Алканы
Первая и самая скучная тема курса. Это несправедливо, потому что химия алканов, на самом деле, весьма интересна и очень важна, прежде всего для промышленности. Но в лаборатории реакции алканов применяются очень редко. Еще раз повторю преостережение с первой страницы семестра – когда мы говорим про алканы, имеем в виду не только простые насыщенные углеводороды, а любые насыщенные алкильные фрагменты в органических молекулах. Из этого как раз и следует, что такие фрагменты – последнее, на что стоит обращать внимание при планировании синтезов. И даже не последнее, а после последнего. Наш курс органики в основном говорит именно о лабораторной органической химии, поэтому и химия алканов в нем представлена довольно убого и никого не увлекает. Тем не менее здесь есть несколько важных тем, которые необходимо обсудить и разобраться. Ниже программа курса по этому разделу. В нем очень много “воды” – общих вещей, которые можно обсуждать сколько угодно в любом разделе курса. Курсивом и цветом выделено то, что действительно важно и в чем стоит разобраться в первую очередь.
Подробная программа раздела
1. Общие сведения, физические свойства, гомология, изомерия.
2. Строение, природа С-С и С-Н связей, геометрия молекулы, межатомные расстояния
3. Вращение вокруг C-C связей. Понятие о конформациях на примере конформаций этана и бутана. Проекции в виде “кóзел”, проекции Ньюмена. Понятие о пространственных препятствиях. Конформации гош-, анти-, заслоненные. Энергетические диаграммы.
4.Физические свойства алканов (кратко). Связь физических свойств и строения на примере пентана и неопентана..
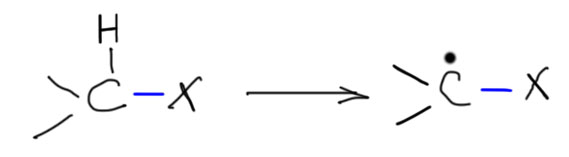
5. Гомо- и гетеролитический разрыв связи.
6. Радикальные реакции алканов. Хлорирование метана. История открытия. Энергетика процесса. Инициирование радикальной реакции (термическое, фотохимическое, с помощью инициаторов). Энергия видимого и УФ света – оценка энергии, необходимой для гомолитического расщепления связи в галогенах. Механизм цепной радикальной реакции: стадии реакции (зарождение, развитие, обрыв цепи.) Энергетика каждой стадии. Выбор оптимального интермедиата. Хлорирование этана.
7. Относительные скорости хлорирования С-Н связей различного типа (первичных, вторичных, третичных) с учетом статистического фактора . Хлорирование изобутана. Строение алкильных радикалов, их стабильность. Гиперконъюгация. Энергия первичных, вторичных, третичных С-Н связей. Избирательность (селективность) хлорирования и бромирования алканов – сравнение, причины различия. Избирательность реакции и температура. О возможности фторирования и иодирования алканов.
8. Другие радикальные реакции алканов. Сульфохлорирование. Нитрование по Коновалову. Чем отличается крекинг термический от каталитическиого? Окисление, горение. Наблюдение образования алкильных радикалов (Панет). Тетраэтилсвинец. Двигатель внутреннего сгорания и дизельный двигатель.
9. Распространение алканов в природе и их источники. Применение алканов.
10. Электрофильные реакции алканов – дейтерообмен, скелетная изомеризация нормальных алканов, образование карбокатионов. Работы Дж.Ола (Нобелевская премия 1994 г) – суперкислотные среды, катион метония. Трехцентровая двухэлектронная связь. Отличие между карбениевыми и карбониевыми ионами. Эта важная тема описана на отдельной странице.
11. Методы получения алканов: 1) реакция Вюрца (практически полностью утратила значение!), 2) использование купратов, 3) гидрирование С=С связи.
12. Методы синтеза алканов которые будут изучены позже: (из реактивов Гриньяра, реакция Кольбе, декарбоксилирование солей карбоновых кислот).
Рассмотрим наиболее важные вещи подробнее (жмите на плюс в разделе, который привлек внимание)
Свободные радикалы
Частицы, имеющие неспаренные электроны. Хотя мы гораздо чаще имеем дело с молекулами и ионами, имеющими только спаренные электроны, ничего особенного в свободных радикалах нет – их полно и вокруг, и внутри нас. Наличие неспаренного электрона никак не свидетельствет о малой устойчивости или высокой реакционной способности частицы. Так как неотъемлемым свойством электрона является спин, о свободных радикалах также часто говорят, как о частицах, имеющих спин.
Для характеристики частиц часто используют число, называемое мультиплетностью, которое очень легко вычисляется из суммарного спина, обозначаемого большой буквой S. Мультиплетность равна 2S+1. Поэтому частицы, имеющие только спаренные электроны, имеют суммарный спин равный 0, мультиплетность тогда будет равна 1, и частицу называют синглет (английский язык знают все, поэтому вопрос о том, как связана единица и слово синглет, можно считать неуместным). Частицы с одним неспаренным электроном будут иметь спин 1/2, и мультиплетность 2, поэтому их называют (довольно редко) дублетами. Все простые радикалы, например, атомы водорода, хлора, метильный радикал – дублеты. Если хотите немного повыпендриваться без особого риска для здоровья, можете ввернуть слово дублет в разговор о радикалах. Еще очень часто встречаются частицы с двумя неспаренными электронами и суммарным спином 1/2+1/2=1, и мультиплетностью 2*1+1 = 3 – это триплеты. Обычные молекулы кислорода, которые числами Авогадро летают вокруг и внутри нас – именно триплеты. Триплет – это синоним слова бирадикал, хотя и не совсем полный, потому что бывают и синглетные бирадикалы.
Простые (дублетные) радикалы имеют только один неспаренный электрон. Такие частицы всегда образуются парами при гомолитическом расщеплении простой связи. Нет ничего проще гомолитического расщепления простой связи – для этого нужно просто обеспечить поступление на такую связь достаточной энергии, которую так и называют – энергия связи. Таблицы энергий связи можно в изобилии найти и в сети, и в книжках, но нам эти цифры даром не нужны – с ними ничего путного сделать не получится. Нужно просто понимать, что разрывается всегда самая слабая связь, а найти ее очень просто в любой молекуле. Во-первых, на эту роль годятся только простые связи, а связи кратные (двойные и тройные) никогда легко не рвутся. Среди простых связей выбор тоже прост. Металлы пока оставим в стороне, а среди неметаллов все просто – чем правее и чем ниже в Таблице Менделеева находятся элементы, составляющие связь, тем она слабее. Самые слабые простые связи поэтому – галоген-галоген, галоген-кислород, галоген-азот, кислород-кислород, кислород-азот, и в меньшей степени азот-азот. Все остальное, с чем мы имеем дело, в том числе связи углерода с любыми неметалами, кроме самого правого и самого нижнего иода, а также связи водорода с любыми неметаллами (кроме того же иода) слишком прочны и просто так не разрываются.
Чем вызывается разрыв самой слабой связи? Самым банальным нагреванием, причем если связь действительно слабая, то нагревание требуется совсем небольшое, – именно то, какое мы и применяем в обычной лаборатории с помощью обычных нагревательных приборов, то есть уж точно не больше 250-300 градусов, а обычно и гораздо ниже. Связи кислород-кислород, например, вполне неплохо распадаются уже при температурах слегка выше комнатной (только это не касается самого простого из таких веществ – перекиси водорода, устойчивость которой выше, а распад очень сложен, и поэтому это доступное вещество никогда не применяют в качестве первичного источника свободных радикалов!). Еще легче распадаются связи галоген-галоген в молекулах галогенов, особенно фтора и иода.
Второй способ вызвать распад слабой связи – облучение светом. Этот способ очень непрост и не рекомендуется для учебных целей. Почему? Потому что мы не изучаем фотохимию (науку о взаимодействии молекул со светом) и не знаем ее законов. Эти законы очень сложны, и у нас на 3 курсе нет шансов в них разобраться даже поверхностно. Наиболее простой подход состоит в том, что окрашенные вещества поглощают видимый свет (собственно именно поэтому они и окрашены). Если в окрашенном веществе есть слабая связь, то можно ожидать ее расщепления видимым светом. Поэтому вполне годится облучение видимым светом для расщепления молекул галогенов. И все. Пожалуйста, не лезьте в ультрафиолет на 3 курсе. Изучите фотохимию когда-нибудь потом – тогда и полезете, если захотите. А сейчас не надо. Жертв и разрушений избежать не удастся.
Поэтому не пытайтесь фотохимически инициировать свободнорадикальные реакции бесцветных веществ ультрафиолетовым излучением. Это ошибка. Ошибка писать hν над стрелкой в реакции, где слева нет молекулярных галогенов, а есть, скажем, HBr или что-то еще, про цвет которого вам ничего не известно.
Инициаторы свободнорадикальных реакций
Перекись бензоила имеет слабую связь O-O и легко распадается уже при слабом нагревании, и дает фенильные радикалы за счет декарбоксилирования первоначально образующихся бензоилоксильных радикалов. Строго говоря, перекись бензоила разлагается уже при комнатной температуре, и реакции запускаются без нагревания, хотя и медленно. Это очень активно используют в быту и технике, и вы без труда найдете, а возможно уже и сталкивались с акриловыми клеями и шпаклевками, для схватывания которых нужно добавить немного вещества из прилагаемого маленького тюбика. Вот в тюбике как раз перекись бензоила, и она самопроизвольно инициирует свободнорадикальную полимеризацию (сшивку) основного компонента, образуя очень прочные склейки и заполнения. А если раствором перекиси бензоила пропитать бумажку, то после высыхания она полежит-полежит, а потом вспыхнет и сгорит (не пытайтесь повторить это дома!) – будет инициирована самопроизвольная реакция с кислородом воздуха.
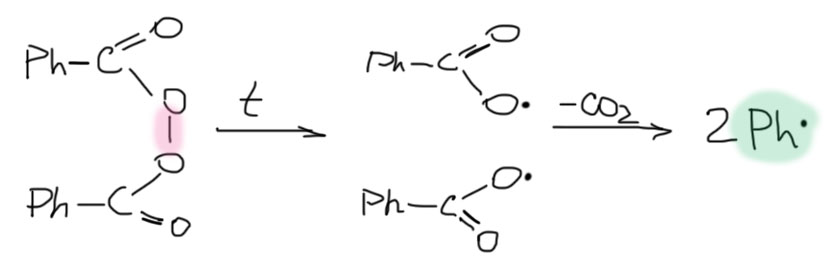
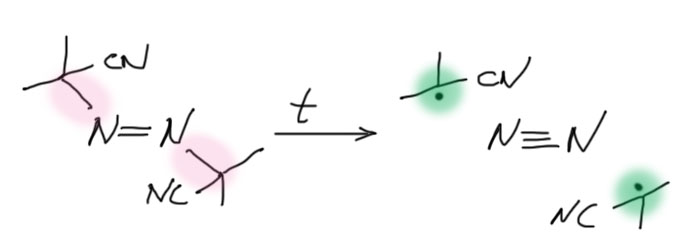
АИБН немного сложнее. Он распадается не потому что содержит заведомо слабые связи, а потому что при распаде получается чрезвычайно выгодная молекула азота, и выделяющаяся при этом энергия компенсирует затраты энергии на распад двух вполне прочных связей C-N. АИБН любят использовать как инициатор, потому что при комнатной температуре он не разлагается, и нужно обязательно вести реакцию при нагревании до 50-60 градусов минимум. Это удобно, потому что реакция не начнется преждевременно, при смешении реагентов, и не пойдет неконтролируемо. Мы сами ее запустим, когда захотим, включив нагревание.
