Гетероциклические соединения: методы и задачи.
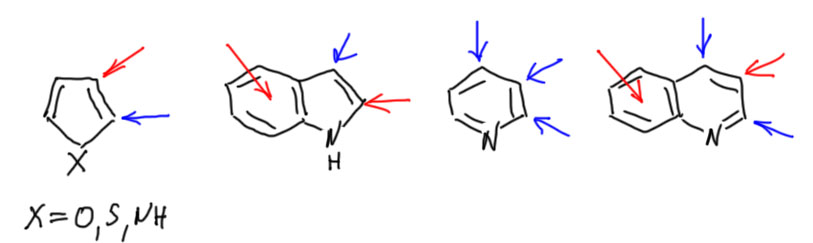
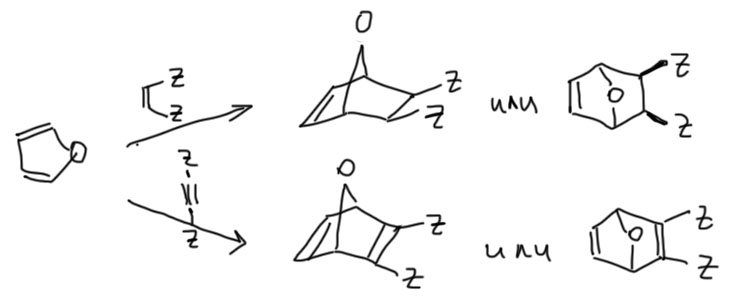
Основная сложность при решении задач из химии гетероциклов – распознать, откуда появились заместители в гетероцикле. Возможных вариантов два – их приделали к уже готовому гетероциклу (назовем этот подход модификацией), или они появились в результате синтеза гетероциклической системы из нециклических исходных. Вот как выглядит самая простая рекомендация для шести гетероциклов, которые мы реально изучаем. Красная стрелка означает – только синтез – группа карточек под заголовком Синтез гетероциклов. Синяя стрелка – в первую очередь модификация, но возможен и синтез.
Последнее обновление:
21 мая 2021 По следам последней контрольной – про прямое литирование пиридина, и почему не надо этого делать, хотя про это и написано в книжке Джоуля-Миллса. И про бромирование тиофена.
7 мая 2020: Добавлено нитрование пиридинов и особые свойства фуранов
Новые C-C связи
Вводим углеродные заместители в готовые гетероциклы
- Выбираем...
- В фуран и тиофен
- В пиррол
- В индол
- В пиридин, положение 3
- Пиридин, положение 2
- Пиридин, положение 4
- Хинолин, положения 2 и 4
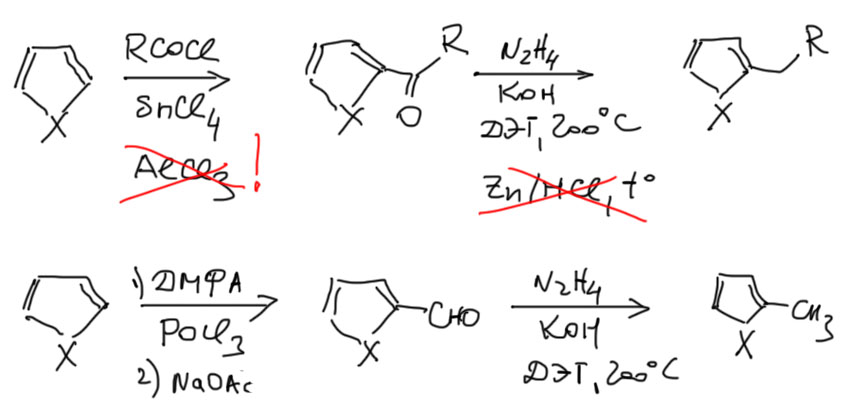
Если у нас есть готовый фуран, тиофен, пиррол, индол, пиридин, хинолин, то мы можем ввести в эти гетероциклы разные углеродные заместители – алкилы, ацилы, карбоксил и т.п в места, обозначенные синими стрелками в схеме во введении. Ни в коем случае не делайте это из общих соображений, используя уже известные методы ароматической химии. У каждого типа гетероциклов свой норов, и ошибок они не прощают.
1. Ацилированием хлорангидридами кислот, но ни в коем случае не используйте AlCl3! Для большинства целей годится хлорное олово. Кроме ацилирования, еще полезно формилирование по методу Вильсмайера-Хаака. Продукты ацилирования и формилирования легко превращаются в алкильные производные восстановлением по Кижнеру-Вольфу-Хуану. Не используйте метод Клемменсена – особенно фураны не любят многочасового кипячения с сильной кислотой и осмоляются без остатка.
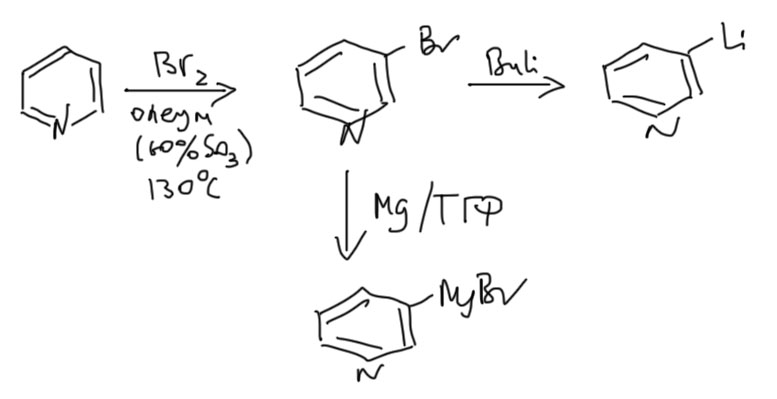
2. Через литиевые производные, котрые получаются прямым литированием. Дальше делаем все, что можно сделать с литиевыми производными – присоединяем к карбонильной группе, или делаем купрат и его используем.
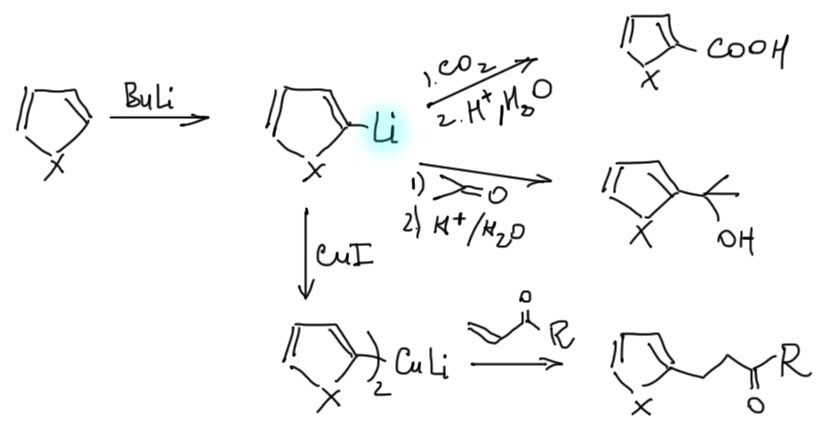
3. Через Гриньяр, который можно получить из бромпроизводного (см. вкладку в разделе Модификация). Этот метод рекомендуется в последнюю очередь, так как его легко заменить прямым литированием, а для дальнейшего синтеза обычно мало разницы между Гриньяром и литий-органикой.
1. Пиррол можно так же, как и фуран и тиофен ацилировать и формилировать.
2. Если пиррол превратить в литиевую или магниевую соль, то их можно напрямую алкилировать (не забывайте про правила SN2!), ацилировать, вводить в присоединение по Михаэлю. Поведение солей с натрием в качестве противоиона смотрим в разделе Модификация.

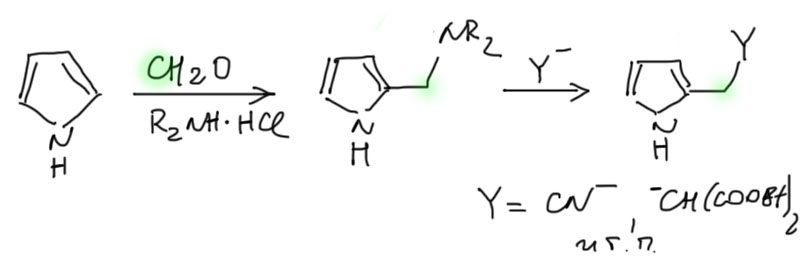
3. Очень донорный пиррол легко вступает в реакцию Манниха с формальдегидом и вторичным амином. Это очень полезная реакция, потому что продукт реакции Манниха легко ввести дальше в реакцию с нуклеофилами (обычно углеродными, такими как цианид-ион, всякие еноляты и т.п.) , причем уходящей группой является амин. Это немного противоречит тому, что мы до сего дня знали про замещение, но вот так устроен пиррол и с этим нужно смириться.
Химия индола практически идентична химии пиррола, с тем важным различием, что все то, что у пиррола идет в положение 2, в индоле идет только в положение 3. В остальном все одинаково – и Манних, и соли. Положение 2 у индолов недостижимо через модификацию и заместители в него попадают только через синтез.
Дальше бром переводят в магний или литий-органику, и шуруют обычными реакциями уже через них.
Нитро-группу можно восстановить до амина и применить обычную химию диазониевых солей. При прочих равных путь через бромирование гораздо лучше, прежде всего потому, что пиридин нитруется очень тяжело и с очень низкими выходами.
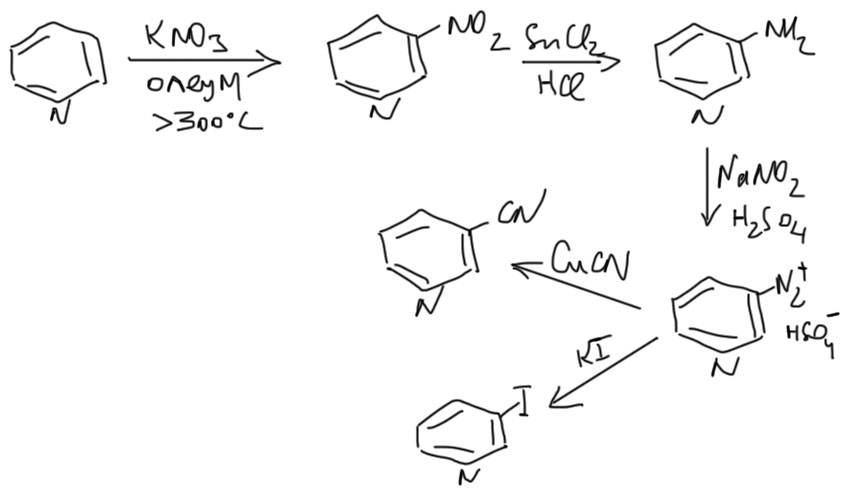
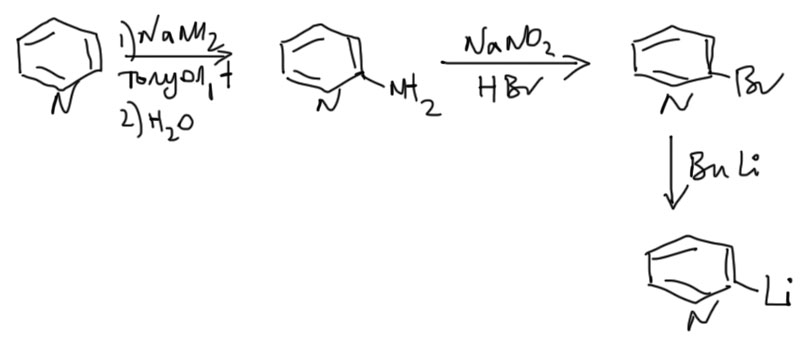
1. реакцией Чичибабина нагреванием с амидом натрия в толуоле. Образуется, после гидролиза реакционной смеси 2-аминопиридин. Далее его диазотируют и непосредственно, не фиксируясь на стадии соли диазония, которая очень неустойчива и немедленно разлагается присутствующими в смеси нуклеофилами. Так получают 2-бромпиридин, его переводят в литийорганическое соединение и далее как обычно с литий-органикой. Еще раз: при диазотировании аминопиридина не нужно писать соль диазония и отдельно ее реакцию с бромидом меди.
2. Реакцией пиридина с литийорганикой при нагревании. Внимание: пиридин не литируется. 2-литиопиридин получают только через бромид.
В этом случае путь всегда один – через N-оксид пиридина. Последовательно нитруем, удаляем оксид, восстанавливаем и переводим 4-аминопиридин в бромпроизводное точно так же, как для 2-изомера – не фиксируясь на соли диазония. Далее литиорганика и все остальное. Внимание: не пытайтесь что-либо изменить в этой последовательности вплоть до 4-бромпиридина – там нет места для импровизации.
