Алкены
Очень “жирный” раздел, просто забитый всякой информацией. Алкены, в отличие от алканов, очень часто используются в лабораторном синтезе – двойная связь имеет разнообразную реакционную способность, и на реакциях двойной связи основаны многочисленные методы. Программа обширная. Сразу обратите внимание на то, что в программе есть много вещей, которые нам сейчас просто “не взять” – сил не хватит (и знаний). Они выделены рыжим курсивом. На коллоквиуме их обсуждать не будем. Возможно, их придется подобрать ближе к экзаменам, и только тем, кто хочет от жизни все и даже больше.
Сразу определимся со словами. Во-первых, алкены часто называют по-старому олефинами. Эти два термина можно считать полными синонимами и использовать попеременно. Никакой смысловой разницы между ними нет. Во-вторых, напоминаю, что в органике алкенами или олефинами называют любые органические соединения, в составе которых есть двойная C=С связь. Поэтому не удивляйтесь, когда увидите что олефином называют спирты, кислоты, кетоны, амины, аминокислоты и все остальное только потому, что где-то в структуре нашлась двойная связь. Так будет всегда и с любым классом органических соединений.
Под программой как всегда разобраны важные вопросы в раскрывающихся блоках.
Обновления
21.10 – небольшое дополнение к гидроборированию, небольшой экскурс в историю, ничего практически полезного
Подробная программа раздела
Программа по разделу – часть первая
1. Электронное строение двойной связи С=С. Гибридизация атома углерода. Строение этилена длины связей, углы). Энергия π– и σ – связи в этилене. Геометрическая изомерия. (цис-транс– и Z/E- номенклатура). Отличие конформеров (алканы) и геометрических изомеров. Относительная термодинамическая стабильность цис- и транс- изомеров бутена-2.
2. Зависимость стабильности алкенов от степени замещения двойной связи (бутен-1 и бутен-2) – оценка относительной стабильности из теплот гидрирования. Факторы, влияющие на энергию двойной связи – гиперконъюгация и стерические факторы (в цис-алкенах).
3. Гетерогенное и гомогенное гидрирование алкенов. Гетерогенные катализаторы на основе соединений металлов платиновой группы. Пример гомогенного катализатора – комплекс Уилкинсона (работает при 20оС, 1 атм). Никель Ренея. Стереохимия гидрирования.
4. Понятие о нуклеофильных и электрофильных и реагентах.
5. Электрофильное присоединение к алкенам. Почему именно электрофилы, а не нуклеофилы легко присоединяются по неактивированной С=С связи? Объяснение “школьное” и с использованием метода МО. ВЗМО и НСМО этилена. Общее представление о механизме реакций, π- и σ-комплексы, ониевые ионы, скоростьопределяющая стадия. Стереоселективность процесса. AdE2 и AdE3 механизмы. Мостиковые и открытые ионы. Когда они бывают?
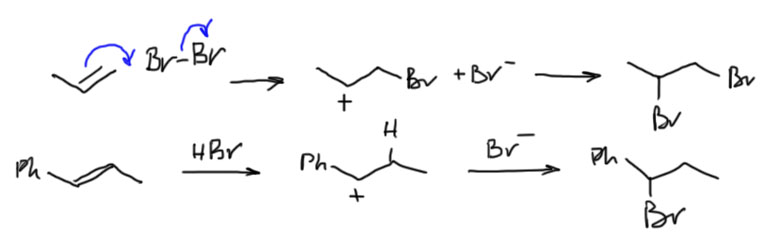
6. Присоединение хлора и брома к алкенам. Механизм, стереохимия, влияние заместителей на стереохимию присоединения (1-фенилпропен). Связь стереохимии присоединения с возможностью существования мостиковых ионов. Пример – присоединение брома к цис- и транс-бутенам-2. Образование мезо- формы (эритро-изомеров) и рацемата (трео-изомеры).
7. Гидрогалогенирование. Региоселективность электрофильного присоединения к алкенам. Правило Марковникова. Примеры. Поляризация исходной молекулы алкена. Стабильность интермедиата (карбокатиона). Влияние заместителей при С=С связи на скорость присоединения. Откуда электрофил “знает” куда ему идти – т.е. какой атом углерода атаковать? Пример применения постулата Хэммонда для стадии присоединения положительно-заряженного электрофила к двойной связи – стабильному катиону соответствует более выгодное переходное состояние. Примеры: присоединение галогеноводородов к стиролу, виниловым эфирам и винилхлориду.
8. Исключения из формальногоправила Марковникова (1)присоединение к электронодефицитным алкенам, (2) присоединение боранов, (3) радикальное присоединение. Различные формулировки правила Марковникова:
9. Электрофилы, отличные от протона. Почему RSCl даёт М+АМ?
10. Выводы по стерео- и региохимии электрофильного присоединения к алкенам. Образование циклических ониевых ионов при электрофильном присоединении к алкенам постулируется на основании кинетических и стереохимических исследований для случаев, когда электрофилом является Br+, Cl+, I+, AcOHg+, RSCl, RSeCl и т.п. В некоторых случаях удается наблюдать образование таких мостиковых ионов спектрально (в случае Br+, RS+, AcOHg+), что, однако, не является доказательством, что подобные “мостики” образуются всегда.
Можно вывести следующие закономерности AdE реакций, проходящих через мостиковые ониевые катионы или ациклические карбокатионы: Симметричный “мостик” → анти- присоединение, региоселективности нет. Несимметричный “мостик” → анти- присоединение, возможно появление син- продукта, региоселективность есть (правило Марковникова). Нет “мостика” (например, для протона) → региоселективность есть (правило Марковникова), стереоселективность есть для AdE3 (анти-), стереоселективности нет для AdE2 (смесь син- и анти-). Мостика может не быть и в том случае, когда возможно образование стабильного карбокатиона, например, в случае присоединения Br2 к 1-фенилпропену – стереоселективности нет, образуются син- и анти- аддукты.
12. Побочные реакции при электрофильном присоединении к алкенам: сопряжённое присоединение нуклеофила (например, образование бромгидринов при взаимодействии олефинов с бромом в водной среде), перегруппировки интермедиатов, карбениевых ионов (гидридные и алкильные сдвиги).
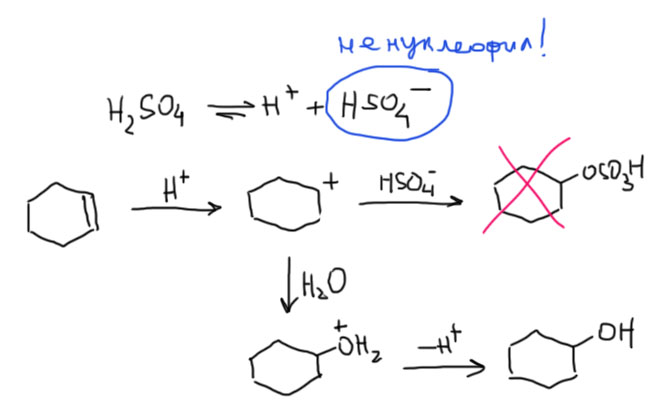
13. Кислотно-катализируемая гидратация алкенов. Условия и ограничения. Альтернативный непрямой метод гидратации – гидроксимеркурирование. Алкоксимеркурирование.
14. Регио- и стереоселективное присоединение гидридов бора (боранов). Региоспецифические гидроборирующие агенты (дисиамилборан, тексилборан, 9-ББН). Какие функциональные группы могут и не могут быть в молекуле алкена, которая подвергается гидроборированию? Превращение борорганических соединений в алканы, спирты, алкилгалогениды. Селективное введение дейтерия как демонстрация возможностей метода.
15. Озонолиз алкенов. Окислительное и восстановительное расщепление озонидов.
16. Окисление алкенов до диолов (гидроксилирование) по Вагнеру (перманганат калия, 1-2% раствор рН 7-8), Криге (тетраоксид осмия, затем NaHSO3, желательно также иметь представление о каталитическом варианте гидроксилирования OsO4 с использованием дешевых стехиометрических реокислителей, например, N-оксидов аминов или красной кровяной соли). Принципы и катализаторы стереоселективного гидроксилирования по Шарплесу. Гидроксилирование через гидролиз эпоксидов по Прилежаеву. Стереохимия гидроксилирования алкенов. Доказательства отдельных стадий син- и анти- гидроксилирования промежуточные продукты при син- и анти- гидроксилировании.
17. Свободнорадикальные реакции: История вопроса, присоединение бромистого водорода по Харашу, инициаторы реакции, механизм. Почему не присоединяются радикально HF, HI? Присоединение H2S, RSH к алкенам. Аллильное галогенирование: получение хлористого аллила из пропилена в газовой фазе, аллильное бромирование по Волю-Циглеру.
18. Молекулярные π-орбитали аллильной системы.
19. Карбены. Методы генерирования. Понятие о синглетных и триплетных карбенах, их строение, гибридизация, углы между связями, Стереохимия присоединения к алкенам. Карбеноиды. Реагент Симмонса-Смита.
20. Полимеризация алкенов (катионная, анионная, радикальная).
Двойная связь
Как устроена молекула этилена (название этилен, а не формальное этен, можно применять совершенно законно – ИЮПАК рекомендует оставить это привычное название, как и многие другие) поняли независимо друг от друга в начале 1930-х Эрих Хюккель и Лайнус Полинг, и именно эта молекула оказалась первой органической молекулой, к которой были применена новая квантовая теория строения молекул. Полинг одновременно придумал и гибридизацию, и тот способ, которым мы и сегодня обычно рисуем электронную структуру этилена.
Молекула этилена плоская и жесткая. Это радикально отличает ее от насыщенных углеводородов, например, от своего ближайшего родственника этана – этан и неплоский, и нежёсткий, вокруг C-C связи происходит практически свободное вращение (об этом подробнее в другом месте). Из того факта, что молекула плоская следует очень важный для химии вывод – у молекулы есть верх и низ, в том смысле что к плоскости можно подойти сверху или снизу (для этилена и большинства простых алкенов, с которыми мы и будем иметь дело, верх и низ это одно и то же, но у более сложных алкенов может быть разница между верхом и низом, этим активно пользуются в так называемом асимметрическом синтезе, но мы не будем – в нашем курсе этого нет).
Когда мы про молекулу говорим “плоская”, понятно, что молекула – всё равно штука трехмерная, а плоскостью молекулы называют только математическую абстракцию, проходящую через центры атомов; слово “плоская” в применении к молекуле означает только эту абстракцию, и то практическое следствие, что у молекулы есть те самые верх и низ, а не то, что на молекулу наехал асфальтоукладчик. Более того, когда мы проводим эту абстрактную плоскость, обычно пренебрегаем атомами водорода, в органической химии играющие роль заполнителя для неиспользованных на что-то более серьезное валентностей. В этом смысле и молекулы пропена, изобутилена (2-метилпропена), 2-бутенов вполне плоские, хотя в них полно атомов водорода, не находящихся в плоскости.
Более того, бывают реакции в которых к двойной связи подходят две молекулы (или части молекул) и тогда может быть две возможности – обе сверху, или одна сверху, другая снизу, и в таких случаях получаются разные молекулы, и эти молекулы по отношению друг к другу являются диастереомерами. Если в какой-то реакции такого типа получается преимущественно один из диастереомеров, реакцию называют диастереоселективной.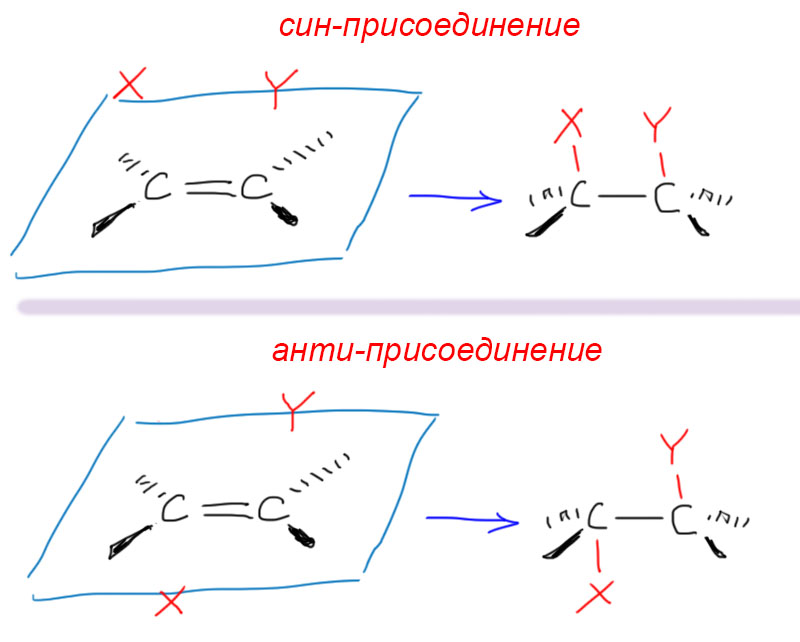
Еще одной важной особенностью этилена, отличающей ее от насыщенных углеводородов, является то, что валентные электроны находятся на двух разных и не взаимодействующих электронных системах: системе σ-связей в плоскости молекулы и π-системе.
Атомы углерода в молекуле этилена находятся в sp2-гибридном состоянии, что означает, что а) у атома углерода три непосредственно связанных с ним атома, в этом случае это два водорода и углерод; б) все четыре атома (их центры) лежат в одной плоскости и это та же плоскоcть, что и плоскость всей молекулы. Для связи в σ-системе используются не чистые атомные, а гибридные орбитали, которые, по Полингу, принято изображать восьмеркой с сильно неравными долями – большой, которая используется для перекрывания и образования связи, и маленькой, которая просто так торчит с другой стороны и ждет, не обратит ли на неё кто-нибудь внимания (увы, никому это и в голову не приходит). Гибридных орбиталей sp2-типа как раз три штуки (из одной s- и двух p-орбиталей получаются три гибридных одинаковых по форме орбитали).
По одной чистой p-орбитали (всего p-орбиталей три, а две из них использованы на sp2-гибридные орбитали) на каждом атоме углерода находятся как раз в пространстве над и под плоскостью, и плоскость является единственным местом, где их нет – это называется узловой плоскостью. В молекуле этилена узловая плоскость совпадает с плоскостью молекулы. Эти орбитали перекрываются, образуя связь, вне плоскости, тоже над и под. Такая связь называется π-связью, главным отличительным признаком которой является то, что перекрывание осуществляется строго вне линии связи – абстрактного геометрического отрезка, связывающего центры связанных атомов. Это очень точный критерий – поскольку эта линия лежит в узловой плоскости, там по определению нет и духа p-орбиталей. И отсюда ясно, что σ-связью называется как раз такая, у которой линия связи находится внутри орбиталей, обслуживающих эту связь. Отсюда понятно, почему используются эти названия – это просто греческие буквы, соответствующие латинским s и p в обозначении атомных орбиталей. Аналогия очевидная – у s-орбитали электронная плотность не равна нулю в центре атома, так же как в связевой σ-орбитали она не равна нулю на линии связи. А у p-орбитали электронная плотность строго равна нулю в центре атома, так же как в связевой π-орбитали она равна строго равна нулю на линии связи. Более серьезно можно было бы использовать орбитальное квантовое число, но это бы означало, что мы хотим вылезти из квантовой химии с весёлыми картинками и лёгким трёпом в настоящую квантовую механику с устрашающими уравнениями и принципиально полным отсутствием любого визуального воплощения. Нам там было бы страшно, а нам там были бы не рады – они вообще никому не рады, даже самим себе, а мы для них глупые. Ну и не больно-то надо, не пойдем мы туда. Нарисуем лучше весёлые картинки вслед за Полингом:
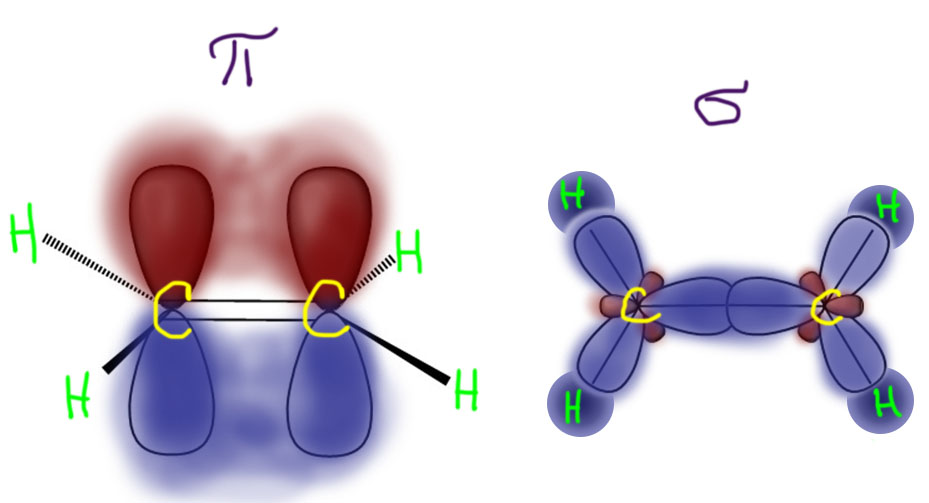
Итак, в молекуле этилена π- и σ-связи полностью разделены и не взаимодействуют, при этом π-связь гораздо более доступна для атаки разными реагентами.
Еще одно важное следствие, на которое часто не обращают внимания – CH-связи в этилене сильно отличаются от CH-связей в алканах.
Это связано с той же гибридизацией атома углерода, sp2 против sp3-в алканах. В sp2-орбиталях больше вклад s-орбитали углерода (совершенно формально этот вклад принимают равным 1/3 – в гибридной орбитали участвуют поровну три атомных, одна s и две p). В sp3-орбитали тогда вклад s-орбитали всего одна четвертая. Фокус в том, что s-электроны находятся в среднем ближе к ядру атома, чем p-электроны, они сильнее связаны, их сложнее оторвать, связи с их участием короче, а значит прочнее. Всё это относится и к гибридным орбиталям с большим вкладом s-электронов. Это не количественная, а качественная картина, в терминах больше-меньше. Но этого вполне достаточно именно для качественных тенденций. Связь, обслуживаемая sp2-гибридной орбиталью короче и прочнее, чем связь за счет sp3-орбитали. Только нужно всякий раз не забывать, что прочность связи играет первостепенную роль в свободнорадикальных реакциях – только в них связи именно рвутся пополам, и каждый атом остается со своими электронами. Прямое следствие этого – атомы водорода на двойных связях практически никогда не отрываются радикалами, нет у этилена такой химии. А у пропилена? У пропилена есть, но там используются атомы водорода на sp3-атомах углерода, и это другая проблема. Свободнорадикальные частицы охотно реагируют с этиленом, но только по π-связи в реакциях свободнорадикального присоединения.
Но CH-связь можно разорвать и по-другому, по другому пути – оставив электроны у более электроотрицательного атома углерода, иными словами, оторвав протон основанием. И здесь уже другие законы: важна не прочность связи, а относительная стабильность карбаниона. Вот от алканов протон оторвать в сколько-нибудь заметной степени невозможно, нет таких оснований на свете. А от этилена трудно, но можно. Это серьезная и важная проблема, и мы обсудим ее подробнее на отдельной странице.
Стабильность алкенов
Исключительно важная вещь. Органическая химия (да и любая другая наука) любит основывать любые рассуждения на небольшом наборе коротких заявлений, имеющих статус постулатов или даже абсолютных истин. Большинство таких заявлений, на самом деле, основаны на безосновательном обобщении на все множество органических веществ и реакций некоторых наблюдений, сделанных для нескольких (одного-двух, но бывает даже 10-15) простых веществ и реакций. Как ни странно, эта сомнительная практика действительно неплохо работает. Но чтобы время от времени не садиться в лужу, полезно помнить о весьма нечетком, относительном характере таких заявлений. С первым таким обобщением – более реакционноспособные реагенты менее селективны и наоборот, – мы уже знакомы. Сегодня к органическому символу веры добавим второе и очень популярное заявление:
Более замещенные олефины более устойчивы!
Учтите, что это всегда звучит только так, и не нужно заменять это на “менее замещенные олефины менее устойчивы” или тем более на “незамещенные олефины вовсе неустойчивы”. Вас не поймут. Или поймут, но неправильно.
Сразу возникает два вопроса: во-первых, что это вообще значит, и во-вторых, “более устойчивы” чем что?
Стабильность алкенов. Продолжение.
Первую вещь нужно понять очень точно – речь не идет о любых олефинах, и более того, речь вообще не идет об олефинах. Бесполезно сравнивать по устойчивости этилен и пропилен, или стирол и циклогексен, или акриловую кислоту и аллиловый спирт. Все эти соединения вполне устойчивы, и говорить тут не о чем. Строгие термодинамические функции энтальпии и энергии Гиббса образования для этих веществ совершенно различны и не показывают никаких осмысленных тенденций изменения. Нам это точно до лампочки.
Вместо этого речь идет об оценке энергии двойной связи, и даже не всей двойной связи, а только ее π-части. Впрочем, это очень важная оценка, так как реакций, в которых двойная связь рвется целиком немного, а в большинстве реакций происходит присоединение и простая связь C-C остается целой.
Проделаем (мысленно) следующий эксперимент. Берем два олефина, которые мы хотим сравнить. Гидрируем их по очереди в калориметре (приборе, позволяющем точно измерить тепловой эффект происходящей внутри реакции), измеряем тепловой эффект каждой реакции в расчете на моль вступившего в реакцию олефина. Полученные величины, даже если олефины разные, будут отличаться не очень сильно, в пределах нескольких килокалорий на моль.
Чем определяется эта разница. При гидрировании разрывается как раз π-часть двойной связи и образуются две связи C-H. Реакция гидрирования всегда экзотермична, то есть энергия образования двух C-H больше энергии разрыва π-части двойной связи. Чем эта двойная связь (ее π-часть) прочнее, тем экзотермичность гидрирования меньше, и тем труднее разорвать эту двойную связь – а олефин с более прочной двойной связью называют более устойчивым.
В нулевом приближении можно считать, что все C-H связи одинаковы (здесь вы конечно же сразу вспомните про различия в энергии C-H связей, ведь мы только что в алканах усердно мусолили эту проблему, но эти различия количественно не очень велики и в контексте гидрирования олефинов этим можно пренебречь). Все остальное в молекулах тоже можно считать неизменным, хотя и это не совсем правда – меняется хотя бы гибридизация этих двух углеродов, а следовательно и длины и энергии связей на них, меняется форма молекулы и всякие взаимодействия между частями молекул (стерическая часть энергии). Но мы договорились про самое грубое приближение, и в нем мы и останемся, и в этом приближении вполне разумна идея о том, что вся разница сидит только в разнице энергий π-частей связей. В этом приближении сравнение энергий гидрирования (не сами эти энергии, а только их разность!) и показывают относительные прочности этих связей.
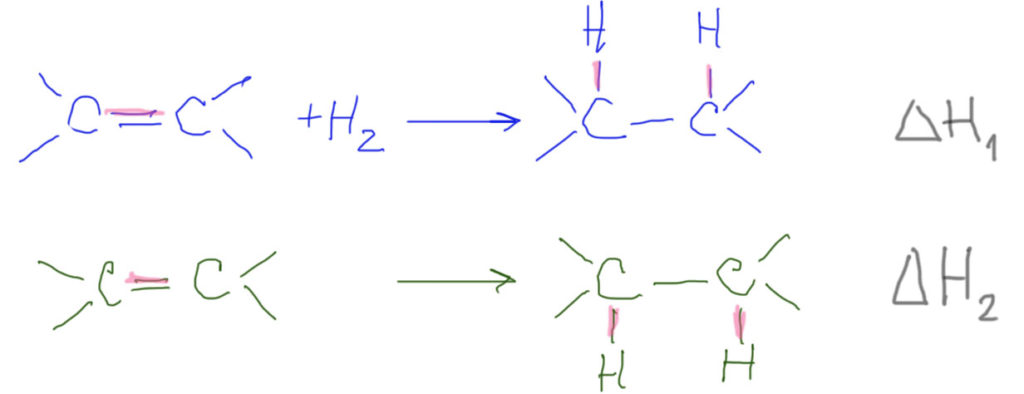
Общая тенденция получается такая – для простых алкенов (этилен, пропилен, бутилены) чем больше заместителей (метилов), тем стабильнее (прочнее) двойная связь. Это экспериментальное наблюдение обычно объясняют с помощью той же самой гиперконъюгации, что и стабильность радикалов. Только здесь придется подумать серьезнее, что с чем сопрягается. Проблема в том, что просто так перенести схему для радикалов на олефин нельзя. Сопряжение π-орбиталей двойной связи с гибридной орбиталью C-H связи рядом выглядит точно так же, как и сопряжение орбитали неспаренного электрона с той же гибридной орбиталью в радикалах. Но – здесь на этой системе 4 электрона (два с одной связи, два с другой), а это, как мы уже однажды видели, приводит не к стабилизации, а к дестабилизации. Выходят из этой ситуации довольно просто – во взаимодействие вовлекают не обе связывающие, а связывающую и разрыхляющую орбитали тех же связей – тогда во взаимодействии 2 электрона (разрыхляющие орбитали пусты). Этот вид гиперконъюгации поэтому является σ-π* сопряжением. Ехидный и недоверчивый человек (эти качества совершенно необходимы любому, кто серьезно решит заниматься наукой, но одних их, увы, недостаточно) непременно спросит, а почему это мы в одном случае (стабилизация радикалов) выбираем σ-π-сопряжение, а в другом σ-π*? Что за произвол – что хотим, то и выбираем. Не совсем так, на самом деле, так как в каждом случае речь идет о балансе всех возможных взаимодействий, и стабилизирующих, и дестабилизирующих. Кто-то проделал эту кропотливую работу за нас, и выяснил, какие взаимодействия вносят наибольший вклад в каждом случае. А мы просто пользуемся результатами этих трудов, выдавая их за глубину собственного анализа. Итак, нарисуем орбитали фрагментов
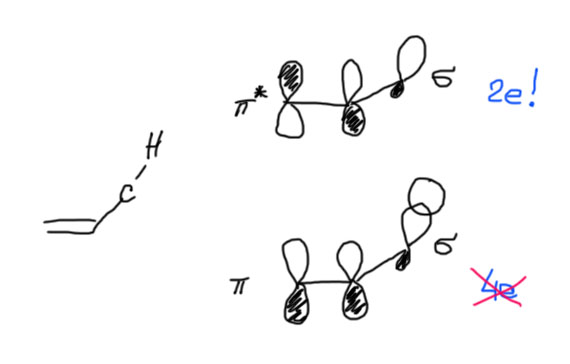
Как и в случае стабилизации алкильных радикалов, и здесь эффект гиперконъюгации невелик, но за неимением других вполне заметен.
![]() Сверху на этот эффект накладывается стерический эффект – если в олефине какие-то заместители на двойной связи сближены, то они отталкиваются, что приводит к дестабилизации. Это очень хорошо видно при сравнении цис- и транс-изомеров – первые всегда менее стабильны чем вторые. Если заместители довольно крупные, то дестабилизация цис-изомеров может быть настолько велика, что перекроет эффект гиперконъюгации. Обратите внимание на смешной значок, похожий на букашку, – так принято обозначать стерическое отталкивание.
Сверху на этот эффект накладывается стерический эффект – если в олефине какие-то заместители на двойной связи сближены, то они отталкиваются, что приводит к дестабилизации. Это очень хорошо видно при сравнении цис- и транс-изомеров – первые всегда менее стабильны чем вторые. Если заместители довольно крупные, то дестабилизация цис-изомеров может быть настолько велика, что перекроет эффект гиперконъюгации. Обратите внимание на смешной значок, похожий на букашку, – так принято обозначать стерическое отталкивание.
Теперь самое главное – зачем нужно знать про “стабильность олефинов”. Ответ такой – в большинстве случаев совершенно незачем. Нет никакой радости в том, что двойная связь в циклогексене (дизамещенном олефине) стабильнее связи в этилене. Как мы уже, надеюсь, поняли, тем более совершенно ни из чего не следует, что циклогексен стабильнее этилена – более того, это не имеет смысла. Когда разберетесь в термодинамике, поймете почему. Но информация о стабильности становится очень важной, когда сравнивают изомерные алкены с разным положением двойной связи относительно заместителей, но с сохранением скелета (изомеры положения). Тогда все остальное в молекуле одинаково (как всегда в некотором приближении), и вся разница кроется только в двойной связи. В этом случае можно в прямом смысле говорить о стабильности алкенов. А стабильность прямо связана с равновесием. Если существует реакция, в которой один изомер превращается в другой, то такая реакция обратима, и положение равновесия смещено в сторону более стабильного алкена (то есть, в большинстве случаев более замещенного). Такие реакции действительно существуют, и называются равновесиями изомеризации. Изомеризация алкенов, например, часто вызывается небольшим количеством молекулярного иода. Очень важно не забывать, что в равновесных условиях изомеризация всегда превращает менее стабильный изомер в более стабильный. Степень превращения редко бывает стопроцентной, или даже хотя бы >90%, чаще получается смесь с преобладанием более стабильного изомера. Для того, чтобы это оценить точно, нужно знать константы равновесия в каждом случае. В принципе, эта информация вполне доступна, если разобраться в термодинамике и в том, откуда берутся значения термодинамических функций для разных веществ. Мы этого делать не будем, поэтому и не имеем права использовать простую равновесную изомеризацию для превращения одного изомера в другой, если это, например, понадобится в какой-нибудь задаче. Есть более надежные неравновесные способы изомеризации, и мы их обязательно изучим.
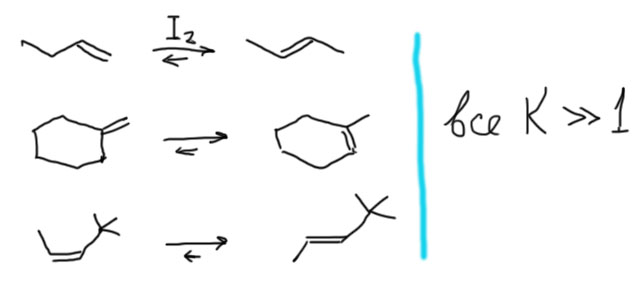
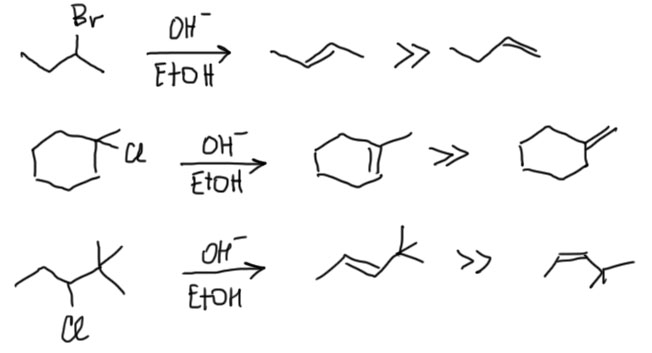
Второе важное применение знания об относительной стабильности изомерных алкенов – предсказание того, какой олефин образуется в реакциях отщепления (элиминирования), например, отщепления галогеноводородов от галогенпроизводных, или воды от спиртов. Подробнее мы это рассмотрим позже, но сейчас отметим, что в большинстве случаев образуется как раз наиболее устойчивый олефин из возможных. Эта важная закономерность называется правилом Зайцева. В отличие от равновесной изомеризации этим можно и нужно пользоваться.
Что такое электрофилы и нуклеофилы?
Множество реакций в органической химии – это реакции между электрофилами и нуклеофилами. В реакциях образуются ковалентные связи, и пара электронов на их образование может быть дана либо поровну каждым из реагентов – это свободнорадикальные реакции; либо пара от одного реагента и пустая орбиталь от другого – это как раз и есть электрофильно-нуклеофильные реакции. Знатоки неорганической и координационной химии немедленно воскликнут – да так же образуется координационная связь, а реагенты, которые ее образуют называются кислоты и основания Льюиса!!! – Зачем новые слова и понятия?!
