Ароматичность, продолжение.
Ароматические ионы
Ни в чем ином ароматичность не проявляется так ярко, как в циклических заряженных частицах, как положительных так и отрицательных. Особые свойства таких частиц настолько очевидны и настолько же невероятны, что просто требуют какого-то яркого объяснения, несводимого к обычной делокализации, просто потому что эффект слишком велик и демонстративен. Это очень важно хорошо понимать.
Для подтверждения теории ароматичности существование и свойства ароматических катионов и анионов гораздо весомее и зримее, чем существование и свойства обычных нейтральных молекул типа бензола или пиридина. То, что углеводород состава C6H6 существует и стабилен – невелико открытие, почему бы и нет, и для того, чтобы показать, что с этим углеводородом что-то не то, мы должны делать много сложных количественных вещей – определять теплоту образования и натужно ее сравнивать с какими-то выдуманными молекулами-фантомами; определять реальную структуру молекулы и радоваться, что в ней все связи одинаковы; добавлять бром и не видеть мгновенного обесцвечивания, и т.п. А вот с катионами и анионами все гораздо проще. Обычно ионы (карбокатионы и карбанионы), и мы это уже отлично знаем – малоустойчивые частицы, образующиеся в особых условиях, часто в неизмеримо малых концентрациях, и не то, что потрогать и понюхать, а часто и спектр-то никакой с них получить не получается. И если вдруг мы видим какие-то яркие исключения из этого – вот прямо нечто кристаллическое, похожее на соль и солью таких катионов или анионов и являющееся, и не то, что все имеющиеся в распоряжении спектры, а и температуру плавления банальную можно часто измерить – значит точно работает какой-то неведомый, но невероятно мощный эффект, требующий нетривиального объяснения, новой теории.
Возьмем, к примеру, трехчленный циклопропениевый катион. Если ничего не знать про ароматичность, то это типичный пример карбокатиона аллильного типа. Стабилизацию аллильного катиона мы легко изображаем двумя граничными структурами, сопровождая это соображениями о том, что реальная структура где-то посредине (то есть плюс делокализован по двум крайним атомам углерода). Циклопропениевый катион в этом смысле должен обладать типичной для аллильного типа делокализацией с одним дополнением – граничных структур будет три, а не две, и плюс делокализован по всем трем атомам углерода. Наверное, это должно было бы дать дополнительный вклад в стабилизацию, но на это накладывался бы наоборот дестабилизирующий эффект очень большого напряжения трехчленного цикла. Вроде бы ясно, что никаких сенсаций быть не должно было бы. Но они есть. Про аллильный катион мы говорим, что он стабилизирован, и что в соответствующих условиях мы можем наблюдать реакции SN1-замещения. В некоторых других реакциях, например, в присоединении электрофилов к 1,3-диенам, мы тоже “видели” катионы аллильного типа. “Видели” – значит объясняли разные особенности таких реакций гипотетическим образованием таких катионов. Мы предполагали, что такие катионы образуются, и хотя живут очень недолго, но достаточно для того, чтобы мы могли их использовать для объяснения разных особенностей. Но мы никогда не видели аллильных катионов в реальности, например, какой-нибудь спектроскопией, даже создав специально особо благоприятные условия. Аллильные катионы остаются короткоживущими, очень реакционноспособными частицами, и поймать их экспериментально по-прежнему непросто. Если бы свойства циклопропениевого иона тоже сводились бы к аллильной делокализации, то с ним было бы как-то приблизительно так же, а скорее всего и сильно хуже, если не забывать про огромное напряжение ненасыщенного трехчленного цикла. Но – этот карбокатион оказался вполне стабилен даже при комнатной температуре и в растворах, и даже в виде твердых кристаллических солей, а некоторые замещенные циклопропенильные катионы устойчивы даже в воде, то есть растворителе с немаленькой нуклеофильностью. Этот факт впервые обнаружил один из самых знаменитых химиков 20 века Рональд Бреслоу, и очень сильно возрадовался, так как существование такого иона предсказывалось теорией ароматичности, и его обнаружение великолепно эту теорию подтверждало. Этот катион и его многочисленные производные обладают высокой термодинамической стабильностью (равновесия смещены в сторону катионов, а не ковалентных форм) и кинетической стабильностью (обладают низкой реакционной способностью по отношению к нуклеофилам). Среди обычных карбокатионов такой выдающейся стабильность обладают только трифенилметильные катионы, и только такие, которые содержат в ароматических кольцах сильнодонорные заместители – вспомните, если забыли, мы уже обсуждали стабильность делокализованных карбокатионов в SN1-замещении.
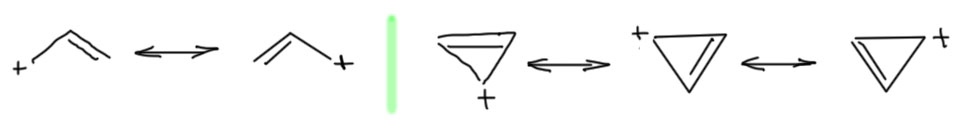
Займемся ароматическими ионами подробнее:
Как считать электроны и отличить ароматический ион от просто иона
Это не так просто, как может показаться. Но если сохранять спокойствие и действовать аккуратно, то все получится. Может быть две ситуации.
1. Структура уже нарисована, все кратные связи и заряды расставлены. Вы должны сказать, ароматический это ион или нет. Внимательно смотрим на структуру. Находим в ней цикл, про который мы должны что-то сказать. Пока что ограничимся только циклами, составленными только из атомов углерода. Обходим цикл кругом и тщательно следим, что каждый из атомов цикла или а) включен в двойную связь (или тройную, но это большая редкость); б) или несет положительный или отрицательный заряд. Если заряд находится на атоме, уже включенном в двойную связь, не обращаем на этот заряд никакого внимания – на ароматичность он не влияет.
