Обновления
Опубликована 23.12.2022
Ацетилен-алленовая перегруппировка
В химии непредельных соединений весьма распространены перегруппировки, связанные с перемещением кратных связей по углеродной цепи. Перегруппировки такого типа встречаются в самых разных процессах, иногда как основное превращение, иногда как побочная реакция. Иногда такие перегруппировки целенаправленно разыскивают и с удовольствием используют, ещё чаще их опасаются, потому что они способны испортить вроде бы тщательно продуманный синтез, в самый неподходящий момент перетащив двойную или тройную связь совсем не туда, куда вы ей определили быть. Приходится волей-неволей разбираться, отчего эти перегруппировки происходят, в чём их причина, есть ли у них какие-то закономерности, можно ли ими как-то управлять и их как-то направлять. Перегруппировки двойных связей в этом смысле особенно зловредны, потому что вызываются самыми разными причинами (кислотами, основаниями, радикалами, переходными металлами, волей богов, и даже просто термодинамикой в согласованных реакциях без внешних причин). Наверное, поэтому мы и не особенно на них обратили внимание в теме алкены: обратишь – сам не обрадуешься. Мы просто тщательно избегаем ситуаций, где они могли бы проявиться, или даже скорее просто делаем вид, что ничего подобного не омрачает наши синтетические упражнения.
А вот перегруппировки тройной связи, как ни странно, гораздо более полезны даже для начальной стадии изучения органического синтеза. У них есть то преимущество, что вызываются они в основном только одним фактором – действием оснований (это не значит, что не бывает кислотно-катализируемых перегруппировок, или какого-нибудь изощрённого катализа, но в случае алкинов эти вещи встречаются намного реже, хотя бы потому что требуют неких особых усилий и реагентов, и просто так на них не нарвёшься), и закономерности их очень понятны и их легко изучить, а затем использовать. Но у них есть и особенность – кроме тройной связи в этих реакциях участвуют и двойные, причём в не очень для нас приятном виде кумулированного диена с двумя двойными связями подряд. Такие диены принято называть по первому члену ряда алленами. Не очень-то мы их серьёзно изучаем, и часто даже не догадываемся о том, что это очень необычные молекулы – такие в некотором роде раздвинутые алкины – две пи-связи алкина как будто выдвинули друг из-под друга и аккуратно разместили на соседних парах атомов углерода, бережно сохранив перпендикулярность. В этом смысле и сама ацетилен-алленовая перегруппировка приобретает несколько комический вид – как будто кто-то последовательно раздвигает и снова сдвигает пи-связи, двигая их так по цепочке. И в конце концов это упражнение привело к появлению одной из самых прикольных реакций в органической химии – реакции ацетиленового зиппера, которую мне очень хочется переименовать в реакцию ацетиленовой молнии, ведь это довольно редкий случай, когда в научной терминологии русский термин может быть образнее и понятнее заимствованного.
Краткое содержание
Разбираем, как вообще устроены равновесия изомеризации, и почему могут превращаться друг в друга ацетилены, аллены и сопряжённые диены на вкладке Общие вещи.
Как и кем была открыта ацетилен-алленовая перегруппировка, и в каких условиях можно ее наблюдать, а также что можно получить в равновесных условиях – на вкладке Ацетилен-алленовая перегруппировка в присутствии оснований. Взаимопревращения простейшего члена ряда, метилацетилена и простейшего кумулированного диена, аллена, а также откуда вообще взялось слово аллен – на вкладке Метилацетилен это не совсем то, что вы думаете.
Механизм ацетилен-алленовой перегруппировки более длинного алкина говорит само за себя.
Дальше займёмся самой интересной частью, находящей очень активное применение в синтезе, сначала с самых общих вещей – можно ли смещать тройную связь из глубины цепи на конец – на вкладке А в обратную сторону можно?
Открытие реакции ацетиленового зиппера, или как бы мне хотелось, чтобы это называли – Ацетиленовая молния.
Разберёмся в механизме, в том числе в заблуждениях, и в том Как это работает и Как это не работает.
Дальше посмотрим в каких случаях применяют эту реакцию, и Что можно подвергать ацетиленовой молнии.
Несколько реальных Примеров использования реакции из оригинальной литературы. И в самом конце немного разберёмся в том, почему перегруппировка ацетиленов и алленов почти никогда не приводит к расползанию двойных связей и образованию очень выгодных сопряжённых диенов, и убедимся в том, что иногда они всё-таки перегруппировываются в такие диены, и это помогает делать большие системы сопряжённых двойных связей.
Общие вещи
Это чрезвычайно интересная и важная реакция, в разных реализациях очень активно использующаяся в органической химии и синтезе. При этом надо очень хорошо понимать разницу между перегруппировкой как таковой и тем, как из этого процесса делают конкретные методы. Может быть много разных способов реализовать перегруппировку, использовать разные механизмы. Вы сами можете придумать новый механизм и попробовать его реализовать – химия бесконечна. Поэтому важно сразу понять, что у любой реакции есть два слоя.
Первый – это термодинамика, энергии и энтальпии образования, энтропия, энергия Гиббса – всё это характеристики равновесия. Из этих величин вы можете для любого процесса получить константы равновесия, а значит и состав равновесных смесей. И важно понимать, что это от вас не зависит, и от реализации не зависит, и даже от механизма не зависит – равновесная смесь при данных условиях (температура, давление, и если равновесие в растворе, то природа растворителя) всегда одинакова. Это общеизвестно, но все почему-то про это забывают, когда начинают разбирать конкретные реакции. Бывают равновесия простые – одна обратимая реакция, вещества слева и справа знака обратимости. А бывают сложные равновесия, когда много веществ соединены обратимыми реакциями, много прямых и обратных реакций. Кажется, что если с первым случаем разобраться можно, то со вторым – врагу не пожелаешь. Напрасно. Равновесие мощно тем, что совершенно не важно, сколько у вас реакций, и прямо или через десяток дополнительных стрелок связаны два вещества – для любых веществ в равновесии можно написать константу равновесия, и это будет именно константа, и она будет одна (при данных условиях).
Второй слой – это кинетика. У всех реакций, входящих в равновесие есть скорости, определяемые константами скорости и концентрациями. И пока равновесие не установилось, состав смеси будет определяться именно скоростями реакций. Это просто увидеть даже на самом простом равновесии – два вещества превращаются друг в друга, есть прямая и обратная реакции со своими константами скорости. В самом начале, если вы стартовали с одного из веществ, второго еще нет или очень мало, скорость его превращения обратно в первое очень мала – состав смеси в это время определяется прямой реакцией, как будто обратной еще нет. Мы отлично знаем эту ситуацию и называем ее кинетическим контролем. Но через некоторое время второе вещество накопится в приличном количестве, и в дело включится обратная реакция – состав смеси начнет приближаться к равновесному. Здесь довольно важно понять еще и то, что даже если константа равновесия велика, и в равновесной смеси законно преобладает одно вещество, это именно равновесие. потому что скорости обратных реакций даже в этом случае часто довольно велики, как только второе вещество в каком-то количестве образовалось. Равновесие установится.
Но, бывают ситуации, когда скорости каких-то реакций в равновесии действительно малы и именно по причине малых констант скоростей. В отличие от констант равновесий, константы скоростей зависят от гораздо большего числа факторов, в том числе от механизма реакций. И тогда, если в каком-то механизме какие-то константы скорости очень малы (даже вообще могут быть равны нулю – ну не обслуживает данный механизм какое-то превращение), то вы в эту часть равновесия с этим механизмом не доберётесь – концентрации соответствующих веществ за этими маленькими или нулевыми константами скоростей останутся ничтожными или нулевыми. При этом остальная часть равновесия могла утановиться, и соотношения остальных веществ пришли к равновесным. Что это такое? Это вполне честное равновесие, хотя его можно было бы назвать частичным, но это просто слово, за которым нет особенного смысла. Так бывает? Конечно, сплошь да рядом. Просто представьте себе любые изомеры, любых соединений, как это иногда дают в начале изучения органики – нарисуйте все изомеры с такой-то брутто-формулой. Стоит взять хотя бы три-четыре атома углерода, как изомеров становится много, а затем – ужасающе много. Открою вам маленький секрет – все изомеры связаны равновесиями изомеризации. И поскольку все энергии Гиббса можно найти (в справочниках, измерить экспериментально, оценить по аддитивным схемам, посчитать – современные методы квантовой химии очень высокого уровня уже вполне доступны и дают отличные оценки термодинамических функций), то и константы равновесия все можно найти. Многие вас удивят – они будут или велики или близки к единице, то есть будут говорить, что какая-то изомеризация вполне выгодна. Но вот оно вещество, которое должно изомеризоваться, лежит в банке сто лет и с места не двигается. Все алканы, например, связаны скелетными перегруппировками. Но чтобы их включить, нужно обеспечить механизм образования карбокатионов, и их обратного превращения в алканы (отщепления-присоединения гидрида), причем, если мы не хотим от алканов перейти в циклоалканы и алкены, нам придется придумать, как исключить реакции элиминирования в таком механизме. Это чертовски сложно. но не невозможно. Работайте, и достигнете. Но пока такой процесс экспериментально не реализован, индивидуальные алканы никуда не превращаются. Константы скоростей равны нулю. Сделаем из этого одно важное заявление:
Равновесие – это возможность превращения; механизм – это способ реализовать эту возможность полностью или, что намного чаще, частично.
Ацетилен-алленовая перегруппировка превращает алкины в смесь алкинов и алленов, но, по крайней мере теоретически, ещё и сопряжённых диенов. Все это изомеры. То есть речь и здесь идёт об изомеризации. Почему это вообще возможно? Как и во всех изомеризациях потому что у разных изомеров часто бывает разная стабильность. Стабильность – это термодинамическое понятие, оцениваемое для близких соединений энтальпиями образования конкретных изомеров. Точнее надо было бы использовать энергии Гиббса образования, то есть еще учитывать энтропию, но у изомеров разница энтропий часто невелика и существенного вклада не даёт, поэтому можно на это забить, и использовать энтальпии образования, которые для углеводородов самого разного строения очень хорошо известны. Нас не интересуют конкретные цифры. Мы хотим на глазок раскидать участвующие молекулы (на самом деле вещества, но на этом уровне это не важно). Это несложно сделать, потому что закономерности того, как разные структурные фрагменты, в том числе кратные связи и заместители, влияют на относительную стабильность, хорошо известны. Важно здесь вот что.
- кратные связи в молекуле углеводорода всегда дестабилизируют молекулу – делают энтальпию образования больше (напомню, что чем энтальпия меньше с учётом знака, тем лучше, тем стабильнее молекула, а точнее вещество, потому что термодинамика работает именно с веществом). Если кратных связей несколько, в первом приближении это во столько же раз сильнее дестабилизирует, но важно знать, как они расположены друг относительно друга.
- тройная связь дестабилизирует намного больше, чем двойная, почти как две двойные, но чуть меньше;
- если две двойные связи кумулированы (идут подряд, такие диены принято называть алленами или 1,2-диенами), то энтальпия образования таких диенов очень близка к энтальпии образования изомерных алкинов, разница редко бывает больше 1 ккал/моль, причем в зависимости от положения тройной связи и двух двойных в цепи разница может быть и в большую и в меньшую сторону, но это очень мало и в общем на это можно не обращать внимания, считая соседние по цепи тройные связи и алленовые фрагменты практически одинаковыми с точки зрения энергии;
- заместители при кратной связи наоборот стабилизируют, и поскольку мы говорим про углеводороды, речь идет про алкильные заместители. Это мы отлично знаем на примере алкенов, но и в других классах ненасыщенных органических соединений то же самое. Иными словами дизамещенный алкин стабильнее монозамещенного. То же справедливо и про алкены, и про аллены (например, монозамещенный алкин немного стабильнее незамещенного аллена, но немного менее стабильнее монозамещённого аллена, и т.п.);
- Сопряжение кратных связей стабилизирует – относительно ситуации, когда кратные связи не сопряжены. Это важный момент – даже сопряженные диены, диины, енины всё равно дестабилизированы, но оносительно меньше, чем похожие, но несопряженные диены, диины, енины. Поскольку мы не собираемся в наши равновесия включать соединения без кратных связей (мы собираемся сохранять степень ненасыщенности), например, циклоалканы, то получается, что в такой системе соединений именно сопряженные диены наиболее стабильны, причем с солидным запасом, и следовательно изомеризации вроде бы должны приводить практически количественно именно к ним. Почему это обычно не происходит, мы посмотрим в самом конце.
Из этого можно получить вот такую картинку. Возьмём цепь в которой можно разместить хотя бы пару кратных связей. В такой цепи возможны изомерные терминальный и внутренний алкин, аллен, сопряженный диен. Чисто качественно нарисуем картинку, как расположены на шкале энергий энтальпии орбразования. Видим очень характерную ситуацию. Аллен (две двойные несопряженные связи, один заместитель) и терминальный алкин (одна тройная, один заместитель) почти одинаковы по стабильности. Довольно сильно лучше внутренний алкин (одна тройная, но два заместителя), в среднем на 3-4 ккал/моль, а это в равновесной смеси дает соотношение более и менее стабильного соединения существенно больше, чем 10:1. И лучше всех в данной ситуации сопряженный диен (две двойные, сопряжённые связи) – это разница не меньше 10 ккал/моль, а это соответствует тому, что в равновесной смеси по всем возможным изомерам содержание сопряженного диена было бы не меньше 99%, а скорее даже больше, до 99,9%. Если бы цепь была длиннее, в ней возникли бы дорполнительные изомеры, алкины и аллены, но с очень близкими энтальпиями с уже изображенным, потому что там ничего нового бы не возникло. Единственное дополнение – несопряженный диен с удаленными двойными связями ничего нового бы тоже не внес – он был бы сопоставим с алленами (две двойные связи, без сопряжения) – ну и плевать на него.
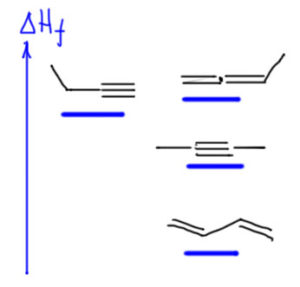
Изомеризацию кратных связей вызывают множество разных причин, в том числе самые разные виды катализа – основный, кислотный, катализ производными переходных металлов, и т.д. У каждого вида катализа свой механизм, и от этого зависит многое. Если мы представим себе универсальный катализатор, способный осуществлять взаимопревращения всех этих непредельных соединений, то что получится, когда равновесие установится? Совершенно очевидный ответ – практически только сопряженный диен (не менее 99%) в равновесии с малым количеством внутреннего алкина (на него придётся оставшийся процент или меньше, если диена больше, чем 99% – потому что разница в энергии (энтальпии) достаточна для того, чтобы константа равновесия была в пользу диена никак не меньше 100:1, а то и более (мы обсуждали это на страничке про конформации, а нет никакой разницы в работе равновесий, неважно что конкретно в них входит, важна разность энергий Гиббса, а это очень часто мало отличимо от разности энтальпий). И только несколько процентов от одного процента, то есть просто жалкие следы, сотые доли процента, – будет приходиться на терминальный алкин и аллены.
Но – далеко не всегда механизм изомеризации обслуживает все взаимопревращения с сравнимыми скоростями. И если представить себе механизм, который хорошо работает только на превращение алкин-аллен, и плохо или никак не работает на превращение алкин-сопряжённый диен, то из состава равновесной смеси исключаем сопряжённые диены, и тогда состав равновесной смеси распределится между внутренними алкинами, алленами и терминальным алкинам, и в равновесной смеси будем в основном внутренний алкин с малой примесью терминального и аллена.
Ацетилен-алленовая перегруппировка в присутствии оснований
Вот ровно такой и является самая первая перегруппировка такого типа, открытая выдающимся российским химиком Алексеем Фаворским в конце 1880-х, когда он был молод и переполнен идеями, а это было очень интересное время в органике – в 1870-х распространилась и стала основной теория структуры органических соединений, органики разобрались в том, как устроены молекулы и начали уже осмысленно конструировать новые, и искать способы их синтеза. Тогда же пошла работа над алкинами и диенами.
Фаворский обнаружил, что при действии на терминальный алкин щелочи или алкоголята при нагревании образуется изомерный внутренний алкин. Но у него же получилось так, что реакция идет и в обратную сторону – если греть внутренний алкин, то получался терминальный, и он даже попробовал установить закономерности, но получилось нечто довольно странное. Поскольку тогда еще представления о скорости реакций и равновесии не были разработаны, можно сказать, что он открыл само явление, но не мог изучить его закономерности. Поэтому если использовать имено результаты Фаворского из его статей 1880-х, то получается картина довольно странная – изомеризация одинаково идет в обе стороны, и если взять внутренний алкин или аллен, то мы можем получить терминальный алкин и наоборот. В реальности так это не работает, и мы сейчас разберем почему. А откуда же Фаворский взял те результаты? Он где-то наврал? Не совсем. В 19 веке так делали химию, причем все учёные без исключения, включая самых-самых легендарных типа Кляйзена, Михаэля, Фишера и т.п., да собственно Фаворский вполне входит в этот круг легендарных основоположников. В статьях того времени часто даже не приводились выходы, и никто не заботился о том, что воздух, вода, не совсем чистые реактивы, и т.д. как-то могут влиять на результаты реакций. Здесь, например, очень даже может влиять, потому что система очень чувствительна к окислению, и многочасовое нагревание со щелочью прямо под воздухом неизбежно даст очень искаженные результаты. Техника эксперимента была очень примитивной, не было, например, никаких мешалок – всё руками крутили – а это в химии очень важная вещь, потому что если вы смесь нормально не перемешиваете, в разных уголках колбы идут разные реакции, и ни о каком нормальном равновесии не может быть и речи. А в роли оснований очень часто применяли металлический натрий, который, конечно, никаким основанием не является, работает через восстановление, в результате которого и образуется основание, так что в таких смесях шла не только и не столько перегруппировка, а в первую очередь именно восстановление. Об этом можно было бы не упоминать, если бы старые результаты самого Фаворского не попали бы в старые еще советские учебники, а оттуда не поехало бы уже в более современные учебники, так что нередко можно встретить отголоски тех старых и не очень достоверных результатов. Поэтому важно только одно – Фаворский установил факт перегруппировки алкинов и алленов друг в друга в присутствии оснований, но закономерности этой реакции он не установил. Ровно то же самое делали все легендарные основоположники химии, и задача довести великие открытия до более высокого уровня осталась для потомков в 20-м веке, а некоторые вещи и до сих пор нуждаются в прояснениях на современном уровне.
