Ароматичность и антиароматичность настоящие и мнимые
В вопросе про тропон, трополон и малеимид заложено минимум две интересных, но часто игнорируемых проблемы. Во-первых, ещё раз пройдём то, что между ароматичностью и антиароматичностью нет симметрии. Это не отражения друг друга в зеркале. Это две принципиально разные истории. Ароматичность – это счастливая история довольных собой молекул, живущих полной и добродетельной жизнью на зависть окружающим. Антиароматичность – это наоборот история несчастных, искорёженных молекул, скрывающихся от всех и от самих себя, мечтающих свести счеты с такой подлой жизнью. Ароматических молекул миллиарды. Молекул, прикидывающихся ароматическими – ещё больше (и не надо здесь тыкать меня носом в известный чудной график, изображающий якобы количество известных органических соединений и говорить, что миллиарда там нет – это туфта, а не график). Настоящих антиароматических молекул вообще не существует ни одной. Молекул, имеющих проблемы с антиароматичностью, как человек может иметь проблемы с нездоровой тягой к просмотру телевизора, довольно много, но это всё равно максимум сотни или тысячи. Молекул, прикидывающихся антиароматическими, немного больше, вот только прикидываются они так топорно, что нет проблем их разоблачить, и отправить обратно к самым обычным, рядовым молекулам без вредных привычек и пороков.
Ароматические молекулы все одинаково ароматические – стабильны как бревно и только и ждут, когда их кто-нибудь пронитрует, потому что ничего интереснее они не умеют (это преувеличение, но не очень наглое). А вот антиароматические молекулы все антиароматичны по-своему, у каждой свой норов и своя жестокая судьба. Следить за ними очень интересно – почти как читать детектив про скрывающегося от правосудия закорнелого преступника, только вот они ничего плохого в жизни не совершали и преследованию подвергаются только по воле злого рока и Рональда Бреслоу, впервые осознавшего, что в хюккелевом счете электронов в 4n заложено какое-то древнее проклятие богов и героев. Прямо эдипов комплекс по-химически. Это интересно – число это следует из формулы Хюккеля, но это очень долго никто не замечал и не придавал значения, в отличие от числа 4n+2, за которое зацепились тоже спустя почти 20 лет после исторической работы Хюккеля, но тогда была война и всем было не до ароматичности. Только спустя ещё более десятилетия великий мудрец Рон Бреслоу заметил, что с молекулами, которым повезло огрести 4n π-электронов, что-то сильно не так, и придумал слово антиароматичность, невольно и создав эту, мудрёно выражаясь, дихотомию – и мы изучаем оба понятия как таких близнецов – родились в один день из формулы Хюккеля, но судьбы их разошлись давно и бесповоротно – кто-то отправился в сияющий мир жить-поживать да добра наживать, а кто-то мыкается с тех пор по притонам. С тех пор все мучаются – что делать с такими молекулами, которых вроде не должно быть, но которые есть, потому что молекулы не уступят в изобретательности тем, кто их исследует – и находят способ как-то приспособиться к странной жизни, представляющей собой непрерывное бегство от своего главного порока – намёка на то, что в одном из циклов может найтись четыре пи-электрона (все остальные цифры антиароматичности – 8, 12, 16 и т.д. уж совсем эфемерны, хотя при очень большом желании некоторые намёки на молекулы с таким счётом тоже можно найти).
В том же вопросе есть и вторая проблема, довольно занятная. Там через запятую перечислены тропон и трополон, и обе эти знаменитые молекулы названы ароматическими. В этом, пожалуй, кроется одно из самых устойчивых заблуждений в теме про ароматичность – путаница между молекулами, которые можно с полным правом назвать ароматическими, и совершенно неароматическими молекулами, в электронной структуре которых присутствует вклад (обычно небольшой) одной-двух граничных структур, удовлетворяющих самому формальному критерию ароматичности. Такие молекулы, в большинстве свойств совершенно обычные, соответствующие тем классам, к которым они структурно принадлежат, в некоторых реакциях проявляют особые свойства, напоминающие настоящие ароматические молекулы. Стоит разобраться в этом получше, чтобы не искать то, чего нет, там, где этого никогда не было. Но и уметь объяснять некоторые аномалии. Более того, осмелюсь предложить такую немного странную идею – именно такие молекулы часто интереснее настоящих ароматических. В каждой из таких молекул скрыта борьба между тем что мешает и что способствует ароматичности, и в каждом случае эта борьба дает уникальный результат со своим особым нравом.
Но я должен в этом месте явно обозначить возможность серьёзных расхождений между тем как разные люди трактуют ароматичность. Есть два основных подхода. Первый, и довольно распространённый, я бы его назвал количественным, работает в основном с разными количественными критериями ароматичности. Их много, и здесь мы не будем на них останавливаться. Это и теоретические величины ЯМР хим. сдвигов, и индексы альтернирования связей, и множественные расчётные оценки энергии стабилизации – в литературе можно найти не меньше десятка только основных таких критериев, и у каждого по несколько разновидностей. И тогда к любой молекуле применяют эти критерии, и если находят хоть небольшую величину пары-тройки этих критериев, говорят о наличии некоторой степени ароматичности (или антиароматичности). В таких работах можно увидеть огромные ряды молекул, выстроенные по этим критериям. Подход вполне понятный, обычно этим занимаются чистые теоретики, но мне категорически не нравится, потому что он, на мой взгляд, совершенно бесплоден – никакого толка от этих рядов никому никогда не было, а комизма ещё и добавляет то, что ряды, выстроенные по разным критериям, сильно различаются – и какой прикажете выбрать?
Моё и не только моё отношение к проблеме ароматичности и антиароматичности другое. Важно уметь разделять соединения с очень существенными признаками ароматичности, и только такие соединения и называть ароматическими. Такие соединения должны быть действительно особенными, их можно видеть по реальной структуре, свойствам, реакциям, а не только по какому-то значению абстрактного расчетного рараметра, часто вообще не имеющего никакого отношения ни к чему реальному. Коротко говоря – они должны иметь право воссесть одесную бензола на пиру молекул.
Отдельно мы должны уметь находить молекулы, которые по большинству свойств, структуре, реакциям нельзя называть особенными, сильно отклоняющимися от ожиданий для аналогов – в общем явно неароматических, но при этом имеющих некоторые особенности благодаря наличию в наборе граничных структур таких, которые соответствуют формальным критериям ароматичности. Эти особенности мы должны уметь заметить и объяснить. Назовём такие молекулы неароматическими с признаками ароматичности. Такой подход практичен, потому что активно использует реальные свойства, структуру, реакции. Именно такой подход успешно разрешает множество идиотских псевдо-парадоксов, которые очень распространены в этой химии, когда некое соединение называют ароматическим по первому подходу, но все, кто с ним работают, никак не могут наблюдать ничего из ожидаемых свойств и особенностей. И тогда начинается канитель с сомнениями – а нужна ли на эта ароматичность, если она ничего кроме бензола и ещё полддюжины известных систем не предсказывает, и всё время попадает впросак.
Краткое содержание
На первой вкладке мы вспомним про циклическую делокализацию пи-электронов как главное условие того, что нам имеет смысл рассуждать об ароматичности или антиароматичности. Мы узнаем как просто находить молекулы с циклической делокализацией, и как находить такие, в которых с этим большие проблемы. И разберём, насколько неприятным препятствием являются экзоциклические двойные связи. И сразу же проверим эти вещи на нескольких интересных молекулах, производных фульвена, и убедимся, что пересилить негативное влияние экзоциклической связи настолько сложно, что не проходят буквально никакие ухищрения, даже из весьма современных работ.
На второй вкладке мы вспомним историю тропона как соединения, с которого фактически и началась современная наука об ароматичности, и попробуем разобраться, насколько ароматическим является это культовое для ароматичности соединение. Возможно, нас ждут сюрпризы, как это часто и случается со всякими дурацкими культами.
На третьей вкладке мы, вдохновленные успехами по возвеличиванию и низвержению тропона с удвоенной силой возьмёмся за его не менее знаменитого родственника, трополон – и он проклянёт тот день, когда к нему привлекли наше нигилистическое внимание. Но сначала мы воздадим ему хвалу, и только на четвертой вкладке начнём курочить.
После четвертой вкладки мы окажемся на пепелище, и не найдя более ничего достойного ниспровержения в области ароматики, с той же дурью ревностью возьмёмся на пятой вкладке за антиароматику. И – не устоит!
На шестой же вкладке мы рискнём отправиться за новыми достижениями в совсем современную химию, и попробуем разобраться, действительно ли антиароматические признаки могут обещать интересную химию. Не буду портить интригу, но, как уже наверное стало ясно из общего настроя этой страницы, с достижениями возможны некоторые накладки. А ещё мы немного познакомимся с тем, как делается современная наука, и возможно, слегка озадачимся. Но у меня нет для вас ни другой химии, ни других химиков, и более того, и мы сами, в лучшем случае, такие же – жаждущие открытий и достижений, и вместо них часто являющие миру нечто почти комическое – и стесняться тут нечего.
Циклическая делокализация превыше всего
Без циклической делокализации нет ни ароматичности, ни антиароматичности
Начнём с начала и вспомним некоторые основы. И сразу поставим все это обсуждение на хорошую основу. Основой и ароматичности и антиароматичности является циклическая делокализация.
Ароматичность (и антиароматичность) могут проявляться только у молекул имеющих циклическую делокализацию π-электронов. ТО есть именно циклическая делокализация первична, ароматичность и антиароматичность это следствия. Если нет циклической делокализации π-электронов, то нет и поводов искать ароматичность или антиароматичность. Была однажды попытка построить такую же историю на циклической делокализации σ-электронов, то есть электронов на σ-связях циклов, но из этой попытки ничего существенного не вышло, хотя это было довольно забавно. Итак, нас интересуют циклы с π-электронами, и мы уже знаем, что это такое – такие электроны водятся на кратных связях, на атомах с парами электронов, и ещё на секстетных атомах, которые предоставляют пустые орбитали (дырки) для делокализации. А как узнать, есть ли в молекуле циклическая делокализация π-электронов. Мы все это обсуждали уже, но напомню: должен быть цикл, на каждом атоме цикла или π-электроны или дырка для них, цикл должен быть плоским (точнее насколько возможно плоским), и это не очень сложное условие, особенно в небольших циклах, потому что π-электроны (или дырки для них) водятся на атомах sp2-гибридизации, такие атомы имеют плоскую тригональную геометрию, и из таких атомов проце всего составить что-то более-менее плоское (можете сами попробовать поиграться с модельками).
Но этого недостаточно. Нужно еще как-то убедиться, что делокализация относится именно к циклу, что это циклическая делокализация. Мы можем очень легко понять, что это такое, если вспомним про обычную делокализацию, где мы знаем, что лучше всего это работает в неразветвлённых цепях сопряжения. Но тогда цикл сопряжения это не что иное как цепь сопряжения, согнутая в кольцо. И опять, как мы знаем, лучшая цепь сопряжения, дающая наибольшую стабилизацию, это цепь между донором и акцептором. Но в циклическом сопряжении у цикла нет начала и конца, поэтому нужно искать новый принцип для наибольшей стабилизации – и мы знаем, что этот принцип давно найден, и это и есть ароматическое число электронов Хюккеля. А как нам легкo найти циклическое сопряжение в молекулах? О, это совсем просто.
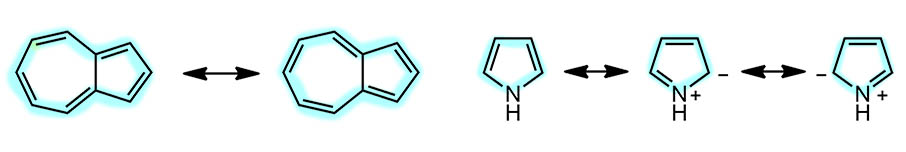
Ключик нам дал сам Фридрих Август Кекуле, который конечно же ничего не знал ни про электроны, ни про сопряжение. Зато придумал для самого главного ароматического соединения, бензола, замечательные формулы, которые с тех пор так и называют структурами Кекуле. После прошло много времени, возникла куча всяких теорий, от Хюккеля до всего-всего, но мы с удовольствием пользуемся именно структурой Хюккеля, для химии, конечно, чтобы реакции писать, но и для того, чтобы было удобно рисовать граничные структуры в тех случаях, когда в кольце бензола есть мезомерные доноры или акцепторы. Из этого мы делаем вывод, что эти структуры отлично передают особенности делокализованной системы. Это хорошее свойство удачной символьной системы – несмотря на очень высокий уровень абстракции, она точно передаёт главную особенность сопряженной системы и то как она служит для передачи взаимодействия. Но есть у этих формул и еще одна приятная возможность применения. Зададим себе вопрос – а что это? Это граничные структуры бензола? Во многом да, хотя есть и возражения (потому я не поставил обоюдную стрелочку), но главную роль граничных структур они улавливают вполне, показывая делокализацию и усреднение. В данном случае усреднение полное – каждая связь это среднее между двойной и простой, потмоу что две структуры равновероятны. И еще – они показывают, что сопряжение установилось (какой глагол использовать со словом сопряжение – поскольку это не действие и не процесс, то нельзя говорить: сопряжение установилось, сопряжение действует и т.п.; сопряжение это результат конкретного расположения атомов и взаимодействия валентных электронов в данной геометрии молекулы) и оно целиком в цикле – участвуют только атомы цикла. Тогда мы думаем, а нельзя ли структуры Кекуле использовать для того, чтобы легко распознавать циклическое сопряжение? Ответ положительный – можно, и это удивительно простой и эффективный способ. В разных разделах теоретической химии это называют немного по разному, мы не будем в этом копаться, тем более, что никакого общеупотребительного способа обозначений выработано не было, но просто констатируем: чтобы распознать цикл с циклическим сопряжением (не ароматичностью! а в более широком смысле) нужно попробовать представить его структурами Кекуле, обычно их будет две, иногда три, поэтому работа сильно непыльная, и если получится, то мы видим цикл с циклическим сопряжением. Вот для примера, пиррол и азулен. Пиррол специально чтобы мы не забывали, что мы можем и должны использовать все элементы сопряжения, и не только кратные связи. В азулене видим внешний цикл (можете ещё поиграться и убедиться, что ни 7-членный, ни 5-членный по отдельности не имеют автономного циклического сопряжения, потому что ни при каких обстоятельствах вы не можете загнать двойную связь на общую между кольцами; а вот в нафталине это делается без проблем)
Этот не просто простой, а издевательски простой приём отлично работает и позволяет искать циклическое сопряжение в самых разных молекулах. Циклическое сопряжение мы ищем для того чтобы следующим этапом подсчитать электроны в найденном контуре сопряжения и определить, что за число – ароматическое или антиароматическое (или нечётное, но это большая загвоздка – что делать в таком случае, проще всего ничего не делать). Чаще всего мы конечно быдем находить ароматические контуры, и рапортовать об ароматичности данной молекулы по причине того, что в ней есть ароматический цикл (или несколько таких).
Возьмём другой случай – когда в молекуле есть экзоциклические кратные связи. Фозьмём фульвен и пара-хинодиметан. Фульвен – вечно многие сомневаются, как в нём считать электроны. Ведь на каждом атоме есть π-электроны. Тогда в цикле пять – фигня какая-то. А если с внешним то 6, так у некоторых и получается, что фульвен ароматичен. Но давайте начинать с начала – есть ли в фульвене циклическое сопряжение? Пробуем погонять кратные связи в цикле, чтобы получить Кекуле-структуры. Э, чёрт, никак не получается сделать так, чтобы не участвовала эта экзо-связь, то ее туда сместить, то сюда. Но так нельзя – раз не получается гонять двойные связи и пары/дырки только в цикле, значит это не тот вид сопряжения.