Обратим внимание на то, что при распаде инициаторов образуются весьма реакционноспособные радикалы, гораздо более активные чем, скажем, радикалы брома (просто потому что радикалы обычно отнимают атомы водорода, и из инициирующих радикалов образуются связи C-H, гораздо более прочные чем связи бром-водород, а следовательно первая стадия продолжения цепи оказывается более экзотермичной и более быстрой, см. обсуждение энергетики ниже).
Свободнорадикальные реакции - основные стадии на примере хлорирования метана
Основной тип реакций алканов – свободнорадикальные. Любая такая реакция состоит из нескольких стадий, часть из которых может повторяться много раз, напоминая цикл в программировании (программирование сейчас преподают всем, начиная с 1 класса, поэтому вряд ли кто-то не слышал, что это такое).
Начало любой свободнорадикальной реакции – инициирование. Как уже сказано, это происходит или за счет одного из реагентов со слабой связью, или специально добавленного инициатора. Инициаторы часто добавляют и в том случае, когда реагент, способный образовывать свободные радикалы есть, но скорость реакции недостаточна, например, в свободнорадикальном бромировании. Дальше начинается собственно стадии, в которых образуются продукты реакции.
Свободные радикалы преимущественно участвуют в одной простой реакции – находят атомы водорода в тех молекулах, с которыми могут встретиться в реакционной смеси (умоляю – не называйте атомы водорода протонами) и отрывают их. На месте оторванного атома водорода образуется новый радикал.
Посмотрим на примере хлорирования метана. Здесь очевидный источник радикалов – слабая связь хлор-хлор. Посторонний инициатор не нужен, так как молекула хлора неплохо диссоциирует на радикалы и под действием видимого света (желательно с коротковолнового конца спектра, то есть синим, но годится и белый дневной свет – в нем есть все длины волн, – но не получится инициировать реакцию при свете красных фонарей), или нагревания. Атом хлора отщепляет водород от метана. Эта стадия определяет энергетику всего процесса – она обведена красным. На этой стадии определяются все особенности радикального хлорирования. Но продукт реакции еще не образовался. Это произойдет на следующей стадии – метильный радикал отщепляет атом хлора от слабой молекулы хлора. Образуется продукт (обведен синим), а новый атом хлора уходит на новый цикл. Такая последовательность называется свободнорадикальной цепью. Если бы в системе не было бы никаких других процессов, то цикл повторялся бы до полного исчерпания метана и для полного превращения хватило бы небольшого количества инициирующих радикалов (в абстрактном идеале – даже одного, но скорость реакции была бы очень мала, так как единовременно в работе был бы только один радикал, и для превращение количества вещества, сравнимого с числом Авогадро, потребовались бы времена, сравнимые с временем жизни Вселенной). Количество повторов цикла называется длиной цепи. В идеале длина цепи очень велика и ограничена только количеством исходного метана. В реальности это не так – радикалы довольно часто гибнут зря, встретившись либо друг с другом (это маловероятное событие, так как радикалов очень мало, и вероятность их случайной встречи ничтожно мала) или со всякими “чужими”, способными реагировать с радикалами (обрыв цепи). Это трудно предсказать и учесть, и в общем не так важно. Из того, что радикалы гибнут впустую, следует только одно – нужно постоянно производить новые, продолжая инициирование. 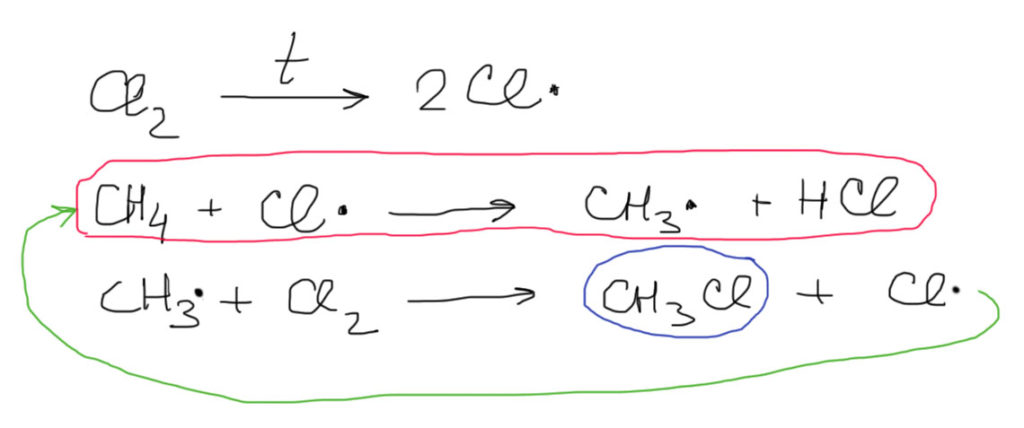
Энергетика цепной свободнорадикальной реакции
Почему свободнорадикальные реакции проще? Потому что нет заряженных частиц, которые сильно взаимодействуют со всем подряд в реакционной смеси (с растворителем, другими ионами и т.п.), и потому что для знания энергетики свободнорадикальных процессов нужно знать только энергии разрывающихся и образующихся на основной стадии связей, а это – легкодоступная информация. Нам даже не нужны цифры, а только знание тенденций в изменении прочностей связей, то есть общехимическая подготовка. В данном случае стоит помнить про то, как изменяются размеры атомов в периодической системе и как считать электроны в валентной оболочке. Напомню, что в первом приближении чем связь короче, тем она прочнее. Длина связи на глазок (с смысле качественного сравнения похожих молекул) оценивается по размерам атомов. На это накладываются еще два противоположно действующих фактора: а) связь еще прочнее, если больше разность электроотрицательностей атомов; б) связь сильно ослабевает, если на обоих атомах есть неподеленные пары (собственно, именно такие связи, как правило, и считаются слабыми) и чем они ближе друг к другу тем хуже. Поэтому, например, связь H-F (самые маленькие атомы плюс разница электроотрицательностей) одна из самых прочных, а связь F-F одна из самых слабых (связь короткая и неподеленные пары сильно отталкиваются).
Посмотрим еще раз на две последовательные стадии продолжения цепи на примере галогенирования метана. Разрывающиеся связи подсвечены красненьким, образующиеся голубеньким. 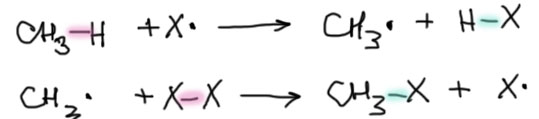 Баланс энергий по двум стадиям (образующиеся связи отдают энергию, разрывающиеся потребляют) определяет общий тепловой эффект цепной реакции. Во многих учебниках так и делают – суммируют все эти энергии с соответствующими знаками и говорят об экзотермичности или эндотермичности обсуждаемых реакций. Но, обращаю ваше внимание, это не очень плодотворный и во многом сбивающий с толку подход. Толку от этого суммарного эффекта не очень много. Причина этого очень проста. Эти две стадии последовательны, и нельзя добраться до второй, если не случилась (или случилась, но очень-очень медленно) первая. Довольно часто бывает так, что первая стадия слабо экзотермична или даже эндотермична, и очень медленна, а вторая как раз сильно экзотермична. Тогда радикалов на первой стадии образуется очень мало, и до второй стадии доходит очень мало частиц. А ведь скорость реакции определяется не только константой скорости (которая, повторю другими словами, в простых радикальных реакциях очень неплохо связана с тепловым эффектом), но и концентрацией реагирующих частиц, а она в этом случае может быть очень мала.
Баланс энергий по двум стадиям (образующиеся связи отдают энергию, разрывающиеся потребляют) определяет общий тепловой эффект цепной реакции. Во многих учебниках так и делают – суммируют все эти энергии с соответствующими знаками и говорят об экзотермичности или эндотермичности обсуждаемых реакций. Но, обращаю ваше внимание, это не очень плодотворный и во многом сбивающий с толку подход. Толку от этого суммарного эффекта не очень много. Причина этого очень проста. Эти две стадии последовательны, и нельзя добраться до второй, если не случилась (или случилась, но очень-очень медленно) первая. Довольно часто бывает так, что первая стадия слабо экзотермична или даже эндотермична, и очень медленна, а вторая как раз сильно экзотермична. Тогда радикалов на первой стадии образуется очень мало, и до второй стадии доходит очень мало частиц. А ведь скорость реакции определяется не только константой скорости (которая, повторю другими словами, в простых радикальных реакциях очень неплохо связана с тепловым эффектом), но и концентрацией реагирующих частиц, а она в этом случае может быть очень мала.
Посмотрим еще раз на эту схему. На первой стадии разрывается связь C-H, и образуется связь H-галоген. В зависимости от природы галогена эта последняя может быть как очень прочной (HF), так и очень слабой (HI), и между этими крайностями есть еще прочная связь HCl и более слабая HBr. Так как слева всегда одно и то же, получаем широкий спектр тепловых эффектов стадий от огромной экзотермичности для фтора, до безнадежной эндотермичности для иода. При этом вторая стадия всегда экзотермична, так как связь C-галоген всегда прочнее связи между галогенами. Единственная радость (или наоборот досада) от второй реакции – она может быть причиной самопроизвольного разогревания реакционной смеси даже в случае, если первоначально реакции идут медленно. Разогревание приводит к ускорению инициирования, образуется больше радикалов, скорость первой стадии возрастает, второй за ней тоже, тепла выделяется еще больше, разогревание быстрее, и происходит очень быстрый разгон, который может закончится взрывом. Ниже еще раз на это посмотрим на конкретных примерах.
Энергия самой важной стадии определяется очень просто – это разность энергий образующейся и разрывающейся связи, что непосредственно видно из сравнения трех реакций: гомолитического распада связи C-H, распада связи H-Cl (точнее, обратной к ней реакции образования молекулы HCl из соответствующих радикалов, и главной стадии цепной реакции. Атом водорода, маячащий и справа и слева, благополучно сокращается.
