Альдольно-кротоновая конденсация
Альдольная конденсация – это реакция присоединения к карбонильной группе (или её аналогу – но не будем пока об этом) нуклеофильной формы енолизуемого карбонильного соединения. В общем виде – это один из самых универсальных и мощных типов превращений органических соединений, позволяющий соединять в новую молекулу два карбонильных соединения, и от нас зависит, насколько широко мы можем трактовать это понятие. Мы пока не будем слишком увлекаться обобщениями, и карбонильными соединениями будем считать только альдегиды и кетоны, и тогда альдольной конденсацией будем называть реакцию двух карбонильных соединений, одно из которых обязательно должно иметь хотя бы один атом водорода на соседнем с карбонильной группой атоме – такое карбонильное соединение принято называть енолизуемым. Если таких атомов нет ни в одном карбонильном соединении, альдольная конденсация в такой паре невозможна. 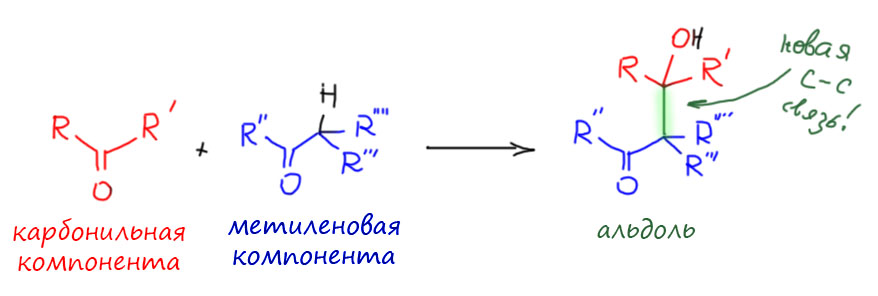
Часто и многим приходит в голову вопрос, отчего у такой важной реакции, как альдольная конденсация нет именного названия – кто-то же её придумал, и это должен был быть великий химик, застолбивший место в вечности. Это так и есть, но проблема в том, что альдольную конденсацию открывали независимо много раз в самом начале органической химии для отдельных кетонов и альдегидов, и только уже в 20 -м веке разобрались в этой химии окончательно, поняли, насколько это универсальный тип превращения. Часть этой удивительной по путанице истории рассказана тут ниже на вкладке про самоконденсацию ацетона. Но если установить, кто открыл альдольную конденсацию невозможно, то сказать, кто придумал слово альдоль вполне можно однозначно – это сделал Адольф Вюрц в 1872 году. Замесив уксусный альдегид с соляной кислотой, он получил вещество, обладавшее свойствами и альдегида и спирта. Вюрц, несмотря на свои немецкие фамилию и имя был французом, французом из того самого Эльзаса, за который Германия и Франция бились во всех войнах, и который переходил от одних к другим, пока наконец после 2-й мировой не закрепился за Францией. В Эльзасе всегда жило и до сих пор живет много французов с немецкими именами, среди них немало крупных химиков и Адольф Вюрц – один из них. Вюрц писал по-французски и написал, что получил альдегид-спирт (aldéhyde-alcool), и сам сократил это в aldol – в те времена любили такие сокращения для образования новых терминов. Именно потому, что это было в оригинале по-французски, мы говорим с мягкими эль – альдоль, а не алдол, как было бы, если бы слово возникло в английском, но английский в те времена не был главным языком химии, главными языками были немецкий и французский.
В альдольной конденсации нуклеофильная форма енолизуемого карбонильного соединения традиционно называется метиленовой компонентой. Это очень древний термин, который неплохо было бы переместить в музей, потому что толком не понятно ни что он значит, ни зачем нам такие древности в современной науке с уже сложившейся корректной и осмысленной терминологией. Но в нашей стране так любят всяческие скрепки, что держатся за эту ископаемую терминологию, как за признак давно ушедшего, скорее мифического золотого века. В англоязычной литературе вы этот реликт не встретите, и никогда не пытайтесь переводить это на английский, если вдруг такая задача перед вами встанет. Более адекватно называть это нуклеофильным реагентом альдольной конденсации, но мы пока оставим исторический термин – метиленовая компонента. И – именно компонента, а не компонент, как стали нередко писать и говорить. В руском языке совершенно законно сосуществуют оба эти слова, но слово компонента имеет более специальный, часто исторический оттенок, оно используется в устоявшихся словосочетаниях и оборотах, филологи такие слова в устойчивых комбинациях называют идиомами. А компонент – вполне современный термин с широким спектром употребления, не имеющий никаких оттенков и смыслов, нейтральный. Ещё раз напомню, что компонента в альдольной конденсации – именно исторический, традиционный термин, и именно ради этого мы его продолжаем использовать, чтобы напомнить в основном себе, что мы тут химией занимались вместе с немцами и французами, когда некоторые нынешние гордецы в прериях буйволов пасли. Пустячок, а приятно. И заменять пахнущее древностью и славой слово компонента на безликий компонент, право, не стоит. Или вообще похороните эту милую архаику в музее истории химии, или не выпендривайтесь, чтите предков.
Электрофильная форма называется гораздо адекватнее – карбонильная компонента, тот реагент, который предоставляет карбонильную группу. С этим можно жить дальше, а можно и называть это электрофильным реагентом конденсации. Оставим пока для симметрии карбонильную компоненту, если уж держимся за метиленовую.
Итак, альдольная конденсация происходит между карбонильной и метиленовой компонентами. Только одна из них обязательно должна соответствовать енолизуемому карбонильному соединению – метиленовая. Мы знаем 4 типа нуклеофильных форм енолизуемых карбонильных соединений: енолы, еноляты, енамины, эфиры енолов. Из них в альдольной конденсации мы никогда не будем применять только один – енамины. В современной органической химии енамины тоже активно применяются в альдольных конденсациях, но мы не будем – это сложно и требует очень точного понимания природы происходящих явлений.
Карбонильной компонентой может быть как енолизуемое, так и неенолизуемое карбонильное соединение.
Альдольная конденсация бывает направленной и ненаправленной. Разница между ними в том, что в направленной конденсации вы определяете роли компонентов. Поскольку карбонильная компонента – это просто карбонильное соединение, и с ним нельзя ничего сделать специально, то в направленной конденсации можно только распоряжаться метиленовой компонентой. В направленной конденсации выбирают такие нуклеофильные формы, которые можно целенаправленно и количественно приготовить из енолизуемого карбонильного соединения. Не ждать у моря погоды, а от Природы милости, не сидеть возле колбы, умоляя уполномоченных божеств послать вам нужную нуклеофильную форму – а буквально взять быка за рога, то есть енолизуемое карбонильное соединение за конкретные атомы водорода, отодрать их нечеловеческим усилием подходящего основания и приготовить эту самую форму селективно и количественно, не оставив ни молекулы исходного карбонильного соединения. Специально можно приготовить не всё, но многое, в первую очередь енолят и эфир енола (енамин тоже можно приготовить, просто мы решили воздержаться от этого).
Конденсации можно делать как в присутствии кислот, так и оснований (мы пока не будем осложнять, потому что есть и вариант, когда используется одновременно и то, и другое). Роль кислот всегда каталитическая: кислотный катализ активирует карбонильную компоненту и ускоряет кето-енольное превращение в том варианте конденсации, когда нуклеофильной формой является равновесный енол (ненаправленной кислотно-катализируемой конденсации).
Основания активируют метиленовую компонету, переводя её в енолят, равновесный в ненаправленной конденсации, или количественно полученный в направленной конденсации. Поскольку енолят – тоже основание, можно считать реакцию с енолятом вариантом активации основанием, хотя в собственно направленной альдольной конденсации с участием енолята в этом случае постороннее основание не используется.
Кислоты активируют карбонильную компоненту. Используют протонные кислоты (кислоты Бренстеда-Лоури) – в этом случае активацию можно представить себе как протонирование карбонила по кислороду, хотя работает и более общий способ активации – образование водородной связи с карбонильным кислородом. Очевидно, что протонирование намного более сильная активация, и именно её обычно представляют в механизмах. Соответственно, в ракциях используют сильные кислоты типа серной, хлористоводородной, толуолсульфоновой и т.п. Не менее часто используют и кислоты Льюиса, соли и комплексы разных металлов. Металлы образуют донорно-акцепторную связь с кислородом карбонила, что приводит к тому же самому, что и протонирование – смещению электронной плотности от карбонильного углерода, он становится беднее, а значит электрофильнее. 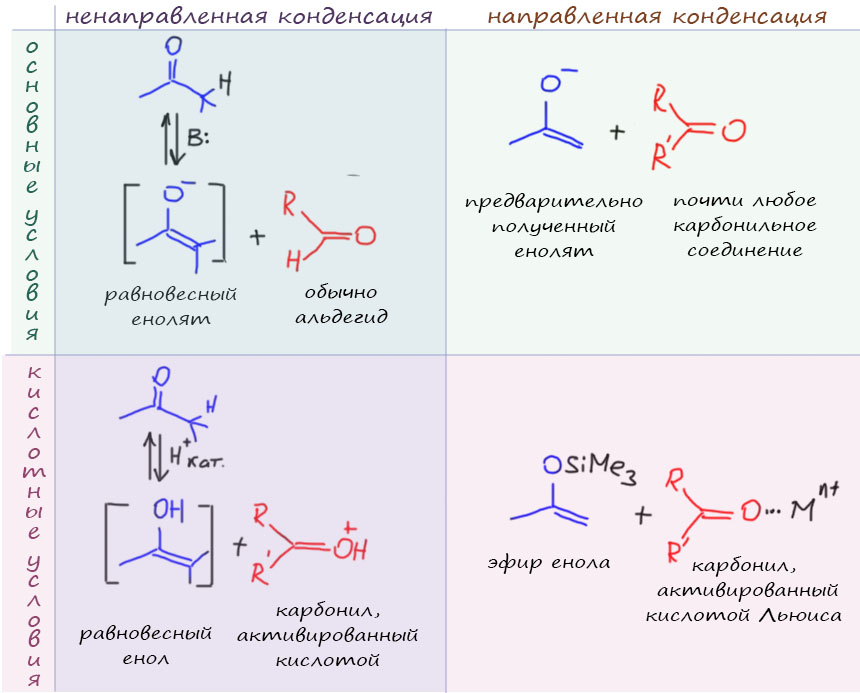
Направленная альдольная конденсация использует енолят или эфир енола.
