Нуклеофильное алифатическое замещение у sp3-гибридного углерода
Третий раздел курса устроен не так как предыдущие или вообще все остальные – речь здесь идет не о классах органических соединений, а о механизмах. И только через механизмы обсуждается, и то довольно кратко и немного невнятно, химия соединений, намертво связанных с обсуждаемыми механизмами – насыщенных галогенпроизводных, спиртов, простых эфиров и специального типа простых эфиров – оксиранов. Сразу смиритесь, что материала здесь много, что он может показаться нудным словоблудием, и что реальная химия этого раздела выглядит довольно бледно и однообразно.
Это связано с чрезвычайной важностью этих механизмов, и даже точнее, типов реакций (под типом подразумевается множество реакций, объединенных общими признаками и механизмом). Реакции нуклеофильного замещения на насыщенном углероде очень часто встречаются в химии самых разных классов. Иногда об этом даже не догадываются. Вот, например, мы только что обсуждали химию ацетиленов и выяснили, что важнейшим и даже единственным общим методом синтеза является алкилирование ацетиленид-аниона. Мы сказали, что реакция эта очень хороша, но ее не стоит применять к вторичным алкилгалогенидам и ни в коем случае нельзя – к третичным. Эти ограничения непосредственно следуют из того, что эта реакция является типичным случаем нуклеофильного замещения у насыщенного атома углерода и подчиняется всем типичным ограничениям этого типа реакций. Кто-то непременно возразит – почему это у насыщенного, там же sp-гибридный карбанион! Да, здесь есть некоторая двусмысленность в выборе реакционного центра – можно считать, что главным реакционным центром в этой реакции является sp-гибридный углерод с минусом и неподеленной парой, а можно считать, что таковым является sp3-гибридный углерод галогенпроизводного. Мы же видим, что в реакции происходит разрыв связи с галогеном и образование новой связи C-С. Это именно замещение – одна группа замещает другую. Так как эти события происходят как раз у насыщенного атома углерода, то нам удобно смотреть на эту реакцию именно так – как на замещение у насыщенного атома, причем раз эта реакция происходит под действием нуклеофила (с классификацией реагентов мы уже познакомились и смирились).
С реакциями нуклеофильного замещения у насыщенного углерода мы будем сталкиваться все время – в каждой теме. И если разберемся один раз, как они идут, что требуют, чего не любят, – то жизнь наша станет лучше и веселее. Нам больше не нужно будет для каждой такой реакции (а я не преувеличиваю – таких реакций будет много – в конце попробуем подсчитать сколько) заново выяснять закономерности. Более того, оказывается, что многие вещи, изученные в этом разделе, помогут нам и в других. Сэкономим кучу времени и сил. Игра стоит свеч.
Подробная программа раздела
1. Что такое нуклеофильное замещение. Типы нуклеофилов (анионы,нейтральные молекулы, растворители). Уходящие группы.
2. Типы реакций нуклеофильного замещения. 1) Y– + RX; 2) Y: + RX; более редкие случаи пока не обсуждали:
3) Y– + RX+, например Вг– + R4N+ = RBr + R3N;
4) Y: + RX+, например R3N + R’SR”2 = R3NR’+ + SR”2.
3. Классификация по механизму.
|
SN1 |
SN2 |
|
SN1, кинетическое ур-ние |
SN2, кинетическое ур-ние |
|
Скорость реакции не зависит от природы нуклеофила и его концентрации |
Скорость реакции зависит от природы нуклеофила и его концентрации |
|
Вид энергетического профиля реакции: двустадийный процесс с участием промежуточного соединения (интермедиата). Скоростьопределяющая стадия – гетеролиз связи с уходящей группой. |
Вид энергетического профиля реакции: одностадийный согласованный процесс. Нет интермедиатов. Переходное состояние – необходимо точно понимать разницу между переходным состоянием и интермедиатом. |
|
Чрезвычайно велика роль растворителя, так как именно этот фактор имеет ключевое значение в процессе разрыва связи с уходящей группой. Наиболее благоприятны растворители, сильно сольватирующие уходящие группы и облегчающие гетеролиз – полярные протонные растворители |
Влияние растворителя зависит от нуклеофила – оно невелико для нейтральных нуклеофилов (типа аммиака, аминов, и т.п.), и очень велико для анионных нуклеофилов, вводимых в реакцию в виде солей. В этом случае наибольшая реакционная способность достигается в апротонных растворителях высокой полярности. |
|
Стереохимия: карбокатион плоский, атака нуклеофила с любой стороны. Стереохимический результат – рацемизация (в реальных системах на стереохимию влияет участие ионных пар, поэтому полная рацемизация встречается редко) |
Всегда атака с тыла. Вальденовское обращение. |
|
Пространственные факторы в первом приближении не важны. Однако переход от sp3 к sp2уменьшает стерич. отталкивание, что сильно чувствуется при очень объемных группах (напр. t-Bu (1) и t-Bu3C (13500), сольволиз |
Пространственные факторы очень важны, см. многочисл. примеры и п.4 ниже |
|
В синтезе используется редко (превращение третичных спиртов в бромпроизводные под действием концентрированной HBr, расщепление трет-бутиловых и других трет-алкильных эфиров) |
Очень широко используется в синтезе. Препаративные реакции этого типа обычно называются алкилированием. |
|
КОНКУРЕНТНО ПРОХОДИТ ЭЛИМИНИРОВАНИЕ, кроме случаев, когда R=Me, Allyl, Bn или нет b–Н атомов |
|
4. Влияние структурных факторов в рядах Me- Et- i-Pr- t-Bu; Et- Pr- i-Bu- Np-. Бициклические субстраты. Винил-, фенил-, аллил- и бензил- галогениды в реакция НЗ. a –галогенкарбонильные соединения.
4-MeO- и 4-NO2-бензилхлориды. Аллильная перегруппировка.
5. Природа нуклеофила. Нуклеофильность и основность. Поляризуемость и сольватация нуклеофила. Мягкость и жесткость нуклеофила. Конкуренция SN и E реакций. a–эффект и супернуклеофилы. “Голые” ионы. Межфазный катализ. Краун-эфиры, криптанды.
6. Природа растворителя. Влияние на SN1 и SN2. Ряд e: диоксан (2.2), Et2O (4.22), моноглим (7.2), ТГФ (7.4), ацетон (20.5), ГМФТА (30), ДМФ (36.7), МеNО2 (38,6), сульфолан (44), ДМСО (49), e > 15 – биполярные, апротонные растворители. Они не должны содержать воды!!!
7. Уходящая группа. Оценка Методы создания хорошей УГ. Протонирование, эфиры сильных неорганических кислот, электрофильный катализ, использование фосфорорганических соединений (Ph3P/CCl4, Ph3P/Br2, реакция Мицунобу).
8. Участие соседних групп. Иприт. Таблица (Беккер с. 198).
9. Кратко, факультативно: Более подробное рассмотрение механизмов НЗ. (“Интимные” (контактные) и сольватно-разделенные ионные пары, солевые эффекты, случай SOCl2, суперкислые среды.
10-11. Важнейшие примеры синтетического использования реакций НЗ.
Разговор о нуклеофильном замещении у насыщенного углерода всегда разворачивается вокруг механизма реакции. Основных механизма два – SN2 и SN1 (именно в таком порядке). Важно сразу понять, что разговор о механизмах – это просто удобный способ обсудить гораздо более практические вещи – как понять, идет или нет та или иная реакция, идет ли она быстро или медленно, что нужно сделать, чтобы медленная реакция стала быстрее, и т.п. Два разных механизма – это, на самом деле, фактически две разные химии. Если мы в SN2, то мы думаем о нуклеофилах, растворителях, подборе реагента под конкретную задачу, старательно избегаем отщепления, и вообще активно управляем событиями в колбе. Место реакций SN2-типа среди методов органического синтеза не просто велико, а в полном смысле этого слова колоссально во всех классах органических соединений. Еще более велико значение этого механизма и связанных с ним процессов в разработке теории органической химии, механизма, выяснения роли среды и других факторов. С другой стороны, SN1 механизм дажеко не так влиятелен. Парадокс в том, что оба механизма возникли одновременно как парные и взаимодополняющие (комплементарные), но развивались совершенно независимо друг от друга. SN1-замещение занимает очень скромное место среди методов органической химии, и реакции этого типа единичны, очень скромны по возможностям и до очень недавнего времени почти не развивались. Львиная доля интереса к механизму SN1 была связана не с разработкой методов синтеза, а с химией карбокатионов, их перегруппировками, структурой, способами стабилизации. В остальном этот механизм и этот тип реакционной способности можно считать одними из наименее полезных в практическом смысле слова во всяком случае, если не выходить далеко за рамки изучаемого курса органической химии. Если мы в SN1, то наш удел – беспомощно наблюдать за тем, как исходный субстрат превращается совсем не в то, что задумывалось, как включаются перегруппировки карбокатионов, и практически никаких способов направить процесс в нужное русло у нас нет. Эти два механизма и связанные с ними реакции необходимо сразу развести как можно дальше друг от друга. Есть случаи, когда они реально могут конкурировать друг с другом в одной колбе, но такие случаи довольно редки, и мы их тоже не оставим без внимания. В большинстве реально встречающихся реакций нет никаких проблем в том, чтобы распознать механизм, и переключить мозги на соответствующий режим.
Начнем мы с SN1-механизма, создающего больше проблем чем решений. Чтобы избегать проблем неплохо понять их источники. Кроме того мы обязательно выясним, где этот механизм все же приносит пользу и служит целям синтеза.
Механизм SN1
Смысл механизма SN1 заключается в том, что субстрат сначала медленно, самопроизвольно и обратимо распадается на карбокатион и уходящую группу. Карбокатион немедленно после образования вступает во взаимодействие с первым попавшимся нуклеофилом, образуя продукт реакции замещения. Альтернативно карбокатион может вступить в реакцию с первым попавшимся основанием, образовав продукт отщепления, и такой путь называется E1-элиминированием, но для этого необходимо, чтобы рядом с карбокатионным центром был еще один углерод с атомом водорода. Из этого сразу следует важный вывод: SN1-змещение и E1-элиминирование – это не два разных процесса, а фактически один процесс с двумя конкурирующими исходами.
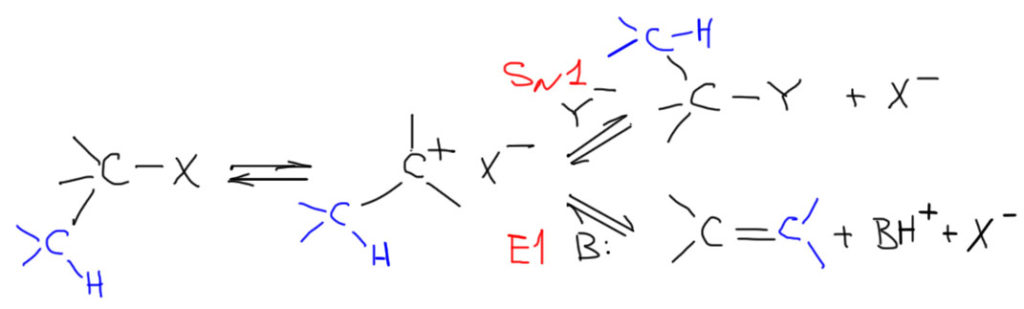
Соотношение продуктов замещения и элиминирования поэтому не поддается никакому контролю извне – проще говоря, мы ничего не можем с этим сделать – и определяется свойствами и составом реакционной смеси. Мы это еще обсудим отдельно, но сразу отметим, что относительный выход продукта элиминирования неизбежно будет увеличиваться, если в реакционной смеси присутствуют молекулы или ионы, обладающие заметной, хотя и слабой основностью. Почему только слабой??? Да просто потому, что сильные основания неизбежно переключают механизм в E2 и берут управление скоростью процесса на себя.