Алкил-производные пиридина и хинолина в положениях 2 и 4 – метиленовые компоненты в реакциях альдольной и сложноэфирной конденсации и присоединения по Михаэлю
- Выбираем
- Аналогия с енолятами
- Алкилирование
- Альдольная конденсация
- Конденсация со сложными эфирами
- Присоединение по Михаэлю
- Винилпиридины и хинолины - акцепторы Михаэля
Синтез гетероциклов
- Выбираем
- Синтез пиррола, тиофена, фурана
- Синтез индола
- Синтез хинолина по Скраупу
- Синтез хинолинов по Дёбнеру - фон Миллеру
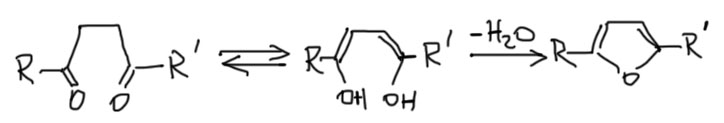
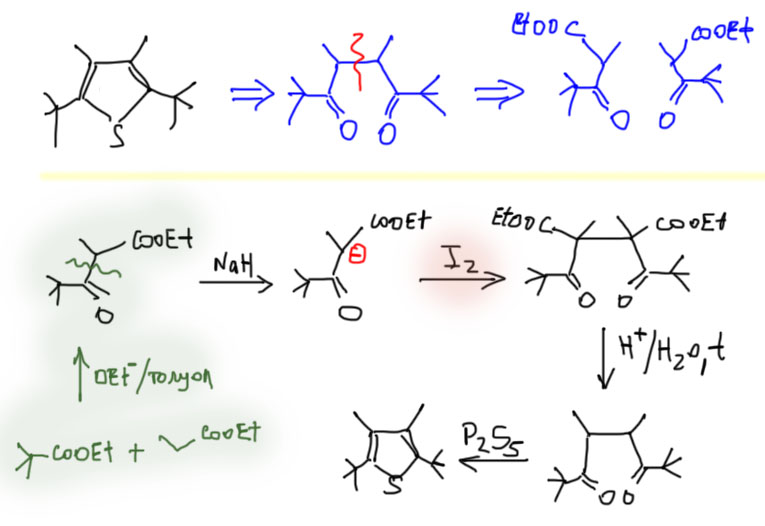
Интересно, что эта реакция обратима, и фураны довольно легко гидролизуются с образованием 1.4-дикарбонильных соединений, или их ацеталей, если реакция идет в спирте. Иногда этим пользуются для превращения фуранов в пирролы и тиофены.
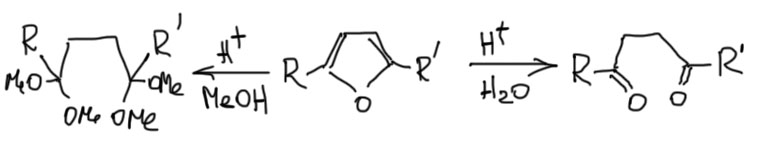
Тиофен в этом смысле просто серный аналог фурана, и получают его соответственно – действием сульфида фосфора на дикарбонильное соединение. Пиррол – двойной енамин того же дикарбонильного соединения? и получают его соответственно действием амина в присутствии небольшого количества уксусной кислоты для кислотного катализа. Если на азоте не нужен заместитель, берут ацетат аммония. Обратим внимание, что когда мы получали обычные енамины в химии альдегидов и кетонов, то удаляли воду, чтобы сместить равновесие. Пиррол – ароматическое соединение и пятичленный цикл, что вдвойне выгодно, поэтому он получается очень легко и удалять воду не нужно. В отличие от фурана циклизации с образованием тиофена и пиррола необратимы.
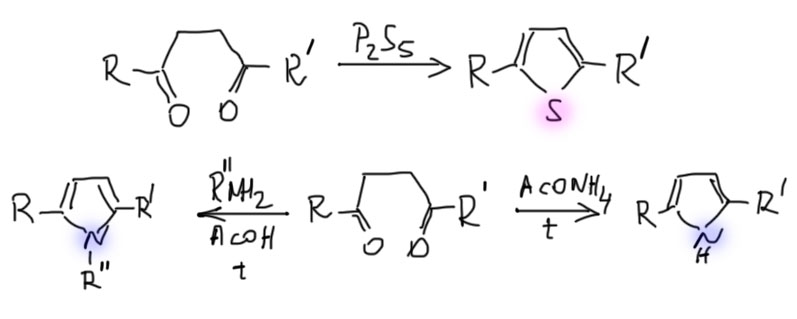
Задачи на синтез этих трех гетероциклов поэтому сводятся к синтезу 1,4-дикарбонильных соединений. Методов довольно много.
- Во-первых, всегда можно воспользоваться обычными методами синтеза кетонов из карбоновых кислот через хлорангидриды
- Во-вторых, можно воспользоваться обычной химией енолизуемых карбонильных соединений, распилив 1,4-дикетон пополам
Получается такая картина.
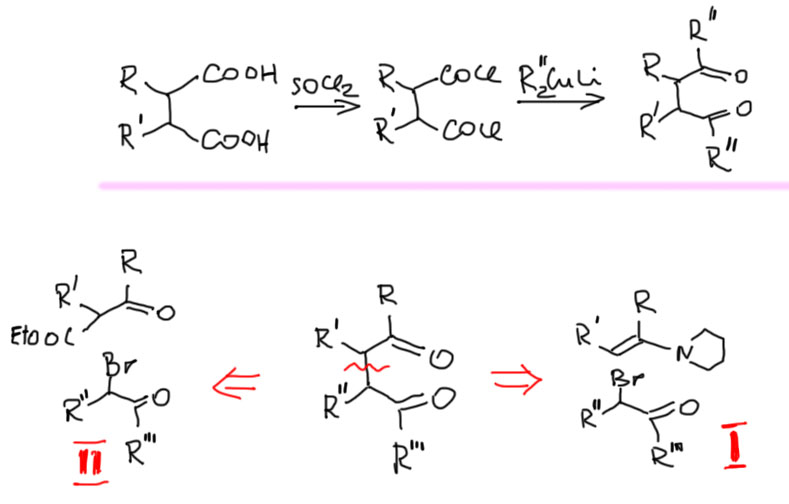
Два пути из распила пополам эквивалентны. Оба используют бромированный кетон в качестве электрофила. Нуклеофильная часть – это либо енамин, о котором мы уже порядком позабыли, или продукт сложноэфирной конденсации (β-кетоэфир), а это малость посвежее. На этом и остановимся. Попробуем на примере. Сложность в том, что распил можно сделать двумя разными способами, поэтому нужно смотреть, что дает более очевидные исходные. В данном примере распил в одну сторону дает ацетоуксусный эфир, очень доступное соединение. На нем и остановимся. Освежите в памяти методы работы с такими кетоэфирами, в частности то, как легко удаляется карбоксильная группа после кислотного гидролиза.
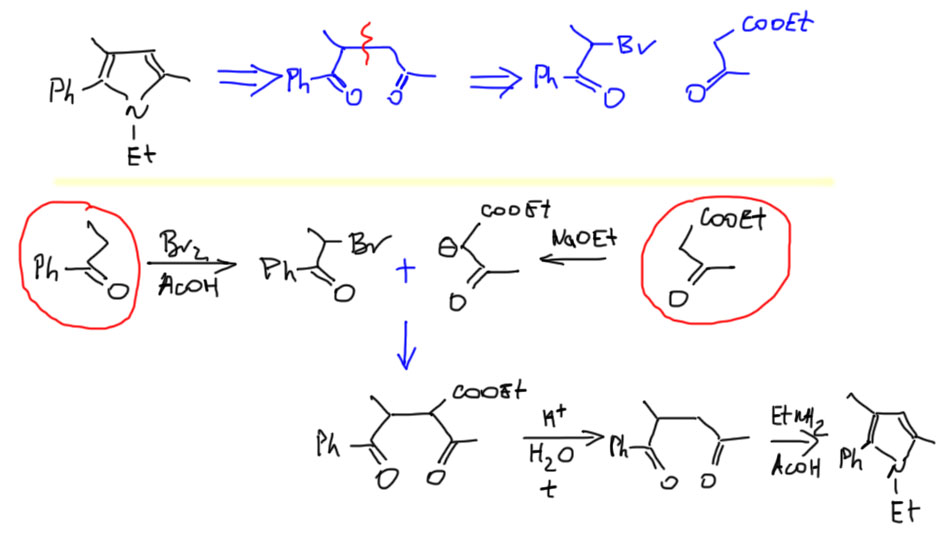
Отдельная история – случай, когда пиррол, тиофен, или фуран симметричные. Тогда их собирают из двух одинаковых половинок, тех же кетоэфиров, но через окислительное сдваивание енолята. Кетоэфир по необходимости получают сложноэфирной конденсацией. Вот пример:
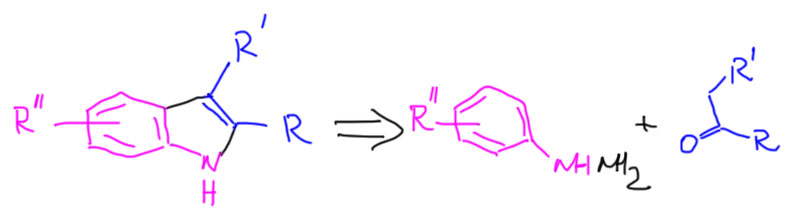
Фенилгидразины получаются из анилинов через диазотирование и восстановление соли диазония бисульфитом натрия или хлористым оловом. Кетоны или альдегиды синтезируют всеми возможными способами. Для реакции необходимо, чтобы в кетоне или альдегиде рядом с карбонилом была группа CH2 или CH3. Если в кетоне такие с двух сторон, и кетон несимметричный, получим смесь индолов. В случае, когда с одной стороны метил, а с другой CH2, реакция в основном пойдет в сторону последнего. Реакция вызывается каталитическими количествами сильных кислот (обозначаем просто H+) или кислот Льюиса типа ZnCl2.
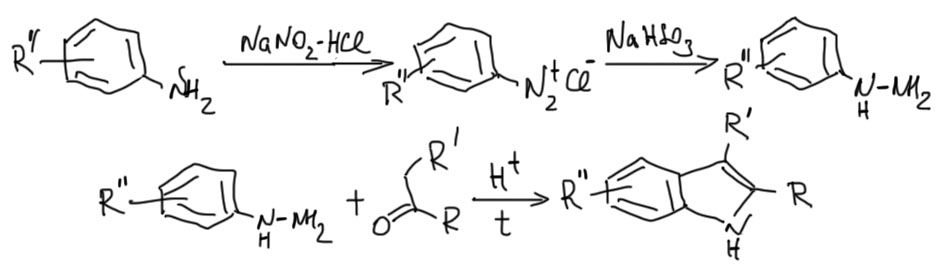

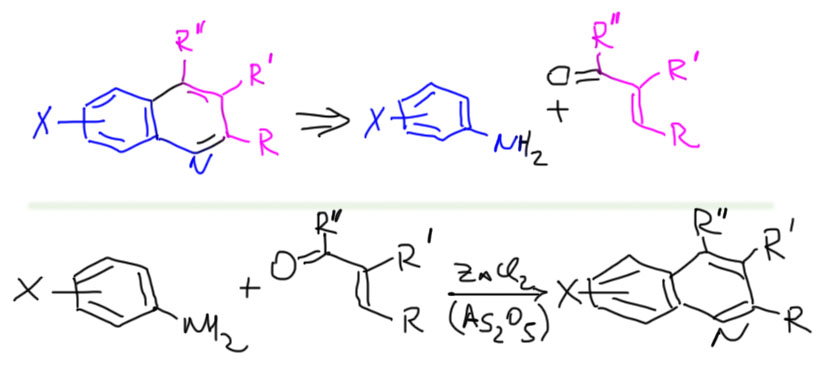
Метод, несмотря на жесткие условия и проблемы с окислителем очень гибок и удобен. С его помощью легко получают, например, все знаменитые хелатирующие лиганды – 8-оксихинолин, фенантролин. Метод вполне неплохо работает не только с анилинами, но и с аминопроизводными гетероциклов. Посмотрим на это в задачах.
Очевидно, что этот синтез сводится к синтезу непредельных карбонильных соединений, как правило, просто альдольно-кротоновой конденсацией.
Разные вспомогательные методы
В реакциях с диенофилами фуран – активный диен, очень похожий на циклопентадиен. Только фуран! Пиррол и тиофен в такие реакции не вступают.