- Енолят – достаточно сильный нуклеофил, чтобы реагировать с карбонильной компонентой как таковой. Это – первый вариант направленной альдольной конденсации.
- Простой эфир енола (обычно триметилсилиловый) недостаточно нуклеофилен, чтобы реагировать с самой карбонильной компонентой. Так как мы уже ничего сделать с его нуклеофильностью не можем, остаётся попробовать увеличить электрофильность карбонильной компоненты. Для этого используется кислотный катализ. Это – второй вариант направленной альдольной конденсации.
Ненаправленная альдольная конденсация использует нуклеофильные формы енолизуемых карбонильных соединений, находящиеся в равновесии с самим карбонильным соединением.
- Равновесный енолят образуется в равновесной смеси в результате действия основания. Он реагирует с самим карбонильным соединением. Такой вариант называют ненаправленной альдольной конденсацией, катализируемой основанием (или в условиях основного катализа).
- Енол находится в равновесии с енолизуемым карбонильным соединением и в условиях кислотного и основного катализа. Но в условиях основного катализа в той же смеси находится и енолят, который заведомо более активен. Поэтому енол работает как нуклеофильная форма только в условиях кислотного катализа. Енол обладает недостаточной нуклеофильностью, чтобы реагировать непосредственно с карбонильной компонентой, но в условиях кислотного катализа происходит активация карбонильной группы (протонирование, водородная связь, координация с ионом металла). Этот вариант ненаправленной альдольной конденсации назовём кислотно-катализируемой альдольной конденсацией.
Итак, у нас образовалось четыре основных типа альдольной конденсации, два направленных, и два ненаправленных.
Разберёмся, когда и что применяют. Для этого сделаем ещё одну вещь. Посмотрим, какие бывают карбонильные соединения.
Альдегиды проще кетонов, хотя бы потому что имеют только один заместитель на карбониле. Высокая электрофильность карбонильной группы делает их карбонильными компонентами в ненаправленной конденсации с енолизуемыми кетонами. С другой стороны, енолизуемые альдегиды – очень капризный тип реагента в альдольной конденсации, что в направленной, что в ненаправленной, из-за избыточно высокой электрофильности карбонила, что создаёт сложности когда мы хотим заставить альдегид быть метиленовой компонентой, нуклеофилом – он всё равно так и норовит вернуться к своей любимой роли карбонильной компоненты, электрофила. Единственный надёжный тип направленной конденсации двух енолизуемых альдегидов – кислотно катализируемая конденсация с простыми (силиловыми) эфирами енолов. Итак, альдегиды бывают
- енолизуемыми (ацетальдегид и т.п.)
- неенолизуемыми (бензальдегиды, триметилуксусный альдегид и т.п.)
- формальдегидом, а это, конечно, тоже неенолизуемый альдегид, но это совсем особая вещь, отец всех альдегидов, и мы его выносим в отдельную вкладку, в альдольной конденсации у него много особенностей.
Кетоны сложнее. Но и спокойнее. Они часто совсем не реагируют, если имеют проблемы со стерическим объёмом заместителей на карбониле, но если уж реагируют, обычно послушно следуют рекомендациям экспериментатора и распределению ролей в паре: сказали быть метиленовой компонентой, будет. У кетона, в отличие от альдегида, две руки – две группы, связанные с карбонилом. Каждая из них может быть енолизуемой или неенолизуемой. Поэтому кетоны могут быть
- полностью неенолизуемые (заместители на карбониле – фенилы, трет-бутил и другие трет-алкилы, и т.п.). Большинство таких кетонов относятся к большим, стерически затруднённым, таким, которые плохо реагируют с любыми нуклеофилами;
- енолизуемые с одной стороны (очень распространены, это, в частности арилалкилкетоны, получаемые ацилированием ароматических соединений)
- енолизуемые с двух сторон, симметричные, с двумя одинаковыми заместителями на карбониле – это очень популярная группа, к которой относятся знаменитые кетоны: ацетон, циклогексанон и другие циклоалканоны;
- енолизуемые с двух сторон, несимметричные (самый простой метилэтилкетон, но вообще таких кетонов очень много) – самая сложная и самая интересная группа кетонов.
Очень коротко свойства этих типов в альдольной конденсации можно свести в такой табличке
| Тип карбонильного соединения | Карбонильная компонента | Метиленовая компонента |
|---|---|---|
| енолизуемый альдегид | великолепная | отличная, но капризная, очень склонны к самоконденсации |
| неенолизуемый альдегид | великолепная | нет |
| формальдегид | лучше всех | нет |
| неенолизуемый кетон | очень плохая, обычно не реагирует | нет |
| енолизуемый с одной стороны | сильно уступает альдегидам | хорошо и удобно |
| енолизуемые симметричные | уступает альдегидам | хорошо и удобно |
| енолизуемые несимметричные | только в направленных конденсациях | только в направленных конденсациях |
Самоконденсация
Направленная и ненаправленная альдольно-кротоновая конденсация рассмотрена на соответствующих вкладках на странице про методы. А здесь пока подробнее поговорим про самоконденсацию. Это, с одной стороны, очень частный вопрос, поэтому не советую на него тратить время, если вы хотите ограничится общими представлениями об органической химии. Здесь это рассмотрено довольно подробно, и предназначено только для тех, кому любопытно, откуда взялись некоторые весьма знаменитые органические вещества. С другой стороны, многие закономерности, характерные для самоконденсации, так же проявляются и в ненаправленной конденсации разных карбонильных соединений (кросс-конденсации). Эти закономерности рассмотрены в основном на первых двух вкладках.
Альдольная самоконденсация
Енолизуемые альдегиды, симметричные енолизуемые кетоны и кетоны, енолизуемые с одной стороны вступают в реакцию самоконденсации, в которой одно и то же соединение является и карбонильной и метиленовой компонентой. Эта реакция требует или основного, или кислотного катализа и происходит в условиях равновесия. Реакцию иногда называют самоальдолизацией. Для этой реакции лучше использовать альдегиды без разветвления на α-атоме. Поскольку это реакция-прототип всей альдольной конденсации, это отчасти объясняет, почему появился термин метиленовая компонента. В старину сначала обнаружили именно самоконденсацию, и не могди не обратить внимание на то, что самоконденсируются только такие альдегиды или кетоны, у которых рядом с карбонилом метильная или метиленовая группа. Намного позже в проблеме разобрались получше, и выяснили что проблемы связаны с обратимостью альдольной реакции. Но термин остался. Альдегиды с разветвлением на α-углероде (прототип – изомасляный альдегид) дают более низкие выходы альдолей, и обычно требуют более тщательной оптимизации условий.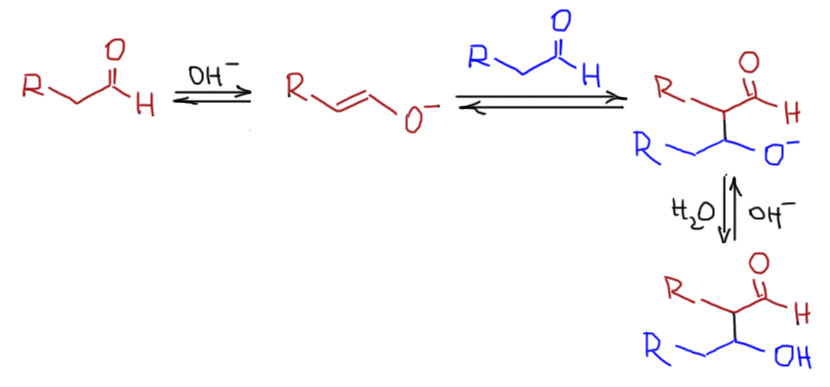
В условиях основного катализа реагирует равновесный енолят с самим карбонильным соединением. Алифатические альдегиды при этом дают альдоли. Самый простой енолизуемый альдегид – ацетальдоль, молекулу-прототип всех альдолей. 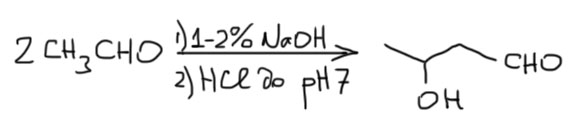 Альдоли простых альдегидов, особенно уксусного и масляного – очень важные промышленные полупродукты для органического синтеза. Катализаторы используются самые разные, это не так принципиально, поэтому всегда можно писать что-то типа щелочей или гидроксида кальция. Выбор конкретного катализатора в этих случаях – чисто техническая задача, и нам это не может быть интересно. После реакции, которая обыно протекает обчень быстро, буквально за несколько минут, смесь очень аккуратно подкисляют расчётнм количеством кислоты (или контролируя pH), так чтобы ни в коем случае не получить кислой реакции, в которой быстро происходит дегидратация альдоля.