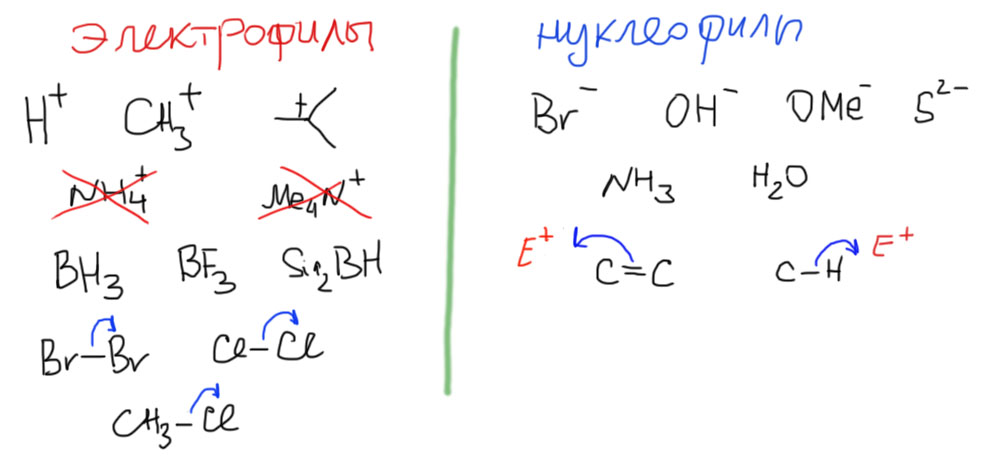
Можно и так посмотреть, но на самом деле, понятия электрофилов и нуклеофилов не сводятся к кислотам и основаниям Льюиса – они шире и удобнее. Итак, как образуется ковалентная связь? Самый простой случай вполне похож на координационную связь из неорганики: катион плюс анион. Катион – электрофил, анион – нуклеофил. Катион при этом должен быть секстетным (посмотрите еще раз вводный концентр, если забыли, что это такое), октетные (они же ониевые) катионы не являются электрофилами. Электрофилом является и самый маленький катион – протон.
Дальше, как это принято в серьезных науках, начинаем обобщать. Электрофил не обязательно заряжен, но может иметь просто вакантную орбиталь. Боран, например, электрофил. Обобщаем дальше – даже не обязательно иметь такую орбиталь, а иметь возможность ее образовать прямо в процессе реакции. Бром или хлор, например, электрофилы, потому что в реакции могут развалится на две части, отдав пару одной половинке. Мы уже видели как они разваливаются ровно пополам в свободнорадикальной химии, но распад может быть и гетеролитическим. Это выгодно тогда, когда эта уходящая половинка радуется приобретенной электронной паре, то есть является элементом с высокой электроотрицательностью или его производным. Здесь не все так уж совсем просто, нужно еще знать много важных деталей, но пока точно сойдет.
Нуклеофил не обязательно анион, но может быть и просто молекулой, имеющей атом(ы) с неподеленной парой. Аммиак или вода, например, нуклеофилы. Не обязательно даже неподеленной парой, но можно и кратной связью – обслуживающие ее пару электронов амбициозный электрофил может вытянуть целиком. Алкены поэтому тоже нуклеофилы. Некоторые, особенно крутые электрофилы могут вытягивать пару не только с кратной, но даже и с простой связи, обычно C-H. Это редкий случай, но мы его знаем. Метан и другие алканы – тоже нуклеофилы.
 Теперь, когда мы успешно разделили все реагенты на две большие кучи, затвердим еще один важный (важнейший!) лозунг органической химии. Он состоит из трех частей – одного разрешения и двух запретов:
Теперь, когда мы успешно разделили все реагенты на две большие кучи, затвердим еще один важный (важнейший!) лозунг органической химии. Он состоит из трех частей – одного разрешения и двух запретов:
Электрофилы реагируют с нуклеофилами
Электрофилы не реагируют с электрофилами
Нуклеофилы не реагируют с нуклеофилами
Пожалуйста, пропитайтесь этим как следует. Неумение отделять нуклеофилы от электрофилов и попытки устроить реакцию между реагентами одного типа – один из важнейших источников глупейших и заведомо неисправимых ошибок в химии. В таких случаях даже спорить не с чем. Реакции всегда бывают только между реагентами разных типов. Это очень похоже на другие священные пары в химии – кислота/основание, окислитель/восстановитель. Позже обсудим это подробнее.
И еще одна важная вещь. В реакциях электрофилов и нуклеофилов обычно (то есть почти всегда) образуются заряженные частицы, катионы и анионы. Гетеролитический распад связей с полным смещением пары на один центр, всегда происходящий в таких реакциях, гораздо сложнее чем гомолитический, с которым мы вроде бы уже лихо разобрались. Это связано с тем, что заряженные частицы очень сильно взаимодействуют как друг с другом, так и с другими веществами, прежде всего с растворителями, и пренебречь этими взаимодействиями нельзя даже в самом грубом приближении. Здесь уже не обойтись простыми энергиями связей, а придется думать еще и о других ионах и о растворителях. Мы пока вообще не умеем это делать, и научимся только в самых общих чертах.
Электрофильное присоединение к алкенам
Механизм самой главной реакции двойной связи алкенов хорошо разобран и в лекциях, и в учебниках, и больших проблем не вызывает. Главное – это точно понять, зачем он вообще нужен. Механизмы изучают и рисуют не просто так, чтобы было что обсудить, а для того, чтобы иметь возможность объяснять и предсказывать реальные реакции, реакционную способность, селективность и т.п. Это нужно точно понимать, и сохранять самообладание, когда на уже понятный механизм начинают навешивать какие-то новые детали или вводить изменения. Всегда спрашивайте себя, зачем нужна та или иная особенность механизма. Если видите какой-то модный наворот, который ничего конкретного не объясняет, смело отправляйте его либо сразу в мусорное ведро, либо на самое донышко памяти, чтобы не путался под извилинами и не мешал.
В самом общем виде механизм формулируется так. Он двухстадийный, первая стадия – присоединение электрофила к двойной связи с образованием карбокатиона. Карбокатион – тоже электрофил, и очень реакционноспособный, он немедленно реагирует с первым попавшимся нуклеофилом. Это важно – какой попадется, с тем и прореагирует. Карбокатион не выбирает, с каким нуклофилом реагировать. Если их в системе несколько, то прореагирует со всеми и приблизительно в тех же соотношениях, в которых нуклеофилы присутствуют в реакционной смеси.
Первая стадия определяет, к какому из двух углеродов присоединится электрофил. Критерий – устойчивость карбокатиона. Нужно посмотреть все электронные эффекты. В вводном концентре мы это делали вполне успешно. Всегда нужно выписать оба катиона и посмотреть эффекты. Здесь часто возникает некоторая неопределенность – один из заместителей в карбокатионе будет той частью молекулы, к которой и присоединился электрофил – а какой эффект у этого куска. Вообще-то это легко прикинуть в конкретных случаях, когда известно, что является электрофилом. Но этим можно и пренебречь – в этой части молекулы редко бывают определяющие эффекты. А вот второй заместитель как раз обычно и является определяющим. Собственно вариантов всего два – если там индуктивный, а еще лучше мезомерный донор, то там и будет карбокатион. Если -I/-M-акцептор, то он будет дестабилизировать, и катион наоборот быдет стремиться убраться подальше от такого подарка.
Разобрались с катионом, переходим к второй стадии и ищем нуклеофил, который заткнет карбокатионный центр. Так как мы договорились, что катион нуклеофил не выбирает, выбираем мы из всех нуклеофильных частиц, которые мы находим в системе. Варианты здесь такие.
- Нуклеофил пришел вместе с электрофилом, и в системе больше ничего нет. Тогда они по очереди садятся, куда положено, и все просто. Заодно посмотрите как рисуются стрелки смещения электронной плотности в сокращенной записи механизма – мы показываем, какие связи рвутся и какие образуются, при этом направление стрелок всегда от нуклеофильной части к электрофильной.
- Второй важный случай. С электрофилом не пришел нуклеофил. Как это???! А очень просто. Возьмем сильную кислоту. Если это соляная или бромистоводородная, то в них есть нормальный нуклеофил – галогенид. А если серная? Анионы сильных кислородных кислот не являются нуклеофилами (точнее, являются, но такими слабыми, что на это можно не обращать внимания). Тогда мы смотрим повнимательнее и находим воду – вода есть даже в концентрированной серной кислоте, а в реакции используют даже немного разбавленную. Затыкаем карбокатион водой, и обращаем внимание, что от первоначального продукта этой реакции нужно для порядка отобрать протон. Это происходит само собой, но иногда требует подщелачивания при обработке реакционной смеси Для этого используют питьевую соду. В схемах реакций допустимо просто писать -H+
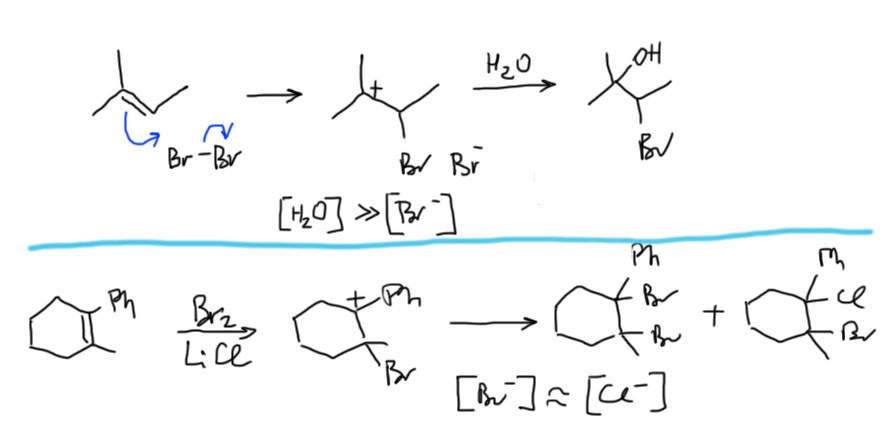
- Третий вариант. Нуклеофил таки пришел с электрофилом, но в реакционной смеси есть еще что-то, например, вода. Очень распространенный случай – используют бромную воду (раствор брома в воде). В таком растворе брома мало, а воды много, поэтому карбокатион будет связывать воду, а не бромид (которого мало потому что брома мало). А могут и специально что-нибудь добавить, например, к галогену галогенид, только другой. Тогда получим или только продукт с посторонним нуклеофилом (если его намного больше), либо смесь, если взяли сравнимые количества. Важно внимательно следить за тем, что куда присоединяется, не забывая об стойчивости промежуточного карбокатиона.
- Четвертый вариант. Нуклеофил обнаружился в молекуле олефина. Так как мы договорились, что олефином называют все, что угодно, если где-то в молекуле есть двойная связь. А еще там может оказаться, например, спиртовая группа. Возьмем хотя бы пент-4-ен-1-ол и присоединим к нему бром. Сначала образуется, как и положено, карбокатион, и будет искать себе нуклеофил. Нуклеофил есть в самой молекуле. Но, казалось бы, в этот раз у него нет никакого преимущества в концентрации. Мы точно можем сказать, что концентрация бромида просто точно равна “концентрации” гидроксила – на каждый образующийся карбокатион в системе есть один бромид и один гидроксил. Тогда, наверное, как мы уже привыкли, карбокатион будет поровну связывать один и другой. Но нет, в этот раз у гидроксила есть огромное преимущество – он в той же молекуле, рядом. Только очень важно понять, что значит “рядом”. По стереохимическим причинам “рядом” означает, что между двумя центрами (в данном случае между карбокатионом и кислородом с неподеленной парой) расстояние равно пяти или шести атомам, включая центры. Тогда внутренний нуклеофил затыкает карбокатион с образованием 5- или 6-членного цикла. В данном случае цикл 5-членный, и это хорошо. На схеме красненьким подсвечен образующийся цикл, и показано, как образуется продукт.
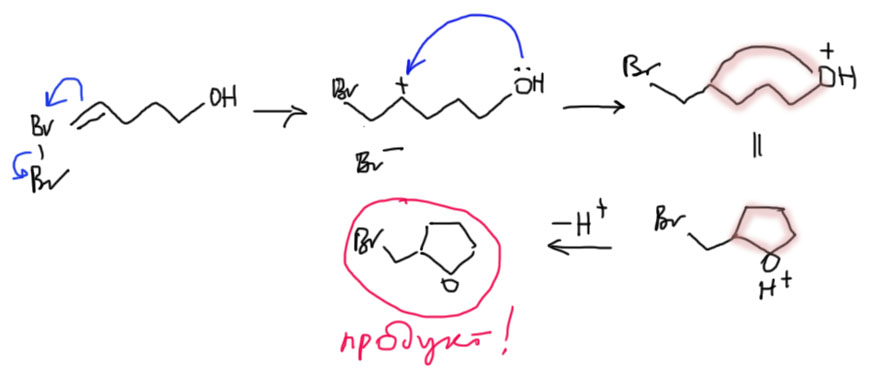
Такая реакция называется внутримолекулярной. Запомним – внутримолекулярная реакция имеет огромное преимущество перед реакциями между двумя разными молекулами (межмолекулярными реакциями), если в ней может образоваться 5-и или 6-тичленный цикл. В остальных случаях преобладают межмолекулярные реакции.
Электрофильное присоединение: проблемы
Но более внимательный подход к этой теме показывает, насколько эта простая картинка далека от реальности. Даже наша программа поднимает как минимум еще несколько вопросов –
- что такое AdE2 и AdE3 механизмы?
- что такое мостиковые ионы и зачем они нужны?
Если же мы попробуем оторваться от учебников и загдянуть в реальную научную и препаративную литературу, мы найдем немало сюрпризов. Один из них – почему так редко в реальных синтезах используются реакции присоединения галогеноводородов? Ну хорошо, про гидратацию нам уже наговорили много гадостей, но уже присоединение HBr-то точно должно быть самой простой из всех возможных реакций. Разве не она использована Марковниковым для открытия своего великого правила, которое уже 150 лет с нами, а мы с ним. Если кому-то кажется, что 150 лет это немного, хотя бы по сравнению с завоеваниями Г. Юлия Цезаря, спешу напомнить, что всей органической химии как науке и есть приблизительно столько лет, ну лет 20-30 можно еще накинуть, и то с трудом. И, да, то есть нет, не использовал Марковников присоединение HBr для правила. Судьба Владимира хранила. Если бы случайно использовал, правило могло бы или совсем не сформулироваться, или быть строго противоположным. Если не верите, посмотрите хорошо известный сборник методик Препаративную Органическую Химию Титце и Айхера, методика Б-2 (стр.65) – синтез 1-бромгептана присоединением HBr к 1-гептену. Никаких перекисей! Условия реакции как нарочно такие, чтобы максимально исключить даже подозрение на свободнорадикальную реакцию – в темноте, атмосфере азота, на холоду.
Это ошибка? Галлюцинация?! Ущипните меня! Сгинь, наваждение!! Ничего подобного, это вполне надежный и проверенный источник методик, и эта реакция с разными простыми 1-алкенами и именно с таким результатом многократно описана в оригинальной научной литературе. Да и в описании методики авторы сетуют на то, что должно бы присоединяться по правилу Марковникова, – а присоединяется наоборот. И ничего с этим не поделать. Эксперимент важнее любых теорий и интерпретаций. Тогда что это? Сенсация?! Правило Марковникова опровергнуто!? Опять ничего подобного. Правило Марковникова на месте. Его вообще невозможно опровергнуть, – оно всесильно, потому что верно, и верно, потому что всесильно, о чем подробнее на отдельной вкладке. А вот с самим гидробромированием действительно есть проблема, которая заслуживает обсуждения.
Далее, что такое мостиковые ионы и зачем они нужны. Нас уверяют, что без них невозможно объяснить анти-присоединение брома и других электрофилов с галогенами или серой к двойной связи. Это не так. Галогеноводороды (опять они!) очень часто также присоединяются анти-способом. Но уж здесь-то мостиковые ионы точно ни при чём. Протон чисто умозрительно мог бы образовать мостиковый ион, но, во-первых, он этого не делает (подробнее почему не делает можно прочитать на вкладке про гидрогалогенирование), а во-вторых, если бы делал, то протон настолько мал, что какое-либо стерическое препятствие создать не может. Значит может быть и другое объяснение анти-присоединению. А если оно есть здесь, то зачем нам мостиковые ионы в бромировании? Ответ простой – они в нем есть, их существование доказано, и не один раз, а пренебрегать существенной научной информацией неприлично. Хорошо, пусть будут. Но они же тащат за собой не только хорошее объяснение анти-стереохимии, но и новую проблему – если они выглядят так, как это обычно и рисуют в учебниках, то почему они присоединяют нуклеофил так, как будто вместо мостикового иона там обычный карбокатион, как у нас и написано в кратком курсе электрофильного присоединения. А почему бы им так и не делать? А потому что бромониевый (или хлоронивый, или иодониевый, или сульфониевый и т.п.) ионы по структуре должны быть аналогичны оксиранам – все это трехчленные гетероциклы с одним гетероатомом. Но оксираны раскрываются нуклеофилами по законам SN2-замещения (это мы все очень скоро будем разбирать), то есть с атакой на менее, а не на более замещенный атом углерода, то есть против правила Марковникова. Но бромониевые ионы почему-то ведут себя не так – нуклеофил раскрывает их по более замещенному атому.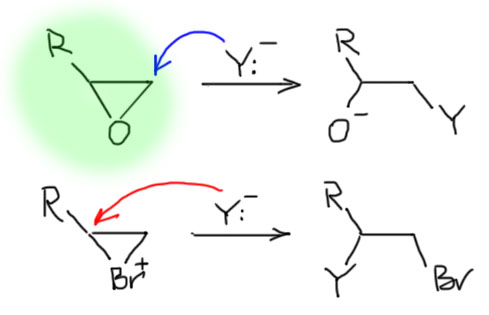
В чем дело? Разберёмся. Это весьма интересная проблема, которая неожиданно стала источником многих интересных результатов в совсем современной органической химии. Так далеко нам лезть не положено, но в основных вещах разобраться стоит хотя бы для того, чтобы один раздел органической химии не противоречил другому.
Два особых типа олефинов - донорные и акцепторные олефины
Для первоначального знакомства с химией алкенов это можно не читать, но если у вас амбиций больше, чем просто сдать коллоквиум и забыть, то очень рекомендую с этим разделом ознакомится – это очень поможет впоследствии и очень скоро, так как таких олефинов в дальнейшей химии будет как грязи осенью.
Как мы договорились, электронные эффекты не изменяют природу явления (например, не делают двойную связь тройной, кислоту основанием, а катион анионом), а только в некоторой, большей или меньшей степени изменяют количественные характеристики, например, оставаясь в теме про двойную связь, мы можем говорить о большей или меньшей реакционной способности по отношению к электрофилам, к появлению частичных зарядов на углеродах, связанных двойной связью, и т.п. Занудство, в общем, страшное, но неизбежное.
Но, как учили нас (к вашему счастью не вас) классики марксизма-ленинизма и пролетарского интернационализма, количество однажды переходит в качество. Так как они это не сами придумали, а просто слямзили у нормальных философов, эта идея не лишена смысла. Вот и олефины остаются олефинами, пока на двойной связи обычные доноры и акцепторы (алкилы, другие углеводородные группы, галогены, и т.п.), но чрезвычайно сильно меняют свойства и даже фактически свою природу, когда заместителями служат или сильнодонорные, или сильноакцепторные заместители с мезомерным эффектом. Получаются два особых класса олефинов, играющие просто неприлично колоссальную роль в органической химии. Оба этих класса прочно и неразрывно связывают химию олефинов с химией карбонильных соединений. Почему – узнаем в свое время, а пока ограничимся простым описанием этого явления.
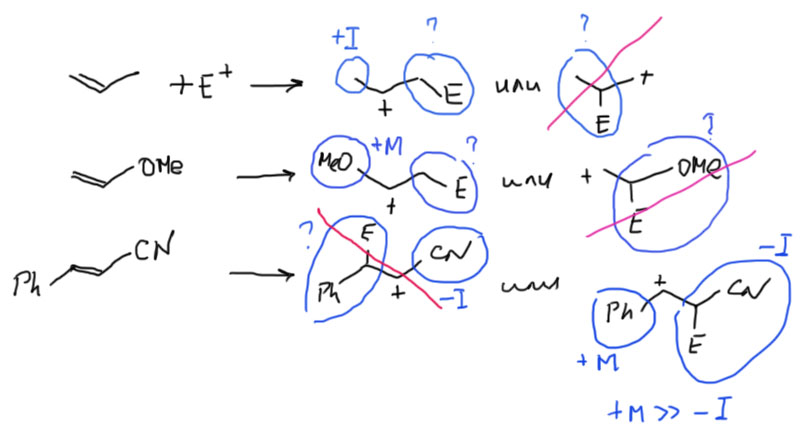
Олефины с сильнодонорными +М-заместителями, они же донорные олефины, они же нуклеофильные олефины на двойной связи несут еще одну (можно больше, но это не так принципиально) +М-группу, например, амин (тогда соединение называется енамин), гидроксил (тогда это енол), алкокси-группу (эфир енола), или даже кислород с отрицательным зарядом (такой ион называют енолят-анионом). Нарисуем просто структуру с обобщенным заместителем, имеющим неподеленную пару, которая легко смещается в сторону двойной связи, а та, в свою очередь, смещается на дальний атом углерода, создавая там приличный отрицательный заряд. Если нарисовать граничные структуры, то в таких олефинах вклад второй граничной структуры с отрицательным зарядом на дальнем углерода и двойной связью на заместитель, всегда значителен, но основной граничной структурой, ближе отражающей реальную струткру олефину остается первая – это все же олефин. 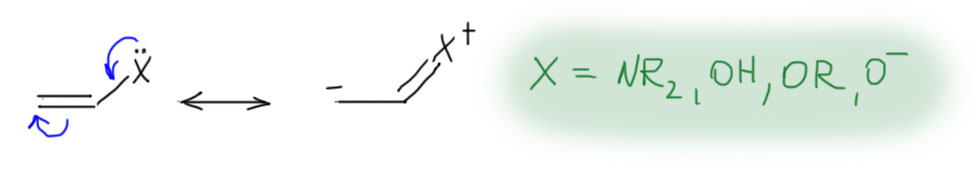 Но главное даже не это, хотя это очень важно, а то, что такие олефины обладают огромной реакционной способностью по отношению к электрофилам, и в таких случаях никогда не идет никаких разговоров ни про какие мостиковые ионы, потому что карбокатион после присоединения электрофила надежно стабилизирован мезомерным донорным эффектом. Более того, это уже и не карбокатион никакой, а просто какой-то аммониевый или оксониевый ион, или даже вовсе нейтральная молекула (если исходный олефин – анион енолята). Такому катиону не обязательно спешить искать себе нукелеофил хоть какой завалящий, а можно просто подождать, когда дядя или тётя над колбой решит, что реакция прошла, и выльет всех в воду – они всегда так делают, мы знаем. В этом случае это обычно заканчивается гидролизом и образованием карбонила, но подробнее мы в этом разберемся в химии карбонильных соединений. Согласитесь, что хотя эта картина в общих чертах тоже электрофильное присоединение к двойной связи, развивается реакция совсем не так, как там – здесь присоединение электрофила быстрое, нет никакого карбокатиона, ни мостикового иона, а нуклеофила, как учил бессмертный Винни Пух, можно совсем не давать.
Но главное даже не это, хотя это очень важно, а то, что такие олефины обладают огромной реакционной способностью по отношению к электрофилам, и в таких случаях никогда не идет никаких разговоров ни про какие мостиковые ионы, потому что карбокатион после присоединения электрофила надежно стабилизирован мезомерным донорным эффектом. Более того, это уже и не карбокатион никакой, а просто какой-то аммониевый или оксониевый ион, или даже вовсе нейтральная молекула (если исходный олефин – анион енолята). Такому катиону не обязательно спешить искать себе нукелеофил хоть какой завалящий, а можно просто подождать, когда дядя или тётя над колбой решит, что реакция прошла, и выльет всех в воду – они всегда так делают, мы знаем. В этом случае это обычно заканчивается гидролизом и образованием карбонила, но подробнее мы в этом разберемся в химии карбонильных соединений. Согласитесь, что хотя эта картина в общих чертах тоже электрофильное присоединение к двойной связи, развивается реакция совсем не так, как там – здесь присоединение электрофила быстрое, нет никакого карбокатиона, ни мостикового иона, а нуклеофила, как учил бессмертный Винни Пух, можно совсем не давать. 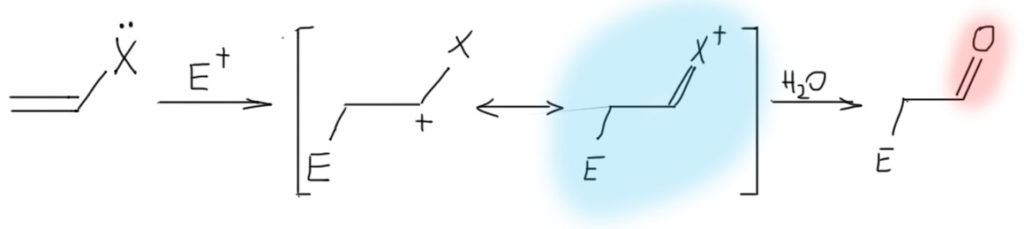
Олефины с сильноакцепторными -М-заместителями, они же электронодефицитные олефины, они же электрофильные олефины, они же акцепторы Михаэля. На втором полюсе находятся олефины с двойной связью, с которой электронную плотность хорошенько стянули. По-английски они называются образнее – electron-poor olefins, но мы не любим простых слов особенно с нежелательным социальным контекстом, и называем их электронодефицитными, что, если вдуматься, довольно смешно и двусмысленно: отчего это олефины вдруг стали дефицитными? ах, это не они дефицитные, а у них связи с дефицитом, то есть недостатком электронов. Ладно, сойдет. В любом случае, это даже более необычный тип олефинов, потому что двойная связь в них радикально меняет реакционную способность. Обычные олефины, в принципе, тоже можно заставить присоединять нуклеофилы, но это требует очень больших усилий, и поэтому такие реакции считаются нехарактерными. Обычные олефины легко присоединяют электрофилы и радикалы, но не нуклеофилы. А эти наоборот предпочитают нуклеофилы, а электрофилы присоединяют тяжело и далеко не любые.
В этих олефинах к двойной связи сопряжены разные -М-группы, чаще всего карбонил (в альдегидной, кетонной, карбоксильной, сложноэфирной и т.п. группах), но может быть и нитрил, и нитро, и много чего еще. В структуре таких олефинов значительную роль играет граничная структура с плюсом на дальнем атоме углерода. Для примера нарисуем именно карбонил. 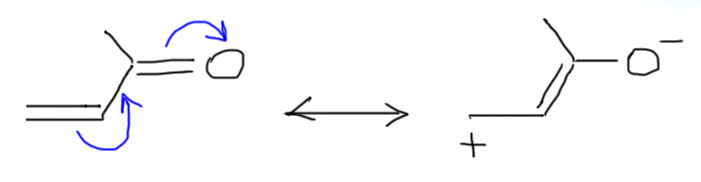
Ясно, что в этих олефинах дальний углерод имеет частичный положительный заряд и поэтому электрофилен, поэтому и предпочитает реагировать с нуктеофилами. Мы подробно будем разбираться с такими реакциями – обобщенно они называются присоединением по Михаэлю, откуда и название акцепторы Михаэля – в разделе про карбонильные соединения. И даже если такой олефин реагирует с электрофилами, то атака электрофила направляется на углерод, ближний к акцепторной группе, поэтому получаются продукты присоединения против правила Марковникова. Более того, скорее всего даже электрофильное присоединение может идти, как нуклеофильное. Подробнее в другом месте.
А теперь более подробно обсудим некоторые важные вещи. Сначала посмотрим на электрофильные реакции. Здесь всё очень подробно, и много лишнего, поэтому это только для особо любопытных. Все основные вещи и так есть на странице про методы.
Гидроборирование
Гидроборирование – одна из важнейших реакций в истории органической химии.
Это также очень важная реакция сама по себе, и мы будем ее использовать очень часто, и использование этой реакции всячески поощряется для селективного гидрирования, получения спиртов и бромпроизводных, а в химии алкинов еще и альдегидов. Ее использование как метода в синтезе мы разберем на страничке про методы. Но здесь поговорим именно о важности этой реакции, ее особенностях, и о том, почему эта реакция, с виду довольно частная, получила в 1979 году половину Нобелевской премии.
Другая любопытная деталь состоит в том, что Браун не открывал собственно гидроборирования. Реакция диборана с олефинами с образованием триалкилборанов была хорошо известна и до него, но о ее применении в органической химии никто даже не задумывался. Браун, во-первых, открыл эффект чрезвычайно сильного увеличения реакционной способности диборана в растворе эфирных растворителей, что делало реакцию удобной и лёгкой; а во-вторых, был одним из первых химиков с работоспособностью мощного бульдозера и хваткой породистого бульдога – открыв реакцию, он за несколько лет исследовал ее вдоль и поперёк, нашёл десятки вариантов её применения в синтезе, исследовал механизм, открыл целый класс производных реагентов, и уже менее чем через 10 лет после начала работ смог написать фундаментальную книгу, посвященную своим работам по применению производных бора в химии, двигая свои работы с целеустремлённостью и энергией, свойственной скорее крупным бизнесменам или политикам. И закономерно получил свою нобелевку не, как это обычно бывает, перед самой встречей с Господом, а в расцвете сил и таланта. Получив все возможные другие премии и почести в десятках стран мира, начиная с почетного рыцарского титула от Е.К.В. Елизаветы Второй, сэр Герберт стал, видимо, первым вип-учёным в истории химии, показавшим, что непреодолимой пропасти между вонючей тягой и сверкающим миром гламура таки нет. После Брауна по этому пути пошли многие. Хорошо это для науки или нет, до сих пор непонятно.
Проблема до Брауна была в том, что боран существует в виде димера, диборана, не имеющего электрофильного атома бора, и поэтому реакция диборана требует довольно сильного нагревания для диссоциации диборана. Димеризация борана в диборан происходит за счет той же самой трехцентровой связи, что и те, что были рассмотрены в разделе про электрофильные реакции алканов, и это не удивительно, ведь боран, BH3 изоэлектронен и изоструктурен метильному катиону CH3+. Но два метильных катиона не могут подойти друг к другу хотя бы по причине ужасающего электростатического отталкивания, а два борана пожалуйста – вот и получается весьма прочный диборан, в котором каждый из электрофильных секстетных атомов бора наезжает на связь B-H второй молекулы, образуя с ней трехцентровую связь. Итого получаются две трехцентровые связи. Диборан обычно рисуют так, как слева, с двумя мостиковыми атомами водорода, и это правильно с точки зрения расположения атомов в постранстве – видно, что у атомов бора sp3-гибридизация и почти тетраэдрическая конфигурация. Но правильнее правая картинка, где ясно показаны трехцентровые связи, склеивающие два борана между собой.
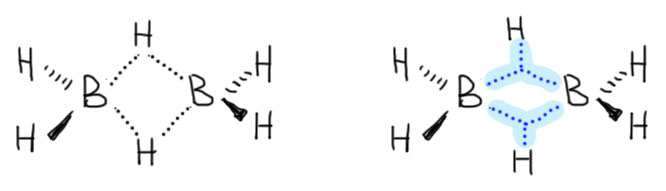
И вот такой димер борана, а это самый простой из устойчивых гидридов бора, как раз и не проявляет реакционной способности по отношению к алкенам потому что в нем, строго говоря, нет электрофильных атомов бора. Единственной причиной электрофильности бора является свободная орбиталь в соединениях типа BX3, и если эта орбиталь использована, например, как в диборане, то нет и электрофильности, ведь на боре нет практически никакого заряда, так как электроотрицательности бора и водорода очень близки (по Полингу, 2.0 и 2.1) и связь B-H практически неполярна. Это место нужно осознать особенно хорошо, так как в некоторых учебниках любят писать откровенный вздор про то, что в боранах эта связь поляризована в сторону водорода, так как водород имеет большую электроотрицательность, и именно это де и является причиной реакции боранов с кратными связями. Эти рассуждения взялись потому что, как ни странно, многие, даже некоторые незадачливые писатели учебников, не понимают разницы между степенью окисления и реальными зарядами на атомах. Степень окисления – чисто формальная величина, и она всегда считается по разнице электроотрицательностей, как бы мала она ни была. В боране степень окисления бора +3, водорода -1. Но это не имеет никакого отношения к реальным зарядам на атомах, для которых важна именно величина разности электроотрицательностей (для простых бинарных молекул) и более тонкий учет смещений электронной плотности (электронных эфектов) в более сложных молекулах. И когда эта разница мала, то реальная полярность связи пренебрежимо мала.
Итак, чтобы заставить диборан реагировать с олефинами нужна довольно высокая температура, вызывающая диссоциацию на две реакционноспособные молекулы мономерных боранов, что делало всю реакцию сомнительным удовольствием, так как сам диборан – весьма неприятный по свойствам газ. Органики вообще не очень любят реакций с газообразными реагентами, когда нужно городить целый прибор для получения газа и его очистки, и подвода в колбу, где идет реакция. И когда газ еще и токсичный, самовоспламеняющийся, то желающих точно будет немного. К тому же, в те времена никому не пришло в голову, что первичные продукты этих реакций, экзотические для правоверного органика бораны, могут иметь какое-то применение в нормальной органической химии.