Аккуратно, без доступа воздуха, с хорошей техникой эксперимента и с полным пониманием того, что в деле замешано равновесие реакцию исследовали много позже. Первая надёжная работа была описана в статье Томаса Джейкобса и сотрудников (T.L.Jacobs, R.Akawie, R.G.Cooper J. Am. Chem. Sос., 1951, 73, 1273), которые собственно и предложили карбанионный механизм перегруппировки и утановили ее равновесный характер. Взяли по отдельности пентин-1, пентин-2 и пентадиен-1,2 и нагревали каждый с раствором щелочи в спирте. Для того, чтобы реакция имела разумную скорость потребовалась температура в 175 градусов, в этом случае равновесие достигалось приблизительно за три часа, после состав смеси практически не изменяется, сколько её ни грей. Во всех трех случаях образуется около 95% пентина-2, и остальные 5% приблизительно поровну на пентин-1 и диен-1,2. Это вполне убедительное доказательство того, что мы имеем дело с равновесием, и что состав равновесной смеси вот ровно такой. В принципе, выход в 95% можно считать почти количественным, и просто утверждать, что и терминальный алкин и аллен практически количественно изомеризуются в 2-алкин.
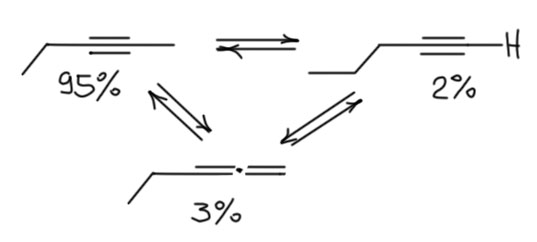
Ещё подробнее разобрались в процессах новозеландские химики Малькольм Карр с сотрудниками (M. D. Carr, L. H. Can and I. Reid, J. Chem. Soc., Perkin I, 1973, 668 и следующая статья номера). Если взять основание сильнее, трет-бутилат калия в трет-бутаноле, то реакция идет быстрее и при умеренном нагревании (около 80º вполне удобная скорость, реакция проходит за минуты вместо часов), но закономерности не изменяются – тройная связь смещается внутрь цепи на один атом углерода, основной продукт – внутренний алкин, соседний с терминальным. Аллен и исходный ацетилен – несколько процентов. Дальше тройная связь практически не едет, буквально следы следующего по цепочке алкина, если его вообще определяли. Еще позже стали использовать трет-бутилат в ДМСО, тогда равновесие устанавливается за минуты при комнатной температуре, но остаётся таким же.
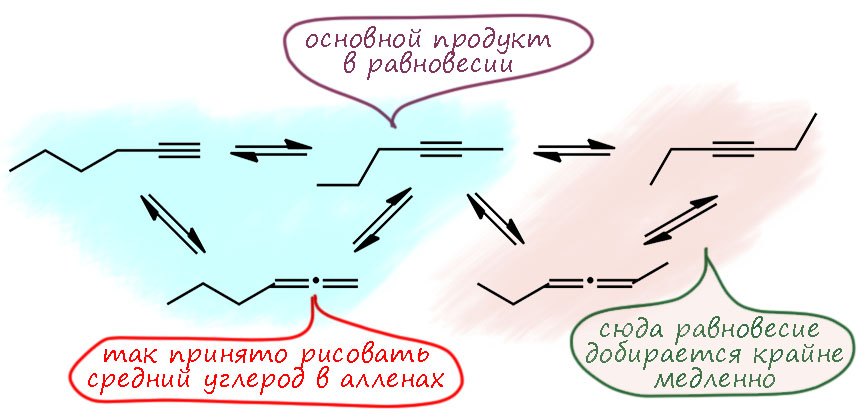
А вот если взять основание сильно сильнее, например, амид натрия в жидком аммиаке, тройная связь едет дальше (если есть куда ехать), и основной составляющей смеси становятся смеси внутренних алкинов, но и здесь движение тройной связи дальше от исходного положения весьма тугое, и каждый следующий шаг в смещении требует больше времени, а полное равновесие, видимо, вообще не достигается, во всяком случае я не видел таких результатов в статьях, терпения ни у кого не хватило, да и смысла в этом большого нет – выяснить, что, например, будет, если взять цепь, в которой может быть побольше изомерных алкинов. Здесь есть немного комический момент – у алкинов нормального строения не так много изомеров: целая часть от деления числа углеродов в цепи на два, то есть, например, у децина и ундецина по пять изомеров. Увеличить число можно или с одного конца нарушив симметрию, проще всего с помощью изотопной метки концевого углерода. Но такого никто не делал, видимо, потому, что результат очевиден, и подтверждать очевидное людям лень. То есть, если взять алкин подлиннее, гексин-1 или ещё длиннее в равновесии должны появиться и алкины и аллены с кратными связями еще глубже уехавшими внутрь. При этом в равновесии уже не будет преобладающего продукта, потому что энергия внутренних алкинов приблизительно одинакова и в равновесии они должны быть поровну. Грубо приблизительно – вместо 95% 2-алкина, здесь должны были бы получить по 45% 2-алкина и 3-алкина. Нафиг такое нужно!!! А если бы взяли гептин-1, то было бы по 30% гептина-2, гептина-3, и гептина-4. Всё остальное – приблизительно поровну между алленами и терминальным алкином. Плохо! Никто не стал бы это использовать – только вещества портить! Разделять такие смеси не будет никто, это чрезвычайно сложно даже в промышленных условиях, где есть чрезвычайно эффективные ректификационные колонны, позволяющие разделить фракционной перегонкой вещества с температурами кипения, различающимися на пару градусов (в лаборатории с обычной перегонкой даже с внушительно выглядящим дефлегматором даже разница в 20° недостаточна для хорошего разделения перегонкой при атмосферном давлении; при вакуумной перегонке разделение лучше, но поскольку все температуры кипения сильно уменьшаются, уменьшается и разница). Промышленная фракционная разгонка на ректификационных колоннах очень энергозатратна, и используется только когда продукт действительно ценен и по-другому его не получить.
Но в реальности всё не так плохо. Если не использовать очень сильных оснований, а ограничиться той же щёлочью или алкоголятом в спиртах при нагревании, то в смеси, образовавшейся через несколько часов будет равновесие, очень похожее на то, что мы видели у пентина – преобладает гексин-2 (гептин-2, и т.п., для любых алкинов с длинной цепью, в которой может быть больше одного внутреннего алкина), и по паре процентов терминального алкина и терминального аллена. Дальше реакция почему-то не едет, а точнее, едет, но крайне медленно. Можно было бы назвать это частичным равновесием, хотя это крайне некорректное выражение, ведь настоящее равновесие всегда одно, какие бы условия и катализаторы мы не применили. Видимо, дело в кинетике: реакции в первой части равновесия идут быстрее, чем во второй, намного быстрее. Поэтому та часть равновесия, которая связывает терминальный алкин, 2-алкин и аллен устанавливается намного быстрее, а вторая настолько сильно отстаёт, что можно в течение некоторого разумного времени вообще не обращать на нее внимания. Но забывать не надо – если сильно затянуть с временем реакции, сильно перегреть, или взять основание сильно посильнее (тот же трет-бутилат в ДМСО) то равновесие поедет дальше и состав смеси неприятно усложнится и станет совсем бесполезным.
Сделаем короткое резюме про то, что практически можно сделать с алкин-алленовой перегруппировкой.
- можно из терминального алкина сделать 2-алкин. Он будет не 100%-но чист, и будет содержать 2-3% примесей исходного алкина и аллена, но эти два компонента можно при желании отделить за счет химических реакций. А часто такой чистоты и так достаточно, в органике 95%-ная чистота реактива часто считается приемлемой. Для этого лучше брать что-то типа трет-бутилата калия в трет-бутаноле (при небольшом нагревании) или в ДМСО (без нагревания). Более сильные основания (типа амида натрия, LDA и т.п.) лучше не применять, так как тройная связь поедет дальше (если это, конечно, возможно) и получится неразделимая смесь внутренних алкинов.
- можно из терминального аллена получить 2-алкин тем же способом и с теми же комментариями, хотя это довольно бессмысленное занятие, так как такие аллены гораздо менее доступны, чем алкины;
- всё, больше ничего нельзя сделать, если иметь в виду только равновесную перегруппировку. Но если мы придумаем, как для какой-то конкретной задачи использовать принцип Ле Шателье и догадаться, как вывести их равновесия один из компонентов равновесия, у нас есть надежда сделать что-то ещё. Есть один знаменитый случай, когда такой трюк получается – так называемый ацетиленовый зиппер – превращение любого внутреннего алкина (или аллена, но так почти не делают) в терминальный алкин. Об этом отдельно.
Метилацетилен это не совсем то, что вы думаете
Если вам когда-нибудь потребуется метилацетилен, и вы решите купить готовый, такой в маленьких симпатичных баллончиках (метилацетилен в отличие от самого ацетилена можно сжижать и так хранить и продавать), вы будете немало удивлены, прочитав в сопроводительной бумажке, что вместо чистого метилацетилена вам продали смесь метилацетилена и пропадиена-1,2, чуть ли не 1:1. Вот жулики, подумаете вы, и будете неправы. Во-первых, скорее всего, это вещество вам понадобилось для того, чтобы сделать метилацетиленид, например, действием бутиллития или амида натрия и использовать его в реакции – ну так из этой смеси ровно он и получится. Это хорошо известное явление – при попытке металлировать терминальные аллены, они очень легко дают именно ацетилениды, даже при охлаждении эта реакция протекает с большой скоростью. Видимо, процесс идёт через дианион, вот как-то так:
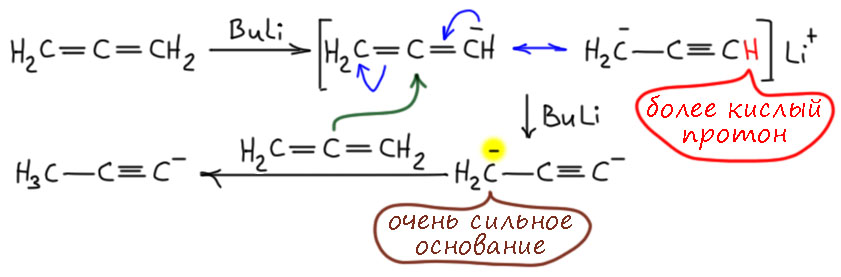
В этой реакции мы не можем оперировать обычной ацетилен-алленовой перегруппировкой, потому что для неё нужен источник протонов – сопряжённая кислота основания или вообще растворитель типа спиртов. Здесь мы просто прибавляем раствор бутиллития к раствору метилацетилена-аллена в чём-то типа ТГФ (если используем були) или жидкого аммиака (если амид натрия) – сначала моментально депротонируется метилацетилен, а затем, намного медленнее, начинает депротонироваться гораздо менее кислый аллен. Поскольку реакция эта медленная, у нас ближе к концу реакции возникает ситуация, когда сильное основание сосуществует с алленильным анионом. Этот анион можно представить граничными структурами, которые хорошо показывают, что там есть второй достаточно кислый протон, менее кислый, чем в нейтральной молекуле метилацетилена, но всё же достаточно кислый, потому что из-за регибридизации в sp-углерод кислотность повышена, и возможно образование короткоживущего дианиона, который уже станет очень сильным основнием, но по другому атому, которые не затронут регибридизацией и остался в sp2-гибридном состоянии (на нём три связи, а не две), и тогда источником протона, который нам так нужен, может просто стать еще непрореагировавшая молекула аллена (напомню, что реакция медленная, и из-за этого в смеси до конца остается немного непрореагировавшего исходного). Вот, мы получили метилацетиленид и еще немного алленильного аниона, которые поедет по этому же пути. Так, терминальные аллены практически количественно металлируются бутиллитием и подобными сильными основаниями в терминальный ацетиленид.
Да и во многих других реакциях эта смесь ведет себя так, как будто это именно чистый метилацетилен. Например, если мы попробуем ввести эту смесь в реакцию Кучерова, получим ацетон (механизм напишите сами).
Вообще это старая история, которая кроме всего прочего объясняет ещё и почему простейший 1,2-диен называется алленом, откуда взялось это слово.
Легкость превращения ацетиленов и алленов в самом начале истории этих соединений долго морочила голову исследователям, ничего, естественно, об этом не знавшем. Когда усилиями Бертло более-менее разобрались с самим ацетиленом, принялись за его ближайший гомолог. Брутто-формула гомолога ацетилена С3Н4 соответствует уже известному тогда радикалу аллилу (название это происходит от лука, по-латински Allium, в луке и похожих едких и острых растениях содержатся аллилсульфиды и похожие молекулы, что и было выяснено очень давно, так и появилось название “аллил”), а это С3Н5 минус атом водорода: уже не раз мы встречались с тем, как органики в начале химии любили все сводить к известным им кусочкам, радикалам в том смысле слова (повторяющаяся часть органической молекулы) и если получалось соединение равное известному радикалу минус водород, к названию радикала прибавляли -ен (этил – этилен, пропил – пропилен и т.д., метил – метилен, фенил – фенилен, мезитил – мезитилен т.д.). А из аллила должен был получиться аллилен. Ровно так это и назвали. Что это? Углеводород С3Н4 который успешно получили, например, отщеплением двух HCl от 2,2-дихлорпропана. Но в 1880-х химия уже зашла очень далеко в понимании возможностей структуры, научились рисовать структурные формулы, и очень интересовалась возможностью изомерии. И среди органиков этого времени, а там что ни имя, то история, именные реакции и методы – возникла живая дискуссия, сколько изомеров у аллилена. Было несколько попыток получить соответствующий диен, но получались смеси, и определенности не было. Например, электролизом соли итаконовой кислоты в 1973 некто Георг Аарланд получил некий газ, но что это было можно только гадать, статьи тогда так писали, что точно установить, что точно было получено невозможно. В 1888 наконец был однозначно получен 1,2-диен. И поскольку под аллиленом к этому времени уже стали точно понимать метилацетилен, этому измеру потребовалось своё название, и получилось оно просто – аллилен сократили в аллен. Вот откуда это странное короткое слово. Самое интересное. что оно прижилось и даже дало называние всему классу кумулированнх диенов, а вот аллилен из химии испарился после того, как возникла органическая номенклатура, после чего все простые соединения стали называть систематическими названиями и необходимость выдумывать отпала.
Чистый аллен впервые получили Густавсон и Демьянов и опубликовали эту важнейшую статью, как тогда было принято, в лучшем международном журнале, то есть по-немецки в Berichte. Но Демьянов был тогда ассистентом у профессора Густавсона в Сельскохозяйственную академию. В СССР она стала называться Тимирязевской или просто Тимирязевкой, и там действительно очень долго была отличная кафедра химии, где было много отличных органиков с очень тесными связями с Химфаком МГУ – вот это оттуда и идет, от Густавсона и Демьянова. Гавриил Гавриилович Густавсон это весьма значительный русский органик, один из первых настоящих профессиональных ученых в этой науке, мы с ним еще встретимся как минимум в циклопропанах, а может и ещё где-нибудь. А его ученик Николай Яковлевич Демьянов так и вообще, на мой взгляд, один из самых интересных и многогранных русских органиков того, классического периода. Демьянов прожил долгую жизнь (1861-1938), но по меркам долголетия многих знаменитых химиков, далеко не самую долгую. Год смерти, 1938, подозрителен – не стал ли он жертвой репрессий? Как ни странно, кажется, нет, хотя это не точно – чекисты умели заметать следы, и быстренько кого-нибудь репрессировав, иногда ещё быстрее реабилитировали, типа, ошибочка вышла, лес рубят, щепки летят – вот вам и довелось стать щепкой. Коммунизм строим, не до щепок. Коммунизм, как известно, так и не построили, но щепок, то есть людей, нарубили – на еще одну большую страну бы хватило. И была бы эта страна намного более развитой, чем оставшаяся, потому что рубили в первую очередь тех, кто чем-то выделялся – образованием, характером, происхождением, талантом, внешностью, совестью, привычками. Но репрессии были страшны еще и тем. что были совершенно безумны – под расстрел мог попасть старый большевик-политкаторжанин, герой гражданской войны, кровавый чекист, рабочий-марксист, да кто угодно, вовсе не обязательно было иметь “неправильное” происхождение или политические взгляды, так называемых “чуждых элементов”. Но происхождение и взгляды сильно способствовали – таких людей уничтожали целенаправленно, а всех остальных – случайно. А Демьянов был совершенно чуждым элементом в СССР. Во-первых, из дворян, даже не просто из дворян, а из настоящей аристократии, из старинного рода, известного с 16 века, с времен Василия Третьего, многие его предки известны по историческим источникам. В западной Европе у них обязательно был бы какой-нибудь громкий титул, маркизов или графов, но на Руси система наследственных аристократических степеней кроме князей из потомков правящих династий Рюриковичей и Гедиминовичей, и ордынских мурз не сложилась. Так или иначе Демьяновы люди были серьёзные, из правящего класса, и родной брат химика стал не много не мало членом Временного правительства после Февральской революции и фактически занимал пост министра юстиции до самого октябрьского переворота. Из России после большевистского переворота ему удалось выбраться за границу, где он впрочем долго не прожил. И вот, брат буржуазного министра советский химик Николай Демьянов жил и работал в СССР с такой биографией и с такими родственниками – и ничего, пронесло. Так много работы было у чекистов, что всех не успели оприходовать. Как говорил товарищ Дзержинский, недоработка вышла у товарищей чекистов, не добили вражину. И Демьянов всю жизнь так и проработал в Тимирязевке. И что самое примечательное, он действительно работал и продолжал делать отличные работы уже при большевиках, а не стал, подобно многим другим дореволюционным большим ученым, выставочным академиком для заседания в президиумах, большим начальником из нового советского дворянства, номенклатуры.
Густавсон и Демьянов получили аллен элиминированием цинком из 2,3-дибромпропена-1, а это соединение легко получается присоединением брома к аллилбромиду. Метод элиминирования цинковой пылью из дибромпроизводных незадолго до этого разработал как Густавсон, получивший этим методом циклопропан, и этот метод мы и называем методом Густавсона.
