Краткие ответы
Фосфор
Пятихлористый фосфор это страшно?
Пятихлористый фосфор это такой старинный реагент из 19-го века, мягко говоря не очень популярный в веке 21-ом. Реагент очень неприятный в работе, легко гидролизующийся, дымящий, часто довольно агрессивный. Современная химия сильно не любит эти экстремисткие реакции из раннего детства химии, когда наоборот их очень любили, потому что не надо упрашивать реагировать – смешал, и уже дым идёт. Мы предпочитаем более мягкие реакции, как раз такие, которые нужно упрашивать с помощью разных видов катализа, а это создает впечатление, что мы реакцией управляем, добиваемся селективности и прочих радостей, и попусту не дымим. Да и экономия не на последнем месте. Пятихлористый фосфор обычно использовался для замещения гироксильной группы на хлор, и тогда из пяти хлоров в продукт переходит только один, а три идут в отвал, причём не так-то просто обрабатывать реакционные смеси после реакции, в них всегда остаётся оксихлорид фосфора, а это тоже очень агрессивное и дымыщее вещество, так что нужно всё делать осторожно, со льдом, тщательным и аккуратным перемешиванием и так далее. Иначе можно устроить зачётное задымление.
Тем не менее, некоторое применение у этого реагента есть и даже сейчас. И если всё делать аккуратно, учитывая его строение и свойства, то можно с удивлением узнать, что сам по себе пятихлористый фосфор – довольно безобидное вещество с весьма умеренной реакционной способностью, которое далеко не всегда реагирует даже с теми веществами, с которым вроде должен, в книжках написано.
Первое, что нужно учитывать, это то, что пятихлористый фосфор очень легко дает HCl при взаимдействии с гироксилсодержащими веществами – не соляную кислоту, а чистый хлористый водород, а это значит, что возникает протонирующая среда, которая может драматически изменить характер наблюдаемых реакций. Гидроксилсодержащие вещества это и остатки воды в растворителе, и спиртовые или карбоксильные группы, и даже енол из енолизуемых карбонильных соединений. Чрезвычайно бурный гидролиз PCl5 связан не с какими-то внутренними причинами – наличием очень активных реакционных центров в этой молекуле, а всего-навсего огромной экзотермичностью превращения довольно слабых связей фосфор-хлор в очень прочные связи фосфор-кислород и аш-хлор. В других реакциях PCl5 редко проявляет высокую реакционную способность, и мы сейчас поймём почему.
Если этого нет, и у нас чистый PCl5, то реакционная способность этого соединения определяется его структурой: напомню (подробнее в страничке про фосфор), что пятивалентный фосфор это гипервалентное состояние, и в этом случае есть всего два способа образовывать связи, и не нарушать правило октета, обязательное для p-элементов. Если связей нужно пять, как в PCl5, то решение находится в конфигурации тригональной бипирамиды: в плоскости три обычные ковалентные связи (экваториальные), а вверх и вниз расположена одна трёхцентровая связь с двумя апикальными атомами – связь эта обслуживается парой электронов (поэтому мы имеем на фосфоре октет: три ковалентные связи и одна трехцентровая, итого 4 пары электронов).
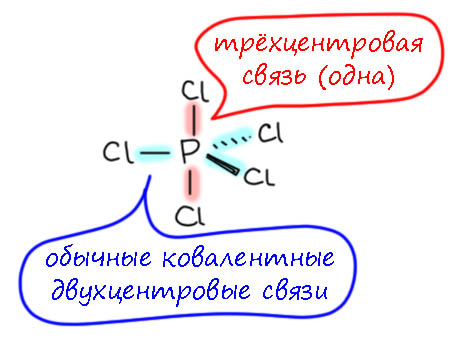
Ещё одна пара электронов делокализована по двум апикальным атомам, поэтому трёхцентровую связь называют 4-хэлектронной, учитывая, что эта вторая пара никогда фосфору не достаётся. Для апикальных положений всегда служат только самые электроотрицательные (относительно фосфора) элементы, а это фтор, кислород, хлор (именно в этом порядке), существенно реже азот, и ещё реже углерод, бром, и совсем редко иод и сера. В случаях типа того же PCl5, когда все пять атомов одинаковы, их неравенство (экваториальные свзаны намного прочнее апикальных) усредняется очень быстрым конформационным движением, псевдовращением Берри, которое с огромной скоростью меняет апикальную пару с двумя экваториальными атомами, поэтому усредненно атомы кажутся равноценными. Но в каждый момент времени они неравноценны. Апикальные – намного более реакционноспособны, и легко обмениваются на нуклеофилы, сильно при этом предпочитая килородные группы (потому что им предстоит включиться в гипервалентную трехцентровую связь). Есть два способа такого замещения – их вполне можно считать аналогиями с SN1 и SN2-замещением. Ограничимся первым, так как нам сегодня не нужно влезать в химию фосфора еще подробнее. Механизм включает обратимуэ диссоциацию именно по более слабой апикальной трехцентровой связи, когда образуется обычный тетраэдр фосфоний-катиона и анион хлорида. Обратная ассоциация в новую трёхцентровую связь дает продукт замещения одного хлора.
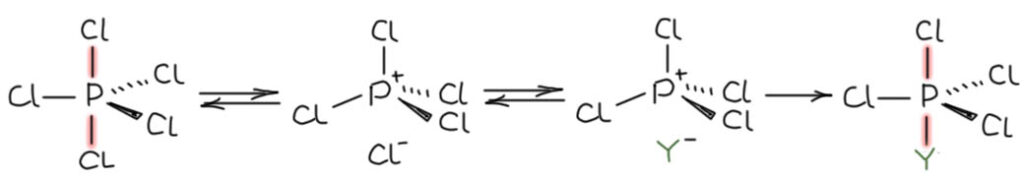
Обратите внмание на то, что фосфониевый катион проявляет электрофильность (реагирует с нуклеофилом). Мы привыкли по примеру аммония считать, что ониевые ионы не имеют электрофильности, потому что плюс не связан с какой-то орбиталью. Но вот у фосфония это не мешает быть электрофилом и именно потому, что за счет гипервалентности нуклеофил подает плотность на разрыхляющую орбиталь связи фосфор-элемент (здесь хлор, но мог быть и другой элемент) с образованием трёхцентрофой связи. Это очень обычный способ реализации электрофильности и кислотности Льюиса у элементов от 3-го периода и ниже. Так, постойте. а разве у азота нет гипервалентности? Есть, конечно, но – у азота нет пятикоординированных соединений, нет тригональной бипирамиды, нет, например, пентафторида азота. И по очень банальной причине – стерике. Азот маленький и связи у него короткие, и пять атомов просто не умещаются. А у фосфора умещаются отлично. Вот, оказывается, в чём дело – в банальной стерике! Это так, но здесь нет ничего банального: стерика – важнейший фактор структур элементов именно 2-го периода, особенно тройки CNO.