Браун к тому же даже не был органическим химиком – он с 1940 года работал в лаборатории знаменитого неорганика Шлезинджера, и искал новые удобные методы получения гидридов бора и их производных, борогидридов. Уже после, став самостоятельным ученым и продолжив эти исследования, он немного случайно обнаружил в 1956, что при взаимодействии борогидрида натрия с разными кислотами Льюиса в растворителях эфирного типа: самом эфире, ТГФ, диглиме (диметиловый эфир диэтиленгликоля) и т.п., получается какая-то форма диборана, обладающая необычно высокой реакционной способностью, драматически отличающейся от свойств диборана в других растворителях. Диборан в эфирных раствориелях становится очень интересным восстановителем (об этом в другом месте), и великолепным гидроборирующим агентом, работающим при комнатной температуре. Быстро разобрались, что в таких растворах это уже не диборан, а просто боран, стабилизированный взаимодействием с растворителем. А можно просто делать это прямо на месте, и тогда не нужно ничего городить – просто прикапывай к раствору олефина растворы борогидрида натрия и эфирата трехфтористого бора. Эфирные растворители, которые являются основаниями Льюиса, стабилизируют мономерный боран в виде достаточно слабых комплексов с собой, и такой боран очень легко реагирует с двойной связью алкенов.
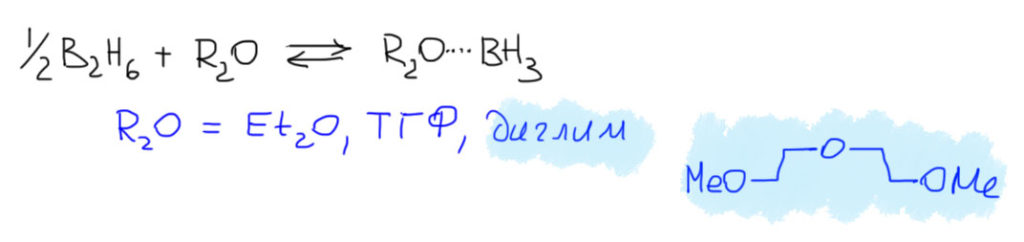
Почему реагирует? Очевидно, потому что такие комплексы диссоциируют на свободный боран легче, чем диборан, и уже при комнатной температуре в растворе есть некоторая концентрация свободного высокоэлектрофильного борана.
Кстати, забегая немного вперёд, с олефинами реагирует не только сам боран, но и любые замещённые бораны, в которых есть хотя бы один атом водорода на боре. И такие бораны тоже при обычных условиях димеризованы в соответствующий диборан. Но и по стерическим, и по электронным причинам (гиперконъюгация стабилизирует секстетный бор точно так же, как это происходит в карбокатионах, изоэлектронных с мономерными боранами такой же структуры) замещённые дибораны намного легче диссоциируют на два борана, и мономера уже довольно много в равновесии и при комнатной температуре и даже при охлаждении. Многие замещённые бораны поэтому замечательно гидроборируют даже без добавления эфирных растворителей. Это было обнаружено Кёстером (Köster, R. et al. Justus Liebigs Ann. Chem., 1964, 672, 1–34) уже после открытия эффекта эфирных растворителей Брауном, поэтому никак не изменило приоритет Герберта Брауна в гидроборировании, и реакции гидроборирования продолжили делать в таких растворителях.
Механизм гидроборирования
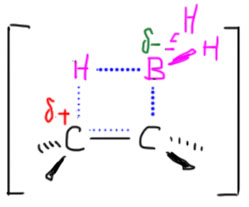
Гидроборирование – это электрофильное присоединение к двойной или тройной связи. И да, и нет. В любом случае, механизм гидроборирования очень сильно отличается от уже привычного нам механизма электрофильного присоединения, в котором сначала присоединяется электрофил с образованием или карбокатиона, или мостикового иона, а затем это уже присоединяет нуклеофил. Боран присоединяется не в две, а в одну стадию, и это присоединение происходит не в согласованном механизме. Еще раз:
Присоединение борана к двойной или тройной связи – согласованная одностадийная реакция.
Напомню, что общим свойством согласованных механизмов является очень сильное влияние стерических факторов. Это совершенно общая идея – если вы имеете дело с согласованным механизмом, то главными будут не электронные факторы (индуктивный и мезомерный эффект заместителей в органической молекуле), а стереохимические, которые всегда упираются в так называемую стерику – минимизацию пространственных препятствий сближению реагентов и оптимизацию орбитальных взаимодействий. Последнее – про орбитальные взаимодействия – неизбежно вызовет у нас ужас, ведь мы ничего в них не понимаем. Это так, но пока успокоимся, возможно, это не так страшно, и многое вполне можно объяснить с помощью очень простых моделей.
Итак, мы решили, что боран, получающийся в небольшой равновесной концентрации диссоциацией комплекса с эфирным растворителем и имеющий пустую p-орбиталь на боре, является электрофилом, и как таковой взаимодействует с двойной или тройной связью. Остановимся пока на двойной. Итак, плоский боран подходит к плоскому же олефину так, чтобы зацепить π-орбитали двойной связи своей вакантной p-орбиталью – это ровно то, что мы называем электрофильной атакой. Это очень похоже на дозаправку самолёта в воздухе: боран в роли самолёта с пустыми баками подлетает так сверху и немного спереди к туше самолета-дозаправщика, то есть алкену, и опускает свой шланг (пустую p-орбиталь) в лоно дозаправщика (в богатую электронами толщу π-связи) и начинает качать топливо, то есть электроны c π-связи на бор. Если бы этот процесс дошел до конца, мы получили бы вот какую картинку: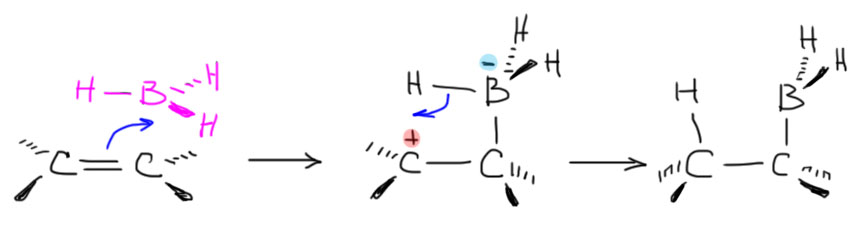
Образовалась бы промежуточная частица с плюсом на углероде и минусом на боре – такие анионы-и-катионы в одной молекуле в химии игриво называют цвиттер-ионами (от немецкого Zwitter – гермафродит, о чем свидетельствует одноименная песня старомодной группы Раммштайн). В таком цвиттер-иона соседствовали бы карбокатионный углерод и гидридный водород на боре, и естественно гидрид немедленно заткнул бы карбокатион. Если бы это было так, а ничего необычного в таком механизме нет, мы получили бы вполне типичное электрофильное присоединение с промежуточным карбокатионом, и стали бы применять обычные наши рассуждения о стабильности карбокатионов и т.п. Все бы сошлось, между прочим. Но вот незадача – все исследования, и экспериментальные, и теоретические, под которыми обычно понимают квантово-химические расчеты пути реакции, говорят, что это не так, и реально происходит нечто не стадийное, а согласованное. То есть начиается все ровно так же – подлетает, цепляет, начинает качать, но одновременно разрыхляется связь бор-водород и водород переселяется на так и не ставший карбокатионом атом углерода. В таких случаях вместо всяких ионов рисуют переходное состояние – умозрительную конструкцию, показывающую взаимное расположение реагентов в самый волнующий момент реакции – на полпути от реагентов к продукту, когда старые связи еще не совсем порвались, а новые не совсем образовались – все это показывают пунктиром, а конструкцию заключа.т в квадратные скобки и помечают значком ≠. И дальше эта конструкция перестраивается в продукт реакции.
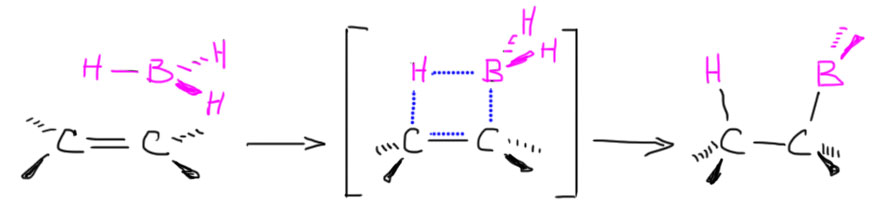
Что следует из этого механизма
Как мы договорились, механизмы нужны не просто так, а для объяснения наблюдаемых особенностей реакции. Что же объясняет этот механизм:
- син-присоединение – бор и водород присоединяются с одной стороны от бывшей двойной или тройной связи. В этом смысле реакция гидроборирования сильно отличается от реакций электрофильного присоединения с участием карбокатионов или мостиковых ионов, которые или имеют неопределенную стереохимию, или идут как анти-присоединение.
- раз в реакции не участвует настоящий карбокатион, то и электронные эффекты заместителей при двойной связи не должны сильно проявляться. Но они все же проявляются, и донорные заместители ускоряют реакцию, а акцепторные замедляют, но их эффект не так силен, как в обычном электрофильном присоединении. Что нужно сделать с механизмом, чтобы он объяснил эту тенденцию?
 Очень немногое – представить себе, что связи рвутся и образуются не строго одновременно – если бы это было так, то на атомах вообще не было бы зарядов, и никакие эффекты заместителей не проявлялись бы. Но связи-то все разные – как они могут так строго синхронно рваться и образовываться? Скорее всего все же не строго синхронно, а одни чуть опережают другие. Механизм от этого не изменится, но если представить себе, что образование связи с бором идет чуть быстрее пересадки водорода с бора на углерод, мы получим небольшой положительный зяряд на том углероде, который в стадийном механизме был настоящим карбокатионом (в немного исправленном переходном состоянии пометим более редким пунктиром более слабые взаимодействия, а более жирным – более сильные). Заместители на углероде с частичным положительным зарядом будут влиять – доноры будут стабилизировать такое переходное состояние, а акцепторы дестбилизировать. Учтем, что там просто небольшой заряд, и нет никаких предпосылок сопряжения – поэтому эффекты имеются в виду только в смысле индуктивном. Скажем, алкилы ускоряют гидроборирование, а галогены замедляют. В гидроборировании нет большого ассортимента заместителей прежде всего потому что всякие сложные группы (карбонилы, карбоксилы и производные, нитро, циан и т.п.) сами реагируют с бораном.
Очень немногое – представить себе, что связи рвутся и образуются не строго одновременно – если бы это было так, то на атомах вообще не было бы зарядов, и никакие эффекты заместителей не проявлялись бы. Но связи-то все разные – как они могут так строго синхронно рваться и образовываться? Скорее всего все же не строго синхронно, а одни чуть опережают другие. Механизм от этого не изменится, но если представить себе, что образование связи с бором идет чуть быстрее пересадки водорода с бора на углерод, мы получим небольшой положительный зяряд на том углероде, который в стадийном механизме был настоящим карбокатионом (в немного исправленном переходном состоянии пометим более редким пунктиром более слабые взаимодействия, а более жирным – более сильные). Заместители на углероде с частичным положительным зарядом будут влиять – доноры будут стабилизировать такое переходное состояние, а акцепторы дестбилизировать. Учтем, что там просто небольшой заряд, и нет никаких предпосылок сопряжения – поэтому эффекты имеются в виду только в смысле индуктивном. Скажем, алкилы ускоряют гидроборирование, а галогены замедляют. В гидроборировании нет большого ассортимента заместителей прежде всего потому что всякие сложные группы (карбонилы, карбоксилы и производные, нитро, циан и т.п.) сами реагируют с бораном. - и самое главное – и региоселективность гидроборирования и скорость (или что то же самое, относительную реакционную способность различных олефинов) определяет энергетика взаимодействия борана и алкена, а это определенно зависит от двух факторов – энергии самой двойной связи и стерического фактора – препятствия сближению борана и алкена. Обсудим это в следующем разделе
Реакционная способность олефинов
Главный фактор – стерические препятствия. Поэтому монозамещенные алкены реагируют существенно быстрее дизамещенных, которые реагируют быстрее тризамещенных, а эти в свою очередь быстреее тетразамещенных. Естественно, это слишком формальное описание, и нужно смотреть еще и на размер заместителей, но когда речь идет об обычном боране, влияние оказывает только близкая стерика – разветвления на самом первом углероде от двойной связи. Дальше может быть что угодно, это никого не волнует. Для больших боранов требования к стерике более сложные. Совсем сильно это нас волновать не должно, так как реально трудно встретить случаи, когда такие различия действительно важны.
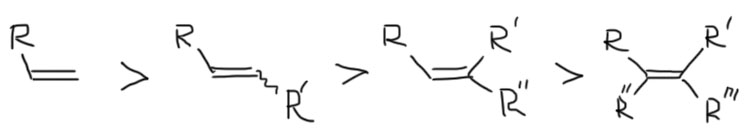
В этом ряду реакционная способность падает довольно сильно, на порядок, а то и на два на каждом знаке >, и это создает возможности для гидроборирования более реакционноспособных двойных связей в присутствии менее реакционноспособных, только для этого надо строго дозировать гидроборирующий реагент, так как в присутствии избытка гидроборирование пойдет дальше. И нужно не забывать, что на бор обычно садится три молекулы алкена (тризамещенных – два, тетразамещенных – одна). 
На этот довольно очевидный и следующий из механизма реакции ряд реакционной способности накладывается несколько занятных эффектов. В принципе, это не очень важно, и воспользоваться этим для селективного гидроборирования не очень получится. Просто для полноты картины, и чтобы было еще яснее, чем гидроборирвоание отличается от других реакций присоединения.
Во-первых, при прочих равных условиях начинает играть роль та самая стабильность двойной связи, которую мы разбирали. Скажем, если брать дизамещенные алкены, то цис-изомеры реагируют заметно быстрее транс-изомеров. Напомню, что в цис-изомерах двойная связь дестабилизирована относительно двойной связи в транс-изомерах, также за счет стерического отталкивания, причем чем больше группы, тем больше этот эффект. С тем же самым связана и повышенная реакционная способность напряженных циклических алкенов, но с напряжениями мы будем разбираться отдельно в теме циклоалканы.
Во-вторых, электронные эффекты все же играют некоторую роль, и акцепторы замедляют реакцию, а доноры ускоряют. Причина этого, как разобрано в предыдущем разделе, в том, что согласованный механизм не предполагает полной синхронности, и некоторый положительный заряд на втором углероде все же появляется, и на этот заряд действуют обычные стабилизирующие и дестабилизирующие эффекты. Обязательно только нужно не забывать, что стерика на первом месте, а эффекты на втором. Тем не менее, скажем, галогены весьма сильно замедляют реакцию. А атом кислорода непосредственно рядом ускоряет, как и положено мезомерному донору. Но эффекты очень маленькие, несравнимые с эффектами в электрофильном присоединении с участием карбокатионов и мостиковых ионов.
И еще одно яркое отличие гидроборирования от других реакций электрофильного присоединения – реакционная способность стирола (фенилэтена). В электрофильных реакциях с участием карбокатиона стирол намного более реакционноспособен чем монозамещенные алкены без ароматичского кольца по очевидной причине стабилизации бензильного катиона. В гидроборировании, напротив, стирол менее реакционноспособен, скорее всего, по тем же стерическим причинам.
Региоселективность гидроборирования
Обычный боран – очень маленькая и компактная молекула. Откуда же там берется стерика?? Все очень просто – стерические препятствия это не следствие абстрактного “размера” молекулы, а следствие конкретной ситуации, в которой молекулы должны строго определенным образом подойти друг к другу, чтобы началось перераспределение электронной плотности, которое и приведет к разрыву старых и образованию новых связей. В согласованных реакциях с участием нейтральных молекул требования к стерике всегда больше, чем в стадийных с участием всяких ионных частиц типа карбокатионов и карбанионов. Это общее свойство согласованных реакций. Разберем, почему это так, где-нибудь отдельно, а пока поверим.
Вот и в гидроборировании – чтобы реакция осуществилась, боран и олефин должны очень близко подойти друг к другу строго определенным образом и почти без допусков (когда мы говорим “подойти определенным образом” то естественно не имеем в виду, что молекулы, управляемые опытными капитанами и штурманами, медленно и филигранно притираются друг к другу, как космический корабль к орбитальной станции – нет, они беспорядочно и с огромной скоростью сталкиваются во всех возможных направлениях, но только те столкновения, которые отвечают оптимальной геометрии сближения приводят, и то с некоторой вероятностью, к развитию реакции). Вот два пути такого сближения. Видим, что путь, в котором атом бора пытается сесть на внутренний атом углерода, имеет больше проблем со стерикой – бор со своими водородами нехорошо сближается с заместителем на углероде. Этого достаточно, чтобы этот путь имел большую энергию активации (преодолевал более высокий энергетический барьер), а это то же самое, что сказать “имел меньшую скорость (был более медленным или менее быстрым). Так это устроено.
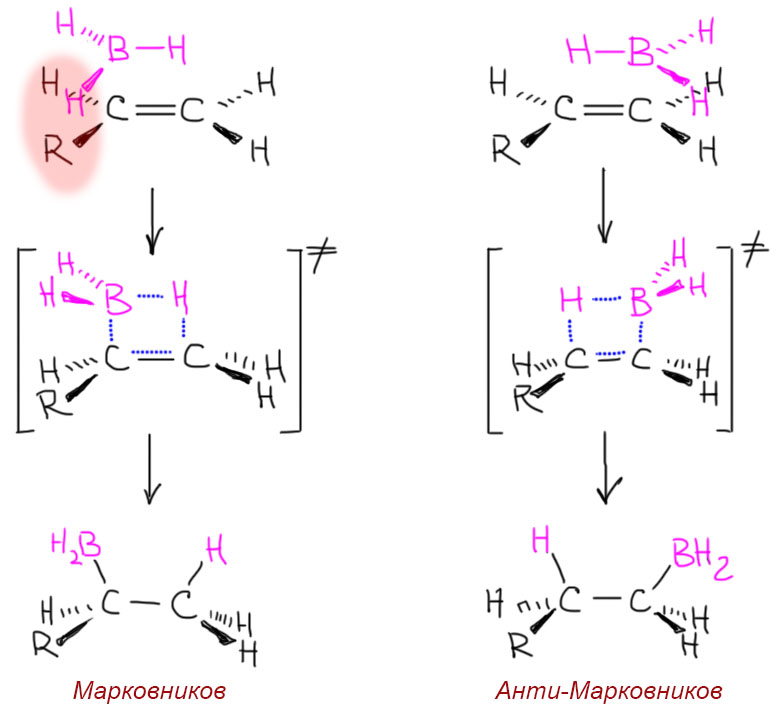
С самым маленьким несимметричным алкеном, пропеном, образуется смесь 90:10 (Singleton, J. Am. Chem. Soc.2009, 131, 3130-3131). С олефинами чуть побольше (нормальными 1-алкенами) доля продукта присоединения по Марковникову снижается до нескольких процентов. Если же алкильный хвост разветвлен, то можно говорить о полной региоспецифичности присоединения. Термин региоспецифичность это то же самое, что 99-100% региоселективность.
Боран присоединяет одну, две или три молекулы алкена
У борана три атома водорода и все одинаковые. Поэтому боран может присоединяться к двойной связи три раза, то есть одна молекула борана присоединяет до трех молекул алкена. Закономерности довольно простые и понятные. Смотрим на количество заместителей на двойной связи.
- если один или два (в последнюю категорию попадают и циклические олефины типа циклогексена) – то все три;
- если три, то присоединяются два алкена, а один атом водорода остается;
- если все четыре, то такие олефины обычно вообще не гидроборируются, если только заместители не маленькие, типа метилов – в этом случае гидроборирование идет, но только с одной молекулой алкена.
Причины такого поведения две. Одна вездесущая – стерика, типа, не лезет больше. Вторая похитрее. Мне так кажется, по крайней мере. Мы уже выяснили, что на боре есть пустая p-орбиталь, точно такая же как на углероде в карбокатионах. Карбокатионы стабилизированы гиперконъюгацией, и бораны тоже. Для боранов это, конечно, не так жизненно важно, как для карбокатионов, но роль свою это взаимодействие играет. Это очень хорошо видно в структурах алкилборанов. Атом водорода на соседнем с бором углероде старается расположиться перпендикулярно к плоскости борана (бор, как и карбокатионный углерод имеет sp2-гибридизацию). Так вот, если алкил разветвленный, это не получается. Поэтому такие бораны менее стабильны, что в частности, выражается в очень легких перегруппировках из-за обратимости гидроборирования (см. ниже и на вкладке про большие бораны про тексилборан).
Откуда берут боран для гидроборирования
Есть довольно много реагентов, используемых для гидроборирования простым бораном. Самый плохой выбор – диборан, B2H6, хотя при большом желании в каталогах реактивов можно найти даже баллоны с этим газом. Но это страшновато и неудобно. Более просто генерировать боран прямо на месте. Есть довольно много предшественников борана. Наиболее часто используют борогидрид натрия вместе с кислотами Льюиса, как собственно и предложил основоположник. Обычно, к смеси алкена и борогидрида натрия в ТГФ или диглиме прикапывают эфират трехфтористого бора. Это самый распространенный прием. Если будете это делать, внимательно изучите методики, чтобы понять, в каком соотношении все это брать и на какое количество борана вы можете рассчитывать. Точно знать количество борана нужно в том случае, если мы хотим селективно гидроборировать одну связь в соединении, где их несколько, но разных по реакционной способности. Или если в соединении есть группы, которые взаимодействуют с бораном, но медленнее двойной или тройной связи, например сложноэфирная или нитрильная, или нитро. Если же ничего такого нет, то избыток борана не мешает и будет после реакции разложен при гидролизе реакционной смеси.
Еще более простая смесь, которую можно легко замутить даже в нашем практикуме – эквимольная смесь борогидрида натрия и элементарного иода в том же ТГФ. Эта смесь должна дать один эквивалент борана. Этот реагент легко применять для простых алкенов, но в более сложных случаях придется подумать о побочных реакциях с иодом.
Если есть немного денег и магазин химреактивов можно приобрести готовые комплексы борана. Достоинством этих реагентов являетс то, что вы точно будете знать, сколько там борана. Самый популярный реагент этого типа выглядит довольно странно – это комплекс борана с диметилсульфидом BH3·Me2S. Вонь страшная. После реакции диметилсульфид нужно аккуратно окислить, чтобы убрать вонь. Но этот реагент исключительно стабилен и очень хорошо хранится. И в литературе вы чаще других будете видеть именно этот реагент в реакциях гидроборирования. Конкурирующий и более простой реагент – комплекс борана с ТГФ в растворе ТГФ, BH3·THF. Не воняет, но хранится плохо, так как боран, как кислота Льюиса и одновременно гидридный восстановитель, довольно быстро ТГФ расщепляет, и концентрация раствора из-за этого постоянно уменьшается.
Гидроборирование: большие бораны
Гидроборирование простым бораном уже дает очень хорошую региоселективность в гидроборировании, образуя почти всегда в основном продукт присоединения против правила Марковникова (анти-марковниковский продукт) и причиной такого направления присоединения является стерика. Но боран все же действительно очень маленькая молекула, и ее стерического объема не всегда достаточно. Главные проблемы простого борана таковы:
- При гидроборировании терминальных монозамещенных олефинов, практически любых, кроме ожидаемого продукта гидроборирования, всегда получается от 5 до 20-25% изомера, продукта присоединения по правилу Марковникова, и ничего с этим сделать нельзя, ни с помощью растворителя, ни используя разные комплексы борана – такая примесь изомера терпима, особенно в простых случаях, и довольно легко устраняется очисткой продукта реакции. Это не очень большая проблема, и для таких олефинов простой боран используют очень часто.
- А вот при гидроборировании ди- и тризамещенных олефинов селективность резко падает – игра на разном размере заместителей (метил против изо-пропила, и т.п.) работает плохо, смеси получаются с содержанием до 30-40% нежелательного изомера. Такие реакции уже не считаются селективными, и такие смеси практически невозможно разделить перегонкой или перекристаллизацией продукта. Это все уже очень скверно. Иногда бывают исключения, особенно с циклическими олефинами, когда региоселективность приемлема, но рассчитывать на это трудно, нужно искать реальные методики в литературе.
- Как правило образуются продукты присоединения трех молекул олефина к одному борану – триалкилбораны. В последующих реакциях образования спиртов, галогенпроизводных, и т.п. участвуют не все три группы, а обычно не больше двух, а одна теряется. Если это ыл простой олефин, то и не жалко, а если сложный, если это какая-то двадцать пятая стадия длинного синтеза, или олефин какой-то драгоценный, то такие потери совершенно неприемлемы.
- С ацетиленами все совсем плохо – региоселективность плохая даже с терминальными ацетиленами, образуются ужасные по структуре аддукты (на странице про алкины подробнее).
Рецепт борьбы с этими недостатками вполне очевиден: если главный фактор в химии гидроборирвоания это стерика, а сам простой боран по размеру все же маловат, то нужно увеличить стерические препятствия – получить и использовать более громоздкие бораны. Возмодность такая вполне заложена в свойствах самого борана, которые мы уже разобрали – боран присоединяет от одной до трех молекул алкена в зависимости от стерических препятствий, которые связаны с количеством и размером заметителей на двойной связи, и старается взять максимально возможное количество алкенов. Это создает возможность сначала присоединить нечто более “рогатое”, и получить или моно- или диззамещенный боран, но он, в свою очередь, сможет присоединить еще один или два менее рогатых алкена, и это второе присоединение должно быть более селективным. Ровно так это и есть. Посмотрим на основные бораны такого типа.
Дисиамилборан
Получается гидроборированием 2-метилбутена-2. Такой олефин присоединяется дважды и остается один атом водорода. Странное название сиамил для заместителей – удобное обозначение для радикала состава C5H11, а такие группы традиционно называют не пентилами, а амилами (сиамил – это сокращенное название несистемно названного радикала втор-изоамила (по-английски – sec-isoamyl). Впрочем, больше это называние нигде, кажется, не употребляется.
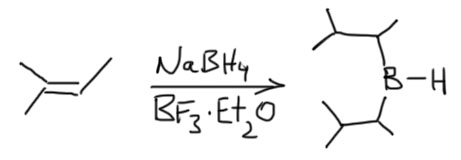
Получают этот реагент прямо перед использованием. Хранится он плохо по причине изомеризации, которая идет уже при комнатной температуре. Это очень популярный гидроборирующий реагент, возможно, самый популярный, прежде всего потому что очень дешев, так как получается из очень простого олефина прямо перед использованием и используется без выделения.
Тексилборан
Получается из 2,3-диметилбутена-2 прямо перед использованием. Название тексил – это такая скороговорка из более полного обозначения этого радикала – трет-гексил (скорее даже это thexyl получается из английского tert-hexyl). В более старой литературе этот боран любили стильно рисовать в виде такой антеннки.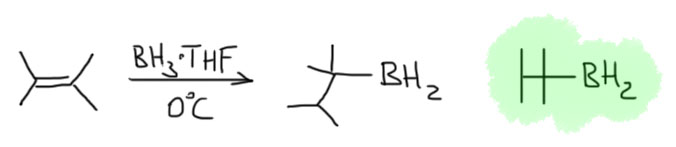
Этот боран еще более неустойчив к обратному процессу, и при работе с ним температура реакции не должна превышать 0º, а часто даже и отрицательных температур. Продукты гидроборирования очень легко теряют тетраметиэтилен, и дальше реакция пойдет так, как будто и не было никакого тексилборана, но при низкой температуре можно получит очень селективно желаемые продуеты гидроборирования.
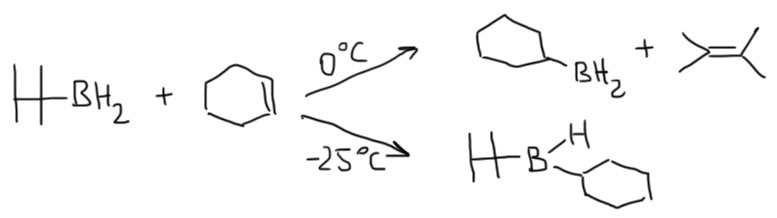
Вывод из этого простой: тексилборан это очень особенный гидроборирующий реагент с очень непростой химией. Не будем для наших несложных целей его использовать вместо Sia2BH. Ограничимся только знанием о существовании такого борана.
9-Борабициклононан (BBN)
Исключительно популярный стерически затрудненный боран. Почему он так странно называется? Это номенклатура ИЮПАК бициклических соединений. Названия строятся от соответствующих углеводородов. Заменяем бор на углерод. Это бицикл – два цикла, бицикло-. Почему не три? Три, но третий цикл получается из двух других – нет смысла его упоминать. Название строится так: в двух циклах всего 9 углеродов в скелете – итого нонан, бициклононан. Но бициклононанов может быть много, поэтому нужно уточнить. Делают это перечисляя количество углеродов в сторонах бицикла (три синих, три зеленых, один розовый), не считая атомов углерода (красных), общих для всех циклов. Все это записывают так: бицикло[3.3.1]нонан. После атомы нумеруют, начиная с одного из общих атомов, направляясь к другому общему атома сначала по самой длинной дороге, потом идут обратно по следующей по длине дороге (если дороги одинаковы, то разницы нет), и в самом конце идут по самой короткой дороге. В данном случае мостик с одним атомом получает последний номер 9. Так можно показать заметители, или замену углерода на другой атом. В данном случае бор. В случае замены атома после названия элемента используют соединительную букву а. Получаем окончательное название 9-борабицикло[3.3.1]нонан.
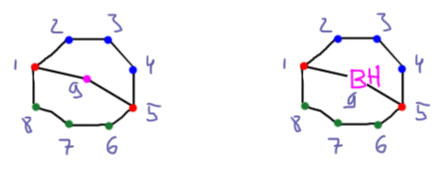
Получают этот боран из очень легкодоступного циклического диена – октадиена-1,5. Эта штука делается в промышленных масштабах каталитической циклодимеризацией 1,3-бутадиена. Гидроборирование идет в 2 стадии.
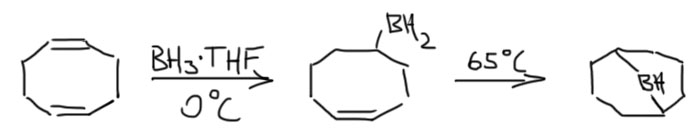
Достоинства BBN многочисленны и неоспоримы, особенно в сравнении с дисиамилбораном и тексилбораном. Это очень стабильное вещество, не имеющее никаких проблем с обратимостью гидроборирования, и перемещениями бора по скелету. Поэтому это вещество можно не получать, а просто купить (хотя и недешево) и спокойно хранить хоть годами. Если залежалось и малость заплесневело, можно очистить перегонкой. Чистый BBN представляет собой бесцветные кристаллы, и в этих кристаллах и неэфирных растворителях он сущесвует в виде димера, точно так же как простой боран, но димер этот существенно менее стабилен и существенно диссоциирован уже при комнатной температуре. Это свойство позволяет применять этот реагент не только в эфирных растворителях.
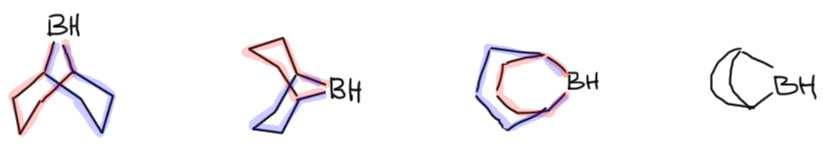
Особая стабильность бэбээна неразнывно связана с его структурой. Два цикла, из которых состоит эта молекула, шестичленные, соединенные через один атом очень аккуратно и точно – оба цикла в такой структуре имеют форму кресла, а это самая выгодная форма, какую только можно себе представить. Вот как это выглядит с нескольких сторон. Первый и второй виды особенно хорошо это показывают – более того, если вы когда-нибудь видели структуру алмаза, или углеводорода адамантана, представляющего собой структурный мотив решетки алмаза, то без труда опознаете это в структуре BBN (только не хватает еще одного мостика). Третий вид – сбоку со стороны одного из циклов, заслоняющего второй – довольно неуклюжий, но он объясняет, почему на схемах реакций BBN, чтобы не вырисовывать соединенные кресла, обозначают таким кокетливым парашютиком.
Пирокатехилборан
И еще один боран часто путается под ногами, хотя и без толку. Это очень простое вещество, получаемое без всякого гидроборирования из обычного борана и орто-дигидроксибензола (известного под тривиальным названием пирокатехин, но по-английски catechol без приставки пиро). В этом боране электрофильность бора очень сильно подавлена мезомерным донорным эффектом от неподеленных пар кислорода, и он имеет поэтому очень низкую реакционную способность. Гидроборировать им можно, но только при нагревании, и только самые реакционноспособные алкены, и большого смысла в этом нет. В современной химии кроме обычной реакции гидроборирования, которую мы изучаем, есть и гидроборирование, катализируемое комплексами переходных металлов типа родия. Вот там этот боран – едва ли не самый главный герой. Но это совсем другая реакция, и по механизму, и по особенностям, и мы ее использовать не будем, так как категорически нельзя использовать непонятные вещи, от этого и случаются чернобыли. Если заинтересуетесь, эта реакция обсуждается в курсе химии переходных металлов в органическом синтезе. 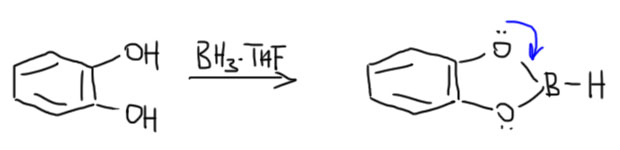
Зачем нужны большие бораны?