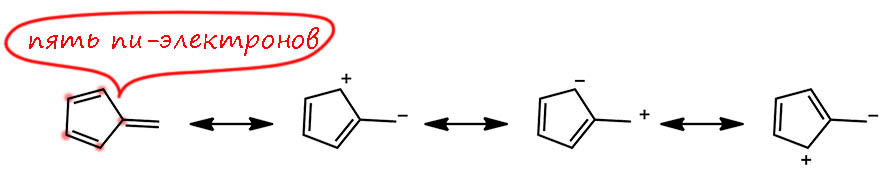
Здесь сделаем остановку и чётко заявим – мы не хотим сказать, что фульфен – несопряженная система! Ещё какая сопряженная, и все атомы углерода участвуют в сопряжении. Но именно это нам и не нужно. Это важный момент – нужно всегда оставаться в рамках той теории, котораяи породила понятие. Ароматичность предполагает циклическое сопряжение. Это непосредственно следует из теории Хюккеля, и всех последующих уточнений этой теории. В основе этой теории так называемые аннулены – то есть как раз модели циклического сопряжения. Если мы добавляет экзоциклические связи, мы разрушаем циклическое сопряжение. И вспомним обычное сопряжение – там тоже нельзя взять и присобачить сбоку что-то с возможностью сопряжения – мы сразу из обычного сопряжения получаем кросс-сопряжение, которое намного сложнее и часто бывает даже дестабилизирующим. Проще не трогать, чем разбираться.
А вот хинодиметан – прототип хиноидной системы. Опять в цикле можно насчитать 6 электронов и возрадоваться – ароматическое! Но вновь поищем циклическое сопряжение, погоняем двойные связи. Смотрите – там в серединке прямо бензольное колечко нарисовалось. И мы ведь еще не все структуры нарисовали. Разве мы не помним мантру – чем больше граничных структур, тем более лучче! Э, да мы эту мантру давно разоблачили как дерьмовый миф. Нужно не только считать структуры, но и оценивать их вклад. И если структур хоть сто, но все они дерьмовые, то и проку от них ноль.
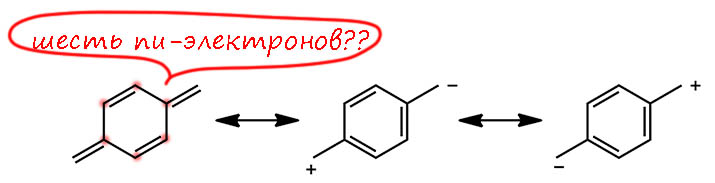
Как же могут быть дерьмовыми структуры с бензолом в серединке?! Почему я так часто стал употреблять производные от слова “дерьмо”? Потому что оно точно соответствует принятому сегодня Государственной Думой Российской Федерации Федерального Собрания Российской Федерации Закону Российской Федерации о чистоте Русского Языка Российской Федерации от чуждых слов нероссийской нефедерации. Слово дерьмо исконное, происходит от древнерусского глагола дерти, в современном языке это превратилось в драть, но немного сузило смысл, в древнем языке он более широкий, можно даже сказать, что это один из важнейших глаголов нашего древнего языка, от которого и произошёл современный язык Российской Федерации, бережно сохранивший исконное слово и его производные нам на радость. Кстати, от того же глагола и дрянь. Будем пользоваться этими исконными словами почаще, точно не нарушим Закон Российской Федерации, и как-то разбавим изобилие заимствованных слов из недружественной номенклатуры ИЮПАК, которую я предлагаю впредь писать маленькими буквами, вот так – июпак – пусть знают своё место, дряни такие.
Так вот, вполне могут быть дерьмовыми граничные структуры с бензолом в серединке. Сейчас разберемся, только сначала скажем, что вновь у нас нет циклического сопряжения, потому что мы не можем гонять двойные связи внутри, не затрагивая атомы снаружи. А теперь – почему структуры никуда не годящиеся? Всегда, когда мы рисуем структуру, мы оцениваем ее выгодность. Вот в середине у нас бензольное кольцо образовалось – это выгодно, мы знаем из книжек и лекций оценку выигранной энергии, что-то около 40 ккал/моль. Но одновременно у нас на одном конце анион, на другом катион. Это дает очень плохой вклад в энергию. Чтобы образовать такие вещи, нужно затратить по около 100 ккал/моль на разрыв связей C-H и еще на ионизацию радикала, хотя это копейки, как и превращение радикала в анион. Но ясно, что это очень высоко по энергии, а значит структуры эти плохие, и вклад дают минимальный, хоть их и 4 штуки. Вы наверняка скажете – откуда такие оценки, ведь никто не рвал связи C-H. Конечно не рвал, но нам надо оценить на глазок относительную энергию граничной структуры, и мы прикидываем то, что даёт существенный вклад в стабилизацию (бензольное кольцо) и дестабилизацию (карбанион и карбкатион) по доступным нам процессам, пренебрегая всем небольшим. Можно нарисовать более точный термохимический цикл, можете сами потренироваться – и увидите, что как ни рисуй, дестабилизация на образовании ионов будет нехилой. Кто-то другой скажет – но вот они и образуют так любимую нами цепь сопряжения донор-акцептор. Э нет, не забывайте, что здесь речь идет не о настоящих веществах, а о граничных структурах, а выгоду от сопряжения мы уже отыграли, сравнивая эти формы с исходным хинодиметаном.
Итак, пора сделать одно обобщение. Экзоциклические связи разрушают циклическое сопряжение. Если вам кажется, что двух структур мало, поищите примеры, когда вам удастся в системе с такой связью (хоть алкеновой, хоть карбонильной, хоть иминовой) получить циклическую делокализацию.
А поскольку экзоциклические связи разрушают циклическую делокализацию, то циклы с экзоциклическими связями не являются ароматическими или антиароматическими. Это просто непредельные соединения с соответствующими конкретной структуре свойствами. Но возникает следующая проблема, неразрывно связанная с этим утверждением. Переходим.
Итак, пусть некая циклическая молекула не имеет циклического сопряжения и не является ароматической. Но среди граничных структур, описывающих делокализацию в такой молекуле, есть такие, которые были бы ароматическими, если бы это была не граничная структура, а настоящая молекула. Что это значит?
Вернёмся к фульвену. Забыли про молекулу в целом и просто нарисовали одну из граничных структур. И взяв уже эту структуру за исходное, спокойно нарисуем структуры Кекуле, соответствующие циклической делокализации. Вот они все, очень хорошо соотвествующие тому, что мы нарисовали бы для циклопентадиенил-аниона, в ароматичности которого не усомнится даже сумасшедший. Единственное но, – у нас немного странный заместиетель – карбокатион. Но он же наверное отлично стабилизирован соседством с таким превосходным донорным центром как циклопентадиенил-анион – это же как бензильный катион но наверное, во много-много раз более лучче, как нынче принято говорить, ведь это должно быть превосходным мезомерным донором.
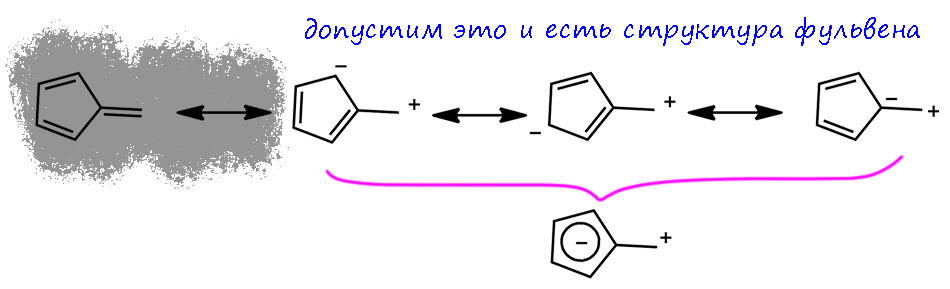
Но – это не работает. Мы уже замечали, что в граничных структурах при обсуждении их относительной выгодности, нужно очень осторожно относиться к электронным эффектам – индуктивные эффекты всегда работают и должны учитываться, а вот мезомерные весьма коварны, потому что когда вы рисуете граничные структуры, вы уже все их учли, ведь вы ровно тем и занимаетесь, что разрисовываете мезомерный эффект. Еще раз повторим – граничные структуры это не настоящие молекулы, это просто символический язык отображения делокализации пи-электронов. И надо дико осторожно относиться к рассуждениям, основывающимся на какой-то с виду весьма привлекательной граничной структуре.
В химии очень важно не забывать никогда, что это экспериментальная наука. И все умозаключения должны строиться на основе экспериментальных данных. Вот вам в голову пришла какая-то мощная мысль, и вот, она уже вами овладела, и вы это уже не вы, а демон/демонесса, одержимый/ая идеей, и собаки лают, не признав в вас привычного хозяина/хозяйку, как в популярном детском стишке С.Я.Маршака. Остановитесь, покормите собак, успокойте их и успокойтесь сами – и спокойно проверьте идею, попробовав, как она соотносится с уместными экспериментальными данными. И если не соотносится, выбросьте её к чёртовой матери, это дрянь, а не идея. Нет ничего плохого в представлении фульвена показанным способом… кроме того, что это не соответствует эксперименту и всем известным свойствам фульвена. В первую очередь тому, что если бы это было правдой, мы бы имели симметричный пятиугольник, может быть слегка искаженный эффектом сильного заместителя. Как у бензола – какие бы мы заместители не навешивали, длины связей отклоняются от тех, что в самом бензоле, очень мало, на пару-тройку единиц во втором знаке. Но у фульвена связи по длине почти точно соответсвуют обычной формуле метиленциклопентадиена – где в ней двойные, там в реальной структуре короткие связи. А гле простые – там длинные. И разница огромна (двойная приблизительно 1.34 Å, простая приблизительно 1.44 Å – это с учетом того, что углероды sp2, и сопряжение есть). Мы уже отлично поняли, почему так происходит – ароматическая стабилизация не перевешивает дестабилизацию, связанную с образованием карбокатионного центра.
Хорошо, но мы отлично знаем, что нужно делать, если стабильности какого-то карбокатиона не хватает. Нужно его еще стабилизировать. Это мы умеем – навесить туда мезомерных доноров, да не жадничать. Навесили два амина, и это, конечно, очень сильно стабилизирует катионный центр. Получили действительно интересное соединение. Во-первых, у него громадный дипольный момент, больше 5 D, намного больше чем у самого фульвена (1.1 D), что говорит о намного большем разделении зарядов, но – и о том, что положительный заряд уехал дальше (в дипольный момент входит и расстояние между центрами зарядов). Во-вторых, это соединение реагирует с хлористым железом, образуя ферроцен с забавными формамидиновыми заместителями в виде соли с тетрахлорферратом(2+), что вроде как говорит о том, что цикл там уже в виде циклопентадиенильного аниона (U. Mueller-Westerhoff, Tetrahedron Lett. 1972, 13, 4639–4642). Но! – посмотрите на длины связей в кольце – они крайне слабо отличаюься от длин в самом фульвене, буквально на 0.01 Å. Что-то непохоже это на циклопентадиенильный ароматический анион. А вот длина экзоциклической связи сильно вытянута – связь явно ослаблена, она больше не совсем двойная (хоть и до простой еще далеко). Но это легко объяснить по-другому. Из-за сопряжения весь внешний фрагмент плоский, и метильные группы стерически наезжают на атомы водорода кольца – это разворачивает две части молекулы друг относительно друга, хоть и на небольшой угол (29°) и соответственно ослабляет сопряжение, что безусловно играет на увеличение веса граничных структур с разделением заряда (“ароматических”).
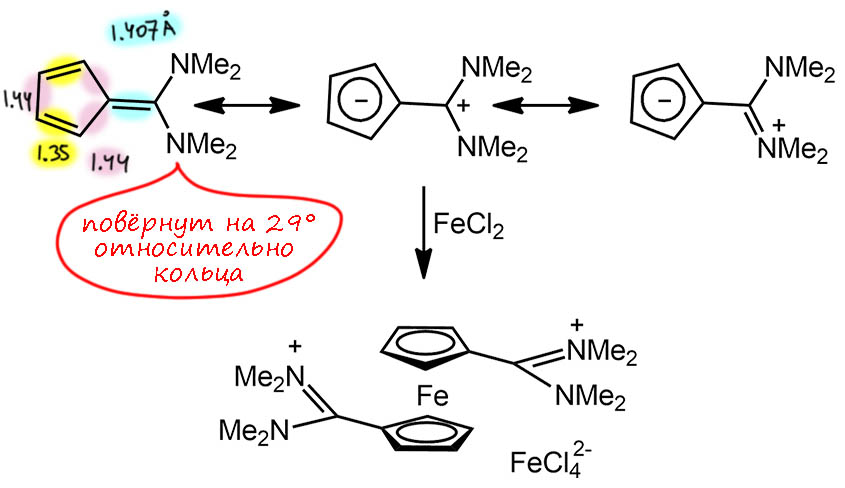
Делаем вывод: даже такие мощные донорные группы не могут изменить природу фульвеновой системы. Вес граничной структры, несомненно, вырос, но мы не можем считать, что такая структура стала основной и доминирует. А ферроцен откуда? Это плохой аргумент, потому что в реакции играет роль энергетика взаимодействия, и образование комплекса с переходным металлом вызывает смещение электронной плотности в сторону кольца и смещает равновесие в сторону очень выгодной ферроценовой системы.
Хорошо. Химики – ребята упорные, и в последнее десятилетие снова нашлись желающие дальше раскачивать фульвен в сторону ароматической структуры. Если не получилось аминами (а не получилось), то ищем нечто еще более стабилизирующее плюс. Находим другую ароматическую систему, и делаем так, что нужная нам граничная структура даст не одно, а два ароматических кольца. Кажется, что игра беспроигрышная – в такой ситуации не должно быть проигравших. Вот эта система, построенная из фульвена и имидазолина немецкой исследовательницей Дорис Кунц с сотрудниками (Kunz, D.; Johnsen, E. Ø.; Monsler, B.; Rominger, F. Highly Ylidic Imidazoline Based Fulvenes as Suitable Precursors for the Synthesis of Imidazolium-Substituted Metallocenes. Chem. – Eur. J. 2008, 14, 10909−10914) В этой молекуле достигнута дополнительная степень выравнивания длин связей (1.37 Å – двойная, 1.42 Å – простая) – и это уже солидно, хотя альтернирование продолжает быть весьма заметным. В более определенных структурах, где циклопентадиенил точно соотвествует ароматическому, например, в знаменитом илиде Рамиреса (трифенилфосфониоциклопентадиениле), разница длин намного меньше (1.38-1.40 Å). Такое дополнительное выравнивание и смещение в сторону ароматической структуры, не очень радикальное по вравнению с просто диамином, можно отнести к дополнительному скручиванию двух колец и-за той же стерики – система более жесткая и отталкивание сильнее, хотя и не драматически. А скручивание уменьшает перекрывание и фульвеновая двойная связь ослабевает, становится почти простой (32 градуса недостаточно для сильного ослабления перекрывания, поэтому назовем ее полуторной), соотвественно, уменьшается влияние экзоциклической связи, и получается более близкое соответствие циклической делокализации, а следвоательно и ароматичности в полном смысле этого слова. Но всё же, признав это, признаем и то, что мы, наверное, ожидали еще более сильного эффекта и полного разделения зарядов в кольцах.
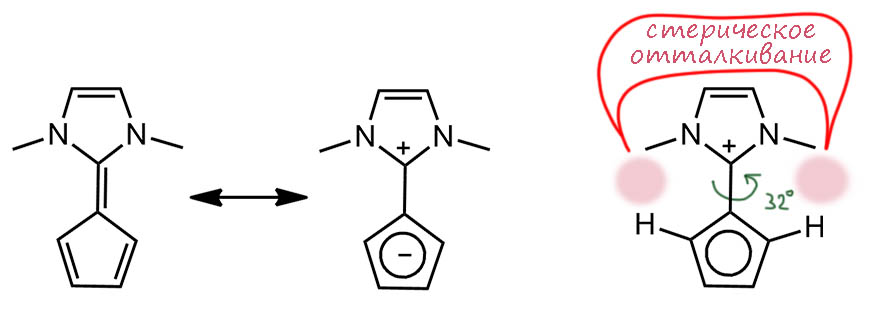
В этом месте нельзя не обратить внимание. что надежда на ароматичность верхнего кольца вряд ли обоснованна. Да, там формально 6 электронов (две на двойной, по паре на азотах и дырка на среднем углероде). Но там вряд ли есть циклическая делокализация. Мы не можем нарисовать структуры Кекуле так, чтобы были задействованы все атомы цикла. Вся верхняя часть остается не у дел. Простые связи никогда не становятся двойными. Верхняя двойная – простой. Нет там никакой ароматичности, и шесть электронов ничего не дают. Нас самом деле, это очень известная история, связанная со стабильными гетероциклическими карбенами, где вечно идут дискусси об ароматичности, которой нет. Но мы это сейчас оставим, просто заметим, что если бы здесь действительно было бы два ароматических кольца, то их стабилизация бы точно передавила бы всё остальное и мы видели бы намного более выровненную систему, похожую на настоящие циклопентадиенильные илиды.