Попробуем такм способом расписать самую простую реакцию PCl5 со спиртом. Сразу видим, что немедленно образуется молекула HCl. Дальше проще всего повторить диссоциацию трехцентровой связи со стороны более слабого атома, а это хлор (кислород связан с фосфором всегда сильнее, и в трехцентровой и в обычной ковалентной связи). Видим, что у нас образовалась отличная уходящая группа, очень похожаю на то, что мы уже много раз рисовали в других механизмах, например, в пеакции Виттига – оксид фосфина. В данном случае это оксид трихлорфосфина, а проще говоря, хлорокись фосфора, что и приводит к SN2-замещению уже на углероде (этого мы уже вдоволь наелись в реакциях типа Михаэлиса-Арбузова и не удивимся тому, что всё везде в химии повторяется, но каждый раз затейливо сочетаясь по-новому)
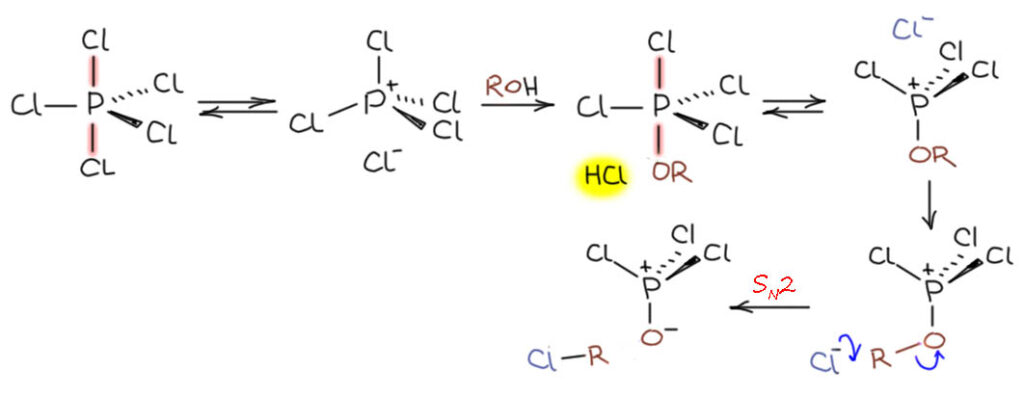
Выглядит очень хорошо. Но проблема в том, что на каждый моль спирта выделяется моль HCl и POCl3 – сильно-кислотных и агрессивных реагента, которые могут вызывать много проблем, если спирт содержит, например, двойные связи, защитные группы, многие другие функции. Этого можно избежать, если проволить реакцию в присутствии какого-нибудь основания, например, пиридина или сухого карбоната кальция, тогда реакция идёт довольно чисто. Самое неприятное в этй реакции, особенно если ее делать на приличные загрузки, это необходимость аккуратно убрать в конце оксихлорид фосфора, не допустив разогревания, сильного подкисления смеси и прочих эксцессов – это не так просто. Поэтому реакцию эту не любят и приименяют очень редко: мы предпочитаем тионилхлорид или другие реагенты замещения гидроксиа на хлор. Но не может не заметить, что по точно такому же механизму идут реакции с реагентами типа трифенилфосфин-хлор или CCl4, но вместо злобного оксихлорида фосфора мы получаем безобидный трифенилфосфиноксид. Единственный недостаток – кусачая цена такого реагента.
Точно так же пойдёт и реакция PCl5 c карбоновыми кислотами с образованием хлорангидридов, только вместо SN2 будет замещение на карбонильном углероде. Но эти реакции еще хуже, потму что получается смесь хлорангидрида и оксихлорида фосфора, и мы не можем просто гидролизовать смесь после реакции. Поэтому ее применяют только в нескольких особых случаях. Один из них – получение оксалилхлорида из щавелевой кислоты: это крайне сложно сделать, потму что не существует монохлорангидрида щавелевой кислоты, и все другие реагенты превращения кислот в хлорангидриды просто разлагают щавелевую кислоту через монохлорандидрид на смесь CO, CO2 и HCl. Только PCl5 пролетает эту стадию, сразу образуя полный хлорангидрид, как однажды обнаружил Штаудингер – но дальше надо искусно разделить фракционной перегонкой смесь оксалилхлорида и POCl3 – это то ещё занятие, требующее просто адской аккуратности и внимательности, зато в случае успеха можно получить и то, и другое (POCl3 запрещён в Российской федерации, поэтому должен быть немедленно уничтожен после получения). Ещё один пример – получение хлорангидрида акриловой кислоты (это совершенно чудовищная дрянь, поэтому если можете без него обойтись, обязательно так и поступите) – он очень летуч, и легко отгонятеся из смеси до оксихлорида фосфора. Ну и наоборот – если хлорангидрид тяжёл и нелетуч, а лучше всего твёрд, то можно наоборот отогнать POCl3 и немедленно уничтожить.
Ещё известна реакция с кетонами и даже альдегидами. В разных учебниках можно найти, что эта реакция вроде бы даёт гем-дихлорпроизводные. В этом месте уже хочется немного удивиться, и спросить, а как это происходит. Прямо целиком карбонильный кислород на два хлора? Но в книжках же пишут, не просто же так. Попробуем представить, как это может произойти. Логичная гипотеза может состоять в том, что PCl5 по тому же механизму, что и со спиртов садится на карбонильный кислород, действуя как электрофильный активатор, это приведет к дополнительной поляризации карбонила, присоединение хлорида, и замещение хлорокиси фосфора на второй хлорид. Как-то так: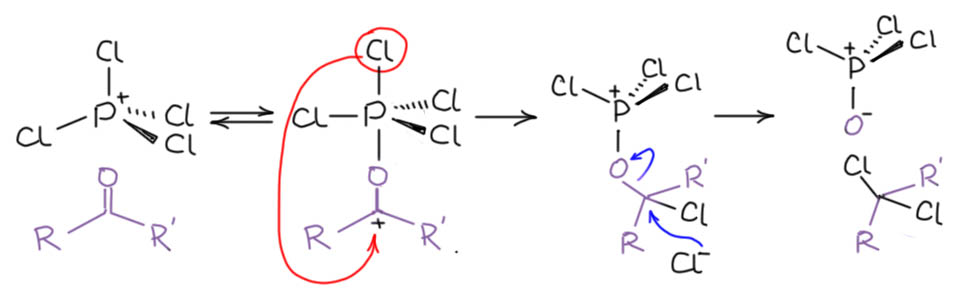
Что здесь не так? Первая проблема состоит в том, что есть очень большое сомнение в том, что активация через гипервалентную связь настолько сильна, что способна активировать карбонил к присоединению хлорида. Второе состоит в том, что стерика может помешать второй атаке хлорида – SN2 всё же, хоть и уходящая группа вполне зачётная. Строго говоря, это неизвестно, хорошо бы кто-нибудь это попробовал хотя бы посчитать. А пока мы выскажем легкий скепсис, но примем, что если такое и возможно, то скорее для неенолизуемых альдегидов. Сделать реакцию енолизуемых альдегидов с AIELVIS s. SEWMAK, GIDEOSF RAENKEALND, WALTER N. KI RN избежать альдольной самоконденсации вряд ли смогли бы даже сотрудники Кириакоса Николау. Вообще, надёжных данных по этой реакции немного, и в основном это старая химия, когда особенно не парились тем, что реакционная смесь почернела и пошёл едкий дым, и довльно часто даже не публиковали выходы. Но в более близкое нам время в самом начале 1960-х реакцией заинтересовался американский химик Мелвин Ньюмен, тот самый, который прославился удобными стереохимическими проекциями. Ньэмен решил изучить механизм реакции, и для начала нашёл, что