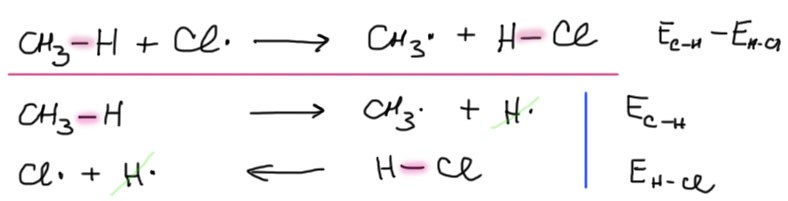
Галогенирование метана
В случае фтора H-F это одна из самых прочных связей, поэтому экзотермичность первой стадии велика. Вторая тоже весьма экзотермична, но это просто подливает масла в огонь, а там и так горячо. В результате имеем не только очень быструю, но и саморазогревающуюся систему. Фтор реагирует с метаном со взрывом в момент смешения при комнатной температуре в темноте. Кто инициирует эту реакцию? Тепло. Для слабой молекулы фтора комнатная температура уже вполне годится. Около 1% молекул фтора диссоциируют на радикалы в этих условиях, что, при большой скорости первой стадии и общей высокой экзотермичности обеих, дает быстрый саморазгоняющийся процесс. При этом будут образовываться все четыре продукта фторирования в смеси. А можно ли провести эту реакцию без жертв и разрушений? Можно, нужно просто быстро отнимать энергию от образующихся частиц, не давая смеси быстро и сильно разогреваться. Например, пропуская смесь фтора и метана, разбавленную инертным газом, в тонкой медной трубке, заполненной кусочками меди. Медь не реагирует с фтором, но эффективно и быстро забирает лишнее тепло просто потому, что плотность атомов меди в металле по сравнению с плотностью молекул в газе огромна (прикиньте на досуге объем моля металлической меди в сравнении с сакраментальным числом 22,4 литра, обозначающим молярный объем любого газа, и сразу станет ясно, что на каждую молекулу газа в маленьком объеме трубки приходятся тысячи атомов меди, плюс еще высокая теплопроводность, из-за которой тепло еще и быстро рассредотачивается по всему объему металла). В результате в таком приборе реакция идет совершенно спокойно, трубка только слегка разогревается. Но все равно образуется не чистый фтористый метил, а смесь всех четырех продуктов фторирования.
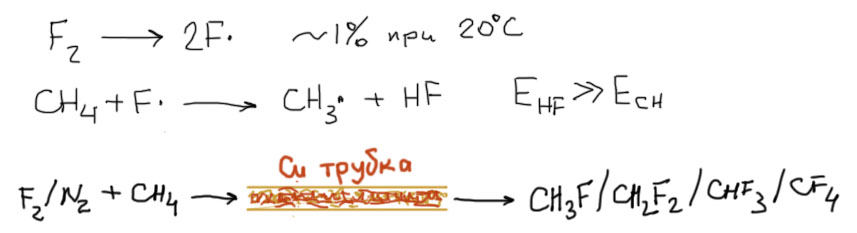
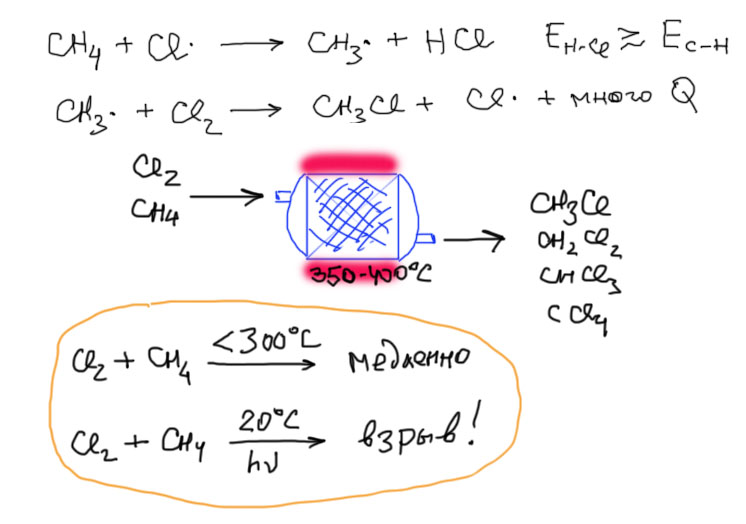
Чем отличается реакция хлора с метаном? Казалось бы ничем – мы же с раннего детства знаем, что смесь хлора и метана взрывается. Но – для этого обязательно нужно эффективное инициирование, как правило, в виде яркого солнечного света. В темноте, да и даже при рассеянном свете хлор и метан практически не реагируют. Первая стадия в этом случае слабо экзотермична – энергия связи H-Cl только немного больше энергии связи C-H. Поэтому первая и самая важная стадия довольно медленная. А значит и вся цепная реакция медленная. При этом вторая стадия сильно экзотермична. Но при комнатной температуре и без яркого света инициирование очень неэффективно, радикалов мало, реагируют они вяло, и до второй стадии мы просто не доходим. Иное дело, когда мы даем реакции хороший импульс в виде быстрого инициирования ярким светом. Много радикалов хлора начинают реагировать, сначала медленно, но тогда и до второй стадии кое-что доходит, выделяется тепло, температура повышается, начинается термическое инициирование, и т.д. – это похоже на сход лавины. Взрыв! Но если нам нужно не красиво бабахнуть, а реально и практически провести реакцию хлора с метаном (это вполне промышленный процесс), делают по-другому. Реакцию сразу ведут при высокой температуре, но эту температуру поддерживают эффективным теплообменом. Тогда и скорость термического инициирования велика, и скорость первой стадии тоже (не забываем, что при достаточном повышении температуры медленные реакции становятся быстрыми), а все лишнее тепло, выделяющееся на второй стадии забирает теплообменник. Никаких взрывов, все спокойно, на выходе ректификационная колонна делит продукты, так как и здесь образуется смесь.
Про бром, наверное, уже ничего говорить не надо – все ясно. Здесь все очень тоскливо. Первая стадия слабоэндотермична, а значит реакция идет чрезвычайно медленно. Вторая стадия здесь вообще не важна (она экзотермична, причем практически с тем же тепловым эффектом, что и соответствующая стадия при хлорировании), до нее просто ничего не доходит. В результате бром с метаном практически не реагирует, и яркий свет ничего не дает. Заметная реакция начинается при температурах выше 450 градусов, но органики не любят такого экстремизма просто потому что мало какое органическое вещество выживает при таких температурах). Поэтому можно просто заявить, что в более мягких условиях, то есть при комнатной температруре или даже сильном нагревании до 100-200 градусов бром не реагирует с метаном с заметной скоростью (значит реагирует с незаметной, но этого просто до сих пор никто не заметил).
Иод. Молекула иода очень слаба (почти так же как фтора), и при комнатной температуре в парах тоже можно найти около 1% атомов иода. При таком хорошем инициировании проблем быть не должно было бы: вспомним фтор, где дело начинается так же, а заканчивается уже через доли секунды. Но в этом случае первая стадия сильно эндотермична (вторая экзотермична и только немного уступает таким же стадиям в бромировании и хлорировании), а следовательно фактически не идет ни с какой измеримой скоростью. Не реагирует иод с метаном ни при каких условиях.
Стабильность алкильных радикалов
Отщепление атомов водорода от алканов разрывает связь C-H, что, казалось бы, должно всегда быть одинаково. Но мы знаем, что в органических молекулах значительное влияние оказывает то, что находится рядом. Как эффекты заместителей могут влиять на прочность C-H связи? Один из способов разобраться в этом – посмотреть на то, как заместители влияют на стабильность образующихся алкильных радикалов. Когда мы рассматривали эффекты заместителей, внимание было сосредоточено только на молекулах и ионах с четным числом электронов – катионах, анионах, атомах с неподеленной парой. Это не случайно – к частицам с неспаренными электронами классические электронные эффекты или вообще неприменимы, или требуют изменений в подходе. Индуктивный эффект, например, вообще не работает. Неспаренный электрон не является ни избытком, ни недостатком электронной плотности – это «свой» электрон, положенный элементу по уставу (напомню, что уставом в химии является Таблица Менделеева и представления об электронной структуре элементов, которые к ней были элегантно прикручены позднее). Смещать его от себя или к себе совершенно не нужно. Есть более хитрый каптодативный эффект, но мы с ним вряд ли встретимся. Мезомерный эффект для свободных радикалов вполне актуален, но работает только тогда, когда в радикале есть пи-электроны. Когда дойдем до алкенов, посмотрим на структуру аллильного радикала. В алкильных радикалах таких возможностей нет. Поэтому в них работает гораздо более слабый эффект, по недоразумению называемый гиперконъюгацией (сверхсопряжением). Да, здесь гипер неожиданно означает не то, что обычно имеют в виду под приставками типа гипер и супер, а то, что мы имеем дело с очень тонким (гипертонким) эффектом, который никогда и не заметили бы, если бы работали «нормальные» индуктивный или мезомерный эффекты. Еще раз – гиперконъюгация это очень слабый эффект, который имеет смысл рассматривать только тогда, когда других нет. В простых алкильных радикалах как раз нет обычных электронных эффектов, но нужно как-то объяснить экспериментальный факт довольно сильного различия прочностей C-H связей в исходных алканах, однозначно и прямо связанных с энергией образования радикалов.
В самом эффекте нет ничего сверхъестественного или непонятного, это просто следствие взаимодействия расположенных рядом орбиталей, практически ничем принципиально не отличающийся от обычного сопряжения. Обычное сопряжение возникает, когда на соседних атомах есть π-электроны, поэтому обычное сопряжение часто еще называют π-π-сопряжением. Такие электроны обитают на орбиталях, которые рисуют как восьмерки, у которых одна половинка беленькая, другая черненькая. Эта картинка наглядно показывает симметрию соответствующих орбиталей. В квантовой науке считается, что орбитали взаимодействуют, если они рядом, и если у них одинаковая симметрия и они не строго перпендикулярны. Еще очень важно, чтобы на взаимодействующих орбиталях не оказалось в сумме четырех электронов – тогда они будут взаимодействовать, но взаимодействие это будет дестабилизирующим, отталкивающим, и это плохо. Если электронов в сумме меньше (три, два), то взаимодействие будет стабилизирующим, и это хорошо. Если посмотреть на алкильный радикал, любой кроме метильного, то мы увидим орбиталь с неспаренным электроном. Эта орбиталь может быть либо обычной p-орбиталью (симметричной восьмеркой), либо гибридной орбиталью (восьмеркой с неравными петельками). Это зависит от реальной структуры радикала и от гибридизации углерода, несущего неспаренный электрон (для краткости часто говорят “спин”). Общего мнения на эту проблему нет, так как реально измерить структуру короткоживущего радикала невозможно, и данные по структуре получают из квантовохимических расчетов, а они не очень точны и не очень надежны. Но это, строго говоря, не важно. Симметрия гибридной орбитали ничем существенным не отличается от симметрии полной p-орбитали.