Вывод из этой стории с фульвенами можно сформулирвоать так – влияние экзоциклической двойной связи на нарушение циклической делокализации столь сильно, что его не удается до конца снять даже очень сильными эффектами, благоприятствующими разделению зарядов. Строго говоря, даже последнюю молекулу лучше назвать неароматической со значительными признаками ароматичности. Сам фульвен – неароматическая молекула со слабыми признаками ароматичности. Пока есть хотя бы остаток экзоциклического пи-связывания, нет и полноценной циклической делокализации.
Историческая молекула и её маленькие секреты
В 7-членном ряду настоящая ароматичность представлена катионом тропилия. Для истории ароматичности это совершенно культовая молекула (ион). Дело в том, что ароматичность конечно неразрывно связана с Эрихом Хюккелем и с его несколькими теоретическими статьями, опубликованными в 1930-х. В этих статьях Хюккель сделал несколько фундаментальных вещей. Во-первых, он отделил π-связи и π-систему от всех остальных связей в молекулах (тогда не было разговора о переходных металлах, поэтому все многообразие связей в молекулах можно точно разделить на пи и сигма, и ничего не забыть). Во-вторых, он предложил очень простой способ обращения с молекулярными орбиталями, доступный уму химика, а не специального квантового физика или крутого математика, причем этот способ предполагал расчет на листке бумаги, не требовавший математики за пределами решения квадратных уравнений. Но – только с молекулярными орбиталями π-системы. В-третьих, он с помощью этого метода нашел общее решение для циклических молекул с общей π-системой (аннуленов) и получил общую формулу для уровней энергии в таких системах. Из этого он нашел что наибольшая стабилизация (то есть, полное заполнение низколежащих связывающих МО) должны получаться для 6 электронов в π-системе аннулена. Поскольку Эрих Хюккель был чистым физиком и в химии разбирался слабо, он спросил своего старшего брата Вальтера, отличного органика, что он знает про такие системы и получил ответ, что эта теория наконец объяснила особые свойства бензола, над которыми человечество ломало голову сто лет. И еще под ту же теорию подпадал открытый Йоханнесом Тиле в самом начале 20-го века циклопентадиенильный анион – Тиле обнаружил, но не мог объяснить необяснимо легкое депротонирование циклопентадиена основаниями, отчего этот диен вёл себя как стандартная метиленовая компонента в конденсации с кетонами. Хотя из той же формулы следовало, что и другие числа электронов могут дать высокую стабильность, этого тогда никто н заметил. Да и других необычных молекул с секстетом электронов тогда не было. Брат Вальтер тем не менее замыслил написать учебник органической химии нового типа, в котором он бы использовал теории своего брата для обяснения свойств органических соединений – и это стало бы революцией в химии. Но вместо революции случилась Вторая мировая война, всем стало не до теорий, и вся история ароматичности подвисла на 20 лет; Вальтер Хюккель таки написал учебник, но самый обычный, про соединения и свойства, и с квантовой наукой ничего не вышло.
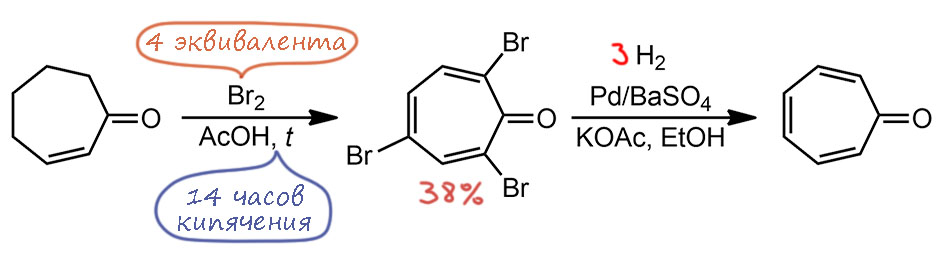
И именно тропон стал тем соединением, которое перезапустило исследование ароматичности. В 1951 году в джаксе выходят подряд две коротенькие статьи, настолько короткие, что у них даже ссылка с одной страницей. Первая так и называется “Тропон” и сделал эту работу молодой американский химик Хип Даубен, который и дальше будет много заниматься ароматической химией (H. J. Dauben, H. J. Ringold J. Am. Chem. Soc., 1951, 73, 876). Вообще-то тропон был известен, потому что химией тропановых алкалоидов очень много занимались большие немецкие химики в конце 19 века – среди этих алкалоидов, как известно, и атропин, и кокаин – два знаменитых вещества с богатейшей историей и настоящим. И это не только очень интересные вещества, но и прямо как по заказу для незатейливой ранней химии – весьма простые, хотя в них есть пара забавных загадок, которые потребовали много времени и усилий самых крутых химиков типа Вильштеттера. И тропон уже получали, длительной процедурой присоединения-отщепления. Но что это такое толком не поняли. Даубен сделал тропон довольно странным образом, который тем не менее многго говорит о том, что это необычное соединение. Сначала он долго нагревал циклогептенон с избытком брома в уксусной кислоте – и неожиданно получил трибромтропон. Попрбуйте сами представить, как это могло быть – очевидно, что происходили многократные присоединения-отщепления, и замещения-отщепления. Выход небольшой, поэтому баланс по брому сойдётся, если попробуете сами представить, как это получилось. Второй стадией Даубен выбрал гидрогенолиз, причем на катализаторе Линдлара, чтобы избежать простого гидрирования, но тщательно следя за объёмом поглощённого водорода – ровно на трех реакцию остановили. Когда через несколько лет это аккуратно повторял японец Нодзоэ, выяснилось, что при гидрировании получается много продуктов в том числе и затрагивающих двойные связи кольца, а тропон просто один из продуктов, но как это часто бывает, такие симметричненькие, полярненькие, плоскенькие, ненасыщенненькие молекулки гораздо лучше пакуются в кристаллы и первыми вываливаются из реакционных смесей, даже если это далеко не самый главный продукт. Так или иначе, Даубен получил тропон и прославился.
Мы из этого видим, что система тропона обладает немалой способностью сохраняться и воспроизводиться в реакциях. Такое поведение мы обычно приписываем как раз ароматичности. Далее было сделано много реакций, хотя и без деталей. Вяснилось, что придействии сильных кислот образуется соль, но структура ее определена не была. С солью диазония образуется некий желтый продукт вроде бы азосочетания, но опять без структуры. А вот раствор марганцовки моментально обесцвечивался, указывая на обычную ненасыщенность. И бром без нагревания просто присоединялся. В общем, было ясно, что соединение занятное, и был сделан намек, что это имеет какое-то отношение к поляризации карбонила и достижению электронной конфигурации, которая была сравнена с бензолом. Даубен действительно был одним из первых органиков, который увлекся квантовыми представлениями и первыми попытками применить теорию молекулярных орбиталей. Еще одну вещь нашел Даубен – вроде бы тропон не дает обычных производных карбонильных соединений (оксима, динитрофенилгидразона), но этот вывод через несколько лет опровергнет один из главных исследователей химии тропона японец Тэцуо Нодзоэ, нашедший, что если не торопиться и работать аккуратно, то из тропона нормально получаются и фенилгидразон, и динитрофенилгидразон, и семикарбазон, и только с оксимом всё немного сложнее из-за таутомерии и побочной реакции, превращающей оксим в аминотропилиден.
Итак, вышла статья Даубена, который получил тропон и отметил его особые свойства, осторожно связав это с аналогией с бензолом, и аккуратно используя термин “aromatic character” – ароматический характер. Но сразу за этой статьей на той же странице идёт статья Уильяма Дёринга и Френсиса Дитерта (28-летний сотрудник Дёринга – ветеран Второй мировой, командир военного катера и участник кровавой битвы с японцами за остров Иводзима, если когда-нибудь видели фильм Письма с Иводзимы Клинта Иствуда) которая называется … Оксид циклогептатриенилия (W. von E. Doering and F. L. Detert J. Am. Chem. Soc., 1951, 73, 876). Я не зря помянул воинскую судьбу соавтора этой исторической работы, с которой начался интерес к ароматическим свойствам тропона и его производных и вообще от них к ароматичности – основное развитие эти исследования в последующие годы получили как раз в Японии в лаборатории Тэцуо Нодзоэ, одного из первых крупных японских химиков. Япония только возрождалась после страшной войны, которую сама же и начала, и война эта была совершенно невероятно жестокой – обе стороны, и американцы и японцы вцепились друг в друга с какой-то первобытной яростью, рвали и дубасили всем, что попадется под руку – морские сражения с участием десятка авианосцев, ковровые налеты бомбардировщиков, сражения за острова до последнего солдата – американцы не отказали себе в удовольствии именно на японцах попробовать ядерное оружие, лишний раз отомстив за вероломный налет на Перл-Харбор, развязавший войну. Казалось, что такая ненависть никогда не может быть избыта, а жертвы не отомщены, и вражда продлится, пока существует хоть один человек с каждой из сторон. Но вот нет – человек так к счастью устроен, что, закончив драться, быстро остывает – желательно при этом еще и осознать, что в пылу драки был утрачен человеческий облик и надо его как-то восстанавливать, а это невозможно и без покаяния, и без сострадания к недавним врагам, и без желания загладить вину, а вина всегда есть даже у безусловно правой стороны в любой войне, – ведь война быстро превращает благородный гнев в звериную ненависть.
Итак, статья Дёринга и Дитерта называлась “оксид циелогептатриенилия”. А что это? А ежу понятно, что это тот же тропон, только записанный в виде структуры с разделением зарядов и ароматическим кольцом. Скандал? Скандал! Две статьи подряд явно говорият про одно и то же соединение, но называют его по-разному. А куда смотрел редактор? Пропустил. Посчитал хорошей научной интригой? Молодец, именно так, публикуя острые и даже спорные статьи по самым важным темам, джакс и стал тем журналом, опубликоваться в котором – достижение само по себе. Он ведь в начале был обычным скромным национальным журналом по химии, ничем не отличавшимся от других национальных журналов по химии. Времена, когда рулили немецкие журналы, прошли (а крайне успешный проект раскрутки Ангевандте ещё далеко впереди). Первенство перешло к журналам Королевского общества – в Британии в середине века завелось много ученых первого ряда и они публиковали в своём национальном журнале чрезвычайно важные работы. Первенство у них пришлось выдирать, на это ушло много времени, но начало было положено как раз в 1950-х, когда джакс стал рисковать, публикуя спорные статьи. Вот это как раз отличный пример.
Почему Дёринг (отдав должное воинской доблести молодого Дитерта, оставим его в покое, он дальше ничего в химии не сделал – науку здесь делает его руководитель Дёринг) так назвал статью, а значит и вещество? Потому что он предложил другой синтез, а предложил он другой синтез потому что именно в это время он приступил к самой главной теме в своей выдающейся карьере – химии карбенов. Он выяснил, что при фотолизе диазометана образуется чрезвычайно реакционноспособная частица, которую он назвал карбеном (правда, он же потом рассказал, что слово было случайно придумано кем-то из троих: им самим, Вудвардом или Уинстейном, когда они весело болтали в ночном такси, выдвигаясь на какую-то конференцию), и что эта частица легко присоединяется к бензолу и его производным, образуя норкарадиены, самопроизвольно перегруппировывающиеся в семичленные циклогептатриены. Когда то же провернули с анизолом, был сразу получен метоксициклогептатриен. Его можно прогидролизовать, как любой эфир енола. И вот что интересно – и получившийся кетон и само это метоксипроизводное очень необычно реагируют с бромом – образуется необычный продукт, который содержит бром в виде ионного бромида, то есть как противоион. Очевидно, что из обоих получается сначала 2-бромпроизводное и выделяется HBr.
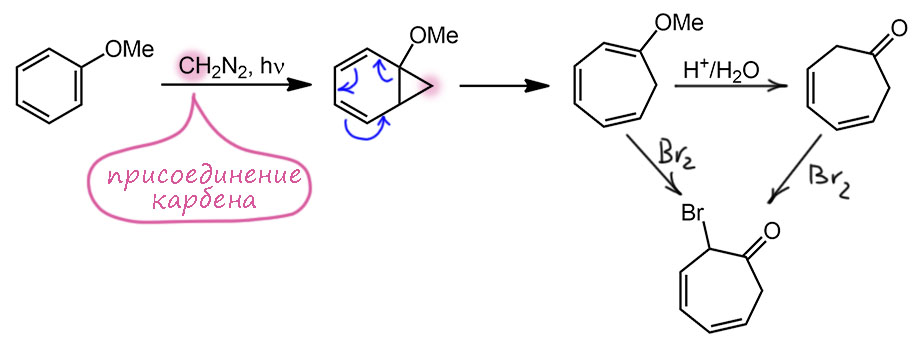
Но этот с виду банальный бромкетон не выделяется, а самопроизвольно элиминирует бромид. Для этого нужно основание, и что здесь является основанием? Видимо, сам продукт этой реакции, поэтому Дёринг и получил именно протонированную форму. В этом случае элиминирование выгодно, потому что создает сопряженную систему (как мы сейчас скажем – с некоторым вкладом ароматической стабилизации), поэтому достаточно основности самого тропона. Сейчас мы еще раз вернемся к этой детали, она занятна. Пока отфиксируемся, и заодно отметим ещё один прикол: мы знаем альфа, бета, гамма-элиминирования. Но здесь расстояние от протона до уходящей группы намного больше, дальше чем дельта и эпсилон – это дзета-элиминирование (дзета ζ ). Итак, при действии брома был получен ионный бромид протонированной формы, которую Дёринг определяет как бромид гидроксициклогептатриенилия (слово “тропилий” будет придумано тем же Дёрингом через 3 года). А из этой соли действием слабого основания, водного бикарбоната, Дёринг получает тот же тропон, только называет его не так (слово только что придумал Даубен), а оксидом циклогептатриенилия, что очевидно – в его трактовке из гидрокси получается окси.
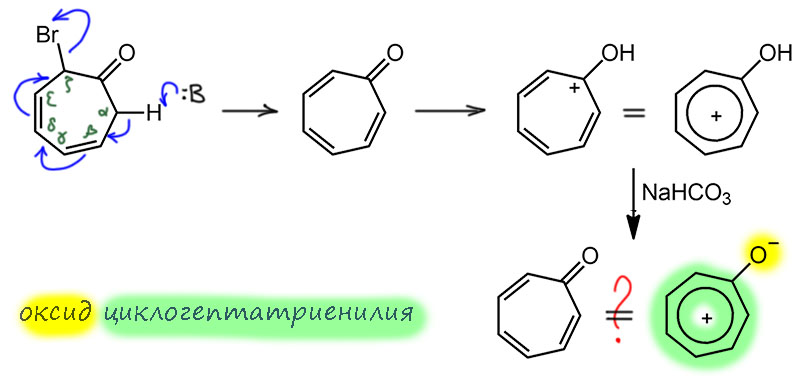
Проследим за ходом мысли Дёринга, учитывая, что на дворе 1951 год, еще жив товарищ Сталин, а из спектроскопии уже в солидных местах есть УФ и ИК, но до ЯМР гораздо дальше, чем до смерти товарища Сталина. Дёринг обратил внимание на необычную лёгкость протонирования и образования соли из – будем называть это как Даубен и мы сами теперь – тропона. На уменьшенную частоту колебания карбонила в ИК, свидетельствующую об ослаблении двойной связи (хотя доводов о том, что это ослабление столь сильно. что это вообще больше не двойная связь у него не было, да и вообще, в приведенном спектре – а тогда приборы были очень несовершенны, вместо настоящего спектра они выдавали отдельные точки, которые потом просто от руки соединялись плавной линией). И, что особенно забавно, на то, что тропон очень полярен, – потому что!!! – он кипит при намного более высокой температуре, чем изомерный ему бензальдегид. Вообще, это хороший, хотя и очень косвенный аргумент, потому что причина более высокой температуры кипения может быть и другой – надо гораздо точнее представлять себе межмолекулярные взаимодействия, чего тогда и в помине не было.