енолизуемые кетоны в этой реакции чаще дают не гем-дихлорпроизводные, а винилхлориды, особенно если реакцию профодить аккуратно, с эквивалентом реагента и при охлаждении, хотя нередко реакция требует нагревания – как уже было сказано, PCl5 в сухих условиях никакой огромной реакционной способностью не обладает. В других случаях гем-дихлорид образуется наряду с винилхлоридом. Занятно, что реакция не дает никаких признаков карбокатионных перегруппировок в тех случаях, когда рядом с карбонилом третичный алкил, а это почти напрочь отметает карбокатионные интермедиаты. Вот, например, что получается из пинаколина (M.Newman, G.Fraenkel, W.N.Kirn J.Org.Chem., 1963, 28, 1851) – никаких признаков ретропинаколиновой перегруппировки, а мы знаем, как чувствительна эта структура к малейшим намёкам на карбокатион:
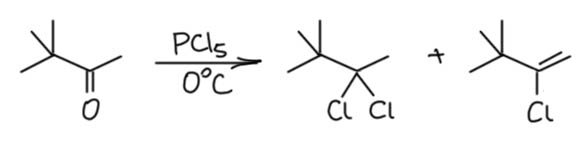
А во многих других случаях гем-дихлориды вообще не образуются, но кетоны, енолизуемые в обе стороны дают оба возможных продукта, как например в этом случае:
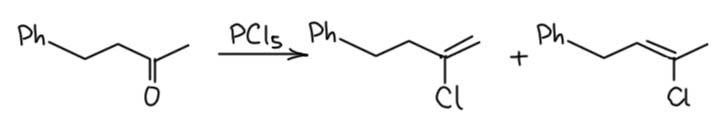
В этом месте мы начинаем немного догадываться. Сам Ньюмен нарисовал механизм, очень похожий на тот, через электрофильную активацию карбонила, хотя он и не использует понятие о гипервалентности – тогда его еще не было. Но я позволю себе предположить, что механизм даже проще, и это очередной пример реакций енолизуемых кетонов через енол, в данном случае равновесный, естественно в виде равновесной смеси двух енолов, менее и более замещённого. Енол реагирует с PCl5 как спирт, с образованием эквивалента HCl, который немедленно присоединяется к эфиру енола, активированному к электрофильному присоединению. Обратим внимание, что из двух енолов образуется один интермедиат. Дальше следует E2-элиминирование, очень легкое из-за отличной уходящей группы, так что годится и такое слабое основание как хлорид-ион – а не забудем увидеть, что здесь хлорид в очень хорошей спортивной форме, потмоу что противоионом оказывается фосфоний, то есть хлорид свободен от сильных взаимодействий с катионом и вполне готов отрывать протон в согласованном элиминировании. Здесь уходящая группа хороша, основание наоборот слабо (несмотря на упомянутую особенность), поэтому согласованное элиминирование должно иметь характер ближе к E1-подобному переходному состоянию, что и работает на образование смеси двух олефинов – в таком механизме полностью не работает правило Зайцева, потму что переходное состояние далеко от олефина.
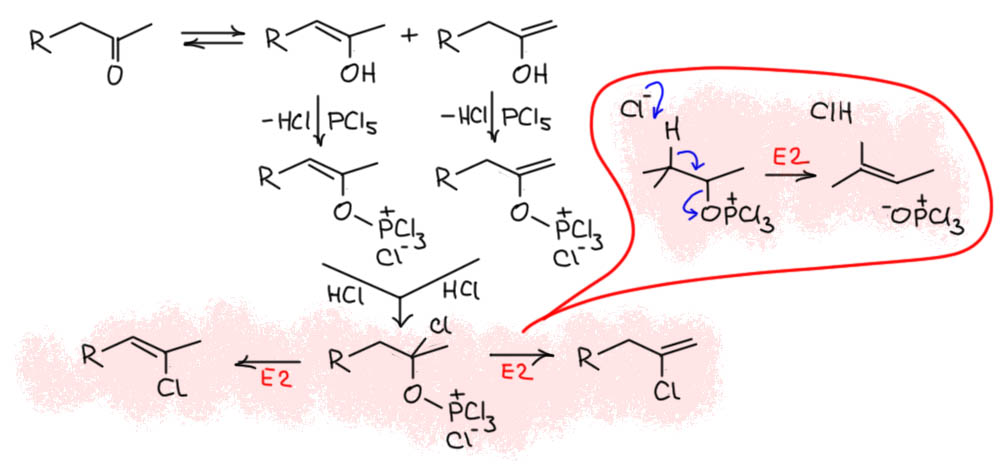
Тогда мы поймём и откуда иногда получаются гем-дихлорпроизводные – это просто присоединение ещё одной молекулы HCl к винилхлоридам. Фокус в том, что винихлориды отнюдь не так активированы к присоединению как еноловые эфиры, и это не может происходить легко, вот и не происходит, если реакцию делают чисто и аккуратно. Видимо, они образуются, если в реакционой смеси есть немного воды (растворители не совсем сухие), тогда HCl активируется к присоединению. Точне сказать трудно, потому что эксперимента хорошего так никто и не сделал, насколько я знаю.
Иногда бывают и более сложные превращения, если есть какой-то очень выгодный путь, и тогда вполне может сыграть и просто активация карбонильной группы в неенолизуемом соединении, как например, у того же Ньюмена приведены примеры реакции, сопровождаемой циклопропилметильной-гомоалильной перегруппировкой. Это настолько легкий процесс (строго говоря, это даже не перегруппировка, а очень хитрый способ делокализации, так что там скорее нужно было не обратимость, а мезомерию нарисовать, но это отдельный вопрос), что явно конкурирует с просто нуклеофильным замещением, и получается такой продукт как основной, хотя это очень старые статьи, и тогда просто могли не бнаружить неперегруппированный изомер. Так или иначе, это очень ясно показывает, что два хлора в продукте реакции кетона с PCl5 возникают на разных стадиях реакции.
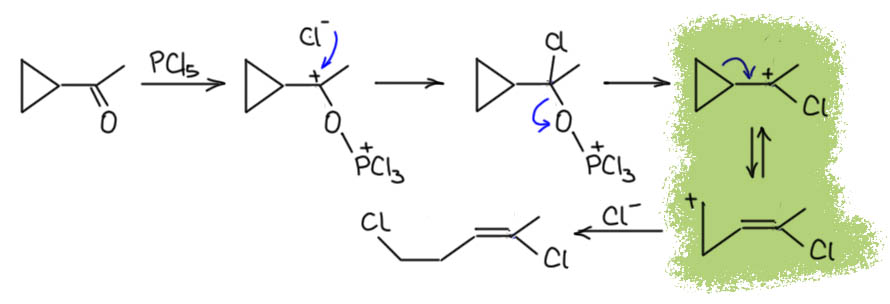
Не менее интересна перегруппировка с ароматизацией. Здесь такая же история – ароматизация тоже очень выгодна, поэтому легко осуществляется сдвиг метильной группы как только произойдёт замещение на хлор, и тогда уже это становится необратимо. В этом случае второй хлорид не понадобится.
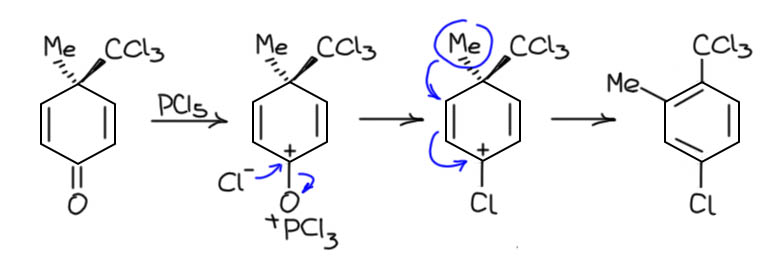
Это уже очень похоже на бензохинон. Или нет? Да, скорее не совсем. Бензохинон, конечно, это кетон, даже дикетон, но очень особенный – это весьма яркий пример кросс-сопряжения, когда два еноновых фрагмента соединены так, что дестабилизируют друг друга. Скорее всего это и есть причина высокой реакционной способности пара-хинонов, которые с огромным удовольствием вступают в реакции, снимающие это кросс-сопряжение, и оставляющие ендион, кстати тоже не подарок, но который к тому же очень легко таутомеризуется в ароматическое кольцо. Из общих соображений можно предсказать, что большого желания реагировать с PCl5 у бензохинона быть не должно – он не енолизуется, и не имеет никаких способов способствовать активации карбонильной группы, так как такая активация только еще сильнее дестабилизирует систему хинона. Да и что мы хотели бы получить? Вряд ли это вообще кто-то пробовал. Поищем, на всякий случай.