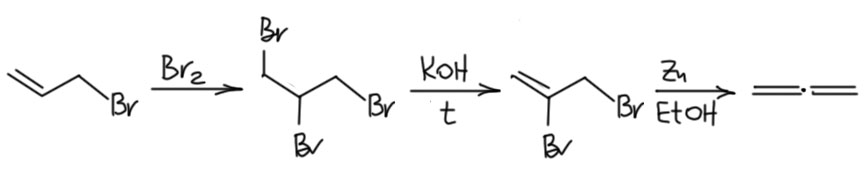
Аллен при этом получается чистейший, без метилацетилена. Это газ, который можно, как Демьянов с Густавсоном собрать в газометр или газовую бюретку, или более по современному, сконденсировать на вакуумной линии и отпаять в ампулу или какой-нибудь современный прибамбас. Аллен без метилацетилена так получают до сих пор, разве что вместо дибромпропена берут такой же дихлорпропен, и это стандартная методика синтеза аллена в лаборатории. И это очень важно, потому что любые методы, включающие элиминирование основаниями или вообще что-нибудь, катализируемое основанием, неизменно дают смесь метилацетилена и аллена. То же самое даёт и гидролиз карбида магния, редкого карбида, который является производным не метана и не ацетилена, а аллена или метилацетилена (в кристаллической решетка карбида магния находятся симметричные линейные тетраанионы C34-, структурно соответствующие именно аллену, но во время гидролиза карбида и ступенчатого протонирования этого тетрааниона мы приезжает в обычное равновесие между алкином и алленом).
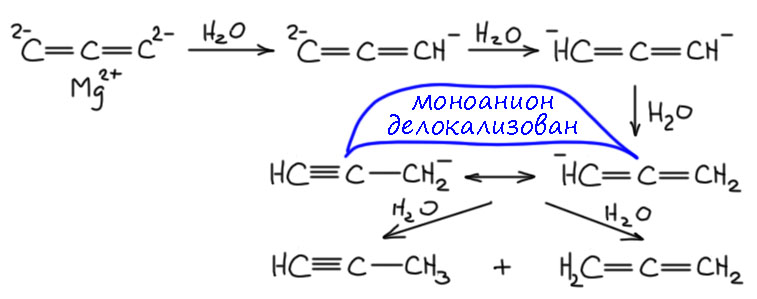
Этот карбид магния довольно необычное соединение из известных карбидов непереходных металлов. Большинство таких соединений это или ацетилениды (большинство карбидов щелочных и щёлочноземельных металлов), или метаниды (карбиды бериллия, металлов группы алюминия и некоторые другие). Магний вообще не образует термодинамически стабильных соединений с углеродом – все известные карбиды магния метастабильны, но это кристаллические решетки с большой энергией и распад карбидов на элементы происходит только при длительном сильном нагревании. Из этого следует, что прямо из элементов карбиды магния получить нельзя, если только не использовать сложнейшие установки для синтеза под сверхбольшим давлением. Ровно так карбид магния Mg3C2 из элементов впервые получили не так давно в совместной работе групп Тимоти Стробеля и Артёма Оганова в США (Strobel, T.A.; Kurakevych, O.O.; Kim, D.Y.; Le Godec, Y.; Crichton, W.A.; Guignard, J.; Guignot, N.; Cody, G.D.; Oganov, A.R; “Synthesis of β-Mg2C3: A Monoclinic High-Pressure Polymorph of Magnesium Sesquicarbide.” Inorg. Chem., 2014, 53, 7020) – несколько часов нагревания около 2000 градусов под давлением в несколько сот тысяч атмосфер. А без этого экстремизма карбид магния весьма высокой чистоты получают действием паров нормальных алканов типа пентана на порошок магния при сильном нагревании, но ниже температуры разложения карбида (около 1000 градусов). Понятно, что это не самое легкодоступное соединение, и практически его никто не использует, тем более что при гидролизе получается ровно та же смесь метилацетилена и аллена, что и при любом банальном элиминировании из дигалогенпроизводных.
В промышленности метилацетилен никто специально не делает. Это очень небольшая примесь в продуктах парофазного крекинга алканов, основном промышленном процессе для переработки всех не очень нужных легких фракций нефти, когда эти смеси смешивают с водяным паром и нагревают до 800 градусов, выше, чем при обычном старом крекинге – при такой температуре реакции расщепления связей идут очень быстро, это позволяет сильно снизить время нахождения углеводородов в горячей зоне реактора и в основном подавить очень неприятный процесс образования сажи, забивающей реакторы. Туже роль играет и водяной пар, работающий как агент теплопереноса от стенок реактора к молекулам алканов, таким образом просто по теории вероятностей снижается частота прямых соударений алканов и стенки, а это и приводит к глубокому расщеплению и образованию сажи. Этот процесс хорошо отладили и он стал основным для переработки бесполезных небольших алканов в смесь этилена, пропилена, бутиленов и бутадиена, обеспечивающую сырьём колоссальную промышленность полимеров. Из-за очень большой температуры этого типа крекинга образуется немного, мене процента ацетилена и метилацетилена с алленом. Смесь разделяют, ацетилен гидрируют в этилен (в промышленности это легко сделать, а в лаборатории нет), а смесь метилацетилена и аллена (называется смесь MAPD – метилацетилен-пропадиен), или иногда с примесью пропана (смесь MAPP – метилацетилен-пропадиен-пропан) сжижают и продают как горючее дял специальной сварки. При совершенно колоссальных объёмах парофазного крекинга доли процентов это всё равно очень много. Такая смесь горит очень горячим пламенем, не таким горячим как сам ацетилен, на несколько сот градусов пониже, но все равно что-то близко к трём тысячам – но при этом безопасна, в сжиженном состоянии не взрывается, и специальных приспособлений для защиты от детонации не требует.
В равновесной смеси метилацетилена немного больше аллена, в полном соответствии с тем, что аллен где-то на 1 ккал/моль менее стабилен, чем метилацетилен. Изомеризация алкин-аллен легче и быстрее происходит при повышенной температуре, в газовой фазе, в присутствии и основных, и кислотных катализаторов, то есть практически всего на свете.
А теперь разберёмся с тем, как происходит перегруппировка метиацетилена в аллен (и обратно) в присутствии оснований. Возьмём метилацетилен и отнимем у него протон. При pK в районе 24-25. В этом месте прямо слышу глас вопиющего от возмущения – неужели для такого важного соединения константа основности не известна с десятью знаками после запятой. Нет, конечно, не бывает констант равновесия с такой точностью, термодинамические величины не измеряются с такой точностью, да это и не нужно. Но даже с точностью первого знака до запятой константу основности измерить невозможно – эти величины очень сильно зависят от растворителя, противоиона, других факторов, и нет ни малейшего смысла оперировать тоными величинами, даже если бы они были измерены в каком-то конкретном растворителе и с конкретным противоионом. В области кислотно-основных равновесий с участием CH-кислот всё очень приблизительно, – мы оперируем диапазонами, верхними и нижними оценками и тому подобными почти совсем качественными понятиями, из которых можно вывести только умозаключения в стиле больше-меньше, может-не-может, будет-не-будет, но ни в коем случае не количественные расчеты.
Итак, мы знаем, что самый кислый протон у ацетилена в терминальном углероде, причём образуется ацетиленид-ион, который никуда не делокализован и обратно протон дожен присоединять ровно туда, где он и был до этого. 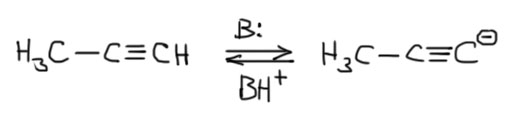
И никакой изомеризации. Мы отлично знаем, как отобрать этот протон и получить ацетиленид, и отлично умеем использовать этот ион в синтезе. И нам никто не говорил, что есть возможность отобрать еще один протон. Точнее, скажем так, говорили, но протон этот можно отобрать только очень сильным основанием, и только после ацетиленового протона. Из этого мы понимаем, что в метилацетилене есть всё же ещё один кислый протон, в метильной группе, и отрыв этого протона, если бы это можно было бы сделать, дал бы нам делокализованный карбанион (такие анионы называются пропаргильными). Хотя и непонятно зачем, запишем, что было бы, если бы такой карбанион можно было получить. Обозначим делокализацию, как обычно, граничными структурами. В очередной раз поучимся корректно выражать происходящее словами: карбанион, получающийся отщеплением протона основанием от пропаргильного положения метилацетилена имеет структуру, находящуюся между чисто пропаргильной с тройной связью, и алленильной с двумя двойными связями, что мы и выражаем с помощью граничных структур и специальной обоюдоострой стрелки. Минус в таком анионе делокализован между крайними атомами углерода. Мы не можем сказать, что он делокализован строго поровну, как в аллильном анионе, потому что граничные структуры неравноценны и следовательно вносят разный вклад в истинную структуру делокализованного карбаниона. Скорее всего, соображения о том, что аллен менее стабилен, чем ацетилен, можно аккуратно распространить и на граничные структуры, сказав, что структура карбаниона несколько ближе к пропаргильной. Но обратное равновесное протонирование сопряжённой кислотой исходного основания даст нам и метилацетилен, и аллен. Если всё это равновесие, то в равновесной смеси, как мы уже выяснили, преобладает с огромным преимуществом метилацетилен, но это не имеет прямого отношения к тому, какое соотношение аллена и метилацетилена получается при протонировании карбаниона. Если бы мы смогли получить такой карбанион в растворе, и после вылили бы это в воду, то получили бы смесь с гораздо большим содержанием аллена, потому что в этом случае протонирование было бы быстрым и необратимым, а скорости протонирования по каждому из углеродов не могут отличаться так сильно.
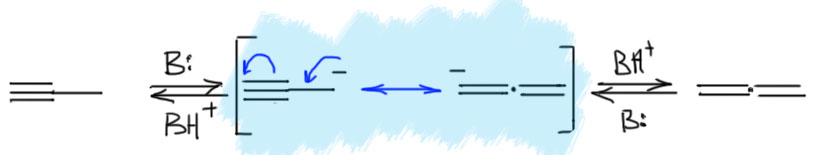
Да это же ровно то, что нужно! С ним всё вышло бы! Да вот беда, он намного менее кисл (кисёл?), чем ацетиленовый. Разве можно из кислоты, у которой два кислотных центра, взять протон у менее кислого центра, оставив в стороне более кислый. Это же то же самое, как если бы у нас была смесь серной и уксусной кислот, а мы бы добавили щёлочь и рассказывали бы, что получили раствор ацетата натрия в серной кислоте. Ересь какая!! Зовите старушку с хворостом, будем жечь еретика (себя что ли? да не доберётся сюда роскомпозор!). Поделом же негодяю, как можно было до такого додуматься!
Спокойно! В химии нельзя действовать так в лоб, потому что есть такая великая вещь как равновесие. Равновесие находит выход из самых безвыходных ситуаций, зажигает свет надежды у слабой кислоты рядом с гораздо более сильной. Фокус в том, что даже если различие констант равновесия (в данном случае кислотности) будет хоть 10 порядков, это все лишь значит, что в равновесии есть и анион сильной кислоты, и анион слабой, но соотношение их и будет приблизительно эти 10 порядков. Для обычной жизни это бессмысленно огромная разница, хотя именно такова разница между имуществом какого-нибудь бессмысленного олигарха-жулика и учителя средней школы. А в мире веществ, где всё измеряется порядком числа Авогардро, и 10 в минус десятой моля – всё равно чёртова туча молекул. И если именно эта мизерная часть вещества способна реагировать и превращаться во что-то другое, а та часть вещества, которой намного больше, ни во что не превращается, то мы видим именно то направление превращения исходного вещества, которое идёт через мизерную, но более активную его часть. Единственное, на что влияет эта разница во много порядков – это скорость реакции с участием вот таких очень малых количеств менее выгодного участника равновесия – это же концентрация в выражениях кинетических уравнений, и даже если константа скорости велика, то при очень маленькой концентрации скорость будет мала. Но мы это знаем – скорость ацетилен-алленовой перегруппировки с обычными основаниями типа щелочей и алкоголятов так мала, то нужна очень большая температура и немаленькое время реакции, чтобы равновесие установилось.
В этом рассуждении всё верно, но есть и проблема – мы бы всё же хотели знать поточнее, какова разница в кислотности терминального протона ацетилена и пропаргильного протона. Увы, хоть и кажется, что в 21 веке всё это должно быть точно известно, но это не так. Кислотность ацетилена по терминальному протону мы знаем, это что-то в разных раствориелях в диапазоне pK от 24 до 28-29, а вот с пропаргильным просто беда, даже оценок приличных нет кислотности в растворе. Есть оценка кислотности аллильного протона в пропилене – очень высокое pK, больше 40, было бы плохо, если бы пропаргильный был близко к этому. Но мы можем считать, что кислотность пропаргильного протона всё же немного больше, хотя бы потому что ацетиленовый фрагмент более сильный акцептор по тем же причинам, связанным с гибридизацией. Скинем 3-4 порядка, и получим что-то около 40, но всё же меньше, может быть 38-39. Есть очень точные оценки кислотности в газовой фазе, и там разность в кислотности очень невелика, и в газовой фазе метоксид-ион элементарно депротонирует и пропаргильный и ацетиленовый, и алленовый протоны. Но как использовать данные по кислотности в газовой фазе для оценки кислотности в растворе? Для этого нужно уметь оценивать сольватацию анионов, и тогда можно было бы попробовать опустить газовые данные в жидкость. К сожалению, это очень сложная задача, для решения которой нужны очень непростые и трудоёмкие расчёты, и для этой пары – ацетиленид-алленид, насколько я знаю, хороших расчетных оценок пока нет. Не устаю напоминать – молодая наука органическая химия, конь ещё не валялся во многих даже довольно простых с виду проблемах, даже сферический, и пока и не собирается валяться. Чисто качественно мы понимаем, что сольватация ацетиленид-аниона гораздо сильнее, потому что заряд локализован на одном атоме углерода, а компактный заряд гораздо сильнее взаимодействует с полярными молекулами растворителя, чем размазанный, делокализованный заряд пропаргильно-алленильного аниона. Поэтому не очень большая разница в газовой фазе и должна очень сильно увеличится в растворе. Десяток единиц pK – хорошая оценка такой разницы, это, ксати, не так много, если перевести в единицы энергии, чуть-чуть больше 11 ккал/моль, для сольватации заряженных частиц вполне разумная величина.
Итак, общая картина того, что произойдёт если к раствору метилацетилена добавить некоторое количество вильного основания, будет выглядеть так: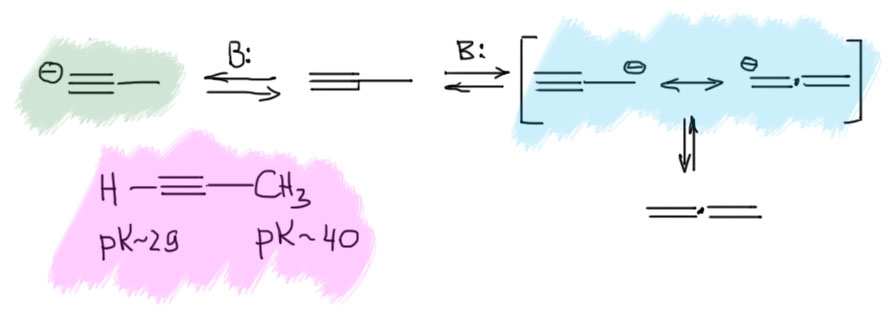
Метилацетилен при действиее основания даёт обычный ацетиленидный анион, но в равновесных условиях в смеси присутствует мизерное количество второго аниона, через который осуществляется изомеризация. Когда равновесие установится смесь будет состоять из равновесных количеств метилацетилена и аллена, а также ацетиленидного аниона, относительное количество которого определяется количеством и силой взятого основания. Если его немного (основание недостаточно сильное, или сильное, но в небольшом количестве) , можно об этом не думать. Если много (основание достаточно сильное и взято в значительном количестве), то этот анион до конца будет присутствовать в смеси, и после погашения водой даст метилацетилен: в этом случае в конечной смеси будет больше метилацетилена, чем было бы в смеси равновесной. Это даёт нам ещё один важный ключ к пониманию сути дела: если основание достаточно сильное и взято в эквимольном количестве по отношению к метилацетилену, то в смеси будет только ацетиленидный анион, и после гашения мы вернём метилацетилен обратно без аллена. То же самое будет если достаточно сильного основания будет избыток. Получим ацетиленид, и после гашения обратно метилацетилен. Ацетиленид-ион включается в рановесие только в присутствии исходного метилацетилена.
У нас есть метилацетилен. Повторим ещё раз:
- Если нам нужен ацетиленид-ион, например, в качестве нуклеофила в синтезе, – берём основание с pK хотя бы на 5 единиц больше, чем у ацетилена, то есть от pK 35 и выше. Поэтому мы берём бутиллитий, амид натрия, LDA тому подобные сильные основания. И количество основания должно быть эквивалентно алкину. Избыток брать не стоит, потому что он иожет помешать той реакции, которую вы задумали с ацетиленидом. И надо строго следить, чтобы основания не оказалось меньше, потому что в этом случае остаток метилацетилена войдёт в равновесную изомеризацию, и это может сильно помешать планируемой реакции.
- Если нам нужно превратить метилацетилен в равновесную смесь с алленом, берём небольшое количество достаточно сильного основания, например, трет-бутилата калия или натрия. Выдерживаем смесь до достижения равновесия. Или можно проделать то же самое в газовой фазе над твёрдым катализатором, или основанием, или кислотой. Кислотный путь изомеризации мы не обсуждали, видимо, он идёт через какие-то катионы, но это не так важно, ведь равновесие не зависит от пути его достижения: любой катализ даст одну и ту же равновесную смесь, и состав её будет зависеть только от температуры, да и то не очень сильно.