Важнейшим выводом в статье была довольно неожиданная вещь – именно там появилось священное выражение 4n+2 (в статье 2+4n). Еще раз напомню, что Хюккель вывел общую формулу для циклических полиенов, но заинтересовался только одним частным решением – шестью электронами. То, что из той же формулы следуют и другие числа подметил впервые именно Дёринг, опубликовав общее выражение для “правильных” чисел, хотя и его заинтересовала тоже только шестёрка.
И в этот момент неизвестен ещё и сам незамещённый циклогептатриенилий, производным которого Дёринг считает и сам тропон и продукт протонирования тропона. Ну ему и карты в руки. И действительно, тропилий был получен именно Дёрингом, но тремя годами спустя. В 1954 году умер товарищ Сталин и родился тропилий. Вернее, скажем так, тропилий первый раз родился ещё полвека назад, в 1901 году бромид тропилия получил довольно известный немецкий химик Георг Мерлинг, но не понял, конечно, что он получил, только описал, как при нагревании продукта присоединения брома к тропилидену (так назывался циклогептатриен) вдруг неожиданно происходит отщепление HBr и образуется кристаллическая масса, что весьма необычно – получалось, что вещество при нагревании кристаллизуется. Тогда статью забыли на полвека, а вспомнил о ней Дёринг потому что уже хорошо знал о поведении при присоединении брома тропона. Повторение эксперимента Мерлинга показало, что тот был прав, но теперь можно понять что это – и это бромид циклогептатриенилия, который был тут же торжественно окрещён тропилием. Сама химия проста, и очень нам понятна. Реакцию с бромом рисуют как 1,4-присоединение, хотя это делается из общих соображений, но это правдоподобно. Продукт более-менее очищают и затем просто нагревают в вакууме, чтобы выделяющийся бромистый водород улетал, смещая равновесие первой стадии – E1-элиминирования. Как только отщепилась первая молекула HBr, получается бромциклогептатриен, существующий только в виде тропилиевой соли – он просто ионизируется, мы прямо впервые воочию видим то, что мы постулировали для SN1/E1-реакций – спонтанное расщеление связей углерод-галоген с образованием ионов. Для всех обычных ионизующихся галогенпроизводных, способных на SN1/E1-реакции это происходит в равновесиях с образованием мизерных количеств карбокатиона в виде ионных пар. А вот здесь благодаря бесспорной ароматической стабилизации тропилия, равновесие количественно смещается, и это же значит,Ю что такой карбокатион как тропилий не реагирует с довольно сильным нуклеофилом бромидом. Это было крутое открытие в 1954 году, и это бесспорно одна из самых важных работ в истории исследования ароматических соединений и вообще самого эффекта ароматической стабилизации. С этого момента все как с цепи сорвались – работы пошли косяком, новые молекулы пачками, бедного Дёринга чуть не затоптали, но поскольку у него было второе отличное занятие – карбены, он как-то справился и не пропал.
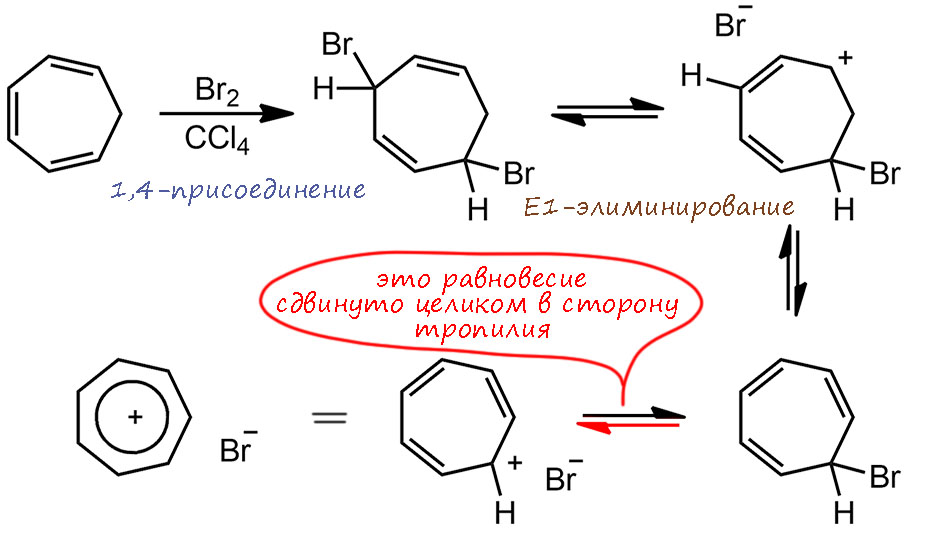 Смотрим на тропилий. Это совершенно симметричный правильный плоский семиугольник. И у тропилия множество производных, сохраняющих этот семиугольник, естественно с некоторыми искажениями, точно так же как в ряду бензола уже у толуола длины связей не точно равны, начинается небольшой разброс, альтернирование, но в очень узких пределах. Тропилий отлично представляется структурами Кекуле, правда их понадобится аж семь штук, чтобы плюсик смог побывать везде. Поэтому как и в случае с циклопентадиенил-анионом, и здесь вполне уместна гайка.
Смотрим на тропилий. Это совершенно симметричный правильный плоский семиугольник. И у тропилия множество производных, сохраняющих этот семиугольник, естественно с некоторыми искажениями, точно так же как в ряду бензола уже у толуола длины связей не точно равны, начинается небольшой разброс, альтернирование, но в очень узких пределах. Тропилий отлично представляется структурами Кекуле, правда их понадобится аж семь штук, чтобы плюсик смог побывать везде. Поэтому как и в случае с циклопентадиенил-анионом, и здесь вполне уместна гайка.
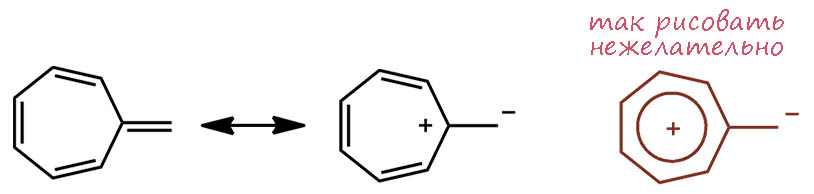
 И точно так же в этом ряду возникает проблема производных с экзоциклической двойной связью. Есть и фульвен – здесь фульвены называются гептафульвенами, и они тоже имеют небольшой вкалад аротаической граничной структуры, оставаясь неароматическими молекулами с сильным альтернированием длин связей. Гептафульвены тоже много исследовали, не так много как самые главные фульвены, которые для справедливости переименовали в пентафульвены. Гептафульвены поляризованы в противоположную сторону в сравнении с обычными фульвенами. И та же аналогия подсказывает нам, что гептафульвен – неароматическое соединение, у которого есть граничные структуры ароматического типа, но из этого нельзя сделать вывод о наличии полноценной циклической делокализации. Иными словами, гептафульвен нежелательно представлять структурой с гайкой и разделением зарядов. Ещё раз повторю почему нежелательно – потому что это вводит в заблуждение относительно реальной структуры. А почему так рисуют – потому что коротко, и по причине общей неразборчивости.
И точно так же в этом ряду возникает проблема производных с экзоциклической двойной связью. Есть и фульвен – здесь фульвены называются гептафульвенами, и они тоже имеют небольшой вкалад аротаической граничной структуры, оставаясь неароматическими молекулами с сильным альтернированием длин связей. Гептафульвены тоже много исследовали, не так много как самые главные фульвены, которые для справедливости переименовали в пентафульвены. Гептафульвены поляризованы в противоположную сторону в сравнении с обычными фульвенами. И та же аналогия подсказывает нам, что гептафульвен – неароматическое соединение, у которого есть граничные структуры ароматического типа, но из этого нельзя сделать вывод о наличии полноценной циклической делокализации. Иными словами, гептафульвен нежелательно представлять структурой с гайкой и разделением зарядов. Ещё раз повторю почему нежелательно – потому что это вводит в заблуждение относительно реальной структуры. А почему так рисуют – потому что коротко, и по причине общей неразборчивости.
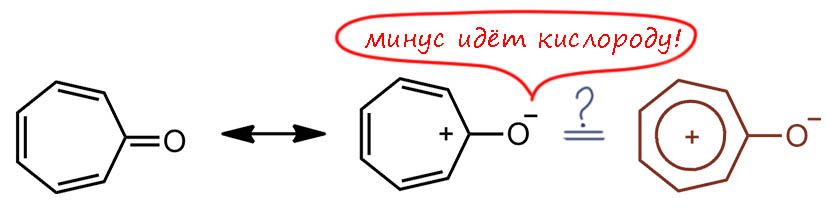
Но теперь мы можем поиграть в ту же игру, что и с обычным фульвеном – попробуем стабилизировать форму с разделением зарядов. На внешнем атоме теперь надо стабилизировать не катион, а анион, для чего нужны акцепторные заместители – но есть же способ намного лучше – зачем нам акцепторы, если мы можем туда просто атом кислорода поставить, уж куда акцепторнее, и получиь оксо-аналог гептафульвена, а это и есть знаменитое вещество, с которым в начале 20-го века возился Вильштеттер, тропон. Это вещество очень легко образуется из тропановых алкалоидов типа атропина, откуда собственно все эти названия (тропон, трополон, тропилий и т.п.) и идут. В тропоне экзоциклическая связь это карбонил, и уже кажется, что может быть очевиднее – такой карбонил наверняка поляризован в сторону кислорода, а вещество поэтому представляет собой такой фенолят тропилиевого ряда – если тропилий рассматривать как бензол семичленного ряда, то так оно и должно быть. Для тропона поляризация выглядит очень привлекательно, даже кажется, что это натолько очевидно, что нет смысла долго мусолить.
Но химия – наука экспериментальная, и как бы нам не казалась очевидной какая-то идея, если её не удаётся проверить в эксперименте, то она не работает. Посмотрим, можно ли действительно тропон представить как фенолят от тропилия, и настоящее ароматическое соединение. Сначала вспомним, как устроен сам тропилий. Это классический хюккелевский [7]-аннулен, точно соотвествующий условиям Хюккеля – симметричный, правильный многоугольник. И молекулярные орбитали пи-системы (их относительно расположение на шкале энергии соотвестует и формуле Хюккеля и визуализации этой формулы – кругу Фроста, или правильнее, Фроста-Мусулина, нельзя же забывать болгарского дипломника Бориса Мусулина, который и нарисовал этот круг на своей дипломной работе, как задание сделать наглядной довольно громоздкую формулу Хюккеля для энергий молекулярных орбиталей [n]-аннуленов). Все орбитали, кроме нижней, полносимметричной, дважды вырождены, как и долно быть для молекулы, имеющей ось симметрии порядка больше двух. Вот они и нарисованы, справа символически, просто обозначая фазы p-орбиталей, соответствующие областям связывания и разрыхления. Слева, как они получаются из расчета (здесь это, просто для справки, DFT B3LYP/STO 6-311+(d,p), но это совершенно неважно, так как картинки орбиталей и их относительное расположение мало зависят и от базиса и от метода расчета) – вид сверху, по сравнению с символическим – тот сбоку. Конкретные энергии нас совершенно не интересуют, а порядо, как видим, вполне соответсвует ожиданиям Хюккеля (хотя в круг они перстают вписываться сразу, как только мы отходим от самого упрощенного из всех разновидностей методов МО, метода Хюккеля – но порядок тот же). Кому-то покажется, что символическая картинка и расчетная непохожи. Но это не так – приглядитесь внимательнее, это просто одно и то же, мы просто в символической форме не пытаемся изобразить “реальные” формы орбиталей, а просто обозначаем цветом фазы (или знак). И не обозначаем веса атомных орбиталей в конкретных МО – это тоже совершенно неважно, особенно для вырожденных, которые можно линейно комбинировать в своей паре любым способом – это будет влиять на веса, но не на симметрию (чередование фаз или цветных лепестков).
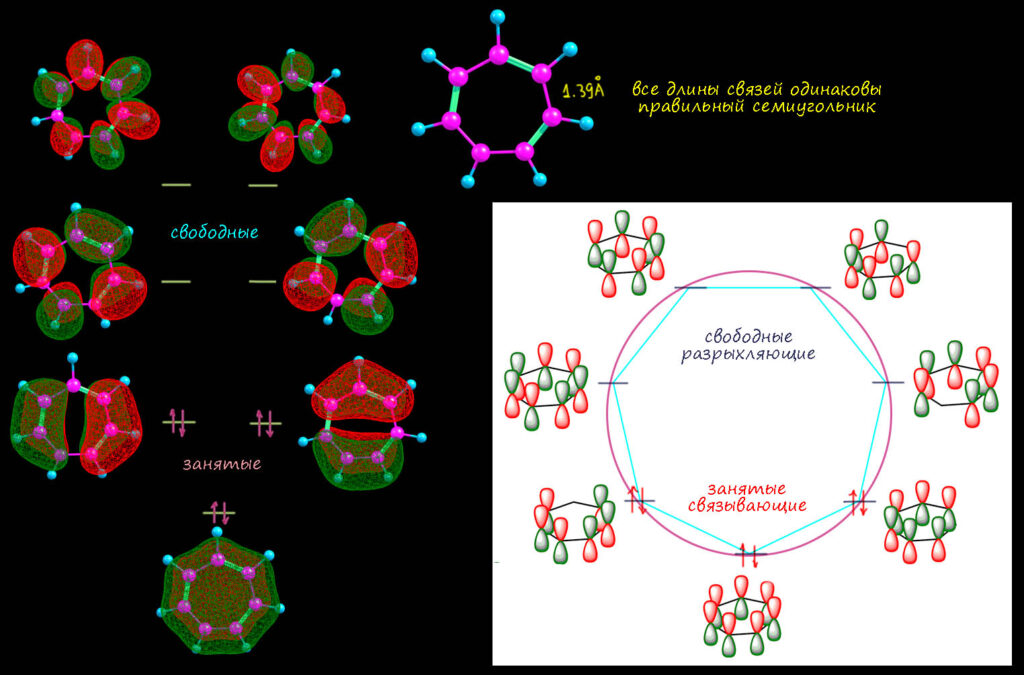
Что нам говорит эта картинка? Где здесь ароматичность? Она нам говорит, что в такой системе есть циклическая делокализация пи-электронов – об этом говорит то, что теория Хюккеля, описывающая такие системы, дает правильные выводы о строении пи-орбиталей и их расположении, и этот вывож подтверждается на любом уровне теории, где еще есть молекулярные орбитали. Уже не сама теория Хюккеля, а небольшая надстройна над ней, из того же выводит и кольцевой ток, а это, как мы знаем, основа самого удобного критерия ароматичности. И второе, не менее важное. В такой системе высшая занятая орбиталь (она не обязательно вырождена, потому что любой заместитель в таком кольце нарушит вырожденность, но не изменит принципиально больше ничего – просто вместо вырожденных пар будут те же орбитали, они будут рядом, одна из них станет чуть выше, другая чуть ниже, но вид их (чередование фаз) не изменится) – всегда связывающая (как об этом судить? – у связывающей орбитали могут быть узловые поверхности, где орбиталь меняет фазу (цвет). И что особенно важно – разность энергий высшей занятой и низшей свободной (тоже орбиталь того же хюккелева набора) – HOMO-LUMO gap (щель ВЗМО-НСМО) значительна по энергии (там есть еще дополнительно несоответсвие этих орбиталей по симметрии, отчего переход становится запрещённым). Мы прямо на глаз это видим, и в этом нам помогает круг Фроста-Мусулина. Вот эта большая разница (и несоотвествие по симметрии) делают дял стабильности таких систем больше, чем всё остальное. Почему – отдельная история, пока оставим, просто усвоим это как важный вывод из теории Хюккеля (а вы будете наверняка смеяться, но в ароматичности ничего лучше никто не придумал, чем этот великий ученый и скромный немецкий профессор, чуть не сгинувший во времена нацизма, но сохранивший и совесть, и достоинство).