А вот, оказалось, что я не прав. Нашлись таки любопытные, и не где-нибудь, а в знаменитой Казанской школе фосфорорганики, берущей начало от главного фосфорорганика всех времён и народов, старшего Арбузова. Да, так в общем всегда и была устроена химия фосфора – попробовать все возможные реакции, и там, где что-то происходит, изучить, и посмотреть, не получается ли что-нибудь интересное. Статью про реакцию бензохинона с PCl5 опубликовали в 1988 А.А.Кутырев с коллегами в довольно известном в те годы специализированном журнале по химии пниктогенов и халькогенов (A. A. Kutyrew, S. G. Gomin, V. V. Moskva REACTION OF PHOSPHORUS PENTACHLORIDE WITH QUINONES, Phosphorus and Sulfur and the Related Elements, 1988, 39, 19-25, DOI: 10.1080/03086648808072850). Как оказалось, реакция всё же идёт, но опытные исследователи быстро разобрались почему. Секрет оказался прост – как бы ни чистили PCl5, в нём всегда остаются хотя бы следы HCl. Если их совсем убрать, просто добавив амин в реакционную смесь, никакой реакции не происходит даже при длительном нагревании. А если оставить, то реакция идёт, и очень просто. HCl очень быстро присоединяется к хинону, а продукт таутомеризуется и в смеси получается хлоргидрохинон.
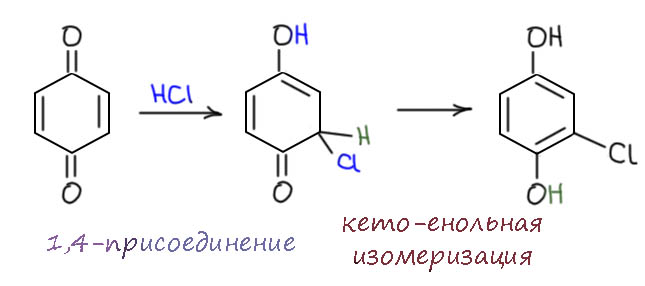
А он уже с удовольствием реагирует с PCl5, но естетсвенно, разорвать связь С-О в ароматическом кольце почти невозможно, и происходит замещение только на фосфоре. Дальше я немного допилю за авторов той работы, так получилось, что химия фосфора очень долго развивалась как чисто экспериментальная, а о структуре и тем более механизмах никто не думал. А нам пора, мы уже в 21 веке, уже нельзя просто всё запоминать. Напомню, что замещение происходит стадийно, по одному хлору и всегда из апикального положения; после каждого замещения псевдовращение Берри переставляет попарно апикальные и экваториальные заместители. На каждом замещении образуется новая молекула HCl и присоединяется к хинону – так понемногу весь хинон превратится в хлоргидрохинон, в этом смысле реакию можно называть автокаталитической – для начал достаточно следов HCl, а дальше оно само образуется, чтобы вовлекать следующую молекулу хинона. Вот так это крутится, отгрызая по одному хлору и каждая стадия создает следующую молекулу гидрохинона.
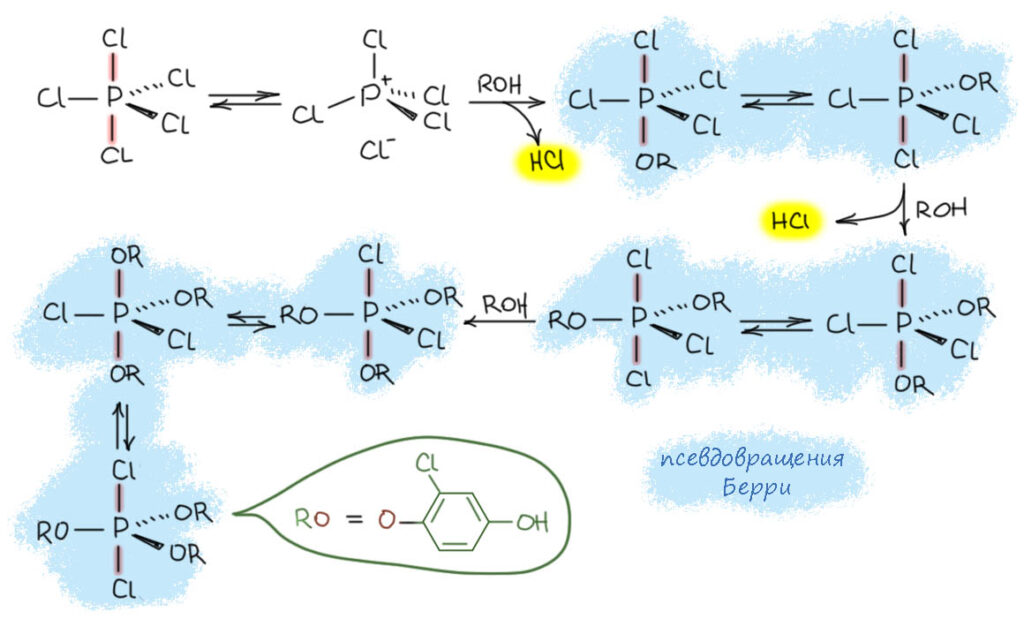
А почему все останавливается на трёх хлорах? Возможно несколько причин. Первая – чисто экспериментальная: такой продукт оказывается малорастворим и выпадает из смеси; так бывает. Вторая – стерическая, остаток гидрохинона довольно объёмистый, а заместители в экваториальных положениях находятся дальше друг от друга (угол 120 градусов), чем экваториальный и апикальный (угол 90 градусов). Третья тоже стерическая – когда накапливается три гидрохинонных группы, замещение еще одного хлора в тетраэдре фосфониевого интермедиата оказывается стерически заблокированным – четвертый гидрохинон просто не подойдёт к фосфору со стороны трех таких групп. А вот гидролиз продукта идёт очень весело и тут нет никаких стерических проблем, а образуется второй тип гипервалентной связи, той самой, которую принято рисовать двойной, а мы не хотим, потому что это неправильно (на страничке про химию фосфора объяснено почему). Конечный продукт – эфир фосфорной кислоты. 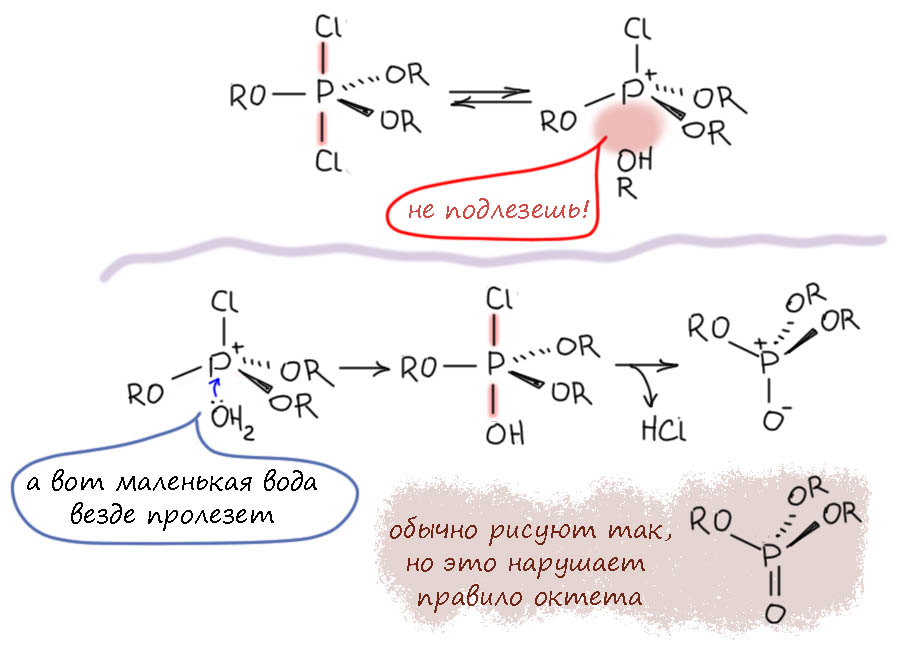
Вот в общем и вся история. Продукт получается не то, чтобы совсем бесполезный, всё же в нём остается открытая фенольная группа, и возможно что делать уже с эфиром фосфорной кислоты. Возможно даже этим кто-то уже занимался, но это уже другая история. Нам же полезно еще раз посмотреть на то, как работает химия фосфора, и понять, что элементарный с виду реагент не так прост, если понимать, как он устроен, и что ничего особо агрессивоного в нём нет, если обходится без воды и других нуклеофилов в форме сопряжённых кислот, так, что начинает образовываться HCl, а это очень сильно поднимает экзотермичность процессов. А когда HCl ет, то всё не просто спокойно, а даже и весьма лениво.