Необходимое условие для SN1 - стабилизированный карбокатион
Механизм SN1/E1 может реализоваться тогда и только тогда, когда карбокатион, который должен образоваться, достаточно стабилизирован. В самопроизвольных SN1 реакциях подавляющем большинстве случаев это или третичный алкильный карбокатион, или карбокатион, стабилизированный соседним мезомерным донором. Обратите на это внимание – вторичные алкильные карбокатионы считаются недостаточно стабилизированными, и самопроизвольно не образуются. Это вполне согласуется с тем, что вторичные алкилгалогениды и похожие на них субстраты не подвергаются сольволизу с измеримой скоростью.
Третичные карбокатионы стабилизированы донорным индуктивным эффектом трех алкильных групп. Это дает неплохую степень стабилизации. При пониженной температуре трет-бутильный катион и некоторые его аналоги были экспериментально обнаружены спектроскопией ЯМР и рядом других методов. Ни в тех же условиях, и ни в каких других ни вторичные, ни первичные катионы никогда и никем не наблюдались несмотря на весьма значительные усилия и очень широкий набор задействованных методов. Это дает дополнительные основания считать, что степень стабилизации третичных алкильных катионов действительно намного выше, и есть принципиальная разница в поведении третичных субстратов и всех остальных.
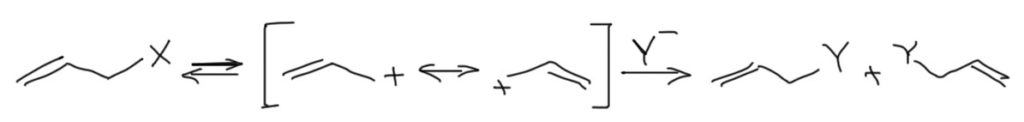
Если не ограничиваться простыми алкилами и допустить разные заместители, то ассортимент стабилизированных катионов существенно расширяется. Любые мезомерные доноры рядом с катионным центром дают существенно большую степень стабилизации. Самый простой такой заместитель – винильный остаток. В таких случаях образуется мезомерно-стабилизированный аллильный катион. Не забывайте, что в таких катионах положительный заряд делокализован, и после его образования два атома углерода становятся или совсем одинаковыми (если аллил незамещенный или симметрично замещенный), или не совсем (если аллильный катион замещен несимметрично). Поэтому в SN1 реакциях аллильных субстратов обычно получаются смеси двух продуктов (если аллил симметричный, то, конечно, эти продукты одинаковы, если только там нет изотопных меток):
В пропаргильной системе (то же, что аллил, только с тройной связью) наблюдается нечто похожее, но несколько более сложное, так как резонансные формы сильно различаются. Нарисуйте это сами. Мы не будем рассматривать пропаргильные субстраты в SN1 реакциях.
Бензильные субстраты также дают стабилизированный карбокатион. Но в отличие от аллильной системы делокализация карбокатиона не приводит к образованию смеси продуктов после реакции с нуклеофилом просто поому, что только в одном случае сохраняется ароматическая система.
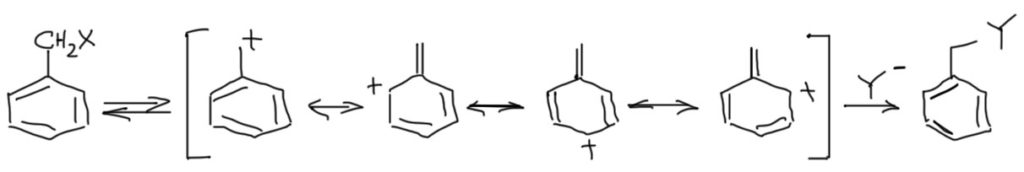
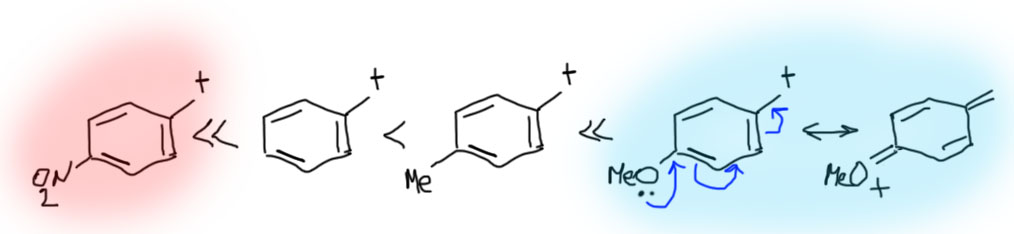
Замещение в ароматическом кольце очень сильно влияет на стабилизацию бензильных катионов. Наличие в орто- и пара-положениях донорных групп дополнительно стабилизирует бензильный катион. Мезомерные доноры, как и положено, сильнее индуктивных. А вот акцепторы, особенно такие мощные как нитро-группа, дестабилизируют бензильные катионы в очень существенной степени, настолько, что в этом случае SN1 реакции можно считать маловероятными. Обратите внимание, что при наличии мезомерного донора в орто- или пара-положениях в делокализации участвует только структура с участием этого самого донора (цепь сопряжения донор-акцептор), а делокализацией в кольцо можно и нужно пренебречь как несущественной (это значит, что обычные граничные структуры для бензильного катиона, как в схеме выше, нарисовать можно и это корректно, но их роль в делокализации пренебрежимо мала, и если нас интересует суть дела, а не только формальные картинки, такие структуры из мезомерной делокализации лучше убирать):
Еще более стабилизированы катионы с двумя ароматическими кольцами – они называются бензгидрильными.
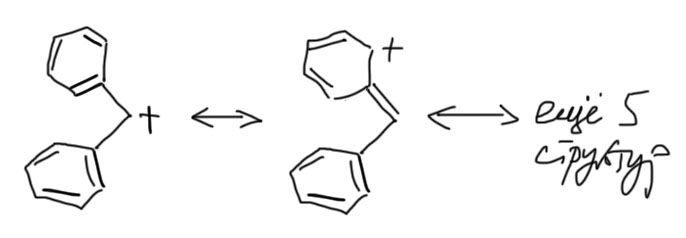
Такие субстраты наиболее хорошо исследованы в SN1 реакциях сольволиза. Фактически все, что написано в учебниках про сольволиз и вообще SN1 реакции было реально сделано на субстратах бензгидрильного типа. Это довольно странная история, но это так – экспериментально механизм SN1 в сольволизе убедительно изучен только для настолько сильно стабилизированных карбокатионов, а все, что мы говорим про третичные алкилы – просто результат обобщения, не подтвержденный в экспериментах. Именно поэтому на счете существует немало серьезных ученых, которые вообще не признают механизм SN1 и считают его очень частным случаем для совсем сильно стабилизированных карбокатионов типа бензгидрильных и трифенилметильных, а общим механизмом нуклеофильного замещения считают только SN2, даже для третичных алкильных, аллильных и бензильных субстратов, и объясняют их особенности так называемым переменным переходным состоянием (эта теория известна, как диаграммы Мор о’Феррала-Дженкса). Я не разделяю эту точку зрения и вам не советую, так как никаких существенных плодов эта теория за прошедшие десятилетия не принесла, а в науке принято орудовать бритвой Оккама, безжалостно отсекая все лишнее и бесплодное, даже если выглядит красиво.
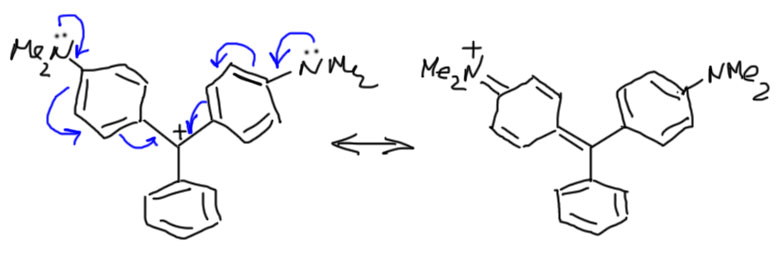
Когда ароматических кольца три, стабилизация становится настолько значительной, что это даже немного мешает. Трифенилметильные катионы очень легко образуются, и настолько стабильны, что нарушают главное правило SN1: карбокатион не выбирает нуклеофила, реагирует с первым попавшимся, скорости реакции с любыми нуклеофилами одинаковы. Трифенилметильные катионы, наоборот, проявляют немалую избирательность, и даже вообще не реагируют со слабыми нуклеофилами. Совсем сильная стабилизация получается, если в каждом из фенильных колец находятся донорные заместители. Самые сильные мезомерные доноры – фенольные и аминогруппы – делают трифенилметильные катионы настолько стабильными, что они спокойно живут в водных растворах и не проявляют никакого желания реагировать ни с водой, ни с многими другими нуклеофилами. Такие карбокатионы обычно имеют яркий цвет и очень широко применяются как красители (это слово имеет простоватый вид, но это очень мощный научный термин, обозначающий вещества, взаимодействующие с видимым или ультрафиолетовым светом, что совершенно необходимо в десятках и сотнях самых разнообразных областей применения). С одним из таких мы все знакомы с раннего детства – это соединение, имеющее название малахитовый зеленый или просто зелёнка. Этот карбокатион стабилизирован всего двумя амино-группами, но этого уже достаточно для практически неограниченной стабильности. Показана только одна граничная структура, вторая такая же на втором кольце.
SN1 реакции бывают самопроизвольными или принудительными
Самопроизвольные реакции SN1 происходят тогда, когда субстрат самопроизвольно и обратимо распадается на карбокатион и уходящую группу. Для того чтобы это было возможно необходимо чтобы
- карбокатион был достаточно стабилизирован, и чем стабильнее, тем лучше;
- уходящая группа была хорошей, и чем лучше, тем еще более лучше;
- растворитель (среда) дополнительно стабилизировали и карбокатион, и уходящую группу.
С карбокатионом разберемся на отдельной вкладке. Хорошие уходящие группы это, прежде всего, атомы галогенов кроме фтора. И это, безусловно, тозилат и другие сульфонаты, но только эти производные очень непросто получить в случае третичных спиртов. Есть еще несколько экзотических групп, которые нам пока не понадобятся. Все остальное – вне закона. В частности, спирты никогда не вступают в самопроизвольные SN1-реакции. Но отлично вступают в принудительные, и об этом поговорим отдельно.
