Обновления
9.04.2023 – Добавлены аудио-дорожки к первой лекции
Комплексы переходных металлов в органической химии
Всем хороша органическая химия, которую мы усердно изучали целый год, кроме одного – она безнадежно устарела. Если открыть любой номер любого научного журнала, печатающего работы по органической химии, за последние лет 10 и посмотреть, о чем пишут, то – не узнаете её. Органику, которую вы изучали, не узнаете. С немалым трудом можно будет найти похожие реакции, но с каждой похожей будут десятки каких-то непонятных, с непонятными реагентами, будут мелькать странные сокращения, производные почти всех элементов Периодической таблицы.
Почему это все, и чем так плоха старая органическая химия. Она совсем неплоха, и дает нам вполне солидный фундамент, особенно если мы не заучивали десятки и сотни реакций, набирая их из какой-нибудь помойки типа книжек Марча или Ларока. Это, увы, тот случай, когда чем больше, тем хуже – получается неразобранная свалка с иллюзией знания. Не забывайте, что знания, как и мусор, нужно обязательно сортировать, иначе будут проблемы, накопится большая-большая куча, станет вонять, отравляя всё вокруг; то, что было положено в кучу первым, погрузится в недра, превратившись в слипшуюся мерзкую неразличимую массу, потечёт омерзительная зловонная жижа, а в конце концов вся ужасающая куча воспламенится от какой-нибудь малой искры, станет гореть, распространяя по округе ядовитый смрад, от которого будут падать в обморок добрые люди. Вот что такое неразобранные сведения, набранные случайно из первых попавшихся мест.
Вместо этого не торопитесь, добавляйте знания понемногу, пытайтесь понять, как работает изученное, и на каких принципах строится. Это-то как раз не пропадет, потому что в новой химии в основных принципах мало что изменилось – кислоты остались кислотами, основания основаниями, и равновесия по-прежнему помогает разруливать Ле Шателье. Но в старой органической химии была одна проблема, которая когда-то не считалась проблемой: все делается не спеша, за много стадий, реактивов не жалеют, а суммарный выход не считается большой проблемой – получить бы, то, что хочется, хоть пару микрограмм. В современной химии вся эта романтическая возня, достойная звезды Мишлена, перестала быть доблестью, прежде всего потому что современная химия гораздо ближе к реальному производству полезных веществ, а там правит бал экономика, а не спортивные достижения типа “получи анилин из бензола за максимальное количество стадий, но не меньше 20”. Нужно не просто получить важное соединение, а сделать это максимально быстро, с наилучшим выходом, вопроизводимо, с минимальными отходами, безопасно, чисто, да еще так, чтобы в конце это можно было поручить не человеку у тяги, а автоматизированному реактору. Поэтому так стали важны высокоселективные методы и методы, основанные на катализе.
Вот, например, важная задача – синтез дифенила с разными заместителями в кольцах. Среди таких молекул очень много важных и полезных веществ. И да, мы умеем их получать, и задачи на эту тему решали. Мы любим эти задачи – там много баллов можно получить, а значит там много стадий. Даже если начинать с нитро-производных, нужно в много стадий сделать азо-бензол, восстановить и перегруппировать, и позаботится о превращении амино-групп. Но, не на бумаге, а в колбе все это тяжелая, грязная, вонючая химия, с кучей отходов, канцерогенными интермедиатами, и низкими выходами.
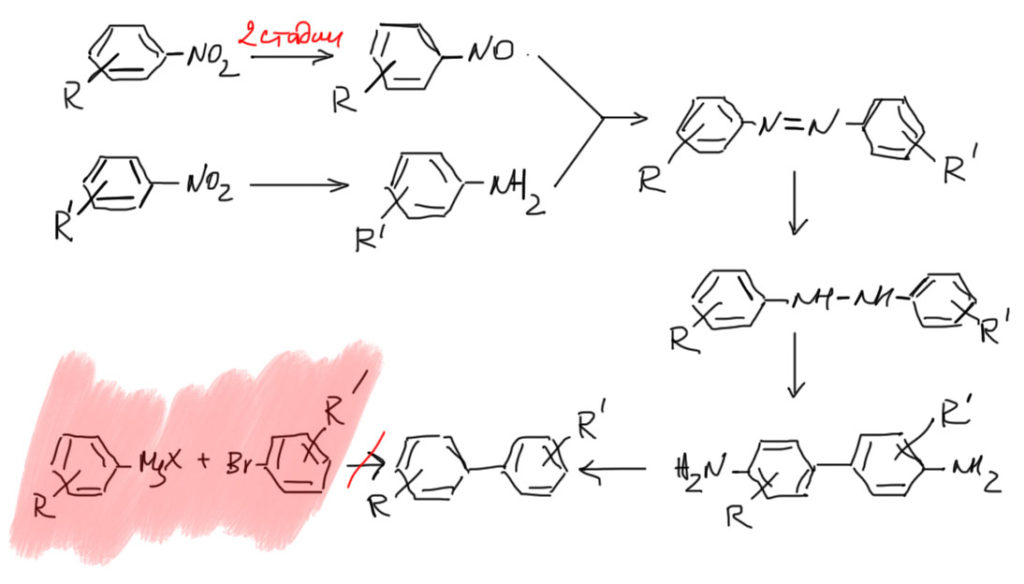
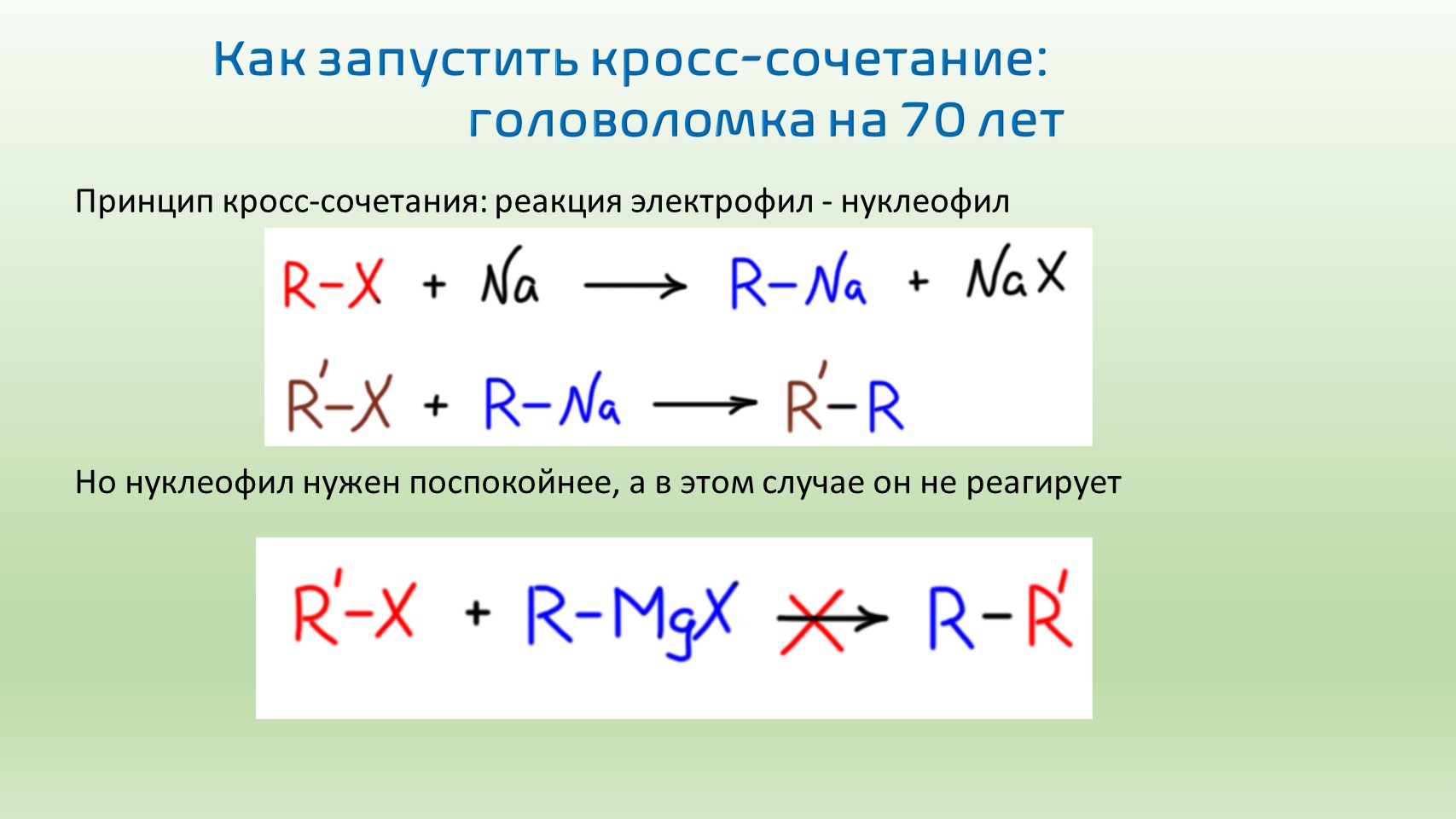
А вот ведь как славненько было бы то же самое сделать в одну стадию – взять гриньяр и бромпроизводное – ведь это нуклеофил и электрофил, должны вроде бы взаимодействовать – и готово! Черта с два, в большинстве случаев просто так ничего не выйдет, эти реагенты либо не реагируют, не хватает реакционной способности, либо прореагируют по своим заместителям, если они содержат функциональные группы, несовместимые с магний-органикой.
Одним из ключевых нововведений в органической химии стало использование комплексов переходных металлов. Эта наука бурно развивается с 1960-х, но в новом столетии скорость ее развития стала совсем головокружительной – любой номер научного журнала из первой двадцатки по рейтингу с гарантией угостит вас парой-тройкой новых методов, а номеров таких в год суммарно выходит под тысячу. Все три последние Нобелевские премии про органическую химию даны именно за реакции с участием комплексов переходных металлов. Эта химия действительно позволила решить множество задач-висяков из старой органической химии, в частности и эту – как легко и эффективно сделать разнозамещенный дифенил. Это теперь называется кросс-сочетание. И теперь мы знаем, как заставить реагировать гриньяр с бромпроизводным. И знаем даже, как сделать это еще лучше, еще чище, еще надежнее, еще селективнее, еще гибче.
Попробуем хотя бы поверхностно разобраться, как это работает. Если заинтересуетесь, на 5 курсе будет более подробный курс про это, и запущен, хотя еще и очень далек от полноты, специальный сайт. На этом сайте можно немного освежить принципы химии переходных металлов, которые вы проходили на 1 курсе, но слегка уже позабыли: там, в частности, объяснено, как считать степени окисления переходных металлов в комплексах, как считать электроны, какие бывают лиганды и прочие полезные вещи. Здесь я не буду тратить на это время.
В экзамене у нас есть пара вопросов на эту тему. Эти вопросы кратко сформулированы на вкладке Программа. А более подробное обсуждение, как обычно, на разворачивающихся вкладках.
Кросс-сочетание: основные стадии
В реакциях кросс-сочетания используют комплексы металлов 10 группы (из длиннопериодной периодической Таблицы) – никеля и палладия. В той же группе есть еще платина, но комплексы платины никогда не используют, и не из-за цены (палладий нынче не дешевле), а из-за очень низких скоростей реакций – комплексы платины слишком стабильны и малореакционноспособны.
Возьмем для определенности палладий. У палладия есть два основных состояния – Pd(0) и Pd(2+). Pd(0) – это просто металлический палладий, тот самый, который мы, например, используем в качестве катализатора гидрирования, нанеся на какой-нибудь твердый носитель – активированный уголь (Pd/C), на сульфат бария или карбонат кальция (катализаторы Линдлара и Розенмунда, которые мы использовали для селективного цис-гидрирования и превращения хлорангидридов в альдегиды) и т.д. Это все – гетерогенные катализаторы. А нам сейчас нужны гомогенные, то есть растворимые, принимающие усачтие в реакциях в растворенном виде. Разница очень понятна – палладий в виде металла представляет собой частицы, в состав которых входит неопределенное количество атомов металла. Даже в мельчайших частицах, размерами в несколько нанометров (наночастицах, коллоидном палладии, зóле палладия) сотни и тысячи атомов металла. В таких частицах в реакциях участвуют только поверхностные атомы, и то не все, и сами эти реакции можно описать только приблизительно.
Соединения в растворах (когда речь идет о металлах, их соединения называют координационными или комплексами) содержат один, а иногда несколько атомов металла, и характеризуются вполне определенным составом и структурой, которую можно установить. Точно так же, реакции таких соединений можно досточно точно понять и описать. Это очень удобно, потому что дает возможность понимать, что происходит, и разбираться с тем, что нужно сделать для того, чтобы происходило получше (с большими выходами, с большим разнообразием исходных, с большей селективностью).
Возвращаемся к Pd(0). Если вы когда-нибудь читали старые учебники неорганической химии (Некрасова, Реми, и т.п. – многие их читали и любили, и даже предпочитали более современным типа Коттона-Уилкинсона), то там очень много про разные соли и комплексы, но нульвалентное состояние считалось присущим только металлу. Идея о том, что металл может иметь производные (комплексы) в окислительном состоянии ноль, довольно свежая, ей всего-то лет 50 с хвостиком, и такие комплексы в свое время всех очень удивили, это считалось сначала таким химическим курьезом – нульвалентный металл(!), в растворе(!!), и не выпадает в осадок (чернью, зеркалом; просто куском, проломив дно). Но когда разобрались, выяснили, что ничего необычного в этом нет, просто нужны лиганды, которые стабилизируют это состояние в основном потому, что не дают таким атомам металла встречаться, взаимодействовать и образовывать частички металла. Самыми распространенными и удобными лигандами, которые решают эту проблему являются
Фосфины
Фосфины – пренеприятнейшие соединения. Исходный фосфин, просто фосфин (по номенклатуре ИЮПАК фосфан) PH3 – газ, жуткий яд (им в следовых количествах травят мышей и насекомых в зернохранилищах, и по надежности – всякий, кто с ним встретится, будь у него две, четыре, шесть или восемь ног – труп, и ему, фосфину, для этого нет замены). Всякие алкильные производные – триметил, триэтил, трибутил и т.п. – тоже очень токсичны и вдобавок еще и горят на воздухе. А вот трифенилфосфин Ph3P гораздо проще – это бесцветное кристаллическое вещество, которое очень легко получить из фенилмагнийбромида и треххлористого фосфора, оно вполне стабильно, взвешивается на воздухе, нелетуче, не самовоспламеняется, почти не пахнет, и отравиться им можно только по большой дури. Недаром, по популярности этот лиганд не имеет себе равных, и просто даже удивительно, как одно-единственное соединение может быть настолько вездесуще.
У палладия координационное число обычно 4, и Pd(0) весьма охотно образует комплекс с четырьмя молекулами фосфина Pd(Ph3P)4 – этот комплекс можно получить восстановлением солей палладия в присутствии трифенилфосфина, и получив, долго хранить в темном холодном месте. Его даже можно купить готовый. Фосфин как лиганд не изменяет степень окисления металла, поэтому в комплексе только с фосфинами степень окисления равна нулю.
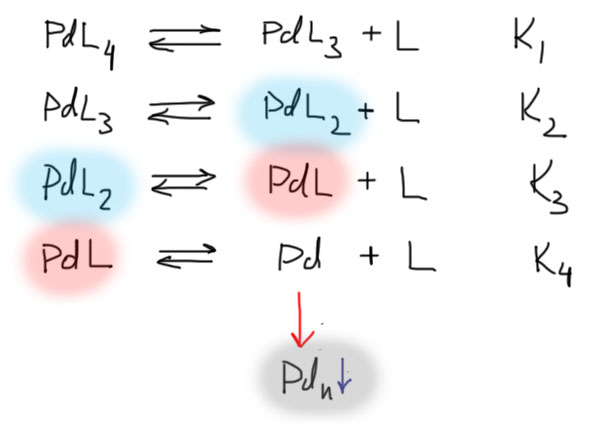
Если этот комплекс растворить в каком-нибудь растворителе, в растворе устанвливаются равновесия диссоциации лигандов. У каждого комплекса есть эти равновесия, а их константы равновесия называются константами устойчивости комплексов (константы равновесия диссоциации, как показано на схеме внизу, называются константами нестойкости, а константы равновесия – это просто величины, обратные константам нестойкости). Первый лиганд уходит довольно легко. На каждой следующей стадии константы устойчивости все больше, поэтому в растворе довольно много комплекса PdL3, существенно меньше PdL2, и очень мало PdL. Если повысить температуру, то комплексов с меньшим количеством лигандов будет все больше. Если переусердствовать, то появится безлигандный Pd(0) – и тут же слипнется, выпадет в осадок металлический палладий, обычно в виде плотного черного порошка, так называемой черни (оцените игру слов – благородный металл превращается в чернь, грань тонка). Это плохо. нужно быть осторожными.
И вот – главный фокус. Комплексы PdL4 и PdL3 в реакциях не участвуют – у атома палладия нет свободного места и валентных возможностей. Участвуют PdL2 и PdL, и чем их в растворе больше, тем лучше идут реакции. И нужно очень аккуратно подбирать температуру реакции, чтобы и реакция шла, и до выпадения черни не допрыгаться. Все еще осложняется тем, что если мы работаем грязно, то воздух в реакционной смеси понемногу окисляет фосфин в хорошо нам знакомый фосфиноксид, и так незаметно лишает палладий лигандной поддержки. Это всегда так выглядит – только что все было нормально, реакция идет, и вдруг разом все чернеет, и до свидания. Почему это так внезапно происходит, и почему “до свидания” – потому что выпадение черни реакция необратимая, и как только она вмешивается, в свои права вступает принцип Ле Шателье, и все сваливается в прямом смысле в чернь.
И еще один фокус. Если взять не трифенилфосфин, а очень похожее соединение с орто-метильными группами – трис(орто-толил)фосфин, то из-за этого метила в координационную сферу палладия четыре лиганда уже не лезут, и вы начинаете с PdL3 – а следовательно до активного PdL2 уже рукой подать – первое же равновесие дает то, что нужно. Поэтому комплексы палладия с таким фосфином часто более активны, даже намного. Увы, с этим лигандом есть свои проблемы, и мы пока не будем его использовать – останемся с трифенилфлосфином.
Окислительное присоединение
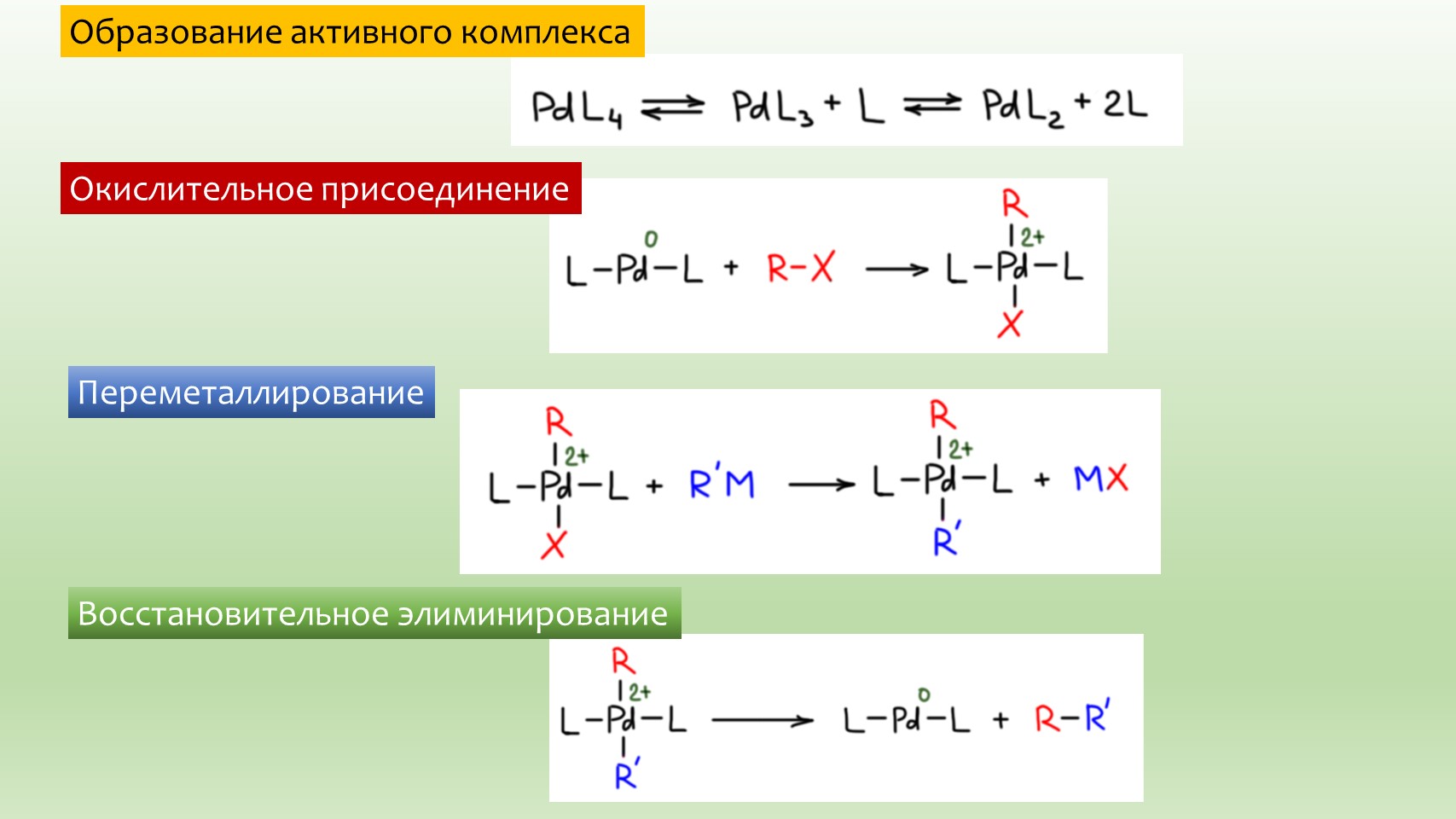
Теперь посмотрим, что будет, если мы комплексу Pd(0) в растворе подсунем какое-нибудь галогенпроизводное, например, бромбензол. Вспомним нашу обычную органическую химию – бромбензол может реагировать с нуклеофилами только в очень жестких или специфических условиях, а просто так – нет, не может. А вот с комплексом Pd(0) бромбензол реагирует очень легко, при небольшом нагревании, нужном, как мы уже знаем даже не для самой реакции, а для того, чтобы в растворе образовалось некоторое количество активного комплекса. Результат реакции – новый комплекс, уже Pd(2+) – палладий вернулся в свое обычное валентное состояние, окислился. А кто его окислил? Бромбензол, ежу понятно. Почему ежу понятно? Просто потому что там больше ничего нет, а раз палладий окислился, значит палладий(0) повстречался с окислителем. Если точнее, то в новом комплексе на палладии висит два отрицательных лиганда (если захочется понять или просто вспомнить, как это устроено, можете посмотреть на другом сайте). А еще, с точки зрения палладия, к нему произошло присоединение, и это типчное присоединение – реакция в которой из двух молекул получается одна, в состав которой входят по частя обе исходные. И вот эта реакция так и называется – окислительное присоединение. Если быть точным, то – окислительное присоединение бромбензола к атому палладия.
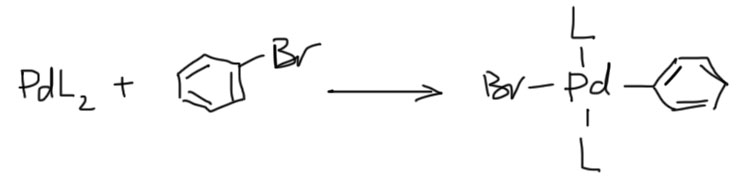
Одного взгляда на эту, якобы, новую реакцию достаточно, чтобы понять, что нам, как водится, опять морочат голову, и что мы это уже видели – разве реактив Гриньяра образуется не так? Для полноты картины вспомним, что и лиганды там есть, только не фосфины, а молекулы эфира. Вот же оно!
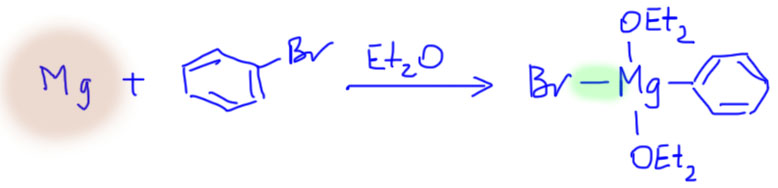
Действительно, как две капли воды. Да, но не совсем. Отличие первое – магний в этой реакции – это именно металл, прямо стружки. А это важно – что металл? Да, невероятно важно, потому что именно из-за использования палладия в виде комплекса в растворе, а не металла, реакции с палладием можно сделать каталитическими. А реакции магния – нельзя. И второе отличие – связи в комплексах магния (а гриньяр – это именно комплекс Mg(2+)) в основном ионные, потому что магний – непереходный металл, у которого просто нет валентных возможностей для образования четырех настоящих связей. Ионные связи ведь отличаются от ковалентных и координационных тем, что связанные атомы не привязаны друг другу ничем, кроме кулоновского притяжения, и в растворе они могут более или менее свободно “гулять”, по капризу меняя партнеров и вообще забыв, кто с кем был связан в момент рождения. Это очень важно, потому что именно тот факт, что в комплексах палладия и других переходных металлом лиганды гораздо прочнее связаны с металлом, определяет, почему они, переходные металлы, с такой легкостью вступают в реакции – энергии образующихся связей в комплексе компенсируют затраты энергии на реакцию, на разрушение старых связей. И еще, из-за того, что связи в комплексах переходных металлов обычно не ионные, следует то, что у них совсем другая реакционная способность. У гриньяра все ясно – и с фенилом связь ионная, поэтому фенил в гриньяре – просто карбанион, сильнейший нуклеофил. А в комплексе палладия связь палладий-углерод вообще почти неполярная, и фенил не проявляет никаких признаков карбаниона. И даже о нуклеофильности мы вспоминаем больше по аналогии, потому что никаких признаков нуклеофильности у фенила в комплексе палладия нет. Комплекс прочен, его легко можно выделить, он не обращает никакого внимания на воду и даже кислоты, и судьба его будет совсем иной, чем судьба гриньяра, который бурно прореагирует с первым попавшимся электрофилом. Еще раз убеждаемся, что в химии внешнее сходство обманчиво, хотя и дает некоторые полезные аналогии.
Переметаллирование
Возвращаемся к комплексу палладия. Теперь этот комплекс может реагировать с органическими нуклеофилами, в роли которых обычно выступают металлоорганические соединения. Самое простое – как раз гриньяр. Обратим внимание, что пока палладий был в форме комплекса Pd(0), он не реагировал с нуклеофилами. Это совершенно естественно, так как полностью соотвествует нашей любимой мантре: “Нуклеофил не реагирует с нуклеофилами”. Pd(0) ведь тоже нуклеофил – это частица с большим количеством доступных для образования новых связей электронов, ведь металл этот находится в 10 группе и обладает полным набором d-электронов, причем в комплексе Pd(0) эти электроны фактически сидят без дела. А вот в комплексе Pd(2+) есть лиганд, галогенид, и есть связь Pd-Br, которая вполне может реагировать с нуклеофилом и образованием нового комплекса, тоже Pd(2+), но уже с двумя углеродными лигандами. ![]()
Такие реакции в химии металлов принято называть переметаллированием. Имеется в виду неочевидный факт – в результате этой реакции из одного комплекса некоторого лиганда R с одним металлом (в этом примере магнием), получается комплекс того же лиганда с другим металлом, в данном примере палладием.
В результате последовательности реакций окислительного присоединения и переметаллирования в координационную сферу палладия входят два органических лиганда. И тогда происходит самое интересноеэ
Восстановительное элиминирование
С реакцией восстановительного элиминирования мы очень хорошо знакомы, даже если понятия о ней не имеем. Она происходит, если на металл в положительной степени окисления садятся два лиганда, и металл оказывается способен их окислить, отнять у каждого по электрону, то есть забрав себе пару и заодно уменьшив степень окисления на два. Лиганды, оставшись каждый с одним электроном, просто образуют связь и новую молекулу. Именно это, например, происходит во всем известной реакции получения хлора из перманганата и HCl: хлориды садятся лигандами на Mn(7+), который их ровно так и окисляет, атомы хлора образуют молекулу хлора, а марганец восстанавливается до Mn(5+), с которым дальше происходят всякие превращения. Палладий(2+) – не марганец(7+), это намного более слабый окислитель. Поэтому он, например, не окисляет галогенид, а ждет, когда он заменится на гораздо легче окисляемый углеродный остаток, и пару углеродных игандов палладий окислить может очень легко. И вот, образуется органический продукт, продукт кросс-сочетания, так как два разных орагнических остатка в результате оказались связаны в новой молекуле.