Альдоли простых альдегидов, особенно уксусного и масляного – очень важные промышленные полупродукты для органического синтеза. Катализаторы используются самые разные, это не так принципиально, поэтому всегда можно писать что-то типа щелочей или гидроксида кальция. Выбор конкретного катализатора в этих случаях – чисто техническая задача, и нам это не может быть интересно. После реакции, которая обыно протекает обчень быстро, буквально за несколько минут, смесь очень аккуратно подкисляют расчётнм количеством кислоты (или контролируя pH), так чтобы ни в коем случае не получить кислой реакции, в которой быстро происходит дегидратация альдоля.
Получить альдоли – ресь здесь идёт именно об альдолях, а не о продуктах их дегидратации, про которые поговорим ниже – из енолизуемых кетонов в условиях самоконденсации очень сложно, почти невозможно. Равновесие самоконденсации кетонов смещено в сторону исходных кетонов. Это точно такая же ситуация, как и многие другие реакции присоединения нуклеофилов: равновесия с участием альдегидов смещены в сторону продуктов, в случае кетонов – к исходным. Чтобы всё же попробовать получить альдоль из кетона в условиях самоконденсации приходится применять ухищрения, достойные хитроумного Одиссея. Одно такое ухищрение хорошо известно и используется для получения альдоля из простейшего кетона, ацетона. По составу любой альдоль является димером исходного карбонильного соединения, а поскольку это очень древняя реакция, относящаяся к временам, когда основным инструментом анализа был элементный анализ и качественные реакции на функциональные группы, этот альдоль прозвали диацетоновым спиртом. Трюк состоит в использовании экстрактора Сокслета (для краткости эту штуку называют просто сокслетом), придуманного в конце 19 века пищевым химиком Францем фон Сокслетом для определения содержания жиров в продуктах. Экстрактор работает так; то, что нужно экстрагировать заворачивают в бумажку или кладут в специальную пористую гильзу и помещают в резервуар. Внизу колба с растворителем для экстракции, ее нагревают, растворитель кипит, пары поднимаются, конденсируются в обратном холодильнике, и растворитель понемногу заполняет резервуар экстрактора, растворяет то, что может растворить. Фокус Сокслета состоит в хитрой трубочке, которая петлёй соединяет резервуар экстрактора и нижнюю колбу. Пока растворитель ниже верхней петли, он накапливается в резервуаре и растворяет то, что нужно. Когда уровень доходит до верха петли, он начинает переливаться вниз, срабатывает эффект сифона и он весь сливается в нижнюю колбу. Дальше весь процесс – кипения, накопления растворителя в резервуаре, экстракции, сливания вниз – повторяется, и так далее столько раз, сколько нужно для того, чтобы экстракция произошла максимально полно, в идеале количественно. Аппарат работает фактически автоматически – зарядил, залил растворитель, пустил воду в холодильник, включил нагревание – и можно заниматься другими делами, эта штука работает сама.
К получению диацетонового спирта это приспособили очень просто. Вместо экстрагируемого вещества в резервуар поместили пакетик с катализатором, в качестве которого используют гидроксид бария (в некоторых разновидностях этой методики гидроксид кальция или даже карбид кальция, также являющийся основанием). В нижнюю колбу наливают ацетон и нагревают до кипения. Скапливающийся в резервуаре конденсат ацетона контактирует с основанием, идёт самоконденсация, но образующийся в резервуаре продукт, диацетоновый спирт, время от времени сливается в нижнюю колбу, а поскольку он намного менее летуч (молярная масса в два раза больше плюс водородные связи гидроксила), то в системе испаряется и путешествует вверх-вниз только летучий ацетон. Итого, в резервуаре продукт не накапливается, а накапливается в нижней колбе, где нет катализатора, при этом он выводится из контакта с катализатором, а значит, из реакции. Можно сказать, он таким своеобразным образом выпадает в осадок, то есть в нижнюю колбу. Равновесие, в полном соответствии с принципом Ле Шателье смещается в сторону продукта реакции. Поскольку катализатор не расходуется, теоретически такая штука может функционировать вечно, перерабатывая килограммы ацетона в диацетоновый спирт, но в реальности содержащийся в воздухе углекислый газ довольно быстро превратит поверхность гидроксида бария в инертный карбонат, да и другие неизбежные процессы будут снижать активность гидроксида на поверхности – реакция-то герерогенная и сильно зависит от качества этой самой поверхности.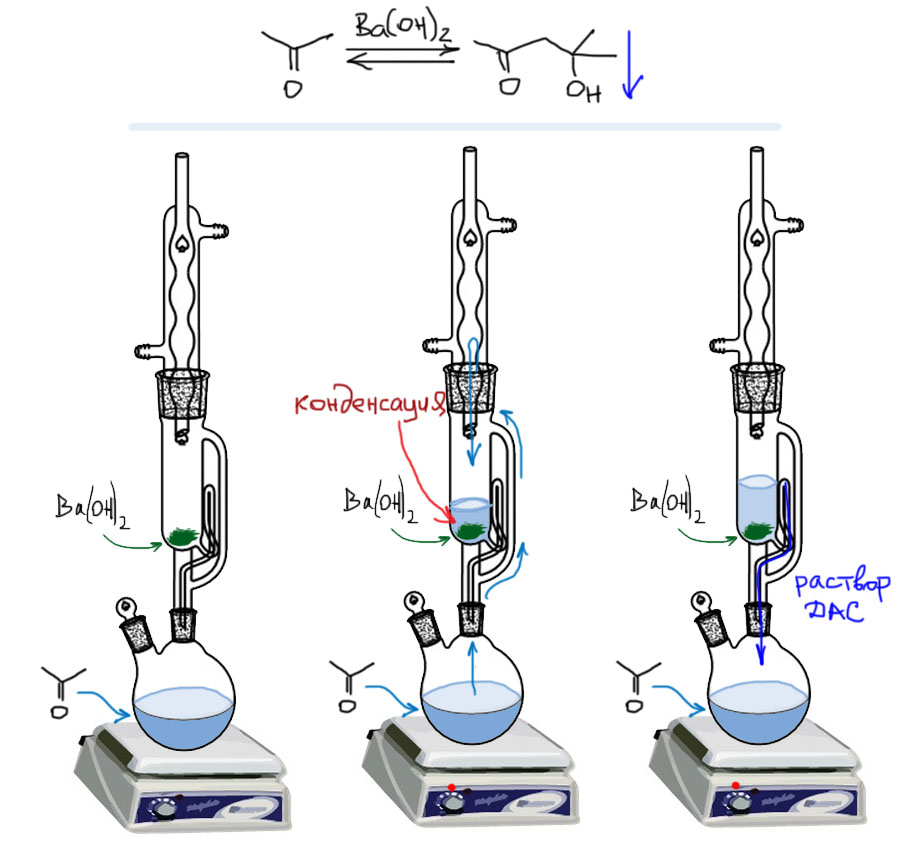
А можно ли сделать само-альдоли из других енолизуемых кетонов? Да, но придётся воспользоваться не самоконденсацией, а направленной конденсацией, превратив половину кетона в силиловый эфир енола или енолят. Тем же прибором воспользоваться не получится, так как все остальные кетоны, которые мы могли бы захотеть превратить этим способом в альдоли, больше ацетона по размеру и кипят при значительно более высокой температуре, и их просто или не удастся довести до кипения при атмосферном давлении, или температура реакции (а резервуар нагревается парами кипящей жидкости) будет слишком велика, что просто приведёт к осмолению.
Альдольно-кротоновая самоконденсация в продукты дегидратации альдолей
Дегидратация альдоля делает саму конденсацию практически необратимой, хотя каждая из стадий принципиально обратима, но равновесие дегидратации/гидратации смещено уже в сторону продукта дегидратации, и это смещает весь процесс в сторону этого продукта. В терминах констант равновесий это означает, что константа равновесия образования альдоля можеб быть больше единицы (для альдегидов), или меньше единицы (для кетонов), но в любом случае это равновесие подвижно и никогда не бывает сильно смещено в сторону продуктов. А вот дегидратация альдоля – это уже равновесие, сильно смещённое в сторону продукта дегидратации (константа равновесия сильно больше единицы), и к тому же, в эту константу в числителе входит еще вода, которую легко удалять (она даже может это делать сама выделяясь в отдельную несмешивающуюся фазу), а тогда общее равновесие образование продукта дегидратации альдоля совсем смещается в сторону этого продукта еще и с помощью Ле Шателье. 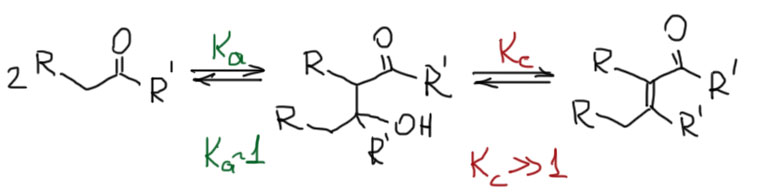
Конденсация с образованием продукта дегидратации – буду называть её кротоновой конденсацией, нужно же использовать преимущества родного языка, ведь очень редко бывает так, что по-русски короче, чем по-английски – требует или кислотного или основного катализа. Обратите на это внимание – речь сейчас идёт только о самоконденсации, не пытайтесь обобщить то, что мы узнаем про самоконденсацию, на конденсацию двух разных карбонильных соединений (перекрёстную или, по-другому, кросс-конденсацию) – там очень много существенных нюансов. В самоконденсации
- кислотный катализ всегда приводит к продукту дегидратации альдоля, иными словами, кислотно-катализируемой всегда является кротоновая конденсация, хотя обратное неверно так как
- основный катализ приводит к альдолю в уже рассмотренных выше случаях, во всех других, а именно в самоконденсации енолизуемых кетонов, образуются продукты дегидратации даже в присутствии оснований, а это означает, что кротоновая конденсация вполне может катализироваться основаниями.