В первую очередь, для исправления недостатков обычного борана. Во-вторую, для каких-то специфических задач – об этом в самом конце.
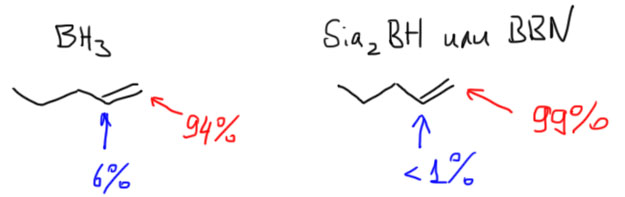
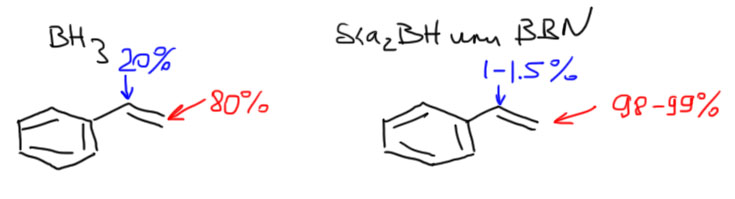
Второй темы мы касаться не будем, это явно за пределами нашего курса. Просто намекну, что это и новые реакции, для которых требуется особый контроль за селективностью. Вот пара характерных примеров. Тексилборан, например, позволяет присоединять два разных олефина с образованием смешанного борана. И такой смешанный боран можно использовать для синтеза несимметричных кетонов (эта реакция забавна тем, что ее сделал нобелевский лауреат 2010 года, начинавший тогда свою научную карьеру в лаборатории нобелевского лауреата 1979 года – Brown, Negishi, JACS, 1967, 89, 5285). Вот как это делается, на одном примере. Сначала собираем боран с тремя разными остаками из трех разных олефинов, первый из которых дает тот самый тексил. Затем этот тризамещенный боран реагирует с CO под давлением и в присутствии воды. И в конце делаем то же самое, как будто хотим превратить боран в спирт, но в этом случае получается несимметричный кетон с очень хорошим выходом. 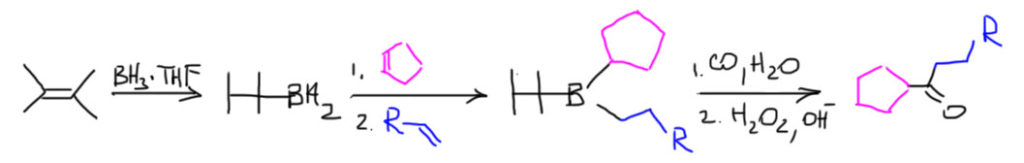
Еще более важным применением боранов с большим заместителем является возможность получения оптически активных продуктов реакции – асимметрический синтез. Для этого, конечно, не подоудут уже известные нам бораны, так как они все не хиральны. Но можно было бы взять оптически активный олефин и гидроборировать его, получив оптически активный боран. Оптически активный олефин? – это, наверное, какая-то чертова экзотика и невероятная ценность! Ничего подобного. Обычный скипидар, если в наше время кто-то еще помнит про существование такой приятно пахнущей жидкости, которую раньше продавали за копейки литрами как дешевый бытовой растворитель. Скипидар получается в больших количества при перегонке из сосновой смолы – это смесь природных углеводородов, терпенов. И главный компонент скипидара – оптически активный олефин α-пинен, очень симпатичная молекула, оптическая активность которой связана, конечно, не с двойной связью, а с обычными стереоцентрами. Оптически чистый пинен легко гидроборируется в полном соответствии с тем, чтобы боран приближался с наименее затрудненной стороны двойной связи. В этом случае это будет означать не только с менее замещенного конца двойной связи, но и с той стороны шестичленного кольца, где нет этой рогатой надстройки. Получается чистый диастереомер в виде чистого энантиомера. D дополнение в двум старым стереоцентрам (красные точки) получается два новых (зеленые), так как и метил тоже получает вполне определенное положение, так как с другой стороны прилепился водород из борана. Получается, что гидроборирование может быть не только региоселективно, но и стереоселективно – точнее, диастереоселективно (давать только один диастереомер из возможных), и энантиоселективно (давать один энантиомер, а не рацемат). Это волнительно и многообещающе само по себе. Получающийся стерически затрудненный и оптически активный боран и называется тоже непроизносимо и загадочно (это у терпенов такие названия, и никто с этим ничего не может поделать) – диизопинокамфеилборан, сокращается Ipc2BH. 
Если мы пустим этот боран в обычную реакцию получения спирта, то получим оптически активный спирт, потому что реакция замещения бора на гидроксил происходит с полным сохранением конфигурации. Итого – мы осуществили получение оптически активного спирта – асимметрический синтез. 
Но это – одна реакция, к тому же довольно очевидная. В исходной молекуле уже была оптическая активность, поэтому неудивительно, что мы получили оптически активный продукт. Но попробуем, вслед за Брауном, применить этот большой и оптически активный боран для гидроборирования нехирального олефина, но такого, в котором образуется стереоцентр, например, цис-2-бутен. Если бы мы взяли дисиамилборан или BBN то получили бы рацемат.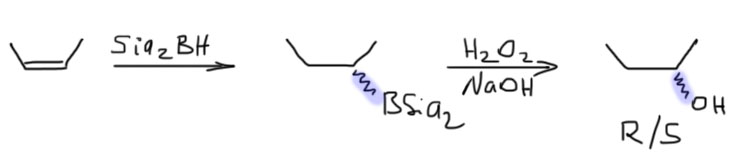
Ничего другого мы и не моли получить, потому что хиральность, оптическая активность не самозарождается – если все реагенты (а также катализаторы, растворители и другие вспомогательные вещества) нехиральные, то хиральности взяться неоткуда. А теперь возьмем вместо дисиамилборана оптически активный диизопинокамфеилборан. Как установил Браун, в этом случае гидроборирование происходит с образованием не рацемата, а оптически активного продукта гидроборирования, который првращается в оптически активный втор-бутанол. Из нехиральной молекулы (цис-бутена-2) получается оптически активный продукт, причем источником хиральности оказалась молекула, от которой в продукте реакции не остается ничего. Как выражаются в этой науке, произошел перенос хиральности с оптически активного борана на спирт.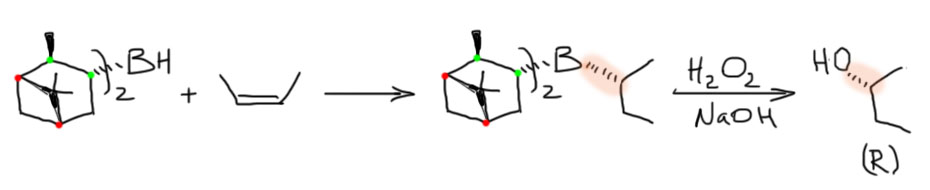 Может показаться, что ничего особенного не произошло, но это не так. Очень важный вопрос – гарантировано ли то, что произошло, обязательно ли при использовании хирального реагента происходит перенос хиральности. Ничего подобного, не только не обязательно, но даже наоборот – это происходит довольно редко и всегда требует очень серьезной работы. Фокус в том, что олефин плоский и нехиральный, верх и низ у него (если расположить его в плоскости) одинаковы. Но когда эта штука лежит в плоскости, при подходе к этой молекуле со стороны двойной связи мы увидим, что слева и справа немного разный пейзаж – с одной стороны виднеются метилы, с другой – ничего. В этом и фокус, если у того, кто подходит, лево и право тоже отличаются. Именно так и устроены хиральные реагенты – это просто следует из определения хиральности: лево ≠ право. Возникают небольшие различия в способе подхода – подойти-то можно в двух сторон. И такие подходы перестанут быть одинаковыми. Другое дело, что разница в энергии между переходными состояниями, ведущими к продукту гидроборировани, не очень велика. Будет ли эта разница серьезно влиять на результат реакции – вопрос более чем непростой, может будет, может нет, может будет, но не сильно. Это очень сложная наука, и мы не будем даже пробовать въезжать в нее серьезнее, просто отметим для себя, что такие реакции есть. В современной органической химии асимметрические реакции невероятно важны, и практически любая органическая реакция известна в асимметрическом варианте. Можно даже сказать категоричнее – если органическая реакция может дать оптически активный продукт, в современной химии уже стало неприличным делать эту реакцию по-старому, на образование рацемата. Извольте искать асимметрический подход и получать оптически активный продукт. Асимметрическое гидроборирование, описанное Брауном в начале 1960-х, было одной из первых примеров эффективного переноса хиральности с вспомогательного реагента на продукт.
Может показаться, что ничего особенного не произошло, но это не так. Очень важный вопрос – гарантировано ли то, что произошло, обязательно ли при использовании хирального реагента происходит перенос хиральности. Ничего подобного, не только не обязательно, но даже наоборот – это происходит довольно редко и всегда требует очень серьезной работы. Фокус в том, что олефин плоский и нехиральный, верх и низ у него (если расположить его в плоскости) одинаковы. Но когда эта штука лежит в плоскости, при подходе к этой молекуле со стороны двойной связи мы увидим, что слева и справа немного разный пейзаж – с одной стороны виднеются метилы, с другой – ничего. В этом и фокус, если у того, кто подходит, лево и право тоже отличаются. Именно так и устроены хиральные реагенты – это просто следует из определения хиральности: лево ≠ право. Возникают небольшие различия в способе подхода – подойти-то можно в двух сторон. И такие подходы перестанут быть одинаковыми. Другое дело, что разница в энергии между переходными состояниями, ведущими к продукту гидроборировани, не очень велика. Будет ли эта разница серьезно влиять на результат реакции – вопрос более чем непростой, может будет, может нет, может будет, но не сильно. Это очень сложная наука, и мы не будем даже пробовать въезжать в нее серьезнее, просто отметим для себя, что такие реакции есть. В современной органической химии асимметрические реакции невероятно важны, и практически любая органическая реакция известна в асимметрическом варианте. Можно даже сказать категоричнее – если органическая реакция может дать оптически активный продукт, в современной химии уже стало неприличным делать эту реакцию по-старому, на образование рацемата. Извольте искать асимметрический подход и получать оптически активный продукт. Асимметрическое гидроборирование, описанное Брауном в начале 1960-х, было одной из первых примеров эффективного переноса хиральности с вспомогательного реагента на продукт.
Гидроборирование: обратимость и перегруппировка более замещенных олефинов в менее замещенные
Строго факультативно: этого нет в программе, только для тех, кому просто интересно
Гидроборирование очень сильно осложняется обратимостью этой реакции. Открытие Брауном сильного ускорения гидроборирования в эфирных растворителях было важно еще и тем, что в этом случае реакция проводится очень быстро и в мягких условиях, при комнатной температуре а часто даже и при охлаждении, и в этих условиях гидроборирование необратимо. И это сильно упрощает дело. И продукты гидроборирования в этих условиях – это продукты кинетического контроля, а соотношение продуктов – марковниковского и антимарковниковского – определяется разницей скоростей присоединения борана, а разница скоростей, в свою очередь, это следствие разницы в энергиях переходных состояний.
Но если реакцию проводить при нагревании, или если нагреть продукт реакции, например, попробовав его очистить перегонкой, то мы столкнемся с обратимостью. Если мы гидроборировали алкен с терминальной двойной связью, то мы почти ничего не увидим, потому что боран будет отщепляться и присоединяться обратно точно так же. Но если алкен был более сложный, то нас ждут сюрпризы, потому что обратная реакция далеко не обязательно пройдет по тому же месту, что прямая. Вот, например, что будет с гидроборированием 2-бутена, если проводить его при нагревании (это просто упрощённая иллюстрация возможных превращений, в реальности с такими легколетучими соединениями такой фокус произвести было бы непросто, и берут что-нибудь подлиннее; и схема упрощена, показан только продукт присоединения одного алкена к борану, не показаны диалкил и триалкилбораны, но это принципиально картину не меняет, хотя в реальной реакции будет много промежуточных форм с разными алкинами на боре). Отщепление борана из первого продукта гидроборирования может дать как исходный 2-бутен, так и 1-бутен, гидроборирование последнего даст в основном продукт с борным остатком на конце цепи. Именно последний будет накапливаться (в виде соответствующего три-н-бутилборана).
Довольно любопытно задать вопрос, почему идет такая перегруппировка. С одной стороны понятно, что если образуется концевой ален, то гидроборироваться он должен именно так. Но этот аргумент имеет кинетическую природу, а здесь именно равновесие – это совершенно непреложный факт, что нагревание боранов с бором внутри цепи приводит к выползанию бора на наименее замещенное место. Значит такой, наименее замещенный боран просто стабильнее более замещенного. Я думаю, что здесь дело в стабилизации гиперконъюгацией. Не забудем, что на боре есть пустая p-орбиталь, точно такая же как в карбокатионах. Поэтому те соображения, которые мы применяли к стабилизации карбокатионов, должны работать и здесь. Пустая орбиталь бора взаимодействует с гибридной орбиталью соседней C-H связи, стараясь встать коллинеарно, как показано на проекциях Ньюмена под боранами (передний атом в проекции – бор, я его обозначил, чтобы было понятнее, хотя обычно мы в ньюменах передний атом буквой не обозначаем). Видно, что в первом боране такой C-H один, а во втором – два (есть два равноценных ньюмена, а это всегда выгоднее чем один). В триалкилборанах, а именно они и будут конечным продуктом этой реакции у бора будет три одинаковых алкила, и у каждого по паре подходящих C-H связей, и там различие в пользу борана с менее замещенными алкилами возрастет еще сильнее.
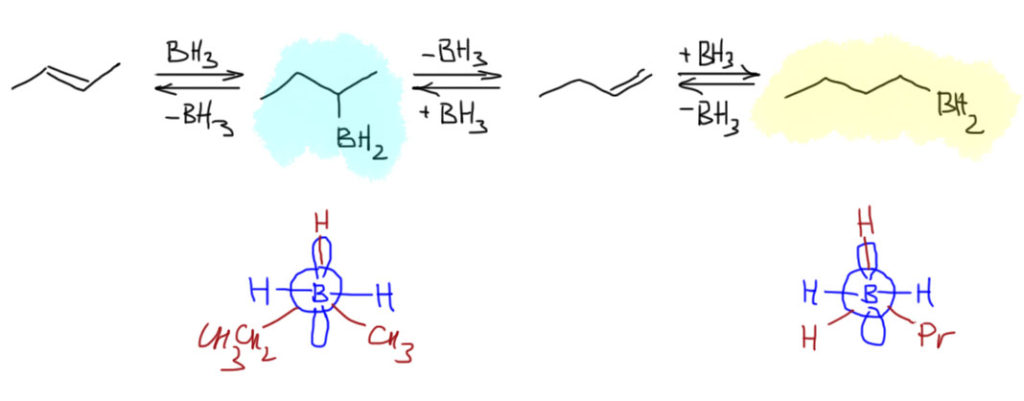
Зачем нужно знать о перегруппировке боранов? В практической жизни – чтобы не совершать ошибок. Чем более замещен боран, который вам нужно получить, тем с большей осторожностью с ним нужно обращаться. Гидроборируют ведь не только концевые алкены, но и замещенные, иногда даже довольно сильно. И тогда нужно очень точно соблюдать методику, не отпускать температуру, не оставлять, как у нас принято, неразделанную реакционную смесь валяться в углу тяги на неделю. Вообще, в химии гидроборирования принято продукт гидроборирования как можно быстрее превращать в конечный продукт реакции – углеводород, спирт, альдегид, галогенпроизводное и т.п. В этот момент все перегруппировки прекращаются.
Интересным следствием из этого свойства явлется малая устойчивость самых известных стерически затрудненных боранов – дисиамилборана и тексилборана. И тот, и другой нужно строго хранить в холодильнике и по возможности недолго, а еще лучше использовать сразу после получения. Очищать перегонкой их нельзя. Иначе они перегруппировываются и теряют значительную долю своей стерической затрудненности, а следовательно и селективности.
Кроме перегруппировки у обратимости гидроборирования есть и другое следствие – если нагреть более замещенный боран с менее замещенным олефином, то бор переедет на новый олефин. Это будет тем более хорошо, если образующийся олефин поменьше и более летуч, и его можно отогнать из реакционной смеси, а олефин-подсадка как раз побольше и менее летуч, так что ни он, ни его триалкилборан не улетят. Браун предложил нормальный 1-децен в этом качестве, но это может быть и что-то близкое – 1-нонен, 1-додецен и т.п. – эти олефины довольно легкодоступны. Это явление обнаружил сам Браун с сотрудниками (JACS, 1967, 89, 567). В таком простом примере, как изображен на схеме, это выглядит немного бессмысленно, – был циклопентен и получился циклопентен. Популярная в этой химии температура в 160º – это просто температура кипения растворителя, диглима (если будете сами что-то делать с диглимом, не забудьте проверить перед реакцией на содержание перекиси и хорошенько почистить от нее, этот растворитель образует перекись не хуже ТГФа, и если про перекись в диглиме забыть, жахнет так, что на этом ваша карьера органического химика оборвется, не начавшись). ![]()
Но вся история становится очень привлекательной, если соединить одно и другое, перегруппировку и пересадку. Берем алкен с глубоко зарытой двойной связью, гидроборируем, греем и перегруппировываем, добавляем 1-децен и греем дальше, отгоняя новый алкен с двойной связью, вытащенной наружу. 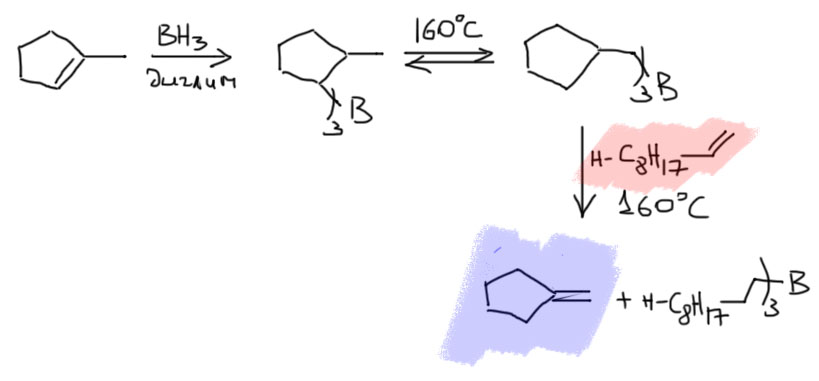 Браун в целях привлечения внимания широких масс назвал это противотермодинамической перегруппировкой, имея в виду то, что из более замещенного, а следовательно, согласно нашей любимой мантре, и более стабильного олефина, получается менее замещенный и менее стабильный олефин. Но парадокса тут нет ни на грош – это просто равновесие, смещаемое, в строгом соответствии с бессмертным учением Ле Шателье, в сторону продукта, удаляемого из реакционной смеси отгонкой. Если бы этого (отгонки) не было бы, получилось бы обычное равновесие, то есть каша из олефинов и боранов.
Браун в целях привлечения внимания широких масс назвал это противотермодинамической перегруппировкой, имея в виду то, что из более замещенного, а следовательно, согласно нашей любимой мантре, и более стабильного олефина, получается менее замещенный и менее стабильный олефин. Но парадокса тут нет ни на грош – это просто равновесие, смещаемое, в строгом соответствии с бессмертным учением Ле Шателье, в сторону продукта, удаляемого из реакционной смеси отгонкой. Если бы этого (отгонки) не было бы, получилось бы обычное равновесие, то есть каша из олефинов и боранов.
Именно это обстоятельство и не делает эту схему общеупотребительной. Если бы мы имели более универсальный метод, получили бы олефиновый аналог зипперной реакции в химии ацетиленов. Увы, это не так, и если задумаете применить ее сами, не забудьте оценить относительную летучесть желаемого олефина, олефина-подсадки (или, как его назвал Браун, жертвенного олефина) и диглима, и если схема не сойдется, например, вместо желаемого олефина полетит диглим (температура кипения олефина окажется сильно выше 160º, а это как раз будет, если в желаемом олефине будет больше 10 атомов углерода). Не забывайте про такие детали, когда планируете применять реакции на практике: химия – наука практическая, а не абстрактная.
Гидратация алкенов
Гидратация алкенов – школьная реакция, которая выглядит совершенно тривиально и не сулит никаких проблем. Увы, это не так. В реальной жизни это весьма непростая и очень проблемная реакция. Я предпочитаю вообще никогда ее не использовать, потому что даже в учебных целях, когда все упрощается, лучше не переходить некоторых рамок и не делать вид, что проблем не существует там, где их выше крыши.
Чтобы разобраться в чем дело, запишем вероятный механизм реакции в самом общем виде. Мы уже договорились, что эта реакция – частный случай электрофильного присоединения к двойной связи, и механизм должен соответствовать общей схеме, где сначала к двойной связи присоединяется электрофил, а затем к образующемуся карбокатиону (или мостиковому иону, но в случае протона это неактуально – атом водорода не может образовать двух связей, и мостикового иона в этом случае быть не может).
Записываем. Сразу напоминаю, что когда над стрелкой пишут плюс протон (+ H+), это не значит, что колбу поставили на пути протонного пучка в Большом адронном коллайдере. Это значит, что в реакционной смеси присутствует сильная кислота. Слово сильная здесь очень важно. Чем отличаются сильные и слабые кислоты (а это качественное, принципиальное отличие, а не просто разница в константах кислотности) можно еще раз вспомнить на страничке про кислоты. Когда над стрелкой пишут минус протон (- H+) или если это подразумевается на обратной стрелке, если над прямой написано плюс протон – это не значит, что частица (ион) плюется протонами и нужно соблюдать осторожность, приближаясь к колбе с такой диковиной, чтобы не получить протоном в глаз – это значит, что протон снимает какое-то основание, присутствующее в реакционной смеси, возможно даже очень слабое, но обязательно наличествующее; во многих случаях таким основанием является растворитель или анион той кислоты, которая протонирует в прямой реакции. Видим несколько важных вещей.
- все реакции обратимы, а следовательно и вся реакция обратима;
- здесь не одна, а две реакции – прямая называется кислотно-катализируемой гидратацией алкенов, обратная – кислотно-катализируемой дегидратацией спиртов по механизму E1;
- в общей схеме есть два кислотно-основных равновесия – протонирование двойной связи с образованием карбокатиона (оно же депротонирование карбокатиона с образованием двойной связи), и протонирование спирта с образованием оксониевого иона с хорошей уходящей группой, водой, вместо плохой уходящей группы, гидроксила.
- в середине присоединение нуклеофила к карбокатиону и обратное отщепление хорошей уходящей группы с образованием карбокатиона;
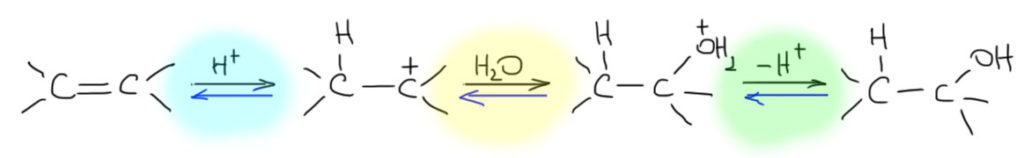
Механизм электрофильного присоединения этого типа – с медленной бимолекулярной стадией взаимодействия алкена и электрофила – называют, как мы знаем, AdE2. B мы, в принципе, знаем, что делать с обратимостью – позвать Ле Шателье. Если мы хотим гидратацию, то должны а) использовать кислоту посильнее – тогда первое равновесие сдвинется максимально в сторону карбокатиона, а карбокатион настолько реакционноспособен, что будет быстро превращаться в продукт присоединения нуклеофила; б) для самых простых газообразных алкенов – использовать давление, но это чисто промышленный путь, там ровно это и делают. В лаборатории все гораздо сложнее. В чем проблемы?
Где карбокатионы, там перегруппировки
Карбокатионы склонны к перегруппировкам. Подробнее мы это явление рассмотрим в другом месте, пока ограничимся простой констатацией этого факта – карбокатионы настолько склонны к перегруппировкам, что жить без них не могут, и даже в тех случаях, когда нам кажется, что перегруппировок нет, они все равно есть, просто нужно присмотреться получше. На практике это означает, что мы можем в результате гидратации не получить тот спирт, который хотели получить. И это еще полбеды. Настоящая беда в том, что перегруппировки редко проходят количественно, и даже вместо ожидаемого перегруппированного спирта мы получим смесь, даже если перегруппировывается менее стабильный карбокатион в более стабильный.
Пример перегруппировки, когда карбокатионы сравнимы по стабильности. Пробуем гидратировать гексен-1. Первоначально образуется вторичный катион, рядом другой вторичный катион, рядом с ним тоже вторичный катион. Перегруппировка пойдет по цепи, и каждый из катионов даст свой спирт. К счастью, второй и третий спирт – одно и тоже, если мы не используем затейливо меченый алкен. Получится смесь двух спиртов. В каком соотношении? А вот это непредсказуемо. Если бы равновесия все установились, предсказать можно было бы, но увы, карбокатионы настолько реакционноспособны и так быстро превращаются в продукты, что равновесие установится не успевает, и соотношение продуктов сильно зависит от реальных условий реакции. Органики очень не любят такие каши с переменным составом. Такую реакцию никто использовать не будет.
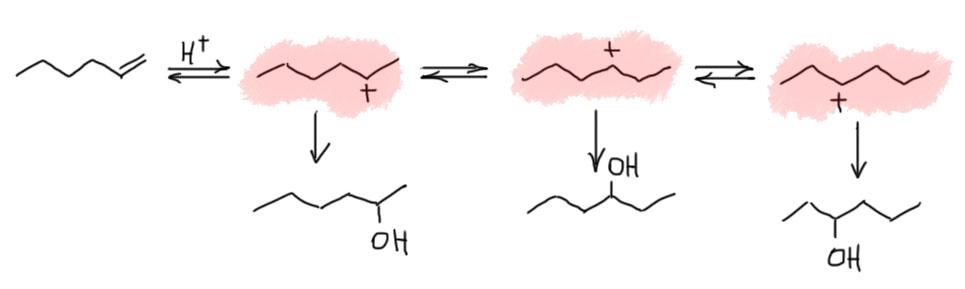
Пример перегруппировки в более устойчивый карбокатион. В этом случае сначала получается вторичный катион, который перегруппировывается в гораздо более устойчивый третичный (особо внимательные обнаружат еще перегруппировку в другой вторичный, но с той же структурой, если не метить – мы это поэтому опустили). И если бы равновесия установились, основным продуктом был бы третичный спирт. В реальности иногда будет и так, но чаще образуется смесь вторичного и третичного, потому что вторичный катион очень быстро превращается в спирт. Иными словами, у вторичного катиона перегруппировка и присоединение нуклеофила – конкурирующие реакции (по английски говорят точнее – competitive pathways – буквально, конкурирующие пути: слово “путь” в этом смысле в русском языке есть, но употребляется не очень часто, русский язык не очень любит слово “путь” во множественном числе). Это довольно разочаровывающий результат, потому что на перегруппировку в более устойчивый катион хотелось бы надеяться, как на главный результат, и так даже иногда и пишут, но это очень далеко от реальности. Эти вещи долго и усердно изучали в прошлом веке, чтобы понять, от чего это зависит, но увы, без толку, и проблему просто забросили. Может займетесь? Пока же практический результат такой – и эту реакцию использовать не будут.
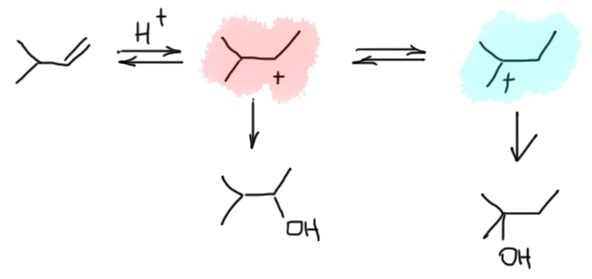
Карбокатионы склонны к побочным реакциям помимо перегруппировок
Карбокатион – очень сильный электрофил, поэтому взаимодействует со всеми нуклеофилами, до которых может дотянуться. Один из них – сам олефин, который подвергается гидратации. Олефин – очень слабое основание, поэтому даже очень сильные кислоты протонируют олефины в очень малой степени, а основная часть олефина ждёт своей очереди в реакционной смеси. Вместо протона олефин имеет шанс дождаться карбокатиона из уже запротонированной молекулы. И это не так уж маловероятно, особенно когда гидратацию делают неаккуратно, смешивая олефин с водным раствором кислоты – тогда из-за малой растворимости углеводородов в водных растворах, олефин образует отдельную фазу (капельки такие масляные будут плавать в растворе), протонирование пойдет на границе раздела фаз, и у катиона будет практически равный шанс поймать воду или олефин). Результат – так называемая олигомеризация. Посмотрим, что будет, например, при гидратации чего-нибудь простого типа пропена: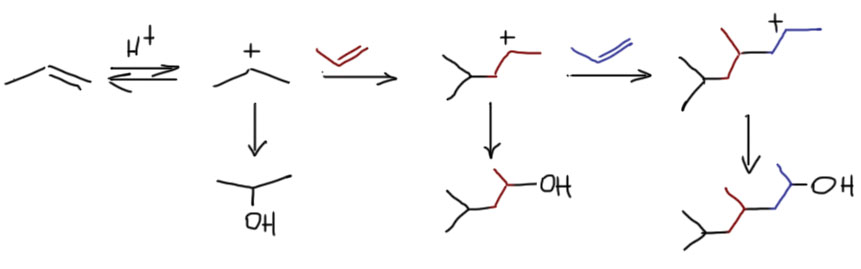
Олигомеризация почти всегда сопровождает реакции с участием карбокатионов, не только гидратацию. К счастью, продукты такой побочной реакции очень сильно отличаются от ожидаемых, потому что в них намного больше атомов углерода, а значит и намного выше температура кипения, и при очистке продукта реакции перегонкой даже в вакууме, они остаются в перегонной колбе (это называется кубовым остатком). Но выход целевого продукта это понижает и довольно сильно. Когда делаете что-то с ожидаемым участием карбокатионов (SN1-замещение, алкилирование по Фриделю-Крафтсу и т.п.), всегда учитывайте такие реакции как побочные, и не удивляйтесь, что при перегонке продукта все, что нужно, уже перегналось, а в перегонной колбе еще полно чего-то жидкого, вязкого, коричневого.
Промышленность, кстати, совсем не имеет ничего против этой реакции и активно использует ее для получения углеводородных хвостов небольшой длины. Если в чем-то промышленном, например, есть слово нонил, то очень часто в структуре вместо элегантной веревочки нормального нонила можно увидеть такой кусок колючей проволоки как на последней схеме, или даже нечто совсем запутанное, что неудивительно, так как на олигомеризацию накладываются перегруппировки.
Реальные реакции гидратации олефинов – это не гидратация
Одна из главных проблем гидратации через образование карбокатиона и его взаимодействие с водой – необходимость высокой кислотности среды, которая вызывает обратную реакцию и все прочие радости плохо контролируемой реакции. Как этого избежать хотя бы частично понятно – нужно избавиться от воды в качестве нуклеофила. Тогда, конечно, образуется какой-то другой продукт, не спирт, но, возможно, его можно будет гидролизовать до спирта. В органике реакции судят не по промежуточным, а по конечным результатам.
Ровно так и поступают. В качестве растворителя используют не воду, а, например, карбоновые кислоты, трифторуксусную или муравьиную. Эти кислоты очень хорошо известны в химии карбокатионов. Считается, что эти кислоты благоприятны для таких реакций. Так как собственной кислотности для протонирования олефина им не хватает, добавляют более сильную кислоту, хлорную или серную к муравьиной, толуолсульфоновую к трифторуксусной. Карбокатион в таких растворителях, как и положено, хватает тот нуклеофил, которого больше всего, даже если этот нуклеофил слаб, а такой нуклеофил это просто кислота-растворитель. Получаются сложные эфиры, которые очень легко гидролизуются в спирты, когда после реакции реакционную смесь обрабатывают водной щелочью. В результате получается спирт. а значит вся реакция с точки зрения конечного результата – гидратация, хотя к олефину электрофильно присоединяется не вода, а муравьиная или трифторуксусная кислота.