Берём тропон. Тропон, это как мы теперь понимаем, такой фульвен, гептафульвен, с заменой внешнего CH2 на сильно акцепторный атом кислорода, что, по нашим представлениям, должно сильно способствовать увеличению веса граничных структур с разделением заряда до такого, что позволит нам говорить о возникновении циклической делокализации и ароматической структуры. Наверняка, такая структра будет отлично видна из молекулярной структуры. Что мы должны увидеть? Во-первых, сильное удлинение связи C-O, ведь она у нас теперь не двойная карбонильная (которая кстати даже в ацетоне довольно сильно поляризована), а настоящая простая, как в феноляте. Мы должны видеть выравнивание длин связей в цикле, может не ровно до тех, что в симметричном тропилии, но что-то в пристойной степени напоминающее. И сам цикл станет близок к правильному семиугольнику-гептагону. Смотрим на расчетную геометрию (метод тот же, и это очень высокий уровень, расчетные геометрии молекул на таком уровне можно считать близкими к реальности с точностью до 0.01 Å и долей градуса; где есть экспериментальная геометрия по рентгену, я проверяю и убеждаюсь в этом). Для сравнения возьмем дигидротропон, молекулу с точно разорванной циклической делокализацией, но в остальном – очень хороший аналог, ведь мы не должны забывать, что помимо возможной ароматичности здесь точно есть сопряжение карбонила с двойными связями. Ба-бах!!
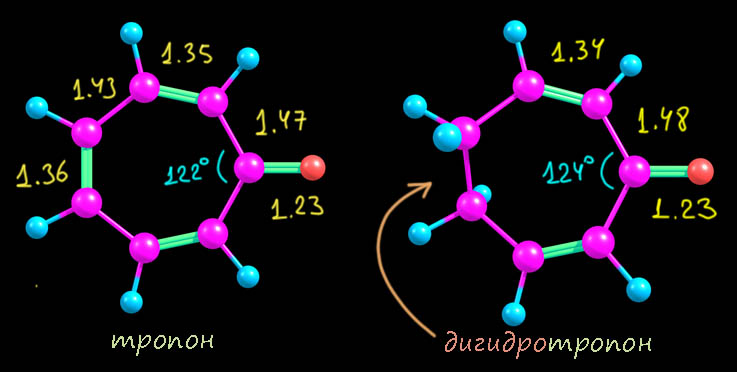
Ой, какая незадача! Это называется, найди пять отличий. Что-то ни одного не видно. А видно то, что связи в цикле альтернированы настолько, что вообще нет вопросов, кто из них двойная, а кто простая. Еще видно, что карбонильная группа точно такая, какая должна быть в еноне, и что карбонильный углерод упорно настаивает на чистой sp2-гибридизации, так чтобы угол был почти ровно 120° и плевать ему на то, что там хочет цикл – перебьётся, подправит свои остальные углы (цикл плоский и школьную теорему про сумму углов многоугольника никто не отменял). На мой взгляд, ловить тут нечего. Очевидно, что никакой ароматичности тут нет, а вклад структур с разделением зарядов мизерен, и в общем это самый обычный фульвен – непредельное соединение, а еще это непредельный сопряженный кетон и все свойства должны соответствовать, например, эта штука должна реагировать с нуклеофилами, и будет там еще и проблема региоселективности; ничего нам это не даст, поэтому мы не будем тратить на это время. Кто-то скажет – а ЯМР, разве это не лучший способ увидеть кольцевой ток! Увы, нет. Это хорошо работает для бензола, но для всяких структур с гетероатомами, мы теряем почву под ногами, потому что там есть химсдвиги, обусловленные не ароматичностью, а обычными эффектами гетероатомов и заместителей. Тропон – это енон, а в енонах сопряжение с карбонилом сдвигает олефиновые протоны к 7 м.д. и даже дальше без всякой ароматичности. Поэтому в современной химии используют не реальные химсдвиги, а расчетные, причем не для ядер в молекуле, а для воображаемых ядер, находящихся над кольцом и не связанных с кольцом – получается величина, называемая nuclear independent chemical shift (NICS) – химсдвиг, независимый от ядра. Этот параметр для тропона (конкретно NICS(1)zz) посчитали и нашли величину -5 мд. Это очень маленькая величина, показывающая какую-то мизерную ароматичность, вполне соотвествующей малому вкладу “ароматических” граничных структур.. Просто сравните – для бензола это -29 мд, и даже для фурана, в ароматичности которого все сомневаются это -27 мд. Для настоящего тропилия это даже больше, чем для бензола (точнее меньше, потому что ароматический сдвиг отрицателен), что вполне соответствует тому, что цикл немного больше по размерам, ведь мы не забываем, что кольцевой ток непосредственно берется из аналогии ароматического кольца и рамки с током в магнитном поле. Любопытства ради рассмотрим молекулярные орбитали пи-системы (опять приведены расчетные и рядом их символические картинки). Здесь уже восемь атомов участвуют в этой системе, поэтому соответствующих МО тоже восемь, чемыре занятых, четыре вакантных. На самые верхние вакантные наплюём, они не нужны вобще ни для чего на свете полезного, оставим 4 занятых и две свободных. Уже видим одну вещь – здесь восемь электронов, и по виду МО мы никак не можем понять, как бы нам удалось поделить их на 6 в цикле и два на кислороде – пи-система едина, и это еще раз говорит о том, что здесь нет условий для циклической делокализации. Это такой типичный случай кросс-сопряжения. И по виду орбиталей мы не видим хорошего соответствия орбиталям пи-системы тропилия. Нет, и по МО это особая непредельная система, которую нет никакой возможности рассматривать с точки зрения ароматичности. Ну и последняя деталь – сильное уменьшение щели ВЗМО-НСМО (у тропона 4.16 эВ, у ароматического тропилия 5.7 эВ – это говорит о том, что система тропона существенно дестабилизирована, скорее всего, тем самым кросс-сопряжением, и это еще один довод против ароматической стабилизации). А то, что это именно дестабилизация можно дополнительно убедиться, если посмотреть на электронную структуру дигидротропона, у которого разорвано сопряжение в цикле: МО мы смотреть не будем, но ограничимся тем, что щель ВЗМО-НСМО у этой системы намного больше, чем у тропона – 4.9 эВ против 4.16 эВ. Чуть ниже мы ещё раз к этому вернёмся, потому что это занятное явление.
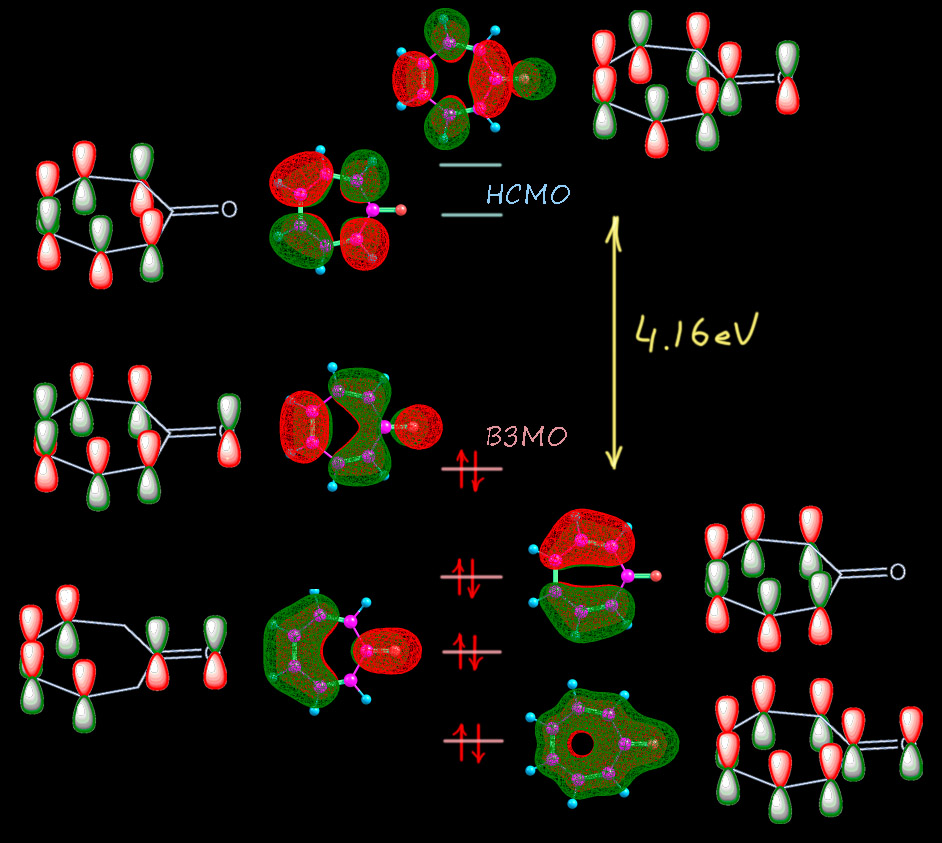
Итак, тропон не является ароматическим соединением в какой-то заметной степени, а с моей точки зрения, как и фульвен – ни в какой. Это типичная неароматическая молекула, с отдельными принаками ароматичности по причине вклада граничных структур, формально соответствующих теории Хюккеля.Самое главное проявление – это способность довольно легко протонироваться. Это свойство драматически отличает тропон от просто кетонов, в том числе енонов, которые очень трудно запротонировать, и это обычно плохо кончается – начинаются реакции конденсации. Тропон отличается от обычных кетонов тем, что довольно легко протонируется. Легко – это не значит, что его можно запротонировать уксусной кислотой, нет, только сильными, pK протонированной формы около -1, что соотвествует достаточно сильной кислоте типа соляной в водном растворе. Смотрим структуру этого иона. О, вот это да! Да это действительно похоже на тропилий просто с заместителем, длины связей выровнялись очень хорошо, в самом тропилии везде 1.39 Å, здесь 1.39 плюс-минус 0.01, вполне в рамках – как я уже замечал, если вы посмотрите на структуру замещенного бензола, длины связей тоже быдут слегка гулять приблизительно в тех же пределах. Углы тоже стали как в правильном семиугольнике около 128°. Повсем признакам это действительно ароматичекое соединение ряда тропилия – такой фенол ряда тропилия.
Теперь посмотрим на молекулярные орбитали протонированного тропона имеено для того, чтобы еще раз увидеть разницу и то, как из тропона получается гидрокситрополон – из неароматической молекулы ароматическая. Опять слева представим орбитали из расчета, и справа – их символическое описание набором из р-АО без учета весов (то есть просто топологию – расположение и число узловых поверхностей), причем покажем, как они строятся из орбиталей двух фрагментов – самого тропилия (см. выше по кругу Фроста-Мусулина) и одной р-орбитали атома кислорода. Соответствующие орбитали помечены цветными галочками. Мы обязаны ее учитывать, потому что это орбиталь π-типа, перепендикулярная плоскости молекулы и имеющая узел в этой плоскости. Поэтому перед нами задача – в тропилии семь МО но в гидрокситропилии должно быть восемь – по числу π-АО, принимающих участие в построении π-МО. Интересно, что в расчетах это всегда получается само собой, просто как результат расчета. А нам надо понять, как это получилось, и здесь вариантов могло быть много, но сработал какой-то один. Он довольно прост. Орбитали на электроотрицательных атомах всегда очень низки по энергии – собственно именно поэтому мы и называем атомы электроотрицательными: они держат свои электроны как голодный последний кусок хлеба. Дальше мы начинаем думать, как орбитали взаимодействуют (лучше сказать – смешиваются, потому что это не реальный физический процесс, а символическое описание чисто математических процедур). Для этого они должны подходить друг другу по симметрии, а лучше сказать по топологии – чтобы доли могла именно смешиваться, а узловые плоскости (поверхности) этому не мешали. Но π-орбитали по определению имеют подходящую топологию, и значит важнее второй фактор – относительные энергии. Сильно взаимодействуют (смешиваются) орбитали близкой энергии; слабо смешиваются – орбитали, сильно различающиеся по энергии. В данном случае мы предположили, что орбитали кислорода расположены глубоко, а значит если и взаимодействуют, что с самыми низшими орбиталями π-системы тропилия. Поскольку занятых орбиталей всего три, и то ничего кроме самой нижней, полносимметричной, не остается. Вот ровно так это и выглядит и прямо по прописям – возникает новая пара орбиталей за счет суммы и разности. Немного странный вид (кто отъел краешек блина, блин!) – это просто потому что нужно построить две орбитали из материала для бывшей одной, приходится немного отъедать у одной и добавлять другой и наоборот, что достигается за счет весов (коэффициентов) АО в МО. Нам это совершенно до-лампочки. А вот что важно это то, что обе новые орбитали содержат по паре, что очевидно. И у нижней область между кислородом и углеродом связывающая, а у второй – разрыхляющая (антисвязывающая). И поскольку обе орбитали заняты, это значит что антисвязывание съело связывание и порядок пи-связи углерод-кислород равен нулю. Мы отлично знаем как это устроено, если еще помним, как заполняются орбитали в двухатомных молекулах из второго периода, с чего обычно начинают и чем заканчивают обсуждение МО в жизни нормального человека. Все следующие орбитали точно соотвествуют исходным орбиталям тропилия с добавлением антисвязывания с орбиталью кислорода там, где на углероде есть вес. Это ничего не дает, кроме небольшой дестабилизации, поэтому (и еще потому что симметрия теперь не ось седьмого порядка. а простая плоскость, и у такой симметрии не может быть вырожденных орбиталей) каждая пара вырожлденных тропилия расходится на две, которые впрочем остаются близко друг к другу. Это еще один признак ароматичности – соответствие орбиталей системы орбиталям аннулена (здесь, тропилия) с сохранением большого расстояния ВЗМО-НСМО (здесь щель оказалась равна 4.83 эВ, обратите внимание на увеличение этого рассоятния по сравнению с тропоном). То, что щель меньше, чем у незамещенного тропилия, тоже объясняется дестабилизацией системы сильно донорным заместителем как в фенолах: повторю, что протонированный тропон – это фенол ряда тропилия.
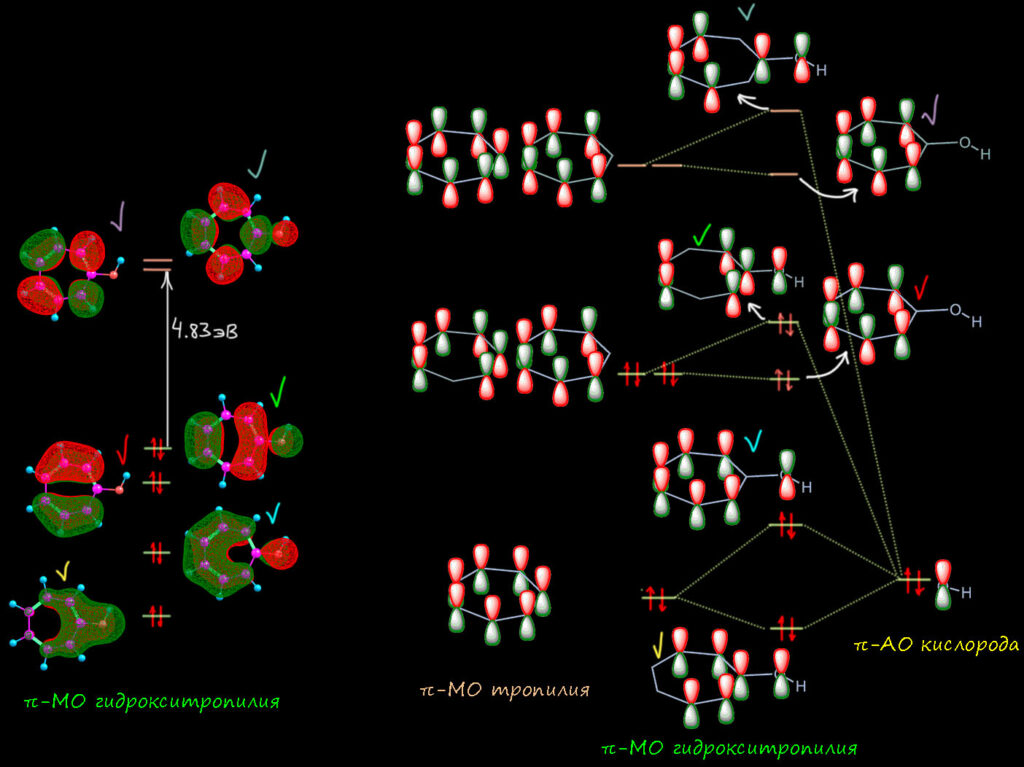
Вот такая картина. Тропон оказался неароматической молекулой, имеющим некоторые неосновные граничные структуры с признаками ароматичности. Но протонированный тропон – полноценная ароматическая молекула, с выровненными связями и правильными молекулярными орбиталями. С чем мы её и поздравляем, и переходим к ещё более интересному случаю – трополону.