- Если вдруг вам понадобилось превратить метилацетилен в аллен с более-менее хорошим выходом, не говоря уж о количественном, у нас проблемы, потому что максимум достижимого – равновесная смесь, а равновесие обмануть невоможно. Ле Шателье позвать тоже не получится, потому что я не вижу способа выведения аллена из равновесия. Может вы придумаете? Если нет, то можно порекомендовать достичь равновесия при более высокой температуре, при 300-400 градусов в присутствии любого твёрдого кислотного или основного катализатора, например, оксида алюминия. Тогда в смеси будет до 20% аллена. Затем вам придётся решить сложную проблему отделения непрореагировавшего метилацетилена, например, путём перевода в нерастворимый ацетиленид серебра или меди. Аллен кладём в копилку. Достаём метилацетилен из ацетиленида, это непросто, но не невозможно – обсудим это в другом месте. Вновь достигаем равновесия. Вновь делим, добавляем аллен в копилку. Повторяем весь процесс столько раз, сколько потребуется для того чтобы накопить нужное количество аллена. Но проще сразу получить этот углеводород по методике Деьянова-Густавсона, см. выше.
Механизм ацетилен-алленовой перегруппировки более длинного алкина
А теперь усложним задачу. Возьмём чуть более сложный терминальный алкин, этилацетилен. И провернём с ним тот же фокус, как с метилацетиленом. То есть добавим немного достаточно сильного основания и дадим им побыть вместе достаточно долго, чтобы установилось равновесие. И что увидим? – почти чистый диметилацетилен с небольшими примесями по паре процентов аллена и исходного, как мы уже и установили, потому что внутренний алкин на 3-4 ккал/моль стабильнее, чем и терминальный алкин и аллен, которые приблизительно одинаковы по энергии (в этом случае аллен будет чуть-чуть, на полкилокалории более стабилен, потоу что он монозамещённый, так же как терминальный ацетилен). Как и в случае метилацетилена, и здесь основание в основном отрывает протон от терминального ацетиленового углерода, но в очень малой доле и от пропаргильного положения тоже, здесь точно так же будет разница в десяток единиц pK, может даже ещё на пару единиц больше, об этом ниже. Этот карбанион делокализован, и при протонировании даёт не только обратно этилацетилен, но и метилаллен. У метилаллена можно оторвать другой протон алленильной системы. Почему? А потому что метилаллен сам образовался обратимо из карбаниона такого же типа, следовательно pK этих протонов должны быть сопоставимы. А почему не из аллильного положения – из метильной группы? Можно и оттуда, но с большими проблемами – и об этом тоже на отдельной вкладке. А пока мы получаем еще один делокализованный анион пропаргильно-алленильного типа, и протонирование этого аниона дает не только обратно метилаллен, но и бутин-2 с внутренней двойной связью.
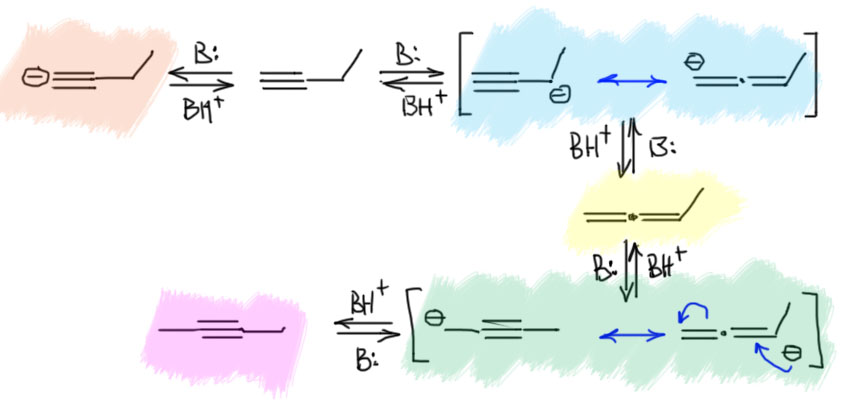
Если мы возьмём цепь ещё длиннее, то возникнет возможность точно так же переместить тройную связь дальше. Но происходит это намного медленнее, как мы уже убедились из опубликованных экспериментальных данных. Почему тройная связь не едет дальше? Точнее едет, но с большим скрипом. Разрисуем участвующие в деле карбанионы. Видим путь к дальнейшему перемещению связи вглубь (помечен голубеньким). Вроде всё то же самое. Поедет? Поедет, но смотрим внимательнее на первый пропаргильный карбанион. Их два – с незамещенной стороны и со стороны, где ещё алкил. Алкил, индуктивный донор, дестабилизирует карбанион. С этой стороны pK будет ещё больше, а кислотность ещё меньше. Разница в одну-две единицы pK невелика, но значительно замедляет движение в эту сторону. Просто можно буквально прикинуть – разница на единицу рК это приблизительно один к десяти – условно, связь поедет в сторону цепи в десять раз реже, чем в обратную сторону (а это просто обратимость первого смещения от терминального положения). А если две единицы рК, то в сто раз реже. Это безусловно грубо, потому что нам нужна оценка констант скоростей, а не равновесий, но в химии есть замечательный постулат Хэммонда, одно из следствий из которого говорит, что для очеь близких реакций термодинамика дает хорошую оценку кинетики с некоторым заранее неизвестным масштабирующим коэффициентом, иными словми, если константа равновесия в сто раз меньше, то и констранта скорости тоже меньше, но не в сто раз, а в некоторую долю от ста – сорок, семьдесят, какая разница, нам не нужны цифры, нам нужны качественные оценки).
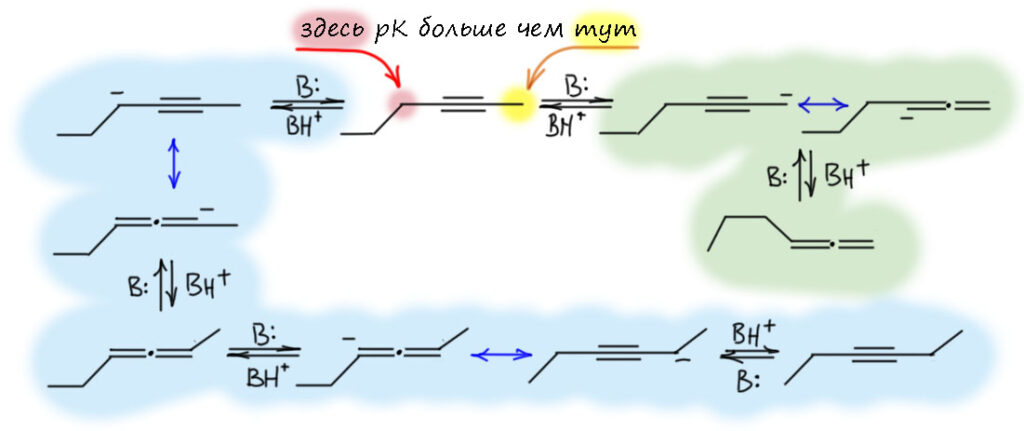
Поэтому очень легко из терминального алкина получить следующий по цепи, примеси будут не более 5%. Лучше делать перегруппировку быстрее, для чего лучше всего подходит сильное основание, соизмеримое по основности с терминальным ацетиленом, а это из тех реактивов, что обычно стоят на полке, трет-бутилат калия. Проще раствор в трет-бутаноле, можно особенно не париться с качеством растворителя, или разок перегнать, или взять как есть, потому что его очень хорошо видно, когда он более-менее чист – при минимальном охлаждении (даже если в комнате просто прохладно) он кристализуется такими длиннющщими острыми иглами. Тогда просто сливаете из банки всё, что не застыло, если жидкость есть, остаток топите тёплой водой и используете. Малая примесь воды ничего не испортит, потому что очень слабо влияет на основность трет-бутилата в своём спирте. Реакцию ведут при небольшом нагревании и через полчаса-час состав станет искомым 95% 2-алкина плюс по паре процентов алкин-1 и аллен и следы следующего алкина и аллена. Если под рукой есть сухой ДМСО хорошего качества, то то же самое можно получить за несколько минут при комнатной температуре. Но с ДМСО работать намного сложнее, чем с трет-бутанолом, поэтому многие предпочтут слегка погреть.
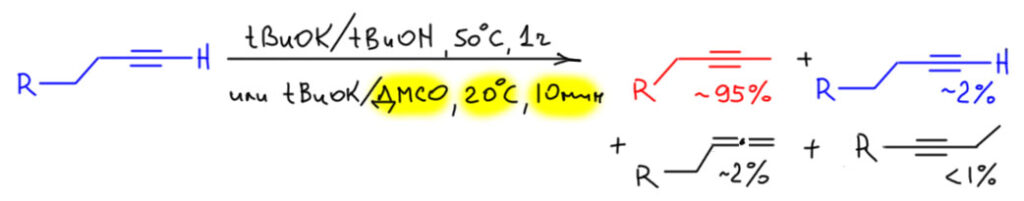
Более сильное основние (например, амиды щелочных металлов) брать не нужно, потому что все реакции ускорятся, в том числе и самые медленные, и тройная связь начнет расползаться по цепи вместе с алленами, и это уже будут плохие, неразделимые смеси. Все, что останется, это вернуть тройную связь обратно в конец, а как это делается, рассмотрим дальше.
А в обратную сторону можно?
Еще раз повторим простую вещь: если у нас есть терминальный ацетилен и мы добавили 1 эквивалент или избыток очень сильного основания, на 10 единиц pK или больше по сравнению с кислотностью концевого протона (при разных оценках кислотности концевого протона в ацетиленах в диапазоне 23-28, это означает взять основание с pK около 40 – это, например, амиды щелочных металлов, бутиллитий или другая простая литийорганика, и т.п.) – мы количественно получим терминальный ацетиленид и более ничего, никакой перегруппировки не будет. И важно понять, что при большой разнице pK депротонирование можно считать необратимым. Эта оговорка очень важна, потому что реакции между кислотами и основаниями принято по умолчанию считать обратимыми всегда. И это безусловно верно, и мы сами только что с успехом воспользовались этой важной идеей, чтобы понять как работает алкин-алленовая перегруппировка несмторя на чрезвычайно неблагоприятную разницу в pK между терминальным ацетиленовым протоном и пропаргильным протоном. В чём же разница? В концентрациях. Если у вас есть терминальный ацетилен и основание, которое оставляет существенную часть не депротонированным (то есть либо это основание, сравнимое по силе с основность ацетиленид-иона в любом количестве, либо существенно более сильное основание, но в недостаточном для полного депротонирования количестве), то возникает конкурентное депротонирование этого ацетилена по двум положениям, что и даст нам мизерное количество прапаргильного аниона, вполне досточное для работы алкин-алленовой перегруппировки. Кстати, если основание изначально выбрано очень сильное, но в недостаточном количестве, то реально в такой смеси будет работать ацетиленид-ион как основание. Разницы никакой нет – получится равновесная смесь, в которой начинается алкин-алленовая перегруппировка.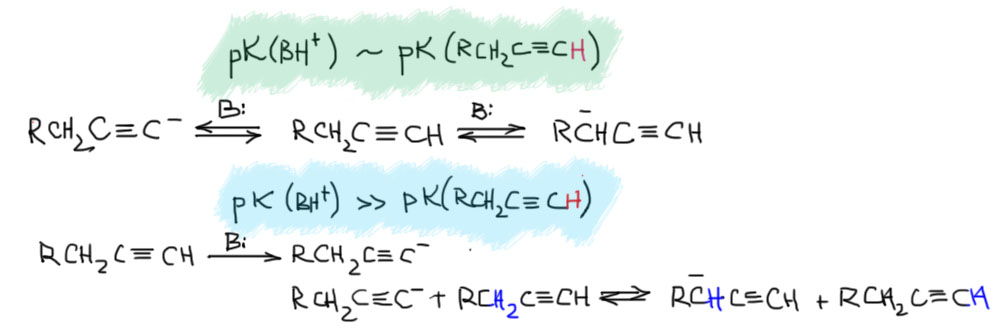
Теперь возьмём очень сильное основание, но эквивалентно по ацетилену. Получим ацетиленид-ион практически количественно, хотя, справедливости ради, не забудем про равновесное количество недепротонированного ацетилена – мизерное количество, так как разница в основностях не менее 10 единиц pK. И хотя через это количество всё равно пойдёт конкурентное депротонирвоание по пропаргильному положению, из-за ничтожной концентрации скорость этого процесса тоже будет ничтожна. И не сомневайтесь – равновесие своё возьмет, равновесие это страшная сила, но на установление равновесия могут потребоваться дни, а может недели, а может месяцы и годы. Столько ждать никто не будет, и поэтому в нормальной шкале времени реакцию можно считать необратимой. Это важная идея: выделим-ка её как очередную мантру (есть у нас ещё свободные бусины на чётках?)
Любое равновесие рано или поздно установится, но обратимую реакцию в течение времени существенно меньшем необходимого для установления равновесия можно считать необратимой.
В реальной лабраторной практике еще и используют избыток очень сильного основания, чтобы еще более снизить остаточную концентрацию исходного ацетилена, и сделать скорость установления равновесия пренебрежимо малой – потребуются годы, чтобы его достичь.
Это всё было, если мы брали исходный терминальный ацетилен.
Хорошо, а что если взять внутренний алкин, ну хоть 2-бутин и добавить к нему такой же избыток того же очень сильного основания, который мы только что обсуждали для реакции с терминальным ацетиленом. Поскольку в данном случае терминальных протонов в начале нет вообще, основание будет конкурировать за протон из пропаргильного положения, кислотность у него сильно пониже, и тогда просто будет запущен механизм алкин-алленовой перегруппировки. Мы войдём в то же самое равновесие, но с другой стороны. И мы знаем законы равновесия – откуда в него не войди, на выходе будет одно и то же. Ну и что, равновесие-то одно и то же, по идее мы должны получить то же самое, а что это, мы уже знаем – равновесную смесь с подавляющим преобладанием внутреннего алкина, а в этом случае он и так взят как исходное. Но в смеси обязательно появится 1-2% концевого алкина и следы аллена, как и диктует на равновесие. Ой, проблема! Как только в смеси появится хоть 1% концевого алкина, он будет количественно депротонирован очень сильным основанием и эту стадию можно считать необратимой – ведь разница pK концевого протона и очень сильного основания настолько велика, что в смеси концевого алкина не останется вовсе (точнее, останется пренебрежимо малое количество). Обратим внимание на этот момент – здесь мы как будто противоречим тому, что если есть равновесие, то оно установится и риведёт к равновесной смеси. Нет, не противоречим, потому что теория равновесий (то есть классическая термодинамика) утверждает, что равновесие превыше всего, и уйти от него в системе обратимых процессов нельзя, и что оно всегда одно (в данных условиях, равновесие зависит от условий, от растворителя, от температуры, но прямо скажем, в разумных пределах, не влияющих на общие тенденции). Это безусловно верно, но теория равновесий ничего не говорит о времени его достижения, а время достижения зависит от скоростей прямых и обратных реакций, водящих в равновесие, а скорости реакций зависят не только от констант скоростей, но и от концентраций. И если концентрация какой-то важной частицы в системе очень мала, то и та скорость, которая связана с этой частицей тоже очень мала и в этой части у равновесия проблемы – на установление потребуются годы. Мы уже видели такую ситуацию, когда пытались понять, отчего тройная связь не едет дальше внутрь цепи, хотя по равновесию должна. Из всего этого следует такая забавная вещь, как возможность частичных равновесий. В систем, где обратимых раеакций много, и связанных этими реакциями веществ тоже много, полное равновесие должно включать все эти вещества, и в настоящей равновесной смеси содержание каждого вещества будет соответствовать его ΔG° то есть быть постоянным, и каждое отношение двух любых веществ из полного равновесия тоже должно быть постоянным и соответствовать константе равновесия (из это в частности следует очень полезная вещь – в полном равновесии, включающем много веществ, вас могут интересовать не все, а только некоторые, и вы можете совершенно честно устанавливать их относительное содержание, наплевав на всё остальное – но только относительное, конечно).
И вот, если переписать без деталей то, что происходит в ацетилен-алленовой перегруппировке – совокупности равновесий между ацетиленами, алленами, карбанионами и неявно – еще и тем основанием, которое всё это затеяло и его сопряжённой кислотой – и подумать, а что будет в тм случае, если это основание очень сильно, намного перекрывает основность терминального ацетиленида, на 5-10 единиц pK (можно больше, но таких оснований очень мало и они сложны в работе) то мы получаем все эти отщепления-присоединения протона, потому что все остальные CH-кислотности в этой системе, как мы уже знаем, как раз единиц на 10 по шкале рК меньше, чем кислотность терминального ацетилена (обращаю внимание еще раз на то, как свободно и как будто произвольно мы оперируем понятиями кислотности и основности в смысле Бренстеда-Лоури, как будто это одно и то же, потому что это и есть одно и то же, ведь основность мы всегда выражаем через ксислотность сопряженной кислоты, и сказав “основность такого-то карбаниона” мы на самом деле говорим “кислотность сопряженной CH-кислоты”). Но стоит в системе с таким сильным основанием образоваться терминальному ацетилену, как он быстро теряет протон, превращаясь в ацетиленид, а вот в обратную сторону реакция практически не идёт, так как ацетилениду надо оторвать протон от намного более слабой сопряжённой кислоты использованного основания. Нельзя сказать, что здесь нет обратимости, но это конкретное равновесие смещено глубоко в сторону ацетиленида именно потому, что здесь неявно присутствует вторая пара кислота-основание.
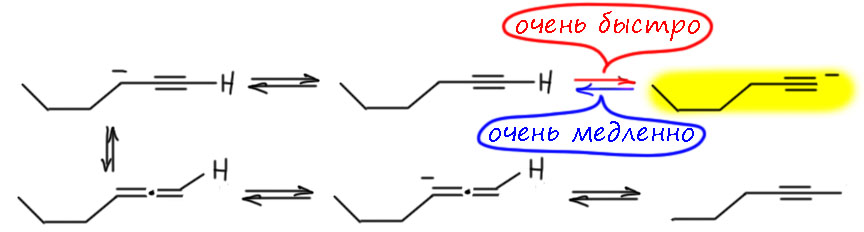
В таком случае, терминальный ацетилен фактически выбывает из общего равновесия между ацетиленами и алленами, общее равновесие старается восполнить недостающую часть себя, делает новый терминальный ацетилен, и он немедленно опять превращается в ацетиленид. Ацетиленид фактически становится такой ловушкой. Получается впечатляющий результат: если ацетилен-алленовое равновесие работает в присутствии очень сильного основания (с рК минимум в 35 единиц, можно больше), оно неуклонно съезжает в сторону териминального ацетиленида и там и остается.