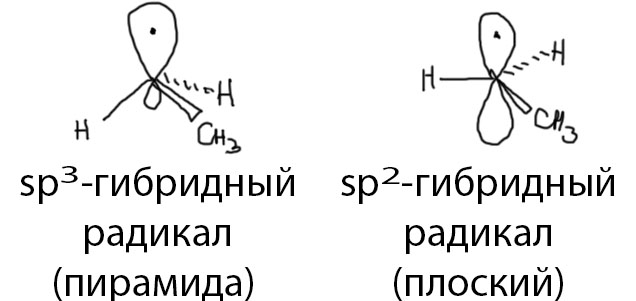
В этильном радикале (и любом другом алкильном) рядом (то есть на соседнем атому углерода) мы увидим C-H связи. Орбитали, обслуживающие простые связи (сигма-связи) – это гибридные орбитали. Размер петелек не важен – важна симметрия, а она вполне восьмерочная (чуть посерьезнее – это такая функция, которая равна нулю в плоскости, где у восьмерки перетяжка, и имеет разные знаки по разные стороны от этой плоскости) и собственно это все, что нужно. Еще раз повторим: гибридная орбиталь обслуживает сигма-связь C-H, но с точки зрения соседнего атома, несущего пи-орбиталь с неподеленным электроном, эта гибридная орбиталь смотрится вполне как пи-орбиталь. Это такие двойные стандарты: смотрим вдоль связи, видим сигма-орбиталь; смотрим сбоку, видим пи. Поэтому такие орбитали (орбиталь со спином, и гибридная орбиталь связи) вполне прилично перекрываются, то есть взаимодействуют. В сумме там будет три электрона (один плюс два). Стабилизация налицо, хотя и довольно слабая. Но другой нет, и такая сойдет. В более-менее современной литературе такие взаимодействия часто называют σ-π сопряжением, чтобы отличать от более обычного π-π сопряжения и отказаться от пафосного и сбивающего с толку термина “гиперконъюгация”, вызывающего просто приступы ярости у многих любителей строгой терминологии. Впрочем, как часто бывает, из огня попадаем в полымя, и новый термин ничем не лучше старого и точно так же сбивает с толку, ведь мы уже разобрались в том, что сопряжение возникает только потому, что обе взаимодействующие орбитали с точки зрения друг друга имеют вполне определенный π-характер, а тот факт, что одна из них на основном месте работы обслуживает сигма-связь, вообще говоря, не существенен. 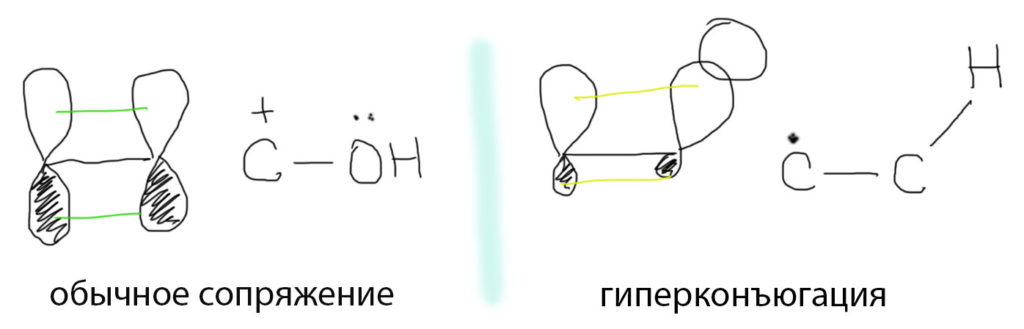 Еще один фокус состоит в том, что мы привыкли, чтобы взаимодействующие орбитали были параллельны. Это – оптимальный вариант, но не обязательный. Обязательно лишь то, чтобы они были не перпендикулярны. Угол между взаимодействующими орбиталями ослабляет эффект, но не уничтожает его. Но мы и так договорились, что эффект слабый. В реальном радикале, даже таком простом как этильный, будет рядом три C-H связи, и эта тренога еще и вращается вполне свободно вокруг C-C связи. Поэтому мы и будем иметь три взаимодействующие гибридные орбитали под разными углами. Углы эти непрерывно меняются, но только один из них иногда может становиться прямым, и в сумме эффект от трех C-H связей будет всегда стабилизирующим. Для порядка неплохо все эти взаимодействия помножить на косинус угла проекции на орбиталь спина, да и проинтегрировать по полному кругу с учетом симметрии. Для красного словца, конечно. Реальная органическая химия никогда не имеет дело с математикой, выходящей за пределы четырех действий арифметики.
Еще один фокус состоит в том, что мы привыкли, чтобы взаимодействующие орбитали были параллельны. Это – оптимальный вариант, но не обязательный. Обязательно лишь то, чтобы они были не перпендикулярны. Угол между взаимодействующими орбиталями ослабляет эффект, но не уничтожает его. Но мы и так договорились, что эффект слабый. В реальном радикале, даже таком простом как этильный, будет рядом три C-H связи, и эта тренога еще и вращается вполне свободно вокруг C-C связи. Поэтому мы и будем иметь три взаимодействующие гибридные орбитали под разными углами. Углы эти непрерывно меняются, но только один из них иногда может становиться прямым, и в сумме эффект от трех C-H связей будет всегда стабилизирующим. Для порядка неплохо все эти взаимодействия помножить на косинус угла проекции на орбиталь спина, да и проинтегрировать по полному кругу с учетом симметрии. Для красного словца, конечно. Реальная органическая химия никогда не имеет дело с математикой, выходящей за пределы четырех действий арифметики.
Еще больше будет эффект в изо-пропильном радикале – там шесть подходящих связей, подставляющихся под взаимодействие с радикальным центром. В трет-бутильном – даже девять. Поэтому и считается, что первичные радикалы стабильнее метильного, вторичные стабильнее первичных, а третичные – вторичных. Соответственно меняется и прочность C-H связей в алканах: самая прочная в метане, слабее – первичные связи в этане и других алканах, еще слабее вторичные, еще слабее третичные. 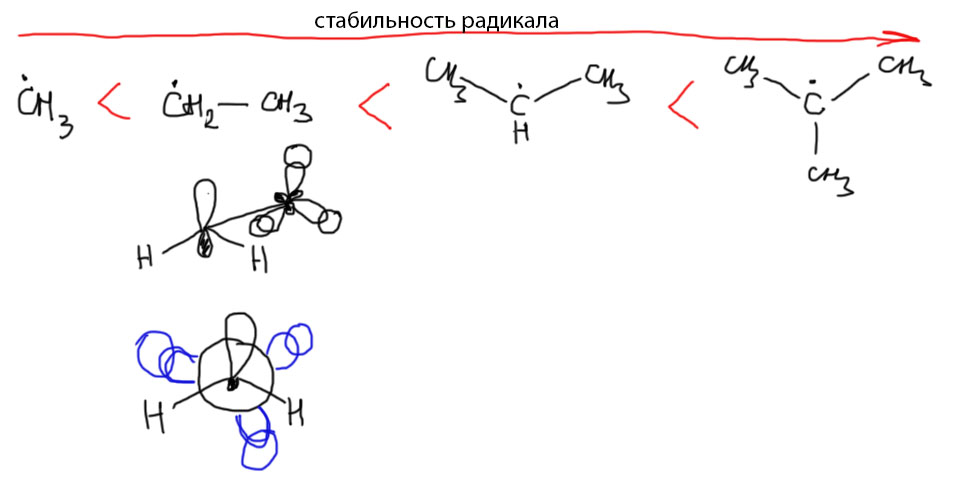
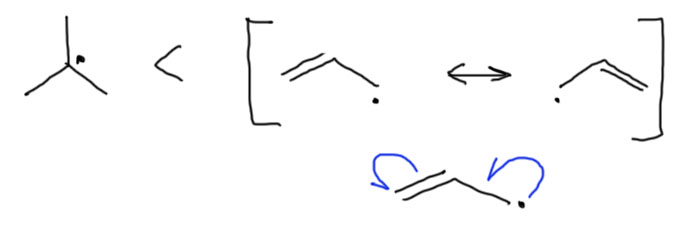
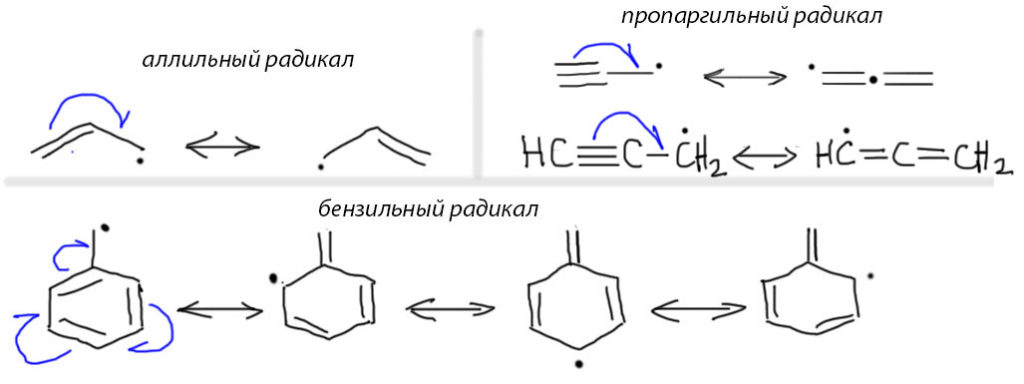
Повторю в 25-й раз – эффект невелик, и даже третичный трет-бутильный радикал, где в гиперконъюгации задействовано аж 9 связей, сильно уступает в стабильности аллильному радикалу, в котором есть нормальный мезомерный эффект, и в сопряжении задействована только одна двойная связь (заодно посмотрим, как корректно изображается мезомерия в аллильном радикале и граничными структурами, и стрелками, которые в этом случае рисуются с половинкой наконечника, чтобы показать, что делокализуется не пара, а один электрон или спин) . Тем не менее, этот эффект имеет огромное значение для свободнорадикального замещения, о чем следующий пост.
Галогенирование алканов. Селективность.
Если взять не метан, а хотя бы этан или даже еще что-нибудь посложнее, ситуация осложняется. Возьмем, например, 2-метилбутан, и посмотрим, что будет, если он встретится со свободным радикалом, способным отрывать атом водорода. В молекуле этого углеводорода много разных атомов водорода, и мы сходу напишем пять радикалов. Немного подумав, поймем, что два из них одинаковы по структуре, и разных радикалов четыре. По структуре эти радикалы различны – среди них есть первичные (обведены зелененьким), вторичные (синеньким) и третичные (красненьким). Из радикалов получатся конечные продукты – четыре разных.
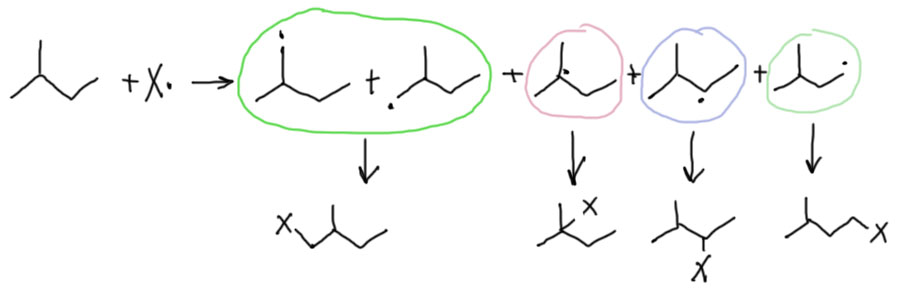
Можно ли предсказать, в каком соотношении получатся все эти продукты (и получатся ли они все, или только некоторые, или только один). Это очень важный вопрос в химии. Если продукт получается один, реакцию называют селективной. Если несколько – неселективной. Это крайний случай. Чаще бывает так, что продуктов получается несколько, но один из них преобладает. Тогда реакцию все равно называют селективной, а случай, когда продукт только один, чтобы не было обидно, переименовывают в специфичную. Потом начинают спорить, что значит “преобладает”. 90%? 80%? 70? А может достаточно уже 50% + хотя бы одна молекула? Из этой комической ситуации выходят просто. Если преобладание действительно очевидно, реакцию называют высокоселективной, А если совсем-совсем слабенькое, то низкоселективной. То есть все равно селективной, но низко.