Почему кислотный катализ приводит к кротоновому продукту довольно ясно. Для простоты запишем механизм для самого простого варианта кислотного катализа – за счёт протонирования сильной кислотой (напомню, что этот вариант кислотного катализа принято называть специфическим). Кислота катализирует как превращение кето-формы в енол, так и собственно конденсацию енола с кето-формой, электрофильность которой повышается при протонировании, что делает ее способной реагировать с енолом. Первоначальный продукт, протонированный альдоль, после переноса протона, обратимо теряет воду и превращается в продукт дегидратации. Последнее равновесие, хоть и настоящее, но практически полностью смещено вправо в сторону конечного продукта, что делает всю реакцию фактически необратимой. 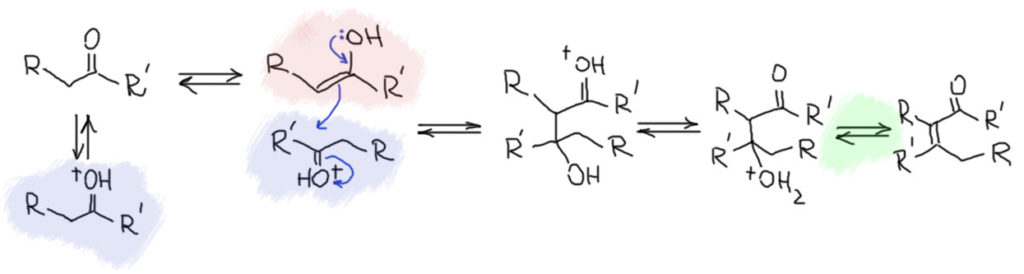 Кроме этого еще бывает вариант катализа слабой кислотой, за счёт активации водородными связями – в кротоновой конденсации это работает всё же редко, поэтому оставим. И ещё есть катализ кислотами Льюиса – всякими солями и комплексами металлов, работающих за счёт координации металла по атомам кислорода: это очень распространённый вариант и самоконденсации и кросс-конденсации, но мы оставим его до обсуждения направленной конденсации.
Кроме этого еще бывает вариант катализа слабой кислотой, за счёт активации водородными связями – в кротоновой конденсации это работает всё же редко, поэтому оставим. И ещё есть катализ кислотами Льюиса – всякими солями и комплексами металлов, работающих за счёт координации металла по атомам кислорода: это очень распространённый вариант и самоконденсации и кросс-конденсации, но мы оставим его до обсуждения направленной конденсации.
А вот почему основный катализ может приводить и приводит к продукту дегидратации. О, это очень просто. Надо просто никогда не забывать про законы равновесия. И помнить, что равновесие одно, от катализа оно (положение равновесия) не зависит, хотя скорость достижения равновесия, естественно, зависит. И даже детали механизма тоже от катализа зависят. И это тоже понятно, механизм влияет на скорость, но никак не влияет на равновесие (положение равновесия или, что то же самое, константу равновесия, если быть точным). Запишем механизм, использовав в качестве основания гидроксид-ион. Можно было бы и другое основание взять, это не принципиально. Итак, при оновном катализе обратимо образующийся енолят реагирует с самим карбонильным соединением, образуя альдоль. И если мы запишем механизм отщепления воды от альдоля, мы в очередной раз устроим себе встречу со старым знакомым. Всё в химии взаимосвязано: отщепление от альдоля это обратная реакция к 1,4-присоединению гидроксид-иона к ненасыщенному карбонильному соединению. Более того, если мы ещё раз прочитаем в обратную сторону 1,4-присоединение, то увидим, что это не что иное как элиминирование по механизму E1cb – помните такой? Отщепление от сопряжённого основания, в данном случае это енолят альдоля, и именно енолят альдоля образуется при присоединении гидроксид-иона к двойной связи карбонильного соединенения. Круг замкнулся. Теперь нам точно понятно, почему в присутствии основания происходит дегидратация точно так же, как в присутствии кислот, хотя и по другому механизму. 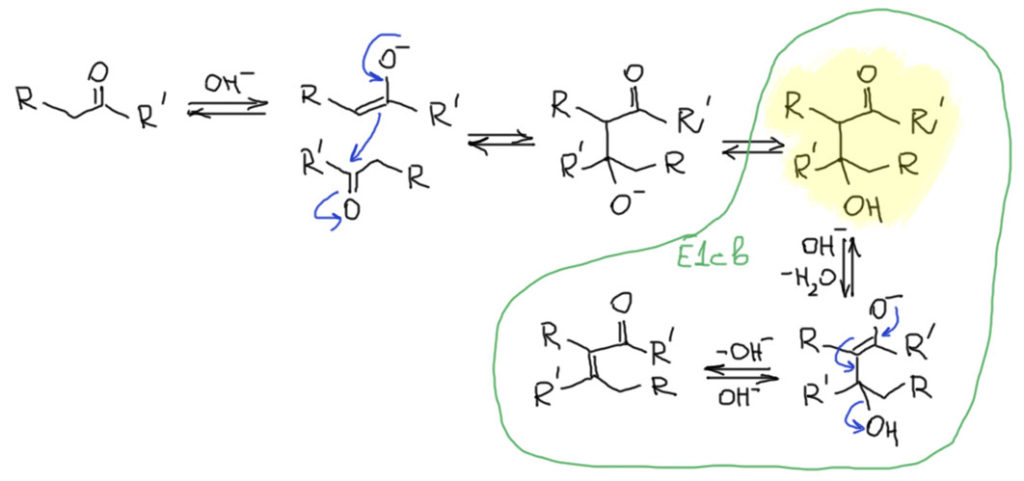
В случае кетонов первое равновесие смещено в сторону исходных кетонов, альдоля в равновесной смеси мало. Поэтому, что бы мы не делали, на альдоле мы остановиться не можем. Но равновесие альдоль-дегидратированный альдоль смещено уже в сторону второго. Понятно почему: возникает сопряжённая двойная связь, да ещё и сильно замещенная – ведь когда реагируют кетоны, все заместители на образующейся двойной связи не водороды. Это тоже стабилизирующий эффект. В принципе, могла бы вмешаться стерика, но до этого дело доходит редко, так как кетоны с большими заместителями вообще не очень охотно вступают в реакции альдольной конденсации, особенно ненаправленной.
На практике кислотный катализ для кротоновой конденсации (самоконденсации!) используют чаще, чем основный. Для альдегидов это почти единственный выбор получить кротоновый продукт сразу, а не дегидратацией альдоля, то есть в две стадии. Для кетонов возможны оба типа катализа. Используют как самые простые катализаторы типа раствора HCl в этаноле (это можно получить очень просто, добавив к этанолу любое вещество, которое с ним бурно реагирует с выделением HCl – хлористый тионил, триметилхлорсилан, безводный хлорид алюминия), так и много численные кислоты Льюиса типа TiCl4 или алкоксилатов алюминия.
Для простых альдегидов кротоновая конденсация идёт легко в присутствии самых разнообразных кислот. Самый простой енолизуемый альдегид даёт кротоновый альдегид в полном смысле этого слова. Кротоновый альдегид настолько реакционноспособен, что очень легко конденсируется дальше во всевозможные продукты и по механизму альдольно-кротоновой конденсации, и через присоединение по Михаэлю, поэтому в промышленности, где очень не любят реакции с большим количеством побочных продуктов, предпочитают альдольную конденсацию в присутствии водной щёлочи, и последующую дегидратацию ацетальдоля. 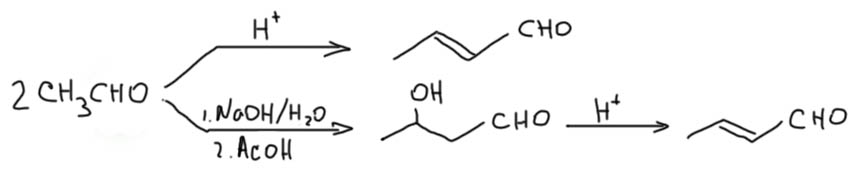 Кротоновый альдегид производят в промышленности в немаленьких количествах, и используют дальше в многочисленных синтезах всяких полезных веществ, в первую очередь сорбиновой кислоты, самого популярного пищевого консерванта (поэтому у неё и её солей международные Е-индексы, Е200-Е203 идут в самом начале раздела консервантов).
Кротоновый альдегид производят в промышленности в немаленьких количествах, и используют дальше в многочисленных синтезах всяких полезных веществ, в первую очередь сорбиновой кислоты, самого популярного пищевого консерванта (поэтому у неё и её солей международные Е-индексы, Е200-Е203 идут в самом начале раздела консервантов).