Фуран и замещенные фураны с донорными заметителями кроме обычных ароматических реакций (электрофильного замещения) часто проявляют свойства диенов. Например, реагируют с некоторыми ковалентными электрофилами (например, бромом или ацетилнитратом) не как ароматическое соединение, а как сопряженный диен по образцу 1,4-присоединения. Но это довольно редкие случаи, не поддающиеся обобщению и превращению в работоспособный метод. А вот [2+4]-циклоприсоединение (реакция Дильса-Альдера) для фуранов очень характерна, и фураны являются одними из любимых диенов в синтезе. Внимание: ни пиррол, ни тиофен просто так в реакции Дильса-Альдера не реагируют даже с самыми страшными диенофилами. Рисуют аддукты фуранов точно так же, как аддукты циклопентадиена, то есть двумя способами, объемным и плоским. Рисуйте, как лучше получается. И, есди вспомните про такую примочку Дильса-Альдера, как эндо и экзо-стереохимия продукта, забудьте об этом в случае фуранов. Фураны очень странно себя ведут в этом отношении, потому что их реакции с диенами часто обратимы, и подчиняются не кинетическому контролю (а именно это является причиной образования эндо-продукта), а термодинамическому. И разбираться в этом нужно долго, и то только в том случае, если это нужно. Забудем и простим.
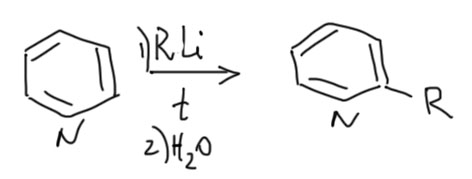
Еше раз напомню – пиридин и простые замещенные пиридины (для нас это значит – любые!) нельзя прямо литировать – то есть получать литиевые производные действием бутиллития (или другого простого литиорганического соединения, применяемого для прямого направленного литирования). Проблема в том, что пиридин активирован к нуклеофильной атаке на кольцо и очень быстро присоединяет литийорганику, а образующиеся аддукты легко превращаются в 2-алкил или 2-арилпиридины. Об этом кратко сказано на вкладке Введение заместителей в положение 2 пиридина.
2-литиопиридин поэтому получают обменом галогена на литий исходя из 2-галогенпиридинов, обычно 2-бромпиридина, который элементарно получается из 2-амиинопиридина.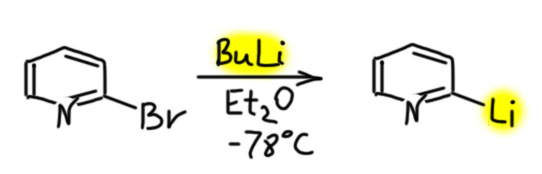
И всё. И больше говорить не о чем.
Но в процессе апеллирования на последней контрольной работе несколько человек таки написали прямое литирование пиридина, а на апелляции предъявили мне ссылку на известную книжку Джоуля и Миллса, где такое литирование описано. Действительно, например, в пятом оригинальном издании (Joule, Mills, Heterocyclic chemistry, Wiley, 2010) эта информация содержится на странице 135. В русском переводе это тоже наверняка есть, хотя он сделан с одного из предыдущих изданий этой замечательной книги.
В чём проблема сейчас разберёмся, но сначала один важный комментарий. Книжка Джоуля и Миллса – не учебник, это специальная монография для профессиональных химиков. Которыми вам еще предствоит стать после защиты диплома. В чём разница между учебником и специальной монографией? Она очень проста -в учебнике за текст отвечает автор(ы) учебника, которая/который/которые переработали материал, отобрали надежные, проверенные, особенно важные, особенно полезные, общеупотребительные, и так далее сведения, все это отсортировали и расклассифицировали – и предложили учащимся в виде надежных основ изучаемой науки. Писать учебники поэтому – чёртов труд и чёртова ответсвенность, и берутся за это очень-очень немногие. Да, не у всех получается одинаково хорошо, но это так всегда бывает. Важно то, что когда вы берете информацию из реукомендованного учебника – вы имеете полное право ссылаться на эту информацию, потому что вся ответственность за ее подбор и надежность на стороне авторов учебника и тех, кто его рекомендовал.
А вот специальная монография (монография – это просто книжка, содержание которой объединено одной темой, обозначенной в названии на титульном листе) это нечто совершенно иное. Монографии в тысячи раз толще и подробнее учебников, и кажутся такими верховными источниками знания. Ни фига подобного! Это заблуждение. Любая монография, кто бы ее ни написал – это просто собрание сведений, полное или неполное, которое ее автор/авторы/авторка/авторки решили туда включить, позаимствовав из оригинальных работ (статей в рецензируемых журналах) в том виде, как это там опубликовано. Авторы монографий никогда или почти никогда не отбирают материал, а в некоторых случаях это просто запрещено правилами научной этики – взялись обозревать область, извольте найти все существенное, что опубликовано, то есть всё, опубликованное в основном корпусе международных научных журналов (в наше время и уже очень давно все существенные журналы имеют статус международных). Не дело авторов монографии как-то дискриминировать чужие данные и результаты, сомневаться в их надёжности. Для пишущих монографии это часто доставляет много проблем, потому что автор иногда видит, что в статье написана какая-то фигня, но подвергать сомнению не имеет права. К науке более чем к какой-то другой деятельности применима пословица: “Что написано пером, то не вырубишь топором”. Вы можете возмутиться, но напрасно, потому что это никакая не трагедия. По очень простой причине – монографиями, в отличие от учебников, пользуются не студенты, а профессионалы, и пользуются для совершенно другой цели – это просто источник ссылок, удобно рассортированный по темам. Многие профессионалы вообще не читают монографии и обзоры (это отдельный вид научной статьи, практически идентичный по целям и способу изложения монографии), а просто быстро находят в них нужные ссылки, и сразу лезут туда – информация там, ответственность там. Другие читают, чтобы понять, что вообще делается в области, но когда находят то, что интересует лично их и нужно не для общего образования, а для работы – лезут в ссылки. Автор(ка, ы, ки) монографии не отвечают за информацию в своей монографии – они просто говорят – во, глядите-ка какую диковину мы тут обнаружили – поглядите, если понравилось, вот ссылка, разбирайтесь сами, может пригодится. Всё.
Вывод отсюда простой – использовать монографии для учебных целей не стоит, тем более, не стоит ссылаться на монографии. Просто потому что вы не сможете перевалить ответственность – просто не на кого. Естественно, если вам интересно и хочется узнать намного больше учебника, вы конечно будете читать монографии, и отлично. Читайте на здоровье. Но – в этом случае, если вы используете информацию из монографии, это ваша ответственность а) разобраться в написанном, понять, как применяется метод или реакция, какие у нее ограничения, почему она решает пробему и так далее; б) если вы решите использовать информацию при решении задачи или на экзамене, вам придется как можно точнее запомнить что конкретно сделано, и откуда вы это взяли. Никто, в том числе ваши преподаватели и профессора, не знают всего. Химия в 21 веке колоссальна и необозрима. Всякие детские считалочки про “а вы тут сидите и не знаете, что…, а ещё назвался профессором, доцентом, академиком, главным научным сотрудником и т.д., ненужное зачеркнуть…” оставьте для обсуждения сортов пива и содержания сериалов. И если вы неточно приводите такие данные, неправильно используете такой неучебный метод или реакцию, не знаете источника информации, извините, – это ваша проблема, не засчитывется, фтопку. Это так устроено, и по-другому это не работает.
А теперь разберемся что там написано у Джоуля и Миллса, и можно ли этим пользоваться. Проблема прямого литирования пиридина, строго говоря, не существует, потому что всех устраивает получение литиопиридинов через обмен литий-галоген. Но на то и исследователи, что бы исследовать. Это ведь довольно занятная проблема. Изучение реакции пиридина с литийорганикой показывает, что литирование идет, но как побочная реакция к присоединению. Присоединение просто быстрее. Отщепление протона далеко не всегда – быстрая реакция. Только тогда, когда основание имеет большой запас по pK. Если не имеет, то скрость переноса протона может быть очень маленькой, и проигрывать другой реакции. Но у реакций переноса протона (а прямое литирование – это именно такая реакция) есть одно очень важное свойство – их скорость намного меньше зависит от температуры, чем скрость более серьезных реакций, в которых образуютсяи рвутся связи между более серьезными атомами, чем атомы водорода. И если температуру снизить очень сильно, то все остальные реакции можно заморозить, а скорость депротонирования останется на приемлемом уровне. Мы отлично знаем этот прием и применяем его при получении енолятов действием LDA на енолизуемые карбонильные соединения, и всегда говорим, что ту реакцию обязательно проводят при низкой температуре – как раз для того, чтобы помешать побочным реакциям. Но – и тут нужно хорошо подумать: реакция просто с бутиллитием или другими литийорганическими соединениями все равно даже при низкой температуре остается в основном присоединением. Причина этого – сильная агрегация бутиллития, что сильно уменьшает доступность карбанионного углерода для отщепленя протона. Нуклеофильность все равно выигрывает у основности. Хорошо – тогда возьмем LDA. Увы, у LDA недостаточно основности – литирование пиридина происходит, но медленно, требуется большой избыток LDA, а это мешает проводить следующие реакции, ведь этот избыток никуда не денется, а это, мягко говоря, не самое инертное соединение.
Проблема зависла. Но в конце прошлого века возникла интересная идея – можно пробовать очень сильно изменять реакционную способность литий- (а также магний-, цинк- и др.) органических соединений с помощью добавок довольно простых реагентов, которые разрушают агрегаты и делают более доступными основные атомы углерода. Это всегда довольно простые вещи, или в основном соли лития, а иногда и соединений других щелочных металлов. Это очень интересная идея, которую стали применять многие исследователи, и это дало очень сильный импульс развитию методов прямого металлирования (литирования, магнезировани, цинкования и пр.). Здесь не будем это обсуждать, как-нибудь в другом месте. У каждого иследователя были свои рецепты таких смесей, для тех конкретных целей, которыми занимались конкретные люди. За пиридин взялся весьма знаменитый в конце прошлого века французский химик Поль Кобер, у которого появился свой рецепт смеси – к бутиллитию он добавил литиевую соль диметиламиноэтанола. Вполне понятная идея – диметиламино-группа является годным лигандом для катиона лития, и такая добавка способствует разрушению больших и ленивых агрегатов бутиллития. И даже более того, Кобер с сотрудниками полагали, что с помощью такой добавки получаются хитрые комплексы с участием пиридина, в которых к искомому протону подтягивают бутильный карбанион. Как-то так: 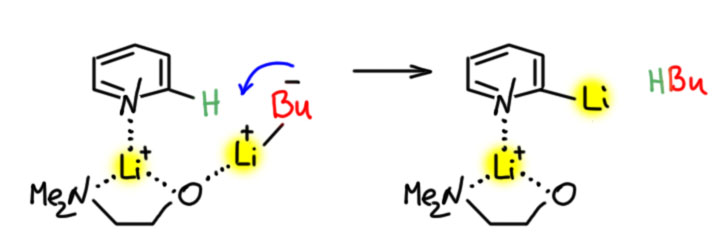
Не надо к таким картинкам относиться слишком серьёзно, это не точное изображение некоего комплекса, а просто принцип действия. Фокус в том, что катион лития очень маленький и очень простой – у него нет никаких валентных возможностей по причине чрезвычайной примитивности электронной стуктуры (пустая s-орбиталь и все), но за счет заряда и маленького размера, он как бешеный притягивает все отрицательное – бутильные анионы, эфирный растворитель, атомы кислорода или азота в соединениях, присутствующих в растворе. Когда ничего нет, катионы лития облеплены бутилами, у каждого бутила всой катион лития, поэтому получается такой комок из катионов лития и бутилов – агрегат, очень ленивый. Когда добавляют что-то типа того, что добавил Кобер, агрегат разваливается и бутилы становятся более доступными, сближаются с молекулой, от которой нужно отодрать протон. Ну и надо это как-то нарисовать.
Итак, такой смесью пиридин (и некоторые замещённые пиридины, например, 2-метоксипиридин) литируются в α-положение (P. Gros, Y. Fort, P. Caubere, J. Chem. Soc., Perkin Trans. 1
1997, 24, 3597). Цель достигнута, и именно это попало в книжку Джоуля и Миллса. Вот как это делают, и что нужно писать над стрелкой, если есть желание использовать метод. 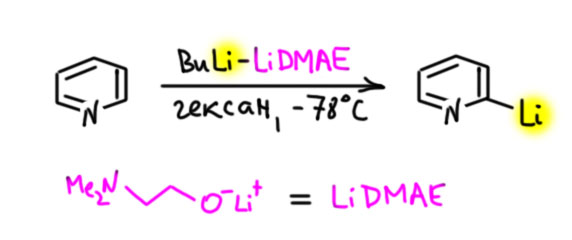
Здесь важно всё, и природа добавки, и растворитель, и температура. Реакция настолько капризна, что любое отклонение кончается крахом. Более того, обязательно нужен немаленький, более чем двукратный избыток були и добавки, этот избыток остается в реакционной смеси, поэтому то, что добавляют потом к литиопиридину, тоже нужно добавлять в большом избытке. Поэтому реакция откровенно неудобна. Решили сэкономить одну стадию по сравнению в получением литипиридина обменом, но получили намного менее удобный метод. Поэтому метод Кобера не прижился в синтезе, его никто не использует, плохой метод. Но с точки зрения свойств пиридина важный и интересный, потому что показывает причины странного поведения пиридина в реакциях металлирования. Для практических целей он, тем не менее, конечно же не рекомендуется.