Обратите внимание, что третье условие не менее важно, чем первые два. Распад ковалентной связи на ионы (гетеролитический распад) невозможен без активного взаимодействия в растворителем (сольватации). Ни в газовой фазе, ни в неполярных растворителях образование ионов не происходит даже в тех случаях, когда образующиеся ионы считаются вполне стабильными (мы уже встречались с этим в очень жесткой форме – даже молекулы галогеноводородов типа HBr не распадаются на ионы ни в газовой фазе, ни в малополярных растворителях, отчего нам и пришлось в теме про присоединение к алкенам изобретать молекулярные механизмы вместо того чтобы просто вешать на двойную связь протон). Это условие жестко ограничивает круг растворителей, в которых возможен такой процесс. Поскольку речь идет о разрыве ковалентной связи с образованием ионов растворитель должен быть, насколько возможно, полярен. Поскольку речь идет об органических растворителях, в этом отношении приходится предъявлять достаточно скромные требования, но и забывать об этом не стоит. Чтобы дополнительно стабилизировать катион, необходимы атомы с неподеленными парами. Следовательно растворитель должен обладать основностью (по Льюису или Бренстеду-Лоури, в данном контексте это не важно тем более что эти два типа основности вполне совместимы). Чтобы стабилизировать анион нужна водородная связь. Следовательно растворитель должен быть кислотой Бренстеда-Лоури. Такие растворители еще называют протонными. Этот мир устроен так, что протонные растворители всегда обладают еще и атомами с неподеленными парами, и очень часто обладают неплохой полярностью, следовательно определение “протонный растворитель” фактически вбирает в себя все три необходимых свойства. При этом, эти свойства дополняют друг друга, и если не хватает одного, может выручить другое. Например, вода обладает огромной полярностью, но не выдающейся кислотностью, и за счет полярности является превосходным растворителем для SN1, хотя эту роль не просто реализовать из-за низкой растворимости органических веществ в воде. Чтобы справиться с низкой растворимостью используют смеси воды со смешивающимися с ней органическими растворителями (метанолом, этанолом, ацетоном, ацетонитрилом, диоксаном и т.п.) – полярность среды от такого смешения снижается, но остается достаточно высокой. Трифторуксусная кислота CF3COOH – малополярная жидкость, но весьма приличная по силе кислота (pK между 1 и 2) и только за счет кислотности является одним из самых выдающихся растворителей для SN1. По той же причине 2,2,2-трифторэтанол гораздо лучше просто этанола (опять кислотность). Муравьиная кислота намного лучше уксусной – уже по причине более высокой полярности при ненамного большей кислотности.
Самопроизвольные SN1-реакции делают просто – растворяют субстрат в протонном растворителе и терпеливо ждут установления равновесия. Можно немного нагреть, но при этом всегда увеличивается доля E1-элиминирования. В таких реакциях всегда получается продукт реакции карбокатиона с растворителем просто потому, что любой такой растворитель является нуклеофилом (в нем есть неподеленные пары), карбокатиону все равно с каким нуклеофилом взаимодействовать, а растворитель всегда под боком. Поэтому такие реакции называют сольволизом, то есть буквально расщепление растворителем (расщепление связи в исходном субстрате). Получаем почти что закон (для особо внимательных замечу, что обратное неверно: сольволиз далеко не всегда является SN1-реакцией, вполне возможен и SN2-сольволиз, и другие есть важные механизмы сольволиза):
Самопроизвольные SN1-реакции – это сольволиз.
Сольволиз – общий термин, в частных случаях используют более конкретные: реакцию с водой называют гидролизом, с этанолом – этанолизом, с уксусной кислотой – ацетолизом, с муравьиной – формолизом, с трифторуксусной – трифторацетолизом, и т.п. Вот пара очевидных примеров:
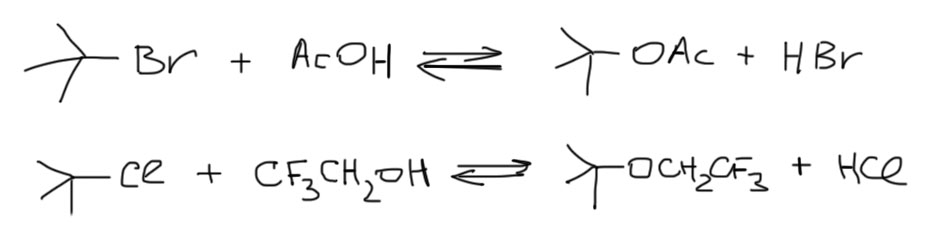
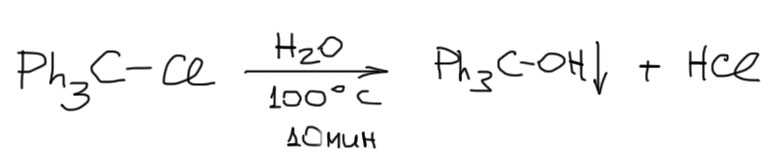
Реакция SN1-сольволиза – всегда равновесие, кроме продукта сольволиза образуется уходящая группа в форме сопряженной кислоты, а это почти всегда – сильная кислота Бренстеда-Лоури, и ее накопление в реакционной смеси вызывает еще и побочные реакции за счет протонирования субстрата, но добавлять в смесь какое-нибудь основание (типа соды) нельзя – сразу получите больше элиминирования. Вывод из всего этого довольно простой – как правило, реакции сольволиза практически бесполезны: их невозможно довести до конца, сместить равновесие, ускорить. Тем не менее, совсем выбрасывать их не нужно, потому что можно найти случаи, когда эти реакции довольно быстры, а отсутствие возможности для элиминирования делает возможным добавление основания и смещение равновесия в сторону продукта, в том числе и за счет банального избытка сольволизующего растворителя, как например, в реакциях трифенилметилхлорида, где все сходится в пользу SN1-сольволиза – и очень стабильный трифенилметильный катион, что обеспечивает высокую скорость, и выпадение продукта в осадок, что обеспечивает смещение равновесия, и отсутстие возможности элиминирования. Но это – только частный случай!:
Принудительные SN1-реакции
Если бы SN1-реакции ограничивались бы только самопроизвольным сольволизом, от этой химии было бы совсем мало прока. Но можно попробовать не ждать, когда связь с уходящей группой разорвется самопроизвольно, а попробовать ускорить это и даже, при некотором стечении обстоятельств, сделать необратимым. Получающемуся таким образом катиону можно попробовать подсунуть какой-нибудь конкретный нуклеофил, и тогда можно ожидать получения желаемого продукта замещения (E1 при этом будет продолжать конкурировать, но и здесь тоже есть кое-какие резервы). Но задача это непростая, ведь придется не только придумать способ ускорить образование карбокатиона, но и обеспечить условия образования только одного продукта. Для этого необходимо будет устроить тщательную зачистку реакционной смеси от конкурирующих нуклеофилов, ведь карбокатиону все равно, с чем реагировать, и выбирать он не будет (это фундаментальное свойство почти всех карбокатионов – они не выбирают, с каким нуклеофилом реагировать, и если есть несколько, всегда будут давать смесь с соотношением продуктов, приблизительно равным соотношению концентраций нуклеофилов (это называется неконкурентным связыванием). Придется и растворитель подобрать, и тщательно посмотреть на все анионы – чтобы, если без них нельзя, остались бы только ненуклеофильные типа тетрафторбората. Это очень важно, потому что карбокатионы очень реакционноспособны и находят нуклеофилы в самых неожиданных местах. Например, исследования Н.С. Зефирова и сотр. недвусмысленно показали, что карбокатионы отлично связывают перхлорат-анион, который до этого считался совсем ненуклеофильным и безобидным.
Посмотрим на основные приемы принудительного SN1. Все они крутятся вокруг одной генеральной идеи – создать прямо в реакции настолько хорошую уходящую группу, чтобы она отвалила от карбокатиона, не помахав даже платочком на прощание. Но из этого следует и один очень полезный вывод – в отличие от самопроизвольных реакций сольволиза, в этих методах мы не ограничены узким набором уходящих групп и можем стартовать с самых разных уходящих групп, даже очень скверных. И в заключение предисловия замечу, что принудительные SN1-реакции очень хорошо развиваются с современной органической химии в новом тысячелетии. Если заинтересуетесь, найдете немало новых методов синтеза, включающих образование карбокатионов за счет целенаправленной помощи уходу уходящей группы. Интересно, что еще недавно SN1-реакции считались узкими и малоперспективными.
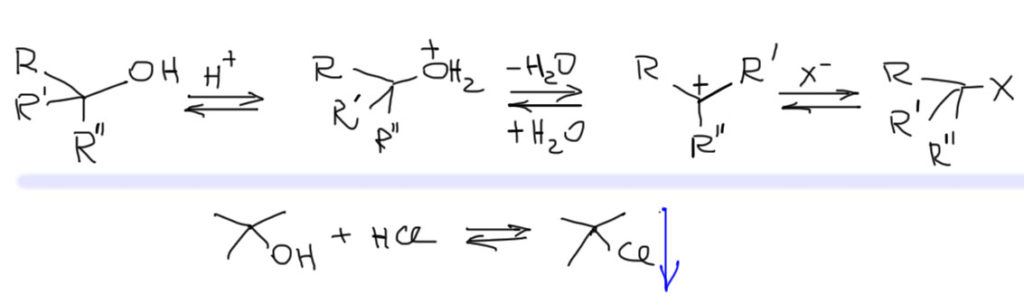
1. Самый простой и древний – протонирование сильной кислотой. Строго этот способ называется специфическим кислотным катализом. Этот способ работает для спиртов и многих других кислородсодержащих производных, например, простых эфиров. Со спиртами сильная кислота протонирует очень плохую уходящую группу гидроксил, переводя ее в отличную уходящую группу воду. Проблема метода – мы не можем избавиться от воды, и карбокатион будет связывать ее обратно, то есть реакция обратима. В выборе нуклеофила мы поэтому очень сильно ограничены, и потому что вода конкурирует, и потому что в сильнокислой среде большинство нуклеофилов протонируются и перестают быть нуклеофилами. Но – работают галогенид-ионы, которые сохраняются в сильнокислой среде. Реакции дополнительно помогает низкая растворимость галогенпроизводных, которые выпадают в отдельную фазу, смещая равновесие. Так получают галогенпроизводные из третичных спиртов. Собственной кислотности галогеноводородов достаточно.
Нечто очень похожее происходит и в реакции Риттера. В этой реакции карбокатион генерируют сильной кислотой из спирта, и подсовывают ему нитрилы, очень слабые нуклеофилы. Но карбокатиону все равно, а образующийся четвертичный катион присоединяет воду в необратимой реакции. Мы еще вернемся к этой реакции в Аминах.
 2. Если уходящая группа галоген, а иногда и в других случаях, используются разнообразные кислоты Льюиса, образующие координационную связь с уходящей группой. И хотя галогены уже хорошие уходящие группы, но самопроизвольный уход их происходит очень медленно. Уход комплексных анионов, образующихся за счет координации галогенида с кислотой Льюиса, ускоряет этот процесс на многие порядки. Подбор кислоты Льюиса весьма широк и приблизительно определяется принципом ЖМКО. Очень приблизительно, на самом деле, но все же это полезно. Так, если у вас легкий галоген – фторид (да, в принудительном SN1 фтор вполне годится в отличие от самопроизвольного сольволиза) или хлорид, то мы будем выбирать жесткую кислоту – BF3, SbF5, AlCl3, и т.п. Бром и особенно иодпроизводные чаще стимулируют мягкими кислотами Льюиса типа солей серебра. В совсем современной химии ассортимент кислот Льюиса для стимулирования карбокатионной химии очень широк, и там встречаются многие довольно экзотические металлы. Дальше карбокатиону подсовывают желательный нуклеофил. Опять нужно не забывать контролировать и не допускать конкурирующие нуклеофилы в растворителях, и даже анионах в солях металлов. Главное ограничение этого метода состоит в том, что приходится думать о том, не будет ли планируемый нуклеофил связываться с кислотой Льюиса легче чем галогенид. И это не очень просто.
2. Если уходящая группа галоген, а иногда и в других случаях, используются разнообразные кислоты Льюиса, образующие координационную связь с уходящей группой. И хотя галогены уже хорошие уходящие группы, но самопроизвольный уход их происходит очень медленно. Уход комплексных анионов, образующихся за счет координации галогенида с кислотой Льюиса, ускоряет этот процесс на многие порядки. Подбор кислоты Льюиса весьма широк и приблизительно определяется принципом ЖМКО. Очень приблизительно, на самом деле, но все же это полезно. Так, если у вас легкий галоген – фторид (да, в принудительном SN1 фтор вполне годится в отличие от самопроизвольного сольволиза) или хлорид, то мы будем выбирать жесткую кислоту – BF3, SbF5, AlCl3, и т.п. Бром и особенно иодпроизводные чаще стимулируют мягкими кислотами Льюиса типа солей серебра. В совсем современной химии ассортимент кислот Льюиса для стимулирования карбокатионной химии очень широк, и там встречаются многие довольно экзотические металлы. Дальше карбокатиону подсовывают желательный нуклеофил. Опять нужно не забывать контролировать и не допускать конкурирующие нуклеофилы в растворителях, и даже анионах в солях металлов. Главное ограничение этого метода состоит в том, что приходится думать о том, не будет ли планируемый нуклеофил связываться с кислотой Льюиса легче чем галогенид. И это не очень просто.