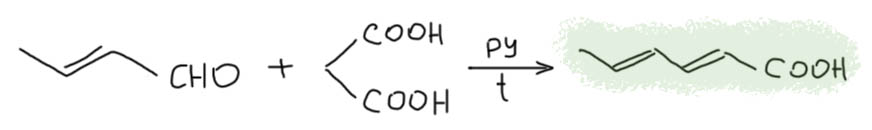
Но самая важная альдольно-кротоновая конденсация в промышленности не получение кротонового альдегида из ацетальдегида, а конденсация н-масляного альдегида (который, кстати, ещё не так давно получали гидрированием кротонового альдегида, но в 21 веке перешли на гидроформилирование пропилена в присутствии родиевого катализатора). Масляный альдегид тоже предпочитают конденсировать в две стадии, через промежуточный альдоль – продукт получается чище. Продукт конденсации в основном гидрируют в 2-этилгексиловый спирт.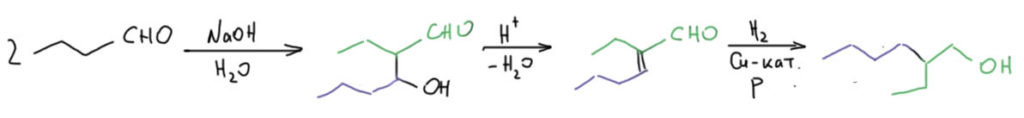 Это с виду странный спирт – один из самых крупнотоннажных продуктов мировой химической промышленности, в год его производят около 5 миллионов тонн, больше чем большинство лругих спиртов за исключением метилового, этилового и бутилового, да и от них отстаёт совсем немного. Если будете заниматься органической химией, особенно такой, которая имеет практическое применение, не раз и не два столкнётесь с этим остатком. Эту штуку используют везде, где нужно придать какому-то веществу или композиции повышенную растворимость в неполярных средах (маслах, жирах, и т.п.), уменьшить вязкость, увеличить пластичность и т.п.
Это с виду странный спирт – один из самых крупнотоннажных продуктов мировой химической промышленности, в год его производят около 5 миллионов тонн, больше чем большинство лругих спиртов за исключением метилового, этилового и бутилового, да и от них отстаёт совсем немного. Если будете заниматься органической химией, особенно такой, которая имеет практическое применение, не раз и не два столкнётесь с этим остатком. Эту штуку используют везде, где нужно придать какому-то веществу или композиции повышенную растворимость в неполярных средах (маслах, жирах, и т.п.), уменьшить вязкость, увеличить пластичность и т.п.
Кротоновая самоконденсация ацетона и вопросы языкознания. И ещё немного истории, местами даже детективной.
Самоконденсация кетонов хоть с кислотным, хоть с основным катализом практически всегда даёт продукты дегидратации, и мы уже обсудили почему. Среди таких продуктов много весьма знаменитых и полезных веществ.
Особенно плодотворен на продукты кротоновой самоконденсации ацетон. Ацетон очень легко вступает в самоконденсацию в присутствии сильных кислот, образуя целый спектр интересных и полезных продуктов. Самый простой продукт носит название окиси мезитила. Почему мезитила, какая к чёрту окись, что за странное название? Это такой исторический след, очень занятный. Ацетон одним из первых получил прусский естествоиспытатель и изобретатель очень модных нынче шарлатанских идей про витальную силу и прочий вздор барон Карл фон Райхенбах при нагревании древесины, перегоняя получившуюся жидкую смесь он выделил промежуточную фракцию, которую и назвал мезит, что как раз и значит “промежуточный”. Серьёзные химики в 1830-х стали разбираться и нашли, что это то же самое, что получается при сильном нагревании солей уксусной кислоты, и по этой причине получило название пироуксусный спирт. Слово “ацетон” тогда тоже уже было, но им называлось не конкретное соединение, а любые продукты, получающиеся при сильном нагревании солей органических кислот. Первый такой продукт был получен из уксусной кислоты, откуда, собственно, и возникло само слово ацетон, только понимали его тогда не как название конкретного соединения, а как такой общий термин – всякое вещество, получающееся при термическом разложении солей органических кислот. В таком смысле продукт из солей уксусной кислоты называли конкретнее, не просто ацетон, а “уксусный ацетон”, и в последующие лет 50 название ацетоны сначала было единственным для обозначения таких веществ, потом возникло понятие структуры и классов органических соединений, параллельно возникло слово кетоны буквально как синоним слова ацетоны (собственно, это просто сокращение слова ацетоны, появившееся сначала у французов – acétone/cétone, но немцы заменили во втором слове це на к, оттуда и пошло всем известное название класса), и только с конца 19 века ацетон стал ацетоном, а названием класса стало только “кетоны”.
Дальше всех продвинулся ирландский учёный и политик сэр Роберт Кейн в конце 1830-х. В те времена органических соединений стало известно уже довольно много, для каждого из них определяли элементный состав и обнаружили, что в органических соединениях часто встречаются повторяющиеся куски, а сами молекулы как будто представляют собой комбинации этих кусков – радикалов. И каждый хотел найти какой-то новый кусок, новый радикал. Вот Кейн взялся за пироуксусный спирт, он же ацетон, он же мезит, и представил его формулу как C3H6O = C3H5-OH, и назвал то, что ему показалось новым радикалом C3H5 – мезитилом. И получилось у него поэтому, что ацетон следует называть мезитиловым спиртом. Тогда никто не вкладывал в слово спирт понятие “представитель класса спиртов”, а просто комбинировали радикал с остатком OH, да и вообще спиртами обычно называли легколетучие органические вещества, получаемые при нагревании какого-нибудь материала (от первоначального смысла слова спирт – дух, нечто летучее). И захотел молодой профессор Кейн найти другие производные радикала мезитила. И добавил к ацетону серной кислоты и стал это перегонять. И получил продукт кротоновой конденсации и определил его состав. Мы его тоже знаем – это два ацетона минус вода, то есть C6H10O, а это просто раскладывается как C3H5-O-C3H5. Сейчас бы мы это могли попробовать назвать эфиром, если бы еще знали структуру и были уверены, что два остатка связаны с одним кислородом, но тогда ничего подобного известно не было, а к анализу – даже не формул, а просто состава – подходили как к паззлу “найди повторяющиеся кусочки и подгони друг к другу”, и в таких ситуациях по аналогии с соединениями элементов использовали слово окись (оксид). Вот и получилась окись мезитила, и живёт с нами уже 180 лет – это одно из самых старых названий органических соединений. Пытался Кейн получить и другие производные мезитила, нагревал ацетон с хлористым водородом, надеясь получить хлорид мезитила – не получил. Ещё разные опыты, с тем же успехом. Но ещё один раз ему повезло. Добавил он побольше серной кислоты, всё почернело и заколдобилось, но из этой мерзкой жижи смог отогнать он соединение, которое мы нынче знаем, как 1,3,5-триметилбензол. Состав его C9H12, но это нам понятно, а в те времена ещё не умели определять молярную массу негазообразных веществ (напомню, что молярная масса газов определяется по относительной плотности по воздуху или другому известному простому газу, это работает для газов и легколетучих веществ, которые можно легко превратить в пар, в частности, плостность самого ацетона в газовой фазе была уже тогда определена очень точно). Поэтому получилось, что состав соотвествует формуле C3H4, а это C3H5 – H. Радикал минус водород тогда называли окончанием -илен (этил – этилен, ацетил – ацетилен и т.п.), вот и получился мезитилен. И это название дошло до нас, и даже узаконено ИЮПАКом. Из всего этого следует и то, что Кейну принадлежит приоритет в открытии альдольно-кротоновой конденсации, но так как в те времена еще никто ничего не знал о том, как идут органические реакции и что такое вообще структура, он не мог должным образом описать своё открытие, и приоритет этот историей не зафиксирован: альдольно-кротоновую конденсацию переоткрыли несколько десятилетий спустя уже с большим знанием дела сразу несколько учёных, среди которых и Бородин, и Кьёцца, и Вюрц, и Кляйзен, и Фиттиг, так что это в некотором смысле совместная ирландско-итальянско-русско-немецко-французская разработка. Про Кейна, впрочем, забыли ещё и потому, что он забросил химию ради политики раньше, чем эта самая химия стала серьёзной наукой с мощным научным сообществом – поэтому он не успел вписаться в это сообщество, и был забыт. Очень жаль, потому что статья про мезитил у него написана в 1839 году так чётко и аккуратно, что местами читается почти как современная.