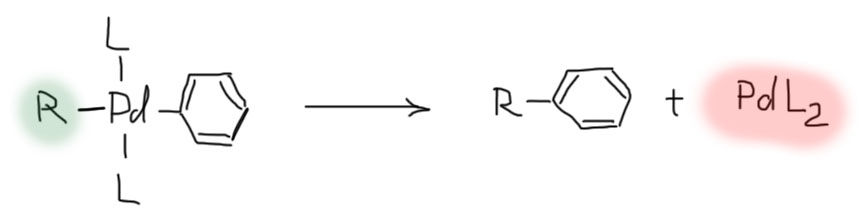
И комплекс Pd(0), как две капли воды похожий на тот, который двумя стадиями ранее вступал в окислительное присоединение. И этот комплекс может вновь вступить в эту реакцию.
И так много раз. Теоретически, пока в реакционной смеси не закончатся органический электрофил и нуклеофил. Практически все-таки намного раньше, потому что всегда есть побочные реакции, в которых расходуется катализатор. Обычно одна молекула комплекса палладия способна участвовать в нескольких сотнях, иногда тысячах последовательных реакций.
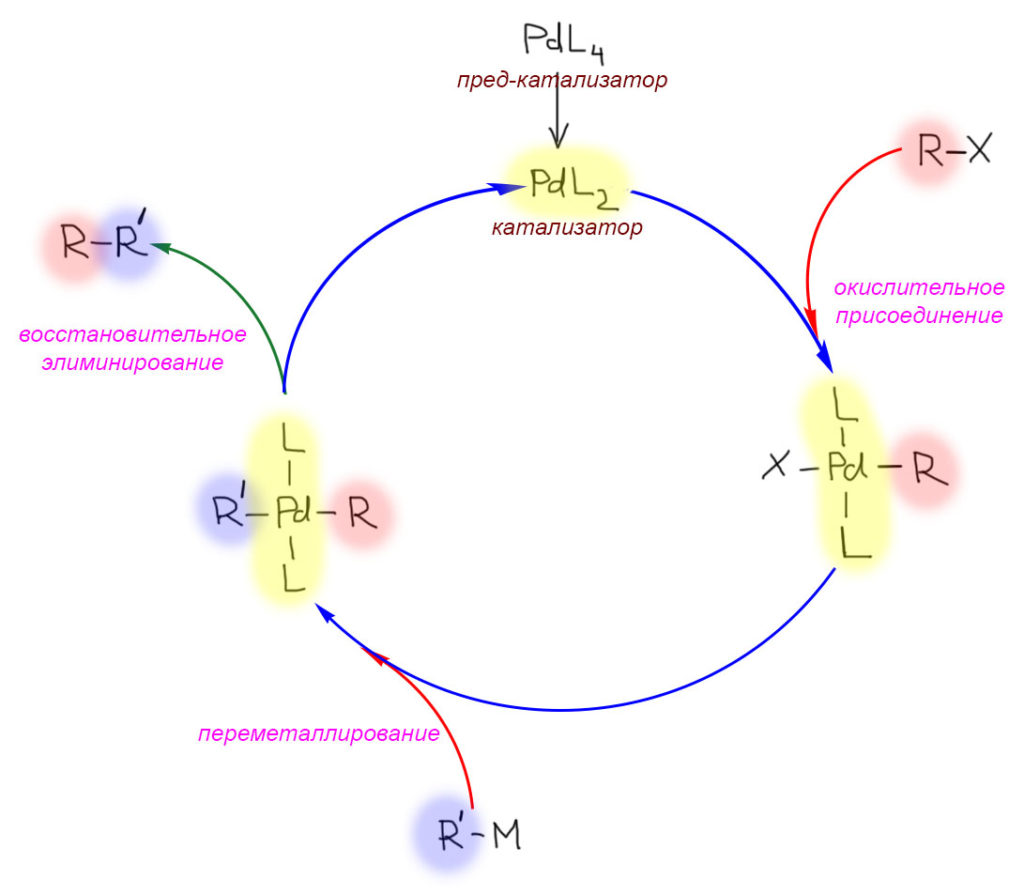
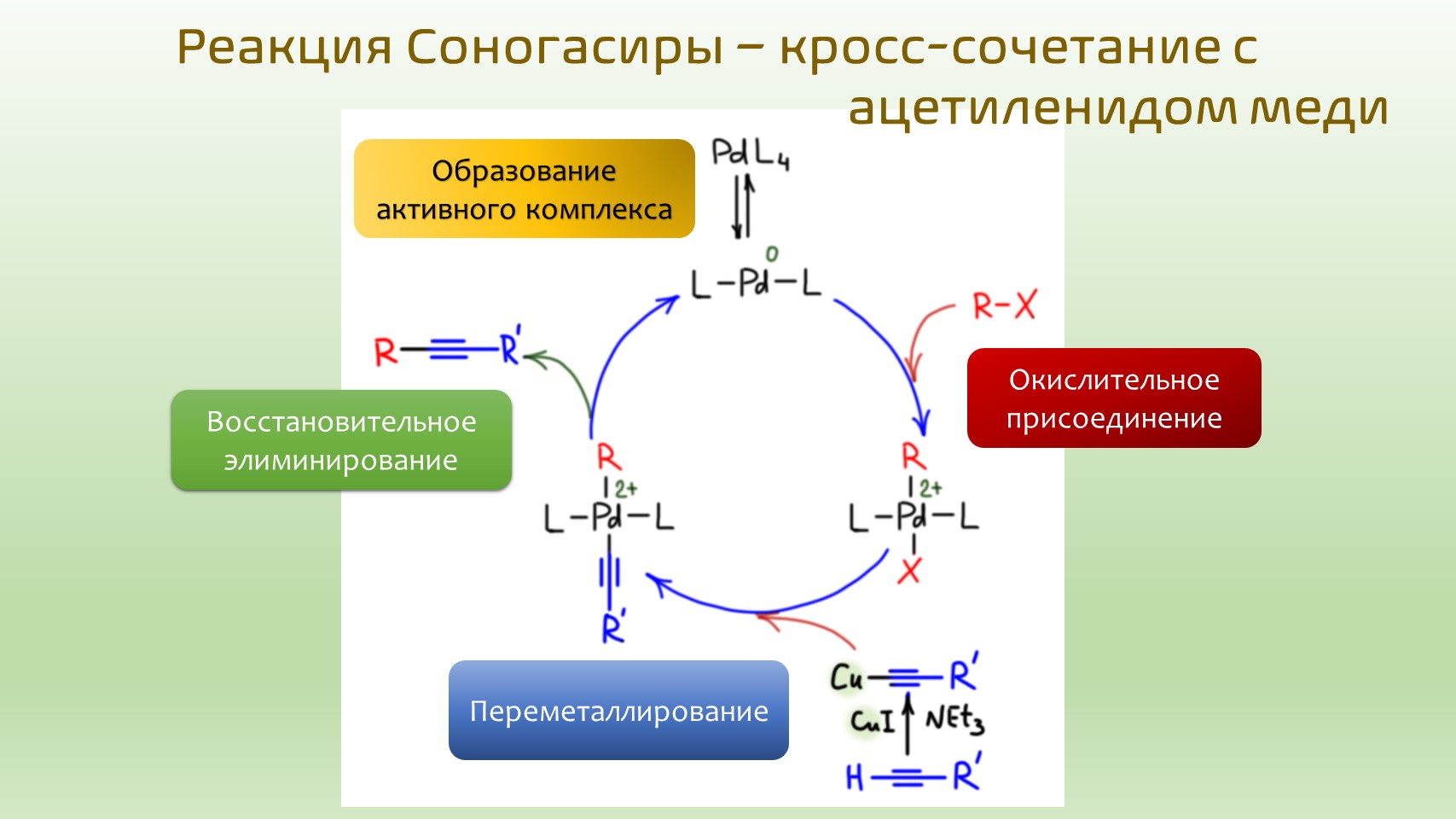
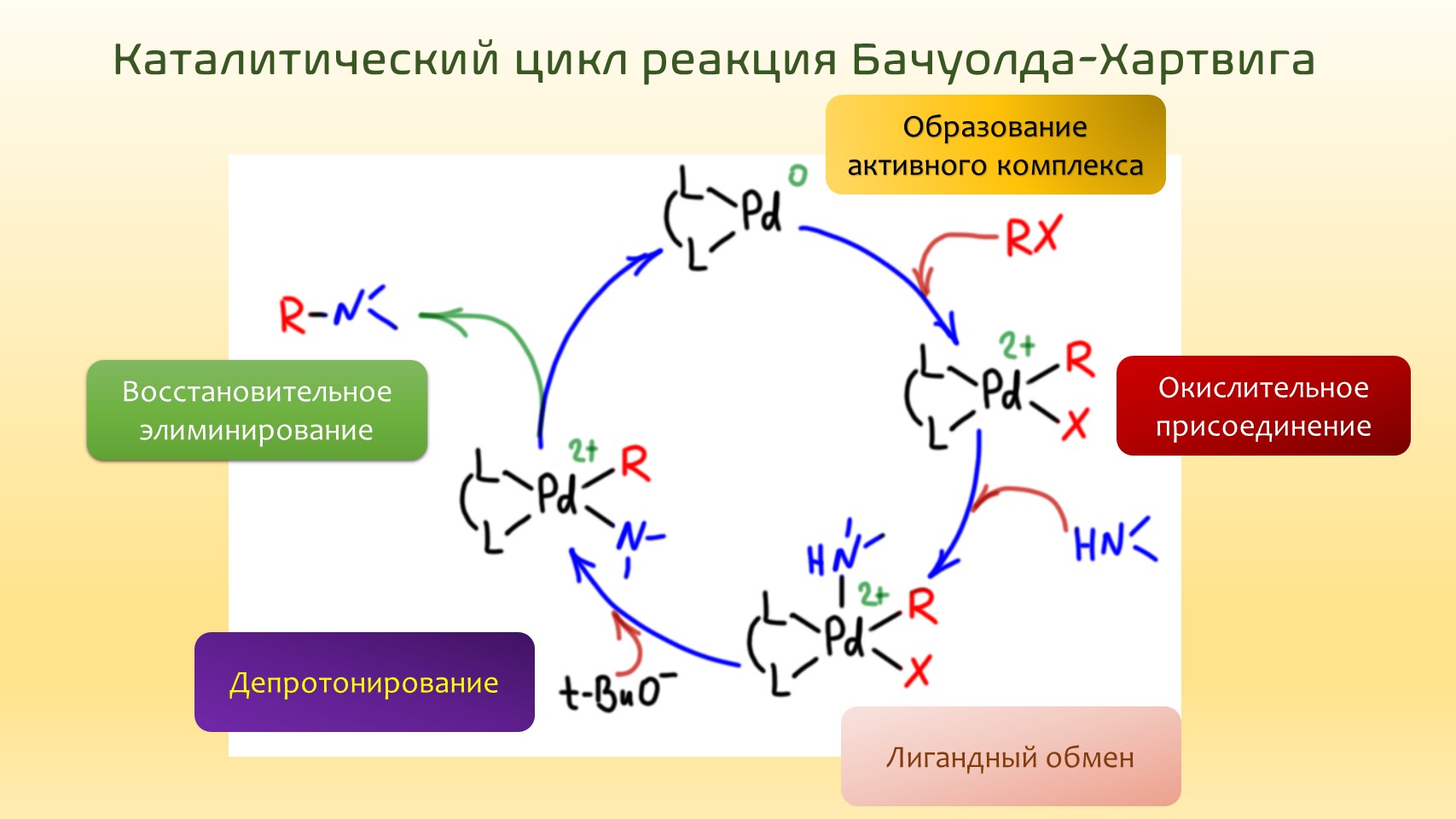
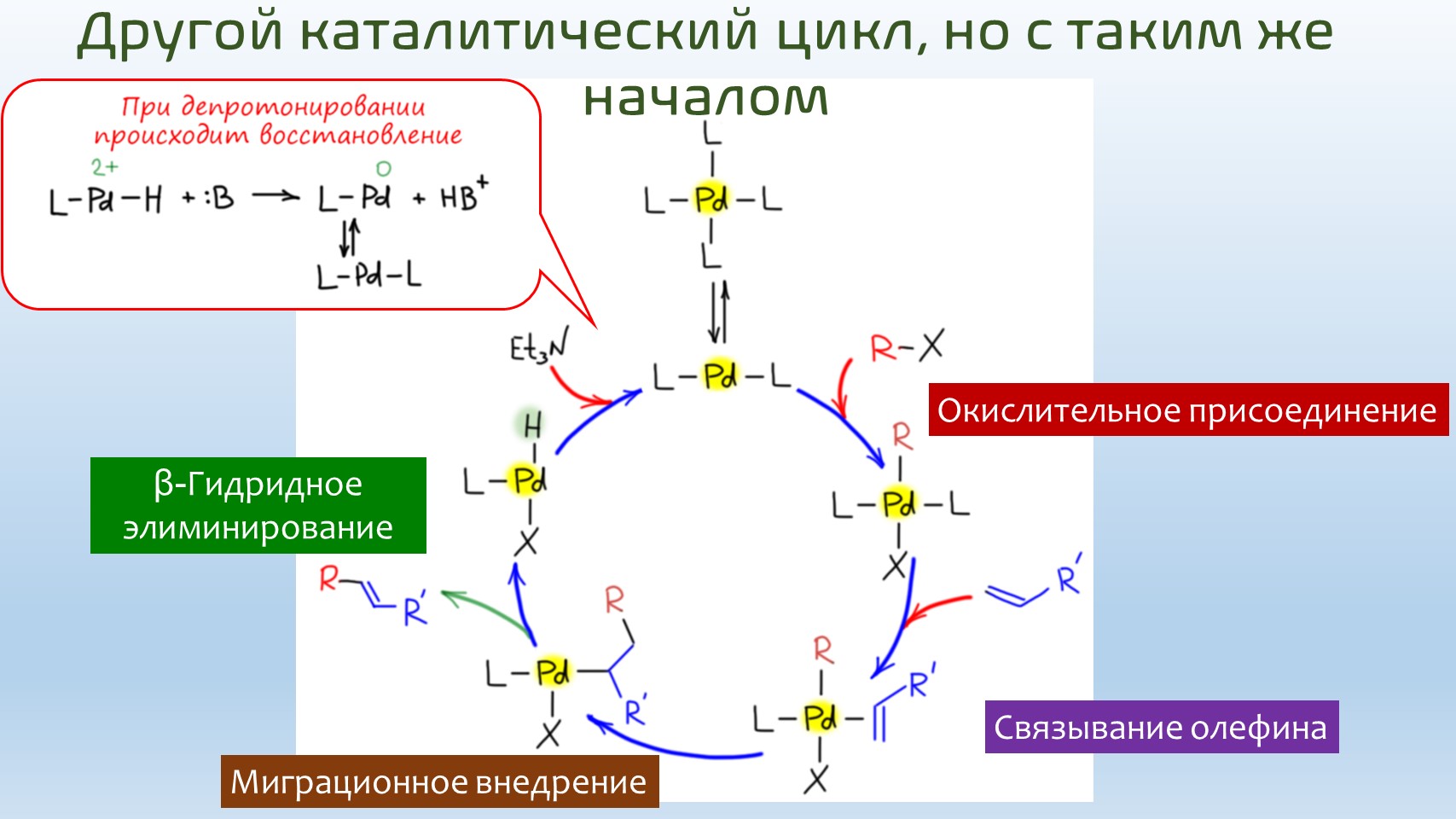
Кросс-сочетание: каталитический цикл
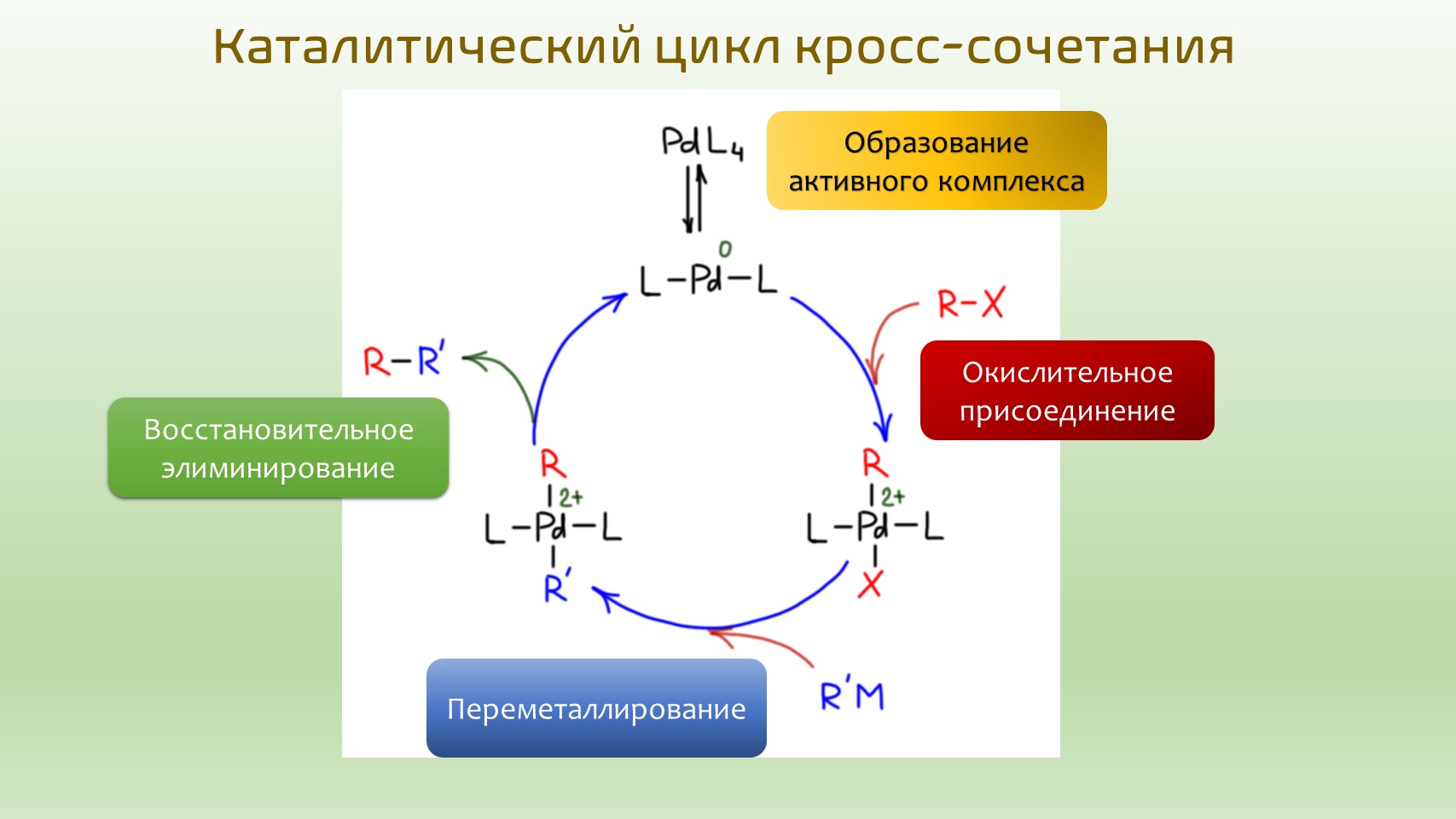
Стадии каталитического процесса принято рисовать не в строчках, а расположив по окружности, показывая катализатор и все промежуточные комплексы. Исходный комплекс, как мы выяснили, для того, чтобы принять участие в каталитическом процессе, должен сначала превратиться в каталитически-активный комплекс. Берем, например, с полки комплекс палладия с четырьмя трифенилфосфинами, и в растворе, при нагревании он самопроизвольно и равновесно превращается в комплекс с двумя фосфинами.
Исходную форму комплекса называют или катализатором, или, точнее, пред-катализатором.
Комплекс, непосредственно участвующий в реакции, называют катализатором или активной формой катализатора. Некоторая путаница в этом есть, но обычно из обсуждения становится ясно, что имеется в виду в каждом случае. Проблема здесь в том, что то, что мы кладем в колбу, заведомо известно. А то, что непосредственно участвует в реакции, известно гипотетически, с некоторой долей сомнений.
Внешними стрелками, идущими внутрь цикла (у меня красными), показывают реагенты, которые вводят в реакцию на каждой стадии. Стрелкой наружу (у меня зеленой) показывают образование продукта реакции.
Вот типичный каталитический цикл кросс-сочетания. Катализатором является комплекс Pd(0). Сначала он реагирует с органическим электрофилом в окислительном присоединении – в координационную сферу входит первый органический остаток. Затем получившийся комплекс реагирует с органическим нуклеофилом в переметаллировании – в координационную сферу входит второй органический остаток. Получившийся комплекс в восстановительном элиминировании дает продукт реакции и возвращает в цикл активную форму катализатора.
Реакция кросс-сочетания между органическим электрофилом и органическим нуклеофилом является каталитической – она использует каталитические количества комплексов палладия или никеля. Считается это так. Допустим, мы взяли по 1 молю органических реагентов и хотим получить 1 моль продукта кросс-сочетания. В реальности мы получим меньше, потому что выход 100% бывает только в статьях некоторых китайских химиков (в современном Китае полно отличных химиков, но до сих пор иногда встречаются любители малость приврать). Просто для примера предположим, что мы получили выход 80% (в этом месте мы предполагаем, что этот выход получился, потому что реакция прошла на 80%, а не потому что она прошла на 99%, а все остальное мы просто потеряли при выделении продукта). И использовали в реакции 0.01 моль комплекса палладия. Это количество катализатора часто обозначают в мольных процентах относительно загрузки основного реагента, определяющего стехиометрию реакции – 0.01 моль делим на 1 моль – это 1 мольный процент (1 моль%). За один оборот каталитического цикла 0.01 моль катализатора произведет 0.01 моль продукта (в каталитическом цикле стехиометрия палладий:RX = 1:1). А всего он, 0.01 моль палладия, произвел 0.8 моль продукта. Значит ему удалось 80 раз (0.8/0.01) пробежать цикл, или можно сказать, что произошло 80 оборотов цикла. По-английски это так и называют – turn-over или просто turnover. А число оборотов называют turnover number, сокращенно TON (всегда большими буквами). Итак, в нашем примере TON = 80. Неплохо, но бывает и больше. Это очень важная характеристика каталитических реакций, и исследователи, разрабатывая новые методы, соревнуются, кто больше оборотов заделает.
Кросс-сочетание: реакции
В реакции кросс-сочетания всегда берут пару реагентов:
- электрофил, обычно органическое галогенпроизводное, и если не прибегать к специальным методам и катализаторам, это или бром- или иодпроизводное; хлорпроизводные в кросс-сочетании реагируют только в очень специальных условиях, а фторпроизводные не реагируют вовсе;
- если не искать себе приключений, то лучше еще и ограничиться только таким бром- и иодпроизводными, в которых эти галогены присоединены к атому углерода в sp2-гибридном состоянии, то есть, или ароматические (в том числе гетероциклические) галогенпроизводные, или галогеналкены;
- sp-гидридный углерод в виде бром- или иодалкинов R-C≡C-X (X = Br, I) тоже годится, но беда в том, что мы не умеем получать такие производные алкинов;
- а вот sp3-гибридного углерода, то есть галогенпроизводных с галогеном на насыщенном атоме углерода бойтесь как огня, и не важно, что там у них еще есть – все это очень специальная химия с кучей отвратительных подробностей;
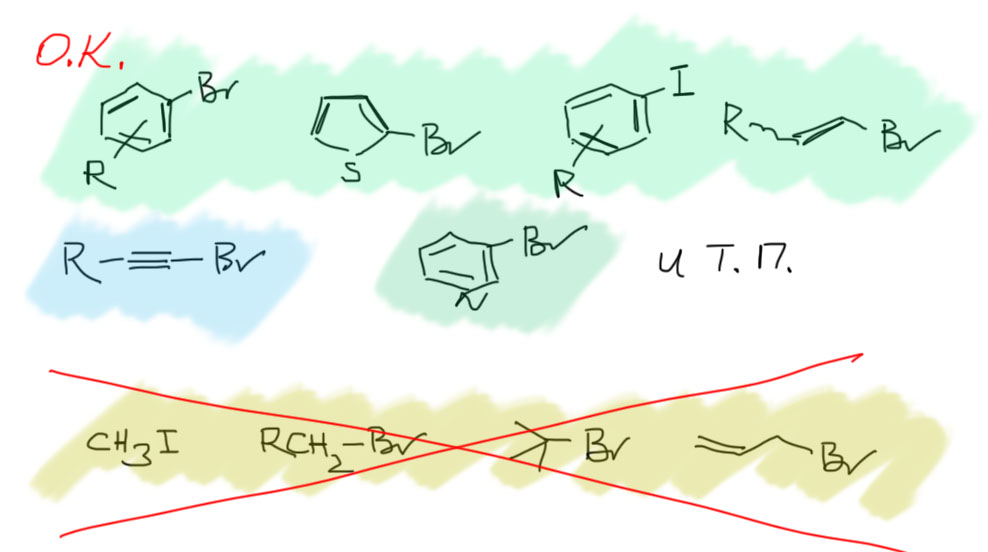
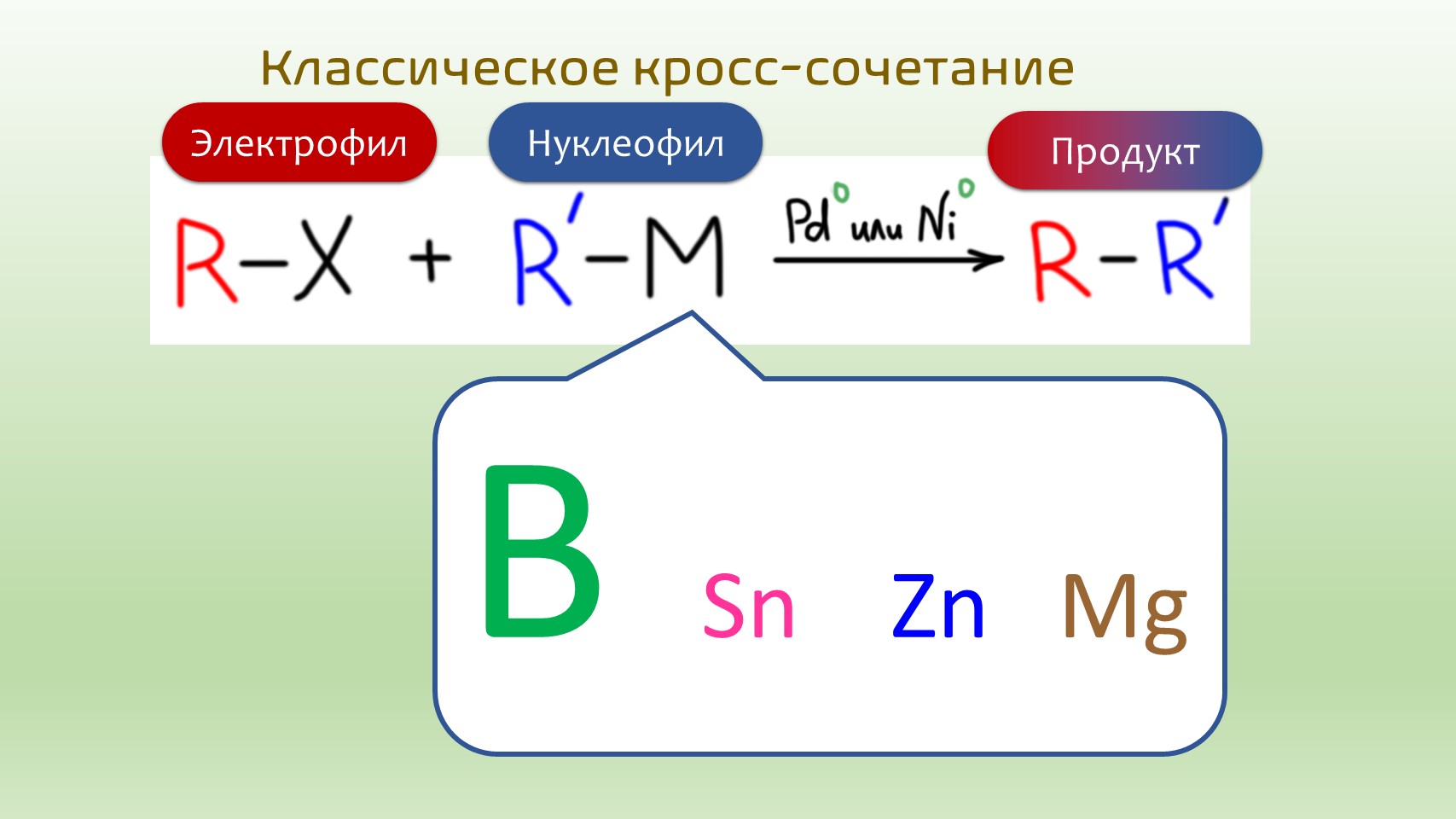
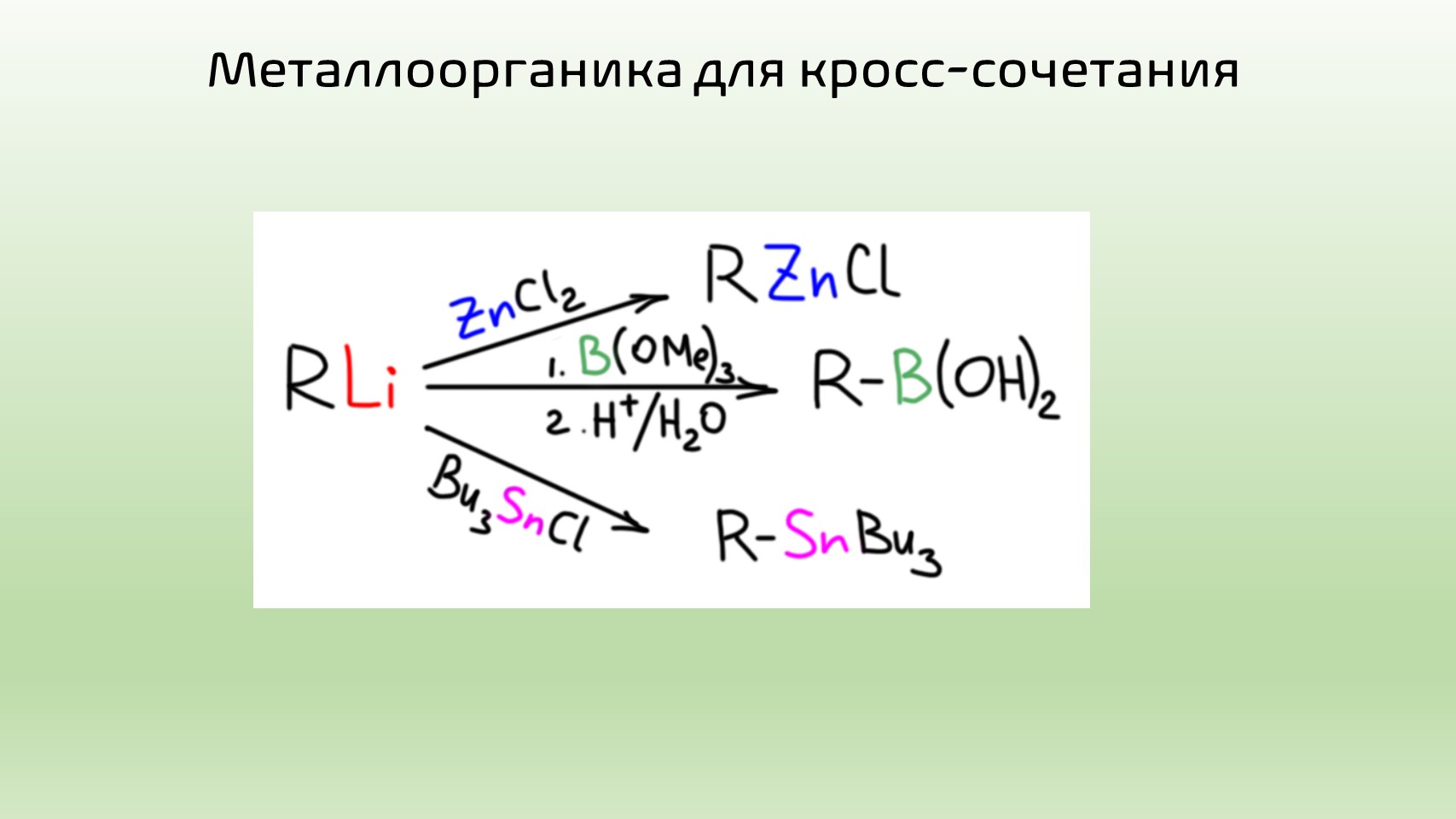
- нуклеофил, в роли которых выступают металлоорганические соединения, обычно производные непереходных металлов и металлоидов, причем эта химия устроена так, что выбирают производные с умеренной реакционной способностью, не слишком активные, и не слишком ленивые. Беда в том, что мы совсем не знаем металлоорганической химии, и оценки эти нам недоступны. Поэтому придется поверить на слово, что картина выглядит так, что
- слишком реакционноспособны литийорганические соединения и то, что мы открыто называем карбанионами и то, что получается прямым депротонированием CH-кислот – все это не годится для кросс-сочетания без использования каких-то особо извращенных методов, которые нам даром не нужны;
- в серединке находятся производные бора, цинка, олова, магния и именно их мы и будем использовать;
- еще очень полезный элемент медь, но его почти никогда не используют напрямую, поэтому мы его использовать не будем, потому что это потребовало бы множества подробностей и специфических знаний;
- все остальное не годится; нам очень трудно будет понять, почему используется один элемент, и не используются его ближайшие родственники, но это так; например, почему бор это хорошо, а алюминий нет; цинк хорошо, а кадмий и ртуть нет; олово хорошо, а свинец и германий нет.