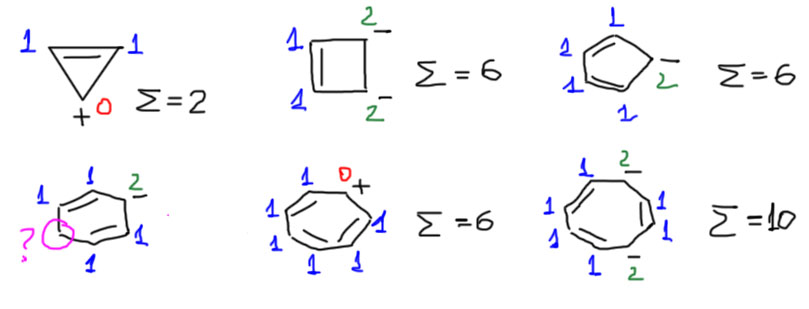
Считаем электроны, обходя цикл. Каждый атом двойной связи дает один электрон . Каждый атом с положительным зарядом – ноль электронов . Каждый атом с отрицательным зарядом – два электрона. Суммируем и считаем – если получится число из ряда Хюккеля (2, 6, 10, 14, 18, и т.п. – но все, что больше большая редкость и обычно просто пустая выдумка), то ион можно считать ароматическим. Оговорка “можно считать” означает только то, что мы должны иметь экспериментальное подтверждение этого предположения, но от нас в таком задании требуется именно прогноз, а не точное знание. Вот шесть структур, в пяти из которых эти условия соблюдаются, а в одной нет – в ней есть один атом углерода, который не входит в двойную связь и не несет заряд. В этом ионе нет замкнутого контура p-орбиталей, и он не может быть ароматическим – это просто мезомерно стабилизированный карбанион.
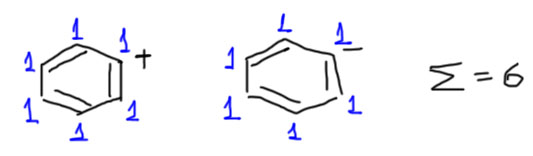
Еще раз – если плюс или минус находятся на атомах углерода, входящих в двойную связь, то нас они не волнуют, но в этом месте нужно быть очень острожными и внимательно смотреть, как конкретно сформулирован вопрос. Вот два типичных иона такого типа – фенильные катион и анион. Оба этих иона содержат ароматическое кольцо, и следовательно на вопрос, ароматические ли это структуры или нет, формальный ответ – да, безусловно. Но ароматичность этих ионов обусловлена только и исключительно наличием бензольного кольца и никак не связана с самими ионами. По стабильности эти ионы мало чем отличаются от других катионов или анионов винильного типа, весьма, как мы хорошо знаем, неустойчивыми.
2. Структура нарисована в виде многоугольника с кружком внутри и зарядом внутри кружка – назовем такой вид изображения условно “гайкой”. Мы тут много раз уже в самой категоричной форме просили не использовать гайки для изображения бензольного кольца, но вот для ароматических ионов гайка – не такая плохая вещь, даже не просто не такая плохая, а в полном смысле хорошая, отличная вещь. (дисклеймер: “гайка” – это жаргон, не пытайтесь использовать это слово в настоящей научной речи или статьях – вас никто не поймет) Лучше пока никто не придумал. Хотя бы потому, что изображение ароматических ионов обычными структурами с двойными связями и локализованным зарядом требует от нас все время помнить, что заряд на самом деле делокализован, и нужно то ли все время рисовать все граничные структуры, то ли просто держать это в голове. Гайка в этом смысле гораздо более инструментальна – она точно говорит нам, что заряд не приватизирован одним из углеродов, а находится в общем пользовании всех атомов, входящих в цикл. В этом, кстати, кроется еще одно важное отличие ароматической делокализации от обычной мезомерной – в последней заряд делокализован не по всем атомам цепи сопряжения, а только по тем, которые принимают заряд в граничных структурах.
Итак, у нас гайка с зарядом. Как посчитать электроны и проверить, а правда ли система ароматическая. Это очень просто, даже проще чем в п.1. Сначала нужно посчитать количество вершин в многоугольнике. Это количество дает нам число “своих” электронов (от каждого атома в делокализации участвует p-орбиталь с одним валентным электроном). От этого числа нужно вычесть (с учетом знака) заряд, указанный внутри гайки. Например, если цикл пятичленный, а внутри гайки минус, то всего электронов 5 – (-1) = 6. Отлично – это ароматический ион. Посмотрим еще примеры.
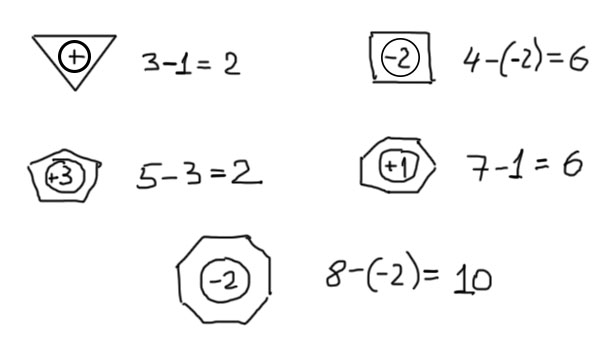
Все эти ионы получаются ароматическими. Все они, сами по себе или в виде производных, реально существуют и действительно проявляют типичные ароматические особенности – выровненность длин связей, аномально высокую устойчивость, слабопольные сдвиги в спектрах ЯМР. Система работает.
3. Как преобразовать гайку в обычную структуру.
