – и таких исходных можно купить сотни и сделать тысячи; и быстро из них методом смешения в маленьких пузырьках наделать огромное количество похожих молекул. Дальше их в таком же полуавтоматическом режиме проверяют на какой-то реакции, и выбирают лучшее. Таким способом делаются почти все современные (современные – за последние 25 лет) работы по подбору новых лигандов или катализаторов. Правда потом, когда что-то путное будет таким образом найдено, серьёзный исследователь начнет уже серьёзное изучение, постарается выяснить, как это работает, и нельзя ли уже осмысленно еще улучшить. Вот и Джейкобсен нашел это страшилище именно таким автоматизированным поиском в библиотеках – в данном случае, если этот лиганд рассмотреть, мы увидим, что это тиомочевина, сделанная из довольно стандарных блоков, с одной стороны это синтетическая аминокислота, с другой легкодоступный диамин, модифицированный с помошью другой простой реакции – основания Шиффа.
Получив такой результат и вбив столбик, обозначивший новую область, Джейкобсен основательно это изучил, нашел, что это можно дополнительно немного улучшить, тиомочевину можно заменить обычной мочевиной и добавить еще немного стерики. А главное – понял, как это работает. Работают оба протона в мочевине или тиомочевине: они так здорово расположены, что цепляют азот имина сразу двумя водородными связями, приблизительно как коршун хватает обеими лапами кролика и тащит его на обед свои детям. Фокус тут в том, что в имине азот sp2-гибридный, а при превращении в продукт он регибридизуется в sp3-гибридный, и вот ровно в этом ему эти два протона и помогают – они как раз сидят там, где азот в продукте хочет иметь два атома водорода. Эти две водородные связи поэтому идеально работают на активацию молекулы имина. И это чертовски важно и вот почему. Проблема в том, что имин и сам реагирует с цианидом, образуя обычный продукт Штрекера, естественно рацемический. И если мы просто добавим в реакционную смесь такой лиганд, часть прореагирует с участием лиганда, часть без него, и мы никогда не получим высокой стереоселективности. Давайте тогда смесь охладим, до такой низкой температуры, при которой свободный амин перестает реагировать. А тот амин, который попадет в когти к лиганду, неплохо активирован и реагировать будет – медленно, чудес не бывает, холодно, – но будет. Охладить пришлось аж до температуры сухого льда, и тогда осталась только реакция имина-в-когтях. Теперь смотрим на сам лиганд. Условно, конечно, но желающие смогут слазить в статью (P.Vachal, E.N.Jacobsen J. Am. Chem. Soc. 2002, 124, 10012-10014), где это описано и посмотреть на настоящую молекулярную модель.
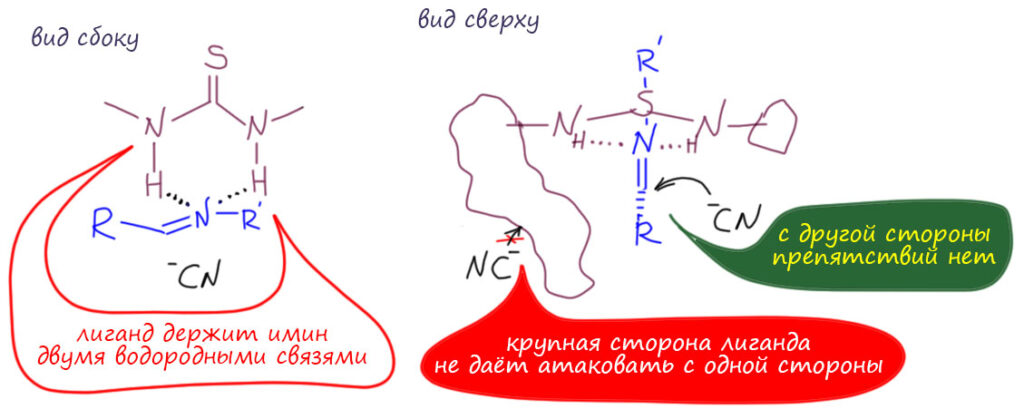
Смысл в том, что если мы смотрим просто на структурную формулу, нам кажется, что с двух сторон остатка тиомочевины болтаются какие-то рогатые штуковины, и мы не может сказать, какая из них крупнее. Но молекулярная модель показывает это очень ясно. Там даже дело не только в размере, а в том, как все это расположено относительно того места, где будет беспомощьно болтаться имин, ожидающий инъекции цианида. Вот та циклогексановая прокладка, которая на первый взгляд кажется какой-то дурной вставкой, типа, ничего другого под руками не нашлось – на самом деле делает большое дело – она направляет то, что там дальше, а там дальше довольно громоздкое ароматическое кольцо с двумя трет-бутильными вениками – прямо в сторону реакционного центра. И эта штука становится прямо щитом. А поскольку молекула в целом хиральна, а это значит , что рядом с этой молекулой право и лево различны, то способность перекрыть одно из направлений атаки нуклеофила и означает появление различия между двумя сторонами до этого плоской, симметричной молекулы имина, попавшего в когти к хиральному чудищу. Появляется дискриминация или дифференциация направлений. Это так и называют, кому как нравится – энантиодифференциацией или энантиодискриминацией, и это и есть причина того, что продукт будет оптически активным с высокой степенью оптической чистоты (до 90% и иногда больше).
А почему не сто? Там же нарисовано, что прям блокирует! Не бывает на 100%, какая-то часть всё-таки прорвётся, а возможно, что реакция неактивированного имина не до конца заморожена, отдельные молекулы еще шевелятся, или еще так может быть, что часть молекул имина ухвачена не жестко двумя когтями, а только одним когтем, второй не дотянулся, и так и болтается и крутится там на одном когде. Это только кажется пустой болтовнёй, на самом деле в энантиоселективных процессах это всегда очень важно – насколько тесно и прочно осуществляется взаимодействие хиральной молекулы, которая является источником асимметрии (осуществляет асимметрическую индукцию, переносит хиральность, обеспечивает энантиодискриминацию – все эти выражения используются) и прохиральной молекулы реагента, которая получит возможность в продукте приобрести хиральность. Если прочного и тесного взаимодействия нет, то почти всегда перенос хиральности неэффективен, оптические выходы невелики. Прочно и тесно – значит в нескольких местах, более прочными связями. Менее прочно и тесно – значит более слабыми связями, в одном месте. Но поскольку в химии все имеет статистическую природу и определяется равновесиями, то и два разных способа могут осуществляться одновременно, с разными вероятностями – один путь высокоселективный и ещё десять путей низкоселективных. И результирующая селективность (оптический выход) определяется конкуренцией всех путей – столько-то процентов по первому, столько-то по второму и так далее. Задача исследователя – оптимизировать, подобрать условия наилучшие для высокоселективного пути, чтобы он выиграл конкуренцию, а остальные сжались бы до нескольких процентов. Но не до нуля, так не бывает, реакции не бывают абсолютно селективными.