Как трополон прикинулся ароматикой и обдурил почти всех
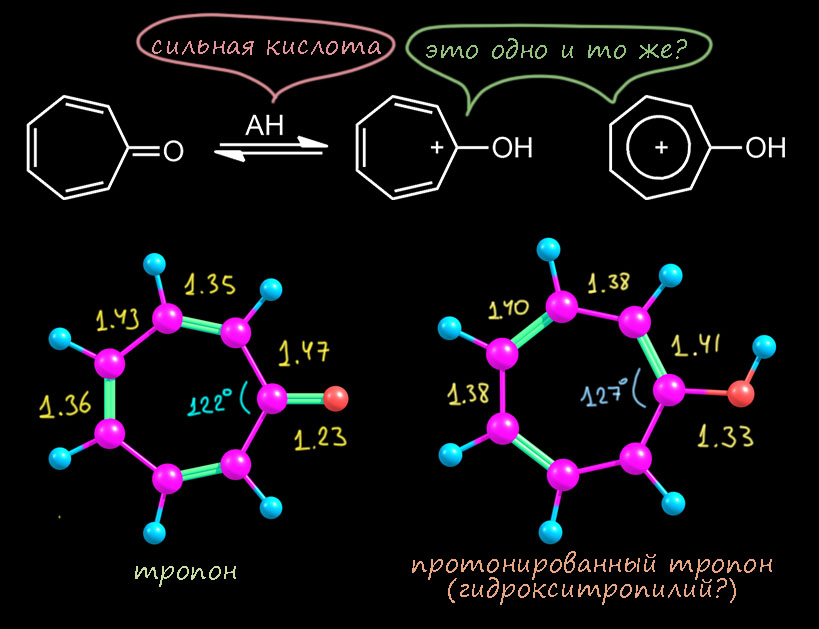
Займемся совершенно культовой молекулой – трополоном. Трополон это 2-гидрокситропон, и эта гидрокси-группа создает множество интереснейших моментов, которых нет в исходном тропоне. Если в ароматичности тропона в общем-то сомневались многие, то трополон шёл всегда как намного более бесспорная ароматика. Но я сейчас попробую показать, что это заблуждение. и что на самом деле трополон еще дальше от ароматичности, чем тропон.
Знаменитость трополона свяана с тем, что это первое соединение кроме бензола и циклопентадиенила, описанных в основной статье Хюккеля, для которого была предположена ароматичность, и сделал это в 1945 году будущий крупный теоретик и один из главных пропагандистов развития квантовой химии Майкл Дьюар. Не для самого трополона, а для одного из природных производных трополона – а в Природе их немало – стипитатовой кислоты (Dewar M. Structure of Stipitatic Acid. Nature 1945, 155, 50–51). Это один из метаболитов плесневых грибов – в годы войны как раз разобрались с пенициллином, за что в 1945-м и дали нобелевскую премию, и стали искать другие полезные вещества в грибах, это одно из них. Но Дьюар верно понял, что самая суть этой молекулы состоит в сочетании карбонильной и енольной группы рядом, а остальные заместиетели не важны, и именно он и там придумал название трополон (-ол и – он) для этой системы. Здесь важно то, что Дьюар видит признак ароматичности не в счете электронов – он вообще про это не говорит, а в взаимопревращении двух форм. Ему кажестя, что именно такая легклсть перемещения и двойных связей и даже протона и есть признак особого характера этой молекулы, причем Дьюар называет это резонансом. Мы сейчас будем немного удивлены такой неряшливости в понятиях, потому что точно знаем, что изображено именно две разные молекулы – два таутомера, и что ни в коем случае нельзя в резонансе или мезомерии или сопряжении трогать атомы, даже такие легкие как водород, даже если мы считаем это не атомом, а протоном, то есть тоже квантовой частицей – но масса протона в 1840 раз больше массы электрона, и поэтому движение (и квантовые свойства) электрона и протона не могут осуществляться в одной шкале времени и энергии. Для электрона протон неподвижен. В этой во всех смыслах исторической статье мы видим, что в 1945 году, всего менее 80 лет назад, теория ароматичности еще не сформировалась, и даже самые передовые умы еще не понимают ни что делать с уравнением Хюккеля, ни что такое ароматичность.
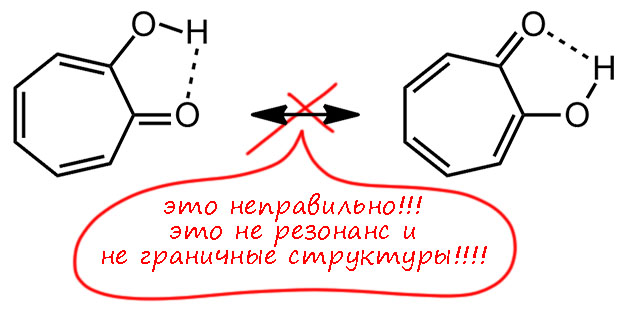
Еще более забавно то, что Дьюар в конце той же статьи делает смелое предсказание, что и гидроксициклопентадиенон должен быть похожей молекулой с похожими свойствами, и даже называет ее по аналогии циклопентадинонолоном (впоследствии придумали более короткое название пенталон, но не ищите его в гугле – сейчас так называется популярный анимешный персонаж). Ой, но мы-то знаем, что тут ароматичностью не пахнет совсем. Более того, здесь сильно пахнет антиароматичностью. Но об этом уж точно в это время ещё никто даже в страшном сне не помышляет. И понятно, что это предсказание совсем не сбылось, а эта молекула до сих пор находится в списке “ускользающих” (elusive) молекул, как и многие другие циклопентадиеноны. Их ловят – а они шасть – и ускользают.
Эта статья Дьюара, как видим, основывающаяся на совершенно неверных гипотезах, тем не менее совершенно историческая. От неё начинается настоящее изучение ароматичности и ароматических молекул. А Дьюар, скорее всго, осознавший что допустил ошибку, быстро развивается в одного и важнейших исследователей квантово-химических методов исследования структуры. До конца прошлого века все, кто делал какие-то квантово-химические расчеты, зависел от развитых Дьаром методов так называемой полуэмпирики, позволявшей на старых и немощных компьютерах прошлого века (самый гигантский из которых, занимавший несколько этажей и потреблявший электричества как целый город, сейчас насмешил бы по характеристикам даже самый скромный смартфон) делать очень толковые расчеты и быстро продвигаться в понимании стуруты молекул и даже вещества.
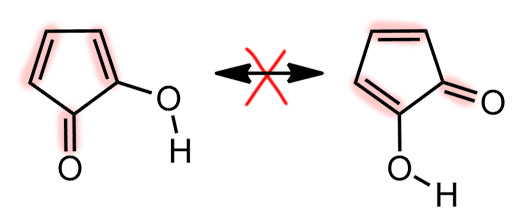
Трополон – исключительно эффектная структура. Оставим пока рассуждения о ее ароматичности, просто восхитимся изяществу системы. Если смотреть на одну из таутомерных форм, она кажется несимметричной, и тогда вроде бы может быть два ряда таутомеров, если в кольце появляются заместители. Но есть одно положение, если считать, как положено, это будет 5, у которого оба таутомера по строению одинаковы. Подчеркну, это по-прежнему два таутомера, и это легкj можно было бы установить, введя куда-нибудь изотопную метку, но по свойствам они неразличимы – это вырожденная таутомерная перегруппировка. Поглядев на эту ситуацию повинимательней, мы не сможем не признать некоторое сходство с пара-замещенным фенолом; только в случае фенола мы имеем дело с структурами Кекуле, которые, как известно, воображаемые, и максимум, что мы можем использовать это срелку мезомерии. А в случае 5-замещенного трополона, это реальные таутомеры, превращение, равновесие.
Кроме того, мы без сомнений скажем, что по природе гидроксильная группа енольного типа. И превращения таутомеров – это кето-енольная таутомерия. Но мы привыкли, чтобы в кето-енольной таутомерии протон переносился из енолизуемого положения, а здесь такого нет, потому что нет дикето-формы. И протон переносится с енола на кето-группу и обратно, тем более, что это происходит через внутримолекулярную водородную связь легко и быстро.
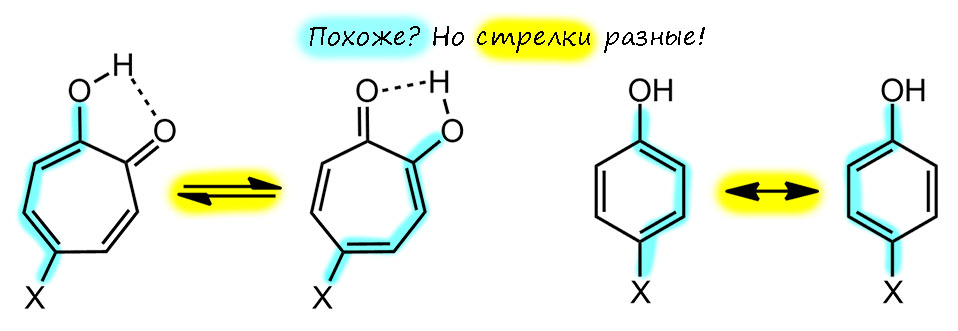
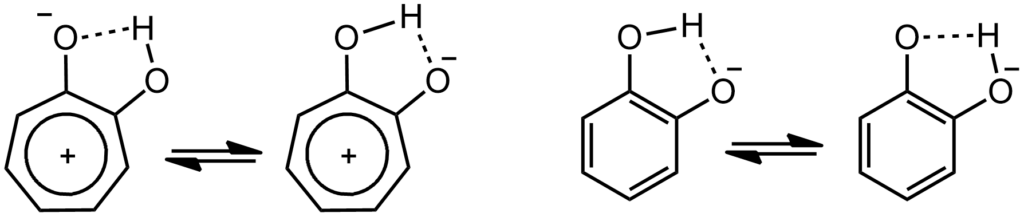
Естественно, про трополон принято говорить, что это ароматическая молекула по той же причине, что и тропон. Но – если это так, то это значит, что доминируют структуры с разделением зарядом и тропилиевой ароматической серединкой. Как только мы это напишем и поверим, то нам придется прийти к выводу, что в этом случае вместо своеобразной, но всё же кето-енольной таутомерии мы получаем нечто другое – водородную связь в моноанионе дигидроксильной формы. И поскольку протон не может болтаться в пространстве чёрти где, а должен быть связан ковалентной связью с одним из атомов, то мы имеем все равно равновесие. И это по-прежнему таутомерные формы. Очень похоже на моноанион 1,2-дигидроксибензола (пирокатехина). В общем, вполне убедительная картинка и мы ее можем встретить во множестве учебников и статей.
Обычно нам еще и говорят, что трополон – более выраженное ароматическое соединение, чем тропон (признавая тем самым некоторую недоделанность ароматичности тропона). Вроде бы это даже можно объяснить тем, что в трополоне два мезомерных донора на тропилиевое ядро, которое все же положительное. а значит, не отказавшееся бы от мезомерной донорной стабилизации. И еще водородная связь выгодна (хотя она есть и в исходной кето-енольной форме) но признаем, что чем больше минус на кислороде, тем вроде бы водородная свзь прочнее.
И есть еще и экспериментальный довод. Как мы знаем, одним из признаков ароматичности является склонность к реакциям замещения, особенно электрофильного. Можно ли нитровать тропон и трополон? А вот здесь большая разница между этими двумя соединениями. Тропон почти не подвергается реакциям электрофильного замещения – электрофилы или присоединяются или не реагируют. Сообщение об образовании продукта азосочетания из первой статьи Даубена было позже опровергнуто. Нитрование тропона идет в довольно жестких условиях и дает не очень понятную смесь продуктов окисления, присоединения, и присоединения-отщепления. Да мы уже вполне поняли, что из тропона можно сделать ароматическое соединение только в условиях допроса с пристрастием или из желания понравиться кому-нибудь важному. А вот трополон дает множество самых банальных продуктов электрофильного замещения и в очень мягких условиях.
Это известно давно. Зададим вопрос – в какое положение кольца должен идти электрофил? Узнать это можно точно так же как в ряду бензола, поиграв с сигма-комплексами. Увы, форма с тропилиевой гайкой нам не поможет, придется вернуться к кето-енольной форме. Здесь нас ждёт одно неприятное открытие. Если мы уже поверили в тропилиевую ароматическую форму, в которой есть фенольный и фенолятный заместители, и спросим себя – кто из них более сильный ориентант? По обычной химии сомнений не останется – фенолятный, конечно, мы же знаем как неотвратимо это работает в химии производных бензола: фенолят сильнее всех, а фенол уступает амину – то есть разница много порядков. Увы, можете сами попробовать порисовать сигма-комплексы.
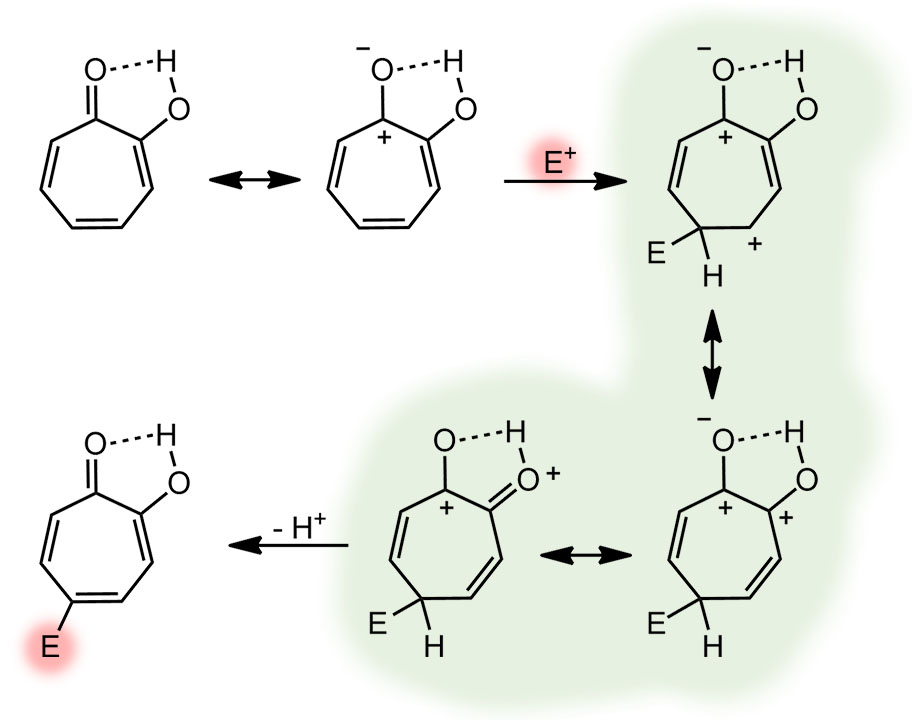
Вы можете сказать – какая ерунда, мы же уже выяснили, что заместителю в этом положении все равно, где какая группа. Да, конечно, но проблема в том, что мы все равно считаем там два таутомера, и как не посмотри, но в момент образования сигма-комплекса положения фиксированы. Да и двойные связи перекидываются в граничных структурах только в таком расположении. В общем, если не обращать внимания на детали, то всё равно, а если обращать, то осадочек остается. Мы очень хорошо видим аналогию с моноанионом пирокатехина, но замещение почему-то идет в пара-положение не к феноляту, а к фенолу. И мы видим еще одну неприятную вещь – заместители заместителями, но если это тропилий, то это катионная ароматическая система, и она должна быть дезактивирована к атаке электрофилов. А что в эксперименте? Этим в основном занимался тот самый японец Тэцуо Нодзоэ, и публиковал работы в забытых японских журналах, но японское химическое общество любезно выложило их в открытый доступ.