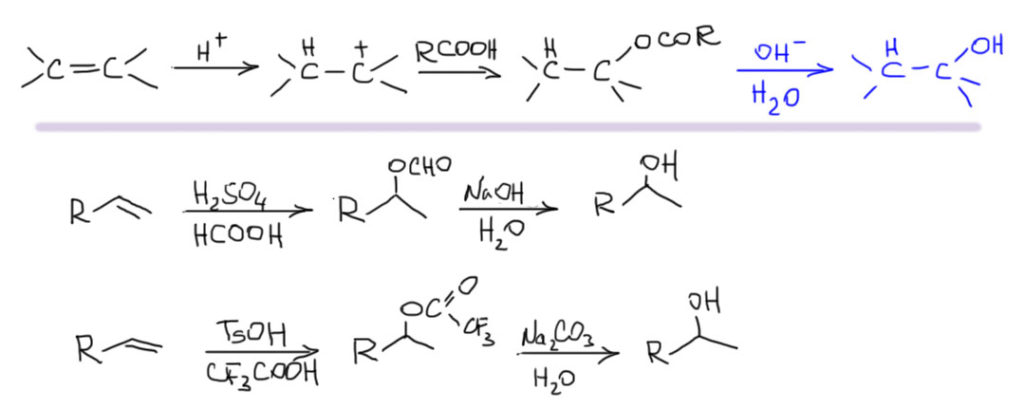
Эта методика позволяет получать спирты из олефинов, но не избавляет от перегруппировок, а вот олигомеризация в таких условиях почти не мешает – все же карбокатиону проще схватить растворитель, которого заведомо больше, чем всего остального. Поэтому это решение хотя и частично реабилитирует этот метод получения спиртов из алкенов, но не спасает его от перегруппировок.
Интересно, что простейшие олефины, этилен и пропен, в лаборатории не додумается гидратировать вообще никто, но в промышленности эти реакции имеют огромное значение. Об этом подробнее ниже, но пока заметим, что в этом случае тоже происходит нечто подобное – к двойной связи присоединяется не вода, а фактически сама кислота. Эти олефины можно заставить реагировать, только растворив под давлением в концентрированной серной кислоте. Карбокатионы в этом случае вынуждены искать нуклеофил совсем в странном месте – ведь в концентрированной серной кислоте вообще практически нет воды. Кто-то наверняка скажет, что ее там 4 массовых процента (концентрированная серная кислота имеет концентрацию 96%), а это соответствует концентрации 4 моль в литре (4 М) – по всем меркам нехило! Это количество точно больше того количества олефина, которое вы можете туда вогнать – значит, вода всегда будет в избытке. Но хорошо известно, что эта вода очень сильно влияет на свойства серной кислоты – кислотность 100%-ной серной кислоты существенно выше кислотности 96%-ной и это очень хорошо видно в самых разных реакциях, зависящих от кислотности. И точно так же, как вода влияет на серную кислоту, серная кислота влияет на воду. Когда будете изучать химическую кинетику, обратите внимание на то, что в уравнение скорости химической реакции на самом деле входят не концентрации, а термодинамические активности. В большинстве реальных реационных смесей разница между концентрациями и активностями совршенно несущественна, поэтому без проблем используют концентрации, и полностью забывают о том, что это не совсем корректно. Но не в такой экстремальной среде, как концентрирванная серная кислота! Активность воды в такой среде практически равна нулю. Это не значит, что вода количественно протонирована, но значит, что та часть, которая не запротонирована все равно очень сильно взаимодействует с молекулами серной кислоты. И что в такой ситуации остается делать карбокатиону? Основное правило жизни карбокатиона мы знаем – встретить первый попавшийся нуклеофил и соединиться с ним, невзирая на то, слаб он или силен. В концентрированной серной кислоте точно есть бисульфат-ион (не может не быть, и потому что протонирование воды неизбежно дает этот противоион, и потому что есть равновесие автопротолиза самой серной кислоты). Значит, в этих условиях образуется не спирт, а сложный эфир с серной кислотой (сульфат). 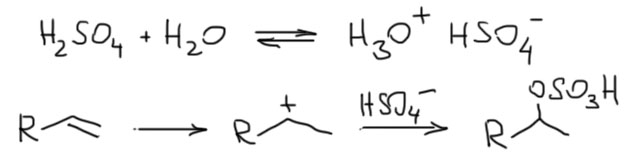 В промышленной системе, где олефин растворяют под давлением и помногу, реакция пойдет даже дальше, катион будет реагировать уже с первоначально образовавшимся сульфатом, и образуется двойной эфир с серной кислотой. Как это выглядит, посмотрим ниже, где про этилен.
В промышленной системе, где олефин растворяют под давлением и помногу, реакция пойдет даже дальше, катион будет реагировать уже с первоначально образовавшимся сульфатом, и образуется двойной эфир с серной кислотой. Как это выглядит, посмотрим ниже, где про этилен.
Этанол из этилена
Гидратация этилена – основной источник синтетического этанола в промышленности (напомню, что этилен – совершенно законное название этого соединения, во-первых, мы не обязаны всегда использовать номенклатуру ИЮПАК, во-вторых, название этилен этой номенклатуре как раз соответствует точно так же как этен – ИЮПАК в явном виде рекомендует продолжать использовать многие укоренившиеся названия простых соединений, придавая им статус альтернативно системных, рекомендация сохранить название этилен дана в правиле А3-1, а вот пропилен и бутилен сохранять не рекомендовано – но мы всё равно имеем право использовать эти названия уже как несистемные). Синтетический этанол – это растворитель и реагент для производства других органических продуктов, весьма важный, и мировое производство поэтому огромно и составляет несколько миллионов тонн в год. Надо сказать, что производство синтетического этанола ни в какое сравнение не идет с производством этанола в виде всевозможного бухла и для косметических и медицинских целей – такого этанода производится минимум на порядок больше – многие десятки миллионов тонн в год – и обязательно процессом брожения из самого разного сырья, содержащего углеводы, от зерна и картошки до продуктов химического гидролиза древесины (в просторечьи такой спирт назывался гидрашкой или табуретовкой, и сильно отдаёт запахом хвои, напоминая по вкусу дешевый джин), но синтетический этанол для всего, что связано с внутренним или наружным применением, не используется.
Производство синтетического этанола налажено с начала 20 века. Основных процессов два. В более старом процессе этилен растворяют под давлением в крепкой серной кислоте с естественным разогреванием до 60-80º, причем получается в растворе смесь этил- и диэтилсульфатов. Эту смесь затем гидролизуют, разбавляя водой и отгоняя этанол, а разбавленную кислоту просто концентрируют упариванием (добавляют немного азотной кислоты, чтобы окислить накапливающиеся побочные продукты олигомеризации и осмоления), и возвращают в процесс. Для современной промышленности этот метод очень грязен и неэкономичен, и поэтому используется уже редко, хотя у него есть одно достоинство – высокая конверсия этилена, следовательно и высокая производительность. 
Большинство этанола в мире получают другим способом – газофазной гидратацией этилена с использованием твердого катализатора. Процесс был впервые запущен компанией Shell в 1947 и с тех пор стал основным способом производства синтетического этанола везде и всюду, тем более что катализатор достаточно прост – обычно это фосфорная кислота, нанесенная на силикагель, весьма немудрёная вещь, которую невозможно скрыть патентами. Этот без преувеличения великий катализатор изобрел выдающийся химик Владимир Ипатьев вместе со своим постоянным коллегой Херманом Пайнсом незадолго до Второй мировой войны, работая в США над процессами олигомеризации алкенов и получения высокооктанового бензина. Сухой силикагель просто пропитывают раствором фосфорной кислоты и все это высушивают и аккуратно прокаливают. В результате молекулы фосфорной кислоты связываются с поверхностными гидроксильными группами силикагеля. Откуда на поверхности силикагеля (диоксида кремния по составу) гидроксильные группы? Поверхность активного силикагеля всегда гидратирована, атомы кремния несут множество гидроксильных (их называют силанольными) групп. Вот с ними и связывается фосфорная кислота, вполне химически. Зачем? Такая поверхностно связанная фосфорная кислота существенно сильнее как кислота, чем фосфорная кислота в водном растворе. Водная фосфорная кислота, даже концентрированная (85%), довольно сильна, но существенно слабее серной, и скорость гидратации этилена с обычной фосфорной кислотой в растворе невелика. Но на поверхности – другое дело. Реакцию ведут при высокой температуре и давлении, очень быстро, при этом в спирт превращается только небольшая часть этилена, но для промышленности это не проблема, непрореагировавший этилен отделяют и возвращают на вход реактора, и так много раз. 
В очень походих процессах получают изопропанол из пропилена, втор-бутанол из смеси бутена-1 и бутена-2, и трет-бутанол из изобутилена. Эти три спирта делают в огромных количествах вполне сравнимых с производством этанола – все это очень важные промышленные растворители и реагенты.
Механизм гидратации без участия карбокатиона (строго факультативно и только для наиболее любопытных).
Гидратация этилена идет в основных чертах точно так же, как и других олефинов, хотя и медленнее и в более жестких условиях. И естественно вызывает сомнения. Реакция по общему механизму AdE2 должна идти через карбокатион. Но здесь карбокатион первичный и почти совсем нестабилизированный. Можно ли вообще предполагать образование такого плохого карбокатиона? Нет ли в этом состава преступления – сознательного обмана и даже мошенничества?
Вопрос этот не так прост, как может показаться. Проблемы устойчивости карбокатионов мы разберем в другом месте, но здесь заметим, что нет никаких данных, которые прямо или косвенно свидетельствовали бы в пользу образования первичных алкильных карбокатионов в какой-либо реакции в растворах (в газовой фазе – пожалуйста, но это не имеет никакого отношения к реакциям в растворах, а мы изучаем органическую химию именно в растворах, и то, что происходит в газовой фазе нам до лампочки). Несколько менее однозначна ситуация с вторичными карбокатионами, но и там корректных данных в пользу их образования маловато. Но в таких случаях почти всегда выкатывают простой аргумент – неустойчивая частица все же образуется, но время ее жизни столь мало, что ее не удается никак “поймать”. ОК, но это уже не наука, а религиозное верование. В науке нужны аргументы более конкретные, и обязательно экспериментально проверяемые. Не можете поймать карбокатион – приведите экспериментальные данные, которые хотя бы косвенно свидетельствуют в пользу. Если нет таких данных, то обязательно нужно рассмотреть альтернативный механизм, и искать доводы в его пользу.
Альтернативны механизм найти довольно просто. Каков бы ни был механизм реакции кислотно-катализируемой гидратации, бесспорно то, что есть обратная реакция – отщепление. Если есть обратимая реакция, то законы химии требуют простой вещи – механизмы прямой и обратной реакции одинаковы, а точнее, есть общий механизм обратимой реакции. Это представление иногда называют принципом микроскопической обратимости, чем сильно пугают ни в чем не повинных людей. Не будем использовать этот чрезвычайно сложный физический принцип во вполне очевидной ситуации, состоящий в том, что если вы предложили и обосновали механизм для какой-то реакции, то стоит вам на каждой стадии вместо прямой стрелочки поставить стрелочку обратимости, то вы автоматически получите уже обоснованный механизм обратной реакции.
Дальше все просто, хотя нам придется забежать вперед. Обратной реакцией к присоединению является элиминирование. Механизмом реакции элиминирования в общем случае является согласованный механизм E2. В этом случае это будет выглядеть так: Элиминирование идет из протонированного этанола, уходящая группа – вода, с другой стороны согласованно отщепляется протон основанием, основанием может являться та же вода. Вода?? Основание??? Блохе??? Кафтан???? Да, безусловно, и одной блохе бархатный кафтан однажды сшили, и вода в подходящих условиях может быть основанием. В жизни много странностей, которые, если рассмотреть поближе, не такие уж странные. Нет ничего странного в том, что даже слабое основание Бренстеда может оторвать протон, если частица, от которой она собирается оторвать протон – достаточно сильная кислота. Никто не сомневаеся в том, что вода это основание Бренстеда, сомневаются только в том, хватит ли у нее основности, чтобы оторвать протон от той или другой конкретной кислоты Бренстеда. Вообще, если бы у нас были константы кислотности и основности для всех более или менее важных молекул и ионов, мы бы просто посмотрели в такие таблицы, и сразу сказали бы, что можно ожидать, а что маловероятно. Увы, в органической химии до сих пор просто беда с количественными данными, и даже для довольно простых реакций количественные оценки малодоступны. Молодая очень наука, что поделать. Мало что уже успела сделать основательно, все почти пока только в набросках, намеках, качественных оценках и прочей беллетристике. Приходится прибегать к таким оценкам “на глаз”, пытаясь сшить общую картину из всяких аналогий и параллелей.
Попробуем так. У серной кислоты вода отлично отнимает протон (или наоборот, серная кислота отлично протонирует воду, что одно и то же), то есть для серной кислоты вода – отличное основание. А у этильного карбокатиона или других алкильных карбокатионов вода может оторвать протон? Скорее всего да – просто подумаем, насколько та же серная кислота может протонировать этилен или другие простые алкены. Откуда это известно? Например, из растворимости алкенов в концентрированной серной кислоте. Известно, что эта растворимость повышена, но вовсе не напоминает, например, растворимость аммиака в воде, при том, что мы отлично знаем, что даже в этом случае процент протонирования аммиака невелик, и в основном аммиак в воде остается в виде молекулы, а не иона аммония. Растворимость алкенов в концентрированной серной кислоте повышена, но не очень велика – мы отлично получаем простые алкены действием серной кислоты на спирты, и алкены выделяются из таких смесей, не остаются в растворе. Из этого и разных других оценок мы знаем, что степень протонирования алкенов в концентрированной серной кислоте невелика. Иными словами, алкены – очень слабые основания по отношению к концентрированной серной кислоте. А следовательно – карбокатионы, сопряженные кислоты алкенов, – очень сильные кислоты, более сильные чем концентрированная серная кислота. А так как серная кислота охотно протонирует воду, значит и карбокатионы тем более легко протонируют воду. Но ведь протонирование воды карбокатионом – это и есть элиминирование! Теорема доказана.
И еще одна проблема. Мы же уже договорились, что у нас нет первичных карбокатионов, таких как этильный. Из этой ловушки химия довольно легко выходит с помощью немного напоминающей ловкость рук концепции согласованных механизмов. В этом случае получится вот что. Катиона нет, но есть движение в его сторону – посмотрите на схему – там от протонированного этанола начинает уходить вода. Это движение привело бы нас как раз к этильному карбокатиону, а это движение приводит, как мы убедились, к драматическому увеличению кислотности протонов на втором углероде. Отлично, вот вода их и подхватит немного раньше, чем образовался бы страшный этильный катион, которого мы как раз и не хотим. Механизм вполне обоснован. В литературе можно найти теоретические модели этого механизма, построенные современными расчетными методами (E.J.Meijer, Angew.Chem.Int.Ed., 2004, 43, 1660), где показано, что это действительно предпочтительный путь реакции, избегающий образования первичного карбокатиона, что крайне маловероятно. 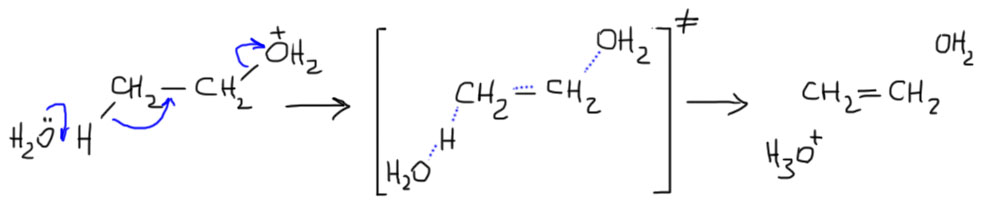
Тот же самый механизм должен работать и для гидратации. Напишем все то же самое в обратном порядке. Ион гидроксония поляризует двойную связь, которая становится доступной для присоединения нуклеофила. Все это происходит согласованно с одним переходным состоянием, и это то же переходное состояние, что в реакции E2. Такой механизм присоединения называют AdE3 потому что в реакции должны встретиться три молекулы – олефин, протонная кислота, нуклеофил.
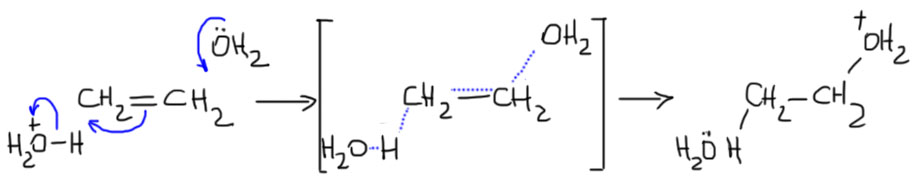
Вот такая картина. Кроме двухстадийного механизма c карбокатионом (AdE2) есть и согласованный одностадийный механизм, общий для элиминирования и присоединения – AdE3/E2.
Гидрогалогенирование: введение
Сразу ограничим себя HCl, HBr и HI. Гидрофторирование невозможно сделать обычным образом, хотя сама эта реакция известна, и, возможно, где-нибудь отдельно мы ее обсудим, так как это довольно поучительно.
Сначала краткое резюме.
- Присоединение галогеноводородов к алкенам идет без участия мостиковых ионов
- Скорость реакции очень сильно зависит от количества донорных заместителей у двойной связи – требуется минимум две штуки, чтобы реакции протекали легко и с значительной скоростью; менее донорные олефины реагируют очень медленно или вовсе не реагируют, хотя если олефин наоборот акцепторный (см. на отдельной вкладке, что это такое), то он тоже неплохо реагирует, хотя и по другому механизму, и при этом получается продукт против правила Марковникова.
- Реакции в органических растворителях протекают таким образом, что электрофилом является не протон, а целая молекула HX, причем реакция облегчается образованием водородных связей между атакующей молекулой HX и одной или несколькими содействующими молекулами HX, которые являются одновременно и источниками галогенида.
- Участие в реакции минимум двух, а иногда и большего количества молекул HX выражается в том, что скорость реакции зависит от концентрации HX в квадрате, а иногда и в более высоких степенях – по-другому это описывают, как второй или более высокий кинетический порядок по HX, а с точки зрения механизма говорят о тримолекулярном согласованном механизме присоединения, сокращенно AdE3.
- Согласованный характер реакции с участием двух или большего числа молекул HX, причем в продукте присоединения H и X оказываются из разных молекул HX, выражается в том, что стереохимией присоединения часто является анти-присоединение, но очень редко этот стереохимический результат проявляется полностью – почти всегда побочно образуется не менее 10-20% син-аддукта. Тем не менее гидрогалогенирование вполне можно считать стереоселективным анти-присоединением.
- Присоединение HBr к малореакционноспособным олефинам с концевой двойной связью очень часто осложняется конкуренцией свободнорадикального пути присоединения (присоединение по Харашу), причем инициаторами могут служить следовые примеси кислорода, перекисей и тому подобных соединений, полностью избавиться от которых невероятно трудно – поэтому реакция с 1-алкенами очень часто неожиданно дает продукт присоединения против правила Марковникова, даже если приняты меры по очистке олефина и исключению кислорода из реакционных смесей. Присоединение HCl и HI с этим никогда не сталкивается. Присоединение HI идёт особенно просто и чисто и всегда по правилу Марковникова.
- Присоединение всех трех галогеноводородов очень сильно ускоряется в присутствии катализаторов, кислот Льюиса и некоторых сильных кислот Бренстеда, в том числе твердых типа силикагеля. В этих условиях присоединение всегда идет строго по правилу Марковникова. Именно этот метод используют в современной химии для получения продуктов гидрогалогенирования олефинов.
Гидрогалогенирование: участвуют ли в этих реакциях мостиковые ионы
Начнем с одной важной общей проблемы. Есть ли мостиковый ион в гидрогалогенировании, или даже шире – в любых реакциях, где электрофилом является протон (то есть, как мы уже договорились, сильная протонная кислота). Забежим сразу вперед и заметим, что в реакциях гидрогалогенирования электрофилом протон не является. Но пока мы этого не знаем, и нам кажется, что является, ведь галогеноводороды – сильные протонные кислоты.
Может ли быть мостиковый ион при взаимодействии протона с двойной связью. На первый взгляд – какие проблемы? Почему нет, тем более что основная причина того, что мостиковые ионы так настойчиво лезут в механизмы электрофильного присоединения – необходимость объяснения, почему в основном образуются продукты анти-присоединения. В реакциях гидрогалогенирования это во многих случаях также наблюдается, хотя это и не так просто показать и наблюдать. В целом, можно сказать, что в реакциях гидрогалогенирования анти-присоединение не так явно выражено, как, например, в реакциях с участием электрофильных галогенов, но скорее является преобладающей тенденцией: в тех случаях, когда это можно доказать, анти-аддукты чаще являются преобладающими продуктами присоединения, хотя чистое (стереоспецифическое) анти-присоединение наблюдается очень редко.
Когда мостиковые ионы стали популярны в органике в работах основоположников механизмов реакций, мостиковый ион с протоном рисовали часто. А потом перестали. Причина этого проста: нельзя просто так рисовать структуры, если мы не понимаем, как они устроены, какова природа связей, причины стабильности и т.п. Попробуем разобраться, как может быть устроен мостиковый ион с протоном в качестве электрофила. Проще всего нарисовать как на первой структуре – просто протон на пунктирчике из середины двойной связи. Так вообще обычно рисуют так называемые π-комплексы с алкенами, когда кислота Льюиса взаимодействует с двойной связью. Если орбитальки нарисовать, как на второй структуре, тоже вроде все нормально, мы берем связывающую π-орбиталь двойной связи, потому что у протона нет своих электронов, а нам нужны электроны для связывания, и взять их можно только с заполненной орбитали. Мы видим, что орбитальные доли одного цвета (одного знака) отлично перекрываются с сферической s-орбиталью протона. Ай, у протона же нет электронов! Электронов нет, а пустая орбиталь есть. Отлично, из этой картинки мы видим, что взаимодействие – химическая связь – действительно может образоваться, только вот что это за связь такая – между тремя атомами и с двумя электронами? Как ее нарисовать? Так, как на третьей картинке, нельзя, потому что на ней изображены две связи от атома водорода, а водород не может образовать две связи. Так как на четвертой картинке можно, но не нужно – картинки такого рода вообще ничего никому не говорят, так как совершенно не ясно, что там за связи, между какими они атомами, где там электроны и заряд – картинки такого типа констатируют, что все взаимодействует со всем, а как и зачем, и что с этим делать, неизвестно.
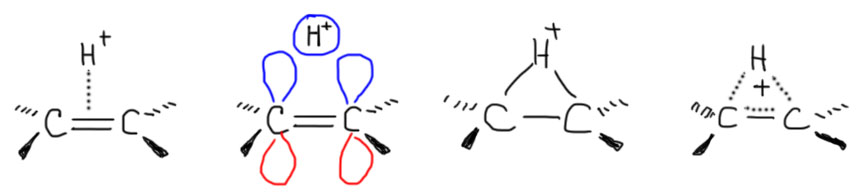
Пока мы не очень продвинулись, но одну вещь установили – в этой системе взаимодействующих атомов три атома и два электрона. Поскольку мы только что обсуждали электрофильные реакции алканов, в памяти еще свежи трехцентровые связи. А это – не может быть тем же самым? Очень похоже, что это разумно, и это хороший след. Правда нужно его немного развить, потому что там у нас такая связь получалась при атаке сильной кислоты, того же протона, на σ-связь C-H. Но никто нам не запрещает распротранить этот подход и на атаку протона на π-связь, поскольку мы видим, что взаимодействующие орбитали не против этого. Нарисуем такую трехцентровую связь так, как мы договорились там – пунктирной звездочкой, соединяющей три взаимодействующих атома – напоминаю, что это не три связи, а одна связь, и обслуживается она парой электронов. И вспомним, что к трухцентровой связи всегда ведут три пути, более или менее равноценных. Поэтому ту же точно связь мы можем получить не только из протона и двойной связи, но и из карбокатиона и соседней C-H связи, и таких пути два: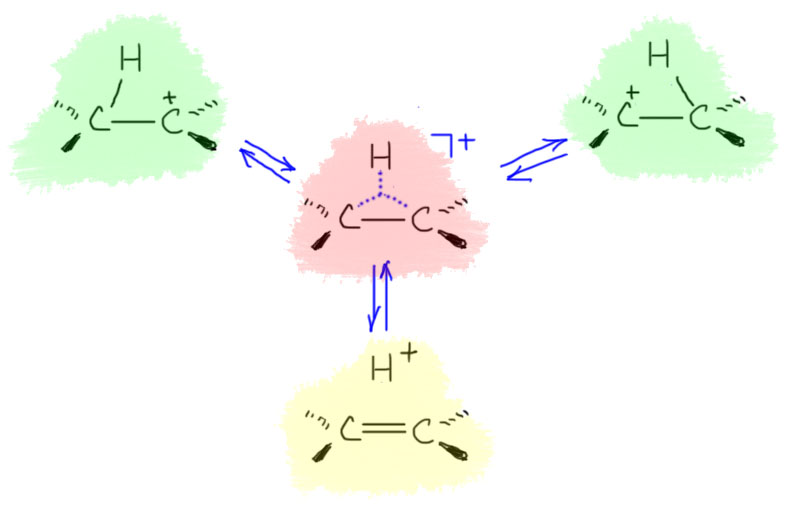
И это очередная демонстрация того, что в химии все взаимосвязано – лезем в одну проблему (механизм, реакцию, процесс) и попадаем в другую, не менее важную, и либо уже рассмотренную где-нибудь в другом месте, либо стоящую на очереди. В данном случае мы смторели на взаимодействие протона с двойной связью, и нечаянно попали… в перегруппировку карбокатионов с перемещением водорода со своими электронами – так называемые гидридный сдвиг. Мало того, что протон вдруг превратился в гидрид, но и карбокатионы всплыли, хотя наша задача состояла как раз в том, чтобы определить, не может ли мостиковый ион занять их место. Так вот этот пока только воображаемый мостиковый ион оказался одновременно и промежуточной частицей по пути перегруппировки карбокатионов. Еще раз – нарисованная схема образования мостикового иона при атаке протона на двойную связь одновременно утверждает, что тот же мостиковый ион должен образоваться и на пути перегруппировки. И если его нет в одном месте, значит нет и в другом – так устроена химия: если у нас есть сложный процесс с участием нескольких частиц, мы не можем произвольно выломать кусок этого процесса, не затронув все участвующие в процессе реакции. Так вот, если мы хотим узнать, может ли быть мостиковый ион в ротонировании двойной связи, спрсим, есть ли он в перегруппировке карбокатионов. К счастью, перегруппировки карбокатионов изучены вдоль и поперек – в исследованиях и дискуссиях по этой проблеме участвовали, кажется, вообще все крупнейшие теоретики органической химии второй половины 20 века – Шлейер, Ола, Уинстейн, Дженкс, и прочие не менее великие, применяя к решению возникающих проблемы все доступные методы эксперимента и теоретических расчетов. И да – нет посредине между двумя перегруппировывающимися карбокатионами мостикового иона как промежуточной частицы. Строго говоря, этот воображаемый ион есть не что иное как переходное состояние между карбокатионами. А раз это переходное состояние между катионами, значит это и переходное состояние при атаке протона на двойную связь – так протон приближается к двойной связи сверху, со стороны максимальной электронной плотности на π-орбитали двойной связи, и непосредственно превражается в один из двух карбокатионов, связанных перегруппировкой. И это наблюдение мы распрстраним и на протонирование тройной связи, хотя в реальности как раз там некоторые вопросы могут оставаться.
Вывод – в протонировании двойной и тройной связи мостиковые ионы никак не проявляются – протонирование непосредственно дает взаимно перегруппировывающиеся карбокатионы.
Гидрогалогенирование: чем отличаются галогеноводороды и почему это важно для реакции с алкенами
Вопрос 2. А что же сложного в реакции галогеноводородов с алкенами? Сложность в том, что это органическая химия. Что такое HCl, HBr и HI для обычного, нормального химика? Это галогеноводородные кислоты, одни из самых тривиальных. Про соляную кислоту знают вообще все. Остальные похожи. Это сильные кислоты, и мы даже знаем, что в группе сверху вниз сила немного увеличивается, хотя все три кислоты относятся к числу сильных, а сильные кислоты не так просто сравнивать по силе – константы диссоциации (кислотности) для них не работают (посмотрите еще раз про сильные кислоты и про то, как их сравнивают). Это верно, но – только в воде, в водном растворе. Увы, реакции органических соединений редко идут в воде, потому что органические соединения в воде почти не растворимы. Есть некоторые, которые хорошо растворимы, но мы сейчас хотим обсуждать углеводороды, алкены. Углеводороды в воде практически нерастворимы.
Поэтому реакции ведут в органических растворителях. Увы, галогеноводороды – весьма агрессивные реагенты и многие хорошие, более полярные растворители (спирты, кетоны, нитрилы, ДМСО и т.п.) с ними реагируют и поэтому для этих конкретных реакций бесполезны. Поэтому приходится использовать более простые, неполярные и малополярные растворители: углеводороды (гексан, гептан и т.п.), хлорпроизводные (хлороформ, дихлорметан и т.п.), эфиры (диэтиловый эфир, диоксан). Еще годится уксусная кислота, но с ней есть другие проблемы, поэтому пока оставим её в сторонке.
Так вот, в малополярных органических растворителях галогеноводороды не такие уж сильные кислоты. И вообще, если вы прочитали страничку про кислоты, то наверняка прониклись мыслью о том, что в таких растворах даже не имеет значения, сильна кислота или нет, если в растворе нет подходящего основания, то протон передать некому и приходится носить его с собой. Если растворить галогеноводороды в малополярных растворителях, они существуют с них в виде молекул и ждут основания. А его нет. А они ждут.
И вот, добавили олефин. Предположим на секунду, что олефин является достаточно сильным основанием для галогеноводорода. Что это значит? Совершенно очевидно, что в этом случае HX передаст протон олефину, а протонированная форма олефина (в терминах теории Бренстеда-Лоури, сопряженная кислота: олефин и соответствующий карбокатион относятся друг к другу, как сопряженные кислота и основание) это и есть карбокатион (возможность мостикового иона мы уже отмели). После передачи протона от HX останется галогенид-анион. Поскольку среда малополярная, далеко он не уйдет, да карбокатион его моментально и съест – получится продукт реакции. Да, это же и есть простой механизм электрофильного присоединения, как мы его уже знаем. Он бимолекулярный – в медленной стадии встречаются две молекулы, олефин и HX. Назовем его поэтому Присоединение (Addition) Электрофильное (Electrophilic) Бимолекулярное, сокращенно AdE2.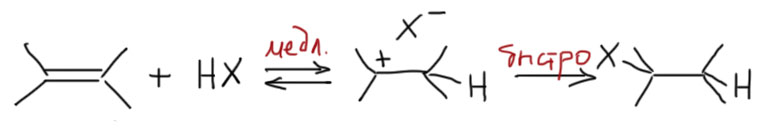
Поскольку кабокатион очень быстро реагирует с нуклеофилом – это даже называется коллапсом ионной пары, чтобы подчеркнуть чрезвычайную легкость этого процесса – равновесие протонирования смещается в сторону продукта реакции, поэтому степень протонирование может быть невелика, никто не просит, чтобы олефин протонировался на значительную глубину. И еще это значит, что время жизни карбокатиона настолько мало, что он может не успевать проявить себя, как положено карбокатиону, и, например, перегруппироваться. Может перегруппируется, а может и нет – перегруппировка ведь тоже реакция, и как у любой реакции у нее есть скорость, и это очень большая скорость, но скорость коллапса ионной пары может быть еще больше или сравнимой. Положим здесь закладку, мы еще вернемся к этому. А пока зададим вопрос, если такой механизм работает, для какого из галогеноводородов мы скорее можем это ожидать. Ответ очевиден – для HI, заведомо самой сильной кислоты не только в водном растворе, но и в органическом растворителе. Сила кислоты, особенно в среде, которая сама не помогает растаскивать кислоту на ионы, зависит от легкости отрыва протона от аниона, а это тем легче, чем анион больше по размеру. Ионы в малополярных апротонных органических средах, в которых неоткуда ждать поддержки, всегда чувствуют себя не в своей тарелке, и чем ион меньше и чем более локализован заряд, тем хуже. И наоборот. Большой иодид-ион (ионный радиус 2.06 Å) относительно неплохо чувствует себя в малополярных органических растворителях, причем одличие даже от ближайшего родственника бромида (ионный радиус 1.82 Å) очень велико, а уж от хлорида (ионный радиус 1.67 Å) – просто разительно. И действительно, иодистоводородная кислота – самая реакционноспособная из галогеноводородов, реакции идут быстро и без осложнений, и всегда получается продукт, соответствующий правилу Марковникова. Что неудивительно – мы уже выяснили, что правило установлено на основе обобщения реакций олефинов с HI. Впрочем, как ни странно, эта реакция довольно малопопулярна, и, насколько мне известно, никогда детально не изучалась.