Почти одновременно с Джейкобсеном похожую работу сделал Кори, и сделал, как ему и положено, красиво. Катализатор какой – не молекула, а загляденье! Это такое хиральное производное гуанидина, а гуанидин это довольно сильное основание, так что такая штука во-первых, сразу депротонирует HCN и живет поэтому в этой реакции в виде дикатиона. Дальше она как-то подхватывает водородной связью имин, активирует его и такая форма присоединяет цианид. Тоже нужен холод по той же причине.
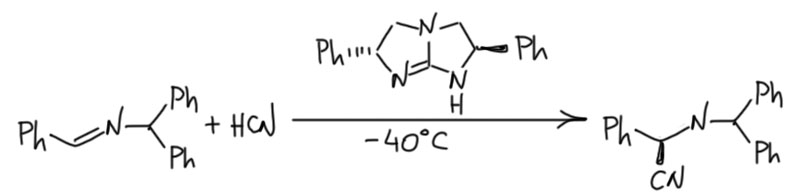
Красивая работа? Да. Кори веников не вяжет (в нашем смысле это значит, что он не пихает по полдюжины трет-бутилов во все места как некоторые). Но из неё видно, что Кори к концу прошлого века отстал от современной химии. Он по-прежнему может сделать красиво, но не может сделать из этого современную работу. Это такая единичная находка, скорее всего сделанная по старому – посидели, подумали, придумали красивую вещь, синтезировали, попробовали, что-то получилось, написали маленькую статейку в новый журнал (Corey, E. J.; Grogan, M. J. Org. Lett. 1999, 1, 157) – нетрудно представить, как редактор журнала упрашивал великого синтетика найти что-нибудь для самого первого номера, и упросил. А как это работает и что дальше? Работает это, видимо, благодаря тому что имин связывается не только водородной связью, но и через ароматические кольца так называемым стекингом, поэтому обязательно в имине должен быть специфический заместитель дифенилметил (бензгидрил), без него оптический выход падает, одной водородной связи не хватает для закрепления. Кори в этот момент и так достиг величия, ему не нужна нобелевка, у него уже есть одна – единственная химическая нобелевка, присужденная не за реакции, методы или теорию, а за красивый трёп, все эти синтоны, ретроны, транспозоны (ой, это, кажется не из этой оперы, но я не уверен). Впрочем, Кори был настолько могучим химиком, что сделал всего на несколько нобелевок, и Нобелевский комитет про это знал, но ему надо в объявлении написать какое-то одно важное достижение. Так что не важно, всё справедливо. И важно то, что химия изменилась.
Работа Кори так и осталась одной короткой статьёй, а сделанное уже на новом уровне исследование Джейкобсена, наделало гораздо больше шума. И сам он, и последователи очень здорово раскрутили хиральные мочевины и тиомочевины как универсальные катализаторы для реакций, где нужна активация водородными связями (общий кислотный катализ). Это стали применять и в промышленных масштабах, французская фармацевтическая компания Rhodia запустила на этой основе производство синтетических хиральных аминокислот, прикупив специально для этого подразделение в Америке. А в новую область – энантиоселективный катализ хиральными кислотами Бренстеда-Лоури – повалил народ несметными толпами и уже через несколько лет там хватало материала на хорошую книгу. Катализаторов этого типа было найдено множество, среди них были и новые соединения, и хорошо известные природные, например, алкалоиды хинолинового ряда, типа цинхонина и хинина.
И этот вид катализа довольно быстро стал считаться разделом органокатализа, и это может выглядеть полной беспринципностью, ведь начинали-то с того, что органокатализ требует ковалентных связей. Но мысль пошла по очень понятному руслу, именно потому что это энантиоселективный катализ. Ведь мы уже поняли, что энантиоселективность возникает тогда, когда взаимодействие катализатор-субстрат достаточно прочно и определенно, чтобы могла быть зафиксирована конфигурация интермедиата. Сначала думали, что кислотно-основных взаимодействий Бренстеда-Лоури для этого недостаточно. Но на рубеже веков поняли, что это не так – не просто достаточно, а и ничем не уступает. И даже выигрывает, потому что не нужно после реакции гидролизовать первичный продукт, чтобы высвободить катализатор, а это далеко не всегда идет быстро и количественно. У водородных связей вообще такой проблемы нет. Ну и тогда решили, а зачем нам ограничиваться – давайте, если есть энантиоселективность, тоже числить это по ведомству органокатализа. Победителей не судят. Получился отличный образец поведения, которое товарищ Ленин называл оппортунизмом, то есть манеры менять убеждения в зависимости от обстоятельств. Товарищ Ленин был самым большим мастером этого жанра, поэтому люто ненавидел всех, кто практиковал то же самое, но не достигал его уровня мастерства. Что бы сказал товарищ Ленин про этих современных мастеров, которые одну и ту же реакцию объявят кислотно-основным катализом, если она не дает энантиоселективности, и энантиоселективным органокатализом – если даёт. Я думаю, что он бы похвалил их и сказал бы своё любимое: “Правильной дорогой идёте, товагищи!” Потому что такой финт действительно заслуживает восхищения. Обратите еще внимание на то, что катализ хиральными кислотами Льюиса никогда не причисляют к органокатализу, потому что жрецам культа органокатализа очень важно внушать своей пастве отвратительность металлов в любом виде, и то, что спасение – в категорическом отказе от металлов, то есть в органокатализе.