Трополон очень легко нитруется азотной кислотой в уксусной кислоте. Азотку берут стопроцентную, но зато без серной кислоты или других активаторов – безводная азотка активирует себя сама, и в растворе уксусной кислоты нитрует не нитроний, а что-то типа ацетилнитрата в условиях кислотного катализа. Это довольно мягкая и аккуратная нитрующая смесь. Нитрование идет быстро, побочно, видимо, получаются динитро и даже тринитропроизводное.
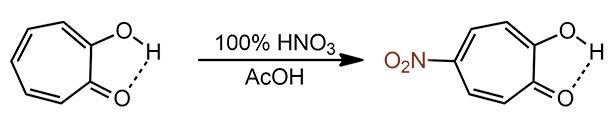
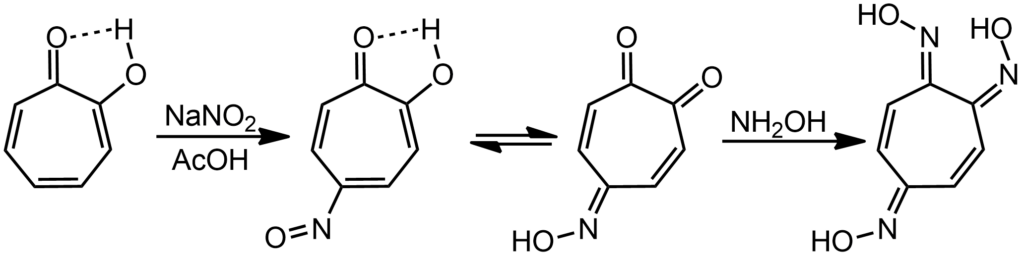
Еще легче нитрозируется. Нитрозирующие агенты – чрезвычайно слабые электрофилы, они реагируют в бензольном ряду только с фенолами и анилинами. Хорошо, у нас тропилиевый фенол – поверили. Нитрозосоединение чрезвычайно легко подвергается нитрозо-оксимной таутомерии, и при реакции с гидроксиламином дает триоксим занятной молекуоы, которую называю тропохиноном.
И легко вступает в азосочетание, правда только в виде аниона, но точно так делают и обычные фенолы. Получаются самые обычные азосоединения, вполне аналогичные таковым из ряда бензола. Можно даже назвать их красителями в специальном смысле, которое есть у английского термина dye – окрашенное вещество, которое может найти какое-то применение именно благодаря этому свойству – поглощению света в видимой области. Применение может быть любое – портки красить не обязательно, годится, наприvер, индикатор или сенсибилизатор.
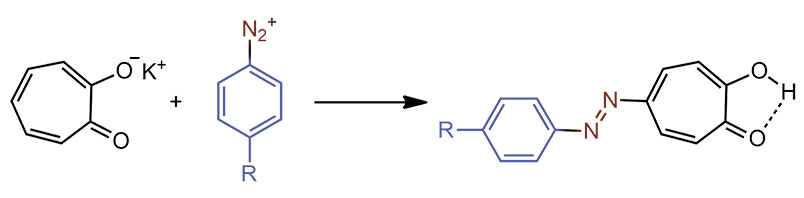
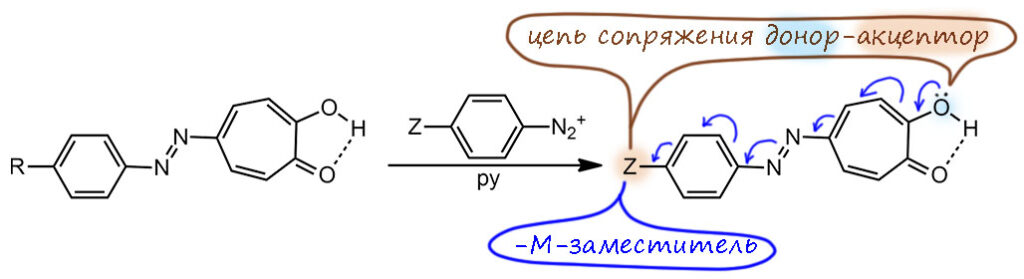
У этих азосоединений есть интересное свойство, отличающее их от азосоединений бензольного ряда – можно заместить один остаток на другой, но с одним условием – замещаемый остаток должен иметь донорный заместитель, а замещающий – акцепторный. Движущая сила такой реакции понятна – увеличение длины цепи сопряжения. Но она же ставит вопрос – а почему это так легко происходит, ведь мы до этого не сталкивались с ипсозамещением азоарильного остатка.
Это – успешные реакции замещения. Но далеко не совсеми стандартными электрофилами трополон реагирует так чисто или вообще реагирует. Напрмеир, ни алкилировать, ни ацилировать его не получится. По понятной причине – есть нуклеофильный кислород и он и станет реакционным центром. Но и галогенировать не получается. Бром к трополону скорее присоединяется, и не очень охотно. Остальные галогены вообще ничего хорошего не дают. Сульфировать конечно тоже нельзя. Дейтерообмен трополона описан, но даёт он довольно невнятные результаты.
Итак, мы вроде бы видим, что трополон ведет себя намного более подобающим для ароматического соединения способом хотя бы иногда. Поэтому в литературе довольно прочно закрепилось мнение, что если с тропоном, возможно, и правда есть большие вопросы, то трополон уж точно ароматическое соединение. Давайте тогда разберёмся со структурой этой молекулы – она довольно занятна и не пожалеет дял нас загадок.
Так ароматичен ли трополон?
Первым делом рассмотрим (не в возвышенном смысле, а прямо глазами) структуры трополона, а также его аниона и сопряженной кислоты – катиона. Я тоже все это посчитал, но можно найти и рентгеноструктурные данные – и трополон и его соли и комплексы изучали методом РСА не раз, и есть очень качественные структуры. Я сравнил с расчетом и убедился, что соответствие отличное, в том числе в тех параметрах, которые нам могут показаться необычными. Поэтому для однообразия я всё же буду использовать расчетные данные. Вот они.
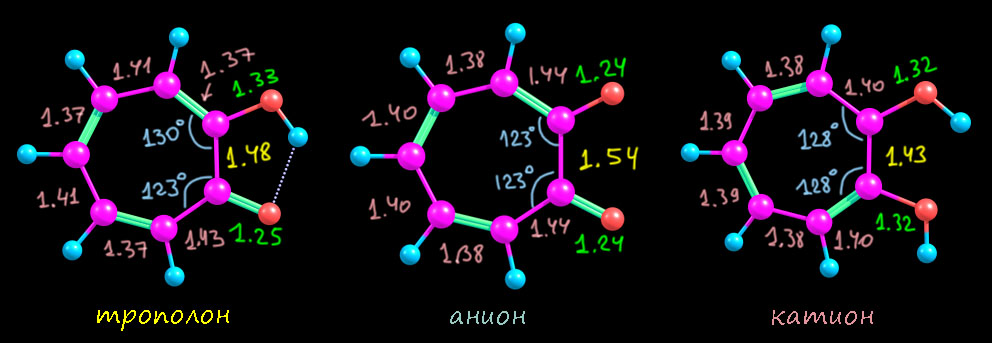
Что мы видим?
- сам трополон, как и тропон существенно искажен, но – намного слабее, чем тропон в том, что касается чередования длин связей везде кроме связи между углеродами под кислородами. Эта связь очень длинная, в общем почти соответствующая простой между sp2-гибридными углеродами. Ещё мы видим, что угол при карбонильном углероде намного острее всех других и почти точно соответствует обычному тригональному карбонильному углероду – это делает цикл искаженным – вытянутым в сторону этого углерода.
- в анионе бросается в глаза совсем странная по длине связь между теми же углеродами под кислородами: 1.54 Å – это, простите, даже не просто простая связь. а отчасти даже еще и немного растянутая. Такое впечатление, что между этими углеродами прямо отталкивание. И – оба угла при углеродах под кислородами острее – здесь кольцо уже совсем нарочито вытянуто. А вот все остальные связи здорово выровнены, почти нет альтернирования.
- а вот катион почти полностью соответствует ожиданиям и похож на такой же катион из тропона – там это был гидрокситропилий, а здесь получается дигидрокситропилий. Кольцо почти правильный гептагон, хотя все равно сотается удлиннённость связи между первым и вторым углеродами, но уже в рамках приличий. Слегка искаженный правильный гептагон
Посмотрим уже по привычке на молекулярные орбитали π-системы. Приведу только расчетные, прикинуть как они выглядят, если их представить символически p-гантельками. Раз теперь два атома кислорода включены в π-систему, МО теперь девять, из них пять занятых (условно шесть в кольце и две пары, или четыре двойные связи и пара), всего 10 π-электронов. Соблазн вскричать – о, это же ароматическое число! – отметём сразу с порога, поскольку мы договорились электроны вне цикла не считать. Разглядывая орбитали, мы можем легко прийти к выводу о том, что во всех трех системах они практически одинаковы – небольшие различия определяются только тем, что анион и катион симметричнее исходного трополона, и это отражается на всешем виде орбиталей, но не на их топологии (числе и расположении узловых поверхностей). Орбитали катиона сильно сдвинуты вниз относительно орбиталей трополона – как в таких случаях говорят, стабилизированы. а орбитали аниона сдвинуьы вверх – дестабилизирвоаны. Ничего странного в этом нет, так всегда бывает при отнятии или добавлении электронов – чем меньше электронов, тем сильнее связаны оставшиеся. И наоборот – чем больше. тем слабее связаны все. Важно то, что это затрагивает весь пакет пи-орбиталей, и они не меняются местами – это свидетельство сильной делокализации. Нас интересует главное – как отражается в молекулярных орбиталях реальная структура этих трёх молекул. Отметим две вещи – в этом случае, в отличие от тропона у протонированной формы орбитали практически такие же как у двух других молекул и мы, в частности, видим области связывания почти во всех занятых – а в протонированном тропоне на связи C-O, как мы помним, пи-связывания почти не было (области связывания и антисвязывания сократили друг друга. Здесь нет, и на связях C-O явно есть пи-связывание. Я бы не стал этому удивляться, ведь гидроксильные группы – очень сильные мезомерные доноры, сопряжённые с кольцом, и даже в ряду бензола начиная с дигидроксипроизводных (двухатомных фенолов), их донорный эффект очень сильно дестабилизирует ароматической кольцо, а когда групп три, тригидроксибензолы вполне ясно ведут себя как соответствующие кето-формы. Тропилиевыю систему, видимо, уже очень сильно раскачать можно двумя группами.
Второе наблюдение важнее. И в самом тропоне, и в дианионе очевидна область антисвязывания на большинстве орбиталей π-системы между углеродами под кислородами – теми самыми, связь между которыми оказалась необычно длинна. И расчетный порядок связи между этими атомами почти не отличается от единицы, хотя между другими парами атомов в кольце это 1.4 – приблизительно полуторный, как и должно быть при делокализации, причем любой, необязательно ароматической.
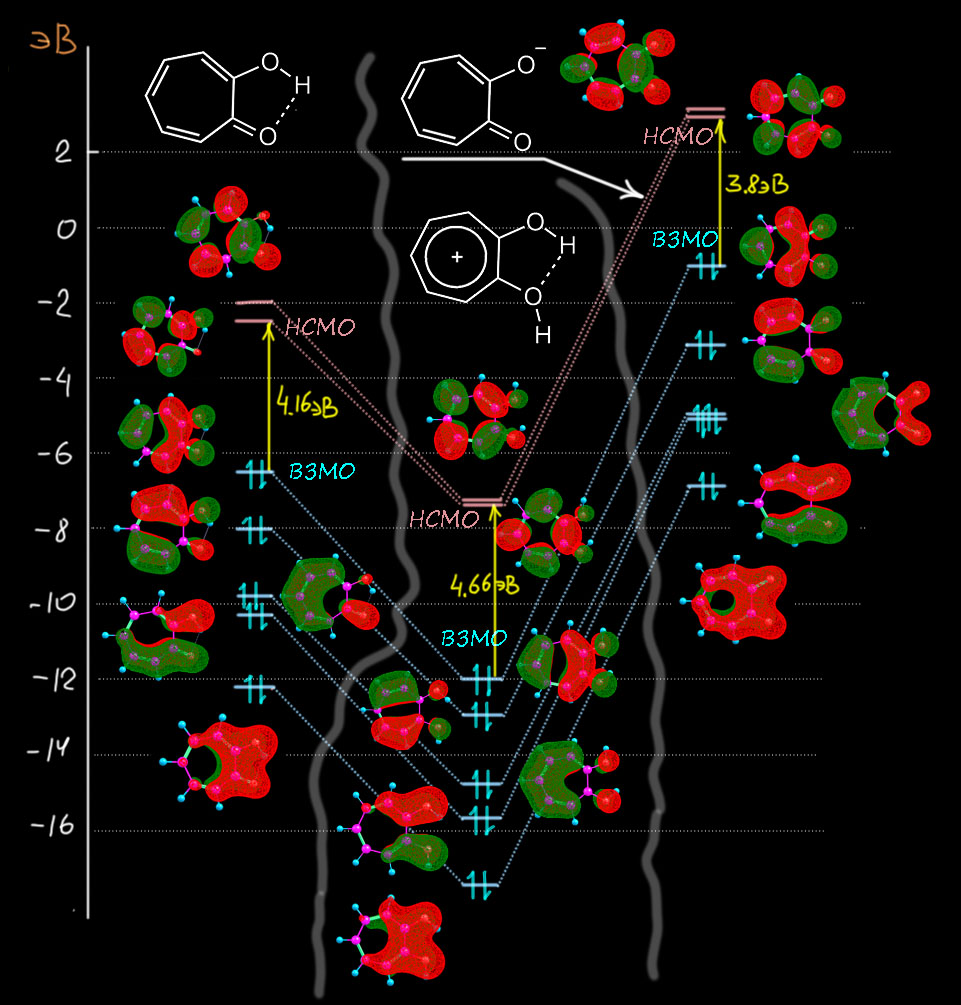
Вот какая история. И, на мой взгляд, единственный способ её разумно интерпретировать прост – и в самом трополоне, и особенно в анионе трополона основной путь делокализации не циклический. Собственно, при расстоянии в 1.54 ангстрёма ни о каком серьезном π-связывании и речи быть не может. Анион трополона поэтому нужно представить очень просто – как линейную цепь сопряжения донор-акцептор вообще без малейшего вклада циклического сопряжения и ароматической граничной структуры.
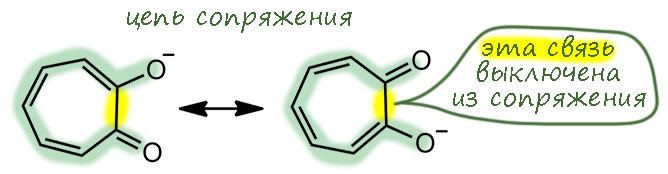
В самом трополоне основная картина делокализации, видимо, та же самая, но в нём, скорее всего, есть некоторый вклад и циклической ароматической делокализации, хотя и небольшой. Всё же длина этой связи 1.48 ангстрём хоть велика, но немного меньше чистой одинарной и небольшая доля двоесвязанности здесь есть. Ещё один довод в пользу небольшого вклада ароматической стабилизации приведу чуть ниже. И напомню, что делокализацию рисую для одного из таутомеров, потому что таутомерия это явление другого порядка – это настоящая реакция, равновесная. А делокализация это явление, а не реакция. Делокализация есть всегда, а на равновесие требуется время.