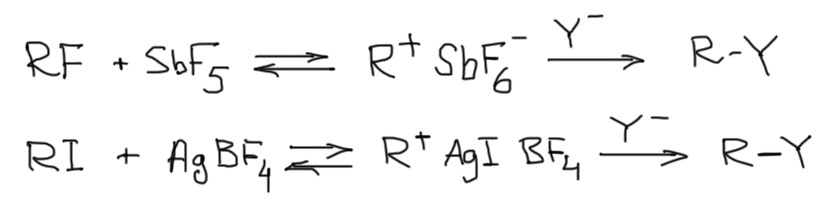
3. Первичные амины можно отлично сделать субстратами для SN1 за счет реакции диазотирования. При этом создается самая выдающаяся уходящая группа всех времен и народов – молекулярный азот. Уход азота фактически необратим (есть доказательства обратимости и в этом случае, но не очень убедительные). Такие реакции называются реакциями дезаминирования. Нуклеофилом обычно является вода, потому что диазотирование ведут в водной среде. Но бывают и более сложные варианты без участия воды и с другими нуклеофилами. Подробнее об этой реакции поговорим в теме Амины.
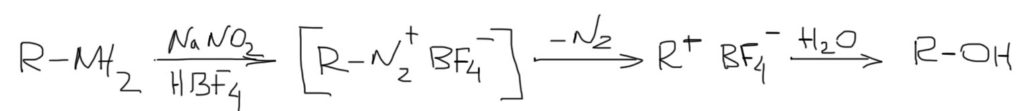
4. Есть и еще один очень необычный метод. Если получить изотопно-замещенный углеводород, содержащий вместо одного из водородов атом трития, то мы получим вроде бы идеальный источник практически любых карбокатионов. Тритий очень быстро распадается за счет варианта β-распада. Атом трития моментально превращается в атом гелия, который вообще не может образовать химическую связь, то есть связь углерода с тритием моментально и полностью исчезает, а на углероде образуется положительный заряд. Вот это уходящая группа! – раз, и нет ее! Прямо исчезающая группа, азот отдыхает! Когда эту штуку придумали, казалось, что все проблемы с карбокатионами и их реакциями можно будет разрешить, хотя и работать с тритием очень непросто. Увы, есть проблема. Когда химическая связь просто исчезает, это ее проблема, но энергия связи исчезнуть не может, она просто целиком переселяется на образующийся карбокатион, а это ситуация, совсем необычная для обычных химических реакций. В любой реакции, с которыми мы имеем дело, каждая молекула или ион всегда, в каждое ничтожное мгновение своей жизни находятся в тепловом равновесии со средой. Это очень важное обстоятельство, которое только и позволяет нам пользоваться обычными понятиями химической кинетики и термодинамики, константами скоростей и равновесий, энергией активации и пр. и др. Мы по-другому не умеем, хотя и не задумываемся об этом. А если какая-то частица откуда-то приобрела избыточную энергию (такие молекулы или ионы называют “горячими”), то именно это для нее и является главной проблемой, и ее поведение очень сильно осложняется. Мы не сможем воспользоваться данными по поведению горячей частицы для того, чтобы разобраться в реакциях обычной. Поэтому если где-то найдете рассуждения о реакционной способности горячих карбокатионов, относитесь к таким данным скептически и критически. А для практических целей это все равно бессмысленно и потому что изотопно чистый тритий чудовищно дорог и требует особых условий работы, и потому что период полураспада составляет около 12 лет, и при желании сделать с ним какую-нибудь практическую химическую реакцию ждать 50% выхода как раз столько лет и пришлось бы, для 75% выхода понадобилось бы 24 года и так далее.

Перегруппировки карбокатионов, когда они происходят
Одним из фундаментальных свойств карбокатионов является склонность к перегруппировкам. Перегруппировки всегда обратимы и очень быстры, а это означает, что всегда будет преобладать наиболее стабильный катион из всех возможных, связанных между собой перегруппировками.
Основной, но не единственный процесс, отвечающий за перегруппировки – сдвиг с соседнего атома углерода водорода или алкила с парой электронов. Это очень важно – перемещающийся атом захватывает пару электронов, поэтому перемещение водорода правильно называть гидридным сдвигом. Но когда перемещается алкил, говорят про алкильный сдвиг. Карбокатионный центр при этом перемещается на тот атом углерода, с которого произошел сдвиг.
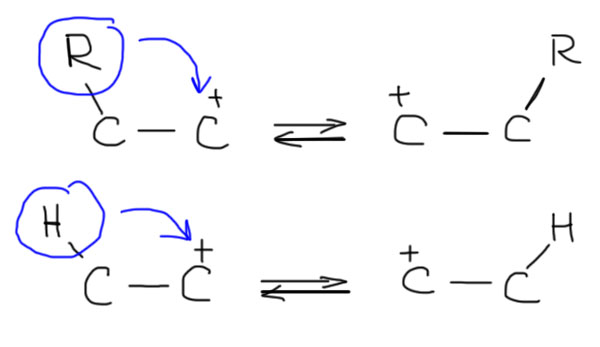
Хорошо, а как узнать, какие перегруппировки происходят в каком-то реальном случае? Это очень просто, потому что а) происходят все возможные; б) они идут строго друг за другом, не перепрыгивая через атомы; в) большинство из них нам даром не нужны, а нужны только те, которые ведут к самому стабильному катиону в каждом случае. Если пока ограничиться только алкилами, то перегруппировки будут всегда идти в сторону третичных карбокатионов. В этом месте нормальный человек застынет в полном изумлении – да ведь только такие катионы и получаются в SN1-реакциях! Откуда там вообще могут взяться не-третичные катионы?? Все верно, но не надо забывать, что SN1-реакции бывают не только самопроизвольные, то есть сольволиз, и в таких реакциях действительно карбокатион или достаточно стабилизирован, или он не получается, и сольволиз не идет. Но SN1-реакции бывают и принудительные, когда мы заставляем уходящую группу уйти, и в этом случае в самом начале катион может получится любой, и тут же вступает в действие цепочка перегруппировок, ведущих к более стабильному катиону. Иногда даже говорят так: если в результате ухода очень хорошей уходящей группы должен получиться какой-нибудь совсем гадкий карбокатион, например, первичный, то перегруппировка вступает в действие непосредственно в процессе ухода уходящей группы, и этот мерзкий первичный карбокатион – глаза бы его не видели! – не успевает образоваться, как тут же перегруппировывается, и наша совесть остается чиста. Ведь не велели же писать первичных карбокатионов! На деле это все не так важно, и разрисовывая перегруппировки, мы вынуждены рисовать и первичные катионы тоже, просто успокаивая себя тем, что у них очень малое время жизни, и они перегруппировываются дальше немедленно. Чуть дальше мы еще вернемся к этой проблеме и увидим, что там есть нечто очень важное.
Вот, посмотрим что будет, если мы возьмем первичный алкил неопентил (неопентил – это очень распространенное название для 2,2-диметилпропила) и попробуем посмотреть, что он будет делать в SN1-реакции. Сольволиза с неопентилбромидом или тозилатом не получится. А вот попытка протонировать неопентиловый спирт вполне возможна. Неопентильные производные и похожие на них субстраты с разветвлением на втором атоме углерода – очень важные молекулы в этом отношении, потому что у них фактически полностью подавлен SN2-путь превращения (почему – разберем в разделе про SN2). И они либо вообще не реагируют в обычных реакциях нуклеофильного замещения, либо твердо встают на путь перегруппировки, то есть входят в SN1-режим. Попробуем, например, взять неопентиловый спирт и попробовать превратить его в неопентилбромид или хлорид так же, как мы делаем с другими первичными и вторичными спиртами – действием хлористого тионила или трехбромистого фосфора. Еще в 1932 году Уитмор и Ротрок попробовали это сделать и увидели, что не получается ничего, кроме немного загадочного частичного осмоления. Эти результаты после много раз пытались опровергнуть, но ничего так и не получилось. Но если взять методику, более подходящую для третичных спиртов, то есть реакцию с концентрированными галоводородными кислотами, то неопентиловый спирт реагирует, правда тяжело и медленно (приходится кипятить смесь несколько дней), и при этом в основном получается третичное галогенпроизводное.
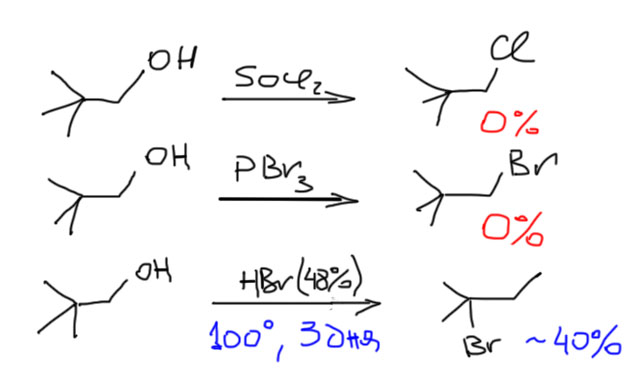
Обратим внимание, что и в этом случае реакция идет “со скрипом” и далеко не полностью. Это понятно – SN2-путь совсем закрыт, а SN1 – тоже далек от счастья, потому что приходится преодолевать очень невыгодный первичный карбокатион. Тот самый, который перегруппировывается. В этом случае путь перегрупировки один – рядом есть только метильные группы, и одна из них шагает на место первичного катиона, оставляя за собой дырку, но уже в более подходящем месте. Дальше можно нарисовать следующие перегруппировки, но они все ведут в менее стабильные вторичные или первичные карбоатионы, а следовательно невыгодны и в общей картине занимают небольшое место. Сразу замечу – это не значит, что такие перегрупировки в действительности вообще не идут, – безусловно идут, ведь мы договорились, что идут все возможные последовательные перегруппировки. И третичный катион обратимо перегруппировывается во вторичный (за счет сдвига гидрида), но в меньшей степени. И даже в первичный (тоже за счет сдвига гидрида) – в совсем малой степени. И продукты соответствующие в реакционной смеси есть, но только как побочные, в количествах от нескольких процентов до 10-15%. И особенно хорошо это видно, если использовать изотопные метки, и с любопытством смотреть, как они скачут в продуктах реакции.

Это одна из неприятных особенностей перегруппировок карбокатионов – они всегда дают равновесные смеси, в которых даже самый устойчивый карбокатион не занимает все 100%. Иными словами, такие реакции никогда не имеют высокой селективности. Поэтому и применяют их для синтеза редко, и предпочитают вообще избегать – делить изомерные продукты никому не нравится.
Итак, первый вывод: основной продукт в перегруппировках алкильных карбокатионов соответствует третичным карбокатионам.