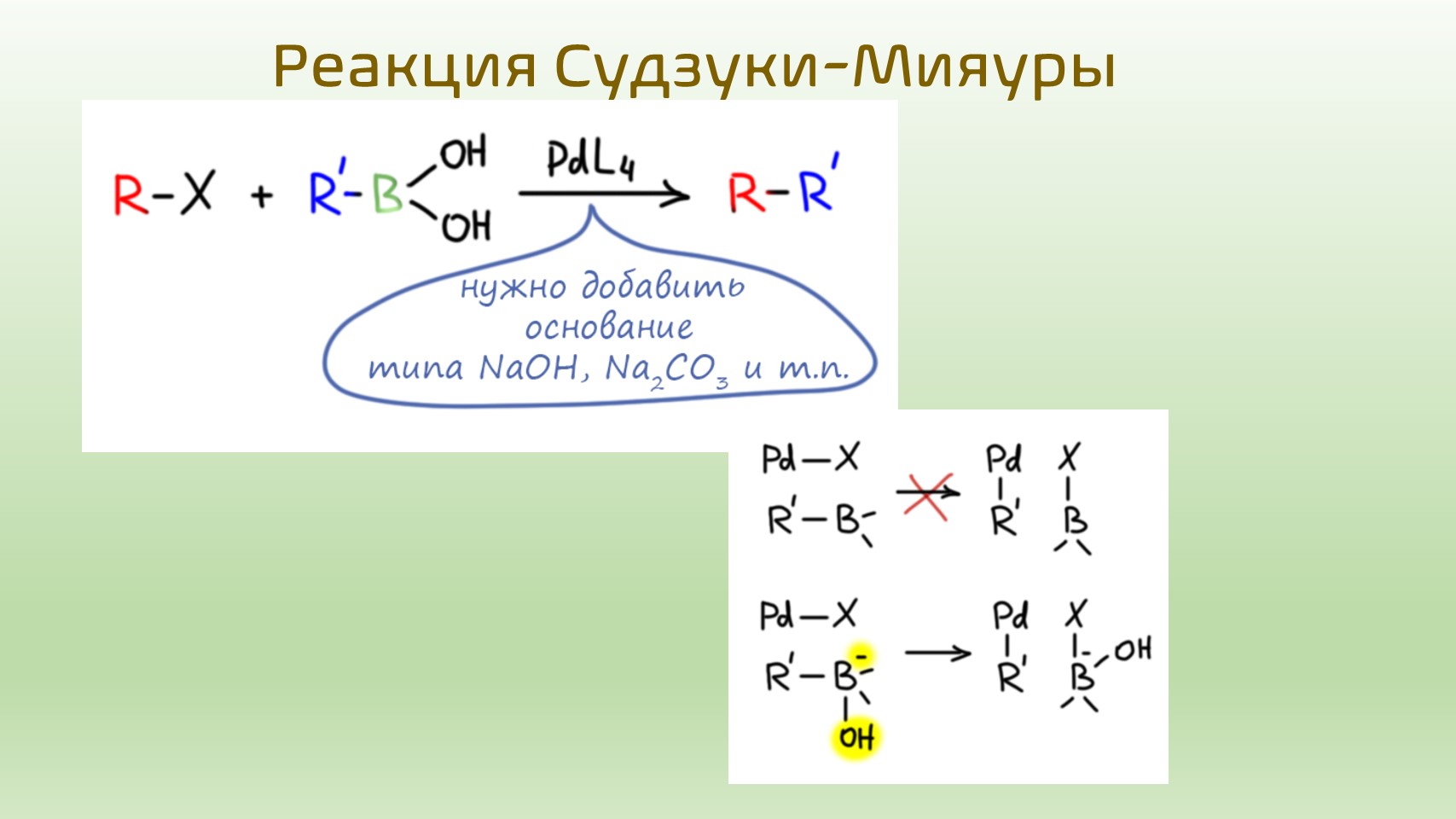
Кросс-сочетание с бороорганическими соединениями: реакция Судзуки
Борорганические соединения – безусловный лидер в химии кросс-сочетания. Их превосходство над всеми остальными металлоорганическими нуклеофилами (нет смысла возмущаться тем, что бор – не металл, а неметалл, его органические производные все равно числятся по ведомству металлоорганики, по крайней мере, в английском языке, хотя в русском языке есть корявое слово “элементоорганические” соединения специально для органических производных металлоидов) не просто подавляющее, а неприлично ощеломляющее. В принципе, все остальное можно даже не вспоминать.
Почему так прославились борорганические соединения в кросс-сочетания. По нескольким причинам.
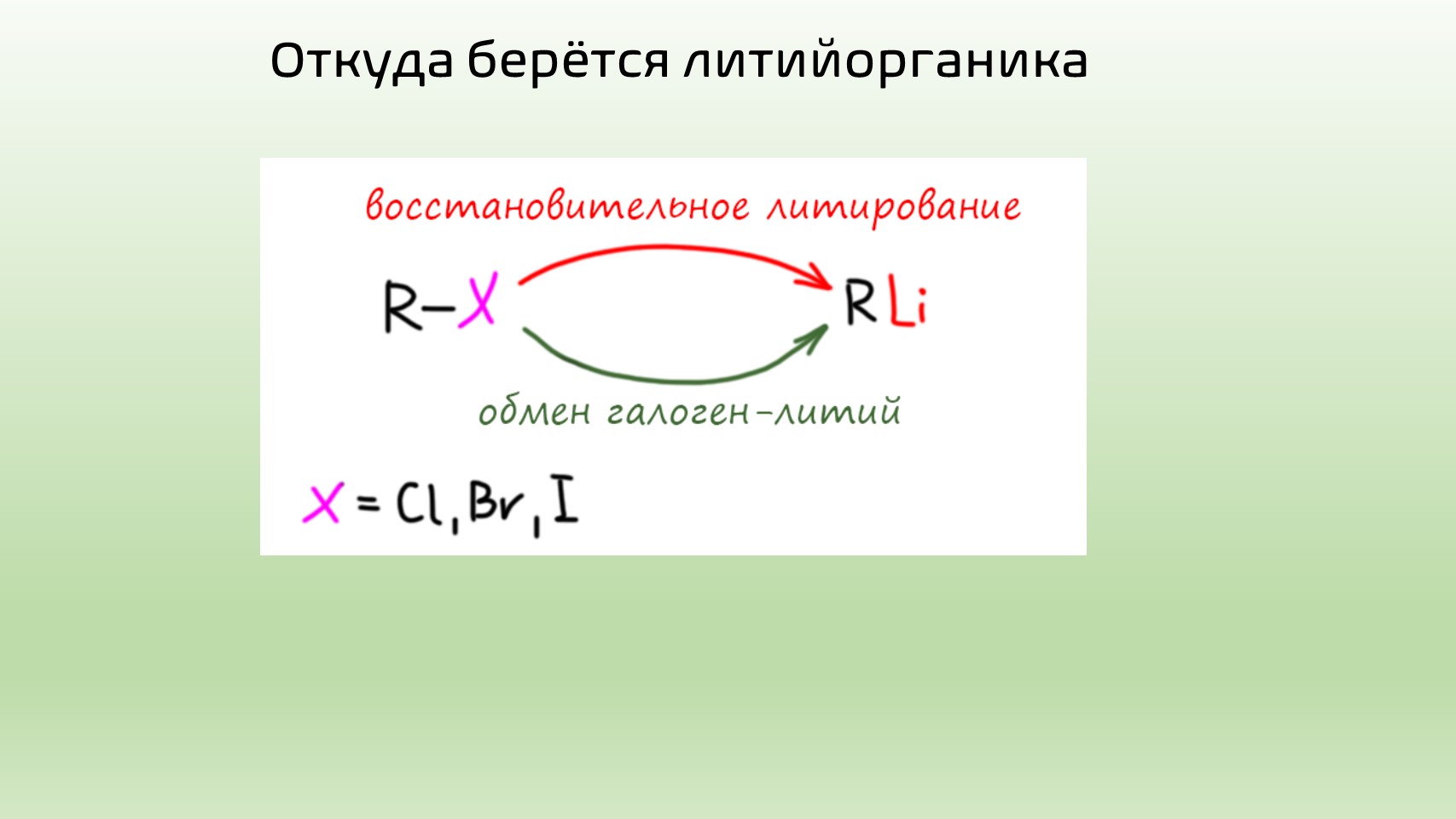
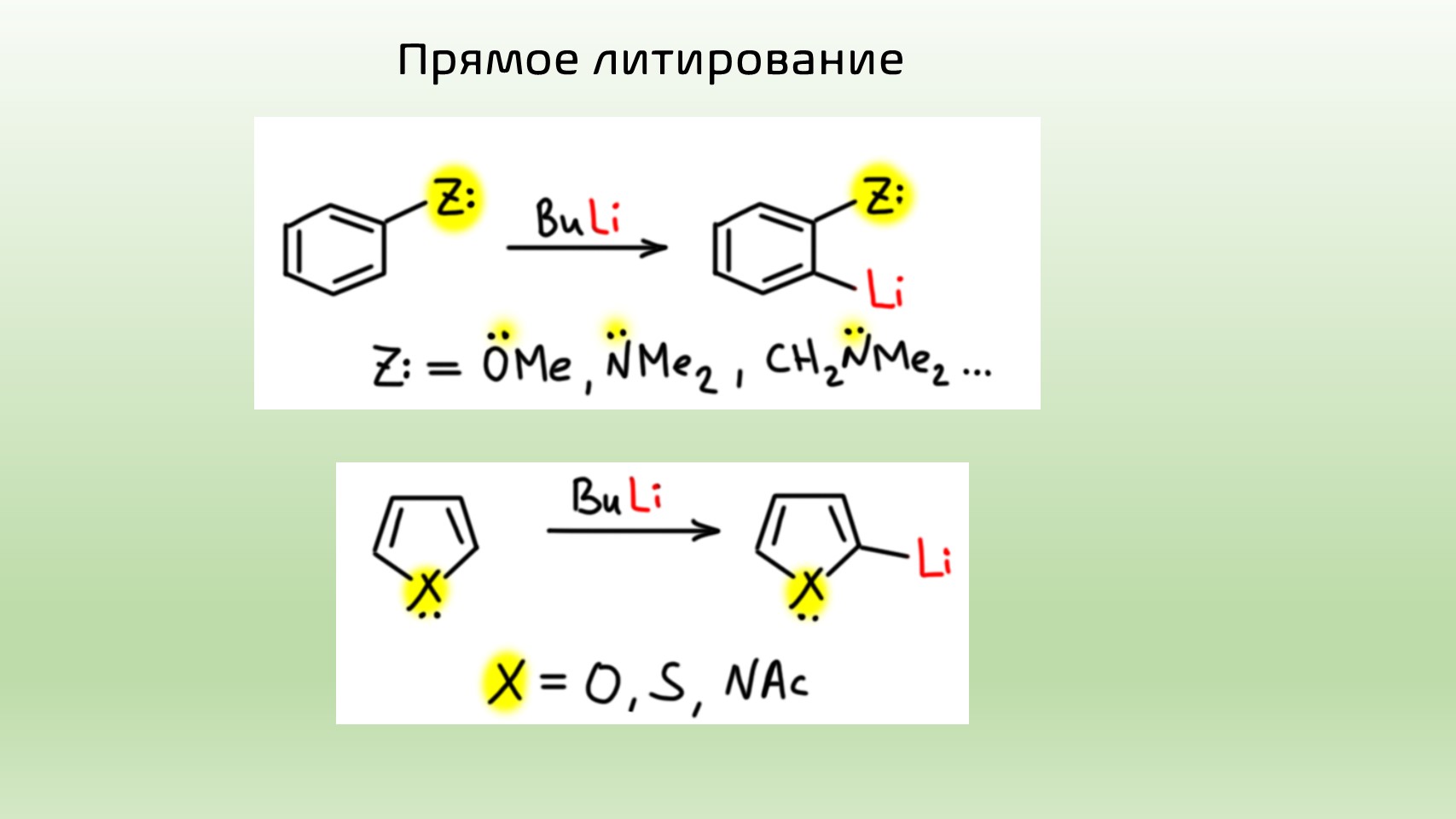
- они легко получаются просто невероятным количеством разных методов буквально из всего на свете. Мы, например, умеем гидроборировать алкены и алкины. Еще более распространены борные (точнее, бороновые) кислоты, которые можно сделать из галогенпроизводных или традиционным методом из магний или литийорганических соединений, или современным борилированием тоже в присутствии палладиевого катализатора (для этого требуется более сложный фосфиновый лиганд бис(дифенилфосфино)ферроцен, сокращенно dppf).
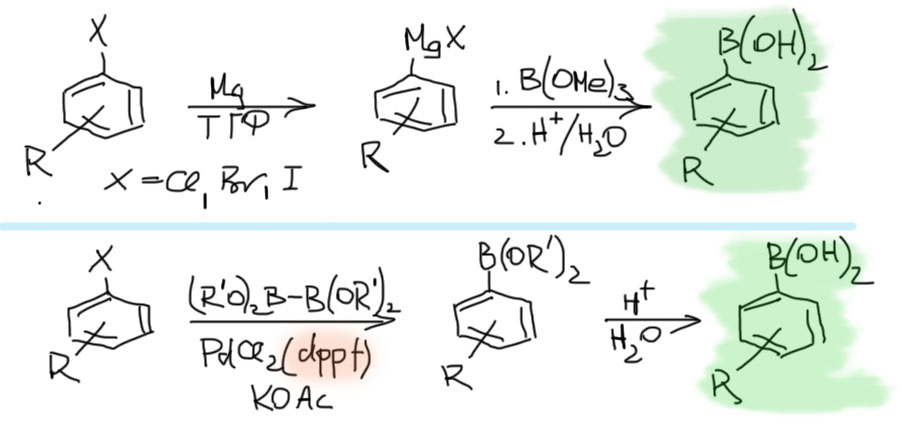
- борорганические соединения могут иметь с молекуле практически любые заместители и функциональные группы, хотя для получения таких борорганических соединений придется подобрать подходящий метод и не допускать ошибок, например, если мы получаем борную кислоту традиционным методом, мы должны соответствовать жестким ограничениям магний- и литиорганических соединений. Современный метод борилирования практически лишен этих ограничений.
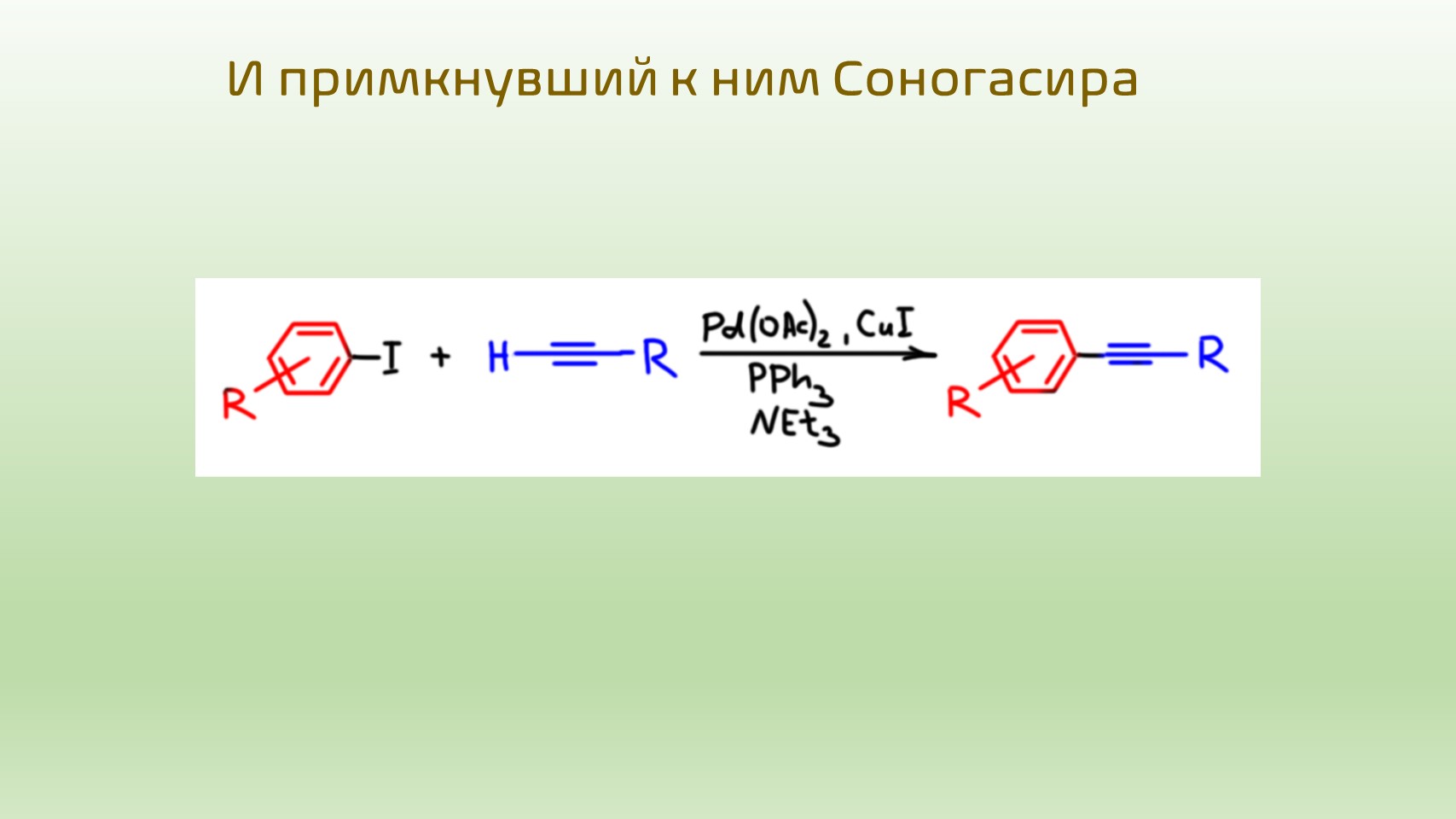
- реакции кросс-сочетания с борорганическими соединениями очень легко идут, и практически не ограничивают заместители и в молекуле галогенпроизводного. Эту реакцию делают довольно просто, и единственно, что требуется кроме электрофила, нуклеофила и катализатора, это очень простое основание типа щелочи или карбоната щелочного металла. Растворитель годится почти любой, и можно по этому поводу вообще не париться. Такая реакция кросс-сочетания называется реакцией Судзуки (именно так! – не Сузуки и не Сузука), а в более свежей литературе (самой этой реакции уже скоро стукнет полтинник) – Судзуки-Мияуры.