Присоединение HCl. Что такое механизм AdE3.
Поведение хлористого водорода HCl в присоединении к олефинам исследовалось гораздо тщательнее и загадки этой реакции много раз вызывали ожесточенные споры. С этой реакцией в некотором смысле произошла неприятная история. Когда в органической химии в 30-х годах прошлого века началось создание теории и изучение механизмов, вначале показалось, что будет все просто и всё быстро упорядочится в очень стройную систему, не хуже чем какая-нибудь теоретическая механика. Ну, может быть какие-то очень сложные реакции потребуют тяжких размышлений и каких-то нетривиальных решений, и это вполне ожидаемо, ведь и механика не очень шустро дает решение задач взаимодействия множества тел. Но уж такие простые и основополагающие реакции, как присоединение галогеноводородов должны были бы стать просто триумфом теории. Но с присоединением и HCl и HBr все оказалось очень непросто, исследования и споры затянулись на десятилетия, и полной ясности нет до сих пор, хотя в современной органической химии на детали механизмов перестали обращать большое внимание – главное, чтобы мы понимали, как поставить конкретную реакцию и получить желаемый продукт.
Начнем с HCl. Этот галогеноводород наименее активен в реакции присоединения к алкенам. Скорость реакции очень сильно зависит от структуры алкена, точнее от того, сколько донорных заместителей висит на двойной связи, причем с увеличением числа заместителей скорость очень сильно учеличивается – есть огромная разница между реакцией этилена, 1-алкенов, и алкенов с внутренней двойной связью, причем каждый переход в этом ряду (от этилена к 1-алкенам, от 1-алкенов к внутренним алкенам) скорость возрастает дале не в разы, а на порядки.
Для многих, возможно, будет сюрпризом, но этилен фактически вообще не реагирует с HCl, точнее реагирует чрезвычайно медленно, потребовались бы месяцы для достижения приемлемых выходов, я даже не уверен, что кто-либо когда-либо смог измерить эту скорость хотя бы минимально правдоподобно. Но ведь даже младенцы знают, что реакция этилена с HCl – промышленная реакция для получения хлористого этила! Да, знать-то знают, но все известные процессы синтеза хлористого этила из этилена и хлороводорода используют катализаторы, а про то, что для присоединения галогеноводородов к олефинам нужны еще и какие-то катализаторы, нам пока что никто никогда ничего не говорил. Что за катализаторы? Кислоты Льюиса, самые разные, а зачем их используют и какую роль они играют мы обсудим ниже. Сейчас же мы обсуждает собственно реакцию олефинов с хлористым водородом без катализаторов. Не с соляной кислотой, заметьте. С соляной кислотой не реагируют почти никакие олефины, так как просто не растворимы и не смешиваются с ней. И если вы просто замесите олефин с соляной кислотой, они так и будут болтаться двумя несмешивающимися фазами: олефин будет в органической фазе, а HCl в водной, и вместе им не сойтись никогда, как писал Р.Киплинг.
Реакции олефинов с хлористым водородом без катализаторов делают или вообще без растворителя – при охлаждении насыщают жидкий олефин сухим HCl и в закрытом сосуде оставляют стоять при охлаждении или при комнатной температуре. Или в растворах неполярных или малополярных растворителей типа гептана или дихлорметана, которые тоже насыщают сухим HCl. Попытка провести эту реакцию в чем-нибудь чуть более полярном, например в эфире или диоксане приводит к практически полной остановке реакции присоединения, вероятно, потому что такие растворители являются основаниями Бренстеда-Лоури и HCl образует с ними комплексы, растрачивая на это всю свою кислотность и электрофильность. Получается такая конкуренция – кислота выбирает более сильное основание, и выбор у неё небогат – олефин или растворитель. Выбирает растворитель, основность атома кислорода эфирного типа точно намного выше, чем основность большинства двойных связей. А с еще более полярными растворителями начинаются проблемы уже другого типа. Не так-то просто подобрать растоворитель для этой реакции. Растворители вообще в органической химии подбирать довольно трудно: Господь, когда создавал всё сущее, зачем-то создал миллионы видов насекомых, но только пару-тройку дюжин растворителей. Лучше бы наоборот, да что ж теперь поделать.
Монозамещенные алкены с концевой двойной связью с хлористым водородом реагируют быстрее этилена, но тоже очень медленно – для достижения высоких выходов требуются недели и даже месяцы. Реакции всегда дают продукт, соответствующий правилу Марковникова, и в случаях, когда можно предполагать легкую перегруппировку карбокатиона, такие продукты действительно получаются, но не на 100%. Самый популярный алкен для теста на перегруппировку – изопропилэтилен. Получаются оба продукта почти 1:1 в условиях, когда олефин реагирует с сухим HCl без растворителя, и это очень медленная реакция (JACS, 1933, 55, 5020). В более современных работах этот результат воспроизводили у же в растворителях типа дихлорметана с почти такими же результатами. 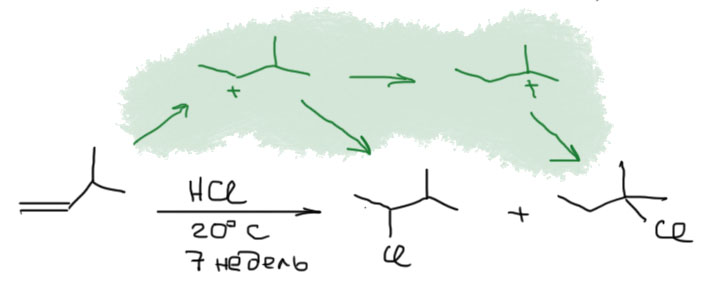
Это очень важный результат, потому что он показывает, что карбокатион в этой реакции образуется всё же вряд ли. Как не образуется – разве перегруппировка не является однозначным признаком карбокатиона? Вон же и на схеме зеленым выделено! На схеме выделен предполагаемый механизм присоединения через карбокатион, как мы его назвали AdE2. Но в анализе реакций всегда нужно анализировать все доступные данные. Фокус в том, что когда такой карбокатион действительно образуется, например, при действии кислот Льюиса на 2-галоген-3-метилбутаны, перегруппировка происходит практически нацело. Это совершенно типичный случай – перегруппировка этого вторичного катиона (его иногда называют втор-изоамильным или, скороговоркой, сиамильным – узнаете этот радикал из дисиамилборана?) в третичный (а этот называют трет-амильным) настолько выгодно, что должно происходить количественно с огромной скоростью. Если не происходит количественно и с огромной скоростью (сама реакция присоединения идет больше месяца!) значит там что-то сильно не так – нет там, видимо, никакого карбокатиона. А, чёрт, но перегруппировка-то идет, пусть и неколичествнно?! Идёт, но перегруппировка не обязательно предполагает образование карбокатиона, она может осуществиться, если реакция идёт по согласованному механизму и только двигается в сторону нестабильного карбокатиона, завершаясь перегруппировкой в более стабильный карбокатион еще до образования нестабильного. Обсудим это подробнее в другом месте.
Еще более странные вещи оказались, когда ученик Хараша Фрэнк Майо взялся изучать кинетику реакции олефина с HCl сразу после Второй Мировой войны. Это было невероятно сложной задачей, особенно если учесть, что оборудование в те времена было еще очень примитивным, и никаких методов быстрого набюдения (мониторинга) концентраций типа всяких спектроскопий или инструментальных хроматографий еще не было. Как точно дозировать газообразный HCl, измерять концентрации, учитывать влияние случайных примесей – всё это было решено кропотливой работой, потребовавшей создать для каждой реакции отдельное оборудование. Результат оказался обескураживающим. Из принятого механизма электрофильного присоединения можно было ожидать кинетики второго порядка, первого по каждому из реагентов, потому что в таком механизме встречаются два реагента и реагируют в медленной стадии образования карбокатиона, как на схеме: 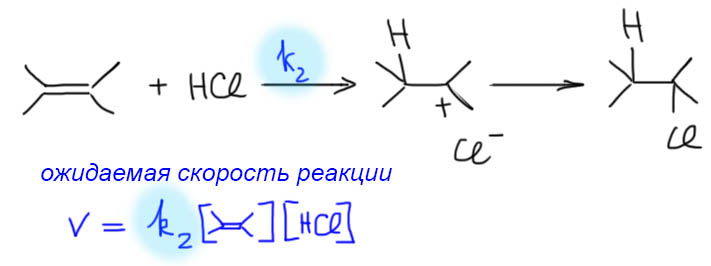
Но в реальности получилась совсем другая кинетика – четвертого порядка, первого по олефину, но третьего по HCl. Это был страшный удар! Только что построили теорию, разрисовали механизмы для основополагающих реакций, и казалось, что органическая химия уже превратилась в строгую науку, и тут такой конфуз! Вообще, четвертый порядок казался вздором – как могут в скоростьопределяющей стадии встретиться аж 4 молекулы – это же практически невероятно. Автора исследования могли объявить неумёхой и неучем, заклевать, вышвырнуть с волчьим билетом до конца жизни из химии. Не на того напали – недаром Майо был не просто учеником великого Хараша, а даже в некотором смысле его правой рукой. Эксперимент был сделан, как полагалось у Хараша – комар носа не подточит, безупречно. Это хороший и поучительный урок – хотите сделать что-то необычное и интересное в науке, придётся работать так, чтобы никто не мог усомниться в качестве исследования. Более того, Майо заметил, что любые примеси – следы воды в HCl, ионы металлов со стенок прибора, ртуть из вакуумного насоса – очень сильно влияли на кинетику, снижая порядок до второго по HCl. 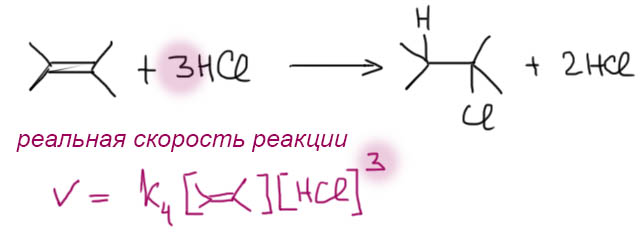
После этой работы кинетику взаимодействия HCl с разными олефинами изучали много раз, и все время получали нечто похожее – порядок по HCl от двух до четырех, не обязательно целый. Что это может значить, и какой механизм соответствует такой странной кинетике.
Во-первых, поймем, что каков бы ни был кинетический порядок по любому из реагентов, сама реакция остается той же – одна молекула олефина реагирует с одной молекулой HCl с образованием одной молекулы продукта реакции, а остальные молекулы HCl как-то влияют на скорость процесса – они необходимы для того, чтобы реакция произошла. Можно даже считать, что они катализируют процесс присоединения. Зачем, и как это происходит?
На вопрос зачем ответить очень просто. HCl в малополярных органических растворителях не распадается на ионы, а остается молекулой. И кислотность этой молекулы недостаточна для протонирования двойной связи алкенов. Это ведь, в принципе, вполне очевидно. Мы только что в обсуждении гидратации обсуждали то, что даже серная кислота делает это с большим трудом, а серная кислота намного сильнее HCl даже в водных растворах, не говоря уж о органических растворителях. Если HCl на ионы не распадается, значит единственный для неё шанс реагировать – делать это прямо целой молекулой. Так прямо подрулить к двойной связи и сразу и связь C-H образовать, и C-Cl. Но при этом получился бы чистый продукт син-присоединения, а в реальности присоединение идёт как преимущественно анти. К тому же, говорят, такая реакция как-то сильно не нравится по квантово-химическим причинам, с симметрией, говорят, там что-то не так. Разбираться не будем, эксперимент важнее, и отсутствие син-присоединения в этом смысле важнее. Да и не забудем, что в этом случае не было бы карбокатиона, реакция была бы согласованной, и тогда скорее всего регулировалась бы так же как гидроборирование стерическими факторами, а значит могла бы давать продукт против правила Марковникова, а это уж точно для присоединения HCl не наблюдается.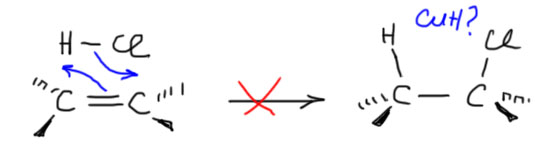
Чтобы спасти ситуацию попробуем построить такую конструкцию. Что если HCl подходит не боком, а электрофильным центром, то есть атомом водорода. Не забудем, что в отличие от борана, в молекуле HCl связь очень сильно поляризована в сторону хлора, и на атоме водорода должен быть немаленький положительный заряд. А нельзя ли просто передать протон двойной связи? Нет, с этим мы уже разобрались. Это не происходит, потому что кислотности HCl не хватает, и потому что в этом случае получился бы настоящий хлорид-ион, а какой, к чёрту, хлорид-ион в малополярной среде. Что он там делать будет? Не его это среда. В переводе на чуть более строгий язык мы скажем, что ионы должны сольватироваться, иначе энергия иона будет непозволительно велика, и это повиснет неподъемным вкладом на энергетике всей реакции. А чем его сольватировать в малополярной среде? Нечем… Тоже не работает. 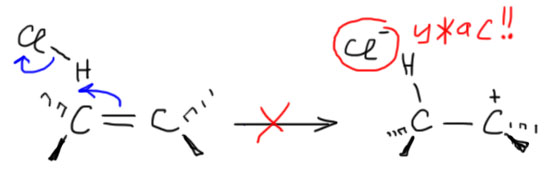
А что если в реакции участвует не одна, а две молекулы HCl. Что это даст? А то, что вторая молекула водородной связью как раз и успокоит уходящий хлорид-ион – среди механизмов сольватации анионов водородная связь на безусловном первом месте по эффективности. Стоп. Сольватация ведь это взаимодействие с растворителем, а какой растворитель из второй молекулы HCl? Не совсем так: сольватация это взаимодействие не с растворителем, а со средой, то есть и с растворителем, и со всем, что в нём есть ещё, если это может помочь больше чем сам растворитель. Тогда HCl вполне годится – это единственный сильный донор водородной связи в малополярной апротонной среде. Вполне соотвествует призыву: Никто вам не поможет, если сами себе не поможете. Более того, нуклеофил как раз и может образоваться из второй молекулы HCl. Попробуем нарисовать что-то такое. Получится такой любимый многими механизм с шестичленным переходным состоянием и перемещением шести электронов. Получится такой любимый многими механизм с шестичленным переходным состоянием и перемещением шести электронов. Лишняя молекула HCl
просто останется и пойдёт дальше соображать на троих с другими молекулами олефина и HCl. Вот такие шестичленные переходные состояния были в большой моде в прошлом веке – их пихали во всё, что ни попадалось на пути, и это считалось верхом химического остроумия. В нашем столетии это больше не носят, или, точнее, носят по необходимости в тех случаях, когда есть неубиваемые аргументы в пользу такого построения молекул, но уж точно не считают самодостаточным объяснением всего на свете. Но дело даже не в том, что это вышло из моды, дело в том, что получится опять син-присоединение. 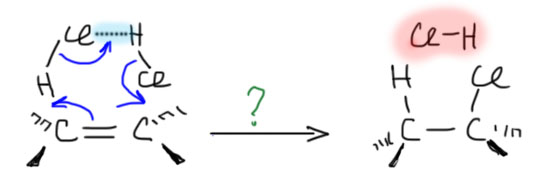
Но нас уже не остановить. Добавим еще одну молекулу HCl. Получится такая цепочка водородно-связанных молекул, и она может достать и до обратной стороны молекулы. Я специально подкрашу эту хлористоводородную чехарду цветом, чтобы не путалась с алкеном. Точнее, придётся достать, потому что с одной стороны такое хозяйство уже не уложится. Ещё одно усилие: 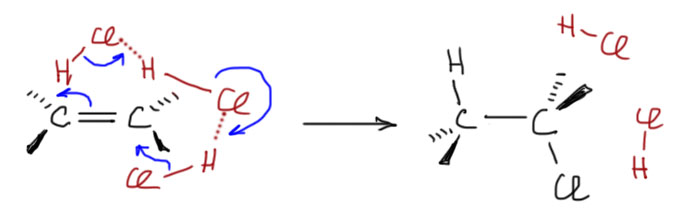
Нарисовали. И хочется спростить – это серьезно? Да, вполне. Во-первых, необходимо объяснить кинетику с зависимостью скорости реакции присоединения HCl от концентрации HCl во 2-й или 3-й степени. Такая модель это объясняет. Во-вторых, современная химия в таких случаях прибегает к квантовохимическому моделированию механизма: производится расчет координаты реакции, рассчитываются энергии промежуточных частиц и переходных состояний. Такие расчеты (последняя статья на эту тему совсем свежая: C.L.Firme, Journal of Molecular Modelling, 2019, 25, 128, и в ней использованы новейшие и очень трудоёмкие расчеты) действительно говорят в пользу реакции с тремя молекулами HCl, в которой эти молекулы, связанные водородными связями выстраиваются во что-то похожее на то, что нарисовано на последней схеме: реакция в этом случае идёт по согласованному механизму без образования каких-либо ионов, ни карбокатиона, ни хлорид-ионов, неуместных в малополярной среде. Механизм, иными словами, заключается в том, что олефин и три молекулы HCl собираются вместе и в такой конфигурации через единственное переходное состояние, перебросив электронные пары от одного из углеродов двойной связи через цепочку молекул HCl сдают вместе с последним хлоридом на второй атом углерода, сразу образуя продукт присоединения и две молекулы HCl. Такие механизмы в присоединении принято называть AdE3 – согласованные механизмы электрофильного присоединения с суммарным кинетическим порядком (то есть числом молекул, участвующих в переходном состоянии) от 3 и более. Конкретизировать, что в конкретной схеме молекул четыре, смысла большого нет.
Вопрос: а почему тогда соблюдается правило Марковникова? Ведь карбокатиона нет, и нет возможности рассуждать о его стабильности. Карбокатиона нет, но механизм является согласованным, но не синхронным: связи рвутся и образуются не совсем одновременно. Если бы был синхронным, то все произошло бы строго одновременно и никакие заряды нигде не возникали и не изменялись бы. Но так быть не должно – связи, участвующие в реакции, имеют разную энергию, разную прочность. Обычно считают (тоже на основании расчетов, а также наблюдаемого влияния заместителей на скорость реакции – в этом случае донорные заместители сильно ускоряют реакцию), что образование связи C-H немного опережает образование связи C-Cl (тоже в повседневном смысле понятно, почему – реакция ведь электрофильная, перенос протона её начинает, а вслед за началом образования связи C-H происходит перераспределение плотности в цепочке атомов, оно просто немного отстаёт, в этом случае на втором углероде успевает возникнуть некоторый положительный заряд, похожий на карбокатион (но не настоящий карбокатион!), и последний в цепочке хлорид садится на этот углерод. В рассчитанном переходном состоянии мы бы увидели более высокий порядок новообразующейся связи C-H, чем порядок другой образующейся связи C-Cl. Тогда и эффекты заместителей будут похожи на их влияние в карбокатионе. Поэтому и прожукт будет тем же, что мы рисовали для воображаемого стадийного присоединения HCl – сначала протон, потом к карбокатиону хлорид.
Вот как непросто, оказывается, идёт такая с виду примитивная реакция. И какая экспериментальная и теоретическая мощь нужна, чтобы в этом разобраться.
А теперь добавим в эту бочку изысканного, сладчайшего и ароматнейшего мёда маленькую ложечку дерьма. Когда я вижу такие красивые картинки переходных состояний, в которых множество молекул выстраивается в выверенную до сотых долей нанометра конструкцию, возникает ехидный вопрос, а известно ли авторам этих конструкций, что такое энтропия. Фокус в том, что как бы ни были сложны и современны расчёты, они дают только энергии частиц или конфигураций на пути реакции. И с энергиями там всё в порядке, скорее всего, но это не так уж и важно. Важно то, что если бы реакция действительно шла через такую изумительно точную конструкцию переходного состояния, за это пришлось заплатить бы очень большой отрицательной энтропией активации. Более просто это можно выразить так: такой путь хоть и энергетически оправдан, но маловероятен, и скорее всего, представляет собой просто одну из возможных конфигураций переходного состояния. Всё, скорее всего, намного проще. В растворе HCl в малополярном растворителе молекулы HCl не плавают поодиночке, а образуют ассоциаты за счет водородной связи: димеры, тримеры, тетрамеры, и т.п., всё это в подвижном равновесии. Олефин в таком растворе будет встречаться с разными ассоциатами, и такая встреча будет время от времени приводить к присоединению по согласованному механизму. У каждой такой ракции будет своя константа скорости. В реальности мы будем видеть просто среднее (взвешенную сумму) из таких множественных путей реакции. И наблюдаемый порядок реакции тоже будет таким средним – и понятно, почему его так трудно измерить и почему он всё время, в зависимости от условий, плавает в диапазоне от второго до более чем третьего. Если нас попросят изобразить механизм такой реакции, мы всё равно нарисуем что-то подобное последней схеме, только скажем, что это один из наиболее вероятных из нескольких (многих) похожих путей, и не будем настаивать, что единственный и точный до десятых долей нанометра. Кстати, именно поэтому не имеет смысла придираться к цифре 3 в обозначении механизма AdE3 – эта цифра более символическая, чем точная.
Итак, резюмируем:
- реакция олефинов с HCl идёт в малополярных средах, без растворителя (растворитель – сам олефин) или в углеводородах, хлорпроизводных угоеводородов и т.п.
- механизм реакции согласованный с участием от двух и более молекул HCl в переходном состоянии, но без образования карбокатиона.
- механизм реакции согласованный, но не синхронный, и электронная плотность с π-связи быстрее смещается в сторону атома водорода молекулы HCl, что создает значительный положительный заряд на втором атоме углерода – поэтому донорные заместители сильно ускоряют реакцию, а способ присоединения соответсвует правилу Марковникова.
- Такой механизм обычно называют AdE3, обозначая этим, что в реакции участует более одной молекулы галогеноводорода.
Еще раз про механизм присоединения: так есть там карбокатион или нет?
После всех этих рассуждений о согласованных механизмах присоединения к двойной связи не может не возникнуть ощущение неудовлетворённости. Классический механизм очень понятен и удобен: найдите электрофил, присоедините к двойной связи двумя способами, получите два карбокатиона, проанализируйте их относительную устойчивость, из этого анализа выберите один более стабильный и следовательно тот углерод двойной связи, к которому и присоединится электрофил, а карбокатион заткните нуклеофилом – продукт готов. И заодно становится понятно, какой олефин более реакционноспособен, а какой менее. И многие другие вещи. Много раз уже сказано, что химии проще судить о заряженных частицах, катонах и анионах.
Но получается так, что в реальности всё происходит не так, и нет никаких карбокатионов, и всё присоединяется в одной стадии А как же тогда судить о том, как идёт реакция.
Эта малоприятная проблема очень часто возникает в органической химии. Как судить о согласованных механизмах? На самом деле всё очень просто, хотя и немного смахивает на мошенничество. К этому приходится прибегать. Современная органическая химия вообще предпочитает согласованные механизмы многостадийным с участием неустойчивых частиц типа карбокатионов. Активным исследованиям механизмов скоро исполнится сто лет, и за это время накопилось множество данных, заставляющих со всё большим сомнением смотреть на возможность образования малоустойчивых и высокореакционноспособных интермедиатов в реакциях. Лет 50 назад рисовать, скажем, этильный катион было вполне нормально, естественно, со всеми оговорками, что это очень активная частица и т.п. Но как иначе присоединить воду к этилену? Сейчас на такую интерпретацию механизма решатся немногие, потому что никаких доказательств возможности даже чрезвычайно кратковременного образования такой частицы получено никем не было. В очередной раз оговорюсь – речь идёт только про реакции в растворах, о газовой фазе мы не говорим, там всё возможно, и метильный катион там не проблема.
Подход к согласованным механизмам может быть таким. В согласованном механизме ключевую роль играеют не интермедиаты, а структура и энергия переходного состояния. В любой согласованной реакции переходное состояние одно и только одно. В переходном состоянии всегда задействованы связи, затрагиваемые реакцией. В присоединении к двойной связи это разрываемая π-связь, образующиеся связи углерода с электрофилом и нуклеофилом, и, возможно, какие-то связи внутри молекул, поставляющих электрофил нуклеофил. Фокусв том, что нигде не написано, что все эти связи образуются и рвутся совершенно синхронно. Если бы это было так, нигде не образовывались бы заметные заряды, ничто не напоминало бы катионы и анионы, а вся реакция развивалась бы с идеальной слаженностью молекулярной машины.
Так бывает? Бывает, но очень редко, в основном во всяких модельных и бесполезных процессах, в которых шило меняется… на шило же. А мыло на мыло. Такие процессы называются вырожденными, их часто используют для большой науки, исследуя или теоретически, или экспериментально, например, с помощью изотопного маркирования, не изменяющего химической природы частиц, но позволяющего их различить. Шило меням на изотопно меченое шило.
В реальных процессах, типа замены шила на мыло, трудно представить себе идеальную синхронность. Но мы можем иметь дело с почти синхронностью: связи образуются и рвутся не строго одновременно, а с небольшими опережениями и отставаниями, но так, что в переходном состоянии всё равно нет заметных зарядов на атомах. На энергию таких переходных состояний (напомню, что с энергией переходного состояния связана энергия активации, а следовательно и скорость реакции – чем ниже энергия, тем меньше энергия активации и больше скорость) слабо влияют все факторы, которые мы использовали бы для объяснения стабилизации катионов и анионов – электронные эффекты заместителей, влияние растворителя или среды. А больше всего влияет стерика – удобство подхода реагентов друг к другу, возможность того, чтобы они расположились друг относительно друга максимально близко и в той ориентации, которая отвечает целям реакции – образованию и разрыву именно тех связей, которые нужны. Как мы уже знаем, именно так устроено, например, гидроборирование.
Еще чаще встречаются согласованные реакции с сильным отклонением от синхронности. В таких реакциях что-то одно происходит быстрее всего остального. Например, в присоединении галогеноводородов, связь C-H образуется быстрее образования связи C-галоген. В переходном состоянии мы увидим что-то сильно похожее на карбокатион, но – не карбокатион. Карбокатион это реальная частица, возможно сильно неустойчивая, но реальная, и если он образуется, у нас есть шансы доказать это экспериментально – дело только в нашем упорстве, владении и обладании доступом к самым современным методам исследования, возможно даже пока еще не существующим, и возможно, шансы есть, но не у нас, а у следующих поколений, но они есть. Всё, что существует, может быть обнаружено. То, что не существует, не может быть обнаружено. Вот переходное состояние как раз и не существует. И не может быть обнаружено. Это точка на потенциальной поверхности. И т.п. Мы всё это уже не раз обсуждали. И если переходное состояние похоже на карбокатион, но не является карбокатионом, мы не можем надеяться его обнаружить экспериментально. Но тогда это просто выдумки, зачем это? Да, выдумки, но полезные. Польза от них в том, что построив модель переходного состояния, мы можем изучать факторы, которые влияют, например, на относительную энергию. Эти факторы в реальной жизни будут влиять на скорость реакции. Связь простая: модель переходного состояния на бумаге или экране компьютера – скорость реальной реакции, и факторы вляиющие на скорость. Если модель предсказывает то, что мы наблюдаем, то это хорошая модель – хвалим ее и считаем работающей, пытаясь на ее основе что-то ещё предсказать. Если не предсказывает, то это плохая модель – ругаем её, топчем ногами, выбрасываем и делаем новую. Как-то так это и делают, раньше просто умозрительно, а ныне и присно, и вовеки веков всё больше полагаясь на расчёты, всё более и более изощренные, но пока ещё довольно топорные, и поэтому не очень убедительные.
Безусловно, у всего этого есть малоприятная сторона: откуда мы знаем, что мы не сможем в будущем обнаружить нечто такое, что не можем обнаружить сейчас? И не может ли то, что мы сейчас считаем переходным состоянием, в будущем стать настоящим интермедиатом, а согласованная реакция превратиться в стадийную? Такая проблема есть, и с чем-то подобным сталкиваются любые исследования в естественных науках, опирающихся на эксперимент и наблюдение. Но в том, что касается химии, есть некоторые вполне объективные пределы. То же время жизни частицы не может быть сколько угодно малым. Если мы не наблюдаем частицу в масштабе пикосекунд (одной триллионной секунды), то дальше ехать особенно некуда – в этом масштабе времени происходят колебания атомов в молекулах, а ничто равновесное не может иметь время жизни меньшее времени одного колебания, и так мы закрываем все нормальные химические реакции в растворах. А если мы дошли до фемтосекунд (квадриллионых частей секунды), в масштабе которых происходят всякие процессы, связанные с перемещением электронов и жизни возбужденных состояний, то мы дошли уже до полного предела наблюдаемому в химии, закрыв уже и химию переноса электрона и фотохимию – дальше мельчить уже совсем нет смысла. А к этим пределам экспериментальная техника дошла уже в наше время, и никто не мешает думать, как ее применить для интересующей именно вас реакции.
По сравнению с электрофильным присоединением, свободнорадикальные реакции присоединения и замещения занимают более скромное, но всё же очень важное место. Самая важная и практически очень полезная реакция этого типа – аллильное бромирование по Волю-Циглеру.
Аллильное бромирование (реакция Воля-Циглера)
Разберемся в этой реакции поподробнее, чтобы понять, отчего она такая уникальная, и какие у нее проблемы, но сначала, как обычно краткое резюме, а потом для желающих подробный разбор. На страничке про методы есть краткое изложение практического применения этой реакции.
- Реакцию проводят действием N-бромсукцинимида в кипящем четыреххлористом углероде с добавлением небольшого количества свободнорадикальных инициаторов, перекиси бензоила или бис-азоизобутиронитрила (АИБН). Инициирование светом (хорошей мощной лампой накаливания или современными светодиодами, лучше синими) возможно, но применяется относительно редко.
- Реакцию можно делать и без инициаторов, но время реакции (а его определяют очень точно – в этот момент уходит желтоватый цвет брома, а осадок тяжелого бромсукцинимида всплывает, превращаясь в легкий сукцинимид) при этом непредсказуемо, выходы меньше, а количество побочных продуктов, в первую очередь, просто продукта присоединения брома к двойной связи – больше.
- Реакция имеет свободнорадикальный механизм, и реальным реагентов в ней является молекулярный бром, образующийся сначала при медленном разложении NBS, а в ходе реакции при взаимодействии NBS и выделяющемся в реакции HBr.
- Реакция протекает через образование мезомерно-делокализованного аллильного радикала, который забирает атом брома у молекулы брома, второй атом брома дальше ведет цепной радикальный процесс.
- Реакция может быть неселективной и давать смеси продуктов по двум причинам – и из-за делокализации неспаренного электрона по двум атомам углерода, которые могут оказаться структурно различными, и в тех случаях, когда исходный олефин имеет несколько разных аллильных положений. В первом приближении эта проблема непреодолима, и таких ситуаций следует избегать, но в реальной жизни аллильное бромирование довольно часто бывает вполне селективно, и есть несложные правила, которые позволяют достаточно точно предсказывать основной продукт реакции.
Откуда взялась реакция Воля-Циглера
Аллильное бромирование называется реакцией Воля-Циглера, и в 2019 году, когда пишется этот текст, ей испольнилось сто лет, всего сто лет или целых сто лет. Это интересный пример того, как в химии появляются именные реакции, и какие имена остаются в истории. Работа Альфреда Воля 1919 года, весьма известного немецкого химика, с которым мы еще встретимся в углеводах, описывает реакцию N-бромацетамида с одним олефином с образованием бромпроизводного аллильного типа. 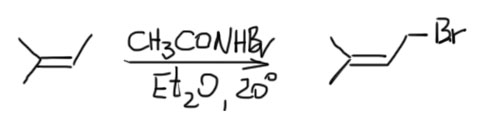
В те времена результаты описывали очень приблизительно, и непонятно, единственный ли это продукт, и даже насколько точно определена его структура. Но это не важно, потому что описанная реакция вообще не имеет никакого отношения к аллильному бромированию – это просто единичный пример электрофильного бромирования с E1-элиминированием и, возможно, аллильной перегруппировкой. Совсем недавно очень похожую реакцию стали использовать для аллильного хлорирования, но об этом где-нибудь в другом месте. И все же – прецедент был создан, показано, что бромирование непредельного соединения может идти не только как присоединение, но и как замещение. И ещё – было привлечено внимание к новому, для того времени, типу реагентов – галогенпроизводным с галогеном на атоме азота, N-галогенамидам. Это очень принципиальное открытие, у которого будет очень плодотворное продолжение не только в виде аллильного бромирования, но и в виде химии так называемых производных “положительного галогена”, переживающей сейчас бурный расцвет. Поэтому работа Воля – характерный пример внешне скромной работы на перспективу. Важно то, что сам исследователь, работающий в свое время и со своими возможностями, редко может догадаться, что его конкретная работа “заиграет” много позже, возможно, когда его самого уже не будет на свете (Воль умер в 1939 сразу после начала Второй мировой войны, а работа Циглера появилась через три года в год двух ключевых сражений Второй мировой войны – Сталинграда и Эль-Аламейна).