Сера
Сера - электрофил
https://doi.org/10.1002/ange.19670792202 – ссылка в Органикуме на статью по получению таких тиализонов.
Вопрос: а чем и как активируется сера в данном случае? Неужели обычный карбокатион может так просто раскрыть S8 или добавление амина (морфолина, пиперидина и т.д.) так способствует?
Я поинтересовался о таком с другой стороны: в одной статье (https://doi.org/10.1021/acs.orglett.7b00819) генерировали дихлоркарбен в присутствии трет-бутилата калия и серы, но выявить наличие тиофосгена у них не удалось, однако изоцианид присоединял серу, стало непонятно, почему изоцианид присоединяет серу, а дихлоркарбен – нет.
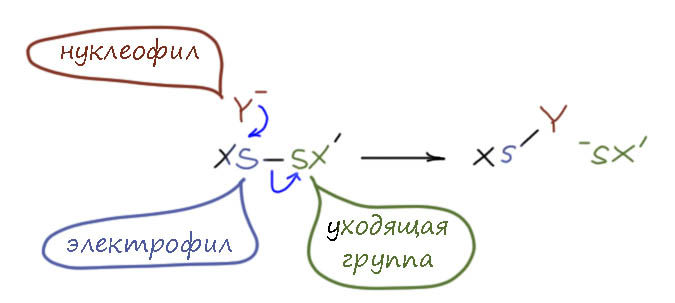
Здесь всё просто, если иметь в виду то, что элементарная сера является электрофилом. Любое соединение со связью S-S может быть электрофилом, точно так же как электрофилами являются перекисные соединения. Здесь нет ничего особенного, потому что точно по той же причине являются электрофилами галогены. Да и вообще любые соединения, в которых есть связи галоген-галоген, галоген-халькоген, халькоген-халькоген являются электрофилами (примем что и кислород в этом смысле халькоген). С азотом и пниктогенами это уже не работает как общая закономерность. Пока оставим вопрос, по какому из атомов в соединении с разными элементами будет электрофильный центр. Сосредоточимся на сере, а дальше это легко будет обобщить, если будет на то желание. Возьмём соединение со связью сера-сера, и какими-то довесками с другой стороны, мы имеем дело с двухвалентной серой. Атака нуклеофила на один из атомов серы может привести к образованию новой связи, и уходу второго атома серы в виде аниона – нуклеофугной уходящей группы. Для серы это вполне естественно, ведь это вполне зачётный неметалл с неплохой электроотрицательностью. Впрочем, точно так же поступили бы и соединения селена или теллура (про полоний не скажу). Если бы соединение было несимметричным, выбор между серами был бы именно по качеству уходящей группы – то, что висит на сере может стабилизировать анион, а может и дестабилизировать (на сере могут быть индуктивные эффекты и гиперконъюгация, но не обычное сопряжение, которое мы оставляем во втором периоде). Это не что иное как нуклеофильное замещение на атоме серы, причем бимолекулярное и дико похожее на наше обычное нуклеофильное замещения, настолько, что мы можем пока без доказательств обозначить этот механизм как SN2(S) – эс-эн-два-на-сере:
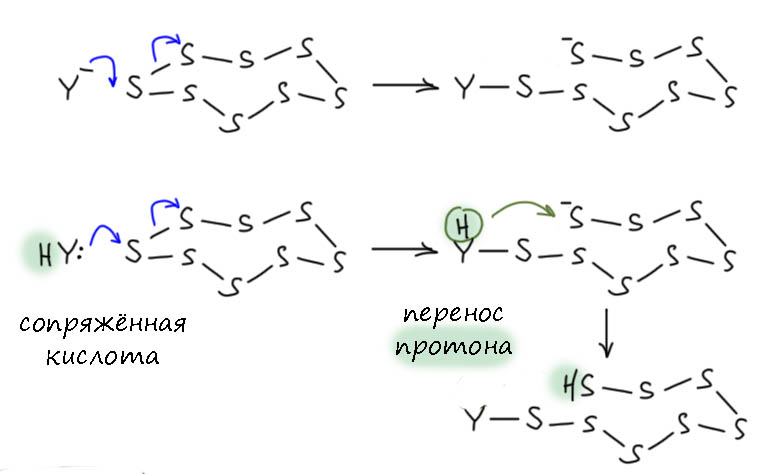
Что такое элементарная сера? Это вещество, составленное из молекул, представляющих собой цепочки атомов двухвалентной серы. Кислород, как известно, не может образовывать цепочки больше чем на 2 атома из-за альфа-эффекта – отталкивания соседних атомов. У элементов 3 периода и ниже этой проблемы нет – городите любые цепочки и проблема только в том, что связи S-S достаточно слабые, и слишком длинные цепочки неустойчивы к термическому гомолизу – поэтому достигается некоторый компромисс между длинной и устойчивостью. Чтобы не думать, куда деть концы такой цепочки, их проще замкнуть в цикл. В обычной сере, с которой мы имеем дело, основная форма это коронообразный конформер цикла S8. Да, собственно, и плевать бы на конкретную форму элементарной серы – пошли бы любые цепочки, но возьмём для определенности именно этот цикл. В нём полно связей S-S, значит и атаку нуклеофила написать легко – все атомы серы в начале одинаковые. Цикл размыкается и на другом конце оказывается сульфид. Но если реакция идёт в протонодонорной среде, то обычным образом можем учесть равновесие нуклеофила с сопряжённой кислотой, и перекинуть протон на конец цепочки, чтобы нас не раздражал отрицательный заряд (он вполне безобиден и никому не мешает, но может оскорбить чувства верующих).