Это не всегда просто и результат получается часто довольно неуклюжий, но это необходимо уметь делать хотя бы для того, чтобы понимать, как в таких структурах работают эффекты заместителей. И не менее важно – это позволяет понять, как хороши и удобны гайки, и от какой невероятной мороки спасают они человека.
Итак, чтобы преобразовать гайку в нормальную структуру, берем заряд из кружка и расписываем его по углеродам кольца, а все оставшиеся без зарядов углероды соединяем двойными связями. Сотворив это, убедимся, что каждый углерод кольца получил или двойную связь, или заряд, и если найдется такой, который ничего не получил – мы сделали что-то неправильно и нужно начинать сначала и более внимательно. Если структура-гайка была опознана как ароматическая, эта процедура гарантировано может быть выполнена, и заряды со связями расставлены без пропусков. Но дальше нас поджидает вопрос – это же можно, как правило, сделать не одним, а несколькими способами, – какой выбрать?? Не беспокойтесь, способ на самом деле один, а все альтернативные структуры, которые вы при этом могли бы получить, являются ни чем иным, как граничными структурами, которые подразумеваются, даже если не пишутся в явном виде. Посмотрим на такой менее тривиальной структуре, как дианион циклооктатетраена. Понятно, как можно и как нельзя расставлять заряды. Из любой годящейся структуры можно получить все другие, просто рисуя обычную делокализацию граничными структурами, перемещая заряд и двойные связи в формально отдельных аллильных фрагментах.
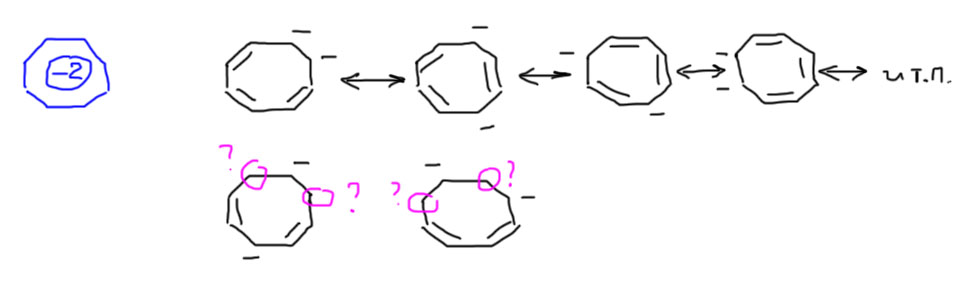
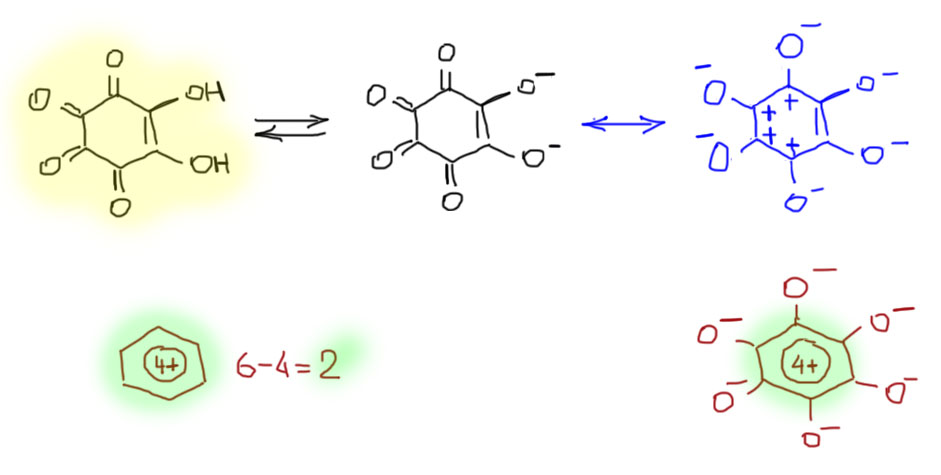
Зачем это может понадобиться? Например, чтобы понимать структуры всяких производных, в которых могут проявляться в виде фрагментов ароматические ионы. Вот, например, родизоновая кислота, известный аналитический реагент на ионы свинца. Эта молекула, с виду не имеющая никакого отношения к ароматичности, обладает необычным свойством – она очень легко теряет оба протона (это очень редкое свойство у двухосновных кислот, которые обычно гораздо труднее отдают второй протон, чем первый – константы кислотности по второй ступени, как правило, на порядки ниже чем по первой, а у родизоновой кислоты обе константы почти одинаковы). Родизоновая кислота так легко теряет оба протона, потому что ее дианион – совершенно симметричное ароматическое соединение, причем с двухэлектронной ароматичностью. Двухэлектронная ароматичность в шестичленном цикле??? Да, именно так, хотя такой незамещенный ион неизвестен, а это был бы, между прочим, тетракатион бензола – продукт изъятия четырех электронов у молекулы бензола! – посмотрел бы я на того, кто попробовал бы это провернуть. Но в родизонате шесть сильнодонорных анионных кислородов в виде заместителей мастерски компенсируют такой страшный положительный заряд внутри кольца и делают весь ион суммарно дианионом. То есть, родизонат это – держитесь покрепче – гексаанион гексагидроксипроизводного тетракатиона бензола. А откуда же там столько плюсов взялось – не вижу в исходной структуре? Из-за известной способности карбонильной группы поляризоваться при необходимости, что отображается резонансной структурой с разделенными зарядами. В данном случае выгодно так поляризовать все 4 карбонила из исходной молекулы именно потому, что в серединке получается двухэлектронный ароматический тетракатион. И вот его преобразования из гайки в обычную структуру (оцените ее неуклюжесть – в такое вообще трудно поверить, чтоб четыре! плюса!! рядом!!! – караул! жулики! так не бывает!! Но во-первых, не рядом, они делокализованы по 6 углеродам, и, во-вторых, по-другому такие странные свойства не объяснить. В трех-, четырех- и пятичленном рядах есть похожие и не менее знаменитые соединения.