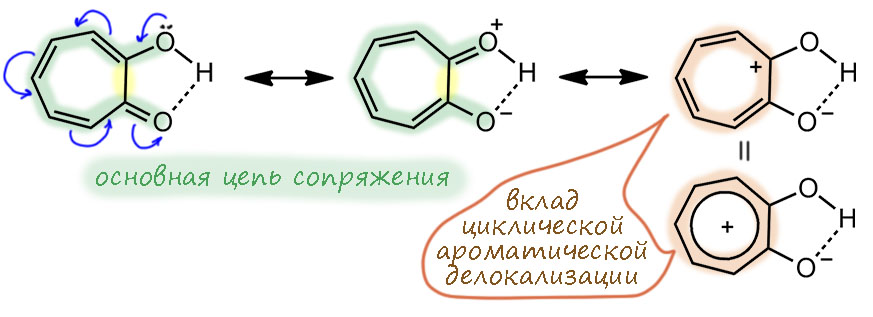
Ну и наконец катион. В катионе наиболее вероятно то, что и цепь сопряжения и циклическая ароматическая делокализация вносят сравнимые вклады в общую электронную структуру. Только это производное трополона можно адекватно называть ароматическим, хоть мы и видим, что оно уже весьма далеко отчаоило и от системы тропилия, и даже от протонированного тропона – гидрокситропилия.
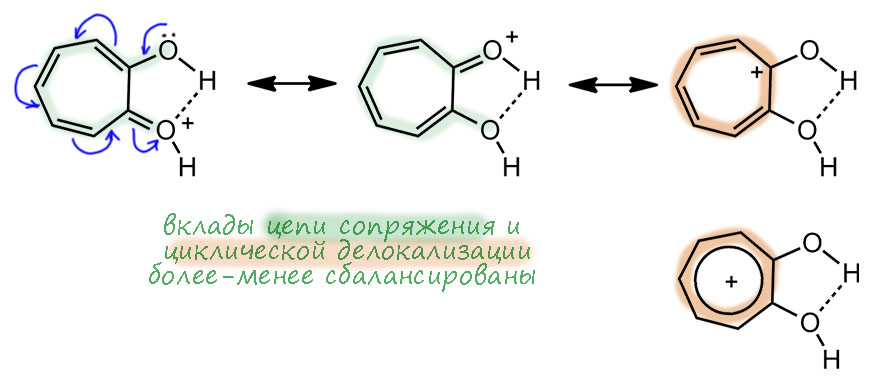
Вы скажете – а как же реакции замещения тропона: нитрование, нитрозирование, азосочетание. Ничего страшного. Химический критерий вообще считается самым слабым и действует только как общий прогноз. Даже обычные олефины нередко в реакциях с электрофилами дают не присоединение, а присоединение-отщепление с возвращением двойной связи. Сопряженные системы и того чаще. Механизмы взаимодействия электрофилов с любыми ненасыщенными системами одинаков, только различаются завершения – элиминированием или присоединением нуклеофила. Но это самая обычная конкуренция в реакциях карбокатионов. Критерий правильно формулировать так – если у какой-то системы в реакциях с разными электрофилами чаще встречается присоединение-отщепление (замещение), то это дополнительный (не основной!!) довод в пользу ароматичности. Главные критерии (признаки) всё же структура и магнитные свойства, а также необычно высокая устойчивость для систем, где это можно хоть как-то доказать.
И вот ровно про стабильность мы и поговорим в конце. Именно это, на мой взгляд, всё же говорит в пользу некоторой ароматической стабилизации в дополнение к стабилизации в хорошей цепи сопряжения. Ведь если мы еще раз посмотрим на тропон, увидим, что это с одной стороны кето-форма, с другой енол. А для аниона увидим, что это делокализованный енолят. И тогда у нас возникнет вопрос – а нециклические соединения такого типа есть? Что нам даёт цикл и даёт ли он что-то вообще. И когда мы зададим такой вопрос, то тут же увидим хорошо известную нам систему – енол и енолят 1,3-дикетона, в который вставили один или несколько винильных удлинителей сопряжения. Такое удлинение называется винилогией.
Понятно, что в таких цепях нет никакой ароматичности, а есть только та самая цепь сопряжения донор-акцептор, ровно такая, какую мы и предполагаем в трополоне и особенно анионе. А ну-ка сравним структуры и длины связей. Обе нециклические структуры возьмём в виде транс-изомера по всем двойным и s-транс конформера по всем простым специально, чтобы не иметь никаких проблем со стерикой. В этой форме получается строго плоская структура, что говорит об очень хорошей стабилизации в цепи сопряжения. Сравниваем, сверху уже известные нам структуры, снизу дважды винилог енола ацетилацетона и его енолят. Пройдитесь по цепочке, сравнивая одни и те же по положению относительно кислорода:
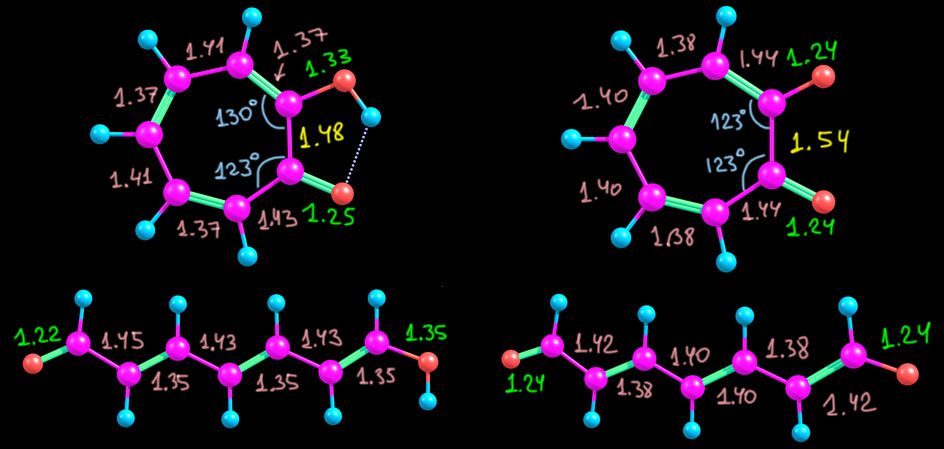
Ну вот. Да это же одно и то же. Тот же характер чередования длин, да и сами величины практически везде отличаются максимум на одну-две единицы во втором знаке. Но учитывая разницу в конфигурациях двойных связей, расхождения неизбежны, удивительно, что они настолько малы. А почему не взять цис-цисоидный изомер-конформер? А потому что он не сможет быть плоским – концы цепи наедут друг на друга и разопрут цепь в разные стороны – сопряжение сильно ослабеет. Это и так очевидно, да я и попробовал – не годится.
Итак, можно сказать, что эксперимент удался. Структуру трополона и его аниона отлично можно передать с помощью простой цепи сопряжения вообще без использования циклической ароматической стабилизации. Тем не менее, для самого трополона небольшая дополнительная стабилизация может объяснять большую устойчивость этого соединения. Всё же эти нециклические формы плохо живут сами по себе и очень любят самоконденсироваться. Трополон же, если в темноте, отлично хранится и не портится. Это не совсем честный довод, потому что виноват может быть и другой фактор: трополон – принудительно плоское соединение. А нециклический дикетон нет, энтропия будет его крутить обязательно, барьеры вращения там не будут велики, сопряжение будет ослабевать, и такая молекула, в которой есть и нуклеофильные и электрофильные места, обязательно начнет кусать сама себя.
В общем, как обычно в химии, стопроцентного ответа – есть ли в трополоне хоть капля ароматической стабилизации, получить не удается. Вот в катионе точно есть. На том и успокоимся. Или наоборот расстроимся. Ведь тропон и особенно трополон вроде положено считать важнейшими молекулами дял теории ароматичности. Не много ли на себя берём? Да нет, на самом деле, если хорошо знать литературу по ароматичности и химии тропона и его производных, то сомнения в ароматичности начались очень рано, и разделяются многими, так что никакой ереси в этом нет, это вполне разделяемая многими – я бы сказал, теми, кто ароматичность не принимает как абстрактную схему, а разбирается с каждой молекулой – так что и нам незазорно. Но анализ структур на этой страничке всё равно чисто мой, поэтому здесь нет ссылок.
А что с антиароматичностью малеимида?
Теперь перейдём к второй части вопроса – а не может ли граничная структура с антиароматическим счётом электронов отравить жизнь какому-то ни о чём не подозревающему соединению. В качестве примера такого кандидата в преисподнюю привели малеимид. Учитывая обычную поляризацию карбонильной группы, вроде бы такая опасность есть:
Но мы теперь немного учёные на своих и чужих заблуждениях. Выяснилось, что когда речь заходит об ароматичности, то такая поляризация вовсе не обчзательно даёт существенный вклад и делает соединение ароматическим. Но там мы хотя бы обсуждали небольшой вклад ароматической граничной структуры и признавали, что небольшой дополнительный вклад в стабилизацию он может давать. Совсем небольшой, труднодоказуемый – но мы так и не решились совсем от этого отказаться.
Но там речь шла об ароматичности, то есть о стабилизации, выгодности. А здесь об антиароматичности, а значит о дестабилизации, невыгодности. Что-то мне говорит, что есть там вклад был невелик, а может и вообще отсутствовал, то здесь его заведомо не будет – какой смысл дестабилизировать.
Но это не совсем верное рассуждение. Молекула своей судьбы не выбирает. Как лягут электроны, так и будет. Мы здесь уже обсуждали однажды проблему антиароматичности и почему только маленькие циклы максимум до 5-членных могут вляпаться в антиароматичность – потому что таким циклам
сложно уйти от плоскостности. И если в них есть циклическое сопряжение и плохой счет электронов, то антиарматическая дестабилизация им гарантирована, и они как-то будут вынуждены с этим жить, по возможности ослабив эту самую циклическую делокализацию (изменением длин связей, выходом хотя бы внешних атомов из плоскости, регибридизацией и т.д.). Но мы уже вполне выяснили и проверили – экзоциклические двойные связи очень эффективно разрушают циклическую делокализацию. А здесь аж две карбонильные группы. В бензольном ряду хиноны или хинодиметаны не проявляют никаких признаков циклического сопряжения и связанной с ним ароматической стабилизации или антиароматической дестабилизации. Вряд ли мы что-то увидим и в малеимиде.
Посмотрим на структуру. И сравним её с амидами малеиновой и фумаровой кислот. С чем ещё можно было бы сравнить? Не знаю, не вижу других молекул для сравнения. Сравнивая с обычными амидами мы, по крайней мере, поймём, есть ли какие-то труднообъяснимые отклонения в структуре малеимида. Попробуем предсказать какие. Антиароматическая дестабилизация всегда приводит к увеличенной степени альтернирования простых и двойных связей и всего другого, позволяющего внизить степень сопряжения и уменьшить антиароматическую дестабилизацию. Это важно понять – если есть условия для такой делокализации и связанной с ней дестабилизации, молекула будет использовать все доступные ей способы для снижения этого эффекта, так что останется только то, чего избежать нельзя.
Длины связей изменить можно, можно за счет регибридизации частично вывести пару из сопряжения – но нельзя согнуть молекулу, если она состоит из небольшого числа плоских фрагментов.
Смотрим, сравниваем. Амид малеиновой кислоты связан внутримолекулярной водородной связью – амиды очень хорошо известны, как эффективные доноры и акцепторы водородных связяй – спросите у белков, они не соврут. Амид фумаровой кислоты не имеет внутримолекулярной водородной связи. Разглядывая молекулы и находя аналогичные связи мы убеждаемся, что никаких серьёзных аномалий нет. Самые большие отличия мы видим в длине амидной связи C-N, которая, как известно, имеет за счет сопряжения весьма высокую степень двоесвязанности, которая её и сокращает. И мы видим, что именно в малеимиде она самая длинная (1.39 Å) в сравнении с 1.35-1.37 Å в других амидах на картинке, и 1.33-1.34 в совсем простых амидах типа ацетамида. Но это объясняется очень легко – двойная связь, сопряжённая с амидной группой создаёт эффект кросс-сопряжения (сопряжены звездой карбонил, азот амида, двойная связь), а это дестабилизирует и снижает обычное сопряжение в каждой из цепей сопряжения (азот-карбонил, двойная связь-карбонил). Из-за этого и небольшле удлинение. Ничего собенного. А почему именно в малеимиде удлинение максимально? Уж не вклад ли антиароматической дестабилизации? Возможно, но отсюда мы видим, насколько он мал, если даже и есть. Но, скорее всего, это просто следствие более эффективного сопряжения в совсем плоской циклической молекуле – в её нецикличесих родственниках из-за стерического отталкивания водородов, молекулы немного скручены и не совсем плоские. Чуть-чуть – но и эффект чуть-чуть.
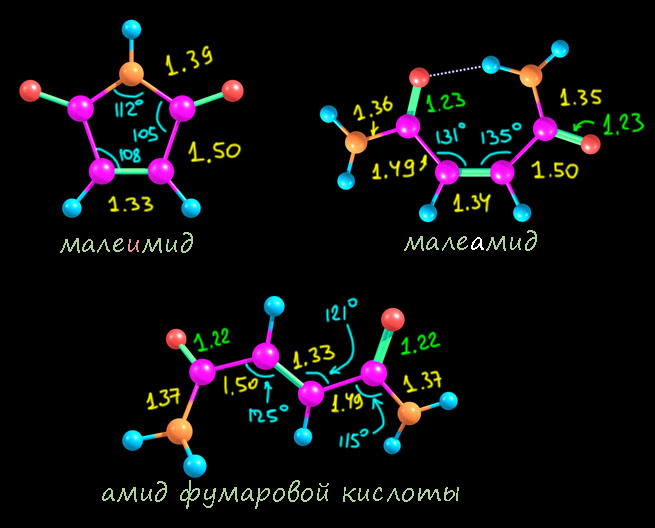
А вот что мы видим очень хорошо, и это, видимо, очень важно, это влияние цикла на геометрию. Сначала – почему это важно. Потому что мы про малеимид знаем одну вещь – это совершенно потрясающий диенофил в реакции Дильса-Альдера. С малеимидом реакции идут всегда и быстро. Гораздо легче и быстрее, чем с малеиновым ангидридом, эфирами и амидами малеиновой и фумаровой кислот. Это один из лучших диенофилов из стабильных веществ (еще лучшие диенофилы это всякие дегидробензолы и прочие эфемерные вещи, но это и понятно). И у нас громадный соблазн – объявить, что эта реакционная способность обусловлена вкладом антиароматической дестабилизации. Я сам любил это объяснение. Оно кажется красивым и неотразимым. И выстраивается в ряды с другими забавными молекулами, в том числе бузусловно антиароматическими – вот циклобутадиен, как только его размораживают из матрицы, немедленно хватает любую другую непредельную молекулу.
Но, давайте всё же почаще орудовать бритвой Оккама. Брат Вильгельм (Уильям, Гульельмо, Виллем и т.п. – никто не знает, какой национальности был этот монах, и все тащат его себе) Оккамский был прав – не надо умножать сущности сверх необходимого. Если мы посмотрим на молекулу малеимида мы увидим гораздо более простое объяснение – угловое напряжение. Это напряжённая молекула и это хорошо видно по сравнению с родственниками. Нормальный угол у sp2-углерода или азота – 120 градусов. Плюс-минус пять градусов – терпимо. Вон у фумарового амида так и есть . У малеинового амида углы увеличены – это эффект водородной связи, которая и распёрла цис-карбоксамиды. Водородная связь же и компенсирует такой распор. А вот в малеимиде все углы сильно уменьшены и это связано просто с геометрией пятичленного плоского цикла – никуда не денешься, формула для суммы углов это вам не ЖМКО – не объедешь ни на какой козе. Уменьшены – значит напряжены и сильно. При присоединении происходит регибридизация в sp3 и снижение нормального угла от 120 к 109 градусам – а значит в пятичленном цикле резкое снижение углового напряжения. Вот вам и причина повышенной реакционной способности (по этой же причине, видимо, маленовый ангидрид лучший диенофил, чем эфиры малеиновой и фумаровой кислот).
Напоследок посмотрим еще на молекулярные орбитали, раз нам так понравилось. Может там есть следы антиароматической делокализации. Сначала только посмотрим, как выгляди настощий антиароматический [5]-аннулен – катион циклопентадиенилия. Рисуем круг Фроста-Мусулина и вписываем туда орбитали. Поскольку электронов 4, а