Неопентильная система очень часто приводится в пример склонности к перегруппировкам, потому что в ней не работает SN2-путь, из-за чего приходится использовать жесткие условия реакций, способствующие включению принудительного SN1. Посмотрим, что нас ожидает, если мы используем не такие специфические первичные или вторичные алкилы. В этом случае (особенно, если алкил первичный) SN2-путь будет основным, и реакции будут идти в основном или полностью по этому пути. И только если мы по недосмотру или специально решим использовать, например, сильную кислоту и достаточно жесткие условия, мы можем вызвать подключение конкурирующего SN1-превращения с неизбежными перегруппировками. Возьмем обычный н-бутиловый спирт, например. Этот спирт отлично дает н-бутилбромид при действии трехбромистого фосфора, строго по SN2-механизму, никаких перегруппировок. Но если мы попробуем погреть его с концентрированной бромистоводородной кислотой, результат будет другим. SN2-путь при этом сохранится и останется основным, но частично пойдет и по SN1-маршруту с перегруппировкой. В этом случае путь перегруппировок более сложен: из первичного во вторичный с гидридным сдвигом, из вторичный произойдет сдвиг метила с образованием изо-скелета, но сначала первичного катиона, который наконец с гидридным сдвигом попадет в третичный. Из-за того, что путь от первоначально образующегося н-бутильного карбокатиона в трет-бутильный включает промежуточные вторичные и первичные катионы, этот путь не очень выгоден и не очень вероятен, но он есть, и при анализе продуктов изомеризованные вторичные и третичные продукты будут обнаружены.
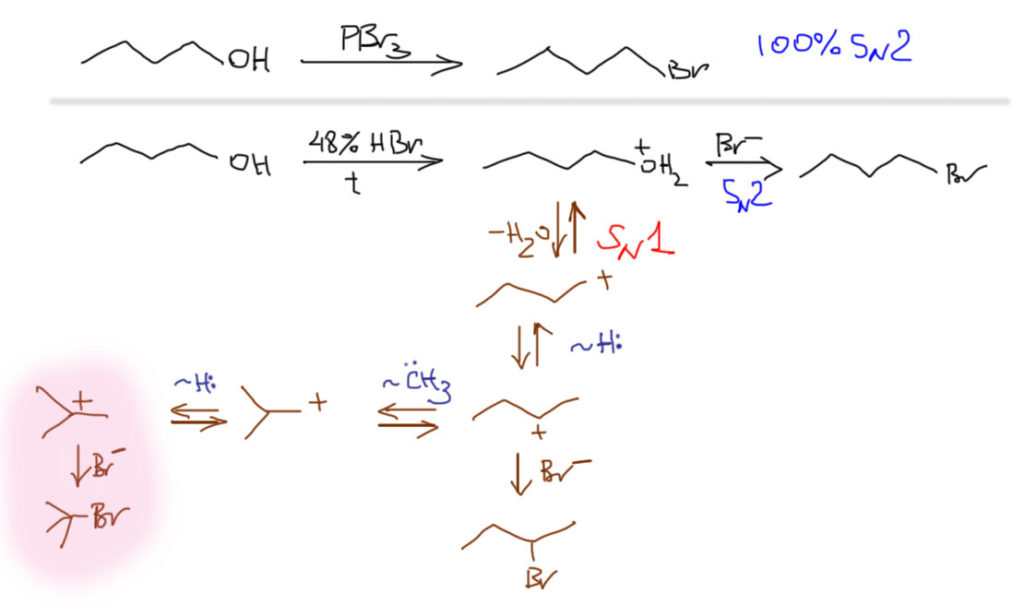
Подведем первый итог: в химии нуклеофильного замещения стоит избегать, насколько это возможно, условий, способствующих образованию карбокатионов (а это, в первую очередь, сильные протонные кислоты и кислоты Льюиса), так как в этом случае можно нарваться на перегруппировки даже в тех случаях, когда этого не ожидаешь. Чем лучше субстрат для чистого SN2-замещения, тем меньше вероятность и вклад перегруппировки, даже если она спровоцирована условиями реакции. Если перегруппировки избежать не удалось, то всегда образуется наибольшее количество продукта, соответствующего третичным карбокатионам, но чем дальше путь от начального карбокатиона до третичного, тем меньше будет вклад перегруппировки.
Перегруппировки карбокатионов, как они происходят
Раз уж перегруппировки почти неизбежны, стоит немного разобраться в том, как они происходят. Есть несколько способов посмотреть на этот процесс. Мы не будем тратить время на все, а посмотрим только один, на мой взгляд, самый наглядный и довольно убедительный. В этом месте человек, не испорченный цинизмом большой науки, может оскорбленно вскричать: “Я не хочу самый наглядный, я хочу единственно верный!!!”. Увы, единственно верный знает только Творец всего сущего (и был еще такой товарищ Ленин, но он уже скоро как сто лет не с нами), а если вы в него не верите, то вообще никто. Нам, смертным людям 21 века, живущим всего менее 200 лет спустя возникновения химии как науки, то есть во времена ее, химии, трогательного младенчества, дано только строить грубые, качественные (не в смысле “очень хорошие”, а в смысле “неколичественные, приблизительные”) модели реальных процессов. В лучшем случае просто наглядные, позволяющие приблизительно понять, как это устроено и делать какие-то очень осторожные гипотезы. Главный критерий того, заслуживает ли внимания то или иное объяснение – полезность. Если гипотезу можно применить, чтобы понять происходящее, обобщить данные по близким реакциям, сделать осторожный прогноз на то, как пойдет реакция, то и хорошо, сойдет, не будем пока беспокоить Творца, выклянчивая что-то абсолютно-верное-ныне-и-присно-и-вовеки-веков, чтоб раз выучить и навсегда. Нет такого. По крайней мере сейчас. Приходите через тысячу или миллион лет, может появится, наука не стоит на месте.
Итак, задача такова. Есть карбокатионный центр, то есть свободная p-орбиталь. Рядом, на соседнем углероде, есть σ-связь, на которой болтается нечто, водород или алкил. Нужно понять, как это нечто переползает на бывший катионный центр, а бывшая σ-связь превращается в свободную p-орбиталь.
Попробуем так. В теме Электрофильные реакции алканов, мы познакомились с гиперкоординированными катионами. Эти катионы возникают тогда, когда сильный электрофил, типа протона или алкильного карбокатиона, нападает на σ-связь, при этом образуется трехцентровая связь, которая быстро распадается по трем различным направлениям. Разве не похоже на наш случай – разница только в том, что электрофил и σ-связь находятся в одной молекуле, рядом. Но это даже хорошо, далеко ходить не надо, а кроме того позволяет понять, отчего это связь, которая в исходной молекуле была отогнута на угол 109°, вдруг начинает пригибаться к катионному атому – вот трехцентровая связь это и обеспечивает. Нарисуем. Напомню, что трехцентровая связь обозначается тремя лучами к каждому из центров.
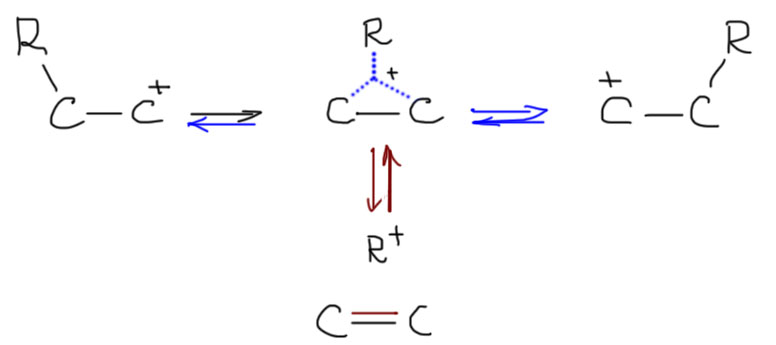
Трехцентровая связь слаба, но достаточна для того, чтобы объяснить, зачем мигрирующий водород или алкил начинает свой неблизкий путь от одного атома углерода к другому. Сама частица с трехцентровой связью при этом вовсе не обязательно должна быть достаточно стабильна для того, чтобы ее можно было реально обнаружить, она вполне может быть или очень короткоживущей, далеко за пределами современных методов, или даже может вообще существовать только на бумаге, то есть быть просто переходным состоянием. Современная химия не сможет ответить на эти вопросы. Строго говоря, это даже не важно. Это просто модель реакции, гипотетический механизм. Он будет полезен только, если позволит что-то понять или предсказать. В этом виде мы видим три пути распада трехцентровой связи: один – просто обратный к исходному катиону, второй – вперед к перегруппированному катиону. И есть еще третий, который нам дает двойную связь плюс мигрирующую группу, ставшую электрофилом (протоном или алкильным карбокатионом). Очевидно, что этот нижний путь самый маловероятный, потому что приводит к образованию частиц с очень высокими энергиями типа свободного протона или метильного карбокатиона. Но нельзя не заметить, что сам по себе этот путь – просто обратная реакция к электрофильному присоединению к кратной связи. Первые два пути просто связывают исходный катион и перегрупированный. И опять – эти пути не обязательно равновероятны, напротив, должен преобладать тот, который дает более стабильный катион.
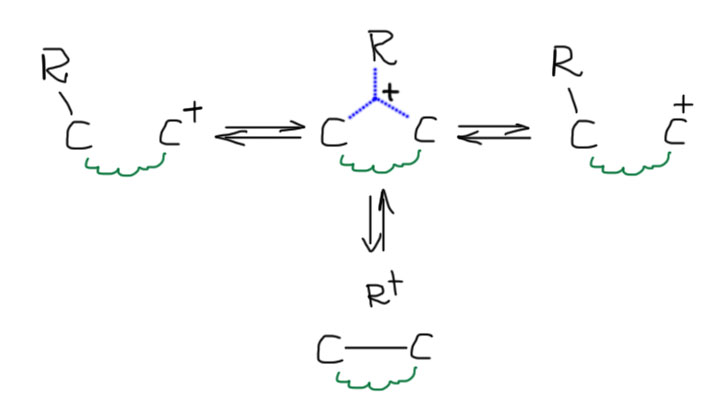
Такое представление о том, как идет перегруппировка легко обобщается на случай, если карбокатион и связь находятся не на соседних атомах углерода. В схеме процесса почти ничего не изменится, просто обозначим пружинкой мостик между атомами углерода. Те же три пути, третий так же маловероятен. Но и весь такой процесс крайне редко осуществляется, просто потому, что трехцентровая связь слишком слаба, чтобы обеспечить сближение и взаимодействие находящихся не рядом катиона и связи. Крайне маловероятен, но не совсем невероятен, и может осуществиться, если молекула сразу так устроена, что эти места сближены. Пока не будем пытаться найти такие примеры, но вспомним про это, когда будем изучать алициклы и так называемые трансаннулярные реакции.
Карбокатионы в присоединении, замещении и отщеплении
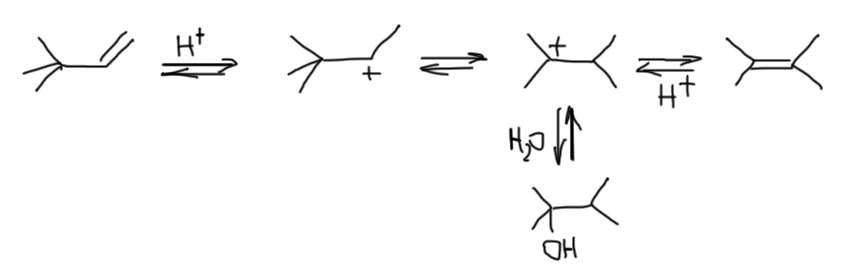
Возьмем самый простой случай и распишем все, что происходит с трет-бутанолом в системе, содержащей сильную кислоту типа HCl. Мы увидим систему равновесий, включающих сам трет-бутанол, трет-бутилхлорид, и изо-бутилен. Все эти равновесия будут связаны одним трет-бутильным карбокатионом.