Точно так же можно прямо литировать и хинолин, но там все еще хуже – избавиться от получения бутилхинолина не получается и всегда оббразуются смеси.
Вот поэтому использовать научную монографию в учебных целях не стоит. А для общего образования – вещь отличная, если разобраться.
Изменения реакционных центров (модификация)
Нуклеофильное замещение в пиридине и хинолине
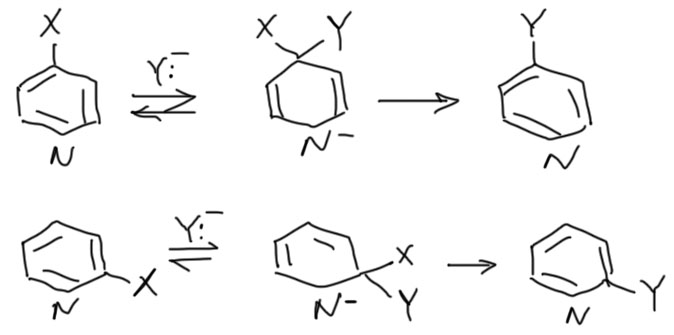
Электрофильное замещение в пятичленных гетероциклах
Важно помнить, что пятичленные гетероциклы не выносят сильных кислот, окислителей, и высокореакционноспособных кислот Льюиса. Применение хлорида алюминия для всей троицы – под абсолютным запретом, немедленно происходит полимеризация и частицы хлорида алюминия покрываются оболочкой мерзкой смолы, полностью блокирующей дальнейшую реакцию. Даже простые ароматические соединения типа бензола и толуола, которые очень часто содержат примеси тиофена, так как все это до сих пор получают из каменноугольной смолы, обязательно очищают от тиофена промывкой концентрированной серной кислотой перед использованием в реакциях Фриделя-Крафтса – иначе и там хлористый алюминий перестает работать.
И не покупайтесь на иногда встречающиеся примеры нитрования или ацилирования производных пятичленных гетероциклов в условиях, очень похожих на реакции обычной ароматики – если приглядитесь, всегда обнаружите в этих примерах дезактивирующие заместители (нитро-, формил, карбоксил, трифторметил и т.п. – такие заместители, как и положено, очень сильно снижают реакционную способность и пятичленных гетероциклов и позволяют немного ослабить бдительность. Это частные случаи! Никогда не обобщайте частные случаи на все множество!
Как уже ясно, нитрующая смесь не годится – там сильные кислоты. Если в молекулах тиофена или фурана уже есть акцепторные заместители, в таких случаях возможно нитрование нитрующей смесью при охлаждении, но нам это знать не обязательно, хотя именно так нитруют, например, фурфурол.
Штатная нитрующая система для донорных гетероциклов – смешанный ангидрид уксусной (или бензойной) и азотной кислот – ацетилнитрат (или бензоилнитрат). Почему это мягкий нитрующий агент? Потому что это ковалентные молекулы, не дающие катиона нитрония, но реагирующие по механизму нуклеофильного замещения на атоме азота, а ацетат (или бензоат) – довольно ленивые уходящие группы. В результате получаются достаточно слабые нитрующие агенты, не являющиеся сильными окислителями, и это именно то, что нужно. Получают их прямо на месте одним из двух способов:
- смешивая безводную азотную кислоту и избыток уксусного ангидрида (для фуранов и тиофенов) при комнатной температуре. Азотная кислота требуется именно безводная, 100%-ная (с плотностью 1.51), потому что в этом случае равновесие практически сдвинуто в сторону ацетилнитрата. Если взять обычную концентрированную с большим содержанием воды, получите более сложную смесь, в которой остается азотная кислота, и она постарается испортить ваш гетероцикл, а заодно и настроение;
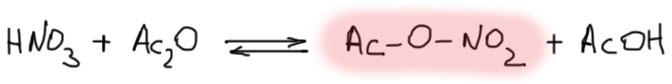
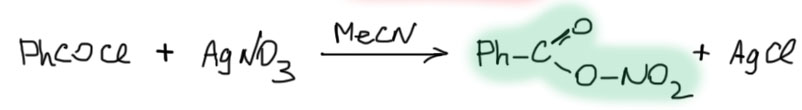
- реакцией ацетил- или бензоилхлорида с нитратом серебра в ацетонитриле. В этой реакции протонной кислоты вообще нет, и это лучше для пирролов, которые даже от тени азотной кислоты из первого метода могут окочуриться без шансов на выздоровление.

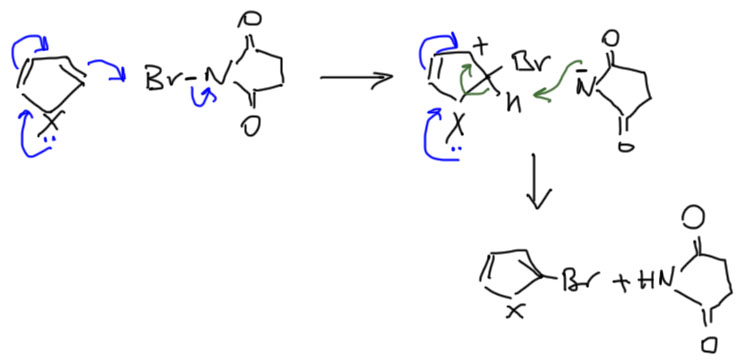
Даже для наименее реакционноспособного гетероцикла из троицы, тиофена, молекулярные галогены – перебор, получаются смеси ди- и тригалогенпроизводных. Пиррол моментально забирает все четыре галогена, даже иода. Тиофен из этой троицы – самый спокойный, и позволяет в точно подобранных условиях бромировать себя даже бромом, но не делайте этого, чтобы не запоминать детали частных методик. Относитесь к всем трем гетероциклам одинаково. Естественно, никаких кислот Льюиса тем более нельзя использовать. Для аккуратного моногалогенирования применяют очень слабые галогенирующие агенты, так называемые производные положительных галогенов, обычно это молекулы, содержащие связи галоген-азот. Остаток с азотом в этом случае работает как уходящая группа в реакции нуклеофильного замещения на атоме галогена.
Типичный реагент такого типа – N-бромсукцинимид (NBS). Мы хорошо знаем этот реагент и считаем его свободнорадикальным бромирующим агентом для аллильного или бензильного бромирования. Но это происходит только в случае свободнорадикального инициирования и только в неполярном растворителе типа CCl4. Сам по себе NBS в растворе, а он растворяется почти во всем кроме совсем неполярных растворителей, – именно электрофильный бромирующий агент, особенно в полярных растворителях (свободнорадикальные реакции с NBS всегда делают в неполярном растворителе четыреххлористом углероде, в котором сам этот реагент нерастворим, и реакция поэтому идет в растворе с малой концентрацией молекулярного брома – вспомните механизм аллильного бромирования в теме Алкены). При желании хлорировать или иодировать можно взять аналогичные реагенты NCS и NIS.
Комментарий к контрольной работе
В последней контрольной работе некоторые использовали странный метод – бромировали тиофен смесью брома и HBr в эфире. Я прямо удивился. Оказывается, это тоже из книжки Джоуля и Миллса. Но – не надо этого делать. Во-первых, посмотрите поточнее, что написано у Джоуля и Миллса – там обозначено дибромирование, а не монобромирование. Во-вторых, авторы плохо дали ссылки – в тех, что они дали ничего подобного нет. Я, право, удивился такой небрежности, обычно всё же книжки больших издательств (а это Wiley), и серьезных авторов делаются поаккуратнее. Если поискать немного получше, можно найти источник этой информации – это статья весьма мощного голландского химика Ламберта Брандсмы хотя и в дрянном журнале (Brandsma, L. Verkruijsse, H. D., Synth. Commun. , 1988, 18, 1763), где использована именно эта смесь, она дает очень быстрое трибромирование тиофена. Не монобромирование!
Здесь расчет именно на скорость – бромировать быстро и не дать тиофену успеть окочуриться. Очень сильная кислота, по идее, должна достичь именно этого, но поскольку смесь двухфазная, кислота и тиофен разведены по своим фазам, и трагического финала удается избежать. Довольно трудно понять, зачем здесь вообще HBr, ведь в реакции и так образуется много HBr. Я думаю, что для активации брома, увеличения электрофильности, а это в свою очередь ускоряет реакцию, и она просто быстро пролетает до трибромпроизводного, которому уже сам чёрт не брат, три галогена здорово дезактивируют ядро и делают его устойчивым к невзгодам сильной кислотности и проч. Так иногда делают особо смелые химики, когда видят чувствительные соединения – надо либо трястись над ними и бояться всего, либо сразу взять, да вдарить чем-нибудь посильнее в надежде, что реакция пойдет так быстро, что исходное не успеет понять, как над ним собираются надругаться. Такие фокусы редко получаются, но когда получаются, химия получает хорошую методику синтеза чего-нибудь сильно полезного. Вот это ровно такой случай. Так или иначе, это просто такая утилитарная методика, никак не общий метод, это ни в коем случае не надо применять к производным тиофена. Сам трибромтиофен очень важное вещество – из него получают восстановлением цинком 3-бромтиофен, и это самый простой способ попасть в химию положения 3 тиофенового кольца, и поэтому метод этот весьма востребован и популярен. 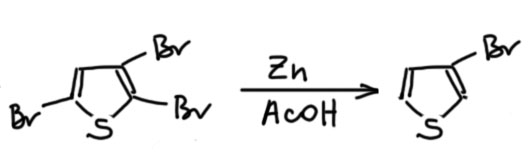
Есть, оказывается и ещё одна статья того же Брандсмы (M. A. Keegstra, L. Brandsma Synthesis 1988, 890) в более серьёзном журнале, специализирующимся именно на всяких препаративных методах, хороших и полезных методиках и тому подобных важных вещах, из которых и состоит органическая химия. В этой статье описаны как раз моно- и дибромирование тиофена с помощью тех же реагентов, но условия обозначены более точно, что в этом случае совершенно необходимо – эта методика получится только если ее точно воспроизвести. Монобромирование получается, если раствор брома в HBr прикапывать при хорошем охлаждении к смеси тиофена, HBr и эфира. Замысел этого метода читается легко – бром прикапывают, его концентрация в реакционной смеси поэтому всегда очень мала. И всегда есть огромный избыток HBr и эфир, а эфир обладает довольно высокой основностью. В таких условиях бром в смеси будет присутствовать в виде трибромид-аниона, намного более мягкого бромирующего агента по сравнению с самим молекулярным бромом. Низкая температура, перевод брома в трибромид, и огромный избыток тиофена (это условие выполняется автоматически, когда используется обратный порядок прибавления – реагент прибавляют к субстрату) вероятно, и делают бромирование селективным. Методика, скорее всего, вполне рабочая, авторитет Брандсмы в препаративной химии весьма велик, и лажей это быть не может. 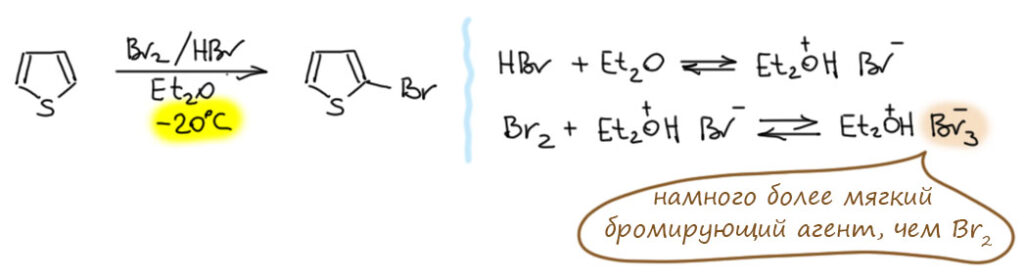
Для дибромирования делают то же, но брома берут два эквивалента, а температуру чуть-чуть повыше (-10ºС).
И еще раз повторю – не пользуйтесь в учебных целях такими частными методиками. В органике их несметное количество и никто не обязан все их знать. Вы с гарантией попадаете в ситуацию непонимания, а ссылки на книжки и тем более на Реаксис не работают, потому что в таких случаях это ваша обязанность – не только подцепить какую-то нестандартную химию, но точно запомнить как она делается, и понять почему.