Ключевая работа Джейкобсена, с которой начинается органокатализ без ковалентных связей, как видим, вышла до первых работ Листа и Макмиллана, да и дальше Джейкобсен не лежал на печи, а мощно и плодотворно работал – это точно один из ключевых персонажей этой области. Пожалуй, если есть несправедливость в выборе лауреатов, то это пренебрежение этим ученым, вполне мог бы быть третьим, а может быть и первым. Даже, наверное, должен был быть. Ведь мы сейчас увидим, что нековалентный органокатализ стал одним из важнейших направлений, более того, возможно, это более плодотворная вещь, чем классика Листа-Макмиллана, которую они сами же быстро забросили. Но вот что он точно не умел делать так, как два лауреата – это подавать свои работы, разводить хайп, из любой реакции раздувать концепцию, красиво рисовать картинки, публиковаться в Nature и Science, ну и всё остальное. Не в упрёк, каждому своё. Ведь правильной же дорогой пошли товарищи, той, что ведёт посуху, да в Стокгольм.
Ну и напоследок посмотрим, куда занесло уже Листа, когда его пролин малость забуксовал, а идея про то, что нековалентные взаимодействия могут работать не хуже, овладела уже им.
ACDC
Первые находки Листа и Макмиллана, из которых быстро выросла новая область катализа, их самих занимала не так долго. Множество других исследователей занялись развитием области, поиском новых органокатализаторов, и необычайно в этом преуспели. До 2010 года вышло минимум три монографии про энантиоселективный органокатализ, каждая с многими сотнями ссылок. Но основоположники были уже довольно далеко. Ни у того, ни у другого практически нет работ по органокатализу в первоначальном смысле этого слова после 2010 года. Макмиллан очень увлекся другим новомодным видом катализа – фотокатализом, и стал в этой области одним из главных светил. Увы, вторую нобелевку ему не дадут, не положено. А еще он очень полюбил сочетать разные виды катализа, комбинировать их в сложных процессах, и сам не заметил, как стал очень широко использовать металлы. Побег от металлов у Макмиллана, прямо скажем, не вышел. Медь, никель, палладий, рутений уже давно в разных видах не вылезают из статей Макмиллана. И правильно – ученый уровня нобелевской премии не может быть догматиком. А Макмиллан, судя по всей его карьере, человек заводной, по-хорошему жадный – влезет везде, куда сможет, и кажется, он решил оставить следы во всех видах катализа, которые только существуют. Не удивлюсь, если скоро он полезет в гетерогенный.
Лист от органокатализа ушел не так далеко, но тоже нашел интересный способ развития. Лист намного спокойнее экспансивного Макмиллана, он предпочел застолбить интересную идею, и явно поставил задачу довести ее до абсурда. А поскольку идея с самого начала была довольно абсурдной, то пришлось из малого абсурда попробовать сделать немалый. Но это интересная работа, хотя её практическую значимость ещё придётся доказать, и возможно. из этого ничего не получится, уж больно несуразно огромны и сложны его последние катализаторы.
Лист заметил одну работу, в которой были получаны доказательства того, что иногда перенос хиральности, причём высокоэффективный, может произойти из совсем неожиданного места – из противоиона. Какого противоиона? Во-первых, хирального. Представьте себе реакцию, в которой участвует прохиральный субстрат и нехиральный реагент, но в виде иона. И у этого нехирального иона есть хиральный противоион (анион – катион, всё как положено). И мы точно не ожидаем серьёзного переноса хиральности от противоиона, который в реакции участвует только в том смысле, что нужно же электронейтральность соблюдать. Мы вообще почти никогда не обращаем внимание на противоионы в реакциях, в которых участвуют ионные реагенты. Нс интересует реагент, а что там при нём, катионы натрия, лития, тетрабутиламмония, анионы перхлората, тетрафторбората, бисульфата, и т.д. – да кого это интересует. Электронейтральность в макро-смысле, всего объёма раствора, смеси – это очевидно. Потому что если не соблюдать электронейтральность, то Майкл Фарадей на кладбище Хайгейт начнет так вращаться в своём гробу, что вызванный этим ток индукции может погубить все человечество. Будем соблюдать. Но больше мы от противоионов ничего не ждём, обычно даже забываем про них, не хотим время тратить, не пишем даже над стрелками часто.
Но это далеко не всегда безобидно. Мы ведь в органике работаем в основном в органических растворителях, а это малополярные жидкости, в которых ионы не могут отойти друг от друга, обычно они составляют ионные пары. В органике очень много эффектов определяется именно противоионами и их поведением, всё связанное с нуклеофильностью, основностью, и это мы даже знаем и обсуждаем. С электрофильностью и кислотностью тоже, но это мы уже редко вспоминаем. А нельзя ли это использовать? То, что иногда что-то получается, обычно в тех случаях, когда в рекции используется большая хиральная кислота Бренстеда-Лоури, которая кого-то протонирует и уже катион участвует в реакции. Катион нехиральный, хиральна оставшийся от кислоты анион, но перенос хиральности иногда замечали. Замечали, но не делали из этого парада тщеславия, обособляя в отдельную стратегию (а может тактику? нет, органики ниже стратегии никогда не опускаются, даже когда делают что-то заведомо немудрёное). Но мы уже видели в этом очерке, что парад тщеславия вполне может быть очень продуктивен, если запускает новые исследования, и эти исследования дают хорошие, бесспорные результаты. Неисповедимы пути познания.
Но здесь нужно пояснение. Если мы что-то протонируем, то большой разницы с тем, что мы уже видели в катализе за счет водородных связей, может и не быть – ведь очень трудно разделить эти две ситуации, да даже если можно, то дело может оказаться в том, что протонированная форма реагента держится водородными связями за хиральный противоион. Нет, здесь нет новой стратегии. Нужно придумать нечто более бесспорное. Чтобы понять, что придумал Лист и что дало ему полное право назвать это новой стратегией, немного вернемся к классике и опять к альдольной конденсации. Да, Лист вернулся к самой главной альдольной конденсации – к реакции Мукайямы. Надо сказать, что все фокусы с органокатализом, иницированные Листом и его последователями, так и не смогли поколебать неколебимое – положение реакции Мукайямы в синтезе было и сталось недосягаемым. Вот мне очень интересно, посмели бы дать нобелевку за органокатализ, если бы Мукайяма не поторопился на встречу с духами предков три года назад.