Вернёмся к химии. Добавление сильных кислот или оснований к ацетону приводит к самоконденсации, но у этой реакции никогда не бывает одного продукта. Первый продукт самоконденсации, окись мезитила, быстро подхватывает ещё одну молекулу ацетона и процесс идёт дальше – образуется смесь. Из этой смеси довольно легко отогнать окись мезитила, она кипит около 130ºС а всё остальное – гораздо выше, но выходы не будут очень высоки. Поэтому препаративно окись мезитила предпочитают получать дегидратацией альдоля – диацетонового спирта, который, как мы знаем, можно получить в чистом виде довольно легко. Эта реакция очень легко происходит при нагревании в присутствии буквально нескольких крупинок иода. Молекулярный иод часто используют как катализатор дегидратации в тех случаях, когда дегидратация очень выгодна и легко идёт, например, образуется сопряжённая двойная связь. Механизм действия иода до конца до сих пор не понятен, но часто считают, что дело в образовании каталитических количеств сильной кислоты, HI, то есть реакция идёт как кислотно-катализируемая дегидратация. В то же время иод не вызывает побочных реакций, по сравнению со специально добавленной сильной кислотой, например, серной, фосфорной или толуолсульфоновой, в присутствии которых окись мезитила, как енолизуемый кетон, самоконденсируется дальше. 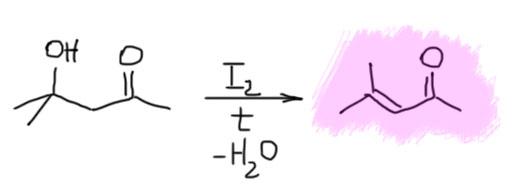
Просто при самоконденсации ацетона в присутствии сильных кислот получается немало второго продукта самоконденсации. Это соединение, которое называется форон, а почему, узнаем ниже, – получается очень просто: это продукт кротоновой конденсации окиси мезитила с ацетоном по свободной метильной группе. Именно ацетон здесь вступает карбонильной компонентой, и мы знаем почему – ацетон это маленький кетон без стерических проблем с более доступной карбонильной группой. К тому же у окиси мезитила карбонил сопряжён с двойной связью, а это снижает электрофильность карбонильной группы. Поэтому в этом случае фактически идёт перекрёстная конденсация двух разных енолизуемых кетонов, но она оказывается довольно селективной. Это не значит, что эта реакция обладает высокой селективностью, в таких конденсациях всегда много побочных продуктов, но основной продукт вполне неплохо получается. Впрочем мы уже скоро выясним, что в этой смеси всё-таки есть ещё один продукт, и он как раз получается, когда ацетон и окись мезитила меняются ролями в кротоновой конденсации. 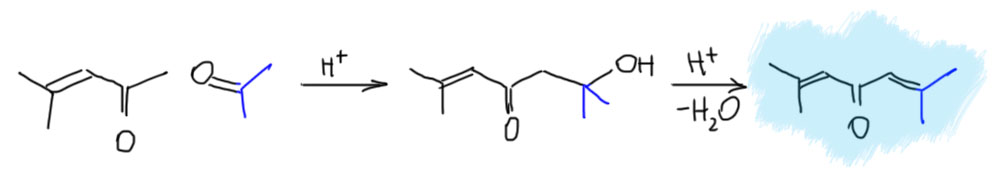
Кислотно катализируемую конденсацию ацетона очень хорошо делают в промышленных условиях, используя всякие кислотные катализаторы, как любят в промышленности, какие-нибудь твёрдые, гетерогенные, чтобы легко можно было удалять катализатор из продуктов реакции. Мы не будем заморачиваться конкретными условиями, а просто покажем обобщённую сильную кислоту – в этом случае это совсем не принципиально, а если вам захочется, в литературе вы найдёте десятки конкретных условий, отличающихся природой кислоты, температурой, временем и прочими условиями, но с приблизительно одинаковым результатом – образуется смесь окиси мезитила и форона плюс немало тёмной зловонной смолы. Всё это перегоняют, выделяют непрореагировавший ацетон, чистую окись мезитила (основной продукт всегда – до 60-70%) и чистый форон (второй продукт, от 10 до 30%). Оба эти вещества весьма востребованы как полупродукты тонкого органического синтеза, из них делают много полезного. 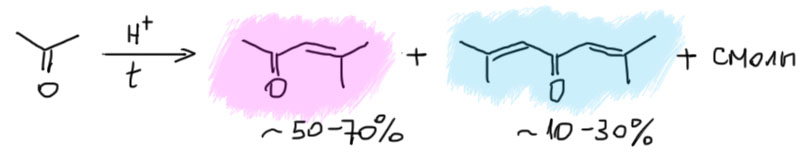
Ещё в глубокую старину в труде ирландца Кейна было известно, что если конденсацию вести максимально жёстко, при добавлении весьма большого количества концентрированной серной кислоты и выдерживания такой смеси в течение нескольких дней, получается омерзительная зловонная чёрная жижа. В этой жиже всё что могло осмолиться уже осмолилось, и не осмолилось только то, что не может просто так осмолиться. Скорее всего там ничего такого и быть не может, ведь и исходное, и все известные нам продукты – весьма реакционноспособные вещества, кетоны, еноны, как они могут не осмолиться в концентрированной серной кислоте, ведь каждый из них способен конденсироваться дальше, до бесконечности – каждый имеет и карбонил и енолизуемые положения, как это можно остановить.. Чтобы завершить славное дело полного осмоления, в конце смесь еще и нагревают, и на всякий случай пробуют из неё что-то отогнать, просто так или с водяным паром. Отчаянная попытка – что там может быть, черна и зловонна бездна, в аду такого, наверно, не встретишь, а если встретишь, то это было бы хорошим местом для некоторых особенно отмороженных злодеев, мы знаем их имена. Мы вообще-то уже знаем, что может выжить в несовместимых с жизнью условиях – простая ароматика, бензол с несложными заместителями типа метилов. Но откуда там это? Посмотрим-ка на возможный продукт кротоновой конденсации ацетона с окисью мезитила, в которой участники поменялись ролями – ацетон стал метиленовой компонентой, а окись мезитила – карбонильной. Вот он, рядом с обычным продуктом, фороном. Красивого названия у него нет, потому что он быстро превращается дальше и из смеси не выделяется.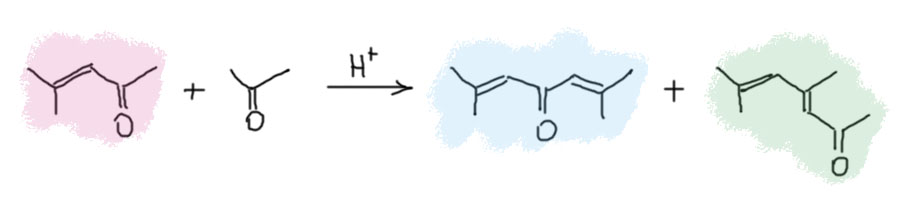 Так мы получили ненасыщенный кетон, у которого есть возможность вступить в ещё одну кротоновую конденсацию, причём внутримолекулярную, но при этом немного необычную. В ненасыщенных карбонильных соединениях возможна енолизация с участием не только α-водородов, но и атомов водорода, отделённых от карбонила двойными связями. Это явление раньше любили называть винилогией, по аналогии с гомологией, но только когда разница между двумя молекулами не метиленовая, а винильная группа. Как мы знаем, в таких случаях такая вставная двойная связь работает как удлинитель сопряжения – проводник мезомерного эффекта. Посмотрим на простом примере, сравнив уксусный и кротоновый альдегиды. Как видим, хотя у кротонового альдегида нет α-водородов, но енол он образует отлично, и этот енол вовлекает в дело очень далёкий от карбонила атом – γ-углерод, который также становится нуклеофильным. Есть некоторый соблазн назвать такой удлиннённый енол диенолом, но так обычно не делают, и так всё понятно. Точно такие же фокусы можно было бы разрисовать и для енолята, и для енамина, и для эфира. Мы не будем слишком глубоко это копать, потому что у таких соединений весьма непростая химия, которая не сводится просто к удлинению – у них возникает дополнительный нуклеофильный центр, а значит и конкуренция, и проблема селективности, в общем мороки на порядок больше.
Так мы получили ненасыщенный кетон, у которого есть возможность вступить в ещё одну кротоновую конденсацию, причём внутримолекулярную, но при этом немного необычную. В ненасыщенных карбонильных соединениях возможна енолизация с участием не только α-водородов, но и атомов водорода, отделённых от карбонила двойными связями. Это явление раньше любили называть винилогией, по аналогии с гомологией, но только когда разница между двумя молекулами не метиленовая, а винильная группа. Как мы знаем, в таких случаях такая вставная двойная связь работает как удлинитель сопряжения – проводник мезомерного эффекта. Посмотрим на простом примере, сравнив уксусный и кротоновый альдегиды. Как видим, хотя у кротонового альдегида нет α-водородов, но енол он образует отлично, и этот енол вовлекает в дело очень далёкий от карбонила атом – γ-углерод, который также становится нуклеофильным. Есть некоторый соблазн назвать такой удлиннённый енол диенолом, но так обычно не делают, и так всё понятно. Точно такие же фокусы можно было бы разрисовать и для енолята, и для енамина, и для эфира. Мы не будем слишком глубоко это копать, потому что у таких соединений весьма непростая химия, которая не сводится просто к удлинению – у них возникает дополнительный нуклеофильный центр, а значит и конкуренция, и проблема селективности, в общем мороки на порядок больше.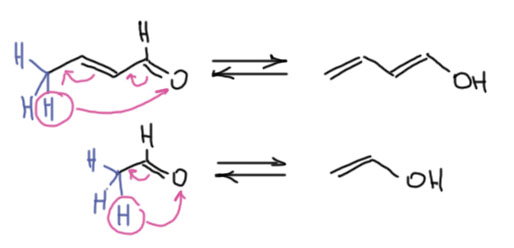
Зато мы теперь понимаем, что в альтернативном продукте кротоновой конденсации окиси мезитила и форона очень даже возможна внутримолекулярная кротоновая конденсация как раз на таком удалённом нуклеофильном центре с образованием того, что мы, а за нами и Природа, так любим – шестичленный цикл, и этот цикл оказывается ароматическим. Перерисуем структуру так, чтобы этот путь был хорошо виден, и получим этот интересный результат – внутримолекулярная кротоновая конденсация может дать продукт, не являющийся карбонильным соединением. Да, нам придётся не просто перерисовать, но и понять, что речь идёт о транс- и цис-изомерах, то есть о настоящей изомеризации, но это не проблема – во-первых, мы ничего не знаем про то, какой изомер там получился, во-вторых, сопряжённые двойные связи слабее обычных и изомеризуются довольно легко, в-третьих, условия у этой реакции достаточно жёсткие. Вряд ли это может быть помехой. Рисуем.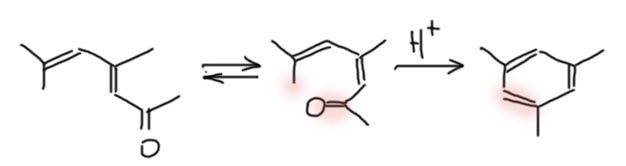
Всё вроде красиво. Но человек внимательный не может не насторожиться. Постойте-ка, скажет она или он, что-то тут не так. Чтобы произошла альдольно-кротоновая конденсация нам нужен карбонил и нуклеофильная форма енолизуемого соединения, пусть енол. Но они нужны одновременно. А поскольку оба этих фрагмента в этом случае находятся в одной молекуле, они зависят друг от друга. И там, где енол, нет карбонила, а там, где карбонил, нет енола. И как же тогда пойдёт конденсация??? Караул, опять жулики и воры впаривают нам туфту, а мы, доверчивые, чуть не купились на эту “красоту”. Где красота, там обман, не правда ли?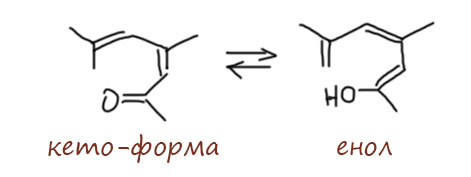
Так, но реакция-то идёт. Продукт циклизации получается. Придётся придумать что-то другое. Возможно, вот что. Уйдём совсем от химии енолов и карбонильных соединений. В химии очень много реакций, в которых что-то интересное происходит, когда встречаются несколько двойных связей в такой ситуации, когда возможно одновременное перемещение шести электронов в циклическом переходном состоянии, что приводит к согдасованному разрыву и образованию связей. Такие реакции называются перициклическими. С одной из них мы хорошо знакомы – это реакция Дильса-Альдера. Есть и другие, например, вот такая: 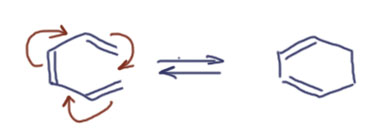
Это называется без затей гексатриен-циклогексадиеновой перегруппировкой. Это довольно редкая реакция, идёт она почти всегда только в сторону цикла просто при нагревании сопряжённого гексатриена. Не будем подробно на ней останавливаться, в программе у нас её нет и не предвидится, хотя одна очень похожая перегруппировка у нас есть и мы ей будем заниматься, может и про эту ещё вспомним. Давайте теперь нарисуем, что будет, если такая реакция произойдёт с енолом, который мы уже нарисовали. 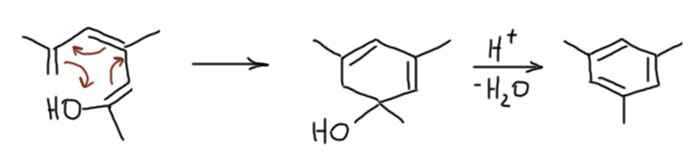 Как видим, всё отлично. Перегруппировка даёт спирт, который безусловно в кислой среде тут же выбросит воду с образованием ароматической системы, и получится тот же мезитилен, только уже без необходимости применять негодный механизм и делать вид, что всё в порядке. Хороша наука химия – какая-нибудь лазейка найдётся всегда, стоит только поискать! И так и должно быть – ведь главное в том, что реакция идёт, это экспериментальный факт, а значит должно быть и убедительное объяснение (механизм) почему.