Присоединение идёт как в 1,3-диенах по схеме 1,4-присоединения. 1,2-присоединение не наблюдается.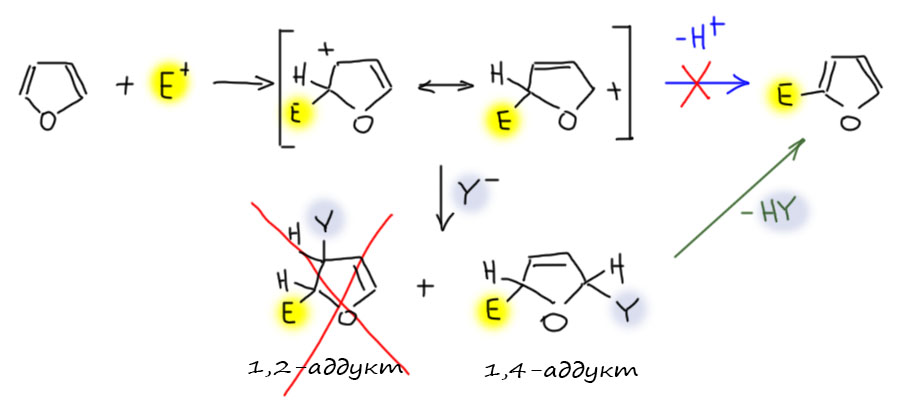
Например, нитрование в уксусном ангидриде идёт через такой аддукт. Его можно выделить, но если при выделении обработать слабым оснванием, как все равно всегда делают в таких случаях, чтобы в продукте не осталась сильная кислота из реакции, образуется обычный 2-нитрофуран.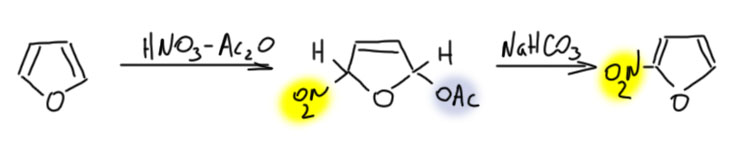
Но чаще всего встречается бромирование, причём в не вполне обычных условиях – в растворе метанола. Происходит обычное 1,4-присоединение, то же самое будет и с простыми 1,3-диенами. Но аддукт этой реакции обладает исключительно высокой реакционной способностью к сольволизу потому что он одновременно и аллильный, и в α-положении к кислороду. Чтобы выход был побольше и не было побочных реакций, прибавление раствора брома в метаноле ведут при низкой температуре и очень быстро. Точно так же можно получить и диэтокси-производное. У таких продуктов возможны два диастереомера, но реакция нестереоселективна и я нарисовал один просто, чтобы этой проблемой не заморачиваться.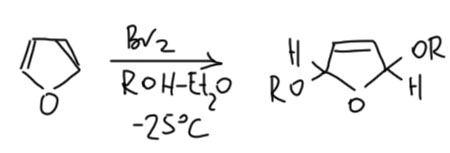
Это по всем признакам SN1-реакция, так как соответствующий катион хорошо стабилизирован. Но с тем же успехом можно написать и SN2-механизм, так как это тот самый случай, когда структурные факторы благоприятствуют и тому и другому. Как же быть?! – Да, наплевать. Это мало кого волнует в наше время, особенно в тех случаях, когда знание точного механизма ничего не даёт (оба работали бы, ну и хорошо), а поди попробуй его установи, не так-то это легко, три четверти 20 века в этих усилиях прошли, и всё без толку (повторю еще раз – эта пессимистическая сентенция касается только случаев, когда факторы одинаково благоприятствуют обоим механизмам, а это бывает нечасто).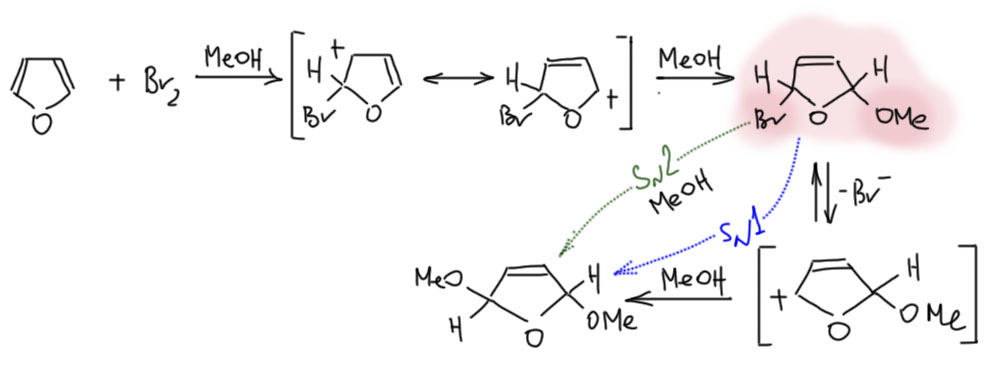
Дальше строго факультативно, для любопытных.
Продукт, который получается при действии брома в метаноле – весьма занятная штука. Если посмотреть на него внимательно мы увидим, что это ацеталь, причем двойной. Разобрав ацетальные группы, мы поймём, что это ацеталь диальдегида малеиновой кислоты, малеинового диальдегида. Реально гидролиз делают быстрой обработкой разбавленной серной кислотой с коротким нагреванием до 80º. 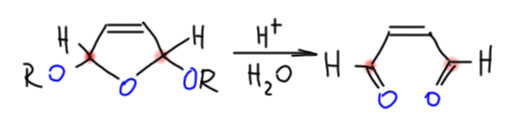
Малеиновый диальдегид – простейший ненасыщенный диальдегид. Это очень реакционноспособное соединение, которое невозможно даже хранить, и его немедленно используют в синтезах, например, в Дильсе-Альдере. 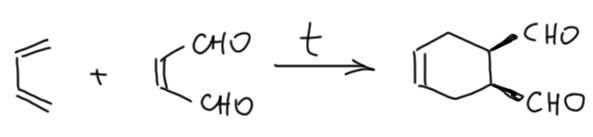
Можно также запустить это в присоединение по Михаэлю, только учтите, что будет непросто предвидеть во что превратится образовавшийся сначала продукт присоединения.
Занятно также и то, что если реакцию фурана с бромом с метаноле провести не так быстро, а чуть отпустить температуру и дать смеси перемешиваться часок-другой, то выделившийся в процессе сольволиза аддукта HBr обеспечит нам кислотный катализ, и при большом избытке метанола мы получим превращение циклического ацеталя в обычный. И при этом произойдёт довольно интересная вещь – мы получим ацеталь не малеинового, а фумарового альдегида (малеиновая – фумаровая кислоты, цис и транс изомеры). Изомеризация цис- в транс- скорее всего происходит как раз при раскрытии цикла. Термическая изомеризация здесь исключена, потому что реакция происходит при -10º. А вот карбокатион, который образуется при кислотно-катализируемом раскрытии цикла, имеет аллильную структуры, следовательно делокализован, следовательно, в нём двойная связь ослаблена, а значит имеет намного меньший барьер вращения и может проворачиваться, образуя более устойчивую транс-конфигурацию. У нее даже будет вторая попытка, когда таким же путем будет полуацеталь превращаться в полный ацеталь на второй группе. 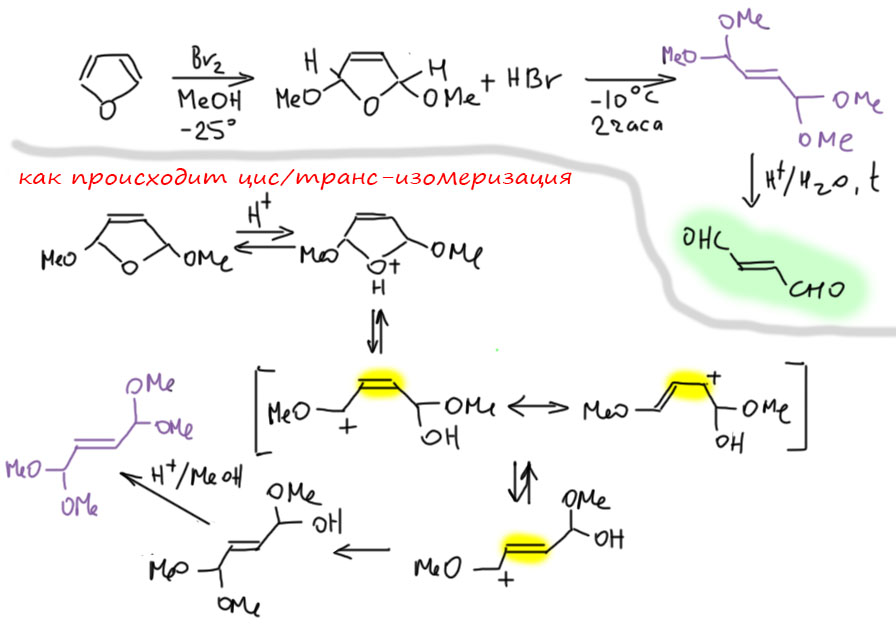
В общем, фуран оказывается удобным источником даже не одного а целых двух весьма привлекательных соединений, пригодных для интересных реакций. И других способов сделать эти два соединения нет. Фуран – очень дешёвое вещество, поэтому это очень привлекательная химия.
Электрофильное замещение в шестичленных гетероциклах
Пиридин обладает невероятно низкой реакционной способностью в реакциях электрофильного замещения. В одном модном учебнике органической химии даже написано, что вообще не вступает в такие реакции. Это – опасный и необоснованный экстремизм. Вступает, и это простой и надежный способ добраться до положения 3- (или β-). Спасает то, что пиридин чрезвычайно хорошо переносит очень жесткие условия в реакциях с электрофилами и кислотами. Поэтому реакции проводят “грубой силой” – в неописуемо жестких условиях. Мы никогда так грубо не обращались ни с одним органическим веществом, даже метаном. Но пиридин просто героически выдерживает чудовищные условия при условии, что чудовища только кислотной и электрофильной природы. К основаниям пиридин относится намного более нервно.
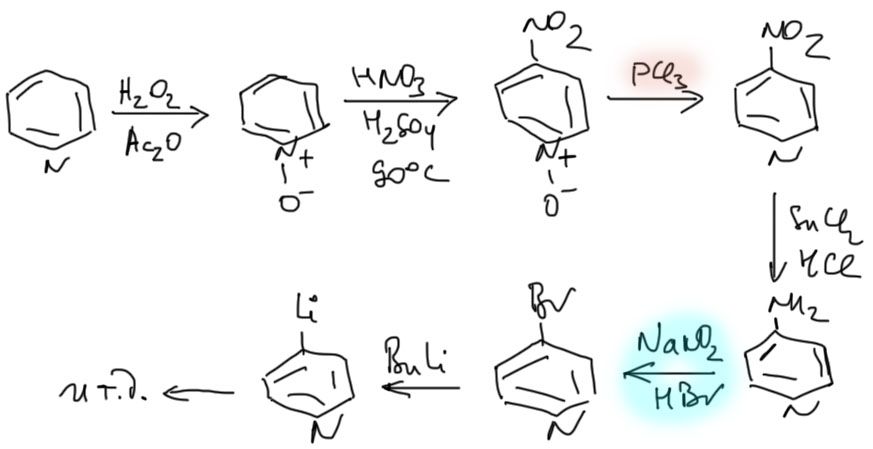
У пиридина есть очень любопытное производное – N-оксид. Это производное обладает особой реакционной способностью в реакциях с электрофильными реагентами. Проблема в том, что этой особостью очень трудно воспользоваться, и есть только одна надежная реакция с N-оксидом пиридина – нитрование. Все остальное (бромирование, хлорирование, сульфирование, ацилирование, и т.п.) нужно строго исключить. Эти реакции или вообще не идут, или идут совсем не так, как нам хочется.
Старая, ещё начала 20 века методика нитрования пиридина состоит в нагревании до температуры выше 300 (трёхсот! – это не ошибка) градусов пиридина в смеси селитры со 100%-ной серной кислотой (это называется моногидрат и делается прямо перед использованием добавлением расчетного количества олеума к обычной серной кислоте – считают содержание SO3 в олеуме и берут его столько, сколько нужно для превращения остаточной воды в концентрированной серной кислоте в чистую H2SO4). Выходы 3-нитропиридина при этом получаются невероятные – иногда даже до 8-10%. Обратите внимание, что на схеме нарисован сам 3-нитропиридин, а не его соль – это потому что в процессе реакции продукт просто отгоняют, а летит, конечно же, свободный нитропиридин, так как все соли азотных оснований при нагревании диссоциируют.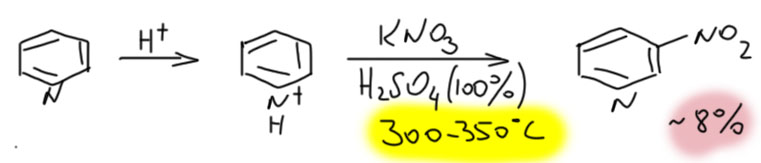
Хотя выход совершенно мизерный, этой реакцией можно пользоваться, если необходимо получать производные по положению 3, хотя лучше для этого использовать бромирование – там всё же выходы побольше, хотя условия реакции не менее варварские.