В равновесиях можно легко найти принудительный SN1 и самопроизвольный SN1-сольволиз, электрофильное присоединение, и E1-элиминирование. Каждый из путей частично пересекается с другими, поэтому разметка цветом не полностью соответствует отдельным путям.
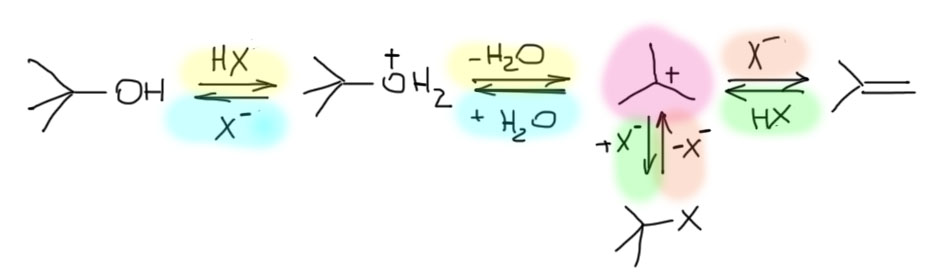
Из этого следует несколько важных вещей. Единственный способ справиться с равновесиями, который изобрело человечество, это принцип Ле Шателье. Применяя его к этой системе, мы можем, например,
- сделать олефин из спирта: добавим сильную кислоту и немного нагреем – более летучий олефин будет удаляться из смеси и равновесие сдвинется в его сторону;
- сделать спирт из олефина: добавим сильную кислоту, не имеющую нуклеофильного аниона, например, серную, и наоборот снизим температуру и увеличим давление;
- сделать галогенпроизводное из олефина: исключим воду из смеси, которая конкурирует за карбокатион, снизим температуру;
- сделать галогенпроизводное из спирта: используем концентрированную галогенводородную кислоту, равновесие сместится из-за низкой растворимости галогенпроизводного в воде.
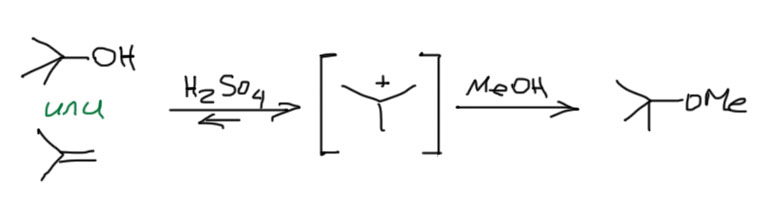
Второй интересный вывод – спирт и олефин играют в одну игру, а следовательно фактически взаимозаменимы во многих реакциях, связанных с карбокатионом. B тот и другой при протонировании дают один карбокатион, которому можно подсунуть нуклеофил и получить один и тот же продукт. Только с точки зрения олефина это будет продукт электрофильного присоединения, а с точки зрения спирта – продукт принудительного SN1-замещения. Но с точки зрения потребителя, который получил нужный продукт, это одно и то же. Так, например, чтобы получить метил-трет-бутиловый эфир, мы берем метанол в хорошем избытке и или трет-бутанол, или изобутилен, и немного концентрированной серной кислоты. Закупориваем сосуд и ставим в прохладное место. Через какое-то время откупориваем и перегоняем. Результат будем почти одинаковый (со спиртом немного хуже, потому что воды немного выделяется, и она конкурирует с метанолом за карбокатион).
Приблизительно то же самое делают и чтобы получить трет-бутиловые эфиры карбоновых кислот. Реакцию ведут с карбоновой кислотой или чистой, или в ненуклеофильном растворителе. Это единственный надежный метод получения трет-бутиловых эфиров самых разных кислот, даже аминокислот.
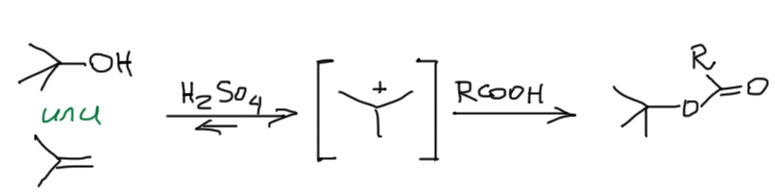
И в уже упомянутой выше реакции Риттера точно так же можно взять или спирт, или олефин. И еще много таких реакций можно найти на просторах органического синтеза.
Если немного обобщить и выйти за пределы трет-бутильной системы, то важно заметить еще одну вещь. В реакциях электрофильного присоединения точно так же часто случаются перегруппировки карбокатионов, и забывать про это не нужно. особенно часто они происходят при кислотной гидратации олефинов, которую именно поэтому считают очень плохим методом синтеза спиртов из олефинов. Так, например, при гидратации 3,3-диметилбутена-1 мы получаем типичное поведение катиона неопентильного типа, хотя и вторичного.
Теперь можно приступить и к главному механизму органической химии – SN2. Насчет “главного механизма” – безусловно преувеличение, но не очень большое. На исследовании этого механизма собственно и появилась эта наука, а найденные закономерности после успешно обобщались на другие реакции и механизмы. Кроме чисто теоретического интереса SN2-замещение используется в огромном количестве практически важных реакций и методов органического синтеза. И везде возникают одни и те же проблемы: как подобрать субстрат и растворитель, как увеличит скорость и селективность, и наконец, как избежать бессмысленных экспериментов в заведомо безнадежных случаях.
Механизм SN2
Нет ничего проще чем механизм SN2-замещения на насыщенном атоме углерода. Вот это дополнение – на насыщенном атоме углерода – очень важно, потому что существует очень много других вариантов бимолекулярного нуклеофильного замещения и на атомах других элементов, и на атомах углерода с другой гибридизацией. Мы не будем их обсуждать, поэтому всегда будем иметь в виду именно бимолекулярное нуклеофильное замещение на sp3-гибридном атоме углерода.
SN2-замещение – это согласованный (по-английски concerted) механизм. Это означает, что по пути от исходных к конечным нет никаких промежуточных частиц (или интермедиатов – это синонимы), которые можно было бы обнаружить экспериментально, с помощью каких угодно сложных методов спектроскопии или еще как-нибудь. Это очень важный момент: речь не идет о том, что сегодня еще нет достаточно точных и быстрых методов, чтобы обнаружить промежуточные частицы, а завтра или послезавтра они, может быть, появятся. Речь идет о том, что там по дороге действительно ничего нет такого, что можно было бы обнаружить. Вот, например, в SN1-замещении с какими-нибудь простыми третичными алкилами, мы утверждаем, что по дороге возникает карбокатион, и карбокатион рассматривается как законная промежуточная частица или интермедиат даже несмотря на то, что экспериментально его тоже обнаружить в реакционных смесях, где идет сольволиз, до сих пор никому не удавалось. Но мы утверждаем, что он там есть, просто время жизни его очень мало. Мы чувствуем его присутствие по разным косвенным признакам (стереохимии, поведении изотопных меток, и т.п.), и поэтому признаем его важность для SN1-механизма. Мы надеемся на то, что когда-нибудь сможем получить в руки настолько чувствительный и быстрый метод, что сможем его и прямо поймать. А в SN2-замещении мы принципиально отказываемся от такой возможности. Поэтому один механизм называют двухстадийным, а другой согласованным. И обратите внимание, что это не имеет никакого отношения к цифре 1 или 2 в обозначении. Эти цифры просто описывают молекулярность (то есть количество взаимодействующих частиц) медленной стадии. В SN2 стадия только одна, и на ней взаимодействует нуклеофил и субстрат. В SN1 стадии две, но медленной является стадия распада субстрата на ионы, и кроме субстрата на этой стадии ничего нет.
Особенности каждого из механизмов принято выражать так называемыми энергетическими диаграммами. О том, что это такое и насколько соотвествует тому, что реально происходит в реакциях, мы поговорим отдельно, а пока просто отметим, что в SN2-замещении (левая, одногорбая кривая) между исходными и конечными веществами только один холм, через который нужно условно перевалить, на вершине холма – переходное состояние, в котором одновременно рвется связь с уходящей группой и образуется связь с нуклеофилом. Показан случай, когда реакция экзотермична, но это не обязательно. Справа для сравнения показана энергетическая диаграмма типичного SN1-замещения. Здесь уже два холма, и на штурм первого отправляется в одиночку только субстрат, результатом является карбокатион, частица с высокой энергией, которая очень легко превращается что в исходную молекулу, что в конечную – и туда и обратно от средней ямки ведут очень низкие барьерчики. Ямка карбокатиона очень неглубокая, что лишний раз напоминает о его высокой реакционной способности и легкости, с которой из этой ямки можно выбраться.
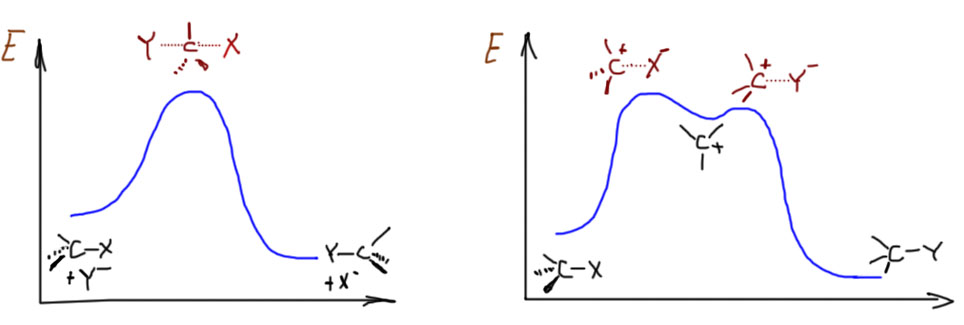
А откуда берутся эти диаграммы? Ни в коем случае не из экспериментальных измерений – не бывает таких измерений. Единственная вещь, которая хоть как-то соотносится с реальностью – это высота самого большого барьера от уровня исходных до вершины, где переходное состояние – эта высота имеет некоторое отношение к экспериментальной энергии активации, которая более-менее легко вычисляется из экспериментально измеренной температурной зависимости константы скорости. В остальном еще недавно это вообще были просто картинки, нарисованные “от фонаря”, просто чтобы показать, как это может выглядеть. Но в связи с очень быстрым развитием квантово-химических методов расчета в последние лет 10 эти картинки стали считать, хотя и очень приблизительно, прежде всего потому что корректный учет взаимодействия частиц с растворителем по-прежнему задача почти нереально сложная. Тем не менее данные из расчетов получаются полезные, которые довольно неплохо подтверждают, что модели механизмов в целом верны, и ничего существенного не упущено. В расчетах получают не все эти никому не нужные загогулины, а только энергии реагентов, интермедиатов и переходных состояний. Получается что-то типа такого:
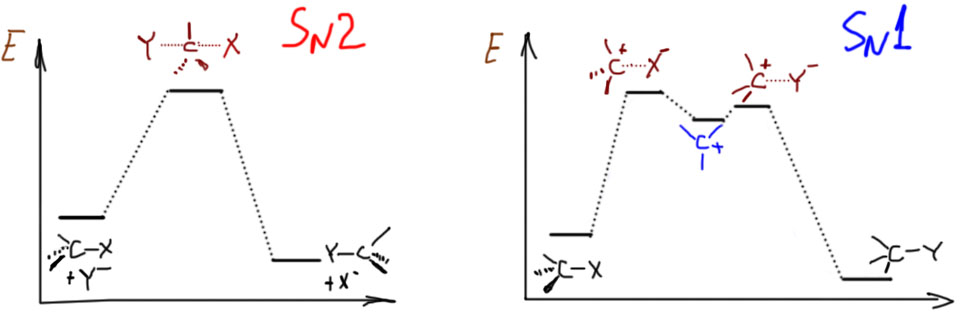
Пунктирчики обозначают связь между последовательными состояниями: если вы хотите проследить, как развивается реакция, нужно строго следовать по пунктирчикам, ничего не перепрыгивая и не пропуская.