С реакцией Мукайямы в последние 20 лет тоже произошло много интересного. Когда счет катализаторов для неё перешел на тысячи, и все металлы периодической системы хоть раз да отметились, вдруг обнаружили, что у неё часто бывает совершенно другой механизм, безметальный. И что этот механизм нехорош, потому что портит энатиоселективность. В каком-то смысле реакция сама себя катализирует. Дело в том, что катализаторы для реакции Мукайямы это обычно красивые комплексы металлов с огромными хиральными лигандами, но у металла должно остаться одно-два координационных места, чтобы цепляться ими за кислород карбонильного соединения. И чтобы эти места обеспечить, у металла оставляют один два аниона очень сильных кислот, которые легко уходят и оставляют координационное место. Эти анионы, обычно это трифлат, но бывают и перхлорат, гексафторантимонат и т.п., имеют нехорошую привычку в присутствии следов воды в реакционной смеси давать мизерные количества сильных кислот – обычный гидролиз соли металла с сильной кислотой, как в школе. Этих мизерных количеств достаточно, чтобы столь же мизерное количество силилового эфира енола расщепилось и в смеси образовался триметилсилил-чтототам, под чтототамом мы понимаем остаток той самой сильной кислоты, например, если был анион трифлата, то в смеси возникало мизерное количество триметилсилилтрифлата. И вот эти соединения кремния не так давно были идентифицированы как невероятно мощные катализаторы реакции Мукайямы, потому что они, оказывается, являются невероятно мощными кислотами Льюиса, причем такими, которые особо любят атом кислорода (это свойство принято называть оксофильностью), а почему, я довольно скоро планирую обсудить на отдельной страничке, потому что это имеет отношение к гипервалентности. И тогда всё, конец энантиоселективности, потому что хиральный лиганд остался на металле, а реакцию катализирует уже не металл, а весьма нехитрое производное кремния, в котором нет никакой хиральности, ни большой, ни маленькой. Причем этот путь настолько быстрее того, что катализируется кислотой Льюиса, что успешно с ним конкурирует, несмотря на мизерную концентрацию этого нечаянного катализатора. Такая конкуренция приводит к сильному ухудшению стереоселективности. Такие ситуации в энантиоселективном катализе иногда называют утечкой хиральности (chirality leak) – в стереоселективном процессе возникает такой обходной путь, по которому образование продукта избегает асимметрической индукции. В самых разных энантиоселективных каталитических процессах есть такие пути утечки, использующие самые разные врождённые дефекты катализатора (точнее, каталитической системы в целом), и это просто бич всего энантиоселективного катализа – только изобретут какой-нибудь красивый процесс и порадуются высоким оптическим выходам, как непременно выясниться, что стоит немного недосмотреть, как идиллия расплывается в мираж, находится путь утечки и оптические выходы падают. Особенно обидно, когда высокоселективную сисему открывают и публикуют, а по опубликованной методике воспроизвести высокие показатели в других лабораториях не получается, или реактивы не те, или руки кривее.
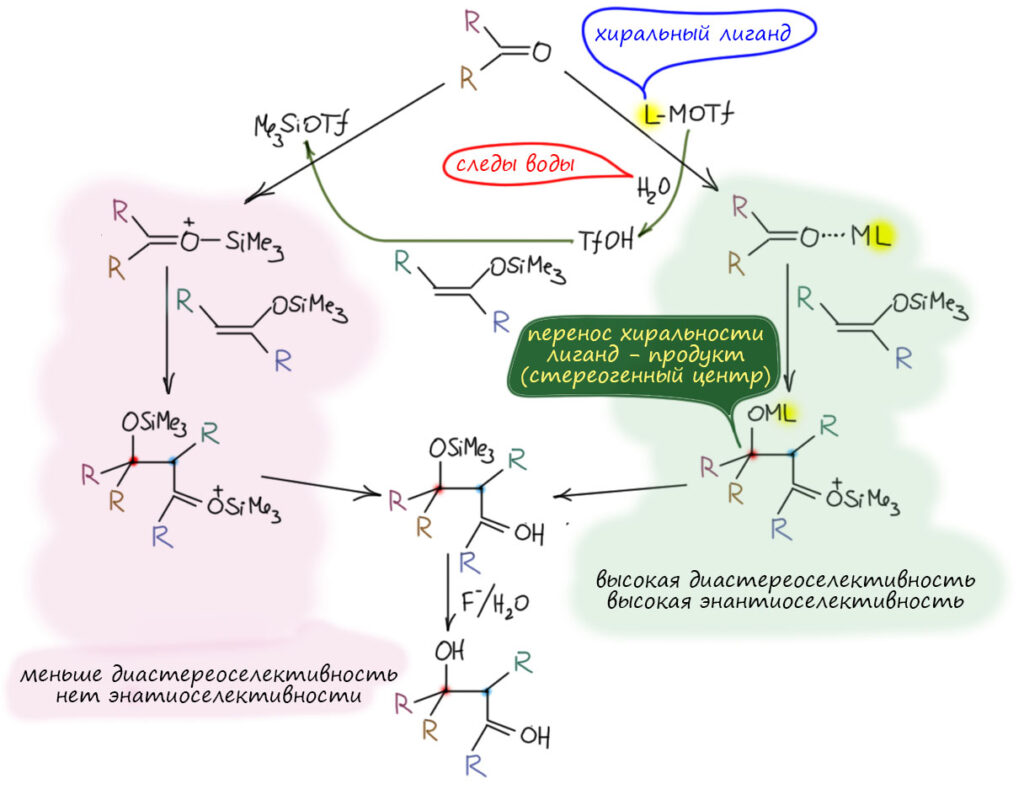
В общем, проблема. С ней удается бороться разными способами, или особой тщательностью эксперимента, или добавлением стерически затруднённых оснований, которые шальные протоны подбирают, а к основному кислотному катализатору не пристают.
А вот изобретатательный Лист, в середине 2010-х, видимо, решивший, что органокатализ как-то стал немного скучной рутиной и больше никого не возбуждает. Надо бы отмочить что-нибудь яркое, чтобы напомнить о себе всем, в том числе и шведскому королю Карлу XVI Густаву, потрясающему реликту европейской аристократии, который одновременно является и потомком наполеоновского маршала Жана Бернадота, одного из победителей битвы при Аустерлице; и по материнской линии ещё и королевы Виктории, а значит он являеся дальним родственником и российских императоров, одного из которых однажды победил его пра-пра-пра-прадедушка. Вполне музейный экспонат, уполномоченный вручать нобелевские премии. Я помянул короля потому что он противоречивостью своей династической истории, объединяющей правящие дома совершенно яростно враждовавших друг с другом государств, а никаких других достоинств у монархов обычно не бывает, – немного напоминает тот трюк, который Лист сейчас провернёт с одной из самых знаменитых реакций.