Нитрование производных пиридина
Заместители влияют на реакционную способность в электрофильном замещении в целом так же, как в бензольном ряду. Но проявляется это влияние немного иначе. Во-первых, пиридины с дезактивирующими заместителями можно даже не пробовать – шансов нет (есть одно занятное исключение, которое мы обсудим в факультативной части).
Пиридины с активирующими заместителями нитровать можно, но активирующие заместители должны быть серьёзные, или несерьёзные, но побольше. Моно-алкилпиридины нитровать бесполезно, условия все равно будут настолько жёсткими, что первым делом эти алкилы и сгорят, а второго дела не будет. Диалкилпиридины уже встретить можно, триалкил тоже, но это все проблемные вещи. А вот алкокси- (особенно если две), гидрокси- и аминопиридины нитровать можно. Обратите внимание на то, что в отличие от бензольного ряда, открытые гидрокси-группы и амино-группы не создают проблем. Посмотрим на примере аминопиридина, почему такое отличие. Пронитруем аминопиридин. Для нитрования активированных пиридинов обычно берут нитрующую смесь, иногда даже покрепче (азотную кислоту берут не концентрированную, которая 67-68%, а так называемую дымящую (95-100%), серную берут концентрированную или моногидрат, и реакцию ведут при пониженной температуре, около 0ºС. Что произойдёт с аминопиридином? Он протонируется. Пиридиновый азот имеет большую основность, чем аминный, потому что пиридиновое кольцо ведёт себя как акцептор, а аминогруппа как донор. После того как пиридиновый азот запротонирован, основность амино-группы еще более уменьшается – амин становится очень слабым основанием, приблизительно как аминогруппа в 2,4-динитроанилине или даже 2,4,6-тринитроанилине (пикрамиде).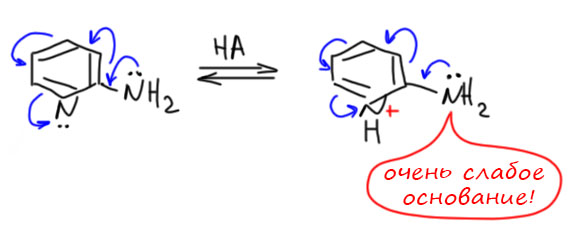
Поэтому даже в нитрующей смеси, то есть в среде с весьма большой кислотностью, аминогруппа остается в существенной степени непротонированной. Но несмотря на очень малую основность, аминогруппа сохраняет и свою способность сильно активировать ядро и ориентацию атаки электрофила (собственно это и есть побочный эффект снижения основности – электронная плотность ушла в ядро). Поэтоу нам, во-первых, не нужно защищать аминогруппу ацилированием, как мы делаем в анилинах (там нам под страхом вечного проклятия запрещали нитровать анилины с ннезащищенной амино-группой). А здесь это можно просто потому что протонированный пиридин ведет себя как такая защита и даже, на самом деле, как более сильная защита чем ацетил. Нитруем спокойно. Ориентация нитрования будет такой же как в бензольном ряду, но в основном получаются пара-изомеры по отношению к амино-группе.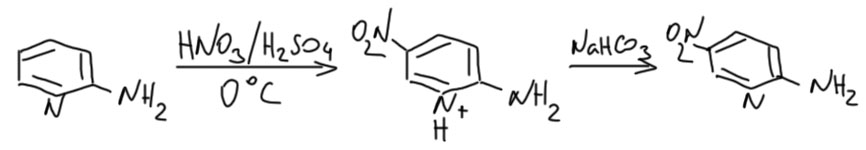
Так же нитруются и другие производные пиридинов с сильными донорными заместителями типа алкокси или гидрокси. Не будем про это писать ничего подробнее, тем более что ничего особенно оригинального по отношению к аминопиридина здесь нет, а с гидроксипиридинами нужно всё равно разбираться отдельно – это особенные соединения, существующие в основном в виде таутомеров с амидной структурой, пиридонов.
А что если ориентант будет не в положении 2-? Ничего особенного – ищите в молекуле относительно ориентанта орто и пара-положения, при этом старайтесь дополнительно соблюдать два простых правила: а) если есть выбор, то больше будет “пара”-изомера по отношению к главному ориентанту, хотя и “орто”-изомеры тоже часто образуются, но в меньших количествах; б) при этом избегайте положений рядом с азотом (2- и 6-) – электрофил туда не хочет, по совершенно очевидной причине – рядом положительный азот, и положительный электрофил не хочет приближаться к положительному азоту. Это не абсолютный запрет и в ряде случаев такие продукты получаются, но в целом это неплохо работает.
Дальше факультативно для любопытных
Во-первых, вернемся к собственной реакционной способности пиридина в электрофильном нитровании. Это всё же довольно интересно, потому что хочется немного понимать, насколько протонирование снижает способность реагировать с электрофилами. На первый взгляд, задача кажется нерешаемой, потому что как, действительно, помешать взаимодействию электрофила с нуклеофильным азотом. Но ответ неожиданно нашелся, немного даже случайно, но и благодаря невероятной настойчивости и педантичности одного из главных гетероциклистов 20 века Алана Катрицкого. Он очень много усилий потратил на изучение электрофильного замещения в пиридинах, пытаясь восстановить шкалу относительной реакционной способности и как-то сшить пиридиновый ряд с рядом бензола. И перебирая заместители в пиридиновом кольце он взял 2,6-дихлорпиридин. Аккуратно измеряя скорости нитрования в зависимости от кислотности нитрующей смеси (кислотность изменяется просто варьированием концентраций азотной и серной кислот, точнее, остаточной воды в кислотах с концентрациями близкими к 100% на страничке про кислоты и основания Бренстеда-Лоури можете вспомнить, что такое индикаторные шкалы кислотности очень сильных кислот в концентрированных растворах, там, где, не работает pH), он обнаружил интересный эффект, который трудно объяснить иначе, чем тем, что 2,6-дихлорпиридин нитруется как свободное основание, а не как протонированная форма. В этом месте можно задать недоуменный вопрос – а что, дихлорпиридин не протонируется? Конечно, это более слабое основание, чем сам пиридин (два серьёзных акцептора рядом с азотом), но концентрированные-то кислоты уж как-нибудь справяться. Конечно протонируется, но вопрос в том, насколько. Если хотя бы доля процента остается непртонированной, и если эта непротонированная форма намного более реакционноспособна, чем протонированная, то реакция пойдёт через свободное основание. Так устроены равновесия, и мы часто сталкивались с такими явлениями. Просто представьте, так, от фонаря, что остался 1% непротонированной формы, но она в 10000 раз более реакционноспособна – реакция без вопросов просто побежит по этому пути. Мы не будем копаться в цифрах (желающие могут подробно изучить работу C. D. Johnson, A. R. Katritzky, B. J. Ridgewell, M. Viney, J. Chem. Soc. B, 1967, 1204), просто качественно подивимся на результат и поймём, насколько он сенсационен. Дихлорпиридин, дезактивированное производное пиридина нитруется обычной нитрующей смесью (там азотка покрепче обычного, зато серная пожиже – итого это не сильно крепче самой обычной нитрующей смеси) при вполне умеренном нагревании. Не забудем, что дихлорпиридин, конечно, тоже очень сильно протонирован в такой среде и свободного пиридина там малые доли процента – и именно они нитруются в общем приблизительно так же, как стали бы нитровать дихлорнитробензол. 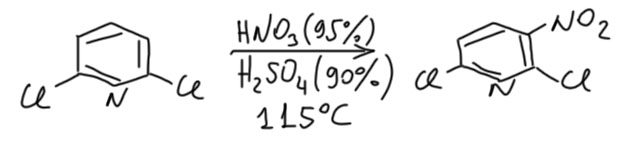
Вывод вполне понятный – если бы можно было нитровать не протонированную форму, а свободные основания пиридинов, то это происходило бы без всякого экстремизма и давало бы приличные выходы. Увы, остальные производные пиридина в условиях нитрования запротонированы практически полностью, и остаточные концентрации оснований настолько ничтожны, что через них нитрование не идёт с измеримой скоростью. А реакционная способность протонированного пиридина чрезвычайно мала. Катрицкий оценил ее в 10-17 относительно непротонированной формы, и эта величина вряд ли нуждается в комментариях.
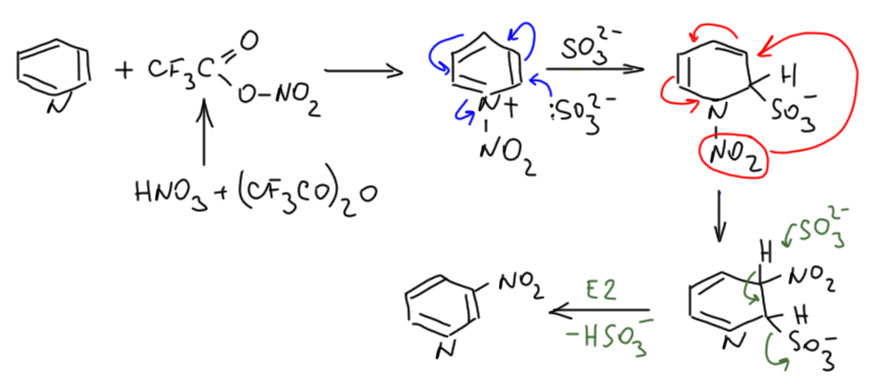
Химия не стоит на месте, и пиридины таки научились нитровать в разумных условиях и с хорошими выходами. В самом конце прошлого столетия это сделал норвежец Ян Бакке (Jan Bakke, Acta Chem Scand., 1999, 53, 356), а Катрицкий с сотрудниками довел метод до удобной лабораторной методики (Org. Biomol. Chem., 2005, 3, 538). Метод Бакке-Катрицкого чрезвычайно странен, но он работает. Сначала на азот пиридина сажают нитроний-катион, сначала это делали с помощью азотного ангидрида, но это нехорошее вещество, более пригодное для полёта на Луну, чем для нормальной лаборатории. Катрицкий взял обычную азотную кислоту или ее соль и смешав с трифторуксусным ангидридом в растворе получил смешанный ангидрид азотной и трифторуксусной кислот – трифторацетилнитрат. Это оединение отлично переносит электрофильный нитроний. Иными словами, на этой стадии произошло то, что можно по другому сделать, если подействовать на пиридин готовой солью нитрония – электрофил сел на азот. Что дальше? Ядро пиридина при этом дезактивировалось точно так же как при протонировании. И даже при избытке нитрующего агента нитрование дальше не пойдёт. Но тут происходит неожиданное – в смесь просто наливают водный раствор сульфита натрия (часто берут бисульфит или метабисульфит), то есть довольно сильный нуклеофил. А пиридин с плюсом на азоте – это превосходный акцептор Михаэля, с двойными связями, сопряженными с сильной акцепторной группой. Нуклеофил присоединяется. И дальше происходит что-то не очень понятное, но результат – образуется 3-нитропиридин с отличным выходом. По дороге температура ни разу не повышалась выше комнатной.
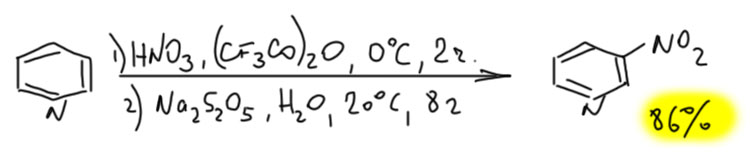
Механизм реакции немного туманен, хотя его и исследовали, но не до конца. Частично уже его обсудили – нитроний сел на азот, присоединился сульфит. Самая важная стадия – перемещение нитро-группы на третий атом. Это может быть или внутримолекулярная электрофильная атака или согласованная реакция ([1,5]-сигматропный сдвиг), но это происходит. В конце происходит просто восстановление ароматической системы за счёт E2-элиминирования сульфита.
Реакция вполне пригодна и для замещённых пиридинов и неплохо работает.