И что, работает? Будет смещение тройной связи изнутри цепи, если добавить очень сильное основание? Будет, и это показал еще Фаворский в 1888. В те времена реактивов было немного, и Фаворский просто нагрел дизамещенный алкин с металлическим натрием. Не устаю повторять, что натрий (а также литий, калий, рубидий и цезий) это не основание. Мне легко рассуждать, от меня до Фаворского всего почти полтора столетия. Но в 19 веке такие тонкости никого не волновали – водород выделяется, если в воду кинуть, щёлочь получается, значит, основание. В реальности большая часть алкина при этом восстановилась, при этом возникли, как побочный продукт этих процессов, натрийорганические соединения, чрезвычайно сильные основания. Это тот же процесс, который происходит при восстановлении алкинов натрием в жидком аммиаке, только там есть хороший источник протонов в виде добавки спирта, а здесь нет, и анион-радикал превращается во что-то ещё достаточно случайное, например, оторвав атом водорода от пропаргильного положения другой молекулы ацетилена:
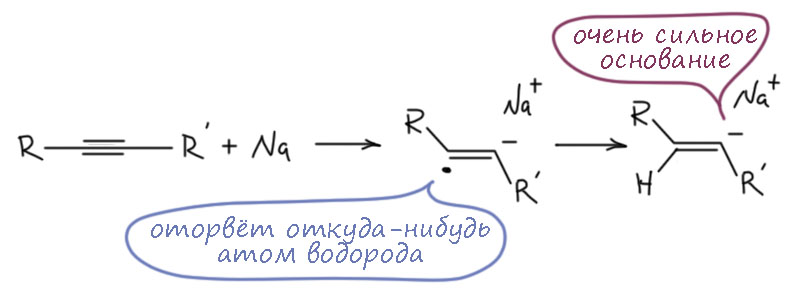
Та часть алкина, которая выжила в этом катаклизме, превратилась в концевой алкин, что и было установлено обычным для тех времён способом – образованием медного или серебряного производного. Поэтому в тех работах Фаворского, откуда и пошла эта химия, было установлено, что тройную связь можно смещать как внутрь, так и наружу, но, конечно, понять почему так происходит и в чём причина, в конце 19 века было принципиально невозможно.
Ацетиленовая молния
В последующие полстолетия с лишним к реакции вытаскивания тройной связи наружу время от времени возвращались, используя более современные основания типа амидов щелочных металлов. Тройная связь смещалась, но и выходы были не очень велики, и скорость реакции невелика, так что пользоваться этой реакцией никто для реальных синтезов не решался. Ждать пришлось до 1975 года – действительно почти сто лет. Это очень поучительная история, показывающая, как важно в химии найти правильные условия для реакции, и насколько сложно это сделать даже если механизм реакции известен. И еще очень важная вещь – эта история показывает, как важно набирать всё больше экспериментального материала, даже если сначала вообще непонятно, что всё это значит. Во времена Фаворского разговоры о сильном основании поневоле вертелись вокруг едкого кали и металлического натрия, а недостаток основности (и вообще реакционной способности решался просто – нагреем посильнее). За сто лет с тех пор арсенал сильных оснований расширился очень сильно, стало возможно подбирать основания целенаправленно. И ещё очень важная вещь – органика была и остается в основном качественной наукой. Если бы у нас были количественные значения килотностей и основностей для большого количества соединений, выбор можно было бы попробовать сделать совершенно осмысленным – знаем, что нужно депротонировать и подбираем основание просто по таблице. Увы, всё не так просто, потому что работает еще и много других факторов, в частности кинетика депротонирования, с которой всё ещё намного сложнее.
Терминальный алкин из внутреннего, то есть менее устойчивый из более устойчивого, это реакция наперекор термодинамике – но ничего странного в этом нет, так как мы отказались от равновесия, сделав одну из стадий эффективно необратимой. Возможно, вы помните, что похожая ситуация у нас уже была в обратимой перегруппировке боранов, которую тоже удавалось сделать необратимой, и таким образом внутренние алкены превратить в терминальные. Ту реакцию открыл Герберт Чарльз Браун, нобелевский лауреат, но существенного применения в синтезе она не нашла, так как оказалась неудобна, ненадёжна, и годилась только для небольшого количества олефинов. Впрочем, в современной химии есть хорошие методы вытаскивания двойной связи наружу, но это сложная химия с участием переходных металлов. А Чарльз Аллан Браун, американский химик, не имеющий никакого отношения к первому, и даже малой толики его амбиций, открыл мощный метод вытаскивания наружу тройной связи, и этот метод поразил органиков мощью и простотой и надёжностью, став просто незаменимым инструментом синтеза, особенно пригодившимся для синтеза соединений, используемых насекомыми для привлечения себе подобных для размножения – феромонов. Феромоны были дико популярны в конце прошлого века, когда компании, занимающиеся средствами защиты растений от вредителей, искали способы заменить инсектициды – токсичные вещества, которыми многократно обрабатывали поля и сады, убивая в них всё живое, в том числе полезных насекомых, таких как пчёлы. Пчеловоды в таких случаях приходят в ярость, и идут громить фермера, по вине котрого погибли пчёлы. Да и продукция с таких полей и садов часто становилась вредна для людей. Искали способы с помощью феромонов привлекать конкретные виды насековых вредителей в ловушки, что делало бы совсем ненужными инсектицидные обработки. И феромон, созывавший на гибель какого-нибудь зловредного пилильщика, вообще никак не влиял ни на пчёл, ни вообще больше ни на кого.
В синтезе феромонов надёжный метод вытаскивания тройной связи наружу оказался просто незаменимым. Феромоны стали делать десятками. Сразу скажу, что по разным причинам этот способ борьбы с вредителями революции в сельском хозяйстве так и не сделал, пестициды не вытеснил, оказался очень дорогим и не очень надёжным, а химия пестицидов за это время сделала новые препараты, намного менее опасные. А многие и вовсе научились обходиться без пестицидов.
Чарльз Браун очень увлекался поиском очень сильных оснований, и стал большим фанатом металлического калия и его гидрида. Калий – злобный металл, работать с ним решаются немногие, потому что если что-то делать неправильно и неаккуратно, калий может решить вас проучить, а жалости он не знает, и с легкостью вынесет вам глаза, изуродует лицо, оставит без руки, или хотя бы так напугает, что решите вы немедленно забыть даже дорогу в химическую лаботраторию и посвятите остаток жизни кулинарии или садоводству. Брауна калий не напугал, экспериментальное мастерство у него и его сотрудников оказалось на высоте, и стали они изучать применение и самого калия и его гидрида и амида, не менее ужасающих веществ, которые начинают гореть от малейших контактов с воздухом. Сам Браун после много раз про это напоминал, когда с интересом наблюдал за тем, как другие пытаются изобрести альтернативные решения, избегая страшного калия и его гидрида, – говорил, что только с этим щелочным металлом система действительно “летит”, а всё остальное, конечно, работает, но так лениво, что даже смешно.
Итак, перебирая сильные основания, которые можно сделать из гидрида калия, Чарльз Браун с сотрудниками открыли, что разные амиды калия хороши, но действительно драматический эффект получается, если в качестве основания взять монодепротонированный 1,3-диаминопропан, а сам этот диамин использовать в качестве растворителя. Собственно, даже не открыл, а просто подметил в одной из ранее вышедших работ по алкин-алленовой перегруппировке (J. H. Wotiz. P. M. Barelski, D. F. Koster, J. Org. Chem., 1973, 38, 489), что сопряженные основания диаминов намного эффективнее в этой перегруппировке, чем просто амиды из моноаминов. И добавил к этому наблюдению свой любимый калий в качестве противоиона. Вот так в химии бывает – наблюдательный и вдумчивый исследователь видит потенциальную новизну вещества или метода там, где другие видели лишь скучную рутину верениц цифр в таблицах.
Именно калиевая соль диаминопропана оказалась решением. Соль, а точнее именно реагент – раствор в диаминопропане – прозвали KAPA (просто перекрестив диаминопропан DAP в АминоПропилАмин APA), потому что это сокращение показалось более благозвучным и прикольным, чем более очевидное KDAP. Найти хорошую аббревиатуру или акроним для нового метода всегда становится не меньшей удачей, чем сам метод, потому что в науке точно так же работают законы рекламы и маркетинга – как назовёшь товар, так его и покупать будут. А броская аббревиатура работает просто отлично, причём каждый язык находит в этом что-то созвучное себе, у кого-то это смешно, у кого-то напоминает имя любимого кота, а иной вообще увидит нечто мистическое.
Строго говоря, и это основание было известно и до этой работы, его время от времени использовали для депротонирования всяких слабых CH-кислот, но никаких чудес оно не творило, просто потому что их никто и не искал. Но здесь оно просто совершило революцию. Это без всяких преувеличений один из самых эффектных реагентов в органике. Внутренние алкины перегруппировывались в терминальные ацетилениды за считанные минуты, даже секунды – нужно было просто догадаться взять избыток.
Почему именно эта система такая эффективная. Общая идея понятна – нужно основание, точнее основная среда, которая имеет некоторый оптимум кислотности и основности, ведь она должна быстро снимать протон и быстро отдавать его обратно. Снимает протон основание, отдаёт обратно его сопряженная кислота. Если основание недостаточно сильное, медленной будет стадия депротонирования. Если слишком сильное, наоборот, протонирования. И то, и другое замедляет процесс прыгания. Нужно оптимальное сочетание основности и кислотности. Вот его, видимо и даёт калиевая соль диаминопропана. Почему? Всё довольно просто. Изначально ясно, что основание нужно сильное, такое как амид щелочного металла. Напомню, что амидами называют не только производные карбоновых кислот, но и сопряжённые основания аминов и их соли или комплексы с металлами. У простых солей такого типа с щелочными металлами pK больше 40. И очевидно, что это перебор – депротонирование идёт легко, а протонирование плохо. Изменим структуру амина так, чтобы основность стала немного ниже, а кислотность, соответственно, немного выше. Диаминопропан – просто идеальный выбор. Гидрид калия быстро депротонирует диаминопропан по одной амино-группе (C. A. Brown, J. Am. Chem. Soc., 1973, 95, 982). Моноанион диаминопропана – это почти такой же амид, как и любой другой амид от первичного алкиламина, но он стабилизирован и внутримолекулярной водородной связью, и хелатным взаимодействием с катионом металла. Стоит только иметь в виду, что это не очень сильные взаимодействия: азот и NH – довольно слабые акцептор и донор водородной связи, а катион калия работает не за счет настоящих координационных связей, для которых у него совсем немного валентных возможностей, а обычной электростатикой, за счёт зарядов на атомах азота и металла. Впрочем, ионные связи не такие уж и слабые, но они очень сильно зависят от расстояния между зарядами, и для довольно крупного катиона калия особой мощью похвастаться не могут. Поскольку ионные связи не имеют предпочтительного направления, в стабилизации аниона могут участвовать и водорожные и ионные связи, вовсе не обязянные толкаться в одной плоскости. 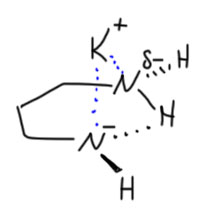
Как мы знаем, стабилизация аниона, то есть сопряжённого основания, отражается на снижении основности такого основания по сравнению с похожим, но не имеющим таких стабилизирующих взаимодействий. Хорошо – это именно то, что нужно, а достаточен ли эффект проверяется по тому, как это работает. Отлично работает, прямо неправдоподобно – попадание в десятку с первой попытки. Победителей не судят за маленькое бахвальство, – победителям справляют триумф, трубят в трубы и славят на все голоса. Забегая вперед, замечу, что с тех пор было предложено множество других реагентов для той же цели, но именно оригинальная KAPA Брауна, получаемая действием гидрида калия на диаминопропан в избытке этого амина, осталась самым эффективным реагентом этого типа, и когда нужна чистота и эффективность, выбирают именно ее. Поэтому славу и трубы, и триумф Браун и сотрудники вполне заслужили.
Браун не ограничился прикольным сокращением для реагента, но и незамедлительно окрестил новый метод ацетиленовым зиппером, вынеся этот броский девиз прямо в заголовок самой первой своей статьи (C. A. Brown, A. Yamashita, J. Am. Chem. Soc. 1975, 97, 891-892). По-русски это стоило бы перевести ацетиленовой молнией, потому что именно так переводится название этой застёжки, но мы боимся двусмысленностей и не любим брать на себя ответствеенность за что-то необычное. А зря! Сама идея Брауна совершенно блестяща – быстрое перемещение тройной связи по цепи действительно здорово напоминает работу застёжки. По английски и американски такую застёжку называют zip или zipper, просто подражая звуку – зззипп, ззззиппп, правда, похоже. По-русски такое же звукоподражание выдало бы что-то типа вжик, вжжжжикк, ну, как-то так, то есть получилась бы какая-нибудь вжикалка. Но мы называем такую застёжку молнией с тех пор, как она появилась в СССР в 1930-е, в этом странное время, когда кожаные куртки носили не только палачи и чекисты, но и весьма популярные авиаторы и полярники. Откуда взялось это замечательное слово, намного более выразительное чем дурацкий зиппер? Это оказалось не очень просто выяснить, и за полную достоверность я не поручусь, но скорее всего, дело было так. Саму застежку этого типа открывали и патентовали много раз, еще с 19 века, сначала в США и Канаде. Застёжку запустили в производство во время Первой мировой войны для военных комбинезонов, особенно лётных, видимо для того, чтобы горящую одежду можно было быстро расстенуть и скинуть. В Европе она появилась позже, и стала быстро распространяться с 1920-х благодаря французской компании Éclair Prym, кажется, существующей поныне. По названию компании застежку и стали называть “застежка Éclair”, а это естественно быстро сократилось просто в éclair, а это по-французски и есть молния. Нам это французское слово, в виде названия пирожного с заварным кремом эклер отлично известно. А это-то тут причём? Словарь французской академии объясняет, что это пирожное так прозвали, потому что оно молниеносно съедается (…parce qu’il est vite mangé). Ну и выдумщики эти французы однако! Так или иначе почти наверняка и как раз тогда это слово попало в другие языки: например, по-итальянски эта застежка тоже называется il lampo – молния. Ну и к нам, или прямо от французов, или через Италию, – чёртовы большевики страну закупорили, но не сразу и не полностью, а мода есть мода, и с большевисткими кордонами не считается. Так мы получили молнию вместо зиппера. Конечно, это слово очень здорово отвечало самому назначению этой застёжки. Тогда невероятно модны были всякие романтические персонажи – первые лётчики, полярники, такие крутые чуваки с заиндевевшими бородами в кожаных куртках, шлемах, и прочем брутальном прикиде. В куртках этих и появилась впервые молния.
Так или иначе, слово дожило до наших дней, хотя многие уже привыкли называть эту штуку зиппером. И отлично можно было бы назвать реакцию в русском переводе ацетиленовой молнией, тем более что это даже точнее передаёт то, что хотел сказать Браун – тройная связь двигается очень быстро, несколько секунд при ноле градусов – молниеносно! Даже эклер не успеете сожрать. Куда лучше оригинального вжика. Вот мне никто запретить не может – назову реакцию ацетиленовой молнией.
Как это работает
Чтобы объяснить работу своей выдающейся реакции, Чарльз Браун не стал ничего выдумывать сам, потому что готовый механизм уже был придуман – ацетилен-алленовая перегруппировка в присутствии натриевой соли диаминоэтана уже изучалась на модельных субстратах. Главный теоретик начальной стадии химии карбанионов Дональд Крэм для перегруппировки алкина в аллен с помошью такого аниона предложил согласованный механизм, который он назвал “conducted tour” – перемещение с сопровождением, или, из туристической жизни, экскурсия с сопровождающим. Чёрт знает, насколько это удачно, на мой взгляд, скорее нет, да и особенно широко этот термин не разошёлся несмотря на огромный авторитет Крэма среди металлооргаников второй половины прошлого века. Имелось в виду вот что:
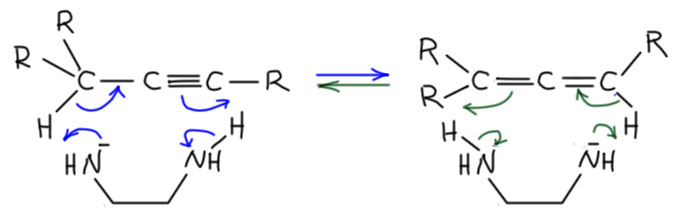
Этот механизм так всем нравится, что его с удовольствием воспроизводят в обзорах и учебниках, тем более что он освящен авторитетом Дональда Крэма, который привёл такой механизм и доводы в его пользу в своей знаменитой книжке про карбанионы, которая в прошлом веке была на столе у каждого уважающего себя органика, исследовавшего механизмы органических реакций. Но не все так падки на красивые картинки. Такие злобные скептики иногда говорят – Too beautiful to be true, – что означает – Ври, да не завирайся. Помни об энтропии – хочется сказать слишком ретивым сочинителям красивых механизмов, в которых молекулы услужливо изворачиваются и выстраиваются в идеальные построения, как команда по синхронному плаванию. Увы, у молекул нет мозгов, и они не подчиняются командам тренера. Любые такие построения дают некоторый выигрыш в энергии, но страшно проигрывают более банальным механизмам в энтропии. Не то, чтобы это было совсем невозможно, – Природа и Жизнь дают нам бесчисленные примеры красивых реакций, идущих явно против энтропии, но вся жизнь собственно это и есть восстание против мерзкого хаоса. Но в колбе нет жизни, и обычно нет и никакого восстания – энтропия там очень влиятельный фактор, перебить который можно только показав, какие взаимодействия заставляют молекулы выстраиваться в красивые ансамбли, – это могут быть водородные связи, какая-то серьёзная электростатика, донорно-акцепторные взаимодействия, ароматический стекинг, – что-то должно быть, извольте найти и предъявить. Когда такое удается сделать, действительно получаются красивые механизмы, и тогда мы видим нечто особенное, например, выдающуюся стереоселективность, эффективный перенос хиральности и т.п. – у нас будет еще много возможностей обсудить такие случаи.