Примеры реакции Судзуки
Посмотрим несколько типичных примеров,
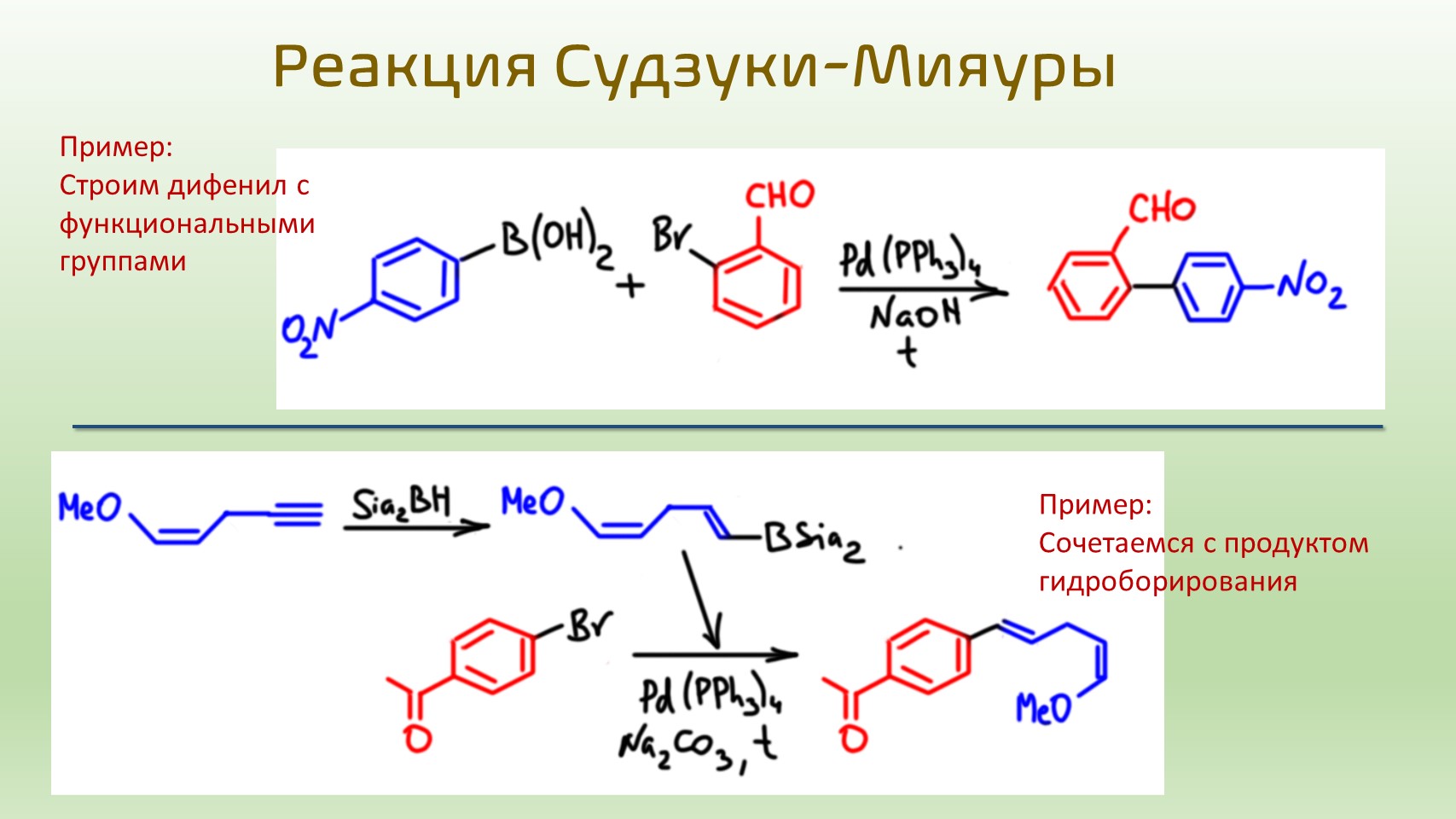
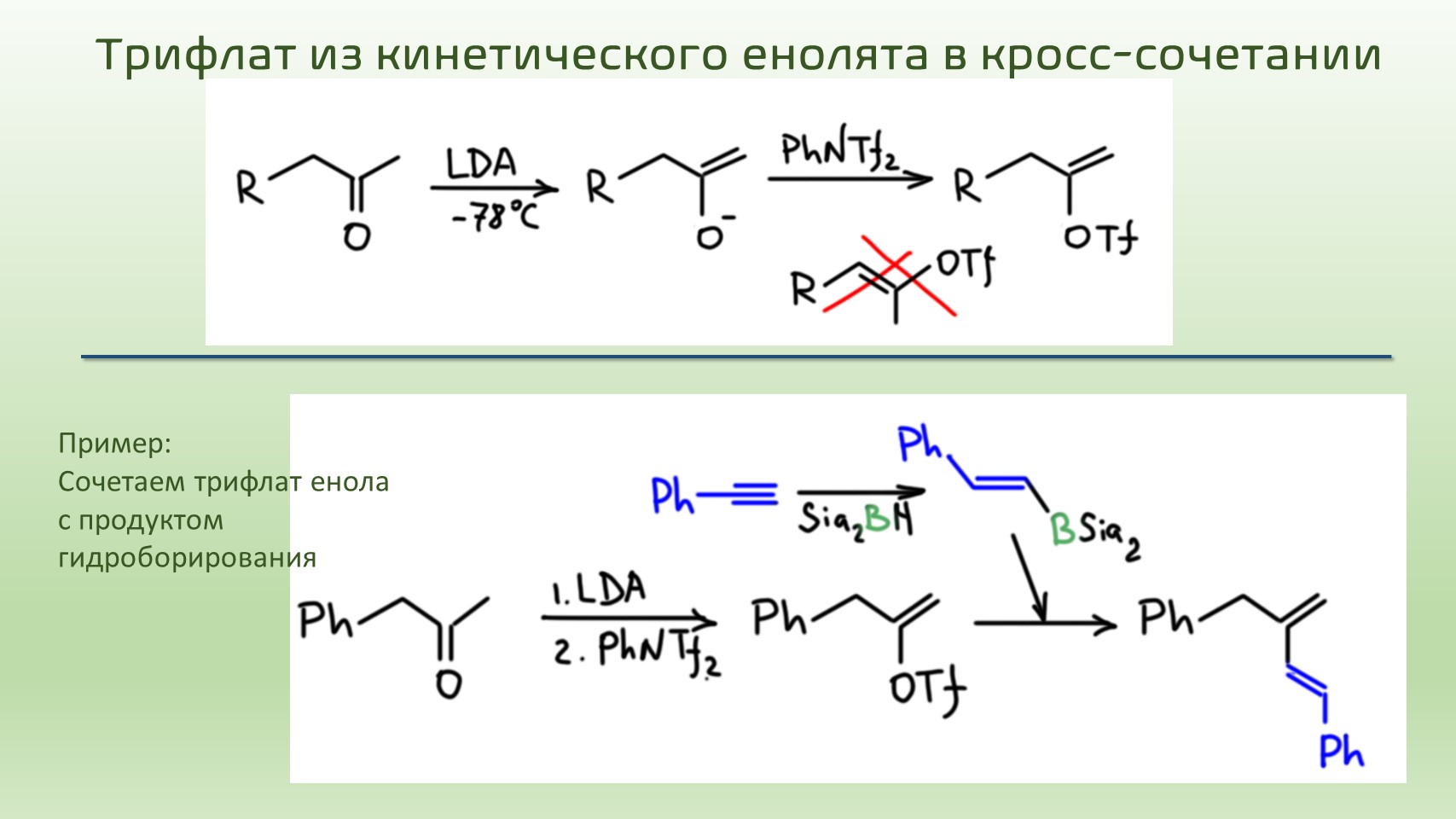
1. Соберем диен, исходя из ацетиленов. Один гидроборируем, второй превращаем в бромпроизводное тоже через гидроборирование. Обращаем внимание на то, что стереохимия исходных сохраняется в продукте, и из продукта гидроборирования 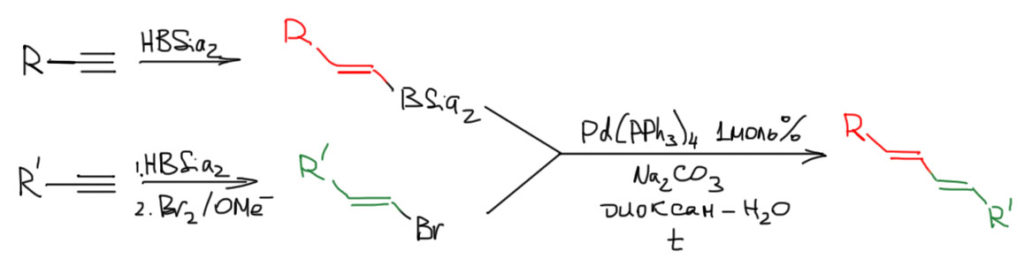
2. Собираем замещенный стирол из ацетилена и бромпроизводного. Для примера используем бромпроизводное с заместителем.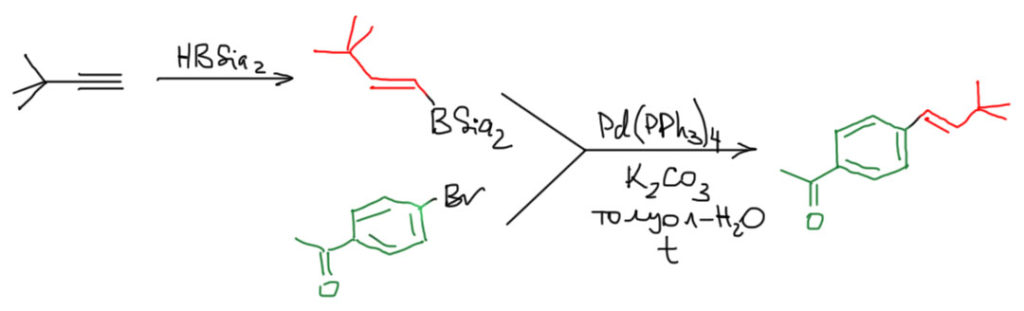
3. Собираем дифенил с двумя разными заметителями. Для этого нам потребуется борная кислота. Сделаем ее из той части дифенила, где нет заместителей, несовместимых с реактивом Гриньяра, в данном случае это метокси, простой эфир. Еще одна распространенная вещь – вместо комплекса Pd(0) используем комплекс Pd(2+). Хотя комплексы Pd(0) вполне доступны, все же они требуют осторожной работы, и при хранении понемногу окисляются. Комплексы Pd(2+) в этом смысле намного проще, это совсем стабильные вещества, которые даже в холодильнике хранить не нужно. А в реакционной смеси они быстро восстанавливаются в Pd(0) теми же борными кислотами, на что потребуется 1-2% этого реагента – потеря, которую никто не заметит. Так почему же их всегда не применяют? Потому что они не всегда хорошо работают, и это очень непростая проблема, а готовые комплексы Pd(0) почти всегда надежнее. Часто бывает так, сначала берут комплекс Pd(2+) и столкнувшись с низким выходом, начинают бегать в поисках комплекса Pd(0). 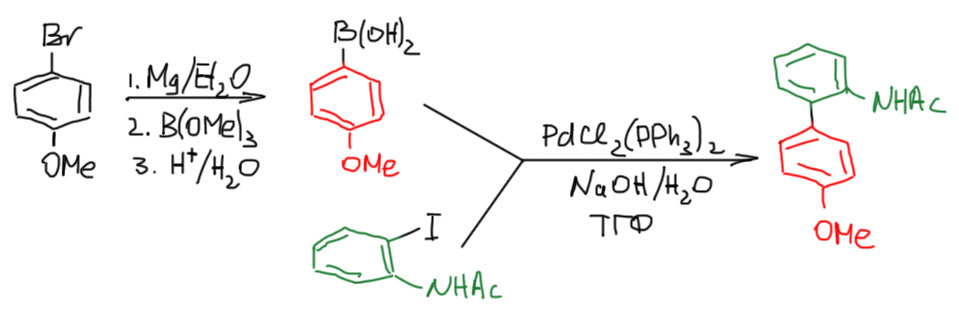
4. Собираем дифенил с такими заместителями, которые не позволят получить борные кислоты через гриньяр или литийорганику. В этом случае мы либо купим соотвествующую борную кислоту в магазине, но это не самое дешевое удовольствие, либо получим ее борилированием. Для борилирования нужен более сложный комплекс палладия, но бонусом будет то, что тот же комплекс будет катализировать и кросс-сочетание, нужно будет просто добавить основание. Такой способ выполнения реакций цепочкой, без выделения продуктов стадий, и с частичным использованием реагентов и растворителей, называют one-pot reaction (реакции в одном горшке), что, увы, совсем не переводится на русский. Это очень удобно, и синтетики очень любят такие фокусы. А почему же всегда так не делают? Потому что это намного дороже, и из-за лиганда dppf, и из-за эфира гипоборной кислоты 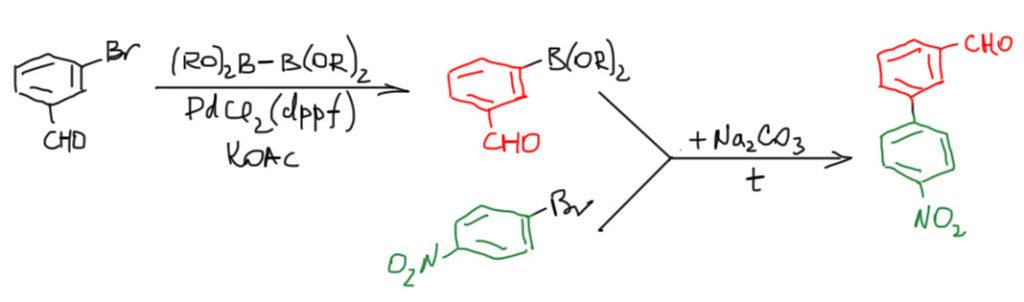
Производные борной гислоты, которые используют в этой реакции называют эфирами борной кислоты. В качестве спирта для этих эфиров обычно используют диолы, очень легко образующие циклические эфиры с борными кислотами. Самый популярный диол в этой реакции – хорошо нам известный пинакон. Поскольку это вспомогательный реагент, не будем рассматривать это подробнее, а если захочется подробностей, их можно найти на сайте transmet.avchem.ru.
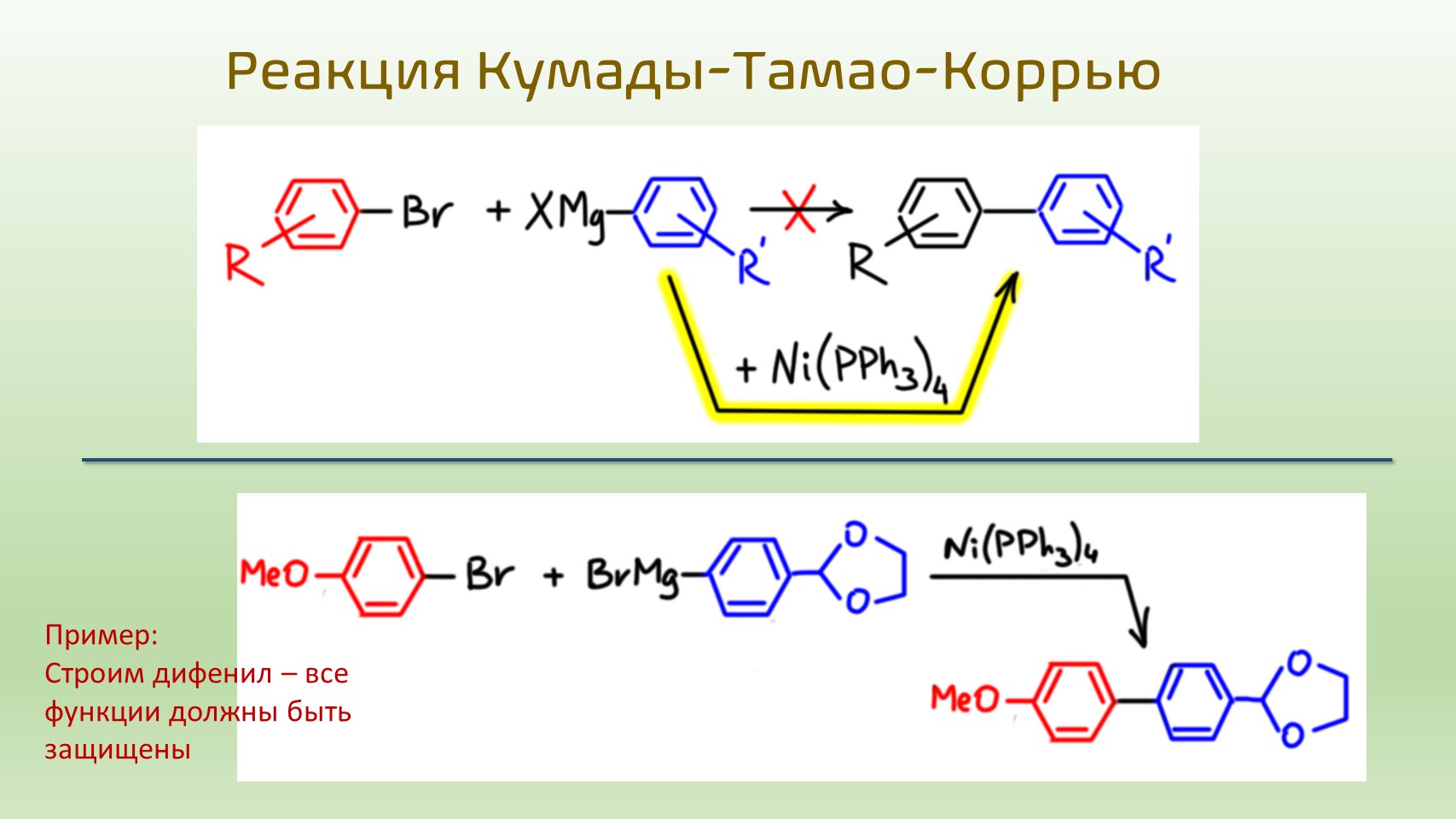
Кросс-сочетание с магний-органикой: реакция Кумады
Это исторически первая реакция кросс-сочетания, ушедшая в тень после открытия реакции Судзуки. Но иногда она удобна, особенно в тех случаях, когда нужно получать борную кислоту через реактив Гриньяра, тогде зачем ее получать, если можно сразу использовать гриньяр. Реакция еще удобна тем, что вместо комплексов палладия можно использовать комплексы никеля, и это сильно дешевле. Для этой реакции не нужны основания. Нужно помнить несколько вещей. Во-первых, не надо забывать, что гриньяр несовместим с большинством заместитей – в обоих реагентах, не только в самом гриньяре, но и в электрофильном компоненте кросс-сочетания. Во-вторых, никогда не заменяйте гриньяр литий-органикой – это не работает в кросс-сочетании.
В качестве примера получим дифенил из компонентов с заместителями, совместимыми с гриньяром – простым эфиром и третичным амином без кислых водородов. 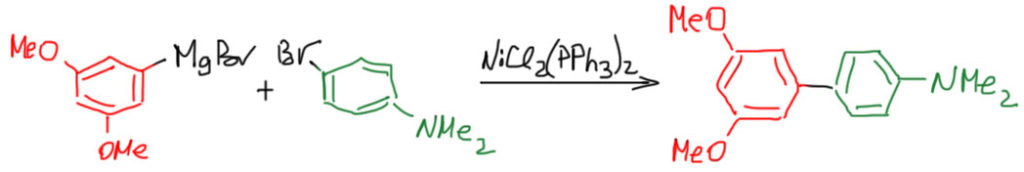
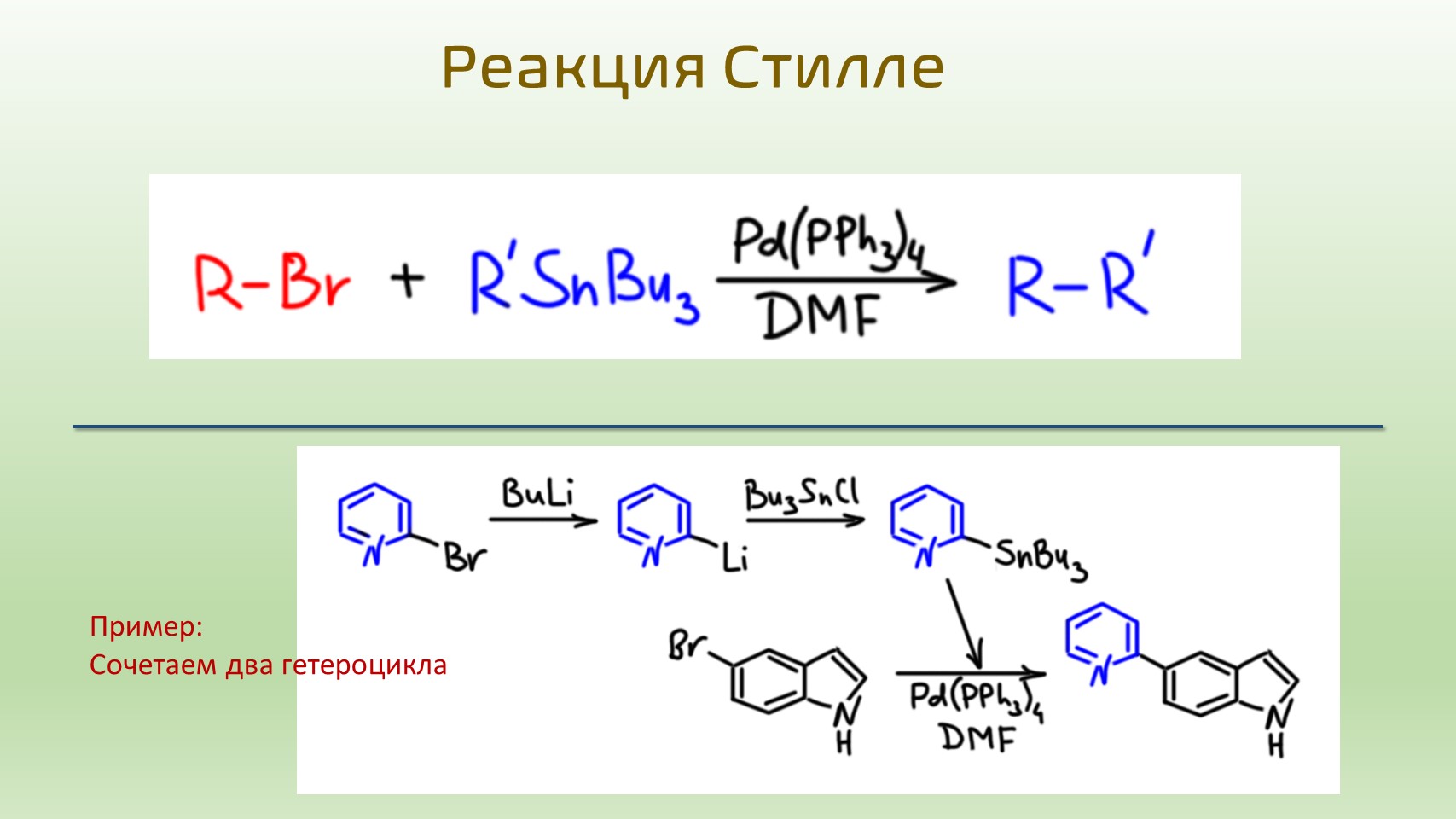
Кросс-сочетание с олово-органикой: реакция Стилле
Олово-органика страшно токсична, работать с ней тяжело и опасно. Но кросс-сочетание с олово-органикой, называемое реакцией Стилле, имеет своb небольшие, но важные области применения. Для синтезов, рассмотренных в кросс-сочетании с борограникой – диенов, стиролов, дифенилов – ее редко используют, но она очень популярна, когда один из компонентов кросс-сочетания – гетероцикл. По разным причинам, борорганика иногда не очень хорошо работает с гетероциклами, и тогда олово приходит на помощь. Олово-органику обычно получают через литий-органику, и используют без выделения. Реакцию еще любят за очень мягкие условия, она редко требует нагревания, в то время как реакции с борорганикой обычно проводят при нагревании. Иногда это важно. Посмотрим на пример: 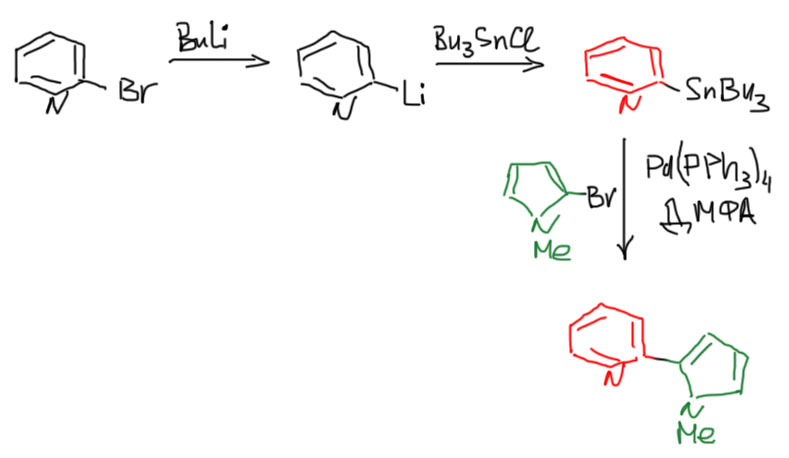
Почему используют производные трибутилолова, а не триметилолова – потому что последние дают более летучие производные, а это очень опасно.
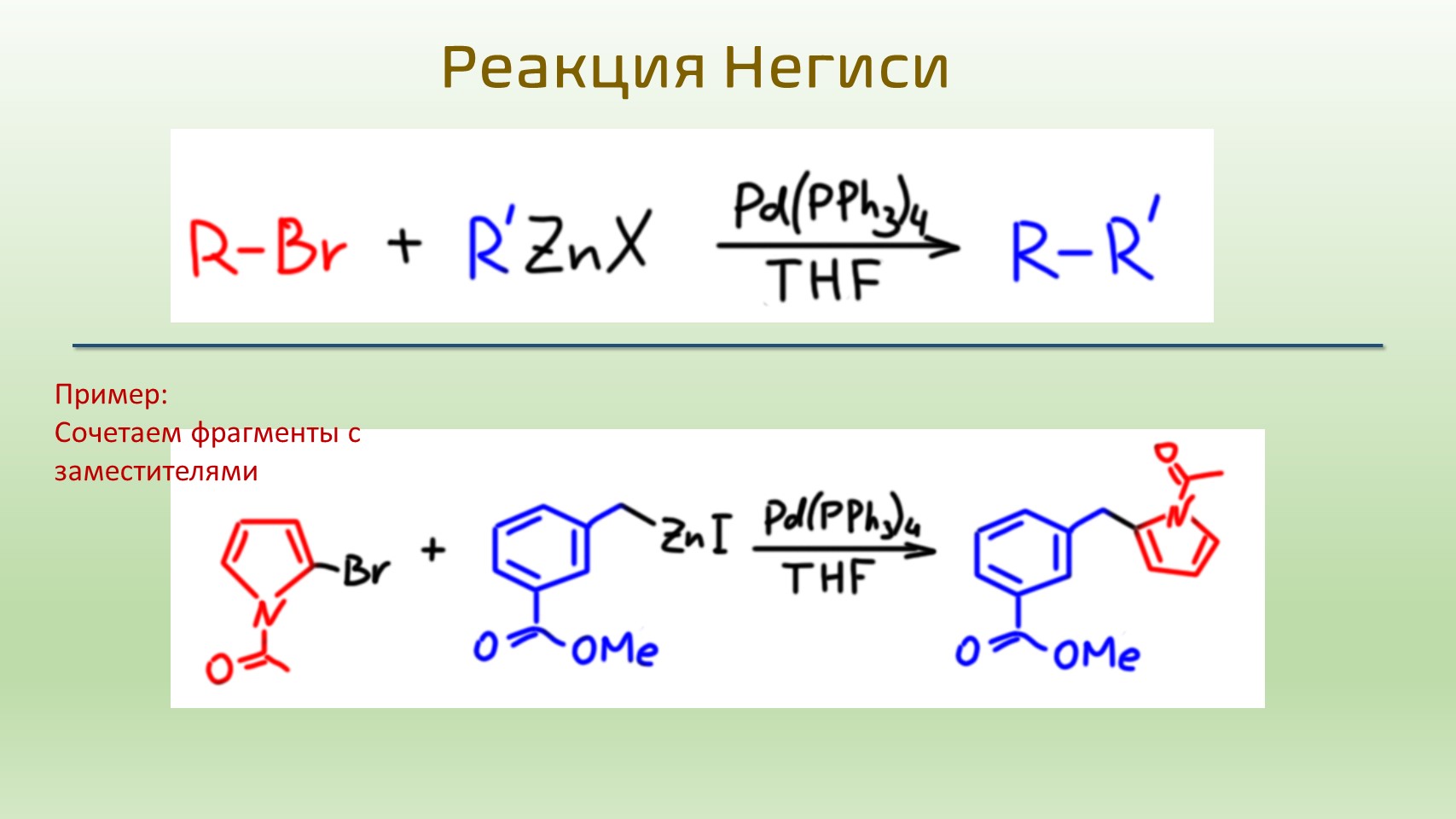
Кросс-сочетание с цинк-органикой: реакция Негиси
Из четырех классических реакций кросс-сочетания эта – самая специальная. На 3 курсе можно было бы совсем ничего о ней не знать, и мы ничего бы не потеряли. С ее помощью можно решить те же самые задачи, что и с теми, что мы уже разобрали. В синтезе ее любят использовать в сложных случаях, потому что она самая мягкая, почти всегда дает высокие выходы, не требует лишних реагентов, хорошо работает с гетероциклическими соединениями, не имеет проблем с токсичностью. Но раз мы сюда уже залезли, подметим одно важное отличие этой реакции от всех остальных – она лучше работает для сочетания через насыщенный атом углерода. Всякие алкилиодиды легко реагируют с активированным цинком: цинк в виде цинковой пыли активируют разными способами для того чтобы раскрыть поверхность металла – самый курьезный способ состоит в быстрой промывке цинковой пыли соляной кислотой – она начинает бурно растворяться, но кислоту быстро смывают водой, воду спиртом, спирт сухим эфиром, и такая пыль очень активна, если использовать ее немедленно после этой вполне спортивной – все нужно делать очень быстро и уверенно – процедуры. Один из занятных примеров применения кросс-сочетания с цинк-органикой – синтез новых аминокислот из природного L-серина, гидроксильную группу в котором заменяют на иод обычной SN2-реакцией, дальше получают цинк-органику и вводят ее в реакцию с самыми разными ненасышенными, ароматическими и гетероциклическими электрофилами. Стереохимия исходной аминокислоты сохраняется, и получаются новые L-аминокислоты. Обратите внимание, что реакция иодпроизводного с цинком – то же самое, что и реакция обарзования реактивов Гриньяра, но в отличие от гриньяров такая металлоорганика вполне совместима и разными функциональными группами – там и амид, и сложный эфир, и кислый водород. В этом смысле отличие цинк-органики от магний-органики особенно ярко и демонстративно.
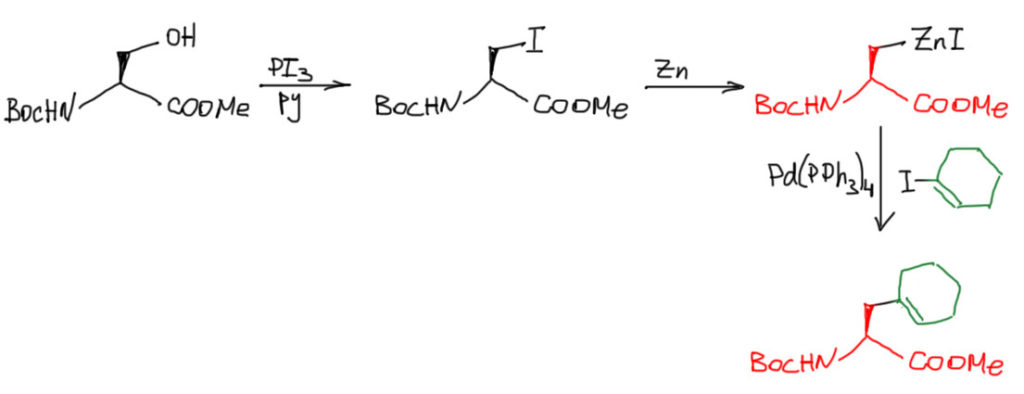
Синтез аминов реакцией кросс-сочетания
Реакции кросс-сочетания способны созавать не только связи между атомами углерода, но и связи углерода с другими атомами. Современная химия кросс-сочетания невероятно обширна, и уже, кажется, не осталось связей, которые невозможно сделать с ее помощью. А что такое вообще кросс-сочетание – не рекламный ли это лейбл, под которым понимают все, что угодно?! Честно говоря, это почти так и есть, но некоторые ограничения фантазии все-таки существуют. Во-первых, кросс-сочетание создает только простые ковалентные σ-связи, не двойные и не тройные, и не ионные. Во-вторых, в кросс-сочетании участвует электрофил и нуклеофил, то есть такие реагенты, которые и так должны были бы реагировать, но им не хватает реакционной способности, и требуется катализатор, чтобы реакция все же состоялась.