Лист придумал вот что. Уж если кремниевый катализ так враждует с металлокатализом и так мешает в альдольной конденсации Мукайямы, надо с ним не бороться, а заставить служить. Возможность такого маневра предоставилась в развитии важной работы японской лаборатории Хисаси Ямамото с сотрудниками (K. Ishihara, Y. Hiraiwa, H. Yamamoto, Synlett 2001, 1851–1854), крупнейших исследователей катализа специальными кислотами Бренстеда, которые обнаружили, что среди силилирующих катализаторов альдольной конденсации особенно эффективны соединения со связью азот-кремний, если возникающий в реакции анион на азоте сильно стабилизирован акцепторами; наилучшим таким реагентом оказался Tf2NSiMe3. Секрет таких реагентов прост – кремний предпочитает связи с кислородом и очень легко пересаживается на кислородные доноры, а имидный анион с двумя сильнейшими акцепторами становится отличной уходящей группой. Такой реагент катализирует альдольную конденсацию Мукайямы в очень маленьких количествах и при сильном охлаждении. Лист на основании этого придумал вот что: он спрятал дисульфоимидный реагент внутри хирального кармана, собранного на основе, наверное, самой знаменитой хиральной системы – 1,1′-динафтила. Эта штука существует в виде двух энантиомерных атропоизомеров, потому что два нафтила скручены друг относительно друга, а перевернуться кольцам мешают два атома водорода в положении 8. У одного такого энантиомера нижнее кольцо вывернуто вперед, у второго назад.
На ее основе сделаны знаменитейшие лиганды, в первую очередь BINAP, добывший в том же 2001 году нобелевку другому знаменитому японцу Рёдзи Ноёри. Тысячи энантиоселtктивных реакций всех типов обслуживаются катализаторами типа BINAP. Лист собрал на таком динафтиле дисульфонилимид с кремнием, и сверху и снизу прикрыл этот центр арильными группами – они так повернуты, что работают немного похоже на такие плоские щипцы, которыми на кухне удобно орудовать, подхватывая всякие ингркдиенты приготовляемых блюд. Но в отличие от щипцов захваты не находятся строго друг над другом, но они сдвинуты, один вправо, другой влево, и поэтому образуют совершенно идеальное хиральное окружение у того, что внутри – право и лево в такой системе не одинаковы. Дальше этот катализатор пускают в конденсацию Мукайямы, она переносит кремниевый остаток на карбонильный кислород карбонильной компоненты. И в этот момент возникает интереснейшая ситуация: во-первых, это ионная пара, и вся вот эта штука – хиральные щипцы становится противоионом-анионом к катиону, силилированному карбонильному соединению. И еще одна важная вещь – реакции делают в малополярных органических растворителях, в которых ионы далеко друг от друга отойти не могут, и это серьёзно. Катион вынужден искать свой анион, и для этого ему приходится залезть внутрь этого хирального кармана. Фантастика! – нехиральная молекула становится хиральной в виде ионной пары с хиральным анионом. В принципе, именно так работают ферменты, в том смысле что они тоже предоставляют такие хиральные карманы для связывания субстратов, но само связывание никогда не достигается только за счет электростатического взаимодействия. Дальше всё просто: реакция с эфиром енола, в которой возникает стереогенный центр, ну и кремний возвращается на азот, чтобы принять участие в следующем цикле.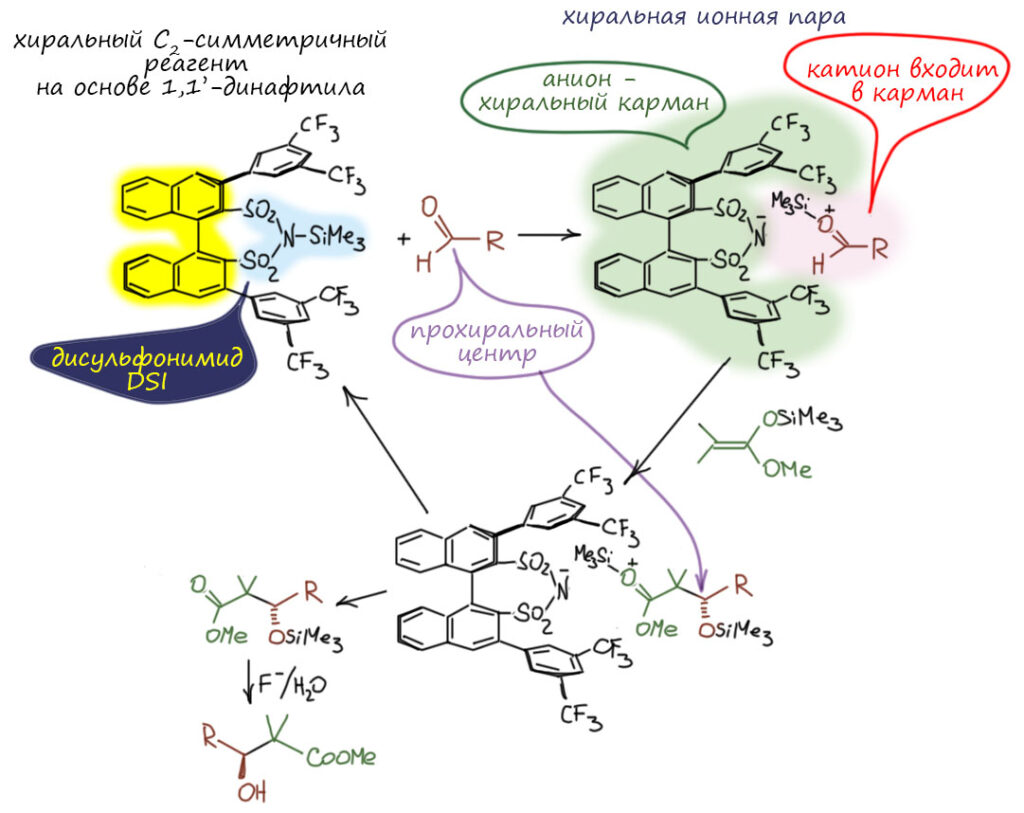
Катализатор оказался хорош, концепция заработала, осталось дать ей красивое название. Молодость Листа пришлась на 1980-е, и хорошее название, броский акроним, нашлось там. Суть метода – катализ, в котором ключевую роль выполняет противоион. Поэтому он и выбрал такое название для этого направления, на которое очень много поставил в последние несколько лет – Asymmetric Counterion Directed Catalysis, сокращенно ACDC. Лист, посвятивший жизнь в науке избавлению катализа от металлов, в обычной жизни, видимо, был большим поклонником хорошего металла в молодости. Я этот выбор не одобряю, я из поколения чуть постарше, и мы тогда довольно пренебрежительно относились к этой команде, считая ее легковесной профанацией настоящего рока типа Led Zeppelin, а AC/DC считали бездарной долбёжкой для тинейджеров. Вот ровно тинейджером тогда и был Лист. Впрочем, возможно Лист имел в виду банальный электрический выпрямитель, но это вряд ли.