Серу раскроет не любой нуклеофил, но только более-менее шустрый, поэтому серу не так просто растворить и приходится подбирать среду. И еще важна прочность образующейся связи: поэтому азотный или кислородные нуклеофилы не раскрывают серу и не растворяют её – связи недостаточно прочные (только не путайте с гипервалентными связями как в серном ангидриде и многих других соединениях), а вот углеродные нуклеофилы отлично раскрывают серные цепочки, птому что связи сера-углерод очень даже ничего себе связи. Реакция с нуклеофилами, скорее всего, инициируется по той же самой причине, по которой и галогены становятся электрофилами – вначале нуклеофил цепляется за сигма-дырку и завязывает слабую, но вполне достаточную для активации там галогенную, а здесь халькогенную связь, это почти одно и то же. О том, что это такое можно посмотреть в моей последней лекции этого нового курса про органику 21 века. Не знаю, смотрел ли кто-то уже на такие реакции с этой стороны, но если не успел ещё, то скоро обязательно кто-нибудь “прозреет” и накатает статейку.
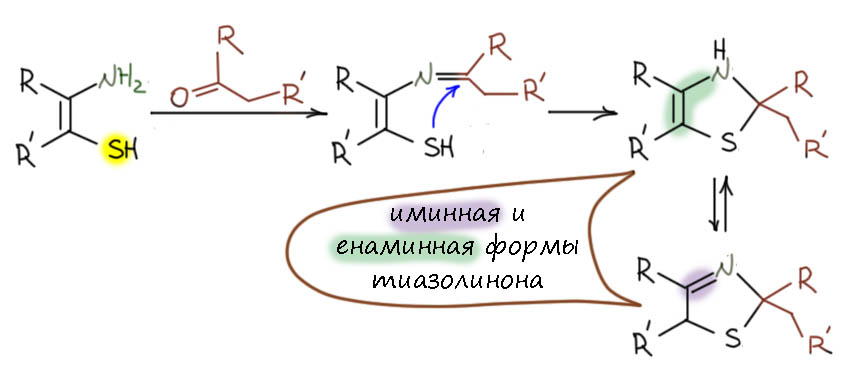
Теперь переходим к интересующей Вас реакции. Есть енолизуемый кетон (или альдегид, но альдегиды в такой системе скорее всего будут давать кучу побочных и низкие выходы желаемого). Поскольку нам нужен для реакции с серой нуклеофил, то это равновесный енол. Но – енола очень мало и он слабонуклеофилен дял реакции с серой без дополнительной активации. Поэтому я думаю, что сначала из кетона и аммиака возникает обычная таутомерная пара имин-енамин, и вот енамин уже вполне нуклеофилен для реакции с серой без активации. Я бы выбрал именно такой путь. На вопрос, а почему мы не получаем енамины из кетонов и аммиака, ответ простой – не остановитесь на просто енамине, реакция пойдёт дальше, для аждого кеона будут свои продукты. Но если мы перехватываем енамин (как, например, мы делаем в восстановительном аминировании), здесь – серой – то такой путь становится не просто реальным, но даже, я бы сказал, вполне очевидным.
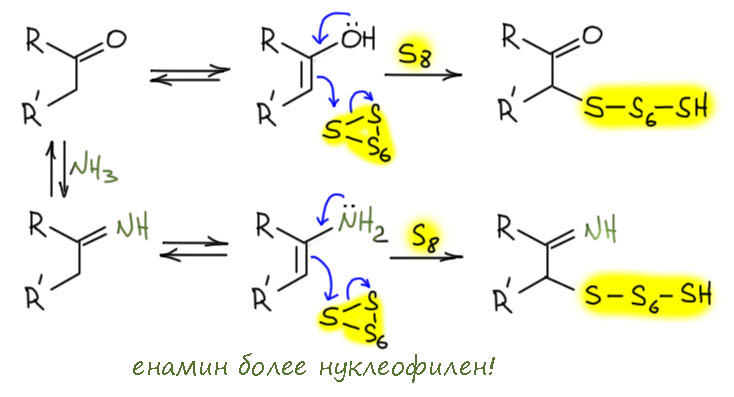
Результатом будет очень реакционноспособный политио-замещённый имин. Цепочка сер в таком интермедиате просит кирпича дальнейшего расщепления нуклеофилами. Где взять? Азот мы уже забраковали, вот углерода бы ещё… А вот у нас оказывается двукратный избыток кетона и есть ещё целый эквивалент. И он тоже наверное – аммиак дуют тоже в смесь от души, не жадничают – находится в равносесии с имином и енамином, вот мы енамином серную цепочку и крякнем. Если сделать это на второй сере, то мы получим в результате еще политиозамещенный имин, но на одну серу меньше. О, сразу становится ясно, что этот процесс – SN2(S) – можно будет продолжать, пока в смеси есть кетон, то есть до конца, и так разобрать цепь сер по одной сере полностью, ведь пока есть связи S-S, есть и электрофильность серы. А уходящей группой (после перекидывания протона) будет тиол-замещенный енамин.
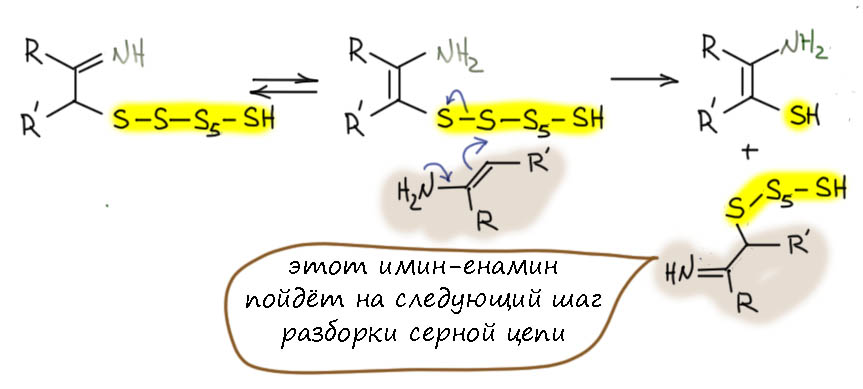 Дальше понятно – обычная гетероциклизация с кетоном, ведь кетон в смеси тоже есть, равновесие образования енамина никогда не бывает количественным. Это самая обычная реакция, похожая на обазование циклических ацеталей с диолами, здесь вместо гидроксилов не менее нуклеофильные амин и тиол, а пятичленная циклизация вытянет любую реакцию. Вот и гетероцикл – с точки зрения последней реакции это циклический диацеталь тио-аминаль. С точки зрения гетероциклов это тиазол с одной гидрированной двойной связью – тиазолин. Это номенклатура такая гетероциклов с азотом: пятичленный ароматический называется по гетероатомам с окончанием – ол, с одной двойной связью – –олин, полностью гидрированный – олидин. Посмотрев на этот тиазолин, мы увидим, что он является по совместительству енамином, ну а значит может иметь в равновесии второй изомер иминной структуры. В реальности это может быть и равновесной смесью, и одной из преобладающих форм в зависимости от конкретных заместителей, но нам сейчас это совсем до лампочки.