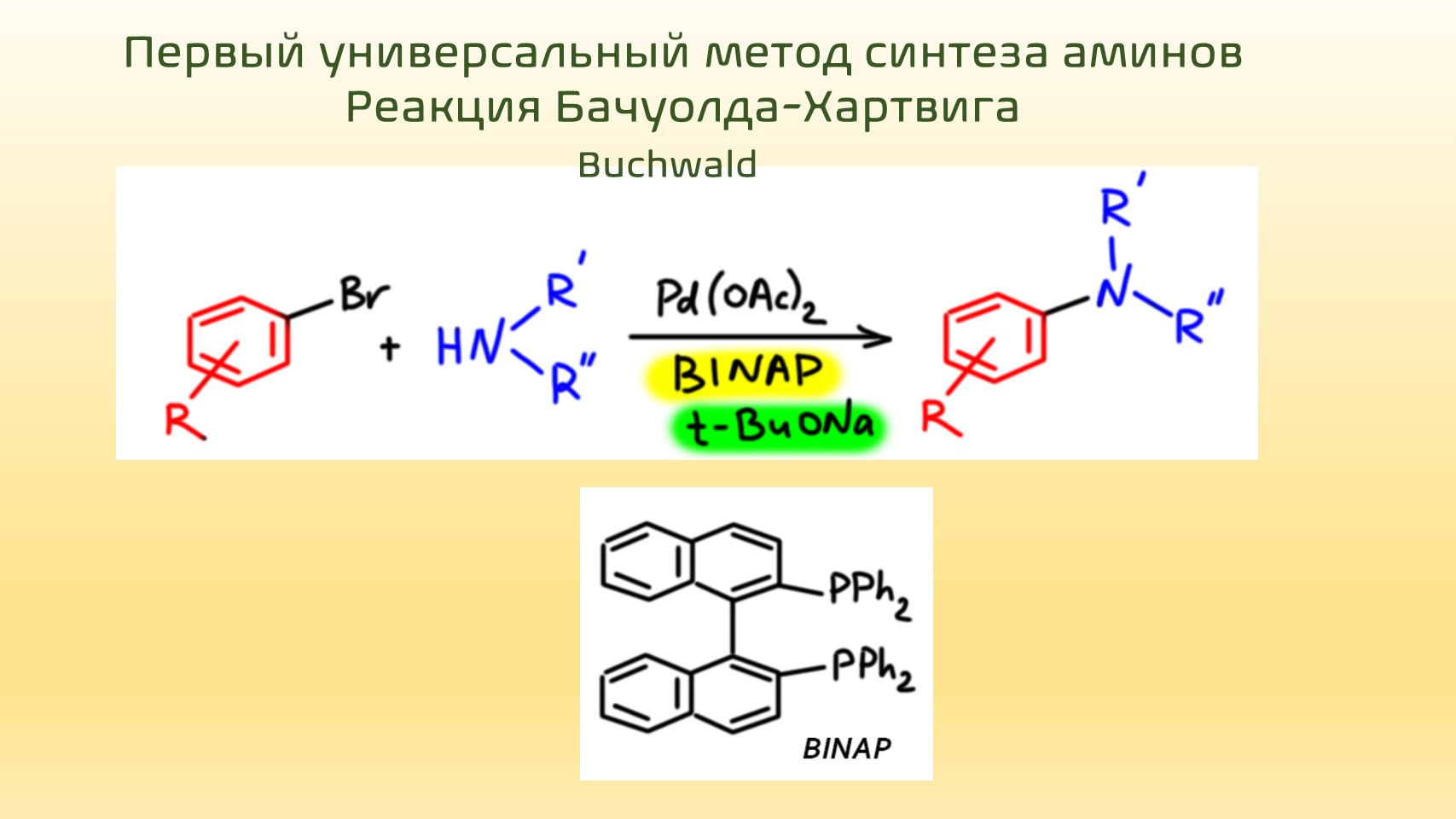
После реакций кросс-сочетания с образованием C-C связей самыми важными являются реакции с образованием связей C-N, которые по-другому еще называют аминированием, амидированием, арилированием аминов и т.п. Почему? В первую очередь потому, что органические соединения азота просто невероятно широко представлены среди важных и полезных веществ современной технологии: лекарств, средств защиты растений, современных материалов и т.п. И если мы вспомним химию аминов, которую недавно изучали, то найдем там много методов, но не найдем, например, методов получения аминов с двумя и тремя ароматическими (или гетероциклическими) остатками И это не потому, что на 3 курсе от нас что-то скрывали, чтобы не перегружать, а именно потому что еще недавно это было тяжелой и почти нерешаемой проблемой. Интересные молекулы с такой структурой получались окольными путями и с мизерными выходами, исследования их свойств показывали, как это интересно, а хороших методов синтеза, чтобы это можно было получать в серьезных количествах, и разумной ценой, так и не было. Но все изменилось в 1995 году, когда была открыта мощная реакция палладий-катализируемого C-N кросс-сочетания, которую часто называют именами открывателей реакцией Бухвальда-Хартвига.
Реакция записывается очень просто.
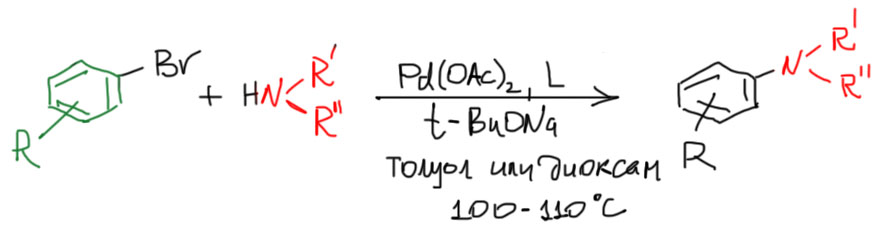
Нам нужен электрофил, точно так же как в обычном кросс-сочетании, лучше всего бромпроизводное. И нуклеофил, в роли которого выступает амин хотя бы с одним атомом водорода (первичный или вторичный). Аммиак использовать можно, но это третует очень специальных условий, поэтому мы этого делать не будем. В качестве катализатора используют комплекс палладия с хелатным дифосфином, например, тем же dppf, с которым мы уже встретились в борилировании. И еще основание, и очень важно подобрать правильное основание, ни слишком основное – из-за этого будут проблемы с преждевременным депротонированием амина, – ни слишком слабое – оно просто не сработает. В результате долгих поисков остановились на трет-бутилате натрия. Именно натрия, а не куда более распространенного калия (попробуйте, как бы в беспамятстве, или приняв пару стаканов, сказать “трет-бутилат” – язык сам машинально выведет “калия” – все так привыкли к этому основанию, что даже не мыслят ничего другого). И что там так уж сильно изменяет какой-то паршивый противоион! Ведь мы с детства знаем, что все щелочные металлы похожи как собаки, и служат только для уравнивания зарядов. Вот это как раз очень глубокое и вредное заблуждение – в реальной химии от противоиона зависит очень многое. Катион натрия намного меньше катиона калия, он обладает весьма значительной льюисовой кислотностью. Посмотрите при случае на банки с простыми солями щелочных металлов – сульфатов, нитратов, ацетатов, и т.п., – обнаружите, что у солей натрия часто бывает кристаллизационная вода, гораздо чаще, чем у таких же солей калия (это не железное правило, потому что вода иногда сидит водородными связями на анионе, а не на катионе, но в целом оно работает неплохо). Эта вода лигандом сидит на катионе натрия. А на катионе калия никто не сидит – слаб он, как кислота Льюиса (рубидий и особенно цезий еще слабее, и этим тоже часто пользуются, когда, наоборот, нужно анион сделать посвободнее). В трет-бутилате натрия катион натрия хорошо связан со своим противоионом, ослабляя его эффективную основность (мы подробно обсуждали этот эффект, когда разбирали межфазный перенос). Кроме того, реакцию всегда проводят в малополярных растворителях типа толуола или диоксана, в которых трет-бутила натрия умеренно растворим и сильно агрегирован (это значит, что ионы там не свободные, а слипшиеся в большие частицы из многих ионов – это, как минимум, резко понижает концентрацию, а точнее, термодинамическую активность, которую, строго говоря, и нужно писать вмсто концентрации в формулах скоростей и равновесий). И это тоже ослабляет эффективную основность. И получается именно то, что нужно.
Почему для этой реакции нужен более сложный лиганд, чем для C-C кросс-сочетания? Вопрос довольно сложный, но одна из главгых причин довольно проста. Механизм этой реакции (каталитический цикл) в общем такой же, как для C-C кросс-сочетания – последовательность окислительного присоединения, переметаллирования (в этом случае на этом месте похожая реакция лигандного обмена) и восстановительного элиминирования. В отличие от C-C кросс-сочетания в этом случае восстановительное элиминирование не идет так легко, потому что палладию нужно окислить не два углеродных остатка, а углеродный и азотный. Но азотный остаток окислить не так просто – азот все же неметалл куда более электроотрицательный, чем углерод, и отнять у него электрон задача посложнее. Поэтому на стадии восстановительного элиминирования нужна помощь, и помощь эта приходит как раз от фосфинового лиганда. Когда он достаточно объемист и занимает много места у атома металла, он просто выпихивает продукт реакции восстановительного элиминирования. Это довольно интересно, потому что в обычной органической химии мы рассматриваем стерические препятствия, стерический объем, как плохие факторы, мешающие реакциям. А в химии переходных металлов стерический объем лиганда очень часто служит неоценимую службу, способствуя выходу продуктов из координационной сферы и ускорению каталитической реакции.
Самый популярный лиганд для реакции синтеза аминов кросс-сочетанием веьма нетривиален – создателю этого лиганда даже дали Нобелескую премию (не только за сам лиганд). Сокращают его BINAP, и это хелатный лиганд очень большого объема (как он выглядит тоже можете посмотреть на слайдах 5 и 6 на сайте).
Примеры синтеза аминов реакцией кросс-сочетания
1. Соберем вторичный амин с двумя замещенными фенилами.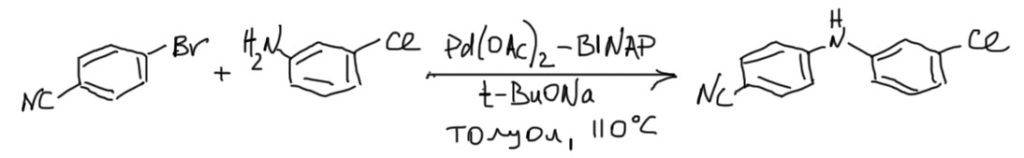
2. Третий фенил добавляется так же, так что можно просто добавить еще одно бромпроизводное.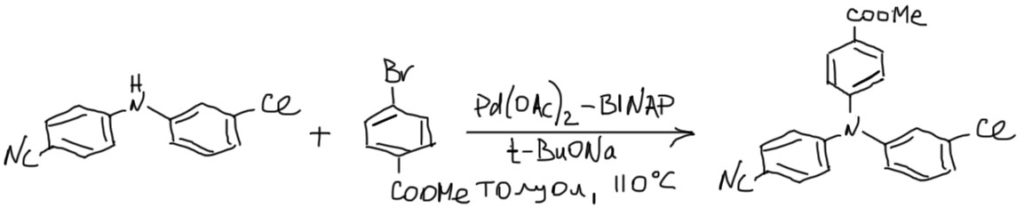
3. Другие амины тоже неплохо получаются, например, можно ввести ароматический заместитель в циклический амин – это тоже непросто сделать обычными методами. Для примера в ароматическое кольцо добавим амидный заместитель – он не будет мешать кросс-сочетанию. 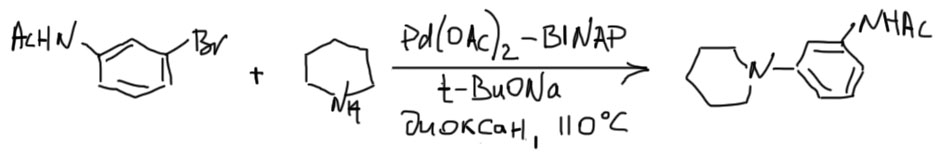
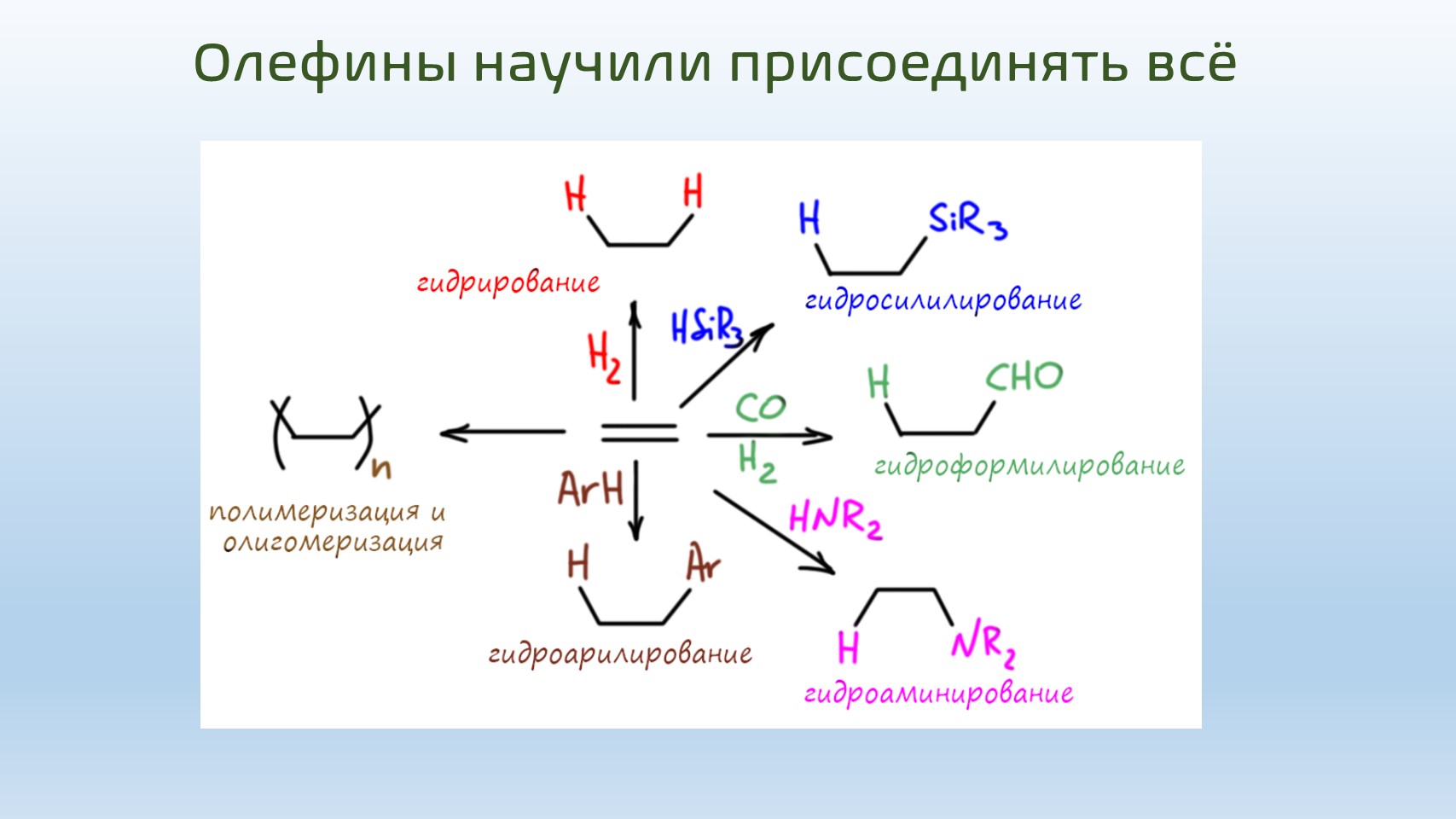
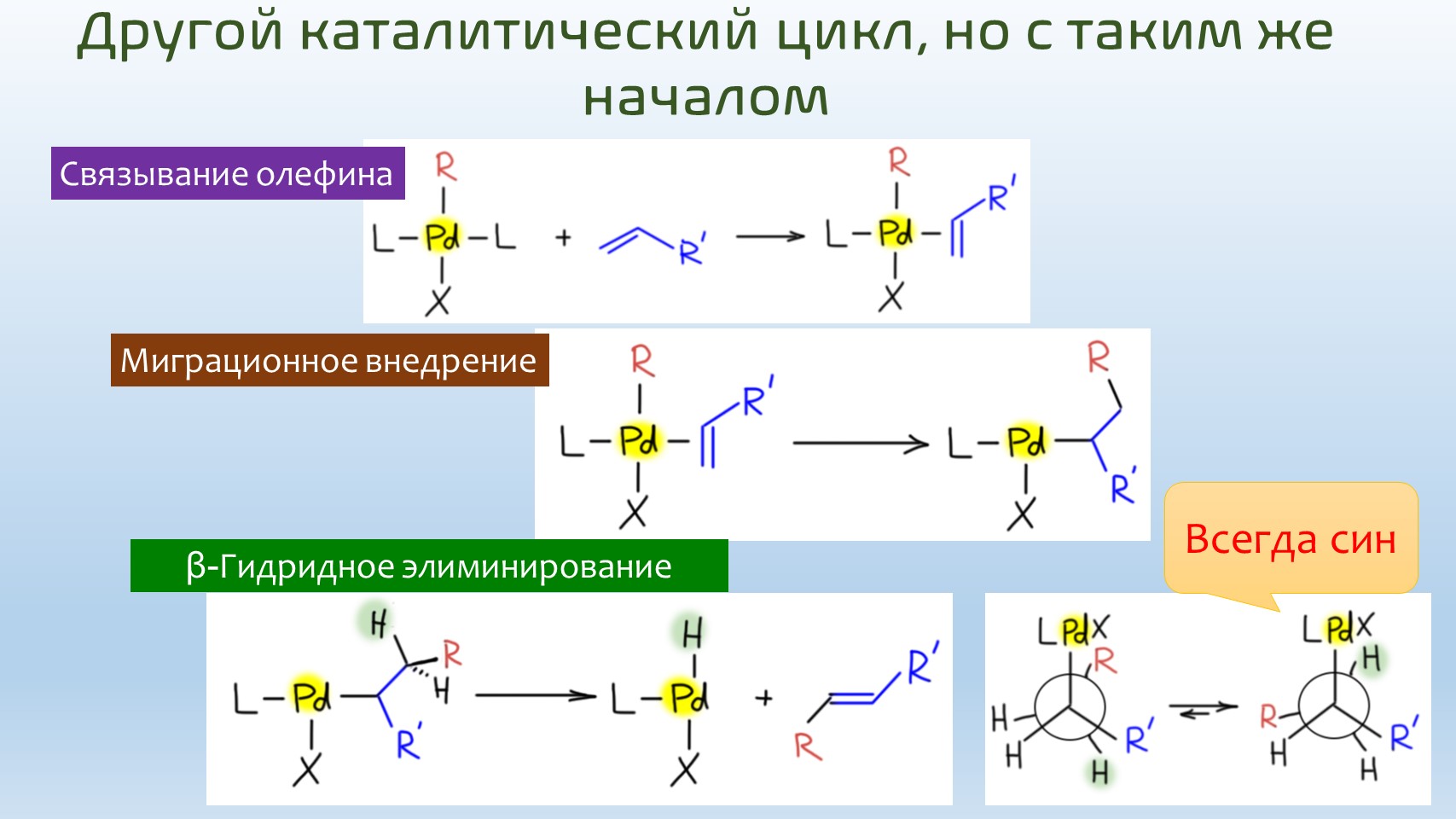
Реакции с олефинами: реакция Хека
Переходные металлы очень любят кратные связи, двойные и тройные, и охотно с ними взаимодействуют. Реакций, связанных с такими взаимодействиями просто невероятное количество, и даже если мы ничего не знаем про такие вещи, по крайней мере про каталитическое гидрирование и полимеризацию Циглера-Натты слышали все.
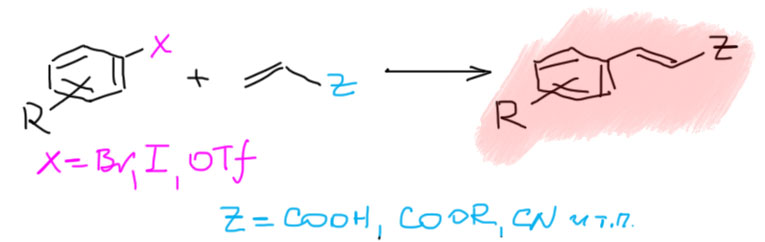
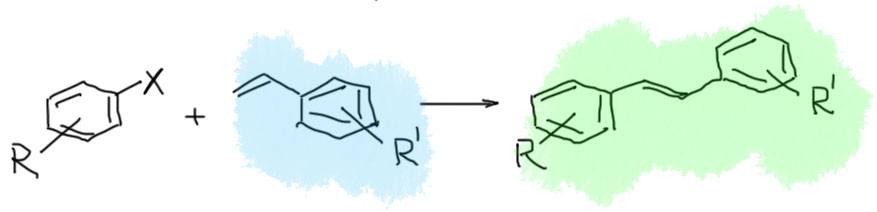
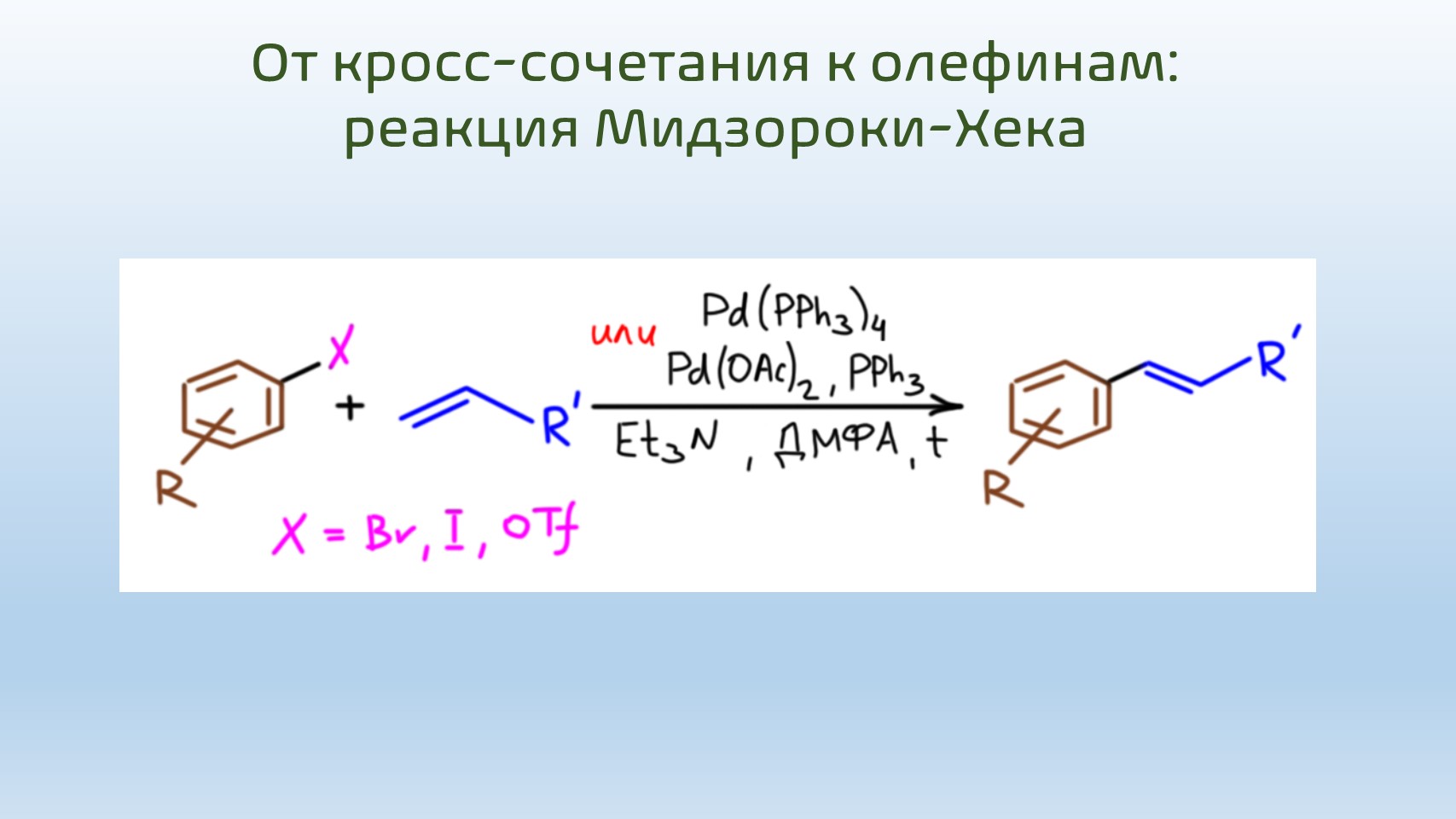
Одной из самых знаменитых и востребованных реакций этого типа давно уже стала реакция Хека (в современной литературе ее чаще называют реакцией Мидзороки-Хека, включив в название японского исследователя, который опубликовал несколько примеров этой реакции на несколько месяцев раньше Хека). С виду реакция весьма непритязательна и очень похожа на еще одну разновидность реакции кросс-сочетания – соединения в новой молекуле олефина и органического электрофила. И хотя сходство это поверхностно, но Нобелевский комитет в 2010 году присоединил Хека к кросс-сочетанию Судзуки и Негиси. Вот как эта реакция выглядит в общем виде. Органический электрофил (бром или иодпроизводное, или трифлат) реагирует с олефином, обычно с монозамещенной терминальной двойной связью в присутствии палладиевого катализатора, фактически точно такого же, как в реакциях кросс-сочетания, и немудреного основания, триэтиламина или ацетата натрия. Результат – дизамещенный олефин.