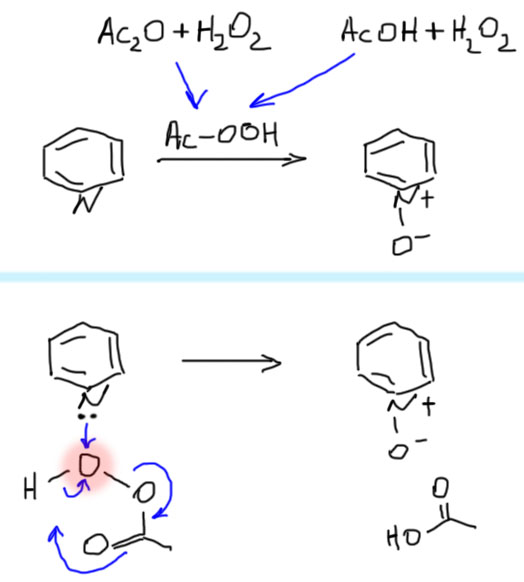
N-Оксид пиридина – типичный представитель N-оксидов соединений азота (третичных аминов, нитрилов, гетероциклов), которые образуются действием на такие соединения пероксокислот. Важно, чтобы у азота не было атомов водорода – в этом случает окисление идет не так и дает другие продукты. Пиридин как раз соответствует этому правилу. По механизму это просто атака электрофильным кислородом (один из кислородов в пероксо-соединениях электрофилен) на неподеленную пару нуклеофильного азота. Как правило, реакцию делают с помощью надуксусной кислоты, получаемой прямо перед реакцией взаимодействием уксусного ангидрида и крепкой (40-50%-ной) перекиси водорода. Если нельзя раздобыть уксусного ангидрида (запрещен в Российской федерации), то надуксусную кислоту можносделать из ледяной уксусной и крепкой перекиси водорода, но это довольно опасная и не очень хорошо воспроизводимая процедура. Еще один вариант – взять мета-хлорнадбензойную кислоту, ту самую, которой мы окисляем алкены в эпоксиды. Если будете делать эту реакцию практически, обязательно проверьте концентрацию перекиси водорода, потому что она понемногу разлагается при хранении, и если концентрация упала ниже 30% ничего не получится. Проверяют концентрацию или измеряя плотность (берете хорошую пипетку, отбираете 1 мл и взвешиваете, повторяете еще 2-3 раза, берете среднее – плотности растворов перекиси водорода можно найти в сети или справочниках). Или титруя иодометрически, если есть бюретка, и еще не забыли аналитическую химию.
Еще раз напомню, что электрофильность кислорода в надкислотах объясняется тем, что этот кислород связан с уходящей группой, забирающей электроны. Одновременное перемещение протона делает уходящей группой просто карбоновую кислоу, а не ее анион. Это выгоднее, и объясняет, почему в таких реакциях треубуется кислотный катализ, который сама же надкислота и обеспечивает (надкислоты заметно сильнее обычных кислот той же структуры).
Почему N-оксид пиридина реагирует с электрофилами в положение 4
Потому что это молекула, как две капли воды похожая на фенолят-ион. Берем фенолят-ион и заменяем один атом углерода на азот. Получаем ровно N-окись пиридина. Из-за необходимости подвести к этоу азоту 4 валентности, атом должен быть положительно заряжен. Это дает в целом электронейтральную молекулу, в отличие от фенолята, который анион. Молекулы, получающиеся из других молекул заменой атомов углерода на другие атомы (гетероатомы), называют гетероаналогами. N-оксид пиридина – гетероаналог фенолят иона.
Зачем это нужно? Для того, чтобы не тратить попусту время. Аналогии для того и существуют, чтобы помогать видеть сходство в поведении и структуре. И для того, чтобы понимать различия. Сходство в том, что обе эти молекулы могут реагировать с электрофилами, и в том, что электрофил атакует эти молекулы в положения 4- и 2-относительно места присоединения кислорода.
А теперь разберемся в различиях. Причина различий проста – вместо углерода, гораздо более электроотрицательный положительный азот. 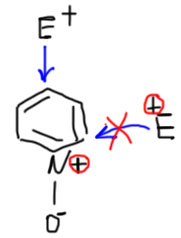 B первое следствие этого – близость положительного атома убирает из соревнования положение 2 – обычно положительный электрофил просто не может подойти близко к месту с положительным зарядом. Эффекты такого рода (их называют полевыми – от эффекта электростатического поля, кулоновского отталкивания зарядов) очень часто встречаются в органической химии – вспомним хотя бы, как сильно уменьшается константа кислотности по второй ступени относительно первой.
B первое следствие этого – близость положительного атома убирает из соревнования положение 2 – обычно положительный электрофил просто не может подойти близко к месту с положительным зарядом. Эффекты такого рода (их называют полевыми – от эффекта электростатического поля, кулоновского отталкивания зарядов) очень часто встречаются в органической химии – вспомним хотя бы, как сильно уменьшается константа кислотности по второй ступени относительно первой.
И второе – этот атом азота в цикле страшно сильно, колоссально дезактивирует – снижает реакционную способность – его по отношению к электрофильной атаке. Но дезактивирует относительно уровня реакционной способности фенолята, а она, как мы знаем, огромна, колоссальна. Эта колоссальная дезактивация фактически съедает весь колоссальный запас реакционной способности фенолята. Встретились два колосса – и сожрали друг друга. И что это значит? Значит только то, что реакционную способность ароматических соединений мы оцениваем относительно бензола. И когда активация и дезактивация съедают друг друга, мы возвращаемся к бензолу. Приблизительно. Мы же на самом деле не знаем насколько активирует фенолят, и насколько точно дезактивирует положительный азот. Гугол вверх – а потом гугол и две морковки вниз. Как-то так. Строго приблизительно, как и все в органической химии. С точностью до третьего знака перед запятой.
Результат такой – реакционная способность N-оксида пиридина где-то рядом с реакционной способностью бензола, возможно, немного ниже. Из этого следует, что условия реакций мы должны выбирать аналогичные тем, что используем для бензола. А бензол нитруют обычной нитрующей смесью при комнатной температуре. Но здесь нас ждет еще один неприятный сюрприз, а точнее, целых два.
Единственная реакция электрофильного замещения, применимая к N-окcиду пиридина – нитрование. И сделать это немного сложнее, чем может показаться на первый взгляд.
Нитрование N-оксида пиридина
Эту полезнейшую реакцию открыл в 1950-м году голландский химик ден Хертох. И с тех пор все делают только так, как он это описал, и не пытаются что-то изобрести. Фокус в том, и ден Хертох это очень хорошо понял, что у N-оксида довольно основный атом кислорода. И с кислотами, даже слабыми, N-оксид дает соли. И если растворить N-оксид в нитрующей смеси, то тем более атом килорода будет запротонирован. И все – прощай гетероаналог фенолята, у нас получается гетероаналог всего-навсего фенола. А фенол – это не фенолят, это гораздо менее реакционноспособная молекула. Мы отлично помним это по химии фенолов. И вся логика того, как колоссы от испуга скушали друг друга, накрывается чашечкой Ай-Петри. Теперь у нас с одной стороны по прежнему плохой колосс – положительный азот, а с другой хороший, но всего-навсего гигант, фенольный гидроксил. Плохой колосс сожрал хорошего гиганта и не поперхнулся. Молекула осталась сильно дезактивированной. И не нитруется.
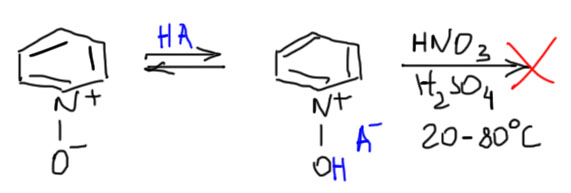
Но если температуру повысить до 90º, а лучше и еще немного побольше, до 110-120º, то нитрование идет, очень медленно, и только в положение 4. В чем дело, зачем температура, почему медленно?
Очень просто. Протонирование обратимо. И как всякая экзотермическая реакция, равновесие при повышении температуры смещается влево – в сторону депротонирования. В смеси появляется очень маленькая концентрация непротонированной, а следовательно реакционноспособной, формы. Которая и нитруется. Медленно, потому что концентрация очень маленькая. Сделать температуру сильно больше смысла нет – опасно это во-первых, во-вторых, начинается динитрование. Так и нитруют; нитрующую смесь составляют из дымящей (то есть, 100%-ной кислоты плотностью 1.52) азотной кислоты и концентрированной серной (никакого олеума!). То есть, условия все же существенно более жесткие чем для нитрования бензола. Причины две – во-первых, видимо, плохой колосс оказался немного крупнее хорошего, и N-оксид ближе не к бензолу, а к таким слабо дезактивированным молекулам, как галогенбензолы (впрочем. как мы увидим очень скоро для хинолина, эта оценка скорее всего слишком пессимистична). Во-вторых, концентрация непротонированной формы в равновесии очень мала, и это плохо отражается на скорости реакции. Иными словами, мы точно так и не знаем, какова реакционная способность чистого N-оксида, потому что в растворах, используемых для электрофильного замещения, всегда есть кислота, а следовательно и N-оксид в основном живет там в протонированной, нереакионноспособной форме.
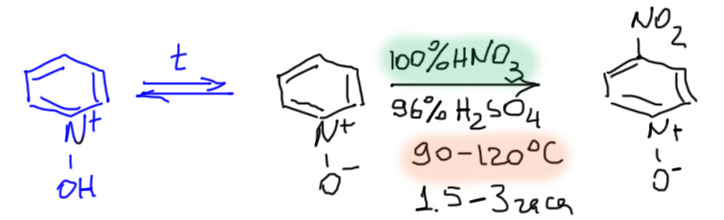
Еще ярче эта ситуация, которую часто называют “вилкой реакционной способности”, проявляется при нитровании N-оксида хинолина. Там есть еще бензольное ядро, вполне реакционноспособное, которое обычно и принимает реакции электрофильного замещения. Вот и здесь так – если мы нитруем N-оксид хинолина при температуре ниже 70-80º, то получаем обычную смесь 5- и 8-нитропроизводных – не пускает электрофил в кольцо пиридина протонированная N-окись. А выше 90º селективность меняется, и нитрование идет уже в 4-положение. И это невероятно здорово – это же значит, что мы не ошиблись, и и что кольцо с N-окисью очень реакционноспособно, что оно все же именно активировано – ведь оно в одну калитку выигрывает конкуренцию у бензо-кольца!
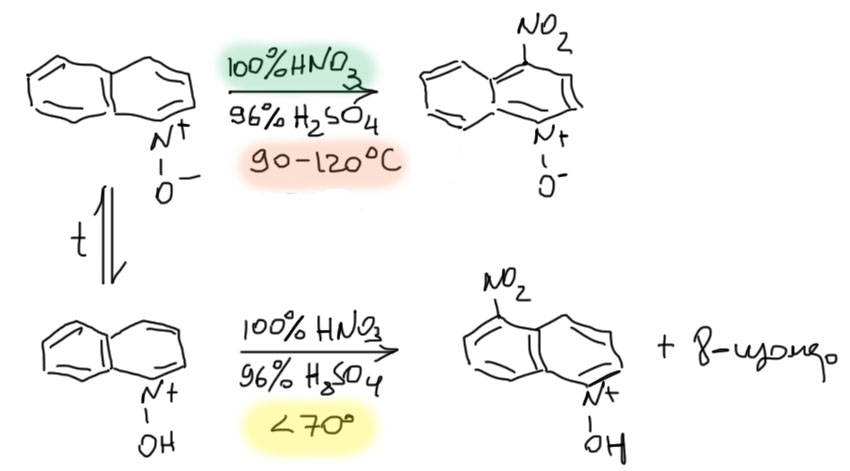
Другие реакции замещения в N-оксидах
Если коротко, то – нет больше никаких реакций, пригодных для использования, по крайней мере, на 3 курсе. Но мне показали книжку про химию гетероциклов, где было указано, что N-оксиды еще можно бромировать.
Поясняю. Нет, нельзя. В этой книжке ее автор не очень добросовестно процитировал одну работу. И создал миф.
Реальность же такова. Если попробовать подобрать условия для бромирования N-оксида, то нас ждут проблемы. Мы не можем использовать обычные кислоты Льюиса (бромное железо, бромистый алюминий и т.п.), потому что они немедленно садятся на кислород N-окиси и, как минимум, убивают ее активирующий эффект (точно так же, как это делает протонирование), а как максимум вообще восстанавивают N-оксид в исходный пиридин (восстанавливает не кислота Льюиса, а бромид-ион, а кислота Льюиса просто оттаскивает кислород. Мы хорошо знаем эту реакцию, потому что как раз и снимаем этот кислород с помощью треххлористого фосфора по очень похожему механизму.
Если использовать протонную кислоту типа олеума, то бромирование пойдет, в очень жестких условиях, почти таких же, как при бромировании самого пиридина. Это известно, но это не то, что нам нужно. При бромировании в концентрированной серной кислоте бромом реакция идет очень плохо, хотя получается несколько процентов 4-бромпроизводного в смеси с 2-бромпроизводным. Если взять олеум, как при бромировании пиридина, то и здесь получается 3-бромпроизводное. Очевидно, что на кислород садится SO3 и дезактивирует, а реакция дальше идет точно так же, как просто в пиридине. Ни та, ни другая реакция никакого применения не нашла.