Теперь зададим вопрос, а что такое совсем неселективная реакция. На первый взгляд, это очень просто. Если, скажем, в реакции могут получится два продукта, то, если их ровно поровну, то реакция совсем неселективная. А если хотя бы 51 на 49%, то уже низкоселективная. Посмотрим на схему вверху с четырьмя продуктами. Поровну – это по 25%? Нет, это не так. Дело в том, что каждый из нарисованных радикалов, а следовательно и продуктов, может образоваться несколькими способами. Первый продукт, например, образуется через отрыв водорода от одной из двух метильных групп, но на каждой по три одинаковых водорода, всего получается шесть. Радикал X с равной вероятностью отрывает один из шести водородов. Если помним азы теории вероятностей, это шесть независимых равновероятных событий, а значит вероятность оторвать любой из этих водородов в шесть раз выше вероятности оторвать какой-то конкретный из них. Для вторичного продукта одинаковых водородов два – и вероятность оторвать любой из них в два раза выше вероятности оторвать конкретный. Для третичного водород один и вероятность одна. Для второго первичного – три, и т.п. Следовательно, если реакция совсем неселективна, это означает, что отрыв водорода из любого конкретного положения равновероятен, а состав продуктов будет 6:2:1:3. Переведем в проценты – первого продукта будет 6/(6+2+1+3) = 50%. Второго первичного – 25%. Вторичного 2/12 = 1/6, т. е. около 17%, третичного всего 1/12 – около 8%. Любое отклонение вверх любой из этих долей (за счет других) будет означать, что реакция проявляет селективность относительно одного из продуктов.
Теперь назовем относительную вероятность каждой конкретной реакции так, как это принято в химии, а не в теории вероятностей. В химии имеют дело с константами скоростей. Они бывают абсолютные, то есть измеренные реально и независимо для каждой из реакций и каждого из продуктов, и это нам не нужно. А бывают относительные, то есть отнесенные к одной из скоростей. И такие константы измерять гораздо проще – они просто равны отношению реальных выходов продуктов с поправкой на количество равновероятных реакций, ведущих к одному и тому же продукту. Такие относительные константы часто еще называют парциальными факторами скоростей. Для совершенно неселективной реакции все парциальные факторы равны единицам. Для селективной – некоторые будут больше единицы, и чем больше, тем селективнее реакция.
Еще раз о том же, но с немного другого угла. Представим себе, что мы узнали, что в какой-то свободнорадикальной реакции 2-метилбутана найдено, что реакционная способность третичного положения в пять раз больше первичного, а вторичного – в два раза больше первичного. Как найти процентное отношение продуктов? Очень просто – это нам и дали парциальные факторы, просто в очередной раз немного другими словами – трет : втор : перв = 5:2:1. Данная реакция, делаем вывод сразу и ничего не считая, селективна по отношению к более замещенным третичным и вторичным центрам, и к третичным больше чем к вторичным. Теперь считаем ожидаемые выходы.
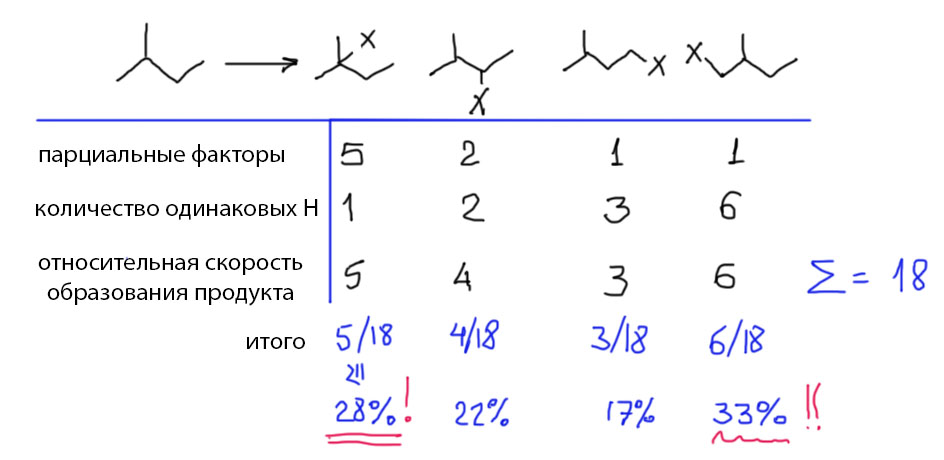
Вот так конфуз! Говорили, что реакция селективна по отношению к третичному продукту, и что он образуется в пять раз быстрее, а выход меньше первичного. Да, все правильно. Потому что действительно быстрее и действительно в пять раз, но не первичного продукта, а одного первичного положения, а таких положений одинаковых шесть, и продукт первичный образуется суммарно из каждого положения с равной вероятностью. Вывод один – к парциальным скоростям, селективностям, и всему с этим связанному нужно относиться очень аккуратно и не забывать о том, что любая реакция затрагивает конкретный реакционный центр (в данном случае это связь C-H), и если есть несколько совершенно одинаковых центров, то фактически мы имеем дело не с одной, а с соответствующим количеством одинаковых реакций, и нужно честно суммировать их результаты. Химия в этом смысле не что иное, как прикладная теория вероятностей.
Галогенирование алканов. Реакционная способность и селективность.
Разобравшись с парциальными скоростями в общем виде, вернемся к реакциям алканов, но уже с конкретными галогенами. Напомню, что ключевой стадией для определения энергетики и оценки скорости цепного галогенирования является первая стадия – отщепление атома водорода. У произвольного алкана водород можно отщепить от разных положений. Так как прочность разрываемых связей различна, то и энергетика, а следовательно и скорости таких реакций должны быть различны. Однако как это обычно и бывает в химии, мало кого интересуют абсолютные цифры эффектов, – с практической точки зрения более важны относительные величины, или даже только их грубая оценка “на глазок”. Разница между энергиями C-H связей в третичном, вторичном, первичном положениях довольно невелика – около 3-5 ккал/моль. Если мы на одной диаграмме приблизительно представим, как выглядят разные связи C-H в сравнении со связями H-галоген по энергии образования, то мы во-первых, наглядно увидим масштаб проблемы, и во-вторых, сможем понять принципиальную разницу между галогенами в цепном галогенировании.
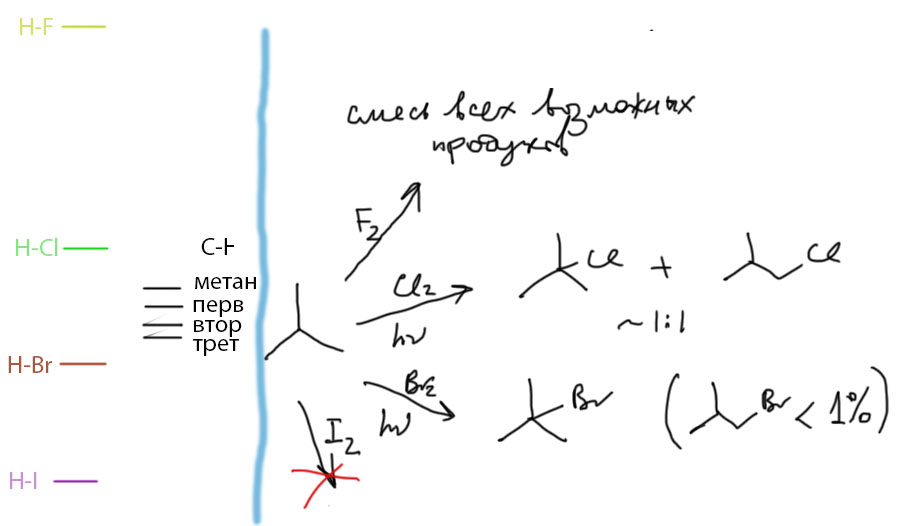
Если мы возьмем реакцию с радикалом фтора, то она очень сильно экзотермична уже для метана, поэтому для других связей экзотермичность будет только больше, но этот привесок в сравнении с общей величиной энергии очень мал. Иными словами фтору совершенно все равно, какой атом водорода отщеплять – на какой натолкнется, такой и съест. С “высоты” чрезвычайно прочной связи H-F разница между прочностями разных C-H связей просто не видна. Этот случай как раз и будет близок к полной неселективности с парциальными факторами близкими к единице для всех положений. В довершение несчастий фтор без проблем откусит водород и от любого из продуктов монофторирования, затем дифторирования и т.п. – в результате мы получим совершенно случайную смесь всех возможных продуктов замещения атомов водорода на фтор. Можете прикинуть, что уже для этана таких продуктов будет очень много. Соотношение продуктов можно немного менять, изменяя начальное соотношение углеводород:фтор и время контакта реагентов, но ничего существенного добиться не получится. Нужна ли кому-нибудь такая безумная смесь, вопрос открытый. Скорее всего, нет. Неселективные реакции редко находят применение.
Для хлора разница уже довольно невелика. Для метана первая стадия слабоэкзотермична, следовательно она тем более будет экзотермична для остальных связей C-H, и разницы будут довольно существенны. Хлор уже довольно неплохо разбирается в сортах связей, предложенных ему для разрыва. Парциальные факторы скоростей (или сравнительные реакционные способности) для первичных, вторичных и третичных связей для хлорирования приблизительно относятся как 1 : (3-3.5) : (4-5). Иными словами, реакция хлорирования проявляет вполне приличную селективность по отношению к третичным и в меньшей степени вторичным положениям. Но, как мы помним, с учетом количества одинаковых атомов водорода в реальных алканах, реальное соотношение продуктов будет более ровным без преобладания какого-то конкретного (как-то так почти всегда получается, что первичных атомов гораздо больше чем остальных). Поэтому с практической точки зрения селективность реакции хлорирования невелика. Хлорирование требует непрерывного инициирования либо ярким светом мощной лампы, либо сильным нагреванием (300 градусов или около того). Заметим, что реакции хлорирования можно проводить и в разных растворителях, которые довольно сильно влияют и на реакционную способность и на селективность. Хотя радикальные частицы не должны в теории сильно сольватироваться по причине своей электронейтральности и отсутствия значимых смещений электронной плотности, реальная химия сложнее простых ожиданий и в ней случаются всякие сюрпризы. Но с этим очень трудно качественно разобраться в рамках нашего простого курса. Просто не удивляйтесь, если где-нибудь всё же встретите реакцию свободнорадикального хлорирования с относительной реакционной способностью третичного положения гораздо выше 4-5, но тогда обратите внимание на то, в каком растворителе и как это делалось.