Дальше понятно – обычная гетероциклизация с кетоном, ведь кетон в смеси тоже есть, равновесие образования енамина никогда не бывает количественным. Это самая обычная реакция, похожая на обазование циклических ацеталей с диолами, здесь вместо гидроксилов не менее нуклеофильные амин и тиол, а пятичленная циклизация вытянет любую реакцию. Вот и гетероцикл – с точки зрения последней реакции это циклический диацеталь тио-аминаль. С точки зрения гетероциклов это тиазол с одной гидрированной двойной связью – тиазолин. Это номенклатура такая гетероциклов с азотом: пятичленный ароматический называется по гетероатомам с окончанием – ол, с одной двойной связью – –олин, полностью гидрированный – олидин. Посмотрев на этот тиазолин, мы увидим, что он является по совместительству енамином, ну а значит может иметь в равновесии второй изомер иминной структуры. В реальности это может быть и равновесной смесью, и одной из преобладающих форм в зависимости от конкретных заместителей, но нам сейчас это совсем до лампочки.
Уф, разобрались, как можно разбирать цепочки сер, каждый раз выхватывая по одной за счёт эс-эн-два-на-сере и используя электрофильность серы в связях S-S. Судя по высоким выходам в этой реакции без использования избытков реагентов, это неплохо и почти количественно происходит, но даже 10-20% зависших на стадии недоразобранных интермедиатов придадут таким смесям неземные ароматы.
Реакций таких, где енолизуемые карбонильные соединения разбирают серу при помощи какого-нибудь дополнителого нуклеофила, немало. В общем, там все должно быть похоже.
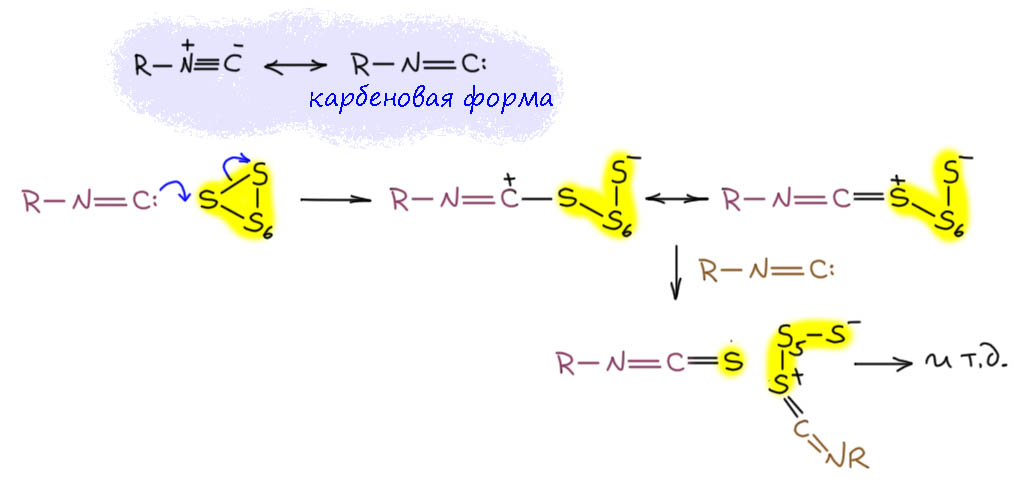
И теперь, конечно же, мы знаем, почему сера охотно реагирует с изоцианидом, но не очень хорошо с дихлоркарбеном. Сера – электрофил и реагирует с нуклеофилами. Изоцианид – это нуклеофильный стабильный карбен (если это не очевидно, опять могу отослать к лекции из цикла про 21 век про карбены). Дихлоркарбен – это электрофильный короткоживущий карбен.
Нуклеофильные карбены охотно реагируют с электрофилами, сера не исключение. Просто как нуклеофилы. Но на углероде тогда остается секстет, а значит мы должны обозначить формальный заряд плюс – это теперь стабилизированный карбокатион. И цепочка разорвана, и если нет протонодонорной среды, минус и заткнуть нечем. Ну и чёрт с ним, не мешает. Приступаем к разборке цепочки как мы уже умеем, выщёлкивая нуклеофилом серу за серой. Аккуратно обозначаем связи и заряды, рисуем одну из граничных структур, а если не лень, то обе. Дальше там просто итеративная процедура, ничего нового. В самом конце, когда останутся две серы, будет немного страшно, но не теряйте веры в справедливость процесса и всё получится.
 Кстати, это хороший способ избавляться от совершенно тошнотворного запаха самих изоцианидов. Только на замену получите не менее тошнотворный запах остатков серных хвостов. Поэтому лучше изоцианид убивать окислителями, особенно пероксидными – это ксатати ровно такая же реакция, только с электрофильным кислородом.
Кстати, это хороший способ избавляться от совершенно тошнотворного запаха самих изоцианидов. Только на замену получите не менее тошнотворный запах остатков серных хвостов. Поэтому лучше изоцианид убивать окислителями, особенно пероксидными – это ксатати ровно такая же реакция, только с электрофильным кислородом.
Дихлоркарбен это электрофил, и нехилый. В принципе, сера обладает и нуклеофильностью, ведь у неё есть пара. Посмотрим, что получится:
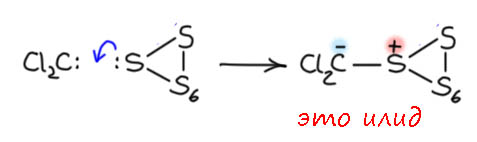
Это илид – карбанион, стабилизированный соседним атомом с положительным зарядом. Но что с ним делать? Мы не видим пути разрыва связей S-S, если только в смеси нет ещё какого-нибудь нуклеофила (сам илид может быть таковым, но мы тогда очень сильно всё усложним). И карбанион надо как-то гасить, протоны искать или другой электрофил. В общем, ничего простого не просматривается. К тому же, реакции короткоживущих частиц с гетерогенными реагентами вообще плохая затея – частица образуется в растворе, до поверхности надо ещё доплыть, а времени жизни отведено мало, по дороге погибнуть успеет. Время жизни и среднее время диффузии могут быть сопоставимы или даже совсем не в пользу частицы. И даже если одной чвастице повезет, интермедиат образуется на поверхности, его надо будет оттуда выковыривать другими частицами, но это уже будет совсем маловероятное событие – второй частице придется доплыть не прото до поверхности, но до конкретного места на этой поверхности.
Вот как-то так. Если что забыл, напомните.
Литирование
На что способно LDA?
https://doi.org/10.1016/j.polymer.2018.11.018 в этой статье его использовали как основание, а бром остался не затронут, поэтому, собственно, и возник вопрос
В самом вопросе есть некоторое недоразумение, и если его разрешить, то всё станет ясно, и вопрос исчезнет. Во-первых, термин “литирование” в органике имеет довольно широкий смысл – получение литиевых производных, а уж как, это дело второе, можно уточнить при необходимости. Поэтому, например, получение литиевых енолятов это тоже литирование, даже несмотря на то, что еноляты нельзя считать литийорганическими соединениями, потому что литий в них координирован по кислороду, а не углероду.
Есть два основных способа литирования: депротонирование CH-кислот с помощью сильных оснований, содержащих противоион лития: a) простой литийорганики (MeLi, PhPi, BuLi, sec-BuLi, tert-BuLi), в эфирных растворителях, и для увеличения эффективной основности в присутствии деагрегирующих добавок; б) амидов лития, самый популярный LDA, но применяются и гексаметилдисилазид LiHMDS, и тетраметилпиперидид; в) гидрид лития. Диапазон достигаемой основности весьма велик, в среднем литийорганика основнее амидов, а они основнее гидрида; всё это можно в широком диапазоне подстраивать координирующими и деагрегирующими добавками. Чтобы что-то депротонировать надежно и не получить равновесную смесь, нужно иметь запас по pK хотя бы 5 единиц; впрочем, поскольку мы в большинстве случаев не знаем эти величины в разных средах, то действовать приходится из общих соображений и по предыдущему опыту.