Откуда берутся ароматические ионы. Для начала, катионы...
Ароматические ионы берутся оттуда же, откуда вообще берутся ионы.
На этом можно было бы закончить. Но тогда не нужно было и начинать. Поэтому все же продолжим. Начнем с общего утверждения, что ионы бывают положительные и отрицательные. И мы вряд ли будем удивлены, что отрицательных ионов больше в самых разных смыслах. Они чаще встречаются, среди них больше знаменитостей, их проще получить, они в целом намного устойчивее и т.п. Это есть простое следствие природы ионов. Ведь для того чтобы получить отрицательный ион нужно присоединить электрон к какой-то частице. Почти всегда этот процесс происходит легко и с выделением энергии. Мы это уже обсуждали: энергия, выделяющаяся при присоединении электрона к частице, называется сродством к электрону и всегда или положительна или равна нулю. Иными словами: анион – это хорошо и довольно естественно. А чтобы сделать катион, нужно отобрать электрон. Для этого всегда энергия затрачивается, и часто немалая. Катион всегда выше по энергии чем соответствующий по структуре анион. И хотя такие простые соображения далеко не все определяют в химии, но они верно предстказывают довольно общую тенденцию – катионы труднее получаются, их сложнее идентифицировать, они почти всегда менее стабильны, склонны ко всяким реакциям, и т.п. Среди ароматических ионов есть два знаменитых катиона – циклопропенильный и циклогептатриенильный (тропилий), и это почти весь ассортимент ароматических катионов, не считая нескольких достаточно экзотических структур. А вот анионов намного больше.
Начнем с катионов.
Ароматические катионы
Катионы можно получить из простых предшественников одним из трех способов: отнять уходящую группу, отнять гидрид, отнять электрон.
Принудительная ионизация
Основной способ получения ароматических катионов нам хорошо знаком по теме SN1-замещение, что неудивительно, так как известные ароматические катионы представляют собой типичные карбокатионы, только сильно стабилизированные. Уход уходящей группы, как мы знаем, может быть самопроизвольный, но если мы хотим реально получить катион в виде настоящей соли, то лучше поторопить события и использовать принудительный уход уходящей группы с помощью какой-нибудь кислоты Льюиса. Фторид обычно просят на выход с помощью трехфтористого бора или пятифтористой сурьмы, а хлорид – пятихлористой сурьмы, хотя есть и десятки других подходящих кислот Льюиса, образующих стабильные комплексные анионы.
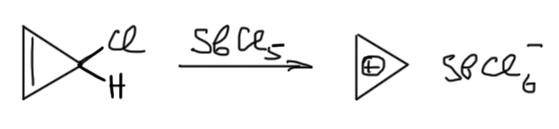
Тот же метод был использован при попытке получить дикатион циклобутадиена из дихлорциклобутена. Но этот ион уже оказался намного менее стабильным, и его пришлось готовить в фирменном растворителе Джорджа Олы, смеси SO2 и SO2ClF, при низкой температуре, используя более мощную кислоту Льюиса пятифтористую сурьму. Ион удалось зафиксировать только с спектре ЯМР, и даже при небольшом повышении температуры он разлагался. Приблизительно то же самое получилось с тетраметил-производным и еще несколькими аналогами.
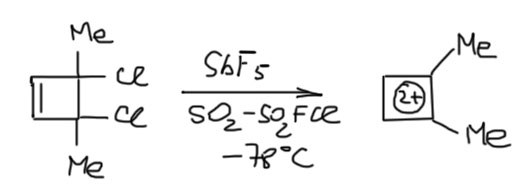
Гидридный перенос
Второй способ тоже имеет прямое отношение к уже знакомой нам химии карбокатионов, и называется гидридным переносом – это когда один катион отрывает от молекулы атом водорода с двумя электронами, то есть гидрид, и превращает ее в катион. Развязно говоря, это когда два катиона устраивают между собой разборку, кому достанется атом водорода с двумя электронами в придачу. Побеждает менее стабильный и более реакционноспособный катион, забирающий себе гидрид. Понятно, что в том случае если одним из катионов является ароматический ион, с плюсом останется именно он. Этот способ очень удобен для получения тропилиевого катиона в виде самых различных солей. В качестве катиона – акцептора гидрида можно использовать трет-бутильный или даже трифенилметильный, делая их прямо на месте из соответствующих спиртов и сильной кислоты. ![]()
Обратите внимание, что в споре тропилия с трифенилметильным катионом, который мы привыкли считать одним из самых стабилизированных из неароматических. В этом месте некоторые обязательно возмутятся – как это трифенилметильный катион неароматический, – там же три фенила, в которые делокализуется плюс, – плюс гуляет по ароматическим кольцам! Очень хорошо, это действительно важный момент, разберем его в отдельном блоке.