А вот бром ведет себя совсем по-другому. Если помните, метан для брома совсем не по зубам – первая стадия эндотермична. Но с учетом меньшей прочности первичных, вторичных и третичных C-H связей, они становятся для брома все более и более доступны. Энергетический зазор невелик, и на его фоне разница в энергиях разных типов связей становится очень существенной. Такое поведение в других науках часто называют усилением слабого сигнала. В результате бром очень хорошо различает сорта связей у углеводородах, и почти безошибочно выбирает третичные. Относительная реакционная способность третичных CH может доходить до 100 и более. Реакции с такими углеводородами идут довольно легко и требуют эффективного иницирования либо ярким светом, либо специальными инициаторами свободнорадикальных реакций типа АИБН. Если в углеводороде есть третичные положения, то образованием других продуктов бромирования можно пренебречь – их очень мало. Если нет – реакция идет очень плохо, требует жестких условий. На практике это просто не применяют. Вывод – реакция бромирования высокоселективна по отношению к третичным положениям что в практическом, что в теоретическом смысле. Этим качеством реакции бромирования очень хорошо пользовались для различения изомеров предельных углеводородов тогда, когда ещё не было никакой спектроскопии. Просто бромировали с облучением хорошей лампой, и если бромирование вообще не шло, делали вывод об отсутствии в молекуле третичных положений, причем с избытком брома можно было пробромировать все третичные положения А если шло, пытались понять, что за бромпроизводное получилось, сравнивая температуры плавления и кипения с известными. Так, например, различили два изомера гексана.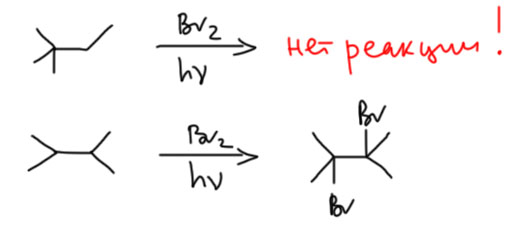
Про иод все понятно без слов. Иод ничего не иодирует, несмотря на очень легкое инициирование (атомов иода полно в парах иода уже при комнатной температуре). Но даже для третичного положения реакция сильно эндотермична. Более того, реакцию можно прочитать в обратном направлении – получится восстановление иодалканов иодистым водородом. Это очень хорошо известная реакция -с ее помощью в 19 веке часто доказывали строение органических соединений кипячением с иодистоводородной кислотой – при этом многие соединения просто восстанавливаются до насыщенного углеводорода, соответствующего углеродному скелету исходной молекулы. Для химии 20 века, не говоря уж о нашем времени это варварский и грубый метод, но не стоит забывать, что с его помощью на заре органической химии великие классики этой науки установили строение многих природных соединений задолго до изобретения всяких спектроскопий и прочих элегантных и точнейших инструментов. Механизм этого восстановления – просто ровно свободнорадикальное иодирование, только в обратную сторону, за исключением только инициирования, вместо гомолитического расщеления HI скорее происходит гомолиз молекул иода, которые всегда есть в иодистом водороде, слабой молекуле, легко распадающейся при повышенной температуре на элементы. Реакция идет в эту сторону по очень простой причине – из слабых связей C-I и H-I получается гораздо более прочная связь C-H. Баланс энергий целиком на стороне восстановления.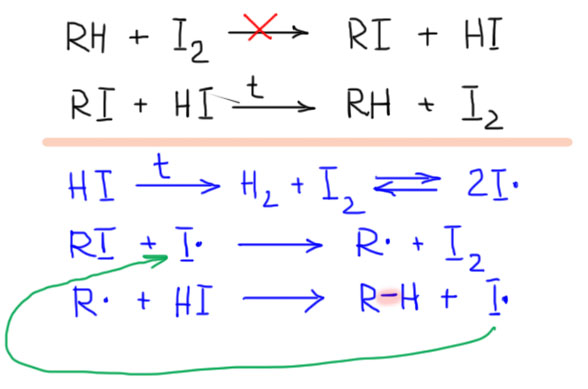
Поскольку сейчас не 19 век, а мы не величественные классики, я не рекомендую использовать эту реакцию для восстановления. Сейчас мы намного больше знаем про реакции, и знаем, что иодистоводородная кислота весьма кисла, и в ее присутствии нередко идут всякие скелетные перегруппировки и прочие радости, с которыми мы еще познакомимся, настолько, что реально предсказать, что получится в процессе такого восстановления, почти нереально. “А как же классики” – спросит удивленный читатель. На то они и классики, чтобы ломиться в вечность. К тому же история так устроена, что сохраняет только поучительные примеры успеха, а куда более многочисленные позорные провалы стыдливо присыпает пеплом забвения.
Итак, бром менее реакционноспособен чем хлор, но более селективен. Хлор менее реакционноспособен чем фтор, но более селективен. Эти простые наблюдения иногда формулируют как общую закономерность.
Более реакционноспособный реагент менее селективен и наоборот
Выглядит очень убедительно, и даже, можно сказать, немного очевидно. Но не стоит обольщаться – это просто обобщение наблюдений над разными реакциями. Это не Закон Природы. Никакого теоретического обоснования у этого красивого лозунга нет (хотя бы потому что нет никакой общепринятой теории о том, что такое реакционная способность). Исключений из этой закономерности полно. Хорошо работает она, как правило, только для очень похожих реакций. Например, для свободнорадикального галогенирования.
Стабильность радикалов с заместителями.
Если с алкильными радикалами все более-менее понятно, хотя эффект гиперконъюгации по какой-то мистической причине всегда вызывает много вопросов, то с радикалами, имеющими другие заместители, дело не так просто. Очевидный вопрос, который возникает в этой связи – действуют ли в рядах замещенных радикалов известные нам электронные эффекты, индуктивный и мезомерный.
Индуктивный эффект на стабильность радикалов в заметной степени не влияет, ни донорный, ни акцепторный. Оговорка “в заметной степени” в данном случае и во всех подобных – признак типичного научного страха не дай бог не сказать что-нибудь, что может быть опровергнуто. Преодолею-ка я этот комплекс и напишу определеннее – не влияют. Имеющиеся в литературе данные по стабильности радикалов, в которых можно видеть проявление индуктивного эффекта, очень слабы, а энергии стабилизации радикалов (а это, в свою очередь, тоже довольно мутная вещь) различаются несущественно. Обсуждать здесь нечего.
Довольно легко понять почему индуктивный эффект так слабо влияет на стабильность радикалов. Индуктивный эффект – это просто небольшое смещение электронной плотности к или от рассматриваемого центра. Неспаренный электрон на атоме принадлежит этому атому по праву, это свой электрон. Атом в этом случае имеет полный набор электронов, положенный ему по его положению в Периодической системе. Эффект разницы электроотрицательностей при этом уже полностью отыгран в той молекуле, из которой получили рассматриваемый радикал, и дополнительное смещение электронной плотности никому не нужно. Поясню, что я имею в виду. Представим себе молекулу и получающийся из нее радикал, и примем, что X – чисто индуктивный донор или акцептор. Связь C-X в исходной молекуле уже полярна, плотность на ней смещена в сторону более электроотрицательного атома. При образовании радикала ничего не изменяется, никакой дополнительной поляризации ожидать нельзя, потому то для этого нет причин, электроотрицательности не изменились. Это в корне отличается от ситуаций, когда образуется не радикал, а анион или катион, имеющие избыток или дефицит плотности, а следовательно и изменившуюся электроотрицательность. Поэтому индуктивный эффект отлично работает для ионов, но совершенно бесполезен в радикалах.
Иное дело мезомерия, эффект сопряжения. Ситуация, когда рядом находятся атомы с π-орбиталями (атомными p-орбиталями, при этом свободный электрон обычно рассматривают как занимающий однократно занятую p-орбиталь). Тогда они взаимодействуют обязательно! Могут быть разные варианты. Посмотрим на них.
1. Самый простой вариант – рядом с радикальным центром кратная углерод-углеродная связь или ароматическое кольцо. Это очень хорошо. Ситуация легко описывается резонансными структурами. Обратите внимание, что в данном случае кратная связь фактически является донором по отношению к радикальному центру, так как на этом самом центре из 7 элетронов становится восемь, а на крайнем углероде кратной связи, наоборот, из восьми семь. Такое смещение одного электрона принято изображать еще и специальной кривой стрелкой с половинкой наконечника – направление стрелки показывает фактическое смещение неспаренного электрона с кратной связи или кольца на исходный центр. Так устроены аллильный, пропаргильный и бензильный радикалы и их многочисленные производные. Эти радикалы поэтому хорошо стабилизированы и очень легко и часто образуются в свободнорадикальных реакциях с участием олефинов, ацетиленов и ароматических соединений с боковыми цепями. Обратите внимание на изображение граничных структур для пропаргильного радикала. Вторая граничная структура имеет две двойные связи подряд (это называется алленильной структурой), и в упрощенной структурной формуле (по-английски такие структуры называют wireframe, то есть прóволочками), когда изображаются только связи и не изображаются атомы углерода, двойные связи подряд слились бы в одну длинную двойную линию. Чтобы избежать двусмысленности вместо углерода рисуют жирную точку. И надо умудриться не нерепутать эту точку и точку радикального центра. Посмотрите на обычные структурные формулы, изображенные под проволочными, и разберитесь, как правильно это делается.
На этом месте можно остановиться всем, кроме тех, кому из чистого любопытства интересно, что еще бывает с радикалами.
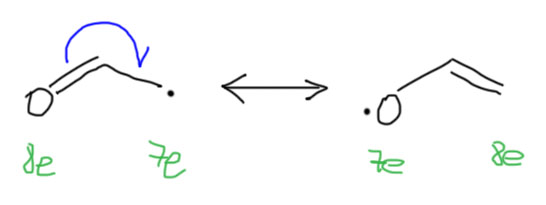
2. Рядом с радикальным центром кратная связь, но с гетероатомом или гетероатомами (азотом, кислородом, серой). Вроде бы этот случай мало отличается от первого, но – посмотрим для примера на делокализацию радикала на соседней карбонильной группе. Почти то же самое, что в аллильном радикале. И здесь нам очень важен вывод, который мы получили для аллильного радикала – двойная связь работает как донор. Но здесь отток плотности будет не от атома углерода, а от атома кислорода – это хорошо видно и по счету электронов: был кислород с октетом, а стал с семеркой. Обидно однако. Безусловно, радикал это не катион, а кислороду остаться с семью электронами не так скверно, как с шестью. Поэтому а) такие радикалы тоже стабилизированы по сравнению с метильным и первичными алкильными; б) но существенно меньше, чем аллильный радикал. Поэтому мы гораздо реже встречаемся с радикальными реакциями в химии кетонов, карбоновых кислот и их производных, нитрилов, нитро-соединений и тому подобных соединений. К счастью, у них есть другие способы реагировать, и мы этим еще займемся.