Структура субстрата в SN2-замещении
Структура субстрата играет самую важную роль в определении возможности SN2-замещения, и относительной скорости реакции. Сразу определимся с последним. Мы уже знаем, и еще подробнее узнаем, что скорость SN2-замещения очень сильно зависит и от нуклеофила, а в некоторых случаях и от растворителя. Поэтому, чтобы разобраться с субстратами возьмем какую-нибудь типичную реакцию с хорошим, но не выдающимся нуклеофилом в растворителе, не предполагающем сильное ускорение реакции для некоторых нуклеофилов. Разные реакции можно взять в качестве таких установочных моделей, например, часто берут реакцию с галогенид-ионом (хлоридом, бромидом или иодидом, причем скорость измеряют, используя изотопы этих элементов) в растворителях типа ацетона или ацетонитрила. Строго говоря, нам это не важно, но просто полезно знать, как делают такие измерения.
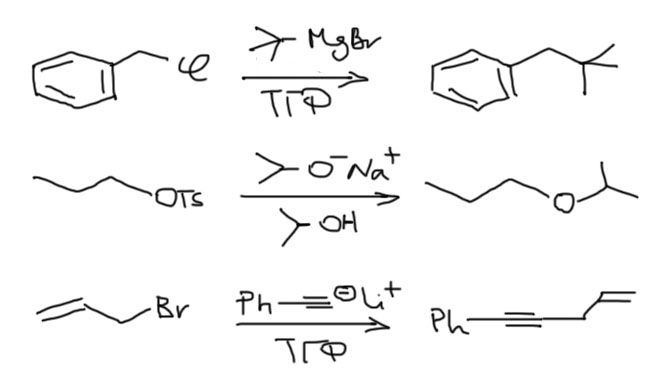
Как обычно, не будем гоняться за числами. Все строго приблизительно. Сначала расположим простые алкилы, разделив их на естественные группы: первичные, вторичные, третичные. И еще метил: метил – это группа из одного представителя, такого маленького, но невероятно важного в SN2-замещении. Получаем такую лестницу вниз. Каждая ступень соответствует изменению реакционной способности минимум на 2-3 порядка (когда так говорят, имеют в виду константу скорости реакции). Звучит это не так впечатляюще, но можно и более наглядно. Если мы начинаем сверху этой лестницы, то если реакция с субстратом на самом верху занимает секунды, то следующая ступень – часы, следующая – месяцы, следующая – столетия. Понятно, что если с третьей ступенью еще можно что-то сделать, увеличив температуру, немного получше подобрав условия, то с последней уже все бесполезно. Можно поэтому считать, что реакция внизу не идет вовсе. Третичные субстраты безнадежны в SN2-замещении.
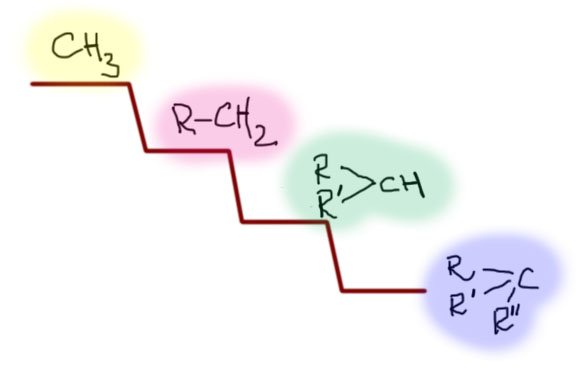
Теперь посмотрим на структурные особенности, которые изменяют положение субстратов на этой лестнице. Первый из таких эффектов – β-замещение, разветвление на втором, считая от реакционного центра атоме углерода. Это – очень сильный замедлитель. Даже самый маленький заместитель на этом месте опускает на одну ступень, а любое более серьезное разветвление, например, два метила – на две ступени. Этот эффект иногда называют неопентильным по названию самого простого и часто встречающегося алкила, страдающего этим недугом. Мы уже обсуждали это в SN1-замещении, где неопентил и его родственники тоже отметились скверным нравом. Понятно, что такое разветвление уместно рассматривать только в первичных алкилах, потому что вторичные уже и так плохи настолько, что дальше портить их бессмысленно. Ну и теперь мы понимаем, что в химическом смысле тоже можно “спустить с лестницы”. Думаю, что понятно, что если на β-углероде висят не метилы, а что-нибудь покудрявее, хотя бы этилы или изопропилы, то с таким субстратом в SN2 можно даже не обращаться. За время, сравнимое с ожидаемым временем такой реакции, обезьяна успела превратиться в человека, а во что может превратиться человек, ожидающий хотя бы 10% выхода в такой реакции, можно даже не гадать.

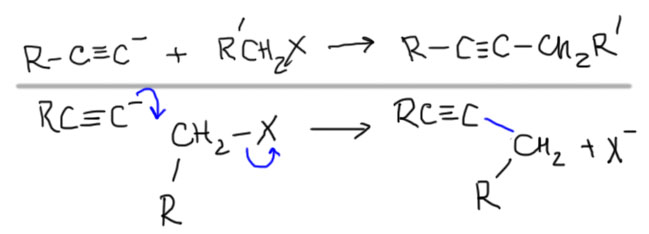
Было бы очень грустно, если бы в такой важной химии, как SN2-замещение верхом совершенства и реакционной способности оставались бы метильные производные. Наверное, есть не только путь вниз, но и путь вверх. Действительно есть. Если совсем все обобщить, то такой путь только один. SN2-замещение очень любит, когда рядом с углеродом, на котором происходит замещение, находится кратная связь, причем фактически любая: двойная или тройная, углерод-углеродная или связь между другими неметаллами. Объясняют это обычно взаимодействием орбитали, вовлеченной в замещение и орбитали кратной связи. Сначала разберемся в том, что и насколько сильно ускоряет SN2-замещение, а после попробуем понять, почему это происходит.
Кратные связи углерод-углерод, а это и двойные связи в аллильных фрагментах, и тройные связи в пропаргильных фрагментах, и ароматические кратные связи в бензильных фрагментах фактически смещают субстрат на нашей лестнице на одну ступеньку вверх, то есть первичные аллильные, пропаргильные и бензильные субстраты по скорости замещения приблизительно равны метильным, и иногда даже немного опережают эти SN2-субстраты par excellence. Вторичные субстраты поднимаются на уровень обычных первичных и становятся вполне хорошими. Только не нужно перебарщивать и не нужно вешать два непредельных заместителя на один центр. Одного достаточно, второй пусть остается обычным алкилом. А что будет, если повесить два непредельных заместителя? Будет много непредвиденных осложнений, в которых мы не сможем разобраться, поэтому лучше не пробовать. Все хорошо в меру.

Но еще больший эффект оказывают кратные связи, если хотя бы один из элементов не углерод, а кислород или азот, то есть более электроотрицательный неметалл, способный образовывать кратные связи. Самой типичной группой этого типа является карбонил C=O, который может быть в составе альдегидов, кетонов, производных карбоновых кислот. Поэтому такие заместители можно все вместе назвать заместителями карбонильного типа. Групп таких бесчисленное количество, и если мы добавим еще и азот, сюда пойдут имины, нитрилы, нитро-группы, да и даже всякие гетероциклические ядра типа пиридина (там есть некоторые сложности, мы еще вернемся к этому, когда дойдем до гетероциклов). Наличие таких групп рядом с центром замещения поднимает субстрат сразу на две ступеньки, то есть в случае первичного субстрата мы с легкостью обходим метил на 2-3 порядка, и на нашей лестнице получаем еще одну ступеньку. И опять, как в случае с углеродными непредельными группами не увлекайтесь и не вешайте два или три заместителя карбонильного типа на один центр – и здесь получите много непредвиденных осложнений. Химия гораздо сложнее простых схем:обобщения в ней полезны, но их нужно держать в рамках.
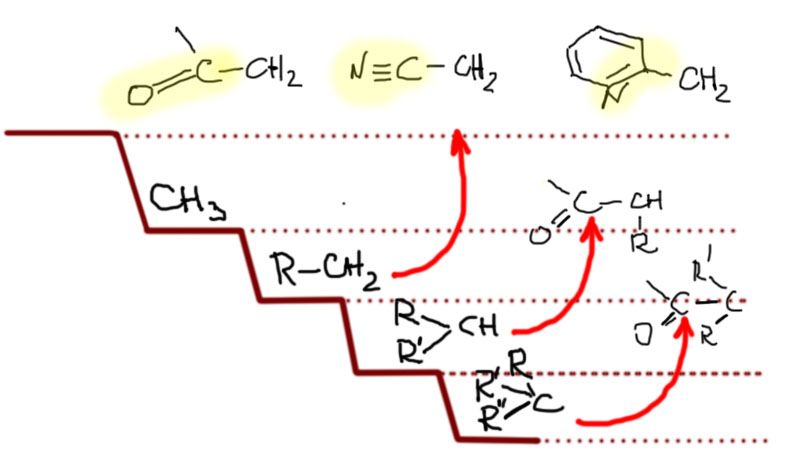
Итак, мы видим, что реакционная способность субстратов в SN2-замещении очень сильно зависит от структуры групп, которые висят на атоме углерода, на котором происходит замещение, и варьируя эту структуру мы перекрываем не менее двенадцати порядков константы скорости, то есть, говоря человеческим языком имеем и очень быстрые реакции, происходящие практически во время смешивания реагентов, и чрезвычайно медленные реакции, идущие со скоростью геологических процессов, то есть не идущие вовсе. Такой широченный диапазон реакционной способности, определяемый только структурой реагирующих молекул, очень характерен для органических реакций. Это показывает, что в органической химии нужно очень осторожно подходить к планированию синтезов, так как во многих случаев оказывается, что реакция, необходимая для превращения одного вещества в другое (например, спирта в бромпроизводное) формально есть, а фактически применить ее нельзя из-за неприемлемо низкой реакционной способности. И все – синтез накрылся! По крайней мере пока мы не найдем замену бесполезной стадии.
На этом месте можно остановиться всем, кроме тех, кому хочется понять, почему кратные связи проявляют такой эффект.
Попробуем разобраться в этой, несколько запутанной истории. Что происходит SN2 в ходе замещения мы знаем из энергетической диаграммы, разобранной на предыдущей вкладке. Здесь мы заметим очень важную вещь: если мы хотим понять, что происходит в ходе какой-то реакции, не нужно пытаться проследить за каждым изменением, происходящем по пути – это все равно невозможно. Вместо этого нужно обратить внимание на переходное состояние, если реакция согласованная, или на промежуточную частицу (интермедиат), если реакция стадийная. Так, для обсуждения влияния разных факторов на SN1-замещение, мы все время ходили вокруг карбокатиона, так как эта реакция как раз стадийная. А в согласованной SN2-реакции мы сосредоточим внимание на переходном состоянии (все равно больше не на чем).