А если не можете, то не выдумывайте, пожалуйста, помните об энтропии. Вот в этом случае не просматривается ничего подобного – что заставляет второй конец аминопропиламида подстраиваться к атому углерода через один от места депротонирования амидным концом? Нет там ни водородных связей, ни серьёзной электростатики. Почему согласованность протонирования-депротонирования так выгодна? На самом деле, если посмотреть на картинку механизма “перемещения с сопровождением” критическим взглядом, то бросится в глаза одна вещь, которой поклонники этого механизма явно пренебрегли: протоны-то может и перемещаются легко, но этот процесс должен сопровождаться регибридизацией двух крайних атомов углерода: один из sp3 в sp2, а другой из sp в sp2, а в обратной реакции в обратную сторону. Регибридизация это не просто орбитальки нарисовать, это очень сильное изменение положения атомов и групп в пространстве, изменение валентных углов и длин связей. Такие изменения обычно требуют немаленьких затрат энергии, а поскольку речь идёт о согласованном механизме, где нет никаких интермедиатов, а есть только преодоление барьера активации, это и должно давать очень значительный барьер – в энергию активации с обеих сторон как раз и входит изменение геометрии молекул по координате реакции.
Сам Чарльз Браун, подобрав механизм “перемещения с сопровождением” у Крэма, сделал еще более радикальное добавление. Во-первых, не забудем, что у Брауна не диаминоэтан, а диаминопропан – ещё более гибкая молекула, которую выстроить нужным образом непросто. Но даже это еще не всё – Браун решает, что бытрота реакции смещения связей определяется даже не единичными перемещениями протонов с помощью аниона диаминопропана, а то, что эта штука действует и дальше тем же способом. Она как будто бежит по цепочке, сопровождая смещающуюся связь. Как она это точно делает Браун толком не пояснил. А мы, если попробуем самостоятельно додумать его мысль сразу натолкнёмся на несколько препятствий. Вот просто аккуратно распишем пару таких смещений, показывая перемещающиеся протоны и анион диаминопропана, который их таскает. Во-первых, мы видим, что для каждого смещения тройной связи нужно перетащить не один, а два протона, и перетаскиваются они между одними и теми же углеродами. Уже это ставит крест на идее про быстро-быстро-бегущие по цепи анионы диаминопропана. Проблема в том, что анион при перетаскивании второго протона должен быть там же, где и на транспорте первого протона. И как он туда попадёт? Простая идея о том, что протон в анионе диаминопропана легко скачет с одного азота на другой – типа, где надо, там минус и будет – довольно спорна и идёт от аналогии с похожими кислородными молекулами (1,3-пропандиолом, например), но кислотность аминов на многие порядки (не менее 10) ниже кислотности спиртов, а водородные связи N-H-N намного слабее кислородных аналогов; поэтому протоны в аминах и их анионах обычно никуда не скачут. Поэтому нам потребуется другая молекула аниона диаминопропана, или та же, но потребуется обычное в жидкостях диффузионное движение – молекулы хаотически перемещаются, и все определяется законами вероятностей. Из этого следует простая вещь – о согласованном движении вдоль цепи (как в живом мире бежит какой-нибудь ферментный комплекс вдоль нитки ДНК) лучше забыть – нет там никаких направленных взаимодействий, и нет никаких даже поводов такие найти, все равно, на каждой паре протонов нужна остановка движения вдоль цепи.
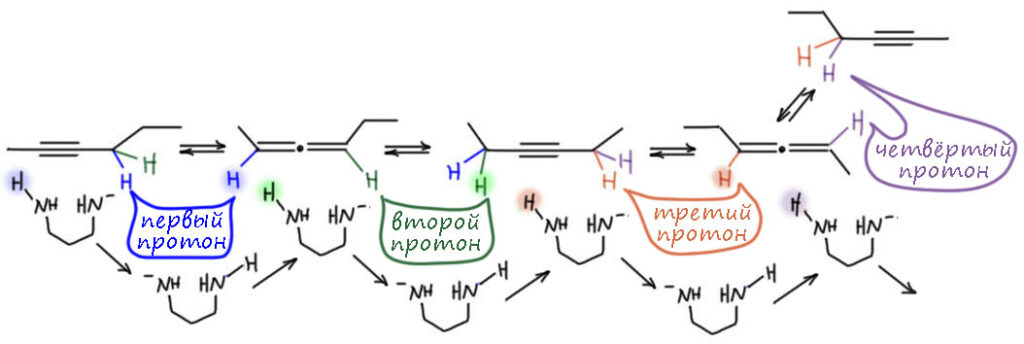
В истории исследования этой реакции было несколько статей, в которых пытались доказать справедливость красивого механизма, увы, и вполне ожидаемо, безуспешно. История понемногу прокисла, так и не придя к победе. Бывает. Но химия и вообще наука так устроены, что однажды нарисованную каким-нибудь крутым авторитетом красивую картинку оспаривать мало кто решается. Так она и живёт, становясь фактом науки, хотя на самом деле является обычным мифом. Люди атеистического склада ума обожают шпынять поклонников религий в том, что они, дескать, верят в выдуманные людьми сказки. То ли дело суровая наука – в ней, дескать, все доказано, проверено и перепроверено. Чёрта с два, выдумок в науках выше крыши, а легковерности учёных и слепому поклонению велеречивым авторитетам позавидовали бы и святые апостолы, некоторые из которых, как хорошо известно, немало сомневались и требовали убедительных доказательств.
А тогда, ну её, эту картинку. Мы бы к ней лучше отнеслись, если бы система работала с эквимольным количеством KAPA, и была столь же эффекивна. Но нет, реакция идёт в присутствии большого избытка диаминопропана, это в оригинальной методике растворитель, а во многих последующих работах просто некоторый избыток. В этом случае вокруг молекулы алкина множество молекул ДАП, и логичнее предположить, что депротонирование и протонирование осуществляют разные молекулы. В этом случае молекулам амина и амида нет нужды так энтропийно невыгодно точно притираться к алкину аккуратной скобочкой прямо к нужным атомам, а каждый акт реакции происходит при банальном бимолекулярном столкновении реагентов. Мы даже не обязаны париться по поводу конформации этих молекул – действительно ли им выгодно выгибаться скобочкой, или гибкая цепь как положено извивается и даже предпочитает обычную для насыщенных цепей конформацию все-анти (она же зигзаг).
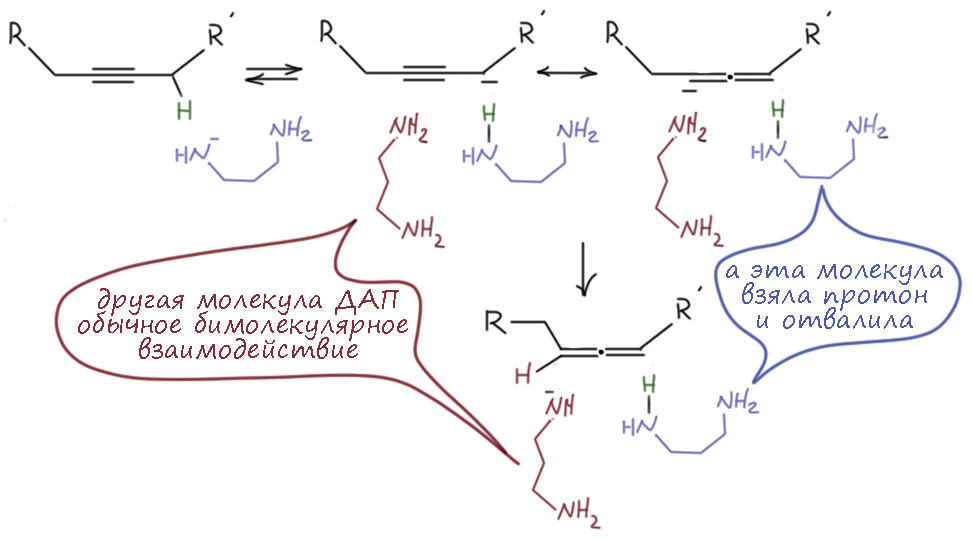
Так почему же именно эта система так хорошо работает? Моё собственное объяснение довольно просто. Во-первых, мы должны обратить внимание, что здесь не одно, а несколько слагаемых успеха. Во-первых, это использование солей диаминоалканов. Лучше всего 1,3-диаминопропана, но с течением времени пробовали разные другие, тот же диаминоэтан, и они тоже работают, вытаскивают связь наружу, но медленнее. Во многих случаях это не важно, и такие системы используют довольно часто, у них много других аббревиатур, но напного менее популярных, чем KAPA. Во-вторых, это всегда использование избытка исходного диаминоалкана, иногда в качестве растворителя, иногда просто добавки, но никогда не берут чистую соль, а это было бы сделать весьма просто. В-третьих, и это последнее соображение, хотя используют три катиона щелочных металлов, и литий (ЛАПА и т.д.) и натрий (НАПА и т.д.) и калий, только калийное производное работает как молния – быстро вытаскивает тройную связь даже из очень длинных цепочек. Остальные производные применяют для более коротких вылетов, но таких реальных случаев немало. КАПА – лучше всех, но и опаснее всех, и часто экспериментатор предпочтет взять куда более простую в работе натриевую или литиевую соль и прекрасно вытаскивают тройные связи, когда длина пути до выхода короче – два-три-четыре звена. Следовательно, в порядке важности надо понять почему диаминопропан и его анион, или близкие аналоги. И во-вторых, почему калий дает особую эффективность.
На первый вопрос ответить легко, если подметить одну простую вещь. Для работы этой системы нужно быстро делать две реакции – депротонирование и протонирование. Если быстро одно, но медленно другое, вся система будет тормозиться. За депротонирование отвечает анион амина (амид – не путайте с отдельным классом производных карбоновых кислот, анионы аминов тоже называются амидами), за протонирование – сам амин, сопряженная кислота амида. Или, если мы возьмем другое основание, его сопряженная кислота. Поскольку за время исследвани ацетилен-алленовой перегруппировки данных накопилось много, можно отлично видеть простую тенденцию:
- если основние недостаточно сильное (гидроксид, алкоголяты), то его сопряженная кислота обладает довольно высокой килотностью. Мы эту ситуацию уже разобрали, и пришли к выводу, что в этих условиях медленно идет перегруппировка в сторону более устойчивого внутреннего алкина
- если основание очень сильное (литийорганика, простые амиды моноаминов, или аммиака), то сопряженная кислота очень слабая. В этих условиях перегруппировка или вообще не идет, или идет крайне медленно, потому что депротонирование идет легко, а обратное протонирование – очень медленно или вообще никак; мы заканчиваем реакцию просто образованием анионов пропаргильного типа, которые в принципе можно использовать в синтезе, только проблема в том, что такие анионы делокализованы и у вас получатся два продукта – ацетилен и аллен.
Ну вот и весь секрет. Диаминоалканы и их анионы просто дают оптимальное соотношение основности и кислотности – депротонирование и протонирование идут со сравнимыми скоростями; никто никого не ждёт. Почему это так? Отчасти это просто следствие, как обычно в химии, кропотливой работы по поиску оптимального реагента – искали-искали, и нашли, и лучший реагент, и славу и благодарность потомков. Так работает прогресс. Отчасти вполне понятная модификация; у моноаминов основность избыточна, а кислотность сопряженных кислот недостаточна, а у диаминов основность на несколько единиц рК меньше (анион стабилизирован и водородной связью, и хелатным эффектом противоиона, и индуктивным эффектом амино-группы), а кислотность диамина соответственно выше (это связанные величины). И вот ровно это явно и оказалось оптимумом. Не забываем и о том, что депротонировать (и соответственно протонировать) нам нужно два положения – пропаргильное и алленильное – и СН-кислотность на этих положениях приблизительно одинакова (анион один и тот же, а дельта-же-образовния ацетилена и аллена очень близки, это мы уже разобрали). Точно так же одинакова и кислотность-основность амина-амида на концах цепочки диаминоалкана, это просто одна и та же группа. Вот и весь секрет – фактически случилась отличная синхронизация депротонирования и протонирования.
Как это не работает
С этой реакцией связано одно очень забавное заблуждение, идущее от самого автора метода. Собственно с ним связано и название реакции – зиппер, молния.
Тройная связь очень быстро вылетает из глубины цепей, легко преодолевая десятки атомов углерода. И это дейcтвительно чудесное свойство, показывающее исключительно высокую эффективность реагента. Исследователи этой реакции замечали, что даже если надо проскочить тридцать атомов углерода, с настоящей брауновской капой (той, которая получена честным депротонированием чистого диаминопропана гидридом калия) это происходит за несколько секунд. Если взять напу, лапу, или нечестную капу – какую-нибудь дешёвую бодягу из диаминопропана и чего-то типа трет-бутилата калия, не удивляйтесь, такое тоже бывает, лень и осторожность на грани с трусостью – профессиональные качества хорошего органика, который хочет не только статью знаменитую сделать, но и дожить до пенсии с полным комплектом частей тела и сохранившимся даром речи – то тройная связь поедет намного медленее и больше трех-четырех атомов за полдня не проедет. Удивительная скорость движения дали этой реакции не только броское название, о чём ниже, но и представление о том, что тройная связь вприпрыжку бежит к нужному концу цепи, ни на мгновение не останавливаясь и не раздумывая. Поэтому и появился тот механизм согласованного движения – скобка диаминопропана, точнее, аниона, бежит по цепи, отрывая и приклеивая, отрывая и приклеивая. Только в одну сторону. Мы уже выяснили выше, что это невозможно даже из общих соображений, но гипотеза оказалась настолько привлекательной, что широко расползлась, тем более что сам первооткрыватель её же фактически и придумал. При это возникает довольно комическая проблема – возможны ведь две ситуации: алкин полностью линеен и вначале тройная связь где-то в глубине цепи, но не обязательно строго в середине; алкин закупорен с одной стороны разветвлением или чем-то ещё – так что бежать нужно в одну строну. но тройная связь опять находится на прямо около упора, а слегка поодаль. В первом слечае вопрос – тройная связь сразу побежит более коротким путём, или это будет случайный выбор (при том, что раз выбрав, бежим со всех пи-связей в выбранном направлении).
Во втором случае ещё интереснее, – тройная связь побежит только на выход, или может сдуру ломануться в сторону тупика и только потом передумать?
Фокус в том, что тройная связь не знает, куда бежать: у нее с обеих сторон совершенно одинаковые CH2 группы. Поэтому тройная связь хаотично мечется между соседними парами углеродов, и только иногда случайно добегает до конца тут же застряв там. Сколько в среднем она делает прыжков для того чтобы добежать до конца очень хорошо знает теория вероятностей: такие задачи о случайном блуждании решают именно в этой потрясающей науке. Типичную задачу такого типа в этой теории называют задачей о блуждающем пьянице – можно ли оценить время, которое понадобится уже полностью потерявшему связь с реальностью пропойце, чтобы дойти до следующей распивочной, если каждый шаг делается совершенно случайно и направления шагов равновероятны. Можете сами попробовать слазить в учебник теорвера и посмотреть, как решаются подобные залачи и заодно оценить среднее время, которое потребуется тройной связи для перемещения в конец цепи. Вообще, если подумать, то задача покажется весьма парадоксальной, ведь кажется, что если соврешенно случайно рыпаться в оба направления, то результатом будет топтание на месте, ведь вроде бы в таком случае нет никакого преимущественного направления. И это, чёрт возьми, именно так и есть, но поскольку радиус топтания на месте не ограничен ничем кроме случайности, все же иногда пропойца быдет попадать в распивочную, а тройная связь – на конец цепи. Ни из распивочной, ни с конца цепи выхода уже нет, поэтому это событие работает как ловушка (кто-то назовёт аттрактором, кто-то стоком (sink), удивительно, но подобные проблемы нередко вылезают в самых разных науках), и именно так это и работает – бессознательное, бессмысленное движение получает цель и работает на эту цель.
Еще более трудоёмкой окажется задача, если добежать можно только до одного конца, потому что с другого есть препятствие, например, разветвление или спиртовая группа. В этом случае тройная связь будет добегать и до препятствия, и как бы отражаясь от него бежать туда, где есть свет в конце тоннеля. – среднее число прыжков в такой ситуации вырастет еще весьма значительно, потому что тройная связь в среднем будет не раз долбиться об этот тупик, и будут обязательно молекулы, в которых тройная связь, добежав до предпоследнего перед концом положения, опять ломанется вглубь, и только основательно намаявшись выберется наружу. А можно ли это как-то увидеть, не изучая ненавистный теорвер. Вполне, надо было бы просто взять дейтерированную капу и проследить дейтериевый след – ведь каждая метиленовая группа, которой повезло участвовать в ацетилен-алленовой перегруппировке, оставит себе на память дейтерий. Тогда можно было бы просто брать пробы из такой смеси и определять распределение дейтерия – сейчас это не составило бы слишком большого труда с помощью ЯМР. Кстати, тогда можно было, например, узнать, что будет, если тройная связь ближе к одному концу длинной прямой цепи – если блуждание полностью случайное, то до дальнего конца связь добираться не должна практически никогда.
Реальность, как всегда, немного скучнее. Такое исследование действительно сделала канадская исследовательница Сюзанн Абрамс, прямо таки собаку съевшая на изучении этой реакции, собственно, кроме неё этим больше никто и не занимался. У Абрамс при этом было тяжёлое неприятие оригинальной системы Брауна – той самой “честной капы”, получаемой из диаминопропана и гидрида калия: мудрой женщине казалось, что даже для самой лучшей реакции не стотит рисковать жизнью и здоровьем, и хорошо бы иметь под руками что-то подобное, но поспокойнее. Поэтому она активно пробовала аналоги с литием и натрием, производные которых тоде очень опасны, но даже близко не напоминают коварный и злобный характер калия и его гидрида и амида. С соединениями лития и натрия надо просто аккуратно и осторожно работать, выполняя все предосторожности, и все будет отлично. Для калия этого мало, и оторвать башку может даже самому аккуратному химику. Абрамс пробовала и лапу, и напу, находида, что они работают, но довольно медленно, и иногда даже требуют нагревания. Лучше работает такой суррогат “честной капы” как смесь напы и трет-бутилата калия, и эта смесь стала впоследствии довольно популярной. Молниеностности капы она не достигает, но работает неплохо и надежно, особенно если вам не нужны рекорды, а просто нужно сдвинуть тройную связь на несколько, но не очень много (редко более шести) связей.