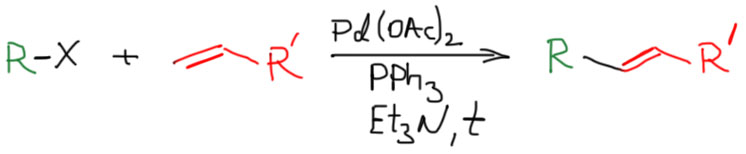
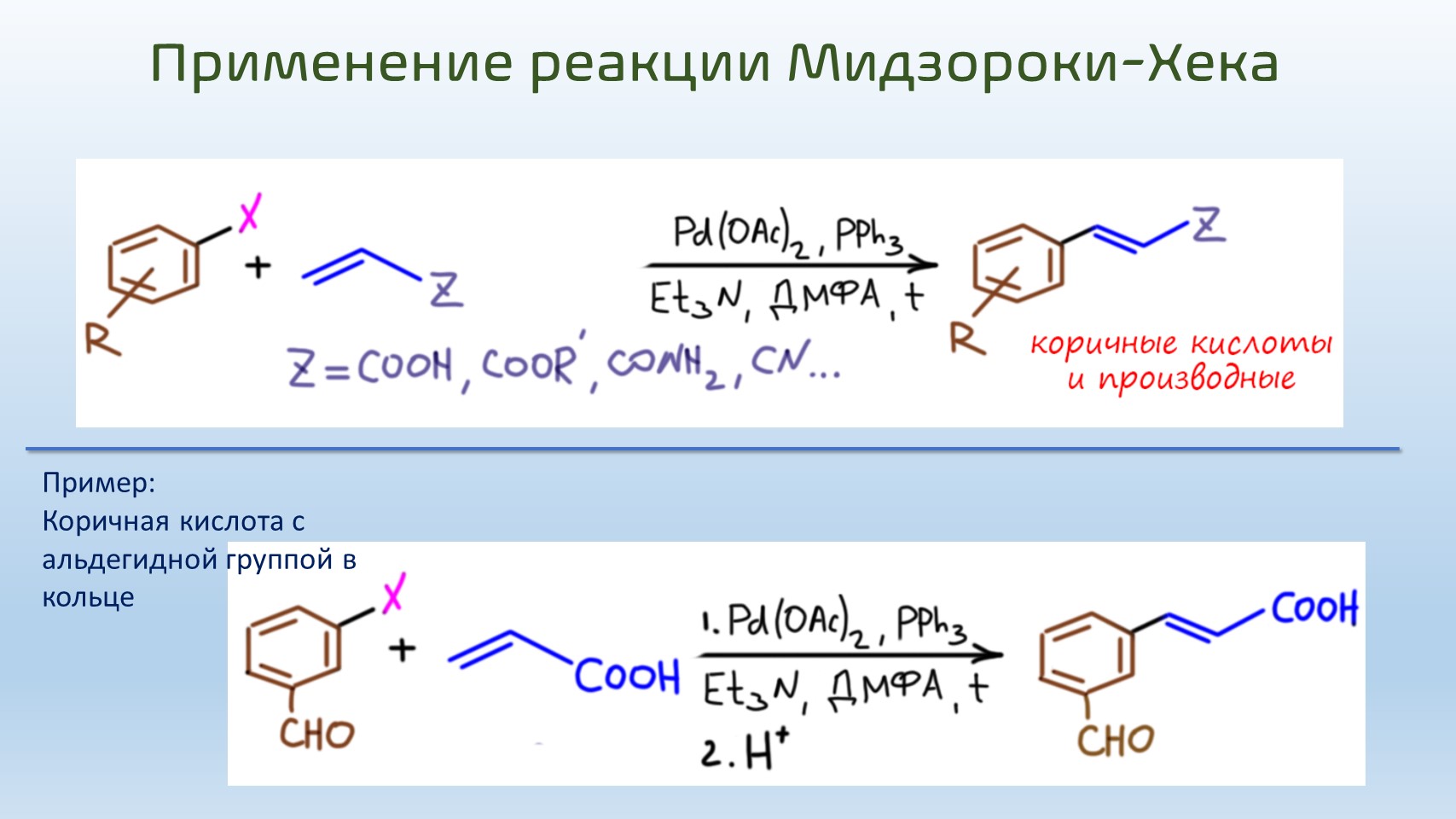
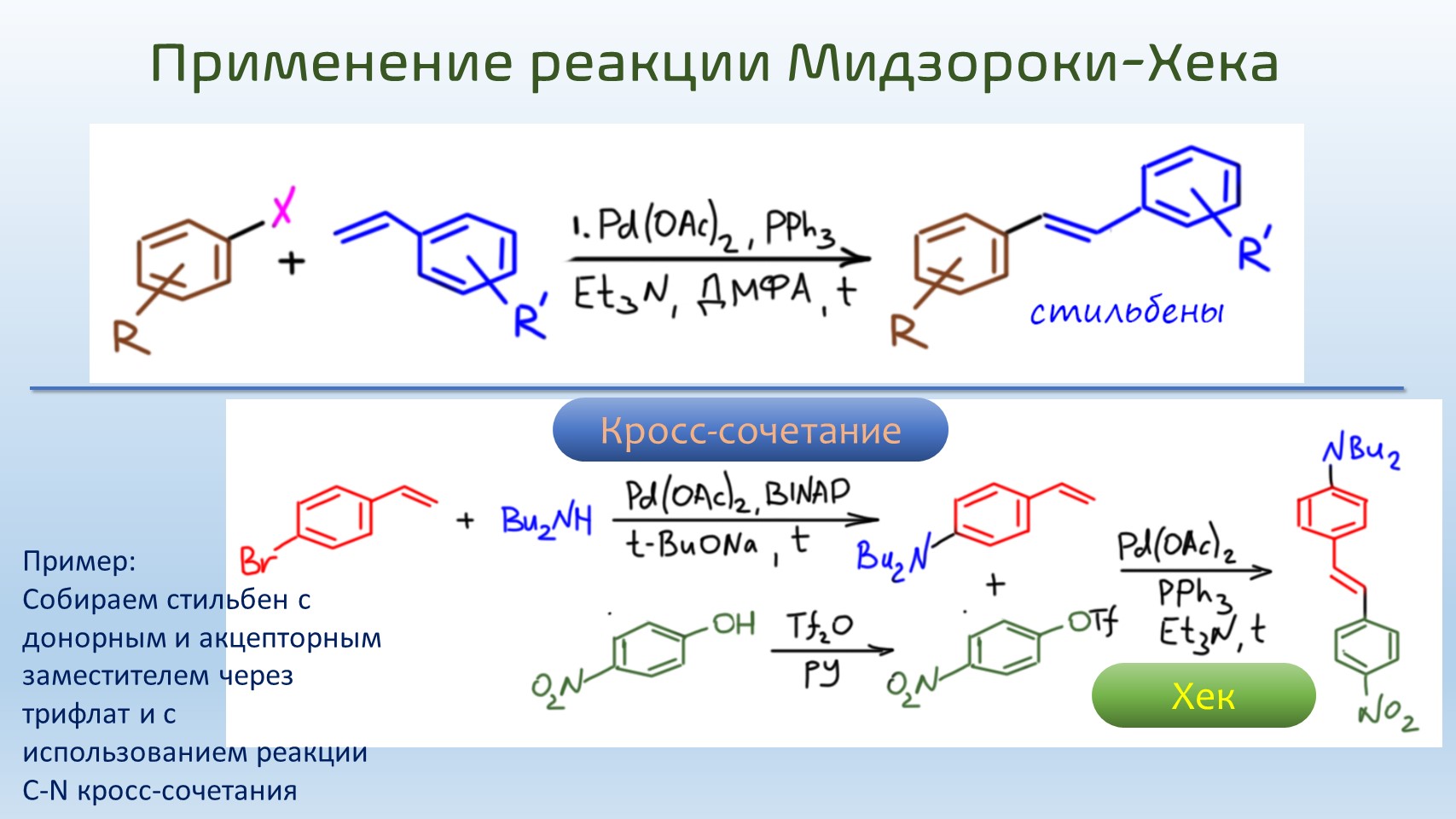
Так как мы уже привыкли к необычайной универсальности реакций кросс-сочетания, реакция Хека нам может показаться довольно скромной. В ней требования к электрофильному реагенту очень похожи на то, что мы видели в реакции Судзуки, но олефин ограничен очень сильно. То, что он почти всегда терминальный с одним заместителем это еще полбеды, но даже здесь годится далеко не все. Не пытайтесь в реакцию Хека всунуть даже какой-нибудь банальный 1-алкен типа 1-пентена или любого другого, хоть нормального, хоть разветвленного строения – получится вместо ожидаемого продукта несусветная каша. Действительно хорошо работают в Хеке два типа олефинов – акриловая кислота и ее производные (эфиры, нитрил, и т.п.) и стиролы, возможно с заместителями в ароматическом кольце. Получаются так называемые коричные кислоты (и их производные: сложные эфиры, называемые циннаматами; нитрилы, циннамонитрилы, и т.п.) и стильбены.
Хотя нас никто не собирается про это спрашивать, все же, любопытства ради, немножко разберемся, как это происходит, и почему это не кросс-сочетание, а совершенно особая реакция. Начинается ведь она точно так же, как кросс-сочетание – электрофил с Pd(0) вступает в реакцию окислительного присоединения. Но дальше все идет не так.
Одним из основных типов реакций комплексов переходных металлов с кратными связями является присоединение. Закономерности реакций присоединения с участием переходных металлов и во многом похожи на известные нам реакции электрофильного присоединения, и в то же время имеют значительные отличия. Любое присоединение в комплексах переходных металлов начинается с того, что алкен (или алкин) входит в координационную сферу лигандом – это часто называют π-комплексом. Дальше происходит замечательная реакция – металл и бывший на нем σ-лиганд (то есть лиганд, связанный с металлом σ-связью, очень похожей на обычную ковалентную связь) садятся на два углерода строго син-способом. Это очень важное отличие таких реакций от обычного присоединения, для которого идет анти-присоединение, но нестрогое, всегда с примесью син-присоединения. А у переходных металлов все очень строго – только син. И совершенно понятно почему – оба присоединяющихся фрагмента (металл и лиганд) и олефин находятся в координационной сфере металла вблизи друг друга и просто не могли бы присоединиться анти-способом. Такую реакцию присоединения к кратной связи одного лиганда самого металла и другого лиганда называют миграционным внедрением (кто там мигрирует, а кто внедряется, можете попробовать разобрать сами, или посмотрите на другом сайте). 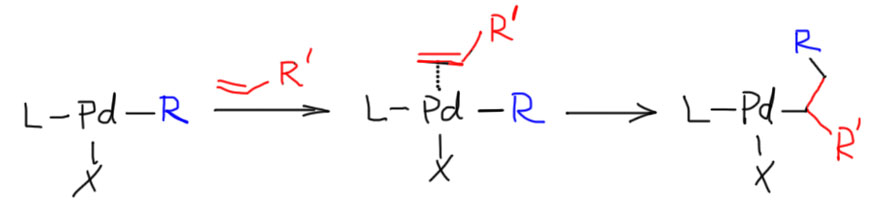
После этого происходит еще одно знакомое нам событие – β-элиминирование. Но в отличие от обычного E2-элиминирования, для этого элиминирования а) не нужно основание; б) оно происходит всегда строго как син-элиминирование – никогда никаких даже следов анти-элиминирования, только и исключительно син. Элиминирует с одного углерода тот же палладий вместе со всеми своими остальными лигандами, а с другого – водород. И мы не конкретизируем, какой водород, протон или гидрид, потому что не знаем этого – это согласованный процесс и стрелки можно направить в любом направлении. Это одна из общих особенностей реакций в химии переходных металлов – они предпочитают именно согласованные реакции без образования катионов или анионов. Реакция называется просто β-гидридным элиминированием. 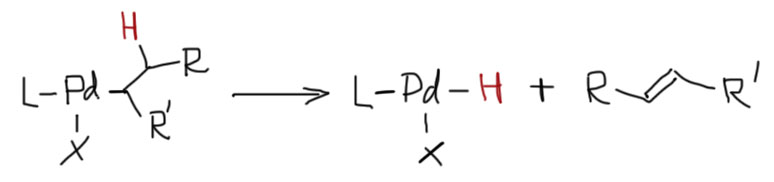
Резонный вопрос – почему в основном получается транс-изомер. Это легко понять, нарисовав наши обычные проекции Ньюмена, ровно так, как мы это делаем в обычном присоединении и элиминировании, и соблюдая син-присоединение и син-элиминирование. Очевидно, что из двух конформеров, тот, который ведет к цис-пробукту менее выгоден из-за сближенности заместителей. Так мы видим, что обычные инструменты анализа стереохимии вполне годятся и для реакций с комплексами переходных металлов.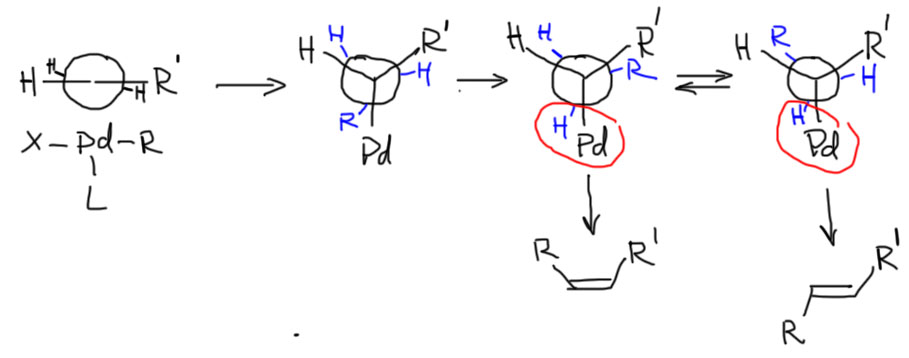
Соберем теперь весь каталитический цикл реакции Хека. Входим, как в кросс-сочетании с комплексом Pd(0), но оставляем на нем только один лиганд типа трифенилфосфина, потому что с двумя нам не хватит места в координационной сфере, координационное число у палладия обычно не больше 4. Это одна из причин, а точнее объяснений того, почему реакцию Хека обычно ведет при более высокой температуре, часто даже за 100º, чтобы сместить равновесия диссоциации исходного комплекса подальше. Дальше входим в окислительное присоединение. Получающийся комплекс подхватывает олефин, происходит миграционное внедрение, и сразу за ним гидридное элиминирование. Все это мы уже разобрали, но на этом дело не кончается, потому что при гидридном элиминировании получается гидридный комплекс Pd(2+) – водород для любого металла, не исключая палладия, лиганд отрицательный: назвался неметаллом, полезай в координационную сферу гидридом, а не протоном. Но тут все так хитро устроено, что и назвавшись гидридом, этот водород может быть удален основанием, как протон. Так вот для чего основание – до этого момента оно лежало без дела. И так как водород из координационной сферы уходит протоном, электроны свои он оставляет палладию – то есть восстанавливает его до Pd(0). Всё, цикл замкнулся – идем на следующий.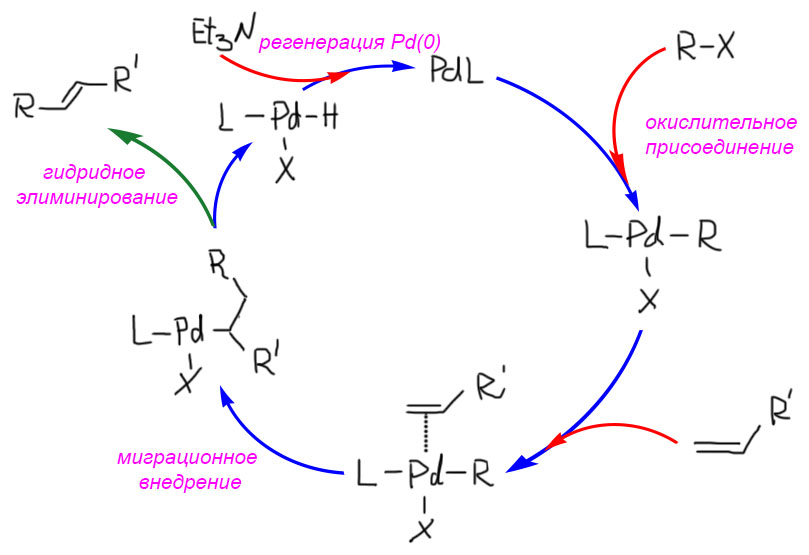
Фенолы и карбонильные соединения в реакциях кросс-сочетания
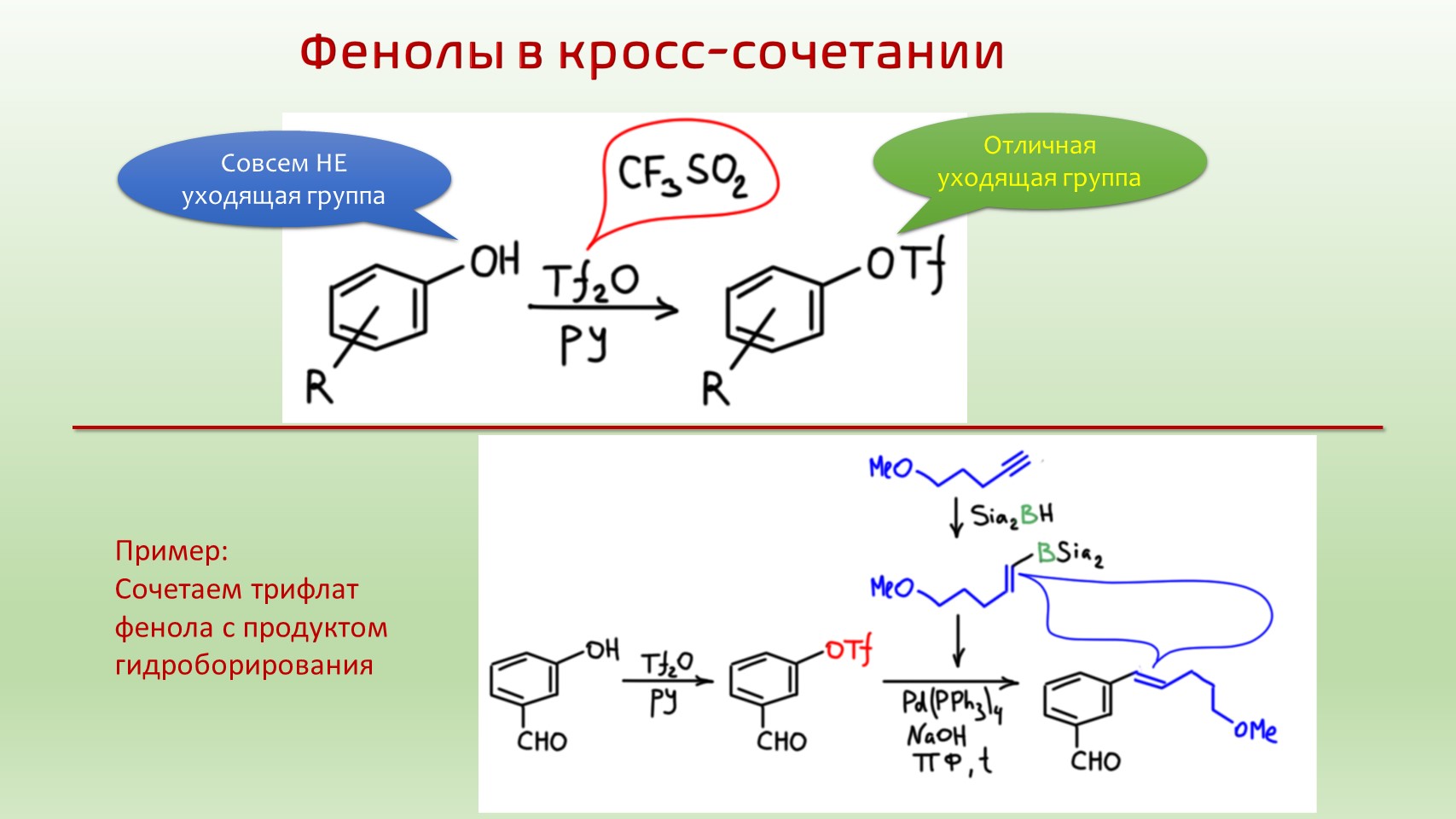
Даже если бы в реакциях кросс-сочетания использовали в качестве электрофилов только иод и бромпроизводные, возможности этих реакций были бы огромны. Но в этих реакциях используют гораздо более широкий круг электрофилов, включая и хлорпроизводные, и соли диазония, и всякие производные серы и многое другое. Но для всего этого требуются особые катализаторы и лиганды, и мы не будем это рассматривать, чтобы не зарыться без надежды выползти. Но есть один класс производных, которые легко получаются, и реагирует ровно в тех же условиях, что и бромпроизводные. Это сложные эфиры трифторметансульфоновой кислоты, трифлаты. В принципе, они очень похожи на хорошо нам знакомые тозилаты (и еще ближе – на мезилаты, как сокращенно называются метансульфонаты, которые применяются практически точно так же, как тозилаты), но мы их никогда в обычной органической химии не применяли. Почему? Потому что это слишком хорошие уходящие группы, и это не обязательно хорошо – в реакциях SN2-замещения это просто неудобно, из-за избыточной реакционной способности получается много побочных продуктов. Зато в химии кросс-сочетания это идеальные уходящие группы. Их используют в реакциях точно так же, как бромпроизводные, с теми же катализаторами и приблизительно в тех же условиях (нюансы есть, но для нас несущественные).
Откуда они берутся? Из фенолов, действием ангидрида трифтометансульфоновой кислоты в присутствии основания.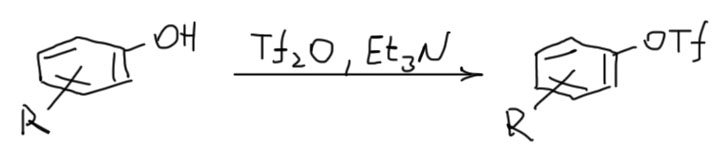
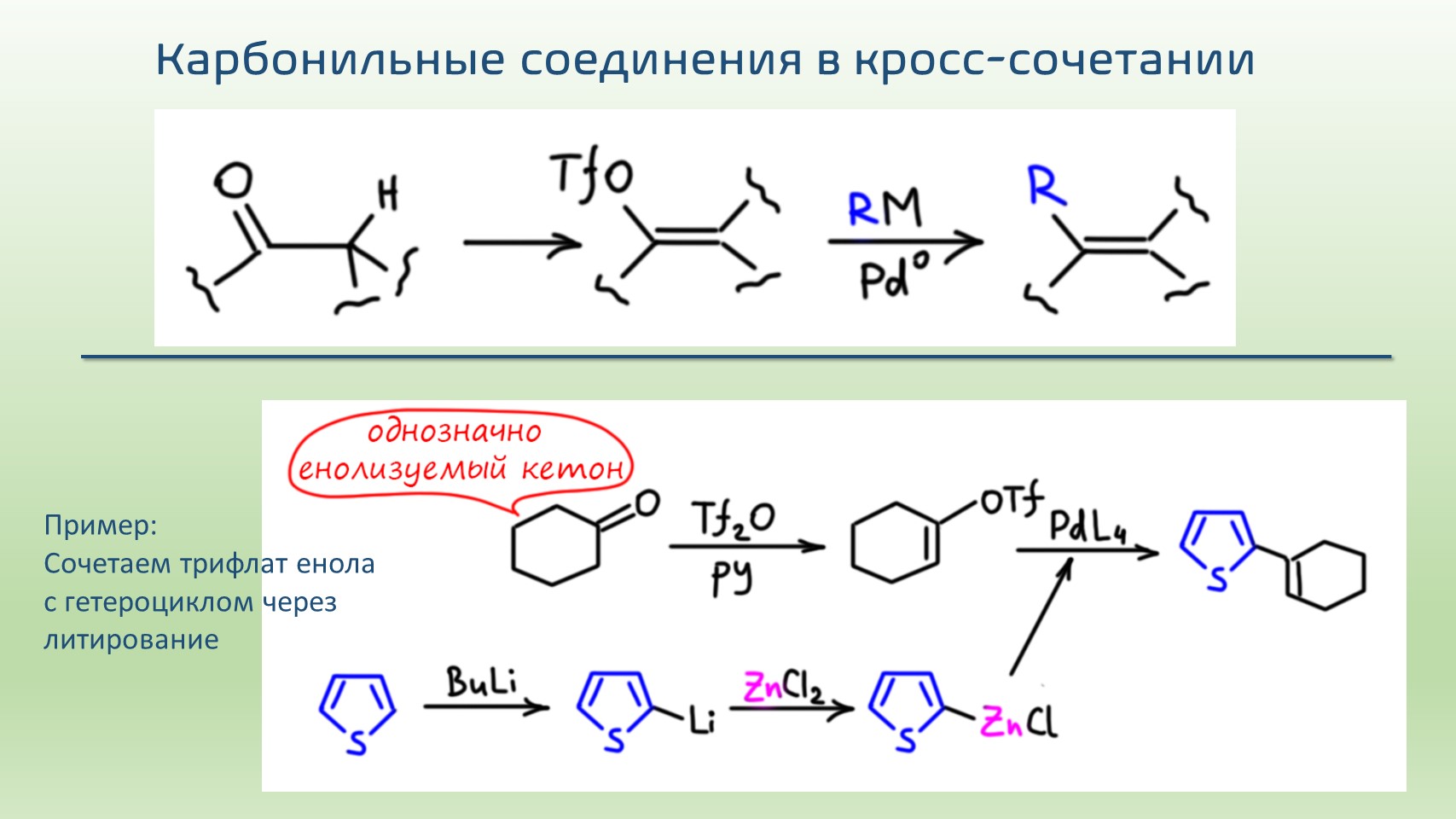
И не менее интересно – из енолизуемых кетонов. Здесь я показываю немного условную схему, чтобы показать сам принцип, а в реальной жизни можно получать самые разные трифлаты енолов и из симметричных, и из несимметричных кетонов, но для этого используются довольно специфические реагенты и основания, если интересно, можете посмотреть на моём другом сайте. 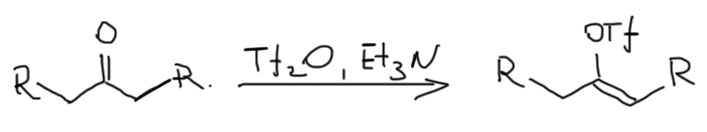
Дальше все это используют в кросс-сочетании. Здесь очень важно обратить внимание, что в обычной химии фенолы в принципе не участвуют ни в каких реакциях с разрывом связи C-O. А здесь участвуют, и еще как! Это удивительно и прекрасно. Да и в енолах мы никогда не делали замещение с разрывом связи C-O, а здесь пожалуйста. Посмотрим несколько примеров.