В переходном состоянии углерод одновременно держит (слабо держит, но держит) и уходящую группу и нуклеофил, располагая двумя валентными электронами. В целом, в этом процессе задействовано 4 электрона, но вторая пара перемещается между нуклеофилом и уходящей группой. Задействованная орбиталь углерода очень похожа на обычную p-орбиталь, и она обслуживает переходное связывание и с уходящей группой, и с входящим нуклеофилом. Вот приблизительно так, как показано на картинке. Если рядом с атомом углерода находится кратная связь, мы получаем рядом π-связь, а все π-связи устроены одинаково – это две p-орбитали, образующие связывающую и разрыхляющую орбитали, одна с парой электронов, вторая пустая. Посмотрим на картинку и убедимся, что форма этих орбиталей подходит для взаимодействия с орбиталью реакционного центра. А раз подходит, значит взаимодействие будет. Вопрос только в том, хорошее (стабилизирующее) или плохое (дестабилизирующее) это взаимодействие. Для того, чтобы взаимодействие было хорошим, как мы уже видели не раз, нужно, чтобы на взаимодействующих орбиталях было в сумме два электрона. В этом и состоит некоторый фокус: нам от кратной связи понадобится разрыхляющая орбиталь, а не связывающая. Тогда взаимодействие орбитали углерода, на котором идет замещение, и орбиталей кратной связи будет хорошим, стабилизирующим – в сумме там как раз два электрона. Именно поэтому на картинке показана разрыхляющая орбиталь кратной связи (доли p-орбиталей связанных атомов попарно противоположны, это показано цветом долей – в разрыхляющей орбитали красненькое наезжает на синенькое). Не обращайте внимание на размеры орбиталей – это может быть по-разному и зависит от природы атомов и заместителей. Нас пока интересует принципиальная возможность стабилизации – и она явно есть.
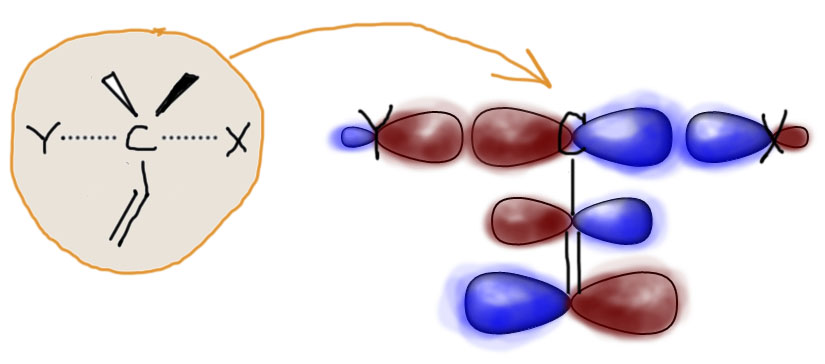
К слову “стабилизация” в этом контексте нужно относиться аккуратно. В полном смысле этого слова стабилизировать (или дестабилизировать) можно только то, что реально существует, то, что имеет время жизни, возможно очень малое, но конечное и принципиально измеримое. В SN2-замещении реально существуют только исходные и конечные молекулы или ионы, а обсуждаем мы сейчас переходное состояние, то есть нечто принципиально неустойчивое, не имеющее времени жизни, – просто наше представление о том, как выглядит вершина перевала между исходными и конечными. Тем не менее это нечто имеет вполне определенную энергию, которую мы можем оценить экспериментально, измерив энергию активации, и можем даже более-менее корректно, хотя и очень приблизительно посчитать, использовав расчетные методы современной квантовой химии. В этом смысле слово “стабилизация” означает не то, что мы делаем более стабильным то, что принципиально абсолютно нестабильно, а то, что мы просто наблюдаем снижение энергии этого переходного состояния, то есть снижение барьера, который преодолевают реагирующие молекулы, чтобы попасть из исходного состояния в конечные, из реагентов в продукты. Снижение барьера – это снижение энергии активации, а следовательно увеличение скорости реакции. Чем больше снижаем барьер (“стабилизируем” переходное состояние), тем быстрее становится реакция. А это именно то, что мы ищем.
Вернемся к картинке. Итак, мы пришли к выводу, что а) переходное состояние “стабилизируется”, если рядом двойная (или тройная) связь; б) стабилизация, вероятно, обусловлена взаимодействием p-орбитали углерода с π-орбиталями двойной или тройной связи; в) наибольший вклад в стабилизацию вносит разрыхляющая π-орбиталь. Последняя идея очень важна, потому что объясняет, почему карбонильная группа и похожие на нее группы дают гораздо более сильный эффект, чем обычная кратная связь углерод-углерод. Вам придется поверить мне на слово, но, если сравнивать π-орбитали углерод-углеродной и углерод-кислородной двойной связи, то можно увидеть, что они а) очень похожи и устроены совершенно одинаково, настолько, что можно не рисовать двух разных картинок; б) но при этом разрыхляющая орбиталь связи C=O ниже по энергии, чем соответствующая орбиталь C=C связи, а следовательно лучше подходит для взаимодействия с p-орбиталью реакционного центра и ее стабилизации; в) именно на углероде C=O связи больше так называемый “вес” атомной орбитали, грубо говоря, там просто крупнее эта типичная p-орбитальная “восьмерка”, а это очень полезно для более эффективного перекрывания и взаимодействия. Эффект, как мы видим из экспериментальных данных по влиянию разных групп с кратными связями, очень ярко выражен.
Нуклеофилы в SN2-замещении
Нуклеофил также очень важен для SN2-замещения. Но важно понимать, что нуклеофил – менее важный фактор, чем структура субстрата. Если субстрат не годится для SN2, нуклеофил не спасет реакцию. Совсем не годятся для SN2 третичные алкильные субстраты, и никакой нуклеофил не может изменить этого печального обстоятельства. Но со всеми остальными субстратами договориться уже можно.
Подробнее про нуклеофилы, нуклеофильность, основность и связанные с этим вещи можно почитать на отдельной страничке. Возьмем оттуда самое важное.
- Самые лучшие нуклеофилы образованы элементами третьего периода и ниже – это галогенид-ионы кроме фторида, производные серы, фосфора, (а также более тяжелых неметаллов, стоящих под серой и фосфором, но это экзотика). Плюс нуклеофилы с α-эффектом, прежде всего азид-ион N3–. Эти нуклеофилы сильны и практически не обладают основностью. Поэтому они реагируют со всеми субстратами, включая такие плохие, как вторичные алкильные и первичные с разветвлением на втором углероде (неопентильные). Скорости реакций с хорошими субстратами велики и очень велики, а с плохими субстратами – вполне терпимы. Никаких мер по увеличению нуклеофильности для таких нуклеофилов не требуется, в том смысле, что применять их не запрещено, но эффект будет невелик, за единственным исключением субстратов неопентильного типа, которые настолько ленивы, что любая помощь реакции принимается с благодарностью и позволяет выиграть денек-другой. Вообще, растворитель в этих реакциях не играет большой роли и подбирается по удобству и по опыту, так что в схемах реакций его можно смело опускать. Вот несколько примеров:
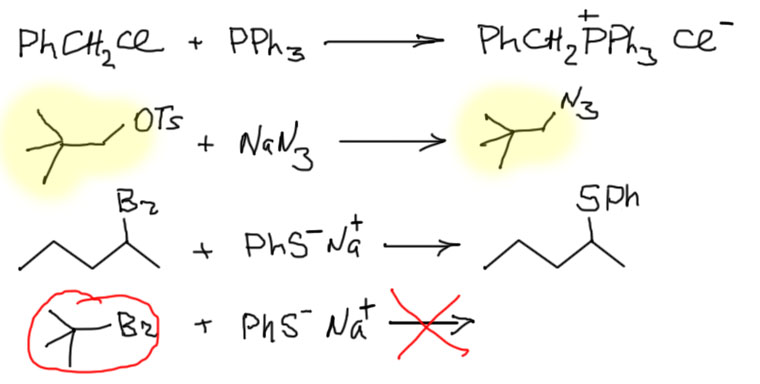
- Анионные нуклеофилы от атомов неметаллов второго периода (углерода, азота, кислорода, фтора), которым соответствуют умеренно слабые сопряженные кислоты с pK в диапазоне приблизительно 2 – 12, а следовательно сами нуклеофилы являются дсотаточно слабыми основаниями. Это фторид, цианид, нитрит, многочисленные соли карбоновых кислот, например, ацетат, формиат, бензоат, и прочие, а также сам карбонат. Не так много, но это все очень важные нуклеофилы. Эти нуклеофилы вполне способны реагировать со всем диапазоном субстратов, включая неопентильные (но не третичные алкилы) и очень любят, а с субстратами похуже (вторичными и неопентильными) даже требуют использования способов увеличения нуклеофильности (спецрастворителей типа ДМСО или межфазного переноса любых типов). С элиминированием у таких нуклеофилов проблем почти никогда не бывает.
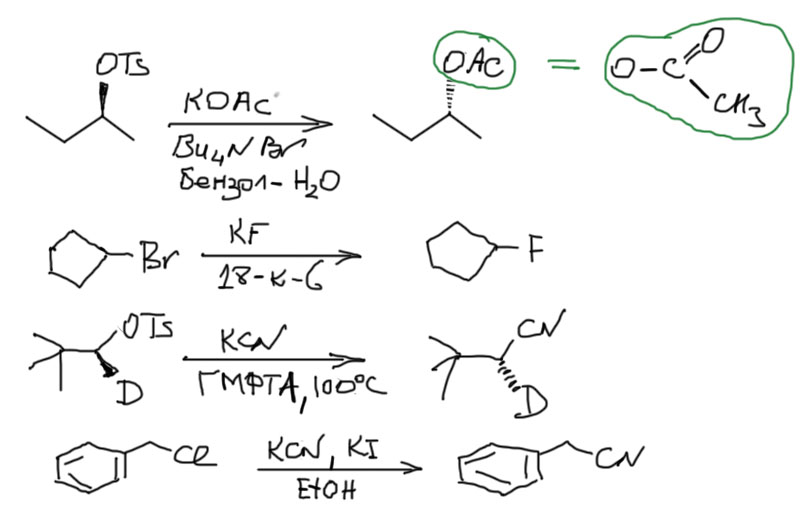 В последнем примере используется очень хороший субстрат, поэтому не нужны спецрастворители, но зато используется добавка небольшого количества иодида. Попробуйте предположить, что она там делает. Без нее реакция тоже пойдет, но существенно медленнее.
В последнем примере используется очень хороший субстрат, поэтому не нужны спецрастворители, но зато используется добавка небольшого количества иодида. Попробуйте предположить, что она там делает. Без нее реакция тоже пойдет, но существенно медленнее.
- Анионные нуклеофилы от неметаллов второго периода (углерода, азота, кислорода), являющиеся сильными и очень сильными основаниями (pK сопряженных кислот от 15 до 50). Здесь и почти все карбанионы, в частности, литий и магнийорганические соединения, ацетиленид-анионы и прочая, амиды (не амиды кислот, а анионы, полученные депротонированием аминов), алкоксид-ионы. В этой области нужно быть чрезвычайно острожными, потому что элиминирование будет ломиться в окна и двери, и нужно сознательно и умело держать оборону. Самое надежное, это когда субстрат не способен к элиминированию в принципе (это метил, бензил и что-то похожее). В этом случае можно делать самые разнообразные реакции и немного расслабиться. Если элиминирование возможно (даже если у нас этил), то лучше ограничится нуклеофилами с умеренно высокой основностью (держать pK сопряженного основания до 30 – то есть ацетилениды годятся, а обычная магнийорганика нет), и с субстратами не опускаться ниже первичных алкилов. С вторичными будет плохо всегда, но небольшие выходы (20-30%) продуктов замещения получить можно (остальное – элиминирование), если деваться некуда (хотя лучше подумать об альтернативном пути синтеза). С субстратами неопентильного типа можно даже не соваться – будет или 100% элиминирования или просто ничего. Еще нельзя использовать субстраты с заместителями карбонильного типа, – они дают с такими нуклеофилами-основаниями совсем другие реакции. Нельзя использовать методы повышения нуклеофильности (спецрастворители типа ДМСО или межфазный перенос) потому то это всегда ведет к 100% элиминированию.