На работу Воля действительно никто внимания не обратил. Единичная реакция это еще не метод, которым можно пользоваться для того чтобы решать свои задачи. Но цель – найти метод введения галогена в аллильное положение алкенов – была очень заманчива, тем более что важность и интерес к очень реакционноспособным аллильным галогенпроизводным была вполне осознана. Циглер решил попробовать разобраться, что нужно сделать для того, чтобы аллильное галогенирование стало настоящим методом, и подошел к делу весьма основательно, получив с десяток разных N-галогенамидов, и всех их испробовав в разных условиях с одним типичным алкеном, циклогексеном. Выяснилось, что ни N-бромацетамид, который был у Воля, ни почти все остальные не дают ничего хорошего, только смеси продуктов в основном присоединения. Только один реагент, полученный впервые Циглером N-бромсукцинимид, и только в одном случае – при кипячении с использованием четыреххлористого углерода в качестве растворителя (температура кипения 77ºС), приводил к образованию аллильного бромпроизводного. Даже очень близкий аналог, N-бромфталимид, работал плохо и неселективно. 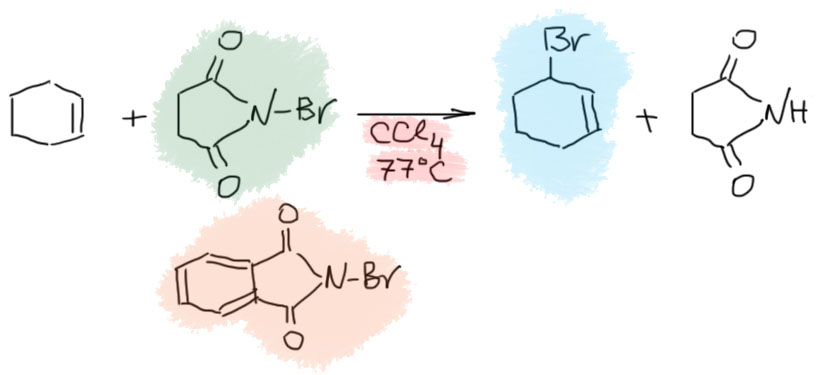
Работа Циглера не может не восхищать. В те времена методов исследования органических соединений было еще очень мало, и в основном все делалось по-старому: реакция ставилась с серьезных количествах, и продуукты выделялись перегонкой или перекристаллизацией и идентифицировались, сравнением с уже известными веществами, или дополнительными реакциями, превращавшими новые продукты с известные вещества. На каждую реакцию уходила пропасть времени. И как в таких условиях (а уже и бомбы начали падать, а студентов и аспирантов пачками отправляли на Восточный фронт в один конец) можно было найти единственный(!) работающий реагент и единственные(!!) условия работы этого реагента?? Невероятно и потрясающе, следствие колоссального опыта и интуиции.
Работа Циглера немедленно привлекла внимание – методом стали пользоваться для решения задач синтеза, попытались разобраться в том, как он работает и нельзя ли его расширить. Уже в 1948 Джерасси опубликовал весьма подробный обзор по применению метода, им же и названного именами Воля и Циглера, на 125 цитированных работ, но понимания того, как работает метод и почему так важно “условие Циглера” – использование одного единственного растворителя.
Механизм реакции Воля-Циглера
Реакция аллильного бромирования появилась как раз тогда, когда создавалась теория механизмов органических реакций, и за нее тут же взялись, как за хороший пример цепной свободнорадикальной реакции. На первый взгляд, здесь все настолько просто, что может служить просто тренировочной задачей на усвоение учебного материала. Реакция свободнорадикальная? Да, значит ищем первичный источник радикалов – способ инициирования. Ищем самую слабую связь, как мы это делали в Алканах. Очевидно, что связь N-Br просто сама просится на эту роль. Отлично, рвём, получаем радикалы брома и сукцинимидоила с точкой на азоте. Думаем, кто из них поведет цепь – кому выгоднее оторвать атом водорода. Тоже очевидно – связь NH гораздо прочнее связи BrH, и ее образование даст гораздо больший энергетический эффект (все эти нехитрые закономерности мы уже рассматривали). Ну а как дальше развивается цепь настолько очевидно, что можно сразу писать. Так и было сделано. Этот очевидный механизм часто называют по имени первого исследователя, который не поленился это записать – механизм Блумфилда (C. F Bloomfield, J. Chem. Soс., 1944, 14) – и опять трудно удержаться от комментария, что в это же самое время соотечественники англичанина Блумфилда усердно ровняли с землей тысячами тонн авиабомб важный промышленный центр Рурской области Мюльхайм, в котором Циглер только что возглавил научный институт – институт, кстати, чудом не пострадал. Война войной, но научные статьи выходили не менее регулярно чем в наши дни. В общем – вот основные стадии механизма Блумфильда (неправильного!). Для краткости остов сукцинимида обозначим как Z. 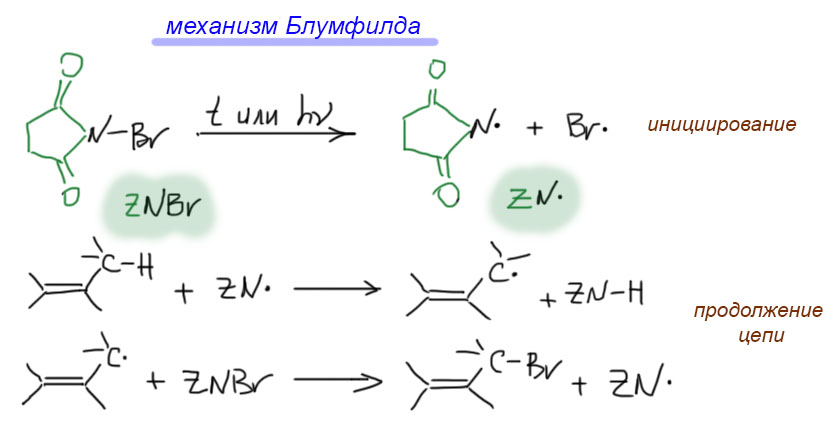
Поскольку механизм вполне очевидный, то с ним естественно согласились, и он стал жить своей жизнью, попав в учебники. Но некоторых проницательных исследователей он не удовлетворил. И мы с ними бы согласились, поскольку тоже не лохи. Мы же настаиваем на одной важной вещи – механизмы пишутся не просто так, чтобы место в учебнике заполнить, а для того чтобы объяснять особенности реакции. И вот с этим сразу стало нехорошо. Вот например, как этот механизм объясняет “условие Циглера”? Да никак не объясняет. А как этот механизм объясняет, почему из дюжины испробованных N-галогенамидов работает только NBS? Любой N-галогенамид имеет слабую связь N-галоген и тоже должен инициироваться теплом или светом. И цепь так же будет выглядеть. Но нет, не работает. Далее, посмотрим на основной радикал, который начинает цепь. Это радикал с точкой на азоте, и такой радикал должен быть очень активен (опять рекомендую посмотреть в теме Алканы обсуждение реакционной способности радикалов в отщеплении атома водорода), а значит не очень селективен. Но реакция Воля-Циглера просто удивительно селективна, об этом отдельно. Это очень непохоже на картину, которая получилась бы при действии радикала на азоте, и много позже это было выяснено экспериментально, – действительно, всё было бы не так, никакой селективности не было бы.
Первую гипотезу о том, как на самом деле идёт бромирование по Волю-Циглеру высказал опять британский кинетик Голдфингер (не имеет никакого отношения к третьему фильму про Джеймса Бонда) в удивительно мутной статье, просто на основании общих соображений о кинетике процесса. Поскольку в дальнейших, уже экспериментальных исследованиях гипотеза была подтверждена сразу несколькими независимыми группами, механизм стал общепринятым, и его так и называют – механизм Голдфингера. Механизм прост как табуретка. Это просто свободнорадикальное бромирование молекулярным бромом при двух важных условиях:
- концентрация брома очень мала, в самом начале реакции это просто примесь брома в исходном N-бромсукцинимиде, она всегда в нем есть даже если он очищен перекристаллизацией прямо перед использованием
- бром в малой концентрации снова появляется из-за очень легкой реакции HBr, выделяющегося при бромировании, с N-бромсукцинимидом
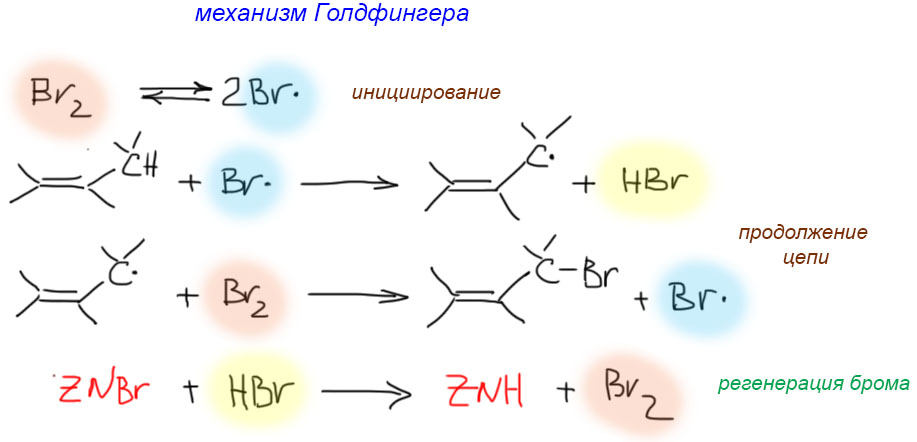
Если внимательно посмотрть на этот механизм, возникает вопрос – а почему бром просто не присоединяется к двойной связи? Ответ простой – концентрация брома очень мала, а растворитель неполярный: в таких условиях скорость реакции электрофильного присоединения просто очень мала. В неполярных растворителях кинетика реакции олефинов с бромом очень сложна и имеет от второго до третьего порядка по брому (подробнее на вкладке про галогенирование, когда она будет написана). Когда порядок больше единицы, зависимость от концентрации становится такой сильной (попробуйте возвести число меньше единицы во вторую или даже третью степень), что при низкой концентрации скорость можно считать неотличимой от нуля.
Второй очевидный вопрос: радикал брома встречается с олефином еще в одной реакции – свободнорадикальном присоединении HBr по Харашу. А почему там атом брома присоединяется, а здесь отрывает атом водорода? А почему там не образуется аллильный бромид? Второе объяснить проще. Там есть олефин и HBr, но нет брома. Поэтому даже если атом брома оторвет атом водорода из аллильного положения, аллильный радикал не может превратиться в аллил бромид. Единственное, что ему остается, это вернуться обратно – отщепление атома водорода атомом брома обратимо.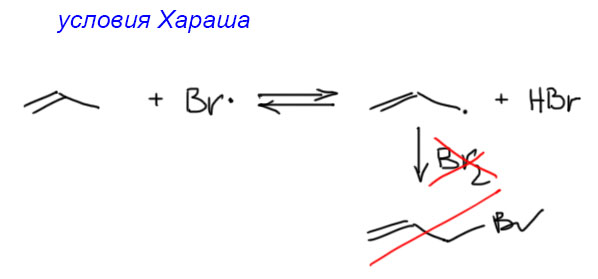
Первое – сложнее. Свободнорадикальное присоединение брома вполне известно. Исследования показывают, что вклад такого способа присоединения в реакции олефина с бромом может быть немалым. И в отличие от электрофильного присоединения, свободнорадикальное не зависит от растворителя и ему не важна полярность, и кинетический порядок по брому вряд ли больше единицы. Как же быть? Очень просто. Скорее всего, эта реакция идёт как побочная. В аллильном бромировании действительно никогда не бывает очень высоких выходов, и образуется много побочных продуктов. В тех случаях, когда в них разбирались (очень редко), действительно видели и продукты бромирования. При выделении продукта аллильного бромирования, особенно перегонкой, такие побочные продукты просто теряются (они значительно менее летучи). Когда будете делать аллильное бромирование и думать про побочные продукты – это очевидный кандидат. 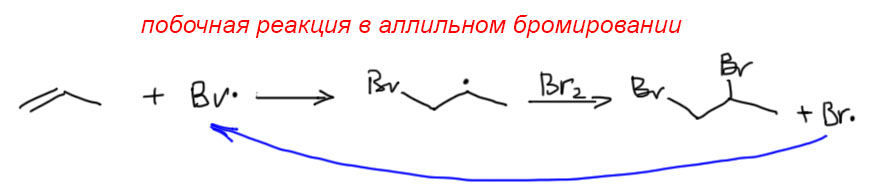
Инициаторы и практическое выполнение реакции Воля-Циглера
В принципе, реакцию можно вести только с термическим инициированием – при кипячении, но это долго и увеличивает долю побочных реакций. Именно так делал сам Циглер в основополагающей работе, и нашел, что скорость реакции очень сильно зависит от олефина – некоторые реагировали у него за 5 минут, а некоторые за 15 часов, причем было совершенно непонятно, от чего это зависело. Это неудобно и очень сильно снижает выход. Но Даубен и Мак Кой однажды совершенно недвусмысленно показали (Dauben, MсCoy, J. Org. Chem. 1959, 24, 1577), что эта реакция совершенно обязательно нуждается в инициаторах, и если их не положить специально, то в этом качестве просто выступают обычные примеси перекисей в исходных олефинах, и естественно содержание таких примесей совершенно случайно, отчего и непонятные времена реакции. Это ровно та же история, что и в присоединении HBr против правила Марковникова – примеси перекисей в непредельных углеводородах очень сложно вычистить полностью и небольшое количество почти всегда присутствует, если не применять совсем уж драконовские процедуры очистки и после этого не допускать даже минутного контакта с воздухом. И этого количества может быть достаточно и для присоединения HBr против Марковникова, и для аллильного бромирования. А может и не быть достаточно. Поэтому результаты такого случайного инициирования предсказать очень трудно, и рассчитывать на них не рекомендуется. В реакции Воля-Циглера всегда советуют использовать добавленный инициатор, а что советуют п присоединении против Марковникова, мы рассмотрим в своем месте.
Поэтому принято добавлять стандартные инициаторы (на страничке про Алканы можно вспомнить что это) – перекись бензоила или АИБН. Также возможно инициирование светом, обычной лампой видимого света, желательно помощнее, причем это еще одно доказательство того, что настоящий реагент в этой реакции – молекулярный бром, а не сам NBS, так как последний сам по себе бесцветен и видимый свет практически не поглощает. Если быть совсем точным, то небольшой хвостик от ультрафиолетовой полосы поглощения в видимой области у NBS все же есть (ChemPhotoChem 2018, 2, 938), и этим объяснятся то, что на свету этот реагент медленно разлагается и окращивается в оранжевый цвет молекулярного брома, но для инициирования этого было бы недостаточно. Если посмотреть реальные методики, то становится ясно, что чаще всего используют один из двух классических инициаторов, по принципу “что под рукой”.
Стандартные условия, которые с тех пор применяют практически без вариантов для осуществления реакции аллильного бромирования: стехиометрическое количество NBS, причём годится как неочищенный реактив “из банки” обычно оранжевого цвета из-за содержания брома, так и бесцветный свежеперекристаллизованный реактив), сухой CCl4 в качестве растворителя (нам хорошо, у нас это расхожий дешёвый растворитель, а вот в США или ЕС вы столкнётесь с тем, что эту жидкость строго контролируют из-за высокой токсичности, просто так вы её не купите, придётся подписывать бумагу, что все последствия своего необдуманного решения вы берёте на себя). Немного инициатора, или перекиси бензоила или АИБН, без разницы. И алкен. Всё это просто перемешивают при кипячении с обратным холодильником и наблюдают за внешним видом. В начале реакции тяжелый NBS лежит на дне и появляется слабая оранжевая окраска брома. Через некоторое время кипячения, реакция завершается обычно довольно быстро, от 15 мин до пары часов, что можно увидеть по двум признакам – осадок всплывает (просто сукцинимид лёгкий, в нём нет тяжёлого брома), и оранжевый оттенок смеси вдруг пропадает. Смесь охлаждают, осадок сукцинимида отфильтровывают, фильтрат упаривают на роторном испарителе, остаток перекристаллизовывают или перегоняют в вакууме.
И самое интересное - что получается в реакции Воля-Циглера
Реакция аллильного бромирования очень часто используется в синтезе, но парадоксальным образом до сих пор никто толком не не понимает, что от нее можно ожидать. Эта важная и популярная реакция парадоксально недоисследована. Споры о механизме, увенчавшиеся принятием механизма Голдфингера в начале 1960х, практически заглохли, хотя до сих пор время от времени находятся желающие отстаивать другие механизмы, но внимания никакого никто на них не обращает. Большая часть примеров применения этой реакции описана в довольно старой литературе 50-60х годов прошлого века, и там не всё хорошо сделано, часто просто выделялся основной продукт, составляющий во многих случаях не более 40-50%, а всем остальным просто пренебрегали. Некритическое отношение к таким данным приводит к тому, что в и в более свежей литературе много откровенно ошибочных представлений о том, как эта реакция идёт, и что даёт. Если в вашей будующей карьере исследователя когда-нибудь найдётся немного времени и возможностей, займитесь этой реакцией с использованием современных методов, и наконец разберитесь с тем, как она идёт и что даёт. Потомки вас не забудут. Пока же мы вынуждены просто пытаться обобщить примеры применения этой реакции в литературе, и поискать закономерности.
Прежде чем углубиться в такой кустарный анализ, повторю простой и практичный совет. На странице про Методы написано, что не рекомендуется применять ее к несимметрично-замещенным олефинам, потому что мезомерно делокализованные аллильный радикалы, а в несиметричном случае их может получится несколько, теоретически должны давать смесь большого количества продуктов бромирования. И я очень советую прислушаться к этому совету, и не искать приключений. На этом месте можно было бы поставить точку, более не злоупотреблять вашим вниманием здесь, а перенести это занятие в другой раздел органической химии.
Но если бы это действительно было так, реакция аллильного бромирования имела бы очень узкое применение, потому что симметричных олефинов не так много. Дело даже не в симметричных олефинах, а в делокализации аллильного радикала. Даже такие простые олефины, как 1- или 2-бутен дали бы по два продукта каждый, например: 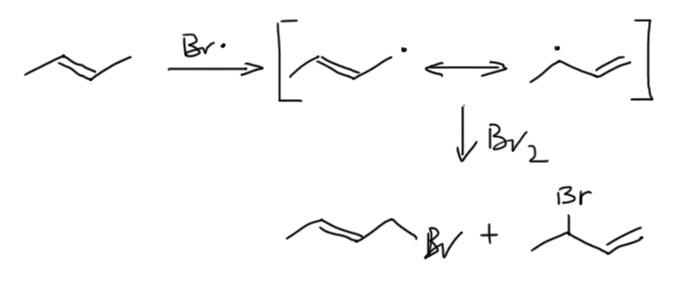
Так это или не так, трудно сказать, потому что этого кажется, никто не делал, скорее всего, потому что кипятить с обратным холодильником раствор бутена довольно трудно – он просто улетит, а под давлением реакцию Воля-Циглера не делают.
Понятно, что если олефин хоть немного сложнее, нас ждут проблемы – много проблем. Попробуем, из общих соображений взять что-нибудь не очень мудрёное, и подумаем, что должно получиться. Попробуем, например, 1-метилциклогексен. В этом алкене три разных аллильных положения, и каждый аллильный радикал мезомерно делокализован. Выпишем все возможные продукты. Атом водорода можно оторвать или от вторичного положения, и таких варианта два, или от первичного. Каждый из этих радикалов делокализован, и у одного вторичного радикала две граничные структуры одинаковы, у второго – разные. И две граничные структуры есть у радикала, полученного отрывом водорода от первичного положения. И при взаимодействии этих радикалов с молекулой брома должны были бы получиться пять разных продуктов – и это было бы совершенно бессмысленно. Смеси 5 продуктов никто никогда не разделяет – это себе дороже. Смертный приговор? 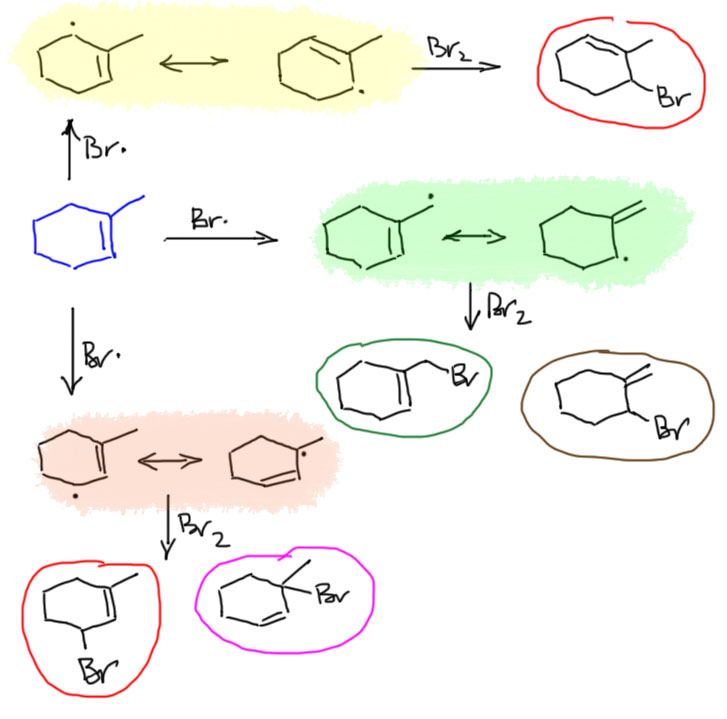
Но получается в реальности только два продукта – тех, что обведены красным. Лучше бы один, конечно, но и два тоже неплохо. Смеси двух продуктов уже можно разделять, это непросто, и нам не по силам, поэтому я везде и всегда в нашем курсе настойчиво призываю такие реакции не использовать. Но в практике синтеза на это часто идут, если во всех остальных отношениях реакция привлекательна – дает большой суммарный выход, легко выполняется, недорога, и т.п.
Попробуем разобраться, почему наши ожидания и предсказания так разошлись с действительностью. Дело, видимо, в двух основных факторах:
- довольно высокой селективности реакции отщепления атома водорода атомом брома. Мы разбирали это в химии свободнорадикального галогенирования алканов, просто напомню, что у этой реакции очень небольшой энергетический эффект – прочности связей C-H и Br-H довольно близки, а так как прочность связей C-H довольно сильно зависит от того, первичный, вторичный или третичный радикал при этом образуется. Метафорически выражаясь, атом брома хорошо различает между сортами C-H связей, предпочитая третичные вторичным, а вторичные первичным. Третичных у нас в этом примере нет, но из этого понятно, почему не образуется первичный радикал и два продукта из него. Пять минус два равно трём. Уже неплохо. И в качестве бонуса, замечу, что это еще и дополнительный довод в пользу механизма Голдфингера, потому что если бы цепь вел не радикал брома, а радикал из сукцинимида, он не разбирал бы между первичным и вторичным положениями, потому что энергия связи N-H намного больше энергии связи Br-H. И еще понятно, что если бы существовал аналогичный процесс аллильного хлорирования, его селективность была бы намного ниже, и первичное положение тоже играло бы.
- стерическим фактором на стадии реакции аллильного радикала с молекулярным бромом: молекула брома весьма тяжелая, неуклюжая и пузатая, а реакция радикала с молекулой брома тоже имеет очень слабую энергетику, и это особенно будет заметно для мезомерно делокализованных радикалов, у которых плотность неспаренного электрона на атомах углерода понижена – он же делокализован, поделен в каких-то долях между двумя углеродами (еще раз напомню, что это не значит, что на атоме углерода сидит какой-то огрызок электрона, это нужно понимать, как вероятность найти неспаренный электрон на каждом атоме). Бром поэтому тоже очень селективен в реакции с аллильным радикалом, и просто не может подобраться к радикальному центру на третичном атоме углерода. Вот мы и убили продукт, обведенный цветом Барби. Всё, пять минус три равно двум. Эти два продукта проходят по всем параметрам, вот они и получаются.
Итак, в аллильном бромировании более сложных олефинов скорее всего получатся те продукты, которые соответствуют отщеплению водорода от вторичных положений, а продукты, соответствующие или отщеплению от третичных, или делокализации на третичные не образуются по стерическим причинам. Учтите только, что если в молекуле нет вторичных углеродов, но только первичные и третичные, то они будут реагировать, если в делокализованных аллильных радикалах в этих случаях есть стерически доступные не-третичные радикальные центры. Так можно упростить многие реакции аллильного бромирования. Попробуйте, например, найти единственный продукт аллильного бромирования а) 2-метилпент-2-ена, б) 1,2-диметилциклогексена; в) 2,3-диметилбут-2-ена.
Есть и еще одно соображение, которое стоит принимать во внимание – стабильность двойной связи в продукте. Если есть выбор, то в продукте получится более стабильная двойная связь – в делокализованном радикале будет преобладать граничная структура с более стабильной двойной связью:
- цис-двойная связь превращается в транс;
- если есть дополнительное сопряжение, то двойная связь будет стремиться к этому сопряжению;
- двойная связь, торчащая из циклов наружу (экзоциклическая двойная связь) будет стремиться заползти внутрь цикла.
Вот примеры таких реакций. Второй пример показывает, как два изомерных олефина дают один и тот же продукт, потому что двойной связи приятнее быть в сопряжении к ароматическому кольцу.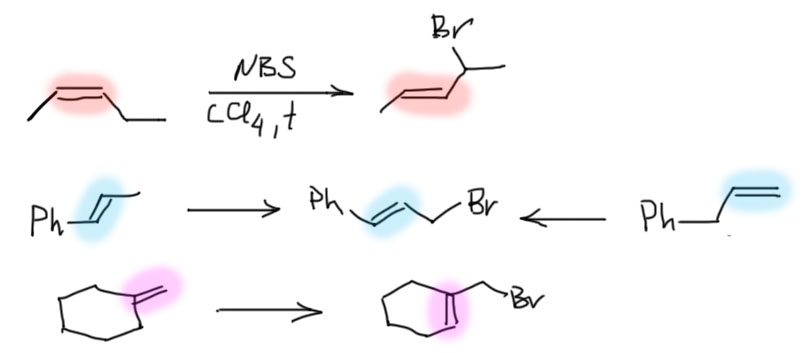
В конце упомяну еще одну засаду, которая подстерегает желающих разобраться в продуктах этой реакции. Эти продукты – бромпроизводные аллильного типа, и мы скоро с ними встретимся еще раз, в разделе про диены. И там выясним, что у них есть неприятная привычка перегруппировываться просто так, при хранении, и особенно при повышенной температуре. В диенах это приведёт нас к анализу кинетического и термодинамического контроля при присоединении галогенов и галогеноводородов к диенам, и там при хранении и при повышенной температуре образуются более термодинамически стабильные продукты того же самого типа – аллильные галогенпроизводные. Почему здесь должно быть иначе? В принципе, именно это и может быть причиной вот этих смещений двойной связи в продуктах, которые приведены на последней схеме. Это стоило бы проверить экспериментально, но, кажется, никто никогда этого еще не делал.
А бывает аллильное хлорирование или иодирование?
Сразу оговоримся – в условиях свободнорадикальной реакции. Электрофильные методы аллильного хлорирования, а также бромирования и иодирования есть, они довольно популярны в современном синтезе, но нам они не нужны и мы не будем с ними разбираться.
В условиях реакции аллильного бромирования по Волю-Циглеру нет, нельзя сделать аналогичное хлорирование или иодирование. Есть полные аналоги NBS – N-хлорсукцинимид и N-иодсукцинимид, вполне доступные реактивы, а также множество аналогов. Но реализовать что-то похожее на аллильное бромирование несмотря на длительные усилия не получилось. С иодированием все ясно сразу, и отсылаю вас к свободнорадикальному галогенированию алканов – энергетика стадий реакции не велит. Как ни инициируй реакцию, атомы иода всегда действуют как ингибиторы радикальной цепи. Разговаривать не о чем.
Другое дело хлорирование. Но и тут все плохо. Дело, скорее всего, в чрезвычайной легкости реакции свободнорадикального присоединения хлора к двойной связи. Поэтому если у нас есть атомы хлора, они предпочтут присоединение, а не отрыв атома водорода, и реакция пойдет по другому пути с образованием продуктов присоединения. В то же время, аллильное хлорирование очень простых алкенов типа пропилена осуществляют в промышленных условиях при высокой температуре. 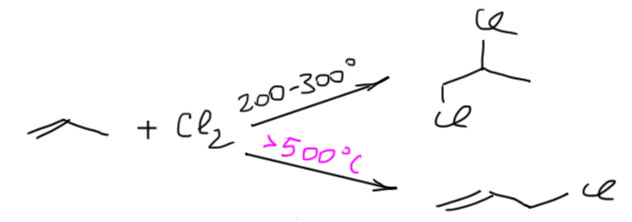
Реакция эта – отличный пример прямого аллильного галогенирования и конкуренции двух путей реакции атома галогена с алкеном – присоединения к двойной связи и отщепления атома водорода. При более низких температурах преобладает присоединение, при более высоких – отщепление. Дальше все идет просто как обычная цепь свободнорадикального хлорирования, ничем не отличающаяся от хлорирования алканов. 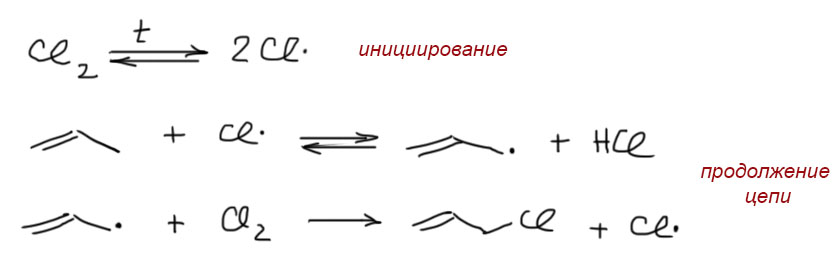
Естественно можно задать вопрос, отчего атом хлора при меньших температурах предпочитает присоединяться, а при высоких – отдирать атом водорода. Ответом на этом вопрос должно быть детальное исследование кинетики и термодинамики обоих процессов, а это очень редко реально делают, так как это чрезвычайно трудоемкая (и немножко бессмысленная) работа. Чисто на словах картина должна быть, скорее всего, такой: у каждой из стадий обоих процессов есть константы скорости, и они зависят от температуры. Температурные зависимости констант скоростей различны – одна может расти быстрее другой, у каждой есть своё уравнение Аррениуса со своими параметрами. Пока оставим это в стороне. Важно то, что если при одной температуре одна реакция быстрее второй, но скорость второй реакции сильнее зависит от температуры, то при повышении температуры вторая реакция рано или поздно обгонит первую. Это обычная история, но в обычной работе мы редко имеем возможность изменять температуру больше чем на 100-150 градусов. А в газофазной реакции в промышленных реакторах температура ограничена только началом термического разложения реагентов. Органика плохо выдерживает температуры выше 250-300º за исключением или очень простых соединений на 3-4-5 атомов углерода, или всякой ароматики, поэтому в этом смысле разгуляться некуда. Так и это высокотемпературное хлорирование максимум, куда можно сунуться с температурой за 500º, это 4-5 атомов углерода. Поэтому нам очень важно понять, что эта реакция совершенно не годится для лабораторного синтеза и никак не обобщается на другие алкены.
В учебниках иногда пишут, что высокотемпературное аллильное хлорирование идет через образование продукта присоединения и термическое элиминирование HCl. Это не так, и это было давно установлено теми же английскими исследователями, которые и обнаружили саму реакцию аллильного хлорирования (G.W.Hearn et al., Industrial Engeneering Chemistry, 1939, 31, 1530). Дело в том, что такое элиминирование HCl из 1,2-дихлорпропана действительно идёт, но не дает хороший выход аллилхлорида из-за образования второго продукта (1-хлорпропена), поэтому в промышленности реакцию специально ведут так, чтобы присоединение не происходило – перед смешиванием нагревают оба газа до 300º и только после впрыскивают их в камеру с 500º – тогда образуется чистейший аллилхлорид почти без побочных продуктов.