1. Диены из кетонов и ацетиленов с использованием реакции Судзуки. 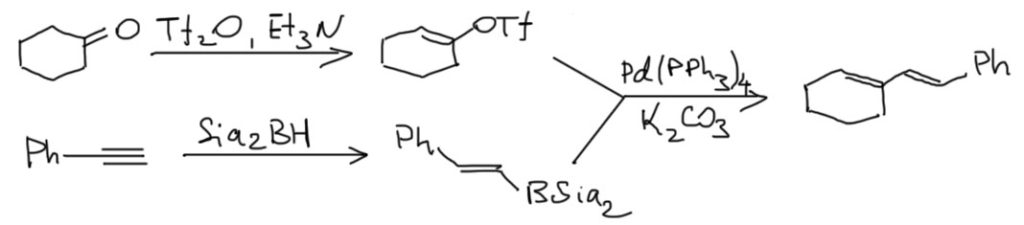
2. Дифенил из двух разных фенолов с использованием реакции Судзуки, причем и борную кислоту получим через трифлат. Напомню, что в этом случае катализатор борилирования справляется и с кросс-сочетанием, нужно только добавить основание.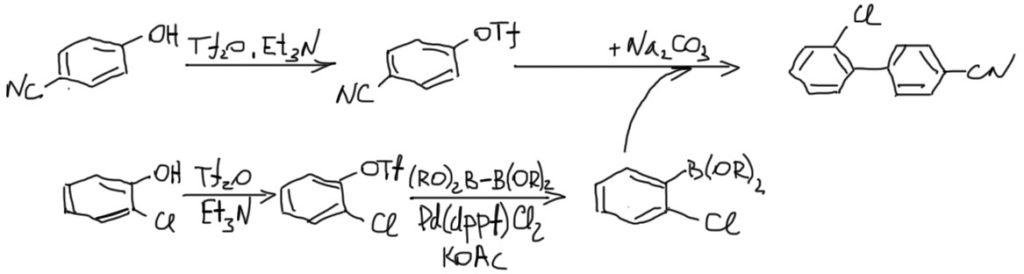
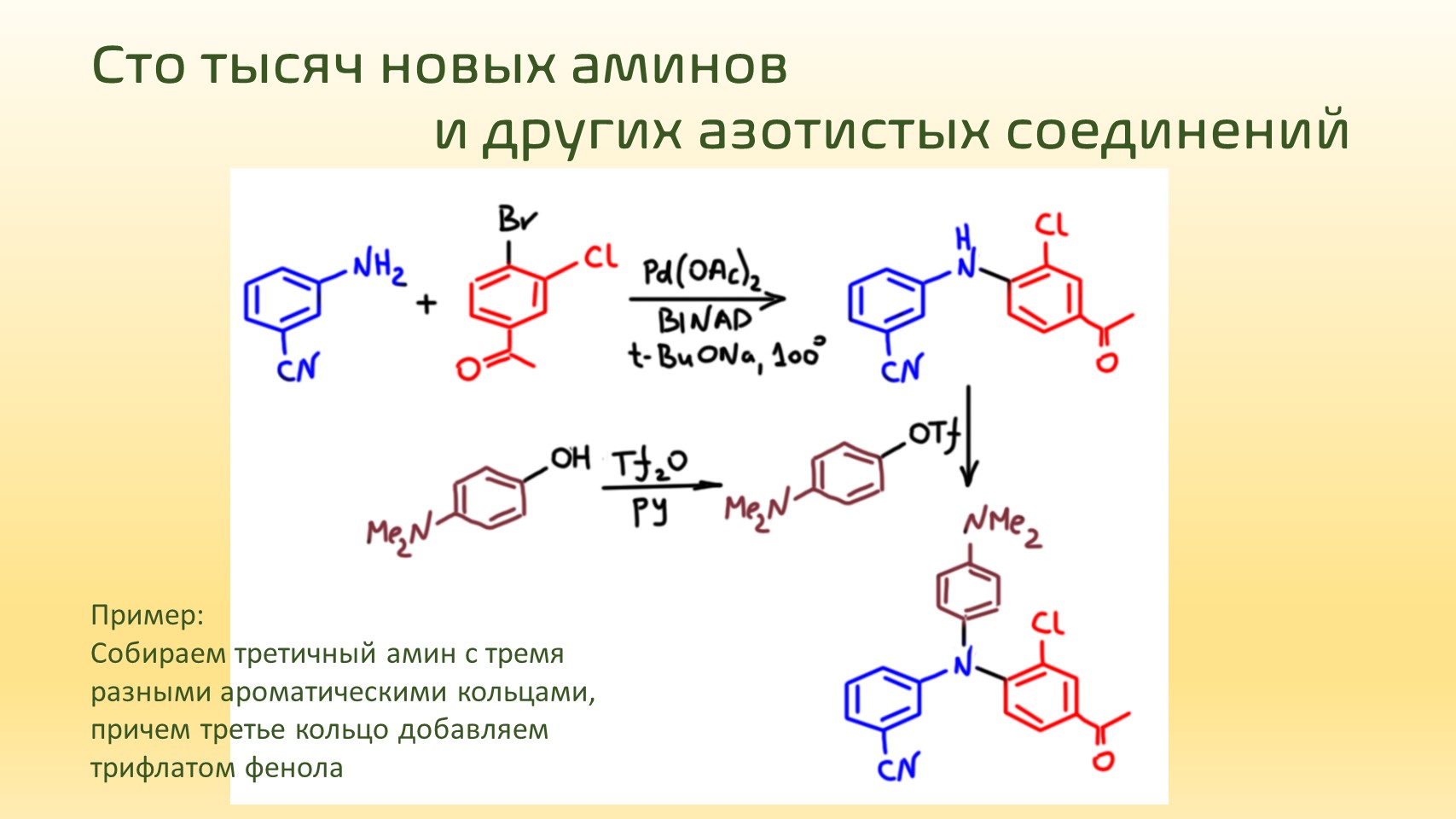
3. Вторичный амин из фенола и анилина. Ради того, чтобы еще раз показать, что вся эта химия совместима с самыми разными заместителями, возьмем анилин с непредельным заместителем (м-аминостирол) и трифлат, полученный (не буду в 10 раз повторять как) из салициловой кислоты.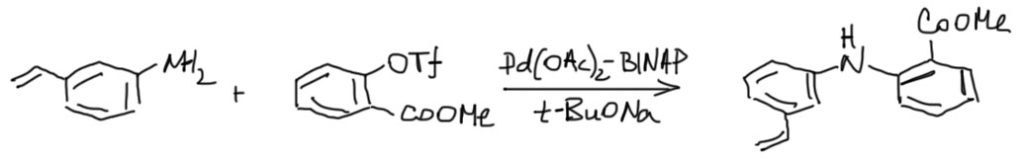
4. Эфир замещенной коричной кислоты реакцией Хека, но из фенола. 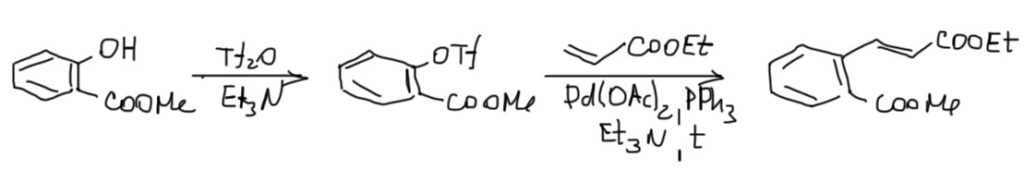
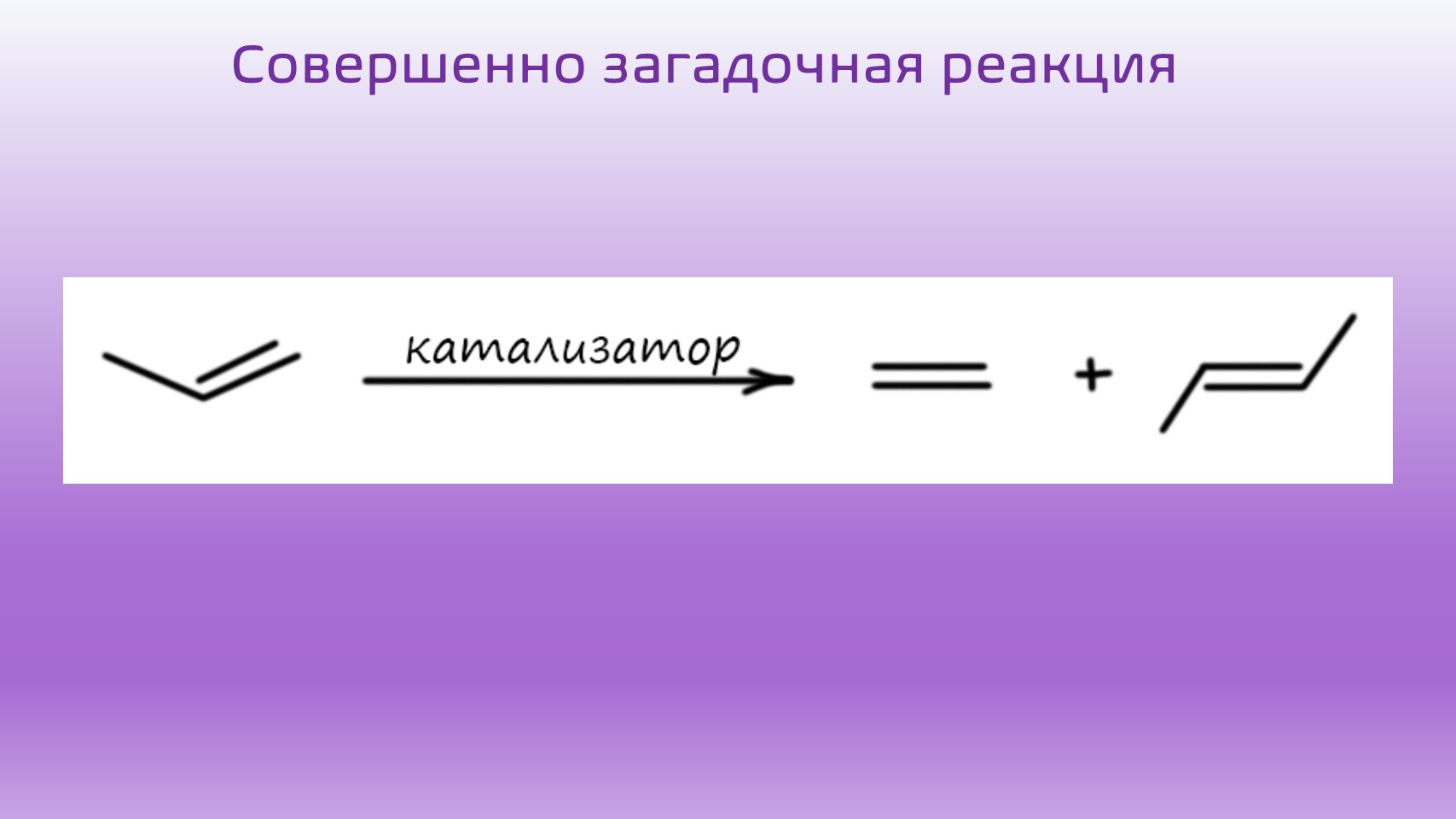
Метатезис олефинов
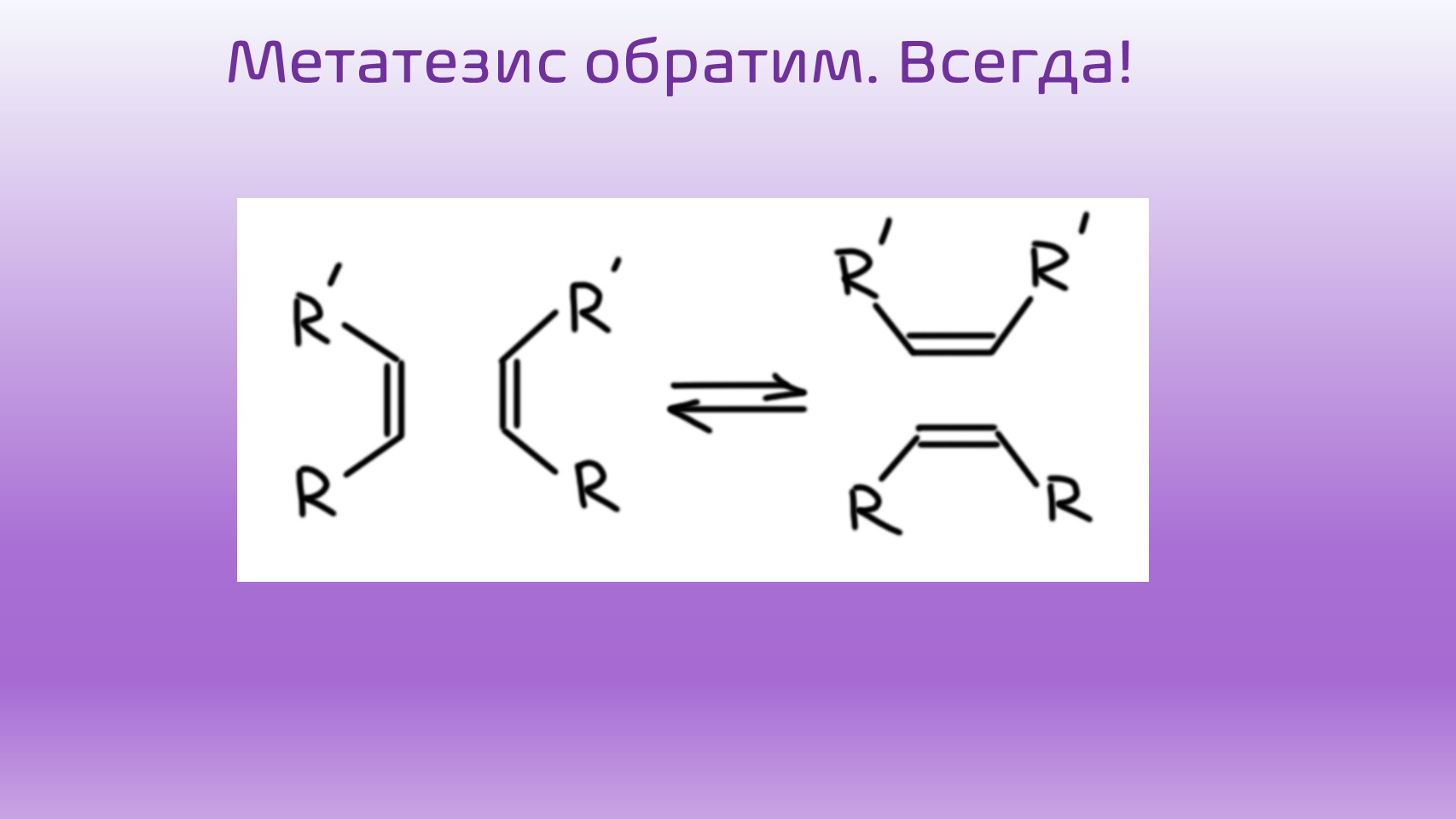
Метатезис – это синоним слова обмен. Реакции обмена, когда из соединений AB и CD получаются AC и BD? хорошо известны в самых разных разделах химии. В неорганике обменом получают из двух солей две новые соли. Реакция обмена всегда и просто по определению обратима. Чтобы сместить ее с сторону какого-то конкретного продукта, нужно использовать принцип Ле Шателье. В неорганике, например, обмен солей становится необратимым, когда одна из новых солей нерастворима и выпадает в осадок.
В применении к алкенам обмен (метатезис) кажется абсурдом. Алкены должны как-то порвать свои двойные связи и обменяться кусками. Как-то так:
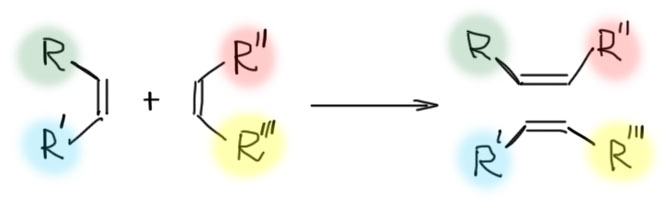
Но такая реакция действительно есть. Ее случайно открыли при исследовании полимеризации алкенов с катализаторами Циглера-Натта, а это всегда какие-то более или менее сложные комплексы переходных металлов 4-й, 5-й, 6-й групп. И долго не знали, что с ней делать, считая ее такой противной побочной реакцией, которая портит чистые алкены. Реакция действительно довольно сомнительная, и это ясно даже из этой простой схемы: ну хорошо, обменялись, а там цис- или транс-алкены? А почему дизамещенные? А тризамещенные можно? А моно? А если один из алкенов на схеме перевернется? А это равновесие? А куда оно смещено?
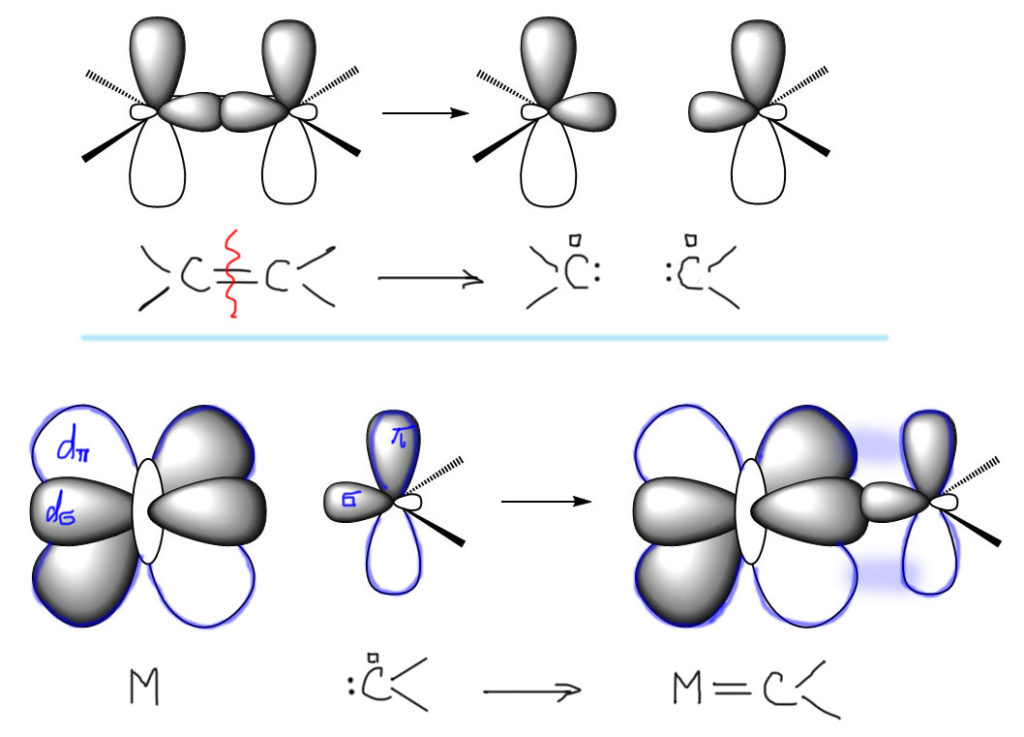
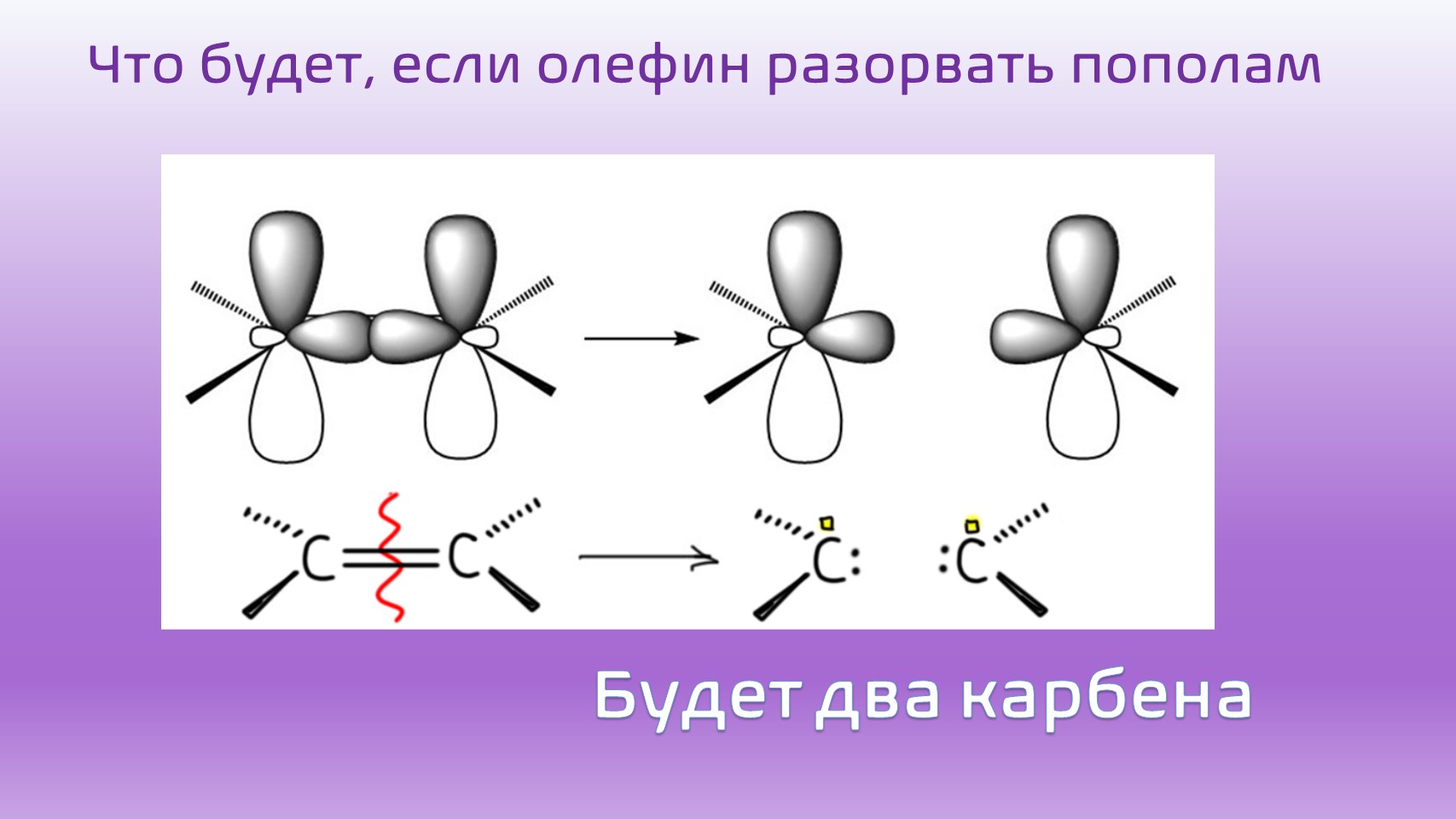
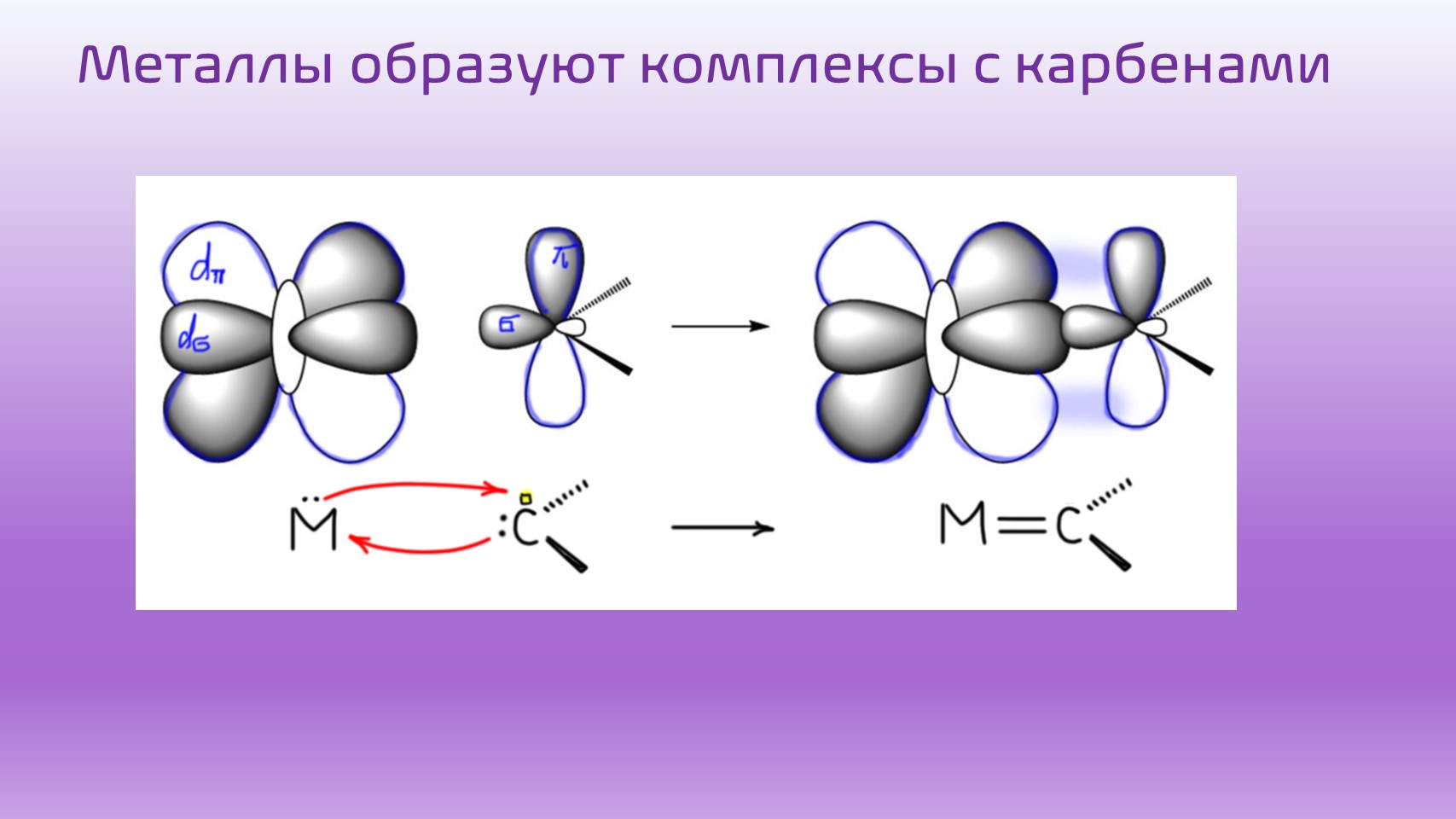
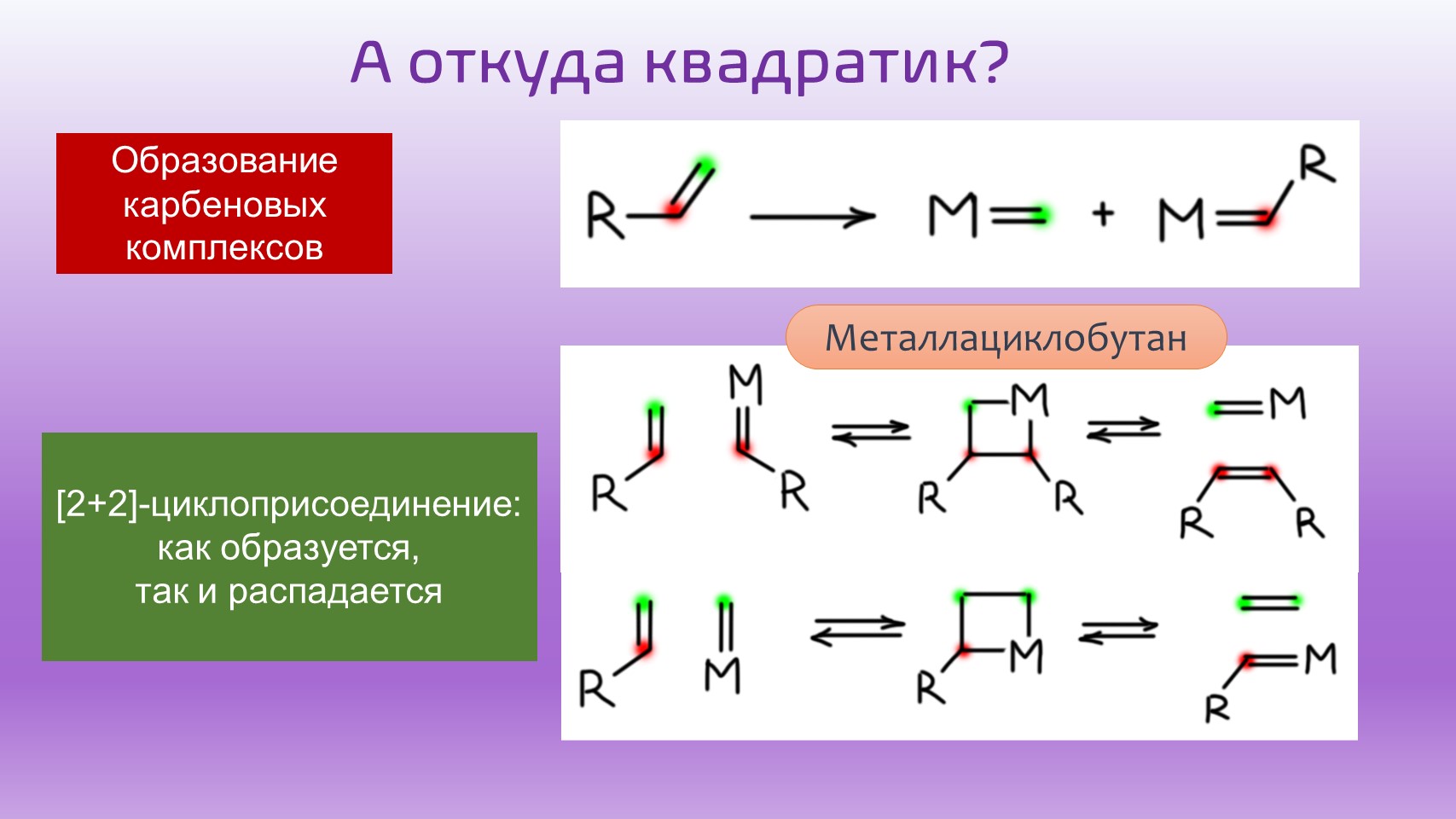
А как вообще это может происходить? Механизм реакции поняли далеко не сразу, но в начале 1970-х его исследовал французский исследователь Ив Шовен, за что ему в 2005 году дали Нобелевскую премию. Для реакции нужен катализатор, комплекс переходного металла с карбеном. Карбен – это как раз и есть половинка алкена, точно так же, как радикал является половинкой алкана. Если порвать двойную связь совсем, то есть не так, как мы делаем, когда присоединяем к алкенам водород или бром, – а совсем, обе связи, и σ-, и π- пополам, то получается карбен. Свободные карбены, как мы знаем, очень неустойчивые, бешено реакционноспособные частицы, которые немедленно реагируют, как только образуются. Если реагировать не с чем, они сдваиваются обратно в алкены. Но переходные металлы сильны тем, что они отлично используют всякие диковины в качестве лигандов, причем в виде комплексов это вполне стабильные молекулы, которые можно получить, хранить, изучать, использовать. Комплексы металлов с карбенами условно обозначают, показывая двойную связь между металлом и углеродом. У карбена (точнее, синглетного карбена) в полном соответствии с тем, что он происходит из разрыва двойной связи, есть заполненная σ-орбиталь (она образовалась из разрыва σ-связи двойной связи алкена) и пустая π-орбиталь (она образовалась из разрыва π-связи двойной связи алкена, обозначим ее пустым квадратиком). Они взаимодействуют с d-орбиталями металла, пустой и заполненной, образуя весьма прочную связь металл-углерод в карбеновом комплексе.