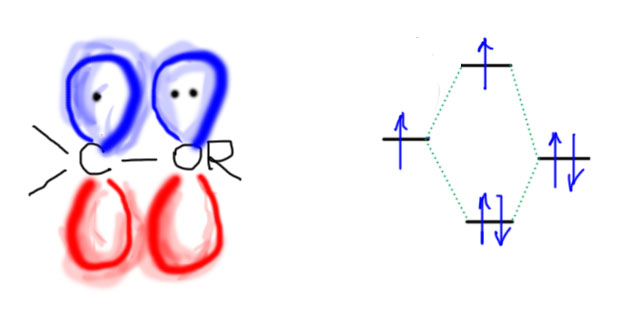
3. И самый мутный, но очень интересный кейс – рядом с радикалом гетероатом (азот, кислород, галоген) с неподеленной парой. Опыт говорит, что часто такие радикалы хорошо стабилизированы и легко образуются. Мы знаем как легко окисляются кислородом в свободнорадикальном автоокислительном процессе эфиры (образуется взрывчатая гидроперекись), амины, альдегиды (в кислоты через надкислоты). Это нужно объяснить, а это не так просто. При попытке нарисовать резонансную делокализацию так, как мы это привыкли делать, получается форма с нарушенным правилом октета, а этого нам не велит делать дух основоположника этого правила Джилберта Ньютона Льюиса, и у нас нет никаких оснований пренебрегать его заветами. Вот как могла бы получиться попытка нарисовать резонансную структуру для делокализации радикала соседним атомом с неподеленной парой. Вроде бы, на первый взгляд, все правильно – по одному электрону с двух сторон образуют новую связь, и на кислороде образуется радикальный центр. Но – мы видим странную вещь: формальный счет электронов дает нам невесть откуда взявшиеся лишние электроны и откровенное нарушение правила октета. Налицо тяжкое оскорбление чувств верующих в традиционные основы химии.
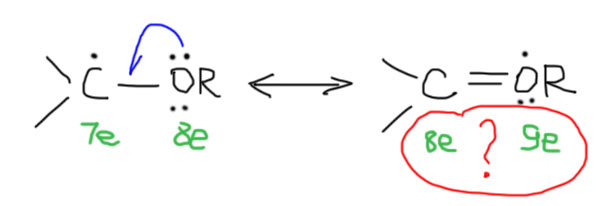
Что же делать? Похоже на провал. Но не будем отчаиваться. Из этого тупика есть несколько вполне симпатичных выходов. Один из них состоит в том, чтобы просто представить себе взаимодействие орбиталей. Там рядом две π-орбитали p-типа, на одной два электрона, на второй один. Эти орбитали одинаковы по форме (симметрии), а следовательно могут и даже должны перекрываться ровно так же, как они это делают в аналогичном катионе. Перекрывание орбиталей порождает связь (это не совсем точное утверждение, но пока сойдет). Если бы там был катион, то связь была бы самая обыкновенная, двухэлектронная (обычная химическая связь, ковалентная или донорно-акцепторная всегда обслуживается парой электронов). Но там радикал. Отлично – это значит, что такая связь обслуживается не парой, а тремя электронами. А так бывает? Конечно. Фокус в том, что трехэлектронные связи очень слабые, намного слабее двухэлектронных. Можно это показать и на диаграмме взаимодействия орбиталей. Из двух исходных получается две новых, одна выше (разрыхляющая), другая ниже (связывающая). Заполняем их тремя электронами и видим слабую связь (сильная связь это когда только два электрона, они занимают связывающую орбиталь, если бы было 4 электрона, то они занимали бы по паре и связывающую и разрыхляющую и давали бы суммарное отталкивание – отсутствие связи, так как такие диаграммы всегда устроены так, что дестабилизация разрыхляющего уровня всегда немного превосходит стабилизацию связывающего).
Хорошо, но громоздко. А резонансными структурами нельзя все же это изобразить? Можно, хотя это и не будет более наглядно. Фокус в том, что резонансной (или граничной) структурой является любой результат перемещения электронов в пределах цепи сопряжения с полным сохранением положений атомов. В данном случае мы не имеем права рисовать полноценную связь между атомами углерода и гетероатома, поэтому просто перемещаем электрон с одного на другой. Вот что получится:
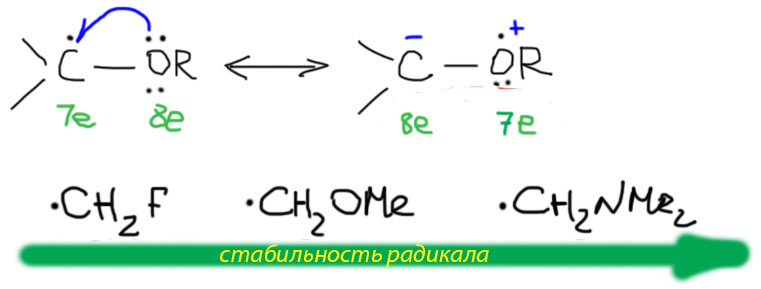
Во второй граничной структуре электрон перемещается с одного атома на другой в пределах цепи сопряжения (здесь она очень короткая и состоит из двух атомов). Насколько это выгодно? Ответ очевиден – это зависит от электроотрицательности атома-донора – чем она выше, тем менее выгодно. Галогенам, особенно фтору совсем неохота расставаться со своим электроном даже только в граничной структуре. Следовательно эта структура невыгодна, поэтому галогены, особенно фтор и хлор почти не стабилизируют радикал. Кислород в этом смысле более податлив, и стабилизация имеет место, что и выражается в относительной легкости образования таких радикалов в радикальных реакциях, например, автоокислении простых эфиров. Еще сильнее стабилизированы радикалы с донорным атомом азота, и это полностью соответствует общеизвестной легкости окисления аминов кислородом воздуха.
4. Капто-дативная стабилизация
Самая сильная стабилизация радикала возникает тогда, когда сочетается то, что описано в пункте 2 и пункте 3. На радикальном центре одновременно висит и что-то типа карбонильной группы, и гетероатом с неподеленной парой. Получается изумительная по красоте система, наглядно показывающая как тесно взаимосвязано все в химии. И если вы читаете это, облачившись в джинсы, то можете и практически оценить ценность капто-дативной стабилизации радикалов, потому что именно ей обязан своим существованием и уникальными свойствами знаменитейший синий краситель индиго, цвет которого носят все классические jeans.
Сначала о термине. Капто – от слова capture, захват, захватывать. Дативный – и так понятно – давать. Почему так, а не просто акцепторно-донорный? Чтобы получился новый термин, и придумавший его снискал кусочек славы. Тщеславие – главный двигатель науки, искусства и политики.
Дальше начинается знатнейшая путаница, но мы в ней разберемся. Проблема в том, что радикалы невозможно стабилизировать акцепторами. Мы это уже обсудили, но еще раз – атом с неспаренным электроном несет семь электронов. Отнимать у него значит двигаться в сторону шести, а это не может быть выгодно. А вот донорный заместитель подает плотность и от семи мы едем к полному октету – это выгодно. Поэтому все типы заместителей, которые мы видели до сих пор на радикалах хуже или лучше всегда работали как доноры, даже карбонильная группа и ее аналоги. И здесь первая засада – нормальному органику мысль о том, что карбонил может быть донором, кажется невыносимой ересью. Но здесь придется смириться – является. Но это – если она в структуре одна. А если мы создадим систему из трех кусков – гетероатома с неподеленной парой, углерода с неспаренным электроном и карбонила (или чего-то на него похожего, см. п.2) то получим вовсе не то, как это часто описывают – типа, радикал с донором и акцептором, один подает плотность, другой снимает. Мы получим полноценную цепь сопряжения, радикальный центр в которой – просто проводник сопряжения. Посмотрим на очень простую аналогию: донор – этиленовый фрагмент – акцептор. Это классическая цепь сопряжения, обеспечивающая прямое взаимодействие между донором и акцептором через двойную связь, работающую как удлинитель цепи сопряжения донор-акцептор. Удлинители, как мы знаем, могут быть и длиннее – это цепи сопряженных двойных или тройных связей или ароматические кольца.
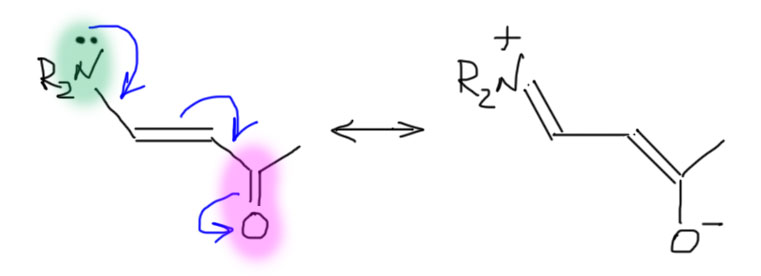
А может быть удлинитель короче одной двойной связи? Сразу хочется ответить, что нет, никогда и ни в коем случае. А почему? Что такое двойная связь, как удлинитель сопряжения? Просто два соседних атома углерода с p-орбиталями, на каждой из которых по одному электрону. Ну так давайте попробуем взять половинку от этого, то есть только один атом углерода с p-орбиталью, на которой один электрон, то есть радикальный центр. Нарисовать такое сопряжение граничными структурами или кривыми стрелками только немного сложнее, и нам поможет уже найденный в п.3 прием (только не воспринимайте это как такую ступенчатую делокализацию, сначала в одной части делокализуется точка, потом в другой – минус; вспомните еще раз, что резонанс это не процесс, а просто способ нарисовать картинку делокализации, и граничные структуры не существуют в реальности).
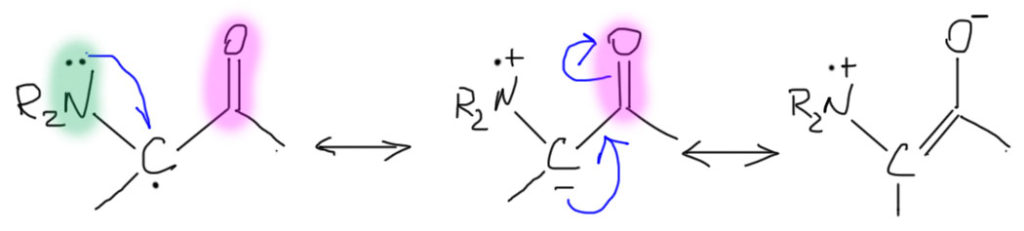
В результате получаем эффективное взаимодействие между донором и акцептором, переданное через один атом углерода. Как бы это ни казалось странно, но мы пытались изобразить стабилизацию радикала заместителями, а изобразили нечто совсем иное и весьма хорошо известное – образование единой цепи сопряжения донор-акцептор, и радикальный центр сыграл здесь чисто вспомогательную роль проводника сопряжения, находясь в ее середине. Впрочем, это не так важно, ведь сопряжение выгодно, а эффективное сопряжение донор-акцептор еще более выгодно. Следовательно и сами такие радикалы выгодны, хорошо стабилизированы, и этот тип стабилизации наиболее эффективен по сравнению с стабилизацией заместителями из пунктов 1-3. Но встречаются такие радикалы довольно редко.
5. Есть еще и пятый тип стабилизации радикалов – стерический. Но этот тип особый, он не связан с энергией образования или энергией стабилизации радикала, а определяется невозможностью для радикала вступить в реакции просто из-за того, что радикальный центр прикрыт объемистыми группами. Такой тип стабилизации (связанный со скоростью реакций, а не энергетикой самой частицы) называют кинетическим, и это особый разговор.
Стабильные радикалы. Реакции с участием стабильных радикалов. Автоокисление.
Но не все радикалы обладают этим свойством. Есть и вполне стабильные радикалы именно в том смысле, что встреча таких радикалов не обязательно приводит к рекомбинации. Такие радикалы могут накапливаться и присутствовать в значительных концентрациях. Один из самых известных радикалов такого типа – обычный кислород. Кислород в основном состоянии (то есть тот кислород, которым мы дышим и т.п.) – это стабильный радикал, точнее бирадикал, но это как раз несущественно.