Но есть еще одна занятная японская работа (Saito, Hamana, Heterocycles, 1979, 12, 475), в которой используют бром и ацетат таллия в очень мягких условиях, причем N-оксид хинолина и правда дает 4-бромпроизводное.
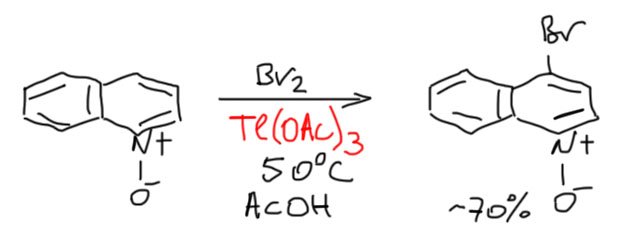
Это очень остроумно, потому что как раз обходит проблему дезактивации по кислороду. Таллий (3+) – хорошо известный электрофил, очень похожий на Hg(2+), и это мягкая кислота Льюиса, кислород она не любит. Происходит электрофильное таллирование, а связь углерод-таллий расщепляется бромом.
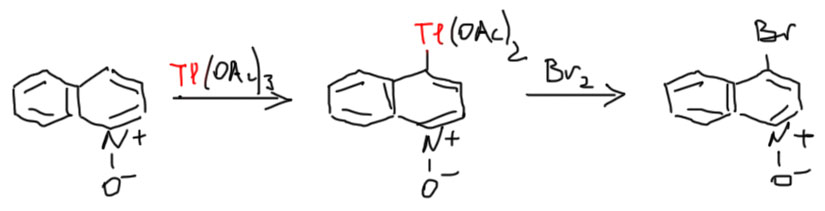 Очень симпатично – но совершенно не годится для реальной работы. Используется 3-кратный избыток соли таллия, а это очень дорого, и исключительно опасно; таллий – один из самых токсичных элементов, работать с ним соглашаются немногие, потому что это смертельно опасно. Представьте себе такую работу в наших практикумах с текущими делительными воронками – шансов выжить при работе с солью таллия нет ни единого. Да и в более лучше оборудованном месте такая работа представляет собой неприемлемый риск. Не удивительно, что эту реакцию, кажется, никто никогда больше не применял (это определяется просто по цитированию – если бы применял, то процитировал бы, а ничего такого не нашлось). Кроме того, эта реакция оказалась неприменима к N-окиси пиридина – он просто не прореагировал в этих условиях. Единственное производное пиридина, с которым этот метод сработал – N-оксид 2,6-диметилпиридина, и то с не очень большим выходом (50%). Почему – неизвестно. Нам это не нужно. Нам нужно понять, что если мы хотим попасть в положение 4 пиридинов и хинолинов, нужно идти через нитрование N-оксидов.
Очень симпатично – но совершенно не годится для реальной работы. Используется 3-кратный избыток соли таллия, а это очень дорого, и исключительно опасно; таллий – один из самых токсичных элементов, работать с ним соглашаются немногие, потому что это смертельно опасно. Представьте себе такую работу в наших практикумах с текущими делительными воронками – шансов выжить при работе с солью таллия нет ни единого. Да и в более лучше оборудованном месте такая работа представляет собой неприемлемый риск. Не удивительно, что эту реакцию, кажется, никто никогда больше не применял (это определяется просто по цитированию – если бы применял, то процитировал бы, а ничего такого не нашлось). Кроме того, эта реакция оказалась неприменима к N-окиси пиридина – он просто не прореагировал в этих условиях. Единственное производное пиридина, с которым этот метод сработал – N-оксид 2,6-диметилпиридина, и то с не очень большим выходом (50%). Почему – неизвестно. Нам это не нужно. Нам нужно понять, что если мы хотим попасть в положение 4 пиридинов и хинолинов, нужно идти через нитрование N-оксидов.
Модификация фурана, тиофена, пиррола, индола
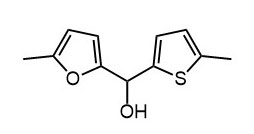
- Из фурана, тиофена, ДМФА и днр получите вторичный спирт
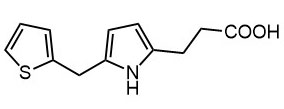
- Из пиррола, тиофена, диметилформамида, формальдегида и малонового эфира получите вот такую штуку
Модификация пиридина
2. Из пиридина и индола получите 2-пиридил-3-индолилкетон
3. Из пиридина, бромбензола, и днр получите 6-фенилпиридин-2-карбоновую кислоту
4. Из пиридина, уксусного альдегида и диазометана получите 4-циклопропилпиридин
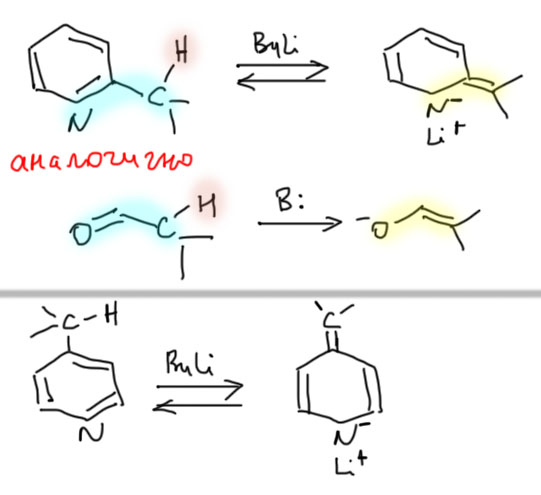
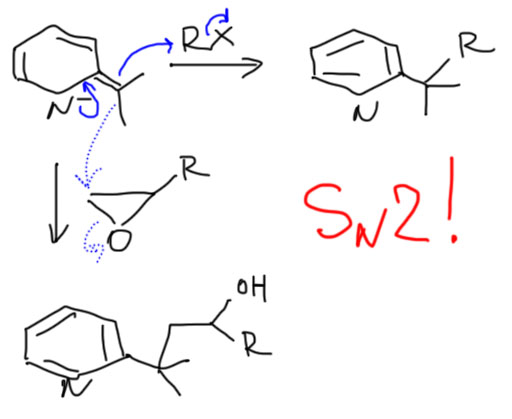
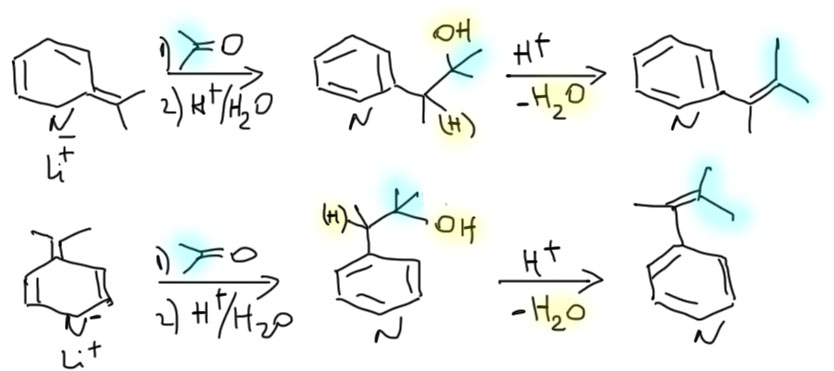
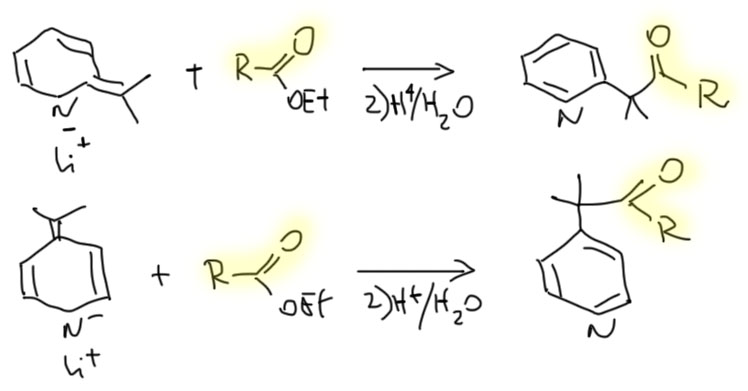
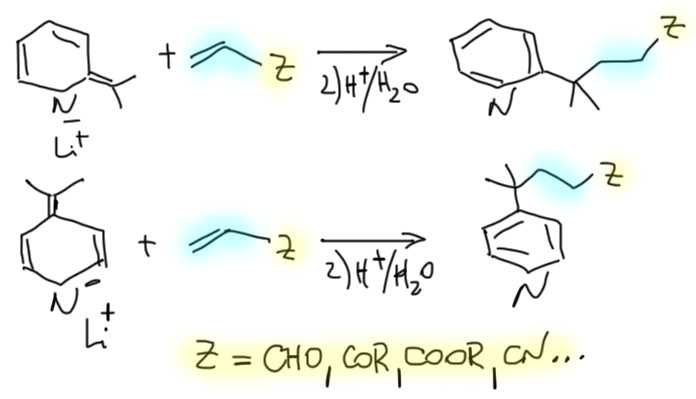
Пиридиновые и хинолиновые "еноляты"
2. Из хинолина, этилацетата и окиси этилена получите спирт (В)
3. Из хинолина, метилиодида и циклогекс-2-енона получите кетон (С)
4. Из пиридина, метилиодида, ацетальдегида и малонового эфира получите 4-(2-пиридил)-3-метилбутановую кислоту
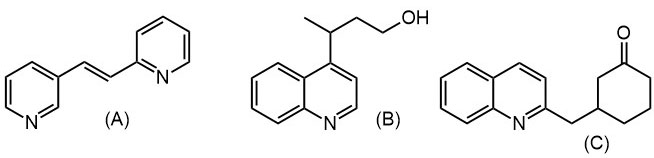
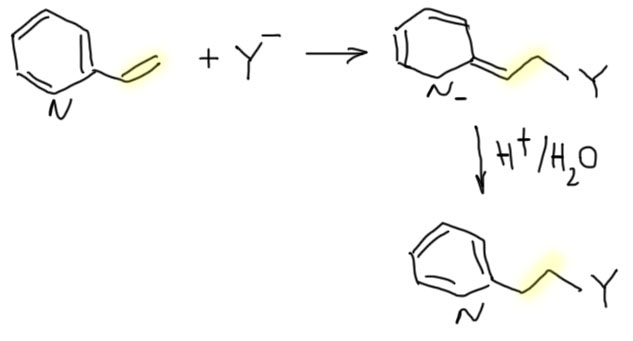
Нуклеофильное замещение в пиридинах и хинолинах
Диеновый синтез с фуранами
Синтез 5-членных гетероциклов
2. Из циклогексанона, ацетоуксусного эфира и анилина получите пиррол (B)
3. Из о-ксилола и днр получите тиофен (C)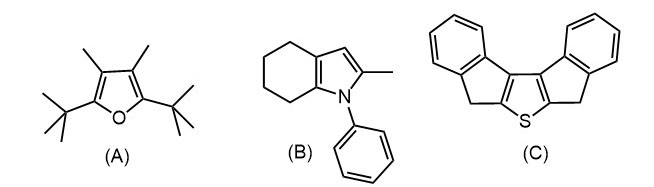
Синтез индола по Фишеру с последующей модификацией
2. Из бензилхлорида, метилиодида, анизола и днр получите N-метил-2-бензил-3-фенил-5-метоксииндол
3. Из терефталевой кислоты, метилиодида и анилина получите 1,4-бис(2-индолил)бензол.
Синтез индола по Фишеру из циклических кетонов
2. Из циклооктина, анилина и днр получите индол (B)
3. Из ацетона, малонового эфира, диазометана, o-нитрофенилгидразина и днр получите индол (С)
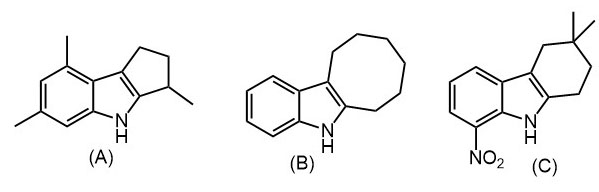
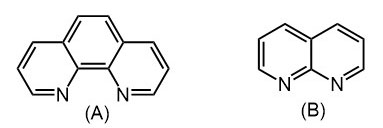
Синтез хинолинов по Скраупу
2. Из анилина и глицерина получите фенантролин (A).
3. Из пиридина и глицерина получите 8-азахинолин (B)
Синтез хинолинов по Дёбнеру - фон Миллеру
2. Из анилина, пиридина, ацетонитрила, бензальдегида и днр получите 2-фенил-4-(3-пиридил)хинолин.