Обозначив новую область, ACDC, Лист бросается наполять ее содержанием. Здесь ему пришлось попыхтеть в основном одному, такие штуки придумывать, это вам не пролин куда ни попадя пихать, на что желающих в своё время нашлось немедленно множество. И чтобы держать область ему пришлось буквально пуститься во все тяжкие. Это вообще частая проблема. Сначала делается красивая работа, придумывается, как здесь, красивый катализатор. Дальше его пытаются пристроить в другие реакции, да вот беда – всё остальное выходит хуже. Тогда приходится первоначальный замысел усложнять, и так шаг за шагом из первой, красивой вещи, выходит нечто монструозное. Оно работает, и получаются новые реакции. Разбирать не будем, хватит уже, но я просто нарисую два следующих поколения катализаторов из концепции ACDC.
Сначала такая динафтильная система была удвоена на остатке фосфорной кислоты, но через амидный мостик (я везде рисую структуры не как у Листа, а как мне нравится, с учетом гипервалентности фосфора, поэтому там много зарядов на атомах). Такое удвоение дает во-первых, увеличение размера кармана, в котором могут помещаться более крупные молекулы. Во-вторых, хоть на бумаге это не видно, но у такой молекулы очень затейливая форма, хирально организующая пространство внутри такой молекулы. Это всё больше и больше напоминает то, что происходит в ферментах, но молекула по-прежнему остается по сравнению с ферментами небольшой. Этот катализатор работает как кислотный, в тех реакциях, где требуется протонирование. Предполагается, что после протонирования тоже образуется ионная пара, катализатор становится анионом, и катион из реагента оказывается в большом хиральном кармане, где и происходит энантиоселективное превращение.
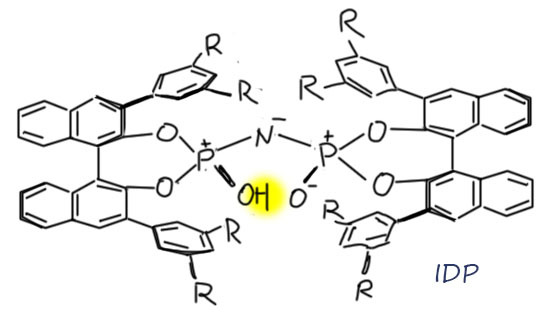
Одна из реакций, которую катализирует эта штука, это образование спиро-циклических ацеталей, точнее кеталей, а это очень симпатичные молекулы, распространенные в Природе, например, как сигнальные молекулы у насекомых. У спиро-соединений есть хиральность, причем для нее нужно меньше, чем для обычной энантиомерии на стереогенных центрах – просто чтобы в каждом из циклов было хотя бы одно отличие, которое уничтожит обе плоскости симметрии. Хиральность такого типа называется осевой, собственно это тот же самый тип хиральности как и у самой динафтильной системы, на которой Лист строит свои новые катализаторы. Но какая бы ни была хиральность, законы ее возникновения одинаковы, нет никакой специфики в том, как обращаются с ней обращаются и как она возникает в реакциях – точно так же нужен перенос хиральности от хиральной молекулы. Вот у этих спирокеталей как раз осевая хиральность. Насекомые отлично разбираются в энантиомерах, и бывает так, что самцам больше нравится один энантиомер. а самкам другой, впрочем это не значит, что насекомые делают чистые энантиомеры – они делают рацемат, а потом каждый насеком выбирает из смеси свой энантиомер. Вот, например, одна из таких молекул, олеан, служащая сигналом у маслинной мушки, которая в теплых странах портит оливки.
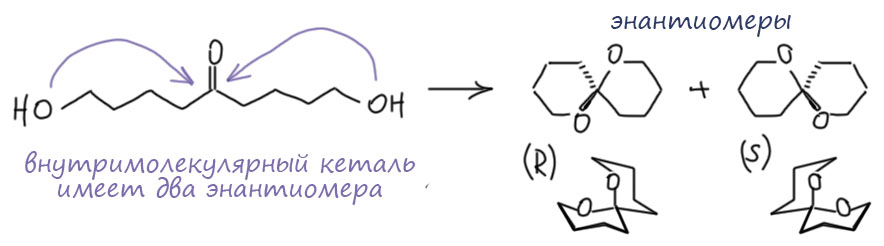
Вы когда-нибудь видели червивые оливки? Если видели, вто это она и есть, эта мушка и ее противные червячки-личинки, а какую красивую химию, сволочь, использует! Один из способов борьбы с такими наскомыми состоит в том, что делают ловушки с такими сигнальными веществами, феромонами. И насекомые туда летят и пропадают, например, прилипают на клей. Должны лететь. Не могу не заметить, что что-то с этим способом защиты растений, бешено популярным в конце прошлого века, – тогда просто все делали какие-нибудь феромоны, – пошло не так, и я всё меньше вижу, чтобы его реально применяли. То ли это слишком дорого, то ли эти феромоны слишком быстро выдыхаются, то ли еще что. Кроме того, Лист в своей статье нарисовал оба энатиомера этой очаровательной молекулы и не преминул написать сказку про то, что особи разного пола используют разные энантиомеры. Увы, это не так, а даже если бы и было так, то зачем бы это могло понадобиться – делать ловушки отдельно для самцов и для самок? Наверное, это бы очень одобрили талибы, если они, конечно, возделывают оливки посреди бескрайних полей опийного мака. Так или иначе, мотивация дурацкая, но химия красивая. Чистый энантиомер для этого делать не нужно, достаточно рацемата, но умение делать чистый энантиомер производит впечатление. Лист применил IDP-катализатор для энантиоселективного замыкания второго кетального цикла, стартовав из нехирального эфира енола. Получилось отлично, потянуло на Nature (I. Čorić, B. List, Nature 2012, 483, 315–319). Здесь работает тот же принцип: сильнодонорная двойная связь протонируется, образуется стабилизированный катион, который и замыкается, причем это происходит в чреве катализатора, где анион и катион образуют ионную пару. Если бы замыкание цикла происходило на просторе, где лево и право одинаковы, то кислород атаковал бы электрофильный углерод равновероятно с двух сторон цикла и образовался бы рацемат. Но в чреве IDP-катализатора, где катион удерживается электростатическим притяжением, довольно тесно, и что самое главное тесно по-разному слева и справа атака становится не равновероятной, и даже сильно неравновероятной, поэтому и образуется один энатиомер с высокой оптической чистотой более 90%. Что сказала по этому поводу маслинная мушка, Лист почему-то утаил, но, надеемся, она осталась довольна.