Гидридный перенос – типичная реакция электрофильного замещения на насыщенном атоме углерода. Мы подробно разбирались в механизме таких реакций в электрофильных реакциях алканов. Электрофил, например, карбокатион атакует связь C-H, образуя короткоживущий гиперкоординированный ион с трехцентровой связью. Эта связь легко распадается по одному из трех направлений, но в нашем случае направление распада будет соответствовать самому устойчивому катиону в системе, то есть именно ароматическому. Результатом как раз и будет перемещение водорода с двумя электронами от одного катионного центра к другому:
![]()
Одноэлектронное окисление
Циклоктатетраен очень легко дает ароматический дианион за счет восстановления, и мы это обсудим на вкладке про анионы. Для дианиона это будет 8+2=10, десятиэлектронная ароматичность. Но можно попробовать и в другую сторону, и электроны отнять 8-2=6, и получить шестиэлектронную ароматичность. Эта идея лежит на поверхности, но реально осуществить ее непросто. Дело в том, что катионы обычно гораздо менее устойчивы и более реакционноспособны, и пока мы будем окислять, стремясь к ароматическому дикатиону, промежуточные монокатионы успеют развалиться, перегруппироваться, заполимеризоваться, нахватать нуклеофилов из окружающей среды и т.п. Нужны специальные условия и нехилый опыт работы с карбокатионами. Поэтому за эту работу впервые взялся классик карбокатионной химии, карбокатионный нобелевский лауреат Джордж Ола (кстати, еще один выдающийся ученый, который вовремя свалил из плохого места, но на этот раз из тоталитарной коммунистической Венгрии в 1956 году, по рождению он венгр, Ола Дьёрдь), первый человек в мире, который видел трет-бутильный катион, и прочая, и прочая. Он нашел, что его любимая кислота Льюиса SbF5 не только кислота, но и нехилый окислитель, причем сразу забирает два электрона, переходя из сурьмы(+5) в сурьму(+3). И все это можно делать в его любимом растворителе, смеси SO2 и SO2ClF, которая остается жидкой при очень низких температурах и не обладает практически никакой нуклеофильностью. И еще в нем сразу можно снимать спектры ЯМР. Ола попробовал целый ряд замещенных циклоктатетраенов (ЦОТ), но осмысленные результаты получились с тремя: 1,4-диметил, 1,3,5,7-тетраметил, и октаметилЦОТ. В каждом из этих случаев при низкой температуре получалось нечто с очень простым спектром, напоминающим о желаемом ароматическом дикатионе. Например, из тетраметилЦОТ получалось нечто с двумя сигналами в протонном ЯМР с соотношением интегралов в 1:3, первый сигнал в слабопольной части при 10,1 мд, второй при 3,6 мд. Слабопольный сигнал уехал так далеко не только потому, что он испытывает типичный ароматический сдвиг из-за кольцевого тока, но и из-за влияния положительного заряда (это общее явление: положительный заряд возникает из-за недостатка электронов и действует как акцептор, смещая сигналы в ЯМР с слабое поле, а анионы – наоборот). Похожая история была в 13C ЯМР. Это позволило уверенно интерпретировать частицу в растворе как искомый ароматический дикатион. Увы, он оказался малоустойчивым и полностью развалился при попытке поднять температуру хотя бы до -20ºС. Больше, насколько мне известно, к этой ароматической системе никто не возвращался (прошло уже без малого 50 лет).
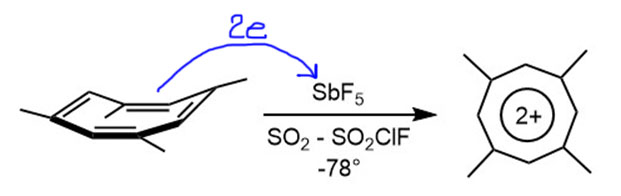
В общем, с дикатионами и циклобутадиена, и циклоктатетраена получается весьма двусмысленная история. Из четырех признаков ароматичности эти частицы более-менее уверенно соответствуют только магнитному в спектрах ЯМР. Со стабильностью у них ничего выдающегося – неароматических карбокатионов, спектры которых можно надежно получить при низкой температуре пруд пруди, достаточно вспомнить только трет-бутильный катион, а сравнение с настоящими ароматичскими катионами циклопропенилия и тропилия они вообще не выдерживают. Реакций никаких с такими ионами никто делать даже не пытался, а структура их остается экпериментально неопределенной, и мы не можем надежно сказать, усреднены у них связи или нет (впрочем, есть несколько приличных расчетов). Придется сделать вывод, что в ряду ароматических ионов дикатионы пока довольствуются весьма спорным местом.
...и анионы
Типичных способов получения ароматических анионов тоже не очень много, хотя самих таких анионов известно намного больше. В отличие от ароматических катионов, ассортимент которых, как мы уже видели, невелик, бывают ароматические моно- и дианионы, и их довольно много с самыми разными размерами циклов. Более того, ароматические анионы нередко встречаются среди больших циклов (от 8- до 18-членных и даже более), там где очень редко встречаются нейтральные ароматические соединения и никогда не встречаются ароматические катионы. Получается, что в больших циклах ароматические анионы часто стабильнее чем нейтральные соединения, которые почти всегда не являются ароматическими из-за неплоской геометрии.
Причина этого довольно понятна – анионы мягче и податливее нейтральных непредельных соединений, их легче распластать в плоский цикл. Нейтральные непредельные соединения содержат двойные связи с атомами в sp2-гибридном состоянии. Такие фрагменты сами по себе плоские, но углы между связями равны 120º или около того. Попробуйте сделать хороший плоский цикл, если вы ограничены углами в 120º. Шестиугольник получается идеальный, но там в ароматике проблем нет. Для всех циклов больше 6 углы больше 120º, и чем цикл больше, тем тупее углы. Это значит, что попытка сложить плоский цикл из максимального количества двойных связей (иначе не получится сопряжения) приводит к искажению углов (это называется угловым напряжением и мы подробно рассмотрим это во 2 семестре), а значит, затратам энергии, это невыгодно, и чем шире цикл, тем невыгоднее. Из-за этого (и не только), большие непредельные циклы складываются в такие неплоские конструкции, сохраняющие немалую подвижность врагментов (это, кстати, тоже выгодно, уже с точки зрения энтропии). В результате, плоских ароматических аннуленов с циклами более 8 не просто мало, а очень мало, и все известные молекулы этого типа используют всякие ухищрения или для фиксации плоской структуры, или для разгрузки напряжения. Но об этом – в другом месте.