Очень хорошее подтверждение того, что аллильное хлорирование при высокой температуре идет как цепная свободнорадикальная реакция получили (G.W.Hearn et al., J. Am. Chem. Soc., 1953, 75, 1392), когда в реакцию ввели вместо пропена, 2-бутен. Получилась весьма типичная смесь аллильных хлоридов, объясняемая мезомерной делокализацией радикала. 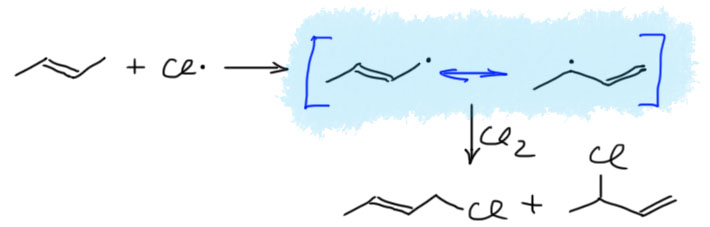
И даже еще лучше, когда попробовали хлорировать изомерные аллилхлорид и 1-хлорпропен – они дали одинаковую смесь двух продуктов, что ясно говорит о том, что радикал по дороге из обоих исходных алкенов получается одинаковый, мезомерно стабилизированный. 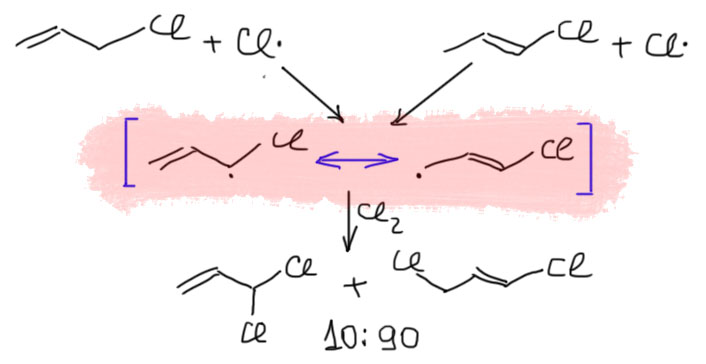
В конце рассмотрим несколько факультативных вопросов. Этого нет в программе, и читать это стоит только тем, кому просто интересно, что еще бывает.
Присоединение HF к алкенам
Бывает ли гидрофторирование по аналогии с присоединением других галогеноводородов? Если бывает, то почему про это почти ничего не слышно?
Фтористый водород драматически отличается от всех остальных галогеноводородов. У него, в каком-то смысле, всё наоборот. В водных растворах это относительно слабая кислота, но это-то нам как раз до лампочки – мы не делаем органических реакций в воде. В вот жидкий безводный фотористый водород – кислота очень сильная, близкая к 100%-ной серной (о том, что такое кислотность сильных кислот и что такое суперкислоты, можно почитать на страничке про кислотность), гораздо более сильная, чем остальные галогеноводороды. Если кому-нибудь придет в голову возражать, говоря, что HF получают действием обычной концентрированной серной кислоты на фториды, призываю не забывать про законы равновесий и про то, что HF – очень летучее вещество. Очень чистый жидкий фтористый водород, тщательно очищенный даже от следов воды и других примесей, как показал неутомимый исследователь сильных кислот Рональд Джиллеспи (тот самый, правилами которого мы пользуемся для предсказания геометрии молекул), даже еще более кисёл – он вполне котируется как суперкислота, почти такая же по силе как фторсульфоновая кислота, которая считается по кислотности начальной точкой множества суперкислот – все, что сильнее фторсульфоновой кислоты, называется суперкислотой (R.J.Gillespie et al, JACS, 1988, 110, 6053). Впрочем, и это тоже не очень важно, так как если его реально использовать, в нем придется что-нибудь растворить, и растворённое вещество испортит чистейшую жидкость, и рекордная кислотность все равно просядет от суперкислоты до просто очень сильной кислоты. В общем, можно спокойно считать, что жидкий HF по кислотности приблизительно эквивалентен серной кислоте.
Так что, если есть желание и возможность работать в жидком фтористом водороде – а это по-настоящему страшно, и требует специального оборудования и недюжинного экспериментального мастерства, – то в протонировании проблем не будет, олефины это точно запротонирует. Но дальше нам понадобится нуклеофил, а вот с этим в среде жидкого фтористого водорода всё очень скверно. Фторид-иона там нет совсем, вместо него там водородно связанные полифториды типа иона HF2–, а это очень слабые нуклеофилы, потому что для того чтобы из такого иона выдернуть фторид, придётся разрушить одну из самых прочных водородных связей и заплатить эту энергию. К тому же, даже таких ионов там очень мало – равновесие автопротолиза фтористого водорода имеет очень маленькую константу равновесия (константа равновесия автопротолиза, как мы знаем из химии воды, называется ионным произведением, и мы знаем эту величину для воды – так вот для HF ионное произведение на много порядков ниже). Если мы к этому месту уже неплохо разобрались с поведением карбокатионов, должен раздаться возмущенный возглас – ну и что?! – карбокатион ест любые и не разбирается: съест, наверное, и этот. Совершенно верно, съест, но всё же нуклеофильность этих ионов настолько мала, что конкуренцию им окажет сам олефин, и конкурентно пойдет полимеризация. Это очень похоже на проблемы в гидратации алкенов, которые мы уже разобрали – хотим получить спирт, а получаем олигомеризацию и перегруппировки. 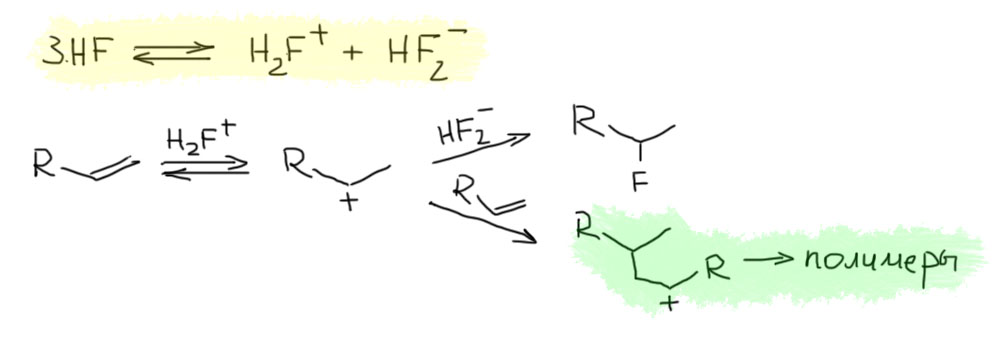
Эту реакцию попробовали сделать еще в 1938 году в США в лаборатории В.Н.Ипатьева, одного из крупнейших русских химиков, основоположника промышленной нефтехимии и катализа, успевшего ускользнуть в 1930 от почти гарантированной страшной гибели в годы террора. Он прожил очень долгую жизнь и до 1917 года был не только крупным ученым, но и целым генералом – и в 1930-х в ССР не избежал бы репрессий. Он эмигрировал в США, вспомнил там, что он профессиональный химик и наконец занялся своим призванием – исследованиями, основав там одну из первых R&D-лабораторий исследования промышленных каталитических процессов. Простые олефины (этилен, пропилен, циклогексен и др.) реагировали с жидким фтористым водородом под давлением в специально изготовленных автоклавах, образуя с небольшим выходом ожидаемые продукты гидрофторирования наряду с полимерами (J. Org. Chem., 1938, 3, 26). С этиленом, кстати, и HF реагирует медленно, и для достижение приемлемых выходов приходилось или греть до 90ºС (представьте себе смертельный эксперимент – смесь этилена и жидкого HF нагревают под давлением много часов!). И точно так же, как для других галогеноводородов, реакция ускоряется кислотами Льюиса, но это приводит только к увеличению доли полимеризации. Немного позднее так же превратили ацетилены с продукты присоединения двух молей HF. Понятно, что повторять такую химию в обычной органической лаборатории никто бы не решился, и так это и оставалось в забвении на десятилетия, а органики оставались в убеждении, что HF присоединять к кратным связям невозможно, не нужно и пробовать, если жизнь дорога.
Блестящая идея, как изменить условия и добиться хороших выходов пришла к главному знатоку кислот и электрофилов Дж. Ола (кстати, тоже беженцу от советского тоталитаризма, только венгерской его разновидности). Так получилось, что Ола взял у Ипатьева очень много и великолепно смог продолжить его исследования и довести их, кроме прочего, до Нобелевской премии. Главную проблему HF мы видим – у нее отличная кислотность, но в ней мало нуклеофила. Как повысить количество нуклеофила, не сильно провалив кислотность? Ответ прост и парадоксален, – нужно добавить основание, достаточно слабое (HF настолько сильная кислота, что все равно запротонирует) и в количестве недостаточном, чтобы депротонировать всю кислоту. Оставшаяся кислота обеспечит кислотность, а количество нуклеофила очевидно возрастет за счет депротонированной части кислоты. Так как HF – штука особенная, то депротонирование не дает фторид-ион, а дает вот те самые полифторид-ионы с фторами, взаимно связанными очень прочными водородными связями. При этом, заметьте, и кислотность все же уменьшается, остается высокой, но уменьшается. Это хорошо, потому что уменьшается скорость образования протонированного олефина, то есть карбокатиона. В чистой HF эта скорость была велика, а нуклеофила было мало, и карбокатиону ничего не оставалось, как реагировать с непрореагировавшим олефином и давать полимеры. В этой систем скорость будет меньше, а нуклеофила больше, и это благоприятно скажется на конкуренции с полимеризацией.
 Ола с сотрудниками перебрали несколько оснований и остановились на смеси пиридина и HF (30:70% по молям). Схема слева дает приблизительное представление о составе этой смеси, хотя в реальности вместо иона HF2–, в нем более сложные полимерные гидрофторид-анионы. Для этого тоже нужен чистый жидкий фтористый водород и оборудование для работы с этим ужасающим веществом, но после того как эта смесь сделана, она представляет собой вполне стабильную жидкость, с которой можно работать с разумными предосторожностями в очень простой и дешевой полиэтиленовой посуде. Более того, эту смесь можно просто купить готовую, что, во-первых, означает, что она спокойно хранится в течение длительного времени в самых непритязательных условиях (иначе никто не стал бы рисковать, предлагая его покупателям), а во-вторых, что им могут пользоваться без проблем в любой обычной лаборатории – это самый обычный реактив, и любой вменяемый химик с ним справится. И этот реактив под названиями пиридиния полигидрофторид или фтористый водород/пиридин, или реагент Олы (Olah’s reagent) стал очень популярен как такой заменитель HF, делающий для этого галоидного водорода рельными многие реакции по аналогии реакциями других галогеноводородов. Смысл этой загадочной фразы в том, что если вам известна какая-то реакция с бромистым или хлористым водородом, и вы ищете, как бы ее сделать в химии фтора, то брать нужно не сам фтористый водород, а именно реагент Олы, или, возможно, какие-то его более современные аналоги.
Ола с сотрудниками перебрали несколько оснований и остановились на смеси пиридина и HF (30:70% по молям). Схема слева дает приблизительное представление о составе этой смеси, хотя в реальности вместо иона HF2–, в нем более сложные полимерные гидрофторид-анионы. Для этого тоже нужен чистый жидкий фтористый водород и оборудование для работы с этим ужасающим веществом, но после того как эта смесь сделана, она представляет собой вполне стабильную жидкость, с которой можно работать с разумными предосторожностями в очень простой и дешевой полиэтиленовой посуде. Более того, эту смесь можно просто купить готовую, что, во-первых, означает, что она спокойно хранится в течение длительного времени в самых непритязательных условиях (иначе никто не стал бы рисковать, предлагая его покупателям), а во-вторых, что им могут пользоваться без проблем в любой обычной лаборатории – это самый обычный реактив, и любой вменяемый химик с ним справится. И этот реактив под названиями пиридиния полигидрофторид или фтористый водород/пиридин, или реагент Олы (Olah’s reagent) стал очень популярен как такой заменитель HF, делающий для этого галоидного водорода рельными многие реакции по аналогии реакциями других галогеноводородов. Смысл этой загадочной фразы в том, что если вам известна какая-то реакция с бромистым или хлористым водородом, и вы ищете, как бы ее сделать в химии фтора, то брать нужно не сам фтористый водород, а именно реагент Олы, или, возможно, какие-то его более современные аналоги.
Вернемся к олефинам. Реагент Олы присоединяет к двойной связи HF строго по правилу Марковникова, причем реакции идут в удобных условиях в разных растворителях (ТГФ, дихлорметан, пентан и т.п.) просто при перемешивании (Synthesis, 1973, 779). Вот несколько примеров. Выходы обычно хорошие, хотя и далекие от высоких, что говорит, что полностью устранить проблему конкурентной полимеризации не получилось, всё же нуклеофильность гидрофторидных анионов слишком мала, чтобы полностью выиграть конкуренцию. И это такой принципиальный момент, с которым совсем полностью справиться невозможно – протонирование-то нам все равно нужно. 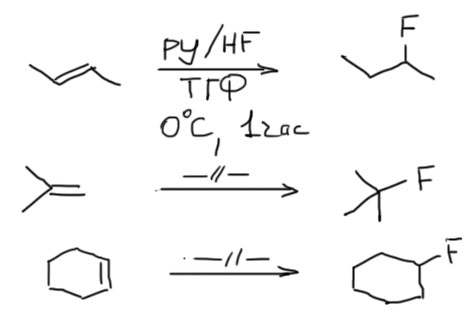
Хотя реагент Олы остается самым популярным, со времени его изобретения было предложено много аналогов, построенных на том же принципе – добавьте к HF немного основания Бренстеда-Лоури, и получите реагент, работающий как сильная кислота и одновременно источник более-менее нуклеофильного фтора. Самый последний писк этого направления можно увидеть в недавней работе шанхайского профессора Бо Сю, весьма плодовитого исследователя в этой области (B. Xu et al., J. Am. Chem. Soc. 2017, 139, 18202). Сю предлагает с одной стороны решение той же природы – замесите HF с каким-нибудь слабым основанием, но с другой стороны весьма неожиданное, потому что он в результате исследования широкого множества кандидатов в основание остановился на довольно парадоксальной вещи – кислом сульфате калия, KHSO4. Основность бисульфат иона это то же самое, что кислотность серной кислоты, и мы вроде бы уже выяснили, что HF серной кислоте по кислотности почти точная ровня, и тогда как же HF сможет протонировать бисульфат-ион? Да, вопрос есть, но Сю и его соратники уверяют, что бисульфат-ион все равно работает как акцептор водородной связи, так что в этой смеси тоже возникают те самые полимерные гидрофторидные анионы H(n-1)Fn–, и дает самый лучший реагент для присоединения HF к двойной связи, приводя огромное количество примеров, в том числе потрясающей селективности, например, не затрагивает тройную связь. 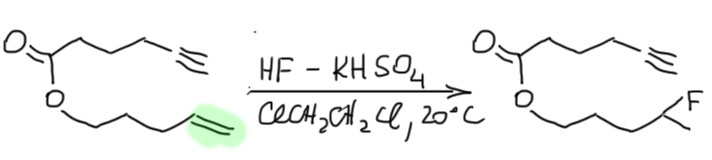
Так что возможно это гениальный реагент, новое слово в химии гидрофторирования. Одна проблема, и это типичная проблема для химии 21 века – новые реагенты испытывают и описывают для реакций на несколько миллиграмм, а реакции делают в малюсеньких пузырьках, определяя результат по спектрам, часто или прямо с реакционной смеси, или по данным автоматизированного анализа. Это позволяет делать огромные массивы испытаний реагентов, условий и субстратов, но не дает никакой гарантии, что когда эту реакцию потребуется реально использовать для какого-то ощутимого количества вещества и с выделением продукта реакции, вообще что-нибудь получится. Поэтому пока держимся классики, которой в этом деле является реагент Олы, но желающие могут пробовать современные идеи.
NBS - это свободнорадикальный или электрофильный реагент?
Правильный ответ – и то, и другое, причем когда мы говорим, что это свободнорадикальный агент, мы не имеем в виду самую знаменитую реакцию – аллильное бромирование по Волю-Циглеру, потому то, как мы уже убедидись, в этой реакции NBS вообще не является реагентом в полном смысле этого слова – алкен с ним вообще никак не соприкасается. Но свободнорадикальные реакции с NBS, как и другими N-бромамидами сделать вполне можно, потому что в этих реагентах есть слабая связь N-Br и ее можно инициировать. При этом мы получим страшно реакционноспособный и поэтому неселективный радикал с точкой на азоте. Такая химия известна, но применения почти не находит. Забудем.
Другое дело электрофильность. NBS и другие N-бромамиды, а также N-хлорамиды и в существенной степени N-иодамиды – вполне типичные электрофильные агенты в условиях отсутствия явного свободнорадикального инициирования – яркого света, излишнего нагревания, и инициаторов. С другой стороны, очень желательно что-нибудь, способное отдавать протоны хотя бы в виде водородных связей – протонные растворители типа спиртов, или даже просто немного воды. Это часто кокетливо называют сырыми (wet) растворителями, имея в виду, что примесь воды может быть небольшой и в некотором смысле даже случайной – в плохой лаборатории все растворители такие, как я люблю это называть “из помойного ведра”. Зачем нужны доноры протонов? Очень понятно. Это типичная история, с которой мы еще столкнемся сто раз – в помощь плохой уходящей группе. Возьмем любой такой агент, хоть NBS, хоть любой другой N-бромамид и обозначим его Z-N-Br. Вспомним, что электрофилы бывают сразу катионами, и это явно не тот случай, и нейтральными молекулами с ковалентной связью, имеющей способность разрываться не гомолитическим способом – по одному электрону на рыло с образованием свободных радикалов, а гетеролитическим с парой электронов, целиком уходящей к одному из атомов, и тогда группа, включающая этот атом, будет называться уходящей группой (подробнее мы это быдем рассматривать скоро в теме про нуклеофильное замещение). Это дико интересный кейс – пара уходит не обязательно к более электроотрицательному атому, даже скажем еще определеннее – способ разрыва связи и то, какой атом окажется электрофильным центром, не зависит от поляризации связи в исходной молекуле. Связи азот-галоген, например, в таких реакциях рвутся так, что пара уходит к атому азота, а не галогена, даже если галоген это хлор (и даже фтор!). Попробуем разобраться, как это работает.
Начнем с самой простой гипотезы. Если просто связь разрывать, то получился бы анион с минусом на азоте, а это очень плохие уходящие группы, даже при том, что мы здесь имеет дело с галогенпроизводными не аминов, а амидов, и минус на азоте стабилизирован делокализацией мезомерными акцепторами. И должен получиться при этом положительно заряженный галоген – катион Br+. Посмотрим на NBS с этой стороны:
Здесь всё очень плохо. Во-первых, уходящая группа плоха. Во-вторых, катион галогена, любого, хоть Br+ и даже I+– очень плохая штука, хотя это не так просто понять, почему – ведь дело не только в том, что отнять еще один электрон от сильно электроотрицательного атома должно быть непросто. Но есть и чисто квантовая проблема, состоящая в том, что у таких катионов 6 валентных электронов, секстет. Попробуйте разместить 6 электронов на одной s и трех p-орбиталях. С s-орбиталью все понятно, она заберет пару. А теперь размещаем оставшиеся 4 на трёх одинаковыхпо энергии орбиталях – они одинаковы, потому что у нас просто единичный атом, шарик, сферически симметричная частица, в системе координат которой все три измерения – x, y, z – одинаковы и неразличимы. Следовательно и три p-орбитали одинаковы и неразличимы. В общем об этом можно даже и не думать, а просто применить правило Гунда, одно из немногих положений квантовой науки строения атома, которое знают все. Оно говорит нам, что мы не имеем права положить две пары электронов на две p-орбитали и оставить одну свободной; вернее, имеем, но это будет вышележащее состояние. А основное состояние выглядит просто – размещаем сначала три электрона на трёх разных p-орбиталях, затем пристраиваем оставшийся на одной из них. Получаем частицу с двумя неспаренными электронами – триплет (мы скоро столкнемся с очень похожей историей, когда будем думать, как устроен карбен – частица с 6 электронами на атоме углерода, и разберемся, почему правило Гунда хорошо работает только для одноатомных частиц). Фокус в том, что при разрыве связи, как, например, на вышепоказанной схеме с NBS, слева ковалентная связь, электроны все спарены. А справа катион Br+, у которого, как оказалось, два электрона неспарены. Почему мы так решили? Потому что, когда мы просто пишем какую-нибудь реакцию, и над или под стрелкой написаны просто другие реагенты, растворители, нагревание или охлаждение, то такие реакции принято называть термическими (это означает, что реагенты берут энергию или отдают энергию, необходимую для того чтобы двигаться в сторону продуктов реакции, только из теплового равновесия с другими молекулами системы и больше ниоткуда – и таковы почти все известные нам реакции), и в таких реакциях не может происходить изменения спина электронов, в частности, спаренные электроны не могут вдруг не только распариться, но и получить два неспаренных электрона с одинаковым спином. По-другому это читается так: в термической реакции из синглета не может получиться триплет, и наоборот. Такие суровые высказывания принято и называть сурово – правилами запрета. В принципе, это почти законы Природы. Слово почти здесь означает, что нарушения запретов все же иногда случаются, но у этого всякий раз должна быть очень веская причина, и она должна быть найдена и разоблачена. В данной реакции ничего подобного не наблюдается, и запрет должен соблюдаться. Итак, мы решили, что просто развалиться на катион галогена и уходящую группу ни один ковалентный реагент не может (кстати, это касается и молекулы брома, и то, что в некоторых учебниках иногда можно увидеть дисоциацию брома на катион и анион, стоит понимать как неполное соотвествие конкрентного учебника функции надежного источника знаний).
Прежде чем пойти дальше, остановлюсь на висящем в воздухе вопросе – а как же гомолитический распад ковалентной связи на два радикала? Никаких проблем – кто сказал, что у образовавшихся радикалов одинаковые спины? Сразу после распада термическим способом (при инициировании нагреванием) два образовавшихся радикала имеют противоположные спины, и в сумме спин равен нулю. Но это никого не волнует, так как дальше каждый радикал имеет свою судьбу, ведет свою реакцию. В сумме по все молекулам системы – мы же никогда не забываем, что, когда мы говорим про реакции, мы никогда не имеем в виду одну молекулу, а только очень много молекул, вещество – спин тоже будет равен нулю. Если бы это было не так, то произошло бы чрезвычайное событие – в реакции возникал бы из ничего магнитный момент, а это совершенно невозможно, так как хорошо известно, что все моменты сохраняются: раз был ноль, значит и будет ноль.
Так, ладно, а какие еще бывают реакции кроме термических? Бывают еще фотохимические, в которых энергия берется из взаимодействия реагентов с электромагнитным излучением, с фотонами. Правила запрета действуют и для них, но поскольку там в дело замешаны возбужденные состояния молекул, то всяких фокусов там гораздо больше. Слава богам, мы не изучаем фотохимии на 3 курсе, а то пришлось бы нам совсем туго, ведь лезть в фотохимию без квантовой науки – курам насмех. Побережёмся.
Боже мой, это ж надо было так далеко уйти от самой обычной реакции! Ну да, паршиво получилось, извиняюсь, но где-то надо было это обсудить.
Итак, возвращаемся к NBS – у нас не одна проблема, а две. Нужно как-то улучшить уходящую группу, и одновременно уйти от необходимости образования Br+. Первое достигается водородной связью от донора протона – воды, спирта, карбоновой кислоты, что есть. Это позволяет уходящей группе смягчить горечь расставания, сделать вид, что она уходит не анионом, а сопряженной кислотой. Образующийся из донора протона анион, скорее всего, тут же пригодится как нуклеофил. От образования катиона уйти еще проще – представим себе, что разрыв ковалентной связи происходит не до взаимодействия с двойной связью, а одновременно, как мы уже научились говорить, согласованно. Получаем очередную разновидность того, что мы уже и так видели, когда обсуждали просто бромирование бромом и тоже обнаружили согласованные механизмы. (На схеме для простоты нарисован карбокатион, но в реальности это такой же мостиковый ион, симметричный или несимметричный, как в обычном бромировании).
Зачем всё это? NBS оказался электрофильным бромирующим агентом. А чем он отличается от просто брома? О, это очень важно. Второго атома брома у него нет, вот чем. Мы уже видели, что двойная связь реагирует с бромом в присутствии воды, образуя продукт сопряженного присоединения, бромгидрин. Но это только на бумаге так красиво. На практике это не так просто сделать, ведь обычная рекомендация взять бромную воду сильно попахивает допотопной химией эдак середины 19 века. Проблема в том, что бром очень плохо растворим в воде. И тем более плохо в ней растворимы олефины. Поэтому если вы смешаете бромную воду и олефин, в гораздо большей степени пойдет другая реакция: нерастворимый в воде олефин образует отдельный слой или капельки, если сильно перемешивать; в этот слой или капельки будет экстрагирован бром из бромной воды, потому что галогены неполярные вещества, лучше растворимые в неполярных жидкостях; ну и в этом слое или капельках пройдет обычная реакция присоединения брома с образованием дибромпроизводного. И даже если немного изменить условия реакции, подобрать растворитель, растворяющий и олефин, и бром, и воду, чтобы реакция стала гомогенной, все равно получим конкуренцию между обычным бромированием и сопряженным.
И вот здесь NBS – просто находка. Сам остаток сукцинимида, в принципе, тоже нуклеофил, но настолько слабый, что почти не конкурирует с другими нукоеофилами. Поэтому, NBS – чистый источник электрофильного брома для образования мостикового иона, а нуклеофил можно добавить другой, по вкусу. NBS – почти идеальный реагент для сопряженного присоединения брома и других нуклеофилов – воды, спиртов, карбоновых кислот – к двойной связи. Этот метод пользуется совершенно ошеломляющей популярностью – можно без труда найти многие сотни а скорее даже и тысячи примеров применения в реальных синтезах.
Когда хотят присоединить бром и воду, используют самые различные растворители, в которых растворима вода – ацетон, диоксан, ТГФ, ацетонитрил, диметилформамид, ДМСО и т.п., иногда в присутствии пары капель серной кислоты, чтобы NBS пошустрее шевелился: любой источник протона в этой реакции работает как кислотный катализатор, но понятно, что сильная кислота гораздо сильнее ускоряет реакцию, чем вода, спирты или карбоновая кислота. Иногда это нужно, но чаще нет. Реакции всегда стереоселективны (это анти-присоединение), а с двойными связями с разными заместителями реакция всегда идет так, как и должна идти в предположении, что бром – электрофил, а вода,
спирт или карбоновая кислота – нуклеофил, как, например, в этих примерах (разберитесь сами, почему получаются именно эти продукты): 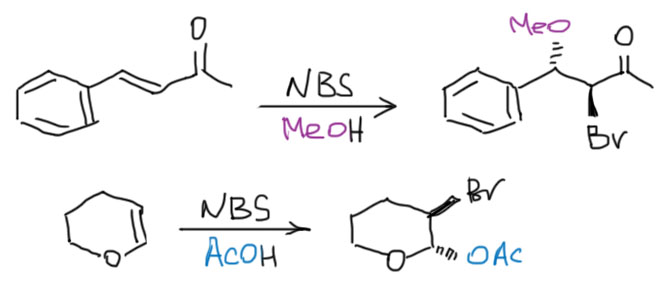
Эта реакция настолько гибка, что может использоваться даже в тех случаях, когда нуклеофил, спирт или карбоновая кислота не так просты. Например, учитывая меньшую реакционную способность тройной связи по сравнению с двойной, можно взять ацетиленовый спирт. 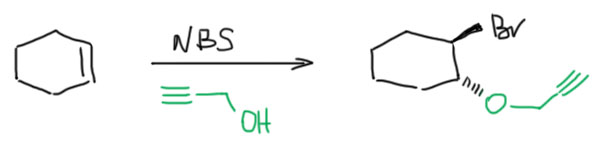
Реакцию можно провести настолько аккуратно, что она затронет только самую донорную, а значит самую реакционноспособную в эдектрофильной реакции связь, как, например, в таком удивительно селективном превращении природного триена мирцена (Zeitschrift fur Chemie, 1980, 20, 293). Кроме просто донорности может быть и другое объяснение, почему не затронуты другие двойные связи – они здесь образуют сопряженную систему диена, а это приводит к стабилизации, а стабилизация часто является причиной пониженной реакционной способности, типа, если и так всё хорошо, зачем суетиться, реагировать с каким-то NBSом. Выберите себе объяснение по душе – в органике часто так бывает, что явление можно вполне разумно и адекватно объяснить несколькими способами, и понять, какое из них верно, очень непросто, а часто и невозможно.
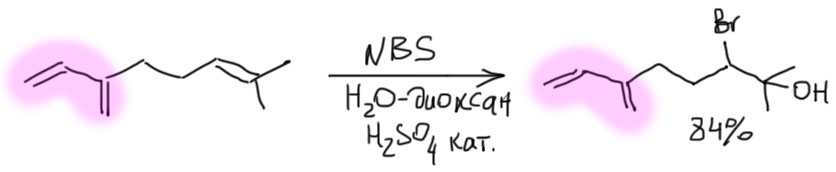
Реакция отлично идёт внутримолекулярно, когда нуклеофил находится в молекуле – получатся циклические простые или сложные эфиры. Циклические сложные эфиры кратко называются лактонами. При этом обычно получаются пяти или шестичленные циклы, так же как и в других внутримолекулярных реакциях. 
Второй пример может вызвать вопрос – откуда бром знает, что ему нужно садиться именно на этот атом углерода. В этом случае двойная связь практически симметричная и вопрос о стабильности карбокатиона не имеет однозначного решения. В первом примере всё ясно – карбокатион предпочтет сидеть под фенилом, а во втором примере такого эффекта нет вообще. Понятно, что второй возможный карбокатион не дал бы замыкания в цикл, но бром-то на двойную связь садится сначала, а нуклеофил атакует то, что при этом получается на второй стадии. Получается, что что-то (или кто-то) направляет реакцию… Мистика, однако. Никакой мистики, если мы все же не будем забывать, что карбокатион образуется в электрофильном бромировании только, если он очень хорошо стабилизирован, а если нет, то образуется мостиковый ион, а если двойная связь еще и симметрична по заместителям (не в геометрическом смысле, а в том смысле, что с двух сторон одинаковые или почти одинаковые по эццектам заместители) – то этот мостиковый ион это симметричный циклический ион бромония. Дальше нуклеофил просто раскрывает ион бромония (по механизму SN2 – скоро дойдем до этой фундаментальной вещи, а пока просто поверим), в данном случае внутримолекулярно, и это можно сделать только одним способом с образованием именно такого продукта. Не забываем про водородные связи, которые используются для активации NBS и вообще все это держат вместе. В этом смысле образование этого продукта можно рассматривать, как еще одно доказательство того, что электрофильное бромирование идёт через мостиковый ион. 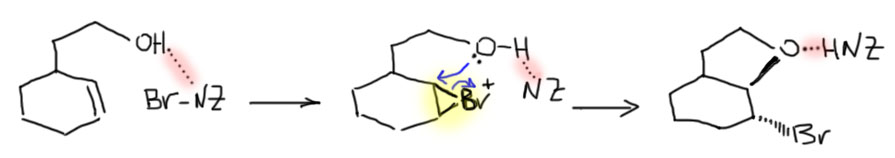
И в современной химии дело доходит до очень дерзких вещей. Нуклеофилом может быть и что-нибудь лихое, как например, в бромфторировании, или азидобромировании. Для бромфторирования нам понадобится фторид как нуклеофил плюс что-то протонное. Мы уже обсуждали тут рядом, как можно совместить эти две плохо совместимые вещи, и знаем, что решение предложил Ола, предлжив смесь безводного HF и пиридина. И если в эту смесь добавить NBS, то получим ровно то, что нужно – электрофильную активацию связи N-Br и источник нуклеофильного фторида. Собственно и это тоже предложил Ола, как впрочем и то же самое с иод- и хлор-аналоганми NBS (J. Org. Chem.1979, 44, 3872). Работает отлично, чистая стереохимия – только анти, и места присоединения фтора и брома полностью соответствуют нашим предсказаниям, например: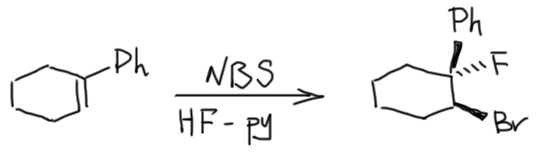
C азидом это работает еще проще, что совсем не удивительно, так как азид-ион – великолепный нуклеофил, не требующий никаких ухищрений. Могла бы быть проблема – нам ведь еще нужен протон, а в солях с азид-ионом протона нет. Но оказалось достаточно плеснуть немного воды. При этом проблем с конкуренцией с водой азид не испытывает – по нуклеофильности дистанция между водой и азид-ионом совершенно астрономическая – азид просто не замечает такого жалкого конкурента. Метод очень популярен и применялся и применяется очень часто, приведем самый простой пример:
Как видим, NBS – очень полезный электрофильный реагент, гораздо более удобный и разносторонний чем сам бром, потому что позволяет использовать очень много разных нуклеофилов в электрофильном присоединении. Всё это очень чисто, аккуратно, селективно – региоселективно (чётко выполняется основная схема – сначала присоединяется электрофил с образованием более устойчивого карбокатиона, затем к положительному углероду присоединяется нуклеофил); стереоселективно (получается только продукт анти-присоединения).