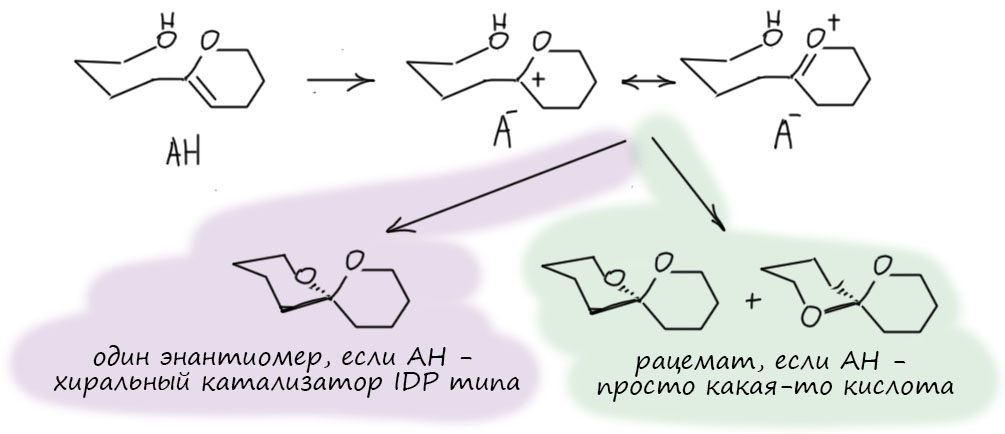
Ну и дальше дело пошло на следующие навороты – было сделано очередное семейство катализаторов, которые были названы IDPi, и это опять NH-кислоты, предназначенные для переноса силильной группы в альдольной конденсации Мукайямы. Они стали еще селективнее, получили возможность расширить синтетический диапазон и всё такое, что в этих случаях обычно говорят, и о чём можно прочитать в работах последних трех лет. Нелишне сказать, что то, что обозначено на этих структурах радикалом R из банальной трифторметильной группы превратилось во впечатляющие надстройки, так что вся молекула, если там ничего не сокращать, перестала помещаться в экран.
Оценим творческий путь Листа от начала до кануна нобелевки – начал с маленького и дешевого пролина и альдольной конденсации, причем это был новый тип альдольной конденсации, органокаталитический, и цель ее была уйти от конденсации Мукайямы, потому что в ней металлы, а металлы это нехорошо. Закончил (пока закончил, мы уверены что впереди много новых достижений) альдольной конденсацией Мукайямы, правда вариантом без металла, но зато с огромными и очень сложными катализаторами, привязка которых к органокатализу осуществляется с помощью концепции ACDC. В этом месте пришлось вмешаться шведскому королю, иначе дело могло принять уже совсем неожиданный оборот – было решено срочно дать нобелевскую премию.
Пара слов о лауреатах
Бенъямен (или просто Бен, как написано на сайте его института, и это тоже новое слово в немецкой науке, раньше не допускавшей панибратских сокращений имён авторитетов и начальников на американский манер) Лист – немец из Франкфурта из очень непростой семьи с долгой историей, среди родственников немало известных медиков, ученых и деятелей культуры. Но к великому венгерскому пианисту и композитору Ференцу Листу отношения не имеет никакого. Прадедушка Листа – химик Якоб Фольхард, которого мы знаем по реакции Гелля-Фольхарда-Зелинского, довольно немудрёной. Прадедушка гордился бы правнуком, если бы смог понять, что тот сделал. Химия проделала огромный путь всего за 150 лет, время жизни двух человек.
Лист начинал свою научную карьеру в США в институте Скриппса, очень мощном биомедицинском исследовательском центре в Калифорнии (оттуда же и лауреат этого года нобеля по медицине Ардем Патапутян, да и раньше там водились другие нобелиаты), и именно там Лист сделал первые и самые цитируемые свои работы по органокатализу. В 2003, как только почувствовал, что дело пошло и он зацепился за нечто жирное, Лист вернулся на родину в знаменитейший институт Исследования Угля общества Макса Планка. Этот институт – главный центр немецкой органической химии, появился еще перед первой мировой войной и тогда назывался институтом Исследования Угля кайзера Вильгельма. Именно угля, а не углерода и не органической химии. Это название красноречиво говорит о том, что корни этого института в главном угледобывающем и сталелитейном центре Германии, Северном Рейне-Вестфалии, в городе Мюльхайм на Руре, и создавали его для того, чтобы разрабатывать химию переработки угля в искусственное топливо, процесс Фишера-Тропша, необходимый Германии для того чтобы воевать за господство с остальным миром. У Германии для этого было много рвения, но мало нефти, нефть предполагалось добыть в сражениях, но немцы – люди предусмотрительные и на тот случай, если что-то пойдёт не так и было учреждено это научное учреждение. С мировым господством и топливом из угля, как известно, ничего хорошего не вышло, но зато институт получился выдающийся, и стал он заниматься органической химией в целом. Долгое время им руководил великий химик Карл Циглер, про которого я на этом сайте уже писал, нобелевский лауреат за открытие полимеризации Циглера-Натты, а мы его лучше знаем по аллильному бромированию, а кто полюбопытнее знает еще, что это один из трёх отцов литийорганики. После разгрома 1945 и всего, что было до этого, великая немецкая химия, некогда главнейшая в мире, давшая до 1940-х львиную долю всех нобелевских лауреатов, вышла сильно пришибленной, и ей пришлось очень долго выкарабкиваться. Еще 30-40 лет назад немецкая органика была весьма скромна, особенно по сравнению с достижениями прошлого. Причиной этого было не только последствия разгрома науки сначала нацистами и затем войной, но и сама природа немецкой науки, очень почитающей авторитетов, чины, заслуги и прочее, и поэтому сильно отставшей от гораздо более динамичной англо-американской. Тем же институтом Угля обычно десятилетиями руководил очередной выдающийся, ну вот так они и подъехали в концу 20 века, с большими достижениями, в основном в прошлом, но малость заплесневевши. В конце прошлого века институтом долго руководил выдающийся химик Манфред Рец. Вот он-то и решил, что пора догонять и возвращать немецкой органической химии былую славу и мощь. И начинать надо со своего знаменитого института. И надо не выпендриваться, а посмотреть, как там у них, в Америке и Англии это делается, раз уж раз за разом нобелевские премии пролетают мимо носа за проливы, моря и океаны.