Если в цикл добавить один-два отрицательных заряда, вместо крепких двойных связей получим много сопряженных аллильных анионов (посмотрите еще раз, как это устроено в предыдущем блоке). В аллильных анионах двойные связи сопряжены, а значит ослаблены (сопряжение снижает эффективный порядок связи), и поэтому более гибки, менее трепетно относятся к поддержанию валентных углов в 120º. Такие циклы можно распластать на плоскости, подогнув внутренние углы под размер цикла. И в компенсацию этого выдать ароматическую стабилизацию.
Основных способа изготовления ароматических анионов два. Выбор довольно прост и зависит от числа атомов в цикле, вернее от того, четное или нечетное их число.
Депротонирование
Если число нечетное, то от насыщенного углерода можно оторвать протон, замкнув цепь сопряжения. Получается это только в тех случаях, когда в исходном цикле было 4n атомов углерода, связанных двойными связями. Тогда после отрыва протона мы добавим пару электронов в сопряжение и получим искомые ароматические 4n+2 электронов. Самый знаменитый ион этого типа – циклопентадиенил, вполне бесспорно можно считать самым знаменитым ароматическим соединением, не содержащим бензольного кольца (небензоидным). Этот ион с самыми разными противоионами легко получается отщеплением протона от циклопентадиена сильными основаниями, причем вполне годится и чаще всего и используется гидрид натрия, а это основание хотя и достаточно сильное, но редко применяющееся для отщепления протона от CH-кислот с высокими значениями pK. И действительно, у циклопентадиена достаточно небольшое pK (всего 19 в ДМСО), существенно меньшее чем у общеизвестных CH-кислот, которые мы и все остальные считают легко депротонируемыми (например, терминальные ацетилены имеют pK около 28, а енолизуемые кетоны типа ацетона 25-26, в том же ДМСО). Относительно высокая кислотность циклопентадиена лучше всего остального говорит о том, что сопряженное основание этой CH-кислоты очень хорошо стабилизировано, и величина этого эффекта стабилизации гораздо больше, чем можно было бы ожидать просто от мезомерной делокализации: pK дивинилметана, CH-кислоты, также формально имеющей две двойные связи рядом с CH-кислотным центром, около 35 – и разницу в 16 порядков между дивинилметаном и циклопентадиеном не объяснить просто образованием цикла во втором случае, что увеличивает число граничных структур с трех до пяти. Поэтому мы имеем право и даже должны вместо резонансных структур в случае циклопентадиенильного аниона изображать ароматическую делокализацию, например, ненавистной, но такой полезной “гайкой” с минусом внутри.
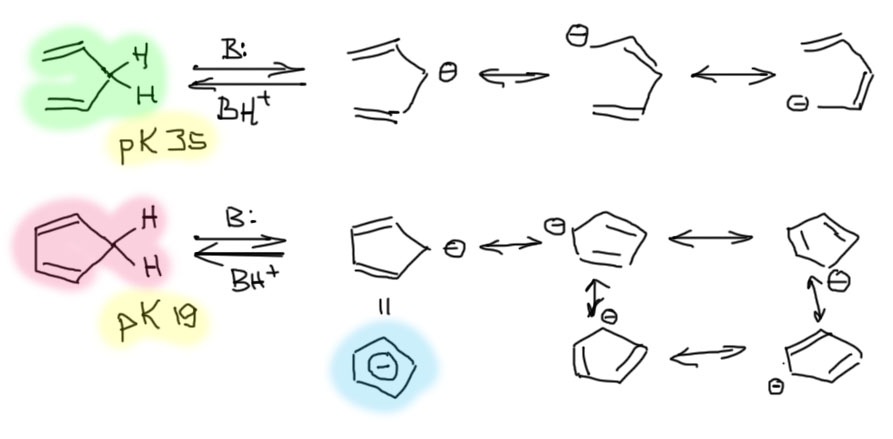
Известно огромное количество производных циклопентадиенильного аниона, также являющихся ароматическими шестиэлектронными анионами, и почти всегда их получают аналогично депротонированием соответствующих циклопентадиенов. Циклопентадиенильный анион (и его аналоги) опять почти бесспорно – самые известные лиганды в металлоорганических соединениях металлоценах, начиная с такой знаменитой молекулы как ферроцен, и в составе комплексов эти лиганды также сохраняют ароматические свойства. Обсудим это подробнее как-нибудь в другом месте.
Кроме циклопентадиенильного аниона нельзя не упомянуть и его 10-электронный аналог, получаемый из 9-членного тетраена также депротонированием.
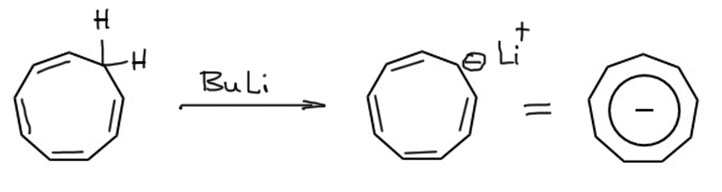
Этот плоский ароматический ион, скорее всего, является самым большим правильным многоугольником среди всех известных ароматических соединений, завершая собой ряд таких структур. Если все такие структуры выписать рядом по увеличению цикла получится такой забавный короткий ряд:
Все известные (насколько я могу судить – возможно я заблуждаюсь, буду признателен, если опровергните это реально охарактеризованной структурой) моноциклические ароматические соединения с циклами от 10 и более имеют более сложную геометрию колец, и не являются правильными многоугольниками. Мы уже обсуждали почему – чем шире цикл в виде простого многоугольника, тем дальше отклоняется угол при его вершине от нормального значения для sp2-гибридного углерода, и в цикле нарастает напряжение, настолько, что выигрыш от ароматической стабилизации уже не может его компенсировать.