Как видим, всё отлично. Перегруппировка даёт спирт, который безусловно в кислой среде тут же выбросит воду с образованием ароматической системы, и получится тот же мезитилен, только уже без необходимости применять негодный механизм и делать вид, что всё в порядке. Хороша наука химия – какая-нибудь лазейка найдётся всегда, стоит только поискать! И так и должно быть – ведь главное в том, что реакция идёт, это экспериментальный факт, а значит должно быть и убедительное объяснение (механизм) почему.
В условиях основного катализа ацетон даёт немного другие продукты. Сначала всё равно образуется окись мезитила. И к ней тоже пристраивается ещё одна молекула ацетона, но поскольку реакция происходит в присутствии основания, то не в виде енола, а в виде более нуклеофильного енолята. А енолят к енону, как мы знаем, присоединяется не по 1,2-пути, а по 1,4-пути. 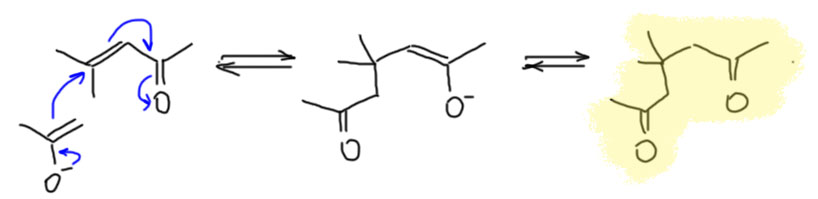 И продукт такой реакции – дикетон, будет иметь карбонил и енолизуемое положение (карбонильную и метиленовую компоненту) на таком расстоянии, что внутримолекулярная альдольная конденсация приведёт к образованию 6-членного цикла, что очень выгодно. Получится такой ненасыщенный кетон, циклогексенон с метилами. Вообще, это не что иное как аннелирование по Робинсону ацетона окисью мезитила. И хотя, безусловно, самокондесация ацетона в основной среде не может быть селективной, и в реальности получается сложная смесь продуктов, но при тщательной оптимизации условий, времени реакции, соотношения реагентов и катализатора, изофорон получается фактически как основной продукт. В промышленности для производства этого производного циклогексенона ацетон конденсируют в присуьствии небольшого количества водной щёлочи, и продукты реакции подвергают аккуратной фракционной перегонке.
И продукт такой реакции – дикетон, будет иметь карбонил и енолизуемое положение (карбонильную и метиленовую компоненту) на таком расстоянии, что внутримолекулярная альдольная конденсация приведёт к образованию 6-членного цикла, что очень выгодно. Получится такой ненасыщенный кетон, циклогексенон с метилами. Вообще, это не что иное как аннелирование по Робинсону ацетона окисью мезитила. И хотя, безусловно, самокондесация ацетона в основной среде не может быть селективной, и в реальности получается сложная смесь продуктов, но при тщательной оптимизации условий, времени реакции, соотношения реагентов и катализатора, изофорон получается фактически как основной продукт. В промышленности для производства этого производного циклогексенона ацетон конденсируют в присуьствии небольшого количества водной щёлочи, и продукты реакции подвергают аккуратной фракционной перегонке. 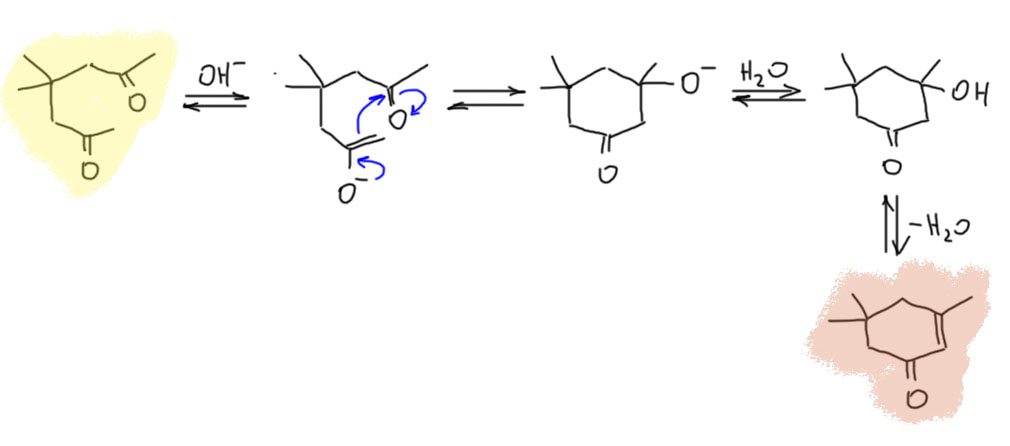
Фокус в том, что этот продукт имеет тот же состав, что и форон, продукт конденсации ацетона в кислой среде, это изомер форона. И очень долго эти два продукта вообще не могли различить – состав у них одинаковый и кипят они почти при одной и той же температуре. Нагревали ацетон кто с кислотами, кто с щелочами, после реакции перегоняли и получали практически одинаковые фракции – и по температуре кипения, и по элементному составу. Почти весь 19 век их считали одним и тем же веществом, и полагали, что уксусный ацетон и в присутствии щелочей, и в присутствии сильных кислот даёт одно и то же – окись мезитила из двух молекул ацетона и продукт из трёх молекул ацетона. Как же он называется?
О, это детективная история – история вещества-самозванца, позаимствовавшего чужое имя. Этой истории больше 150 лет и она до сих пор не закончена – это можно легко увидеть, если посмотреть статьи в Википедии про соединение, называемое “форон”. Что за форон такой? Читаем википедию: это продукт самоконденсации ацетона – тот самый, который получается в кислой среде. Хорошо, это мы и так знаем, а почему всё таки название такое загадочное – почему форон? А потому что какие-то древние французы в глубокой старине, когда было принято все новооткрытые органические кислоты в виде кальциевых солей как следует нагревать, получили это соединение нагреванием кальциевой соли камфорной кислоты. Сначала они назвали это соединение камфорилом, а чуть позже вспомнили, что такие вещества стало принято называть ацетонами, то есть это по признаку реакции, в которой он получен, камфорный ацетон. Чуть позже сократили в камфорон, и довольно быстро появилась и совсем краткая форма – форон, эти названия стали употреблять вперемешку. Через некоторое время это вещество сравнили с продуктом самоконденсации настоящего ацетона (уксусного ацетона, по тем понятиям). Состав у них оказался одинаковый, кипят они очень близко, по тем временам можно было сказать, что одинаково. И хорошо нам знакомый Рудольф Фиттиг (реакция Вюрца-Фиттига никому не нужна, но исторически очень важна, а ещё Фиттиг открыл пинакон и пинакон-пинаколиновую перегруппировку, правда не смог в ней разобраться) предположил, что это одно соединение. Это было почти ровно 150 лет назад. И судя по главе в Википедии, оказался прав, потому что она этот вывод добросовестно повторяет. И не одна, а вмесие с еще немаленьким количеством всяких книжек и справочников.