Ну, в общем, очень просто. Надо первым делом упразднить единоличное руководство, нанять на должности административных руководителей не ученых, а нормальных современных менеджеров, а научное руководство разделить по направлениям и поручить пяти молодым и жаждущим славы и достижений директорам, активно работающим в науке. И нашел он такую команду, что только держись. Металлорганикой, например, стал заведовать швейцарец Алоиз Фюрстнер, невероятно плодовитый и творческий химик последнего 20-летия. Не удивлюсь, если он тоже вырулит на нобелевку в следующие 10 лет, потому что как мало кто умеет делать блестящие работы и красиво их подавать.
А катализом позвал заведовать Листа. В великом институте, гордости великой немецкой науки, выбрать совсем молодого человека (35 лет), непатриотично свалившего в Штаты (своих что ли рядом не было), да и в общем пока только написавшего несколько статей по еще не очень понятной, но с виду какой-то простоватой химии. Лист согласился (вряд ли можно было возразить патриарху немецкой химии) и вернулся на родину, и с тех пор так и работает в знаменитом институте, ну и мы знаем, что Рец не ошибся. Правда Лист чуть не подвел старика – отправился на радостях и чтобы как следует зарядиться перед большой работой почти сразу после назначения с семьёй в Таиланд и попал там в жутчайшее цунами (поинтересуйтесь в сети про цунами 2004 года – это совершенно апокалиптическая трагедия, в которой погибло очень много людей в нескольких странах Юго-Восточной Азии, в том числе множество туристов в Таиланде). Всю семью Листа и его самого смыло огромной волной от бассейна, разметало по всему Таиланду, но чудом никто из семьи не погиб, хотя младшего сына доставали практически с того света.
Дальше была только работа, достижения и успех. Да и вся немецкая наука очень оживилась в новом веке, реформы помогли, и мы видим все больше первоклассной современной химии из Германии. Лист – первый современный нобелевский лауреат по органике из Германии. До войны Германия дала больше половины всех нобелевских лауреатов, и органика была на одном из первых мест, вспомнить хоть сразу двух Фишеров, Эмиля и Ганса. После войны немцам дали еще несколько нобелекок, но это были немцы старой, довоенной школы: Карл Циглер, Отто Дильс (это вообще персонаж из 19 века) и Курт Альдер, Георг Виттиг. И только один молодой, третий Фишер, Эрнст, но это все же не органика, а металлоорганика. И всё, почти на полвека. Оскудела земля немецкая нобелиатами по органике. И вот случилось, Германия снова рулит в органической химии!
Лист обладает очень необычной внешнстью – он похож скорее не на профессора, а или на актера с апмплуа злодеев, или на футболиста, тренера или полузащитника. И судя по огромному количеству фото в сети, любит фотографироваться в острых ракурсах, выявляющих яркую фактуру невероятно выразительного лица. В общем, звезда во всех отношениях. Ученый нового типа – любит успех и славу, умеет это создавать и поддерживать, при этом оставаясь отличным исследователем. Один из забавных штрихов, выдающих характер – всегда раньше ставил себя впереди в списке авторов, и не забывал ставить звездочку, даже если в списке был один. Много раз уже тут писал – скромность ученого не украшает, и Лист очень хорошо показывает справедливость этого принципа. Но в последние 10 лет остепенился и стал следовать обычным нормам этики – начальник последний, он.
Его коллега по премии Дэвид МакМиллан, моложе Листа на 2 месяца. Он шотландец, из простой семьи из рабочего городка в Шотландии. Уверяет, что простое шотландское детство закалило его характер и дало ему ощущение важности в любой ситуации добиваться поставленной цели. Между строк ясно читается – иначе из этого захолустья не выберешься. Выбрался, выучился и уехал в Штаты. Ученик Ларри Овермана, одного из главных синтетиков прошлого века, виртуоза стереоселективного синтеза, и судя по работам, не последний ученик. И как только зацепился за свою науку, работает почти все время в одном месте, в Принстоне. Все, кто интересуется современными достижениями в катализе не могли пройти мимо его работ – он чрезвычайно плодовит и при этом разносторонен, все время что-то изобретает, часто методом высасывания из пальца, иной раз просто возмутительного по наглости и малообоснованности. Но мало кто не согласиться с тем, что он очень яркий химик, всё время ищущий новое. Его невозможно представить копающимся в одной теме много лет. Последнее десятилетие он больше интересуется не органокатализом, а новомодным фотокатализом, в котором тоже уже напридумывал кучу всякой чуши, от которой меня просто натурально колотит, но это мои проблемы, а работы интересные, их интересно читать просто подряд. Кстати, тоже поучительно – не бойтесь выдумывать, пробовать разное, не надо киснуть в одной теме. Не бойтесь ошибаться – если вы работаете в конкурентной науке, найдётся кому поправить, и это не страшно, если, конечно, вам действительно есть, что сказать в науке. Времена, когда люди десятилетиями копались в одной реакции безвозвратно прошли. А робким больше не дают нобелевских премий.