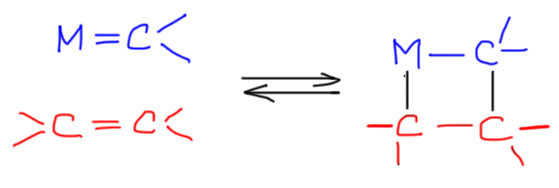
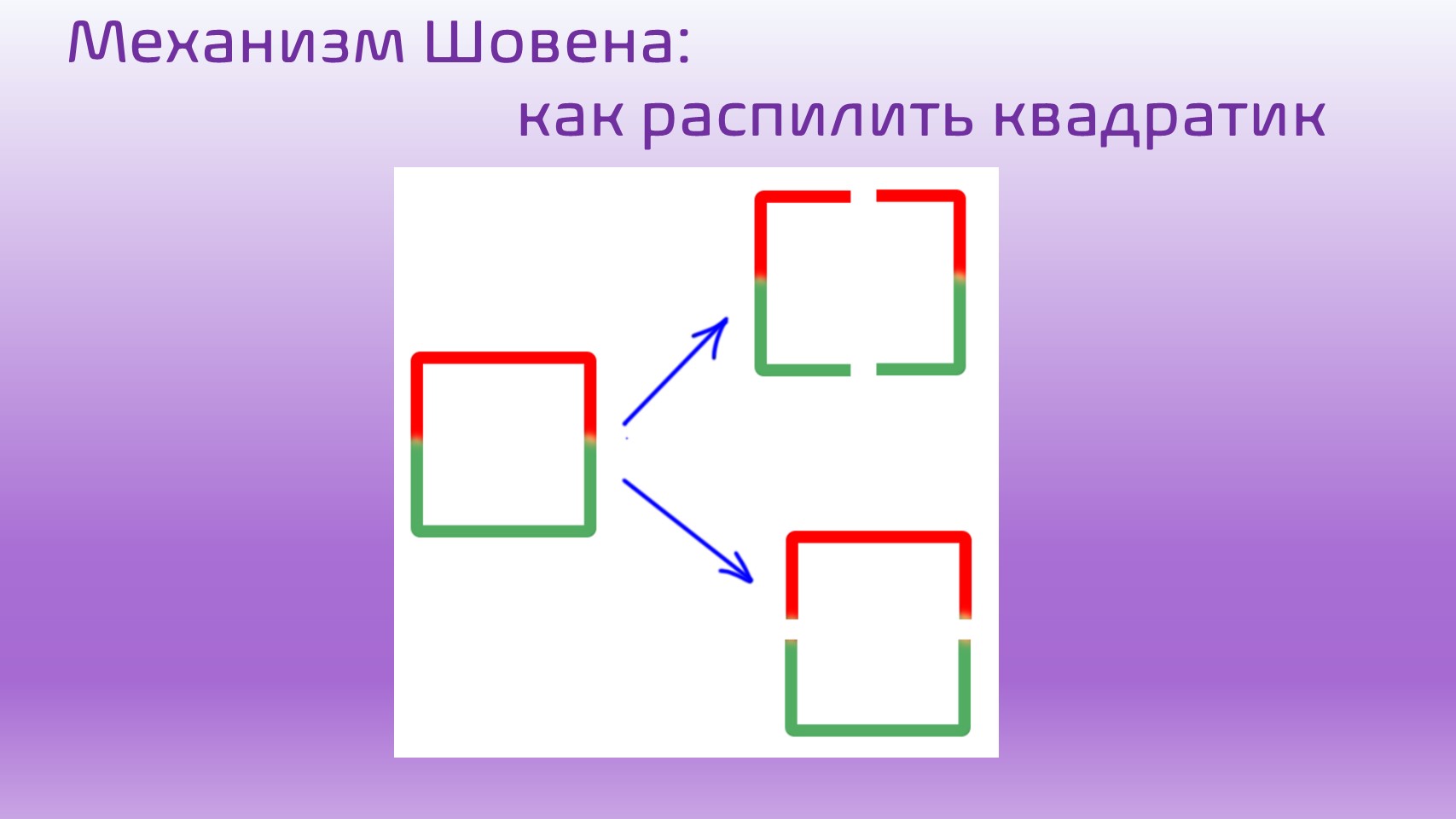
Комплекс переходного металла с карбеном не зря обозначается с двойной связью – его реакции немного напоминают реакции олефинов. Одна из известных реакций олефинов – образование 4-членного цикла в реакции, называемой [2+2]-циклоприсоединением. Для олефинов это реакция редкая, обычные алкены так не реагируют (точнее, реагируют, но только фотохимически, под ультрафиолетовым излучением), но олефины с сильно различающимися по донорности и акцепторности заместителями реагируют вполне охотно. Карбеновый комплекс металла можно рассматривать как такой своеобразный олефин, легко реагирующий с обычными олефинами с образованием нового комплекса металла, который выглядит как четырехчленный цикл. Такие циклы называют металлациклами. В этом случае можно даже точнее сказать – металлациклобутан. Буква -а- в названии, связывающая металл и цикл, означает, что один из атомов цикла земещен на металл.
Образование цикла обратимо. Но обратная реакция не обязательно разорвет те связи, которые образовались – цикл может порваться и по другим двум связям. Дополним схему:
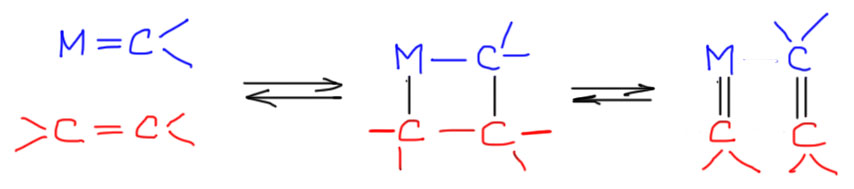
Вот это и есть основная реакция, объясняющая метатезис олефинов. Попробуем расписать, что получится, если взять какой-нибудь олефин, хотя бы монозамещенный, и добавить к нему каталитическое количество карбенового комплекса металла.
Катализаторы метатезиса
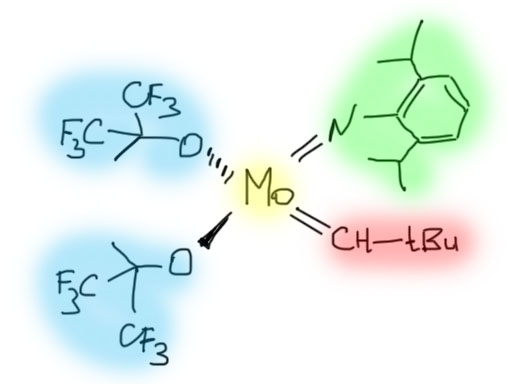
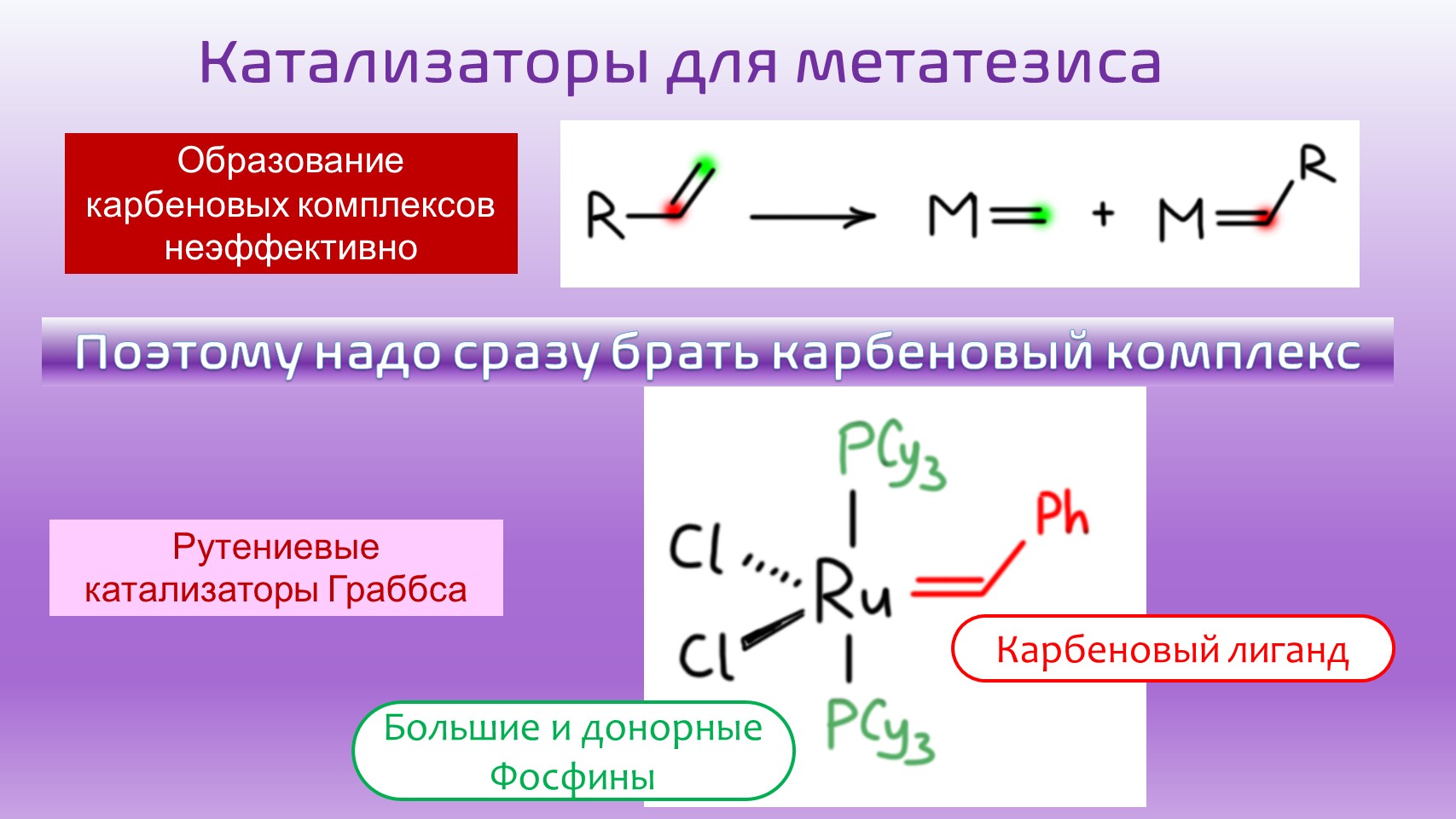
Пока мы условно обозначили катализатор метатезиса как карбеновый комплекс какого-то переходного металла. В реальности разработка катализаторов метатезиса оказалась очень сложной задачей, над которой два десятилетия бились десятки самых мощных исследователей. Битва увенчалась ошеломлющими успехами и победами, и два профессора из США, Ричард Шрок и Роберт Граббс, получили за это Нобелевскую премию в 2005 году. Созданные ими катализаторы представляют собой как раз карбеновые комплексы. Шрок был первым, он разработал целое семейство комплексов молибдена и вольфрама. Катализаторы Шрока мощны и универсальны, но довольно капризны и требуют высокого мастерства и тщательной работы от экспериментатора.
Посмотрим, как выглядит самый популярный катализатор Шрока (его обычно в схемах реакций сокращают просто в Schrock-F12, имея в виду, что в состав входят 12 атомов фтора). Выглядит довольно устрашающе, но все более-менее понятно. Во-первых, он содержит карбен – это самый главный лиганд, который и будет активно участвовать в реакции. Карбен специально выбран с трет-бутильной группой – она большая и занимает много места, и поэтому ее с удовольствием заменят другие карбены, когда этот комплекс прореагирует с первой молекулой олефина. Все остальное выполняет две важные роли: во-первых, создает нужную степень окисления металла, а для реакции метатезиса нужен Mo(6+). Во-вторых, создают толкотню и давку в коодинационной сфере, поэтому они все такие рогатые и объемистые. Там находится объемистый нитрен (нитрен – это азотный аналог карбена), и пара более банальных алкоксидов. А толкотня и давка (по научному, стерическое напряжение) ускоряют реакции в координационной сфере, создают условия, чтобы лиганды, участвующие в реакции (карбены и металлацикл) не засиживались, а быстро-быстро меняли друг друга, были лабильными. Если этого нет, то лигандам нет смысла во что-то превращаться, образуются стабильные комплексы и так и сидят в реакционной смеси. Собственно именно подбор таких вспомогательных лигандов, которые создают эту высокую реакционную способность, и является результатом многолетних исследований. Здесь нельзя не перебрать, ни недобрать – слишком загроможденная лигандами координационная сфера просто не будет впускать туда новые лиганды. Нобелевка ушла по делу – на то, чтобы понять устройство таких сложных коплексов ушли многие годы упорнейшего труда.
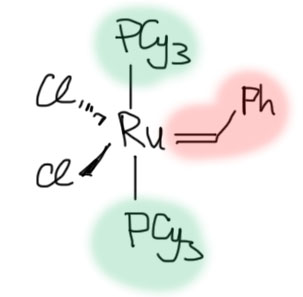
Катализаторы Шрока обладают непревзойденными характеристиками, но слишком сложны в работе, а для повседневной жизни нужно что-то попроще, для того чтобы реакцию метатезиса мог поставить каждый, без излишней мороки, сверхчистых растворителей, инертных газов и т.д. Такие катализаторы создал Граббс на основе комплексов рутения. Рутения не нужно пугаться, это самый дешевый платиновый металл, намного более дешевый чем палладий. Поэтому катализаторы Граббса стали невероятно популярны. Посмотрим на самый первый из катализаторов Граббса, его так и сокращают над стрелками схем – Grubbs-I. Это комплекс рутения(2+), который выглядит гораздо проще и понятнее катализатора Шрока. Главное и тут – карбен, и здесь он попроще. Толчею и давку в координационной сфере создают два фосфиновых лиганда, то не трифенилфосфина, как мы привыкли, а трициклогексилфосфина. Почему такая замена? Трициклогексилфосфин больше по размеру, чем трифенилфосфин, и это понятно, потому что вместо плоского фенильного кольца взяты циклогексановые кресла, которые еще и не стоят на месте, а, как им и положено, все время выворачиваются как свежепойманные рыбы – это и создает значительный эффективный стерический объем. Дополняют сферу два банальных хлорида, создающих степень окисления Ru(2+). Популярность катализаторов Граббса (этого и еще нескольких похожих комплексов) в метатезисе совершенно ошеломляющая. Это почти всегда первый выбор, чтобы попробовать метатезис. Когда используют катализаторы Граббса, то есть почти всегда, над стрелками пишут либо Grubbs-I и т.п., либо сокращенную форму комплекса [Ru=CHPh].
Как используют метатезис?
Прежде чем разобрать, как метатезис используют, еще раз скажем как его не используют. Если коротко – метатезис не используют в тех случаях, когда точно не известно, что его можно использовать. Ни при каких обстоятельствах нельзя действовать, как мы любим, из так называемых общих соображений. Либо я знаю, что в данном случае метатезис используется, либо я его не использую.
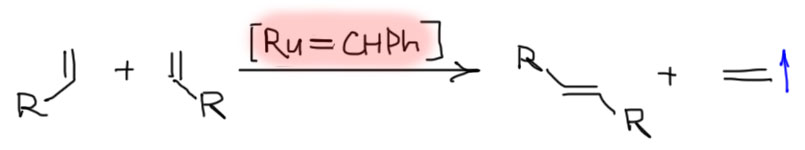
Главная проблема метатезиса, повторим еще раз, обратимость этой реакции. Обратимость приносит гораздо больше проблем, чем просто сложностьи в получения выхода продукта, превышающего величину, определяемую константой равновесия. Если мы возьмем два разных олефина, хотя бы и самых простых, монозамещенных, то первый продукт метатезиса будет реагировать с каждым из исходных олефинов, в результате получится смесь всех возможных продуктов. Такие процессы в химии часто называют рандомизацией – то есть образованием смесей всех возможных комбинаций, задача таким образом из химической становится вероятностной. Нам такой метатезис не нужен – мы всегда хотим чтобы реакции были селективными и давали высокие выходы.
Наиболее перспективный случай – когда мы берем один монозамещенный олефин. В этом случае мы получаем симметричный дизамещенный олефин и этилен. Этилен – газ, он улетает, равновесие смещается. В схеме реакции продукт нарисован в транс-форме, но метатезис, увы, не стереоселективен. Поскольку реакция обратима, в смеси будет преобладать более устойчивый олефин, то есть обычно транс, но, увы, разность энергий цис- и транс-форм обычно не так велика, чтобы цис-изомера в равновесной смеси не было вовсе или было мало. Еще раз напомню, что хотя методов получения двойной связи мы знаем очень много, и присоединили к ним еще и модный метатезис, но если нужны чистые цис- или транс-изомеры, единственный надежный метод – гидрирование ацетиленов.
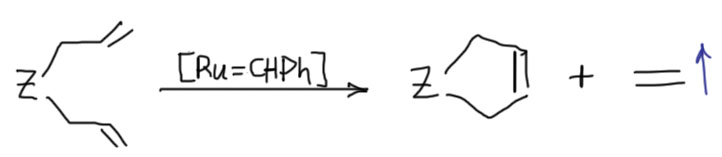
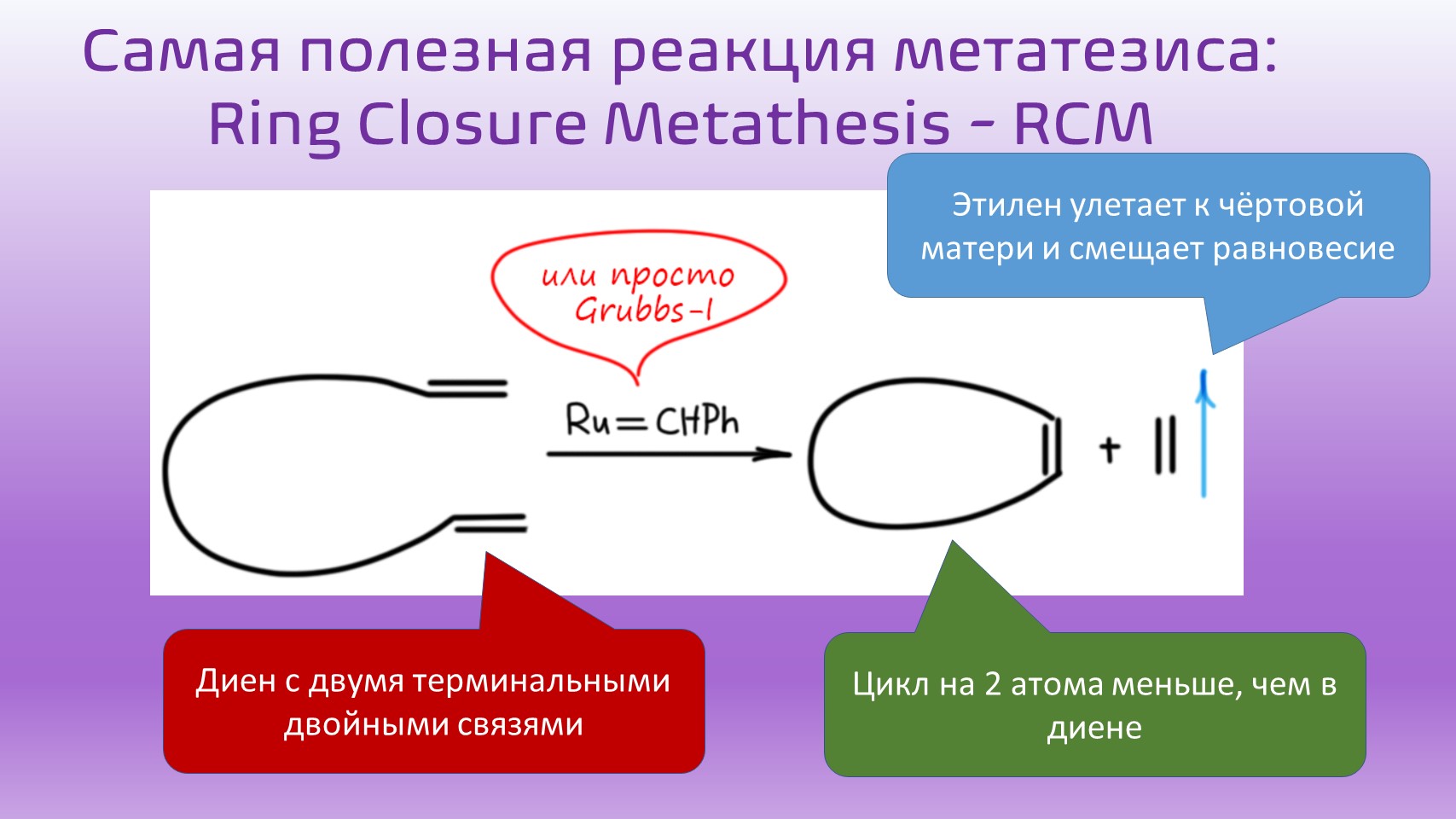
Самая полезная и важная разновидность этого метода – циклизация, внутримолекулярный метатезис в диенах, когда двойные связи находятся на концах цепи.
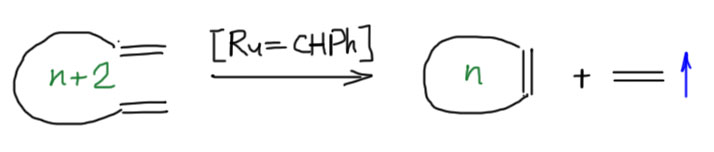
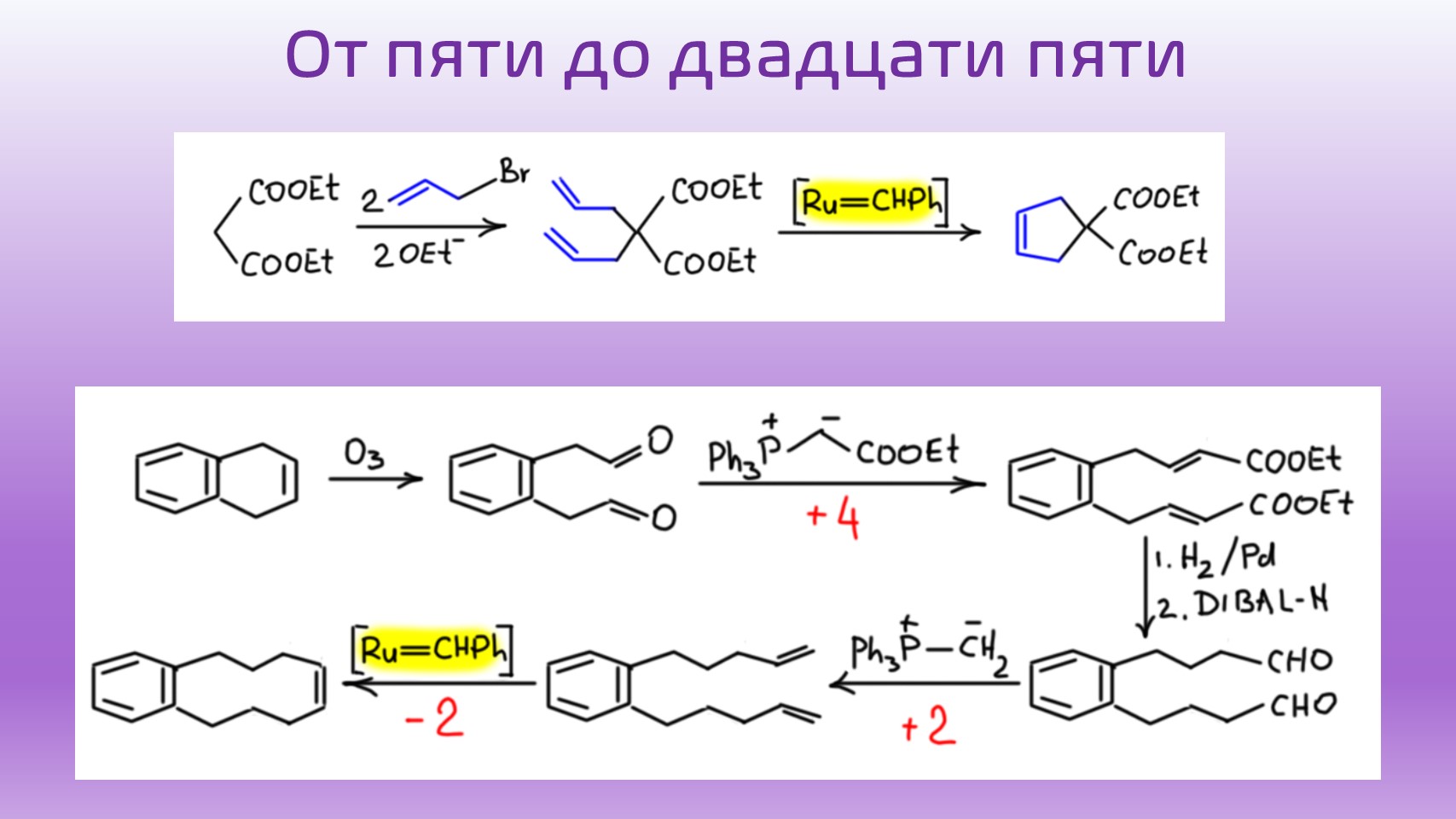
Циклизация метатезисом (часто сокращают как RCM – ring closure metathesis) стала просто невероятно популярным методом получения циклических олефинов. Так получают и 5/6-членные циклы, и циклы большего размера вплоть до больших макроциклов, и не только карбоциклы, но и гетероциклы. Пяти и шести-членные циклы чаще всего собирают из аллилов, который очень легко вводить реакциями SN2-замещения и другими методами (перегруппировкой Кляйзена, например).
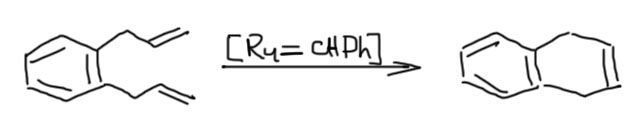
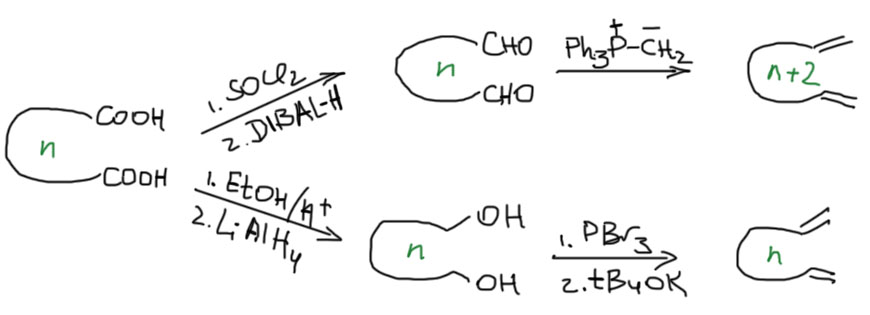
Большие циклы делают из длинных цепочек с двойными связями на концах. Такие диены можно легко получить известными нам методами.
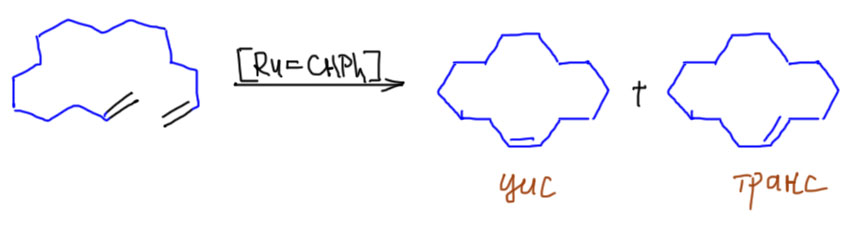
К сожалению, так как для больших циклов двойная связь может быть и цис- и транс-, начиная с 8-членных циклов, в этой реакции нельзя добиться образования чистого диастереоизомера, и с 10-членных циклов всегда образуются смеси. Например, для 14-членного цикла получится смесь:
Как ни странно, наш старый добрый метод построения больших циклов ацилоиновой конденсацией, в этом смысле более гибок, так как позволяет нам чисто получать по необходимости и цис-, и транс-изомеры. Но – для этого потребуется несколько стадий.
Первая лекция
Обе лекции теперь имеют аудио-дорожки, и можно послушать типа лекции. Но, как я и предупреждал, первая лекция – это лекция, прочитанная самому себе, а это самое тоскливое мероприятие, которое только можно придумать. Поэтому, не обессудьте, это невероятно скучно. Обязательно, если решитесь это слушать, обзаведитесь чашечкой хорошего кофе или хотя бы чая. И никогда не слушайте в одиночестве – рядом должны быть родные или знакомые, которые смогут вас спасти, если внезапно овладевший вами глубокий сон приведет к падению и травмам, потере пространственной ориентации, дыханию Чейн-Стокса и другим неожиданным проблемам. Попросите каждые 15 минут проверять зрачки и пульс.
Если всё это готово, можно приступать.




Вторая лекция
Вторая лекция сделана немного по-другому. Я догадался её записать, поэтому там слайды с голосовым сопровождением, можно послушать. Жаль, что не догадался записать первую, возможно, как-нибудь прочитаю её сам себе и запишу, но не обещаю, потому что это тоска смертная самому себе читать лекции.

Программа раздела
Понятие о каталитической реакции с участием комплексов переходных металлов на примере реакции кросс-сочетания. Основные стадии реакции кросс-сочетания. Роль фосфиновых лигандов в каталитическом цикле.
Сравните синтез дифенилов через бензидиновую перегруппировку и реакцией кросссочетания.
Использование фенолов в реакции кросс-сочетания.
Олефины в реакциях с участием комплексов переходных металлов: Реакция Хека галогенпроизводных с олефинами и метатезис олефинов.