В этом ряду есть два дианиона. Посмотрим, откуда они берутся.
Двойное одноэлектронное восстановление
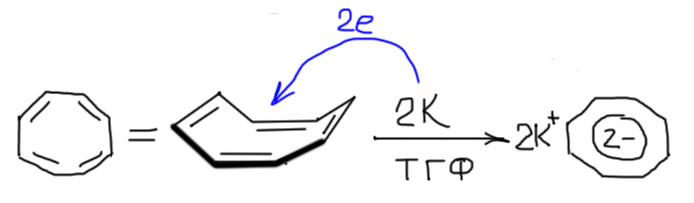
Циклы с четным числом углеродов, но не имеющих сразу ароматического числа электронов, а это 4, 8. 12 и т.п. могут набрать это число, если либо возьмут два дополнительных электрона, или отдадут два электрона, сохранив уже имеющуюся систему сопряженных связей. Мы уже договорились о том, что при прочих равных условиях анионом быть приятнее и выгоднее, чем катионом, поэтому сосредоточим внимание на приобретении двух электронов (дикатионы на самом деле тоже бывают, но это всегда нечто очень эфемерное и не до конца определенное, поэтому можно с чистой совестью их проигнорировать). Откуда органические молекулы могут взять электрон-другой мы уже не раз выясняли – от металлов, быстрее всего от щелочных. Наиболее близкий аналог – восстановление бензола по Бёрчу натрием в жидком аммиаке. Но там мы исходим из ароматической молекулы и портим ее двумя лишними электронами. А здесь мы возьмем неароматическую молекулу и попробуем добавить ей недостающие электроны. Это довольно легко происходит, и известно много примеров такой восстановительной ароматизации. Самый, наверное, известный и яркий – восстановление циклооктатетраена (ЦОТ). Это действительно очень легко происходит, настолько лешко, что реакцию эту открыли независимо друг от друга еще в 1950-е годы сразу несколько исследователей. Наиболее чисто реакция идет в присутствии металлического калия в растворе ТГФ – образуется дианион ЦОТ с проивоионами калия, имеющего в качестве лигандов молекулы ТГФ. Сразу обратим внимание на два важных обстоятельства:
- электроны от щелочного металла всегда переходят по одному, но мы не останавливаемся на продукте переноса одного электрона (анион-радикале ЦОТ), а добавляем к нему еще один до дианиона, и не берем третьего – лишнего не надо, ароматическое число электронов достигнуто, и это хорошо:
- исходная молекула ЦОТ неплоская, сильно неплоская, молекулы такой формы часто называют “ваннами”, хотя в этом случае речь идет не о конформации, а о настоящем стереоизомере с четырьмя двойными связями в цис-конфигурации. Такая форма обуcловлена тем, что каждая двойная связь имеет на углеродах углы в 120º и соединить четыре таких фрагмента можно только таким способом. Но получив два электрона она выпрямляется и превращается в почти идеальный правильный плоский восьмиугольник. Что ее выпрямило – видимо, та же ароматичность, которой по определению легче живется в плоских структурах, обеспечивающих максимальное перекрывание p-орбиталей.
Получающийся дианион может быть выделен в виде кристаллической соли, и неплохо хранится при отрицательной температуре и без доступа воздуха.
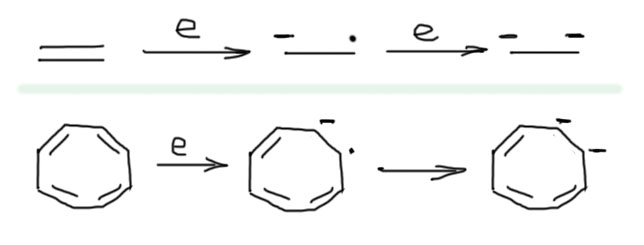
Понятно, как это происходит. Любая двойная связь может присоединить сначала один электрон, затем второй с образованием дианиона. Обычная изолированная двойная связь однако делает это с большим трудом, потому что электроны рядом отталкиваются и не хотят набиваться в таком количестве в тесноту. Но если есть несколько сопряженных двойных связей, это происходит намного легче, потому что оба электрона делокализуются, а в данном случае еще и не просто делокализуются, а с эффектом ароматической стабилизации. Один из способов это изобразить мы рассмотрели на вкладке выше, а здесь формально остановимся на первой попавшейся граничной структуре.
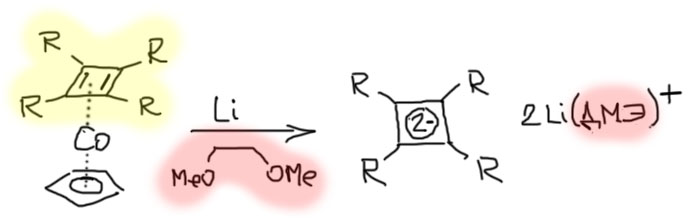
Таким же способом можно было бы получить дианион циклобутадиена с 6 электронами, но есть большая проблема – исходный циклобутадиен неустойчив и не может быть использован в качестве исходного для восстановления. Проблему решили совсем недавно японские химики Секигути и др., использовав любопытную идею: сам циклобутадиен неустойчив, но его комплексы с разными переходными металлами вполне устойчивы и относительно легко получаются. Восстановление одного из таких комплексов щелочным металлом в другом эфирном растворителе диметоксиэтане, который очень хорошо взаимодействует с катионом лития, помогая реакции) и дало искомый ароматический дианион, заодно освободив его от переходного металла. Для дополнительно стабилизации цикл несет объемистые заместители (в данном случае это SiMe3, этот заместитель очень любят металлоорганики в основном для повышения растворимости громоздких молекул), но на структуру ароматического ядра они не влияют, поэтому вполне можно считать, что ряд достроен полностью.