Разновидность депротонирующего литирования является направленное литирование, если есть производное бензола с заместителем, в котором есть гетероатом с неподеленной парой, обычно это атомы азота, кислорода или серы. Или гетероцикл с таким гетероатомом. Тогда такой атом участвует в координации с катионами лития литирующего агента, способствуя сближению протона и основного центра агента. В реальности там всё сложнее чем простой комплекс, который обычно рисуют, – образуются сложные агрегаты с несколькими молекулами литирующего агента, который соственно и сам представляет собой агрегаты, даже если использованы деагрегирующие агенты. Это неважно, а важно то, что это очень эффективный способ литирования по таким положениям, причем так преодолевается очень неблагоприятная разница в рК.
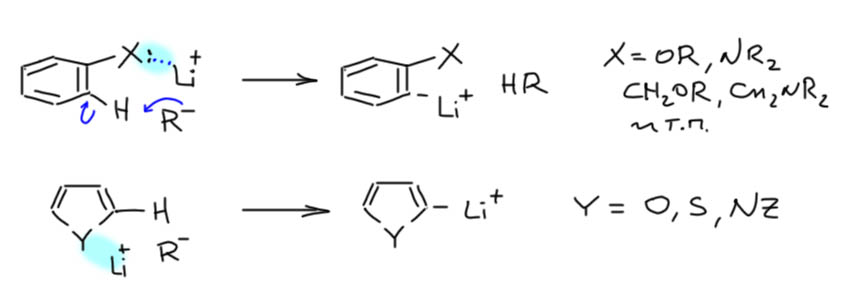
Второй способ литирования – обмен литий-галоген, почти всегда с галогенами на sp2-гибридном углероде. Литирующий агент обычно алкиллитий, что более-менее понятно даже чисто из общих соображений: если литийорганика это как бы карбанион в первую очередь, то винильный, арильный или герероарильный карбанион стабильнее алкильного по общим правилам, поэтому направление реакции понятно. Здесь не может играть роль основность, потому что нигде не переносятся протоны, но может и должна играть роль стабильность аниона, хоть он и не свободный, а в сложном агрегате с противоионами и несколькими другими карбанионми.
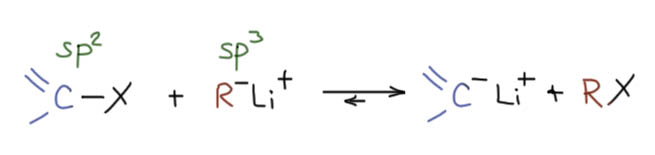
Механизм этой реакции, скорее всего, нуклеофильное замещение на атоме галогена (SN2(X)), тогда реакция должна идти через переходное состояние, предполагающее возможность как прямой, так и обратной реакции, и тогда ясно, почему так важна стабильность карбаниона в литиорганике. В упрощённом виде так, хотя ясно, что во всём, что касается литийорганики, в реакциях участуют не отдельные молекулы или тем более ионы, а агрегаты, но это и способствует реакции, потому что ионы не образуются, что всегда невыгодно, а из одного агрегата переезжают в другой.
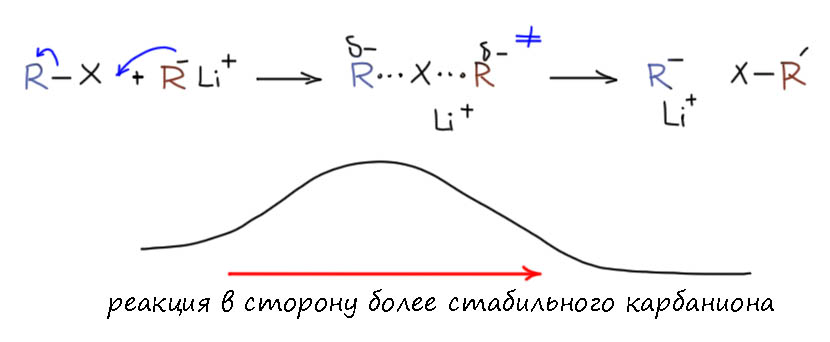
В реакции обмена галоген-литий невозможно использовать амиды лития, ни LDA, ни другие, собственно, мы уже видим почему – атака азотного нуклеофила на галоген невыгодна сразу по куче причин: во-первых, азотный анион всегда стабильнее углеродного просто из-за электроотрицательности; во-вторых, потому что связи азот-галоген намного слабее связей углерод-галоген из-за отталкивания электронных пар на соседних атомах, поэтому даже сама по себе атака амида на галоген должна иметь высокую энергию активации и быть крайне невыгодной. Ну и наконец – просто представьте, что реакция всё же пошла и в смеси начал образовываться галогенамин и вместе с литийорганикой – просто пойдет окислительно-восстановительная реакция. В общем, сплошные обломы.
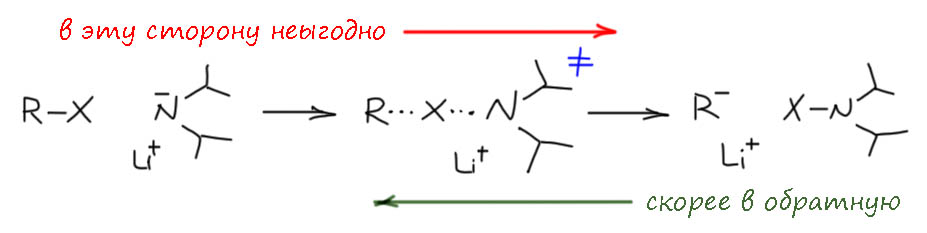
Поэтому LDA и другие амиды лития могут участвовать только в депротонирующем литировании, в том числе направленном, а в обменном литировании не могут.
Смотрим теперь пример из упомянутой статьи. 3-бромтиофен – совершенно зачётный кандидат в направленное литирование, причём соседний бром только дополнительно улучшит условия депротонирования, потому что увеличивает кислотность соседнего протона. Да, в первую очередь работает направленный процесс, но скидкой в пару единиц рК тоже не разбрасываются, тем более, что LDA не самый сильный литирующий агент.
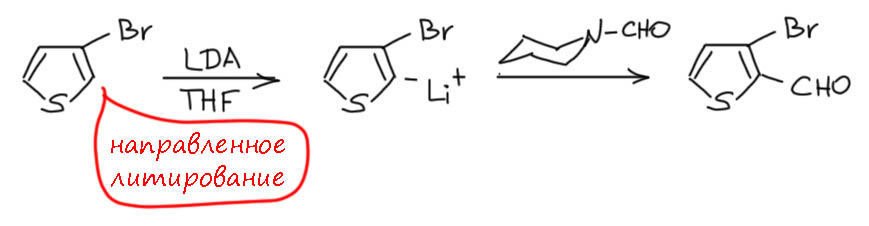
А почему тогда не взяли були? Постому что в этом случае всё же могла быть конкуренция направленного литирования и обменного, а так мягче и чище. Впрочем, ещё чаще дело в том, что конкретным химикам приятнее методика, всё есть под рукой, уже пробовали и понравилось, и так далее. Но вот чего тут точно не могло быть, так это чистого обмена, тем более с LDA. Кстати, на очень похожем субстрате, п-броманизоле, Георг Виттиг и обнаружил впервые направленное литирование.