Нам остаётся или поверить, или всё же попробовать понять, что на самом деле. Просто потому что камфорная кислота как-то совсем не вяжется с кродуктами кротоновой самоконденсации ацетона. И вот что получается. Камфорная кислота образуется из природного кетона, камфоры, как говорили в старину, энергичным окислением. При этом сгорает та часть камфоры, в которой находится карбонильная группа, а всё остальное – насыщенный цикл без двойных связей и легкоокисляемых групп остаётся нетронутым. 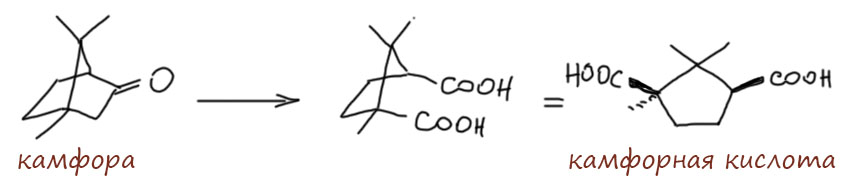
А теперь попробуйте представить себе, как из камфорной кислоты может получиться форон. При нагревании кальциевых солей обычно получаются кетоны. Это одна из древнейших органических реакций, известных ещё в 1830-х, когда всё делали совсем наощупь, не имея никаких методов исследования кроме определения соотношения элементов. И, как уже было замечено, вначале продукты таких реакций называли ацетонами, а слово кетон появилось много позже, когда уже более-менее стали представлять структуру. Итак, некие французы на заре органической химии нагрели кальциевую соль камфорной кислоты и получили нечто, названное камфорным ацетоном, или камфороном. И как на беду, состав этого соединения оказался таким же как у продукта самоконденсации настоящего ацетона (в те времена известного как пироуксусный спирт или мезитиловый спирт или уксусный ацетон, в зависимости от того, каким методом он был добыт), и температура кипения тоже (что не удивительно, изомеры, особенно соединений одного класса кипят очень близко). Это заблуждение царило весь 19 век, хотя время от времени кто-то начинал сомневаться, что продукт из камфорной кислоты – то же самое соединение, что и продукт самоконденсации ацетона. Но до открытия современных методов исследования структуры почти единственным способом разбираться, что происходит в реакциях было выделение продуктов, которые подвергались простым превращениям – гидрированию, окислению, получению простых производных – с буквальным сравнением внешнего вида и физических констант получаемых соединений. Объем данных быстро накапливался, сравнения становились всё более точными и безошибочными, но и ошибок оставалось довольно много – физические константы для близких по структуре соединений, тем более изомерных, часто очень близки.
Если мы посмотрим современными глазами на то, что может получиться при пиролизе солей камфорной кислоты, получим, как из других двухосновных кислот, циклический кетон. У этого кетона будет небольшая проблема – он получится бициклическим, и один из его циклов окажется четырёхчленным, то есть достаточно неустойчивым, сильно напряжённым. Поэтому неудивительно, что в весьма жёстких условиях пиролиза такой цикл лопнет, образуются два радикала, которые обменяются атомами водорода, и получится конечный продукт довольно простого строения – производное циклопентанона. Такой кетон тоже был получен независимо из других исходных, а в начале 20 века Луи Буво, замечательный французский химик, с которым мы ещё не раз встретимся, получил его перекрестной кротоновой конденсацией метилциклопентанона и ацетона (до открытия направленной конденсации такие перекрёстные реакции делали, используя всякие специфические трюки – сейчас мы этого повторять не будем). 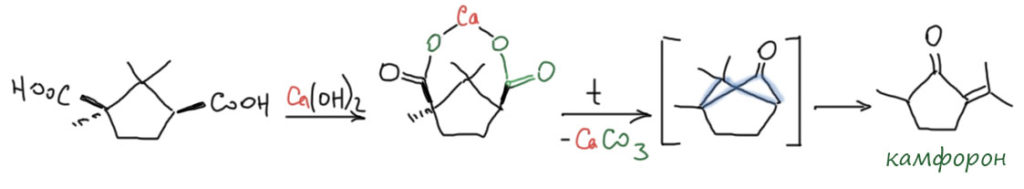
И действительно, именно это и получается. Исторический камфорон не имеет никакого отношения к форону, и это стало ясно уже на рубеже прошлого и позапрошлого веков. Но традиция переписывать старые ошибки настолько сильна, что до сих пор легко встретить утверждение, что форон и камфорон – одно и то же соединение. И приблизительно тогда же окончательно разобрались и в том, что продукт самоконденсации в кислой среде и основной среде – разные соединения. Второй стали называть изофороном. Вот они все три рядышком.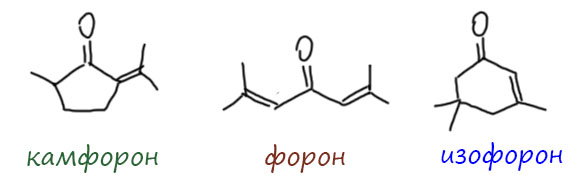
Из всех трёх самое большое значение имеет именно изофорон. Его производят в весьма больших количествах в промышленности, потому что это и очень ценное сырьё для тонкого органического синтеза, и весьма полезный специальный растворитель. Когда мы дойдём до фенолов, узнаем, что основной метод промышленного синтеза фенола – кумольный метод – производит кроме фенола и ацетон. Фенол гораздо более востребован рынком,чем ацетон, и излишки ацетона часто перерабытывают именно в изофорон.
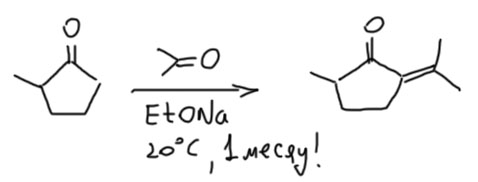
Кротоновая самоконденсация других кетонов
Кротоновая самоконденсация других простых кетонов тоже хорошо известна и даёт много полезных и интересных продуктов.
Очень легко подвергаются самоконденсации два типа кетонов:
- симметричные, у которых в енолизуемом положении метиленовые группы
- енолизуемые только с одной стороны, и желательно, чтобы там просто был метил.
Наиболее популярными производными первого типа являются циклические кетоны – циклоалканоны. Они действительно очень легко самоконденсируются хоть в присутствии кислот, хоть в присутствии сильных оснований (щелочей, алкоголятов), в основном образуя очень симпатичные продукты, например: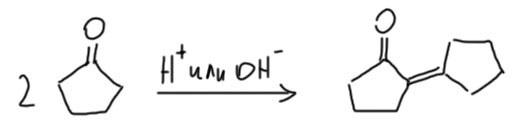
Реакции в кислой среде часто дают большие выходы потому что в присутствии сильных оснований первоначальные продуты легче втупают в разные реакции, например, в ещё одну конденсацию. Лёгкое оратимое образование енолятов приводит и к тому, что двойная связь между циклами норовит заползти внутрь одного из циклов, где ей намного уютнее (это общее свойство так называмых экзоциклических – торчащих наружу из цикла – двойных связей).
Таким продуктам очень легко найти применение в синтезе. Учитывая то, что простые циклоалканоны очень легкодоступны, а реакция кротоновой самоконденсации очень проста в выполнении, хотя и никогда не даёт высоких выходов из-за изобилия побочных реакций, такие бициклические ненасыщенные еноны очень привлекательны для органического синтеза.
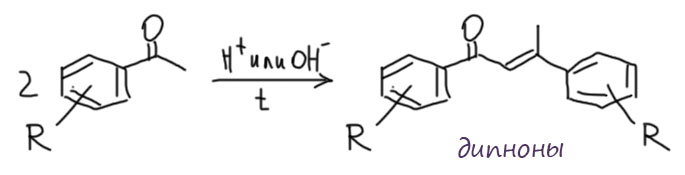
Из второй группы очень часто встречаются хорошо нам знакомые ацетофеноны – продукты ацетилирования ароматических соединений. В таких кетонах кабонильная группа сопряжена с ароматическим кольцом, что делает её ну просто очень ленивой. Из-за этого ацетофеноны гораздо устойчивее в присутствии кислот и оснований, и совсем неохотно ступают в кротоновую самоконденсацию. На самом деле, это хорошо, а не плохо, потому что именно это позволяет нам получать эти соединения ацилированием по Фриделю-Крафтсу – в условиях этой реакции получающиеся ацетофеноны находятся в смеси в присутствии очень сильной кислоты, например, хлорида алюминия и хлористого водорода, а такая комбинация имеет все признаки даже не просто кислоты, а суперкислоты. При обработке реакционной смеси мы тоже не очень церемонимся – льём соляную кислоту. И ничего, с ацетофеноном ничего не происходит. Какой-нибудь ацетон или циклогексанон в таких условиях быстро сконденсировался бы до состояния хорошего битума. А ацетофенону – как с гуся вода. И тем не менее ацетофенон и замещённые ацетофеноны вполне можно заставить вступить в альдольно-кротоновую самоконденсацию и в присутствии кислот (труднее), и в присутствии оснований (легче), хотя во всех случаях приходится запастись терпением и вести реакции при нагревании. Результаты такой конденсации – вполне полезные ненасыщенные кетоны, часто используемые в синтезе. То, что эти вещества интересны и важны можно понять хотя бы потому, что у них есть сокращённое название – дипноны. Это просто такое речевое упрощение более длинного названия 1,3-дифенилбут-2-ен-1-оны (diphenylbutenones – dypnones). Никто не парился бы придумывать краткое название для соединений, которые даром никому не нужны. На схеме в качестве катализаторов условно показаны символы кислоты и щёлочи, в реальности используют множество разных катализаторов как кислотных (HCl, тионилхлорид в спирте, трет-бутилат алюминия и т.п.), так и основных (этилат натрия).