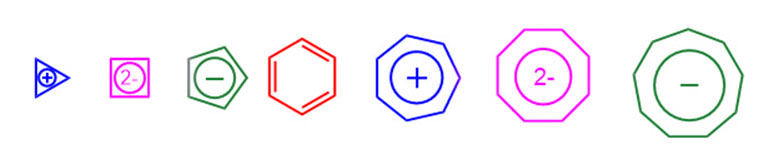
Повторим еще раз этот занятный ряд известных ароматических систем, представляющих из себя правильные плоские многоугольники от треугольника до девятиугольника:
Ряд получается весьма любопытный, настолько, что трудно удержаться от некоторых обобщений специально для любителей конспирологически-нумерологически-метафизической галиматьи (серьезным людям читать такое не рекомендуется, поэтому выделяю мелким шрифтом: у серьезных людей обычно плохое зрение, и они это не прочтут).
Точно в середине ряда бензол, признанное верховное божество ароматического мира, без которого сама теория ароматичности никогда не появилась бы на свет. Только бензол в этом ряду нейтрален, все остальные – ионы. Ошююю и одесную (сиречь слева и справа) бензола ровно по три (по три!) ароматических иона в четком порядке катион–дианион–анион. С дианионов каждый раз начинается переход к следующему числу ароматических электронов 2 – 6 – 10. И наконец, непосредственно рядом с царь-бензолом два самых важных ароматических иона – циклопентадиенид и тропилий. Получается такой деисусный чин ароматической религии, да не будет этим оскорблен ни один верующий, а если будет, то заранее извиняюсь. В теории ароматичности, как и во многих других научных концепциях, основанных на глубокомысленном обобщении экспериментального материала, действительно есть нечто от религиозной доктрины. В ароматичность можно верить, или не верить, и если серьезно заинтересуетесь ароматичностью, без труда найдете вполне серьезных ученых, которые считают, что это – целиком выдуманная конструкция, без которой можно спокойно жить. Я не рекомендую увлекаться этими явно экстремистскими взглядами. С ароматичностью точно жить проще, чем без нее. Это весьма симпатичная и удобная теория, и в наше время она переживает весьма серьезный расцвет – новые ароматические системы сыпятся как из рога изобилия.
Тропилиевый и трифенилметильный катионы: ароматическая и мезомерная стабилизация
Еще раз спокойно обсудим особенности двух сильно стабилизированных карбокатионов, имеющих отношение к ароматичности. Почему мы считаем тропилий ароматическим катионом, а трифенилметильный катион – не считаем ароматическим катионом, а считаем карбокатионом, стабилизированным мезомерным эффектом ароматических колец. Разница здесь довольно тонкая, вроде бы очевидная. но не совсем. И в одном, и в другом плюс “гуляет” по ароматическим кольцам (слово “гуляет” ни в коем случае не принимаем буквально, но просто как просторечный синоним слова “делокализован”). И там, и там это определяет повышенную стабильность иона. Оба этих иона могут быть получены в виде устойчивых солей, которые можно хранить без особых предосторожностей. При этом, мы знаем, что тропилиевый катион намного стабильнее трифенилметильного, потому что перенос гидрида осуществляется в сторону последнего.
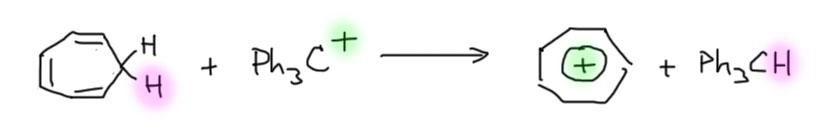
Уже отсюда мы видим, что тропилиевый тип стабилизации сильнее трифенилметильного. При этом, если мы просто будем считать граничные структуры в делокализации, мы не сможем понять причину такой сильной дополнительной стабилизации. В тропилии у нас будет семь совершенно одинаковых структур, в которых плюс обойдет все семь углеродов кольца. Это, безусловно, очень хорошо – теория мезомерии говорит нам, что чем больше одинаковых или близких по энергии граничных структур, тем более лучше. Но тогда в трифенилметильном катионе – еще более, ведь там девять одинаковых (почти одинаковых) структур. А явная разница в стабильности целиком на стороне тропилия, ведь нарисованная выше реакция это равновесие, нацело сдвинутое в сторону тропилия. Это очень наглядно подверждает то, что в тропилии есть вклад какой-то особой стабилизации, которая не сводится просто к делокализации и мезомерии. Этот вклад – ароматичность, ароматическая стабилизация. И разница именно в этом:
- При образовании тропилиевого катиона ароматическая стабилизация возникает именно тогда, когда катион образовался – в исходном циклогептатриене ее не было.
- При образовании трифенилметильного катиона ароматическая стабилизация не возникает – уже в исходном трифенилметане ароматичность наличествовала в полноценных бензольных кольцах, и то, что в катионе в эти кольца запустили “гулять” плюс, то есть, как говорят в физике, “дырку” эту стабилизацию как минимум не увеличивает, а возможно даже немного уменьшает, ведь для ароматичности нужно, чтобы в кольце было шесть электронов, а когда кольцо сопрягается с электронодефицитным катионным центром, электронная плотность на нем суммарно снижается и становится меньше шести.
Итог этих рассуждений прост: ароматическими ионами по примеру тропилия мы будем называть не все ионы, в состав которых входят ароматические кольца, даже если эти кольца активно участвуют в стабилизации, а только такие, стабилизация которых обусловлена возникновением ароматической системы при образовании иона.
И еще один важный вывод: ароматичность не сводится просто к делокализации, и хотя для ароматической системы можно вполне честно рисовать граничные структуры, суть ароматичности не в них, а в образовании особой устойчивой конфигурации электронов в π-системе.