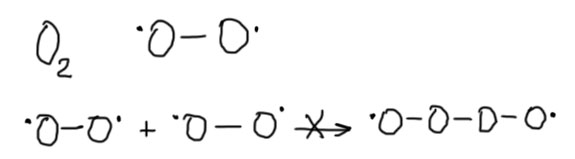
Молекулы кислорода не рекомбинируют. Это общеизвестный факт. Если вам сама постановка такого вопроса кажется бессмысленной, вспомните химию ближайшего аналога кислорода в Периодической системе – серы. Для серы такой процесс хорошо известен и происходит в парах этого элемента. Кислород может присутствовать в реакционных смесях в любых концентрациях. И ждать встречи с любым другим радикалом и рекомбинировать с ним с большим или меньшим удовольствием. При этом, так как концентрация кислорода может быть велика, вероятность такой рекомбинации с другим радикалом тоже велика. “Чужие” радикалы, встретив кислород, гибнут. Поэтому кислород обрывает цепные реакции – является ингибитором цепных реакций. Вывод – стабильные радикалы, неспособные к рекомбинации с себе подобными и поэтому могущие присутствовать в значительных концентрациях, являются ингибиторами цепных реакций. Кислород – пример таких молекул. Есть и другие, но о них поговорим как-нибудь в другой раз.
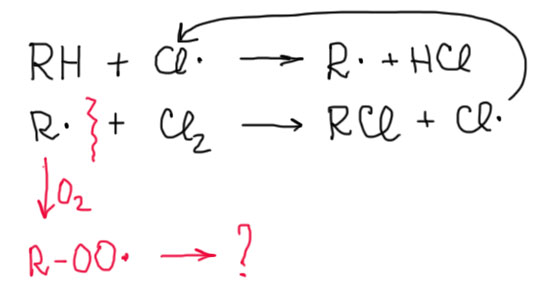
Вот идет, например, цепное хлорирование алкана. Стадии продолжения цепи много раз повторяются. Редко радикалы гибнут, встретив друг друга. Редко, потому что они живут недолго и концентрация их мала. Теория вероятностей ясно говорит нам, что встреча двух редких частиц маловероятна. Но стоит туда попасть кислороду, как вероятность встречи алкильного радикала с кислородом резко возрастает. Алкильные радикалы гибнут и не могут продолжить цепь.
Возникает новая проблема – во что превращаются продукты рекомбинации кислорода с алкильным радикалом. Оставим пока в покое хлорирование и посмотрим, что будет если у нас есть только алкан и кислород. Реагируют ли они, Все знают, что да – а что, собственно, происходит в пламени горелки или двигателе автомобиля? Но чтобы такая реакция началась, нужна искра, пламя спички или зажигалки. А без этого? Без этого, алкан в присутствии кислорода (или воздуха) вступает в медленную реакцию. Инициирование для этой реакции не нужно – кислород уже радикал. Как и положено радикалу, и этот отщепляет атомы водорода. Энергетика этой стадии определяется прочностью связи O-H в перекиси водорода, существенно более слабой чем очень прочная связь O-H в воде. Поэтому эта реакция довольно селективна – кислород предпочитает отщеплять атомы водорода от менее прочных C-H связей – что-то среднее между хлорированием и бромированием. Обратите внимание, что поскольку кислород бирадикал, одним отщеплением дело не ограничивается, и сразу же образуется гидроперекись в результате рекомбинации радикалов. Если ничего больше не предпринимать произойдет одно – в алкане медленно накапливаются гидроперекиси. Этот процесс называется автоокислением. А если нагреть – поднести спичку и т.п., то реакция образования гидроперекиси, как и положено, пойдет быстрее; уже в ней слабая связь O-O разорвется, образуются два очень шустрых радикала, каждый из них поведет свою, уже сильно экзотермичную и совершенно неселективную (водороды будут отщепляться из любых положений, а не только самых слабых) цепь, что, как мы уже не раз видели развивается лавинообразно, в лучшем случае в виде пламени, в худшем – взрыва. 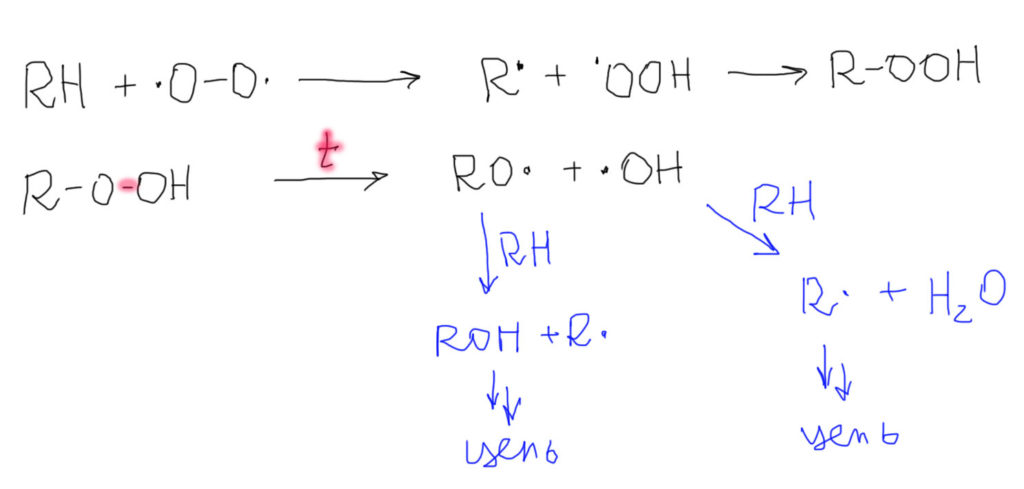
Свободнорадикальная, но не цепная реакция. Реакция Коновалова.
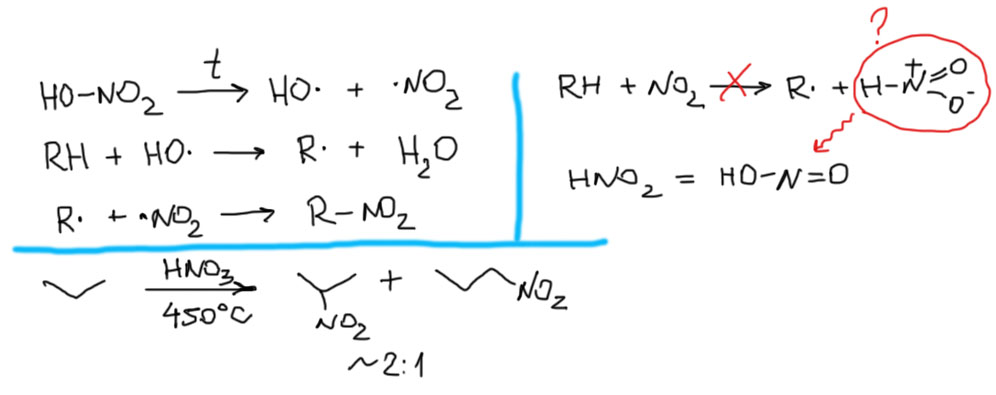
Посмотрим еще на одну реакцию – парофазное нитрование алканов, реакцию Коновалова. Смелый человек был профессор Коновалов – взял да хорошенько нагрел углеводороды с азотной кислотой! Керосин с азотной кислотой – это хорошо известное ракетное топливо. Но важно то, что в топливе азотная кислота стопроцентная, а у Коновалова разбавленная, и довольно сильно. Но все равно это опасный эксперимент. В лаборатории этот метод не используют, но в промышленности он имеет некоторое применение, так как низшие нитроалканы – важные специальные растворители, обладающие очень редким свойством растворять не только органику, но и неорганические соли. В методике, предложенной Коноваловым смесь углеводорода и разбавленной азотной кислоты запаивали в толстостенную стеклянную трубку и нагревали ее в печи до 100 с небольшим градусов. Выходы в такой реакции очень низкие, превращению подвергается только малая доля углеводорода. В принципе, это небольшая проблема, все реагенты дешевые, а после открытия трубки исходный алкан просто улетает и остается перегнать продукты. Но в промышленности это не годится, поэтому реакции ведут в проточном стальном реакторе при температурах около 500ºС, быстро охлаждая продукты. Тогда и выходы более практичные и опасности никакой. Попробуем разобраться в этой реакции, использовав все, что уже обсуждалось (спойлер: у нас ничего не получится, но мы хотя бы попробуем).
Вопрос первый – как она инициируется. В литературе есть две идеи. Одна такая – при нагревании азотной кислоты, она разлагается с образованием двуокиси азота – всем известного рыжего газа, который по природе является стабильным радикалом. Рекомбинация диоксида азота происходит, но только при низких температурах, а при нагревании этот радикал не рекомбинирует и накапливается в значительных концентрациях. Идея о том, что диоксид азота может отщеплять атом водорода, жестко наталкивается и на энергетику, и на то обстоятельство, что при этом должна была бы получиться малоустойчивая изомерная форма азотистой кислоты с пятивалентным азотом. Это не так страшно с химической точки зрения – протон быстро перемещается и такая частица быстро изомеризовалась бы в нормальную азотистую кислоту, но энергия связи N-H в такой неустойчивой молекуле явно маловата для разрыва связи C-H в алканах. Вторая гипотеза кажется более плодотворной. В молекуле азотной кислоты легко найти самую слабую связь так как мы это договорились делать – это связь O-N. Термически эта связь должна рваться более-менее легко с образованием двух радикалов – стабильного ⋅NO2 и очень шустрого HO⋅. Этот радикал не что иное, как продукт отрыва атома водорода от молекулы воды, а связь OH в молекуле воды – одна из самых прочных, прочнее связи H-Cl. Поэтому такой радикал будет рвать атом водорода от любых мест любых алканов с очень низкой селективностью (вспомним про фтор и хлор, а это должно быть где-то посредине, то есть по отношению вторичный к первичному не больше 2). Увы, здесь нас ждет неприятный сюрприз – все опубликованные данные по такому нитрованию показывают весьма неплохую селективность, например, при нитровании пропана даже при 450ºС первичный и вторичный продукты получаются практически в соотношении 1:2, то есть существенно лучше чем при хлорировании. Поэтому эта реакция до сих пор остается далекой от понимания того, как она идет на самом деле, и в первую очередь, что является истинным инициатором свободнорадикального процесса. Так как реакция Коновалова большого практического значения не имеет, эти загадки никто особенно решить и не пытается. Но делать вид, что их нет, неприлично.
Так или иначе, но как бы ни образовался алкильный радикал, он тут же будет рекомбинировать с ⋅NO2 с образованием нитроалкана. Реакция не имеет цепного характера точно так же как автоокисление. В этой реакции требуется постоянное инициирование, но, как мы уже выяснили, мы точно не знаем, как оно происходит.
Задачи и вопросы к коллоквиуму по теме алканы
Инициирование
Селективность
Свободнорадикальная, но не цепная реакция
Вопрос второй. Хорошо извествно, что реакцию Коновалова можно ускорить и повысить выход целевых нитроалканов, добавляя в реакционную смесь немного хлора или брома. Попробуйте написать механизм в этом случае. Какую селективность нитрования пропана можно ожидать в присутствии брома (опять воспользуйтесь результатами предыдущих задач).