На результатах это сказалось только в том, что время реакции стало измеряться часами, а выходы перестали быть количественными, но для сути понимания процесса это, может быть, даже полезно. Абрамс не делала промежуточных измерений, что не позволит нам увидеть след смещения тройной связи в динамике (таких исследований вообще с тех пор не делал никто, а зря – если кому-то нечего делать, я бы посоветовал покопаться в этой реакции современными методами и с помощью современных вариантов сильных оснований – мне кажется, что там ещё много занятного осталось), но и окончательное распределение дейтерия вполне информативно. В одной из первых работ (Abrams, S. R. J. Org. Chem. 1984, 49, 3587) исследовательница просто гоняла тройную связь по цепям и смотрела, какие части цепей дейтерируются. Во всех случаях она брала спирты, просто потому что это действительно один из самых частых случаев использования реакции смещения двойной связи. После реакций смесь разлагали обычной водой, поэтому все кислые гидроны (гидрон – это общай термин для протона, дейтерона и ядра трития) обменены обратно на протон. И вот мы видим, дейтерируются все метиленовые группы по дороге следования тройной связи. А под гидроксил ничего не попадает, потому что водород под гидроксилом намного менее кислый (карбанион дестабилизирован из-за альфа-эффекта).
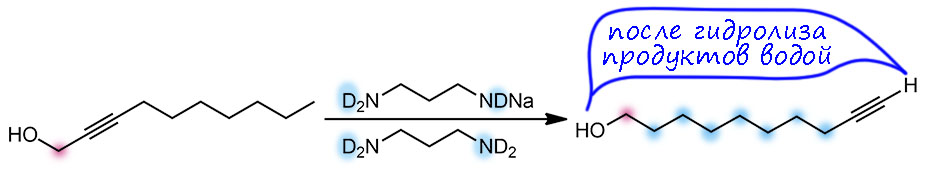
Если тройная связь в серединке, а с одной стороны тупик, то все равно дейтерируются все метиленовые группы в обе стороны – так мы однозначно видим, что тройная связь действительно не движется целенаправленно на выход, а беспорядочно мечется. Ну и, хотя это и так ясно, если и тупик далеко от конца, по другую сторону от тупика ничего не происходит.
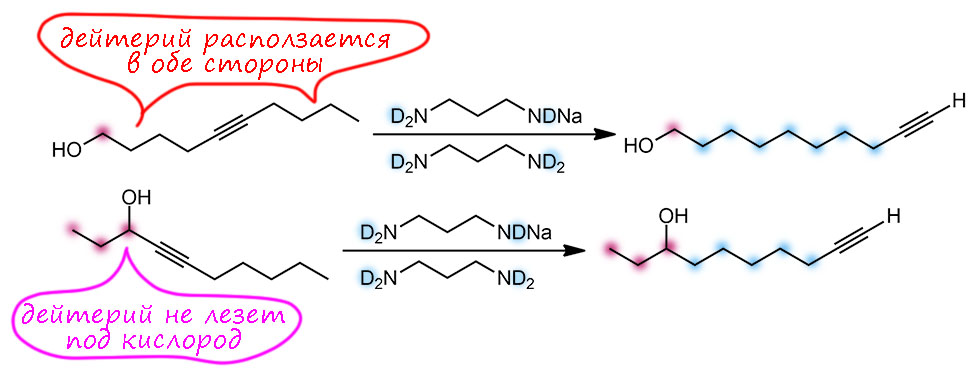
С гидроксильной группой это очень важно показать экспериментально, потому что иначе, из общих соображений нельзя было отрицать возможность того, что под гидроксил подползет алленовая двойная связь, а это могло бы закончиться куда более серьезными проблемами, потому что это уже был бы енолят, который мог бы вообще переключить все в химию карбонильных соединений, но этого не просходит. Ну и хорошо, потому что гидроксил и алкоксил часто используют в реакциях смещения тройной связи – он и сам никуда не девается, и путь запирает.

Надо сказать, что эту работу не задумывали как просто развлечение – типа тройную связь гоняем по цепи как блоху – но ей придумали отличное применение. Попробуйте продейтерировать длинную алкильную цепь, и ничего у вас не выйдет за пределами кислых протонов. А так можно просто исчерпывающе прогнать дейтерий даже по очень длинным цепям, ведь у реакции практически нет ограничений. Гидроксил дальше можно окислить и получить, например, пердейтерированную жирную карбоновую кислоту типа стеариновой, а это очень даже может пригодиться, например, в химии липидов или поверхностно-активных веществ.
Эту историю довели до забавного курьеза: что будет если молнию применить к циклическому алкину (Tetrahedron Lett., 1985, 26, 3431). Вообще, тройную связь непросто запихать в цикл, мы это ещё обсудим в своём месте, но это можно сделать, если цикл побольше, минимум 8-членный, но лучше ещё пошире. Поскольку у кольца нет конца, тройной связи деваться некуда. Если такую реакцию поставить с циклическим алкином, тот же алкин и будет получен, ну разве что несколько процентов аллена ещё образуется. Но если взять дейтерированный вариант молнии, станет ясно сидит ли тройная связь на месте: если нет, то протоны в цикле будут потихоньку замещаться на дейтерий. Вопрос только – где, только вблизи тройной связи, или и подальше тоже. На деле оказалось, что замещаются все протоны, происходит практически количественный дейтерообмен. А это значит, что несчастная тройная связь в бесплодных поисках конца мечется, как ненормальная по всему циклу – не забываем, что она не бегает в одном направлении, а именно мечется, ровно так, как учил один незадачливый вождь – шаг вперёд, два шага назад. Ну, или два шага вперёд, один назад. Но не один шаг вперёд, один назад – в этом случае тройная не стронулась бы с места. Как видим, не просто стронулась, а побывала в каждом уголке кольца. Наверняка возникнет вопрос, почему для этого эксперимента Абрамс использовала не капу, а лапу. Просто, чтобы реакция шла помедленнее, и за ней удобнее было следить. 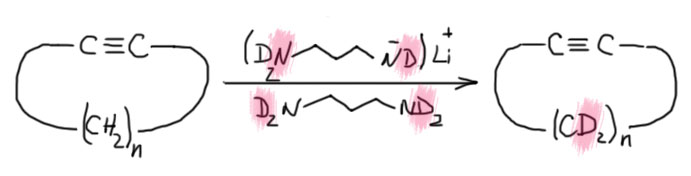
Что можно подвергать ацетиленовой молнии?
Для того, чтобы молния сработала, у тройной связи должен быть путь по цепочке нормального строения, длина цепочки может быть очень большой: насколько мне известно, получить такой длинный алкин, чтобы в нём не произошло смещение связи в конец, пока никому не удалось, хотя стоит и сказать, что в органической химии крайне редко имеют дело с цепями длиннее 20 атомов углерода, а всё что больше 30 атомов и вовсе представлено единичными работами. 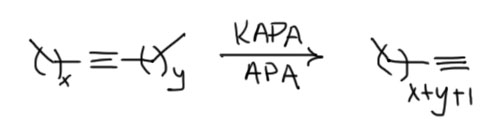 Речь идёт, естественно, об индивидуальных соединениях, а не о полимерах. Любое разветвление цепи блокирует движение тройной связи за этот узел, и тройная связь бежит только в одну строну.
Речь идёт, естественно, об индивидуальных соединениях, а не о полимерах. Любое разветвление цепи блокирует движение тройной связи за этот узел, и тройная связь бежит только в одну строну.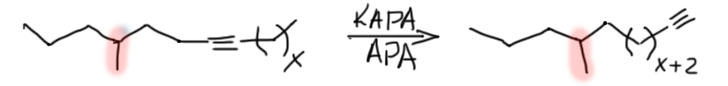
соотвественно, если такие разветвления есть с двух сторон, тройная связь не выберется, но будет метаться в пределах выделенного ей кусочка прямой цепи, что приведёт к потере индивидуальности и образованию смеси возможных алкинов, включая исходный, с небольшими примесями алленов. Такая реакция никому не нужна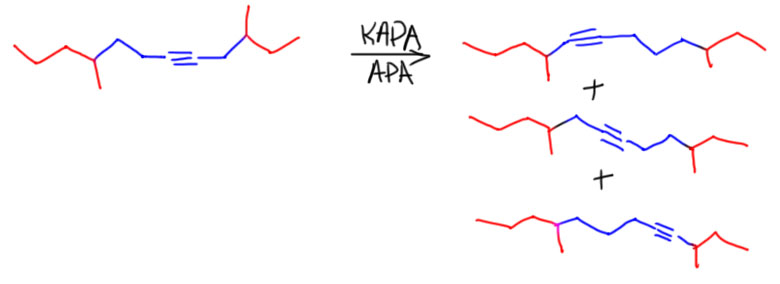
Кроме разветвлений алкильными группами с одной стороны цепи может быть фенил. Тройная связь все равно бежит наружу и не прилипает к фенилу, хотя в этом случае она была бы сильно стабилизирована сопряжением и, из общих соображений, не должна была бы оттуду уходить. И так тройную связь можно вытащить из-под этого сопряжения, что довольно удобно, потому что много таких алкинов легко сделать из легкодоступного фенилацетилена: 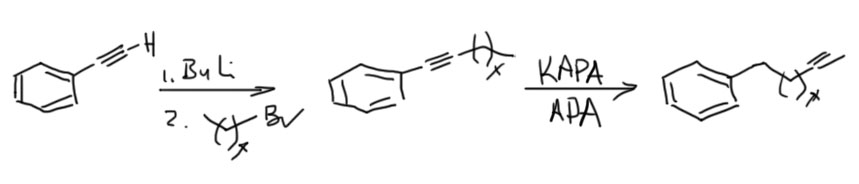
Самым популярным применением ацетиленовой молнии было и остаётся смещение тройной связи в спиртах. Объяснение просто – исходные спирты исключительно легко получаются по реакции ацетиленидов с карбонильными соединениями или эпоксидами. После этой реакции мы легко смещаем тройную связь на конец цепи и продолжаем что-то делать и с ней, и со спиртовой группой. Множество интересных стуктур было получено с использованием этого приёма. В органическом синтезе этот прием – настоящий хит, использованный в огромном количестве реальных синтезов и продолжающий до нашего времени пользоваться популярностью, настолько он отлажен и надежен, а ничего так не любят синтетики как надежных, хорошо проверенных инструментов. Это ведь только кажется, что каждый синтетик мечтает попробовать новые методы, ведь вон их сколько каждую неделю публикуется. Синтетик мечтает получить намеченную молекулу, и покороче. Конечно, одна из стадий, желательно ближе к концу должна быть поярче, посовременнее, чтобы статья получилась покруче. Но все остальные стадии, а в хорошем синтезе их может быть пара десятков или больше, должны быть попроще и понадёжнее, чтобы уже сотни других химиков это попробовали на примерах поближе. И ацетиленовая молния – реакция из золотого фонда надежнейших синтетеических приемов, отполированная до блеска тысячами предшественников. О том, почему тройная связь бежит от спиртовой группы, как чёрт от ладана, поговорим отдельно. 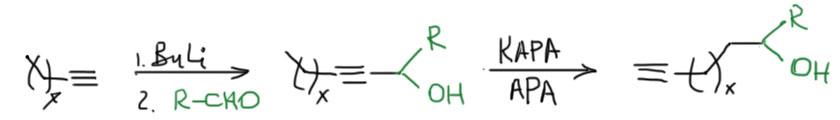
Кроме спиртовой группы еще возможно присутствие алкокси (метокси и т.п.) групп. Вести себя это будет приблизительно так же, как и спирт. Поскольку такие простые эфиры мы всё равно стали бы получать из спирта, большой надобности в такой разновидности метода нет. Собственно, это всё.
А теперь обсудим, чего не должно быть в исходном ацетилене.
- Галогенов. Ну, это понятно. В присутствии сильнейшего основания получите элиминирование. Даже фтор не очень надёжен в таких условиях, а всё остальное строго запрещено.
- Любых мезомерных акцепторов: карбонила, карбоксила, их производных, нитро-группы, циан-группы, и т.п. Тоже понятно почему. Эти группы делают очень кислыми протоны на соседнем углероде, и именно эти протоны будут отщепляться сильным основанием раньше чем всё остальное. При этом обычный карбонил, особенно альдегидного типа еще можно спасти, превратив его в ацеталь (подробнее в теме про альдегиды), и тогда это работает, как обычный простой эфир.
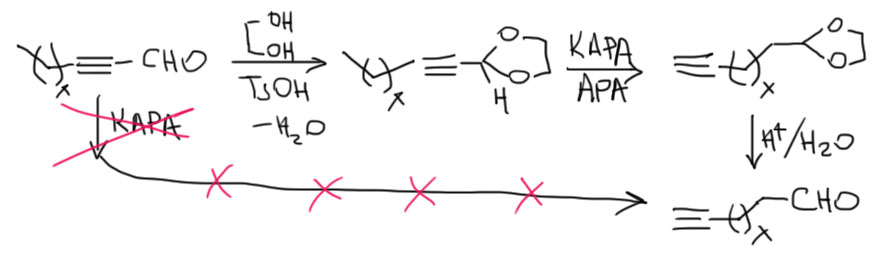
Бегство от гидроксила
Самое популярное применение ацетиленовой молнии – сочетание реакции Фаворского – присоединения ацетиленидов к карбонильным соединениям с последующим смещением тройной связи в конец цепи, что позволяет на этом конце продолжить построение сложной молекулы. Этот приём бы активно использован в бесчисленных синтезах сложных молекул. Метод отлично работает не только, если карбонильное соединение – кетон, когда понятно, что тройной связи деваться некуда: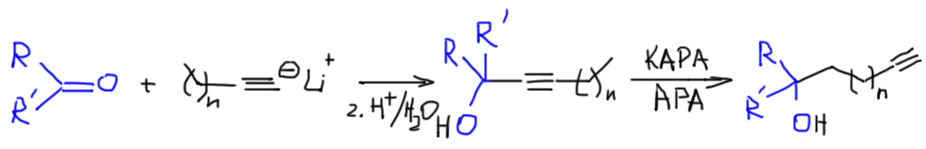
Если взять альдегид, из общих соображений можно было бы ожидать проблем – а что будет, если карбанион заедет под гидроксил, что могло бы привести к образованию соответствющего аллена с гидроксильной группой на двойной связи, а это, как мы скоро узнаем, енол, почти немедленно перегруппировывающийся в карбонильное соидинение, что в условиях большой основности неминуемо привело бы к катастрофе – образовался бы непредельный кетон, реагирующий вообще со всем, что попадётся под руку: 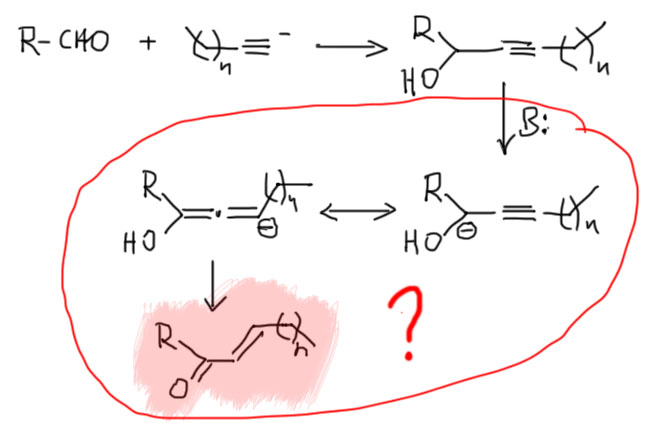
Но ничего подобного не происходит. Почему? Потому что в присутствии очень сильного основания гидроксильная группа количественно даёт сопряжённое основание, а возникновение карбаниона рядом с кислородом с отрицательным зарядом практически невозможно – такой карбанион будет сильно дестабилизирован отталкиванием электронных пар на соседних атомах – это называется α-эффектом, и мы с этим уже сталкивались в обсуждении нуклеофильности. Строго говоря, даже под нейтральной гидроксильной и алкоксильной группой карбанион сделать непросто, так как α-эффект работает и в этом случае, но там хотя бы он несколько уравновешивается интуктивным акцепторным эффектом, и такие карбанионы, хотя и очень нестабильны, но принципиально возможны, что и проявляется в одной очень интересной реакции, о которой в другом месте. Но под отрицательным кислородом это совсем плохо, так как в этом случае даже индуктивного акцепторного эффекта нет, и дестабилизирующий α-эффект нечем компенсировать. 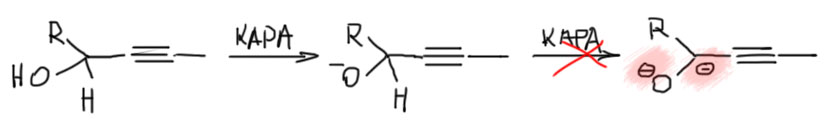
И еще одна деталь, на которую можно было бы не обращать внимания, но трудно этого не заметить – в эксперименте почему-то вместо KAPA использован ее (или его?) натриевый аналог, который можно назвать NAPA – это просто так или со смыслом? Это просто потому что Абрамс страшно боялась гидрида калия, который нужен для настоящей капы, и положила жизнь на то, чтобы заменить его чем-нибудь побезопаснее, например, чем-то с натрием или литем. Это несложно понять, ведь калий действительно и сам гораздо активнее, и его производные типа гидрида или амида требуют намного более тщательной работы, иначе беда. Это обычный эффект противоиона – катион калия намного крупнее катионов нитрия и лития, поэтому кристаллические решетки калиевых солей слабее, они легче и быстрее разрушаются, на это тратится меньше энергии, поэтому твердый гидрид калия быстрее и с большим тепловым эффектом реагирует с водой или кислородом. Гидрид лития можно быстро взвешивать на воздухе, гидрид натрия уже нельзя взвешивать просто так, но под подушкой аргона или использовав продажный гидрид натрия, покрытый парафином, уже можно. А вот с гидридом калия работать без камеры (glovebox) с инертным газом почти совсем невозможно – воспламеняется на воздухе почти моментально, дальше дело может принять совсем плохой оборот. Существует фирменный реактив – гидрид калия в парафиновой оболочке, точно такой же как гидрид натрия. С таким гидридом калия вроде бы работать удобнее и его даже можно взвешивать на воздухе. Но – это все равно требует полной осторожности и недюжинной сноровки – эта суспензия на воздухе, особенно влажном, начинает разогреваться просто за счет диффузии килорода через оболочку; разогревание быстро эту оболочку разрушает и запросто может начаться горение, и тогда это уже не остановить, потому что оболочка из защиты становится дополнительным топливом. В общем – решитесь работать с гидридом калия, отнеситесь к этому как саперы относятся к разминированию.
Поэтому разумные люди боятся этого реагента как огня – ведь без огня и не обойдёшься. Или нужна работающая проверенная камера с атмосферой сухого азота, в ней всё делается медленно, работать надо в неуклюжих перчатках, все заносить в камеру через шлюз заранее, это очень дорогое удовольствие. Вот поэтому люди и пытались найти замену капе, и даже капу пытались делать без гидрида калия, а хотя бы с самим металлическим калием, что тоже не подарок, но с гидридом не сравинить. Но когда пара десятилетий этих усилий закончилась и пришло время подводить итоги, выяснилось, что в отдельных реакциях заменять можно, но в тех случаях когда важно время, максимальный выход, или нужно подвинуть тройную связь на очень большое расстояние, замены настоящей капе из гидрида калия нет. Это еще один удивительный пример того, что первоначальная версия какого-то важно реагента оказывается непревзойденной.
Все замены капы, а их очень много, в том числе и лапа, и напа (до рапы и цапы никто, к счастью, не додумался) вполне себе работают, но без того блеска, как настоящая капа. Реакция идёт довольно медленно, и если бы сам Браун начал с этих вариантов, он никогда бы не решился назвать реагент зиппером – это больше похоже на улитку. Но сути дела это не меняет – все эти реагенты смещают тройную связь в конец, просто медленнее, и выходы бывают меньше.
Примеры использования реакции
Реакция ацетиленовой молнии очень популярна в органическом синтезе, и ее можно найти довольно часто. Посмотрим несколько реальных примеров из оригинальных работ. Обратим внимание на несколько практических вещей. Во-первых, “честную капу” применяют редко, что очень хорошо говорит нам о том, что настоящие химики попусту рисковать не хотят, и это правильно. Применяют обычно суррогаты капы, описанные уже упомянутой Сузанной Абрамс, но в принципе это не что иное как разновидность знаменитых оснований Лохманна-Шлоссера – сочетания литий- или натрийорганических соединений или амидов с алкоголятами калия (чаще всего с трет-бутилатом просто потому что это самый легкодоступный алкоголят калия). Эти смеси часто работают так, как будто там прямо в смеси произошёл обмен и образовалось калий-органичексое соединение или амид калия, а катион поменьше переехал на более жесткий кислород. Зрительно это выглядит так, что первоначально полученная литевая соль, выпадающая в осадок в виде белой суспензии, растворяется при добавлении трет-бутилата калия, и даже внешний вид этой системы очень похож на “чкстную капу” – чистый слегка оранжевый раствор.

Такие смеси поэтому обладают гораздо более высокой оснвоностью, чем исходные производные лития или натрия, и при этом не используют никаких опасных штучек с калием. И они работают. При этом про молниеносность реакции можно забыть – времена реакций в таких случаях это часы, многие часы. Но системы работают и все довольны. Современные исследования таких систем показывают, что там происходят намного более сложные процессы, чем просто обмен, а их эффективность связана не с тем, что там образуется калиевой производное, а с весьма изощренным взаимодействием калийных и литиевых или натриевых комплексов, включающих несколько молекул лигандов. Здесь мы на этом останавливаться не будем.
Пример 1. В синтезе феромона зловредного жука суринамского рисолюба (S.Hötling, B.Haberlag, M.Tamm, J.Collatz, P.Mack, J.L.M.Steidle, M.Vences, S.Schulz Chem. Eur. J. 2014, 20, 3183 – 3191) авторам понадобилось сместить тройную связь не на конец цепи, а на предыдущее положение. Поскольку одной такой реакции нет, они вывернулись просто – сместили в конец, а затем сделали смещение на один атом внутрь, ровно так, как мы это и обсуждали – действием трет-бутилата в ДМСО. Обратим внимание на то, что и в этой статье не хотят брать настоящую “честную капу”, а вместо этого месят напу с трет-бутилатом калия – это очень популярный прием в химии активной металлоорганики – когда вам хочется посильнее, с калием, но рисковать жизнью не хочется, вы берете производные лития или натрия и добавляете трет-бутилат калия в надежде, что произойдёт обмен катионами. Этот приём называется основаниями Лохманна-Шлоссера и мы его обсудим как-нибудь отдельно, а здесь просто отметим, что смесь все же оказалась слабой, поэтому пришлось много частов даже нагревать, что очень непохоже на настоящую молнию. Но результат получен и глаза целы. Синим помечена связь, куда надо было сдвинуть тройную.
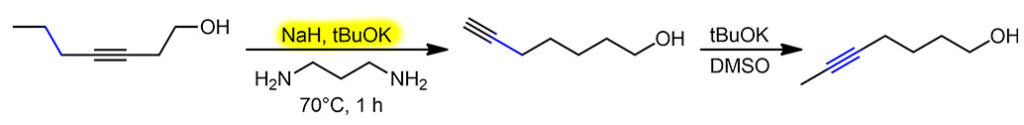
Пример 2. В синтезе циклических диенинов тоже потребовалось сначала сделать длинные алкинолы, что и было достигнуто с помощью системы из литиевого производного диаминопропана (LAPA) и трет-бутилата калия. (H. Hopf, A. Krüger Chem. Eur. J. 2001, 7, 4378). Реакции заняли 2 часа при комнатной температуре.
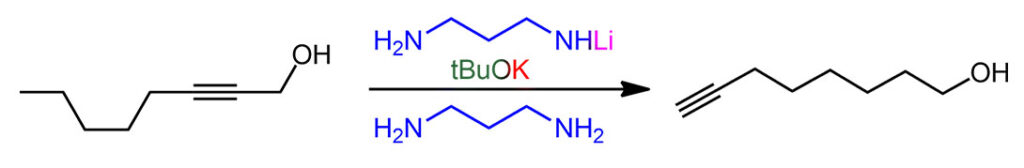
Пример 3. Индийским исследователям макроциклических активных веществ из морского гриба потребовалось для полного синтеза одного из таких соединений сдвинуть тройную связь, не испортив оптической чистоты вторичного спирта, что в полной мере и удалось, так как мы уже выяснили, что карбанион никогда не залетает пол спиртовую группу, а следовательно рацемизации можно не опасаться (S. Pabbaraja et al. Tetrahedron Lett. 2018, 59, 2570–2576).
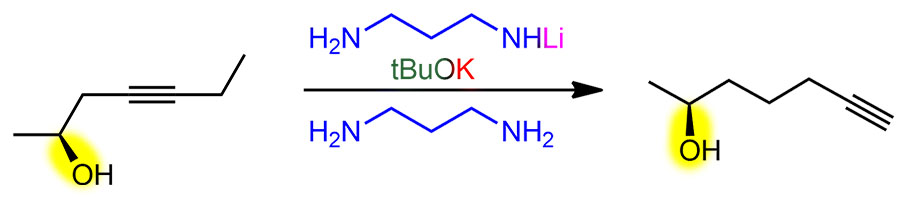
Если ацетилены и аллены такие нестабильные, почему они не перегруппировываются в сопряженные диены? Или всё-таки перегруппировываются?
Возьмём бутин, любой. Изомерами бутина являются бутадиены, алленового типа 1,2-бутадиен, и сопряжённый 1,3-бутадиен. И алкин и аллен, как мы уже знаем, довольно нестабильны, причём аллен немного хуже алкина, особенно внутреннего. А вот сопряжённый диен – очень неплохая штука, за счёт сопряжения он намного стабильнее алкина, минимум на 10 ккал/моль. Вот как выглядят относительные энтальпии образования этих изомерных углеводородов на одной диаграмме: чем выше полочка, тем хуже. Мы уже обсуждали эту картинку.
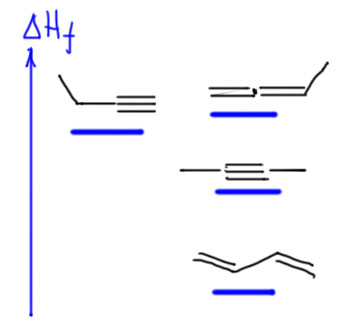 Это огромная разница, и в терминах константы равновесия (для этого нужна, конечно, не энергия или энтальпия образования, а свободная энергия, включающая ещё энтропию, но и этот вклад играет на стороне 1,3-диена, молекулы с большим количеством степеней свободы, чем аллен) эта разница соответствует практически количественному переходу и аллена, и алкина в сопряжённый диен. Так почему же это не происходит? Или происходит, а мы просто не знаем.
Это огромная разница, и в терминах константы равновесия (для этого нужна, конечно, не энергия или энтальпия образования, а свободная энергия, включающая ещё энтропию, но и этот вклад играет на стороне 1,3-диена, молекулы с большим количеством степеней свободы, чем аллен) эта разница соответствует практически количественному переходу и аллена, и алкина в сопряжённый диен. Так почему же это не происходит? Или происходит, а мы просто не знаем.
Попробуем разрисовать возможный механизм изомеризации алкина в диен. Поскольку мы уже разобрались в изомеризации алкин-аллен-алкин, опустим уже известные нам стадии, и покажем только путь в 1,3-диен. 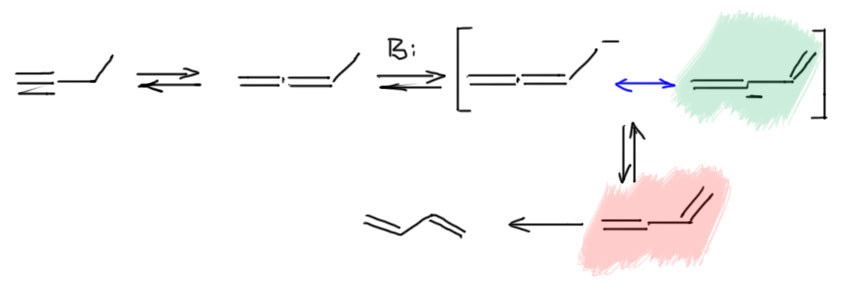
Что мы видим? Во-первых, то, что путь в 1,3-диен начинается из 1,2-диена, а не прямо из алкина: нам пришлось оторвать протон от метильной группы, возможность этого определяется тем, что это положение на первый взгляд, является аллильным, и карбанион будет сопряжён с двойной связью, и это должно быть выгодно. Тогда сначала мы раскладываем тройную связь на две двойных, и только после оттаскиваем одну двойную связь от другой. Из этого следует первый плохой для этого процесса вывод: путь в самый стабильный в этом множестве 1,3-диен ведёт через самый нестабильный в этом множестве 1,2-диен.Но аллен это ещё полбеды на пути в 1,3-диен, настоящая беда ждёт нас дальше. Если мы отрываем основанием протон от 1,2-диена, получаем резонансно делокализованный карбанион. Нарисовав его граничные структуры и хорошенько их рассмотрев, мы сначала увидим радостное – двойные связи расползлись точно так, как нам нужно, но едва забрезжившую на наших лицах улыбку быстро сменит гримаса боли и разочарования – граничная структура-то нехорошая! Ведь мы же знаем главное правило рисования граничных структур – ни в коем случае не трогать атомы, они должны оставаться на своих местах. И тогда вторая граничная структура будет иметь такую же структуру, как и первая – структуру аллена с линейным расположением связей, а это совсем не то, что мы ждём от конечного диена. Получается, что две двойные связи не сопряжены, вместо этого мы видим Из этого следует, что структура это плохая, вклад её в делокализацию будет мал, и вероятность протонирования по этому атому углерода будет очень мала.
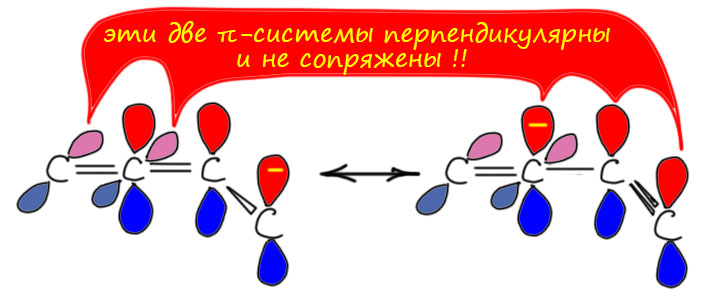
Добавим к этому еще и то, что если такое протонирование и произойдёт, то поскольку протон всегда переностся очень быстро, и диен получится прямой, как кочерга, очень далёкий от структуры настоящих конформеров бутадиена. И это тоже означает, что путь этот на всех стадиях должен иметь высокие энергии активации, а значит быть медленным и маловероятным. Прежде чем мы попадём в очень стабильный 1,3-диен, мы вынуждены переть по кочкам всё время верх и вверх. Химия не любит таких процессов. 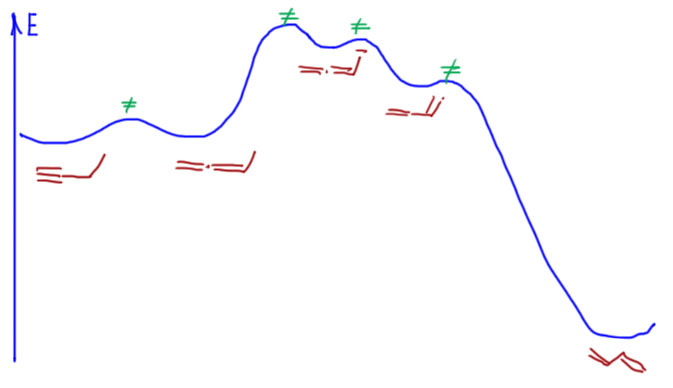
С такой ситуацией очень хорошо знакомы люди верующие: чтобы попасть в рай, нужно много поститься и ещё больше молиться, и вообще делать много не самых увлекательных дел, постепенно увеличивающих внутреннюю энергию (в тех терминах это называется благодатью) кандидата в праведники. Поэтому в рай попадают немногие, возможно даже вообще никто; проверить это, увы, невозможно – путь в рай строго неравновесен и ведёт, если вообще ведёт, только в одну сторону. Один печально знаменитый неудачник правда предлагал более простой, даже в чём-то массовый путь в рай, но верить этому источнику смысла нет, так как он обычно сам не знает, что получится из его прожектов.
Возвращаясь из рая в химию алкинов и алленов, мы увидели, как анализ механизма объясняет не только то, что происходит, но и то, почему что-то не происходит. Как правило не происходит: в большинстве примеров алкин-алленовой перегруппировки образования сопряженных диенов в значительных количествах не отмечено. Тем не менее, есть одно интересное исключение из этой тенденции, обнаруженное Францем Зондхаймером (F.Sondheimer и сотр., JACS, 1961, 83, 1682), основоположником исследования ароматических аннуленов, с которым мы еще встретимся в ароматической химии. Он обнаружил, что если в молекуле есть своеобразная “затравка” – еще одна двойная связь, то алкины легче дают сопряжённые диены в присутствии сильного основания типа трет-бутилата. Эта двойная связь сопрягаясь с новой двойной связью дополнительно стабилизирует все интермедиаты, от карбаниона до диена с неправильной структурой, и делает весь процесс выгоднее. 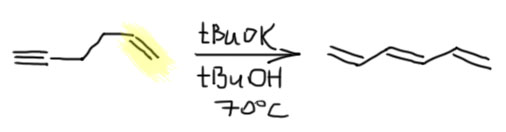
Зондхаймер на этом не остановился, и развил метод для получения сопряжённых полиенов с большим количеством двойных связей. Стоит сказать, что хотя некоторые такие полиены с множеством двойных связей хорошо известны в природе – это важнейщие оранжевые пигменты растений, каротин и его аналоги, каротиноиды (массовое проникновение шарлатанов привело к тому, что в литературе, особенно массовой, а про такие вещи обожают рассуждать всякие спецы по здоровому питанию и БАДам, у этих пигментов понемногу приживается название каротеноиды, от английского термина carotenoids, хотя в русском языке испокон веков существует название каротин, а от него именно каротиноиды). В бета-каротине, оранжевом пигменте морковки и осеннего леса, а на самом деле, вообще всех растений, сопряжённых двойных связей целых 11 и это вполне устойчивая молекула, но в ней кроме двойных сязей еще много насыщенных групп и циклов. А вот простая цепь из двойных связей и более ничего не очень устойчива – сказывается энергетическая избыточность двойных связей, только частично компенсируемая сопряжением, причём в длинном сопряжении выигрыш на каждую двойную связь гораздо меньше той стабилизации, которую мы видим в 1,3-бутадиене. В результате такие цепи долго получить не удавалось, и с задачей впервые справился именно Зондхаймер с его методом разбегания тройных связей на затравке двойных. Его максимальное достижение – простой декаен, углеводород с десятью двойными связями, оранжевый как морковка. И сделал он его из продукта окислительного сдваивания (по красной связи) двух ацетиленов поменьше, а затем разогнал четыре тройные связи на заранее припасённые четыре места, получив желаемое соединение. 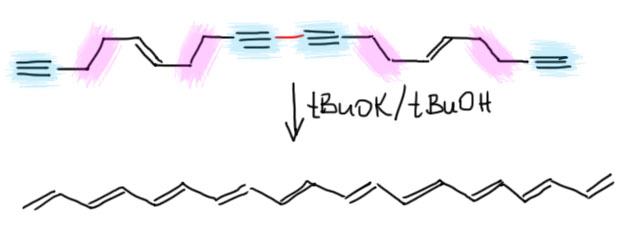
Более крупных полиенов получить всё же тогда не удалось, но к этой химии вернулись через несколько десятилетий, когда выяснилось, что полимер такого строения, который можно получить полимеризацией ацетилена, из-за чего он и называется полиацетилен, является органическим проводником, молекулярной проволочкой, по которой можно пустить бежать электрон. В 2000 году электрон добежал до Нобелевской премии, и за него получил эту премию японец Хидеки Сиракава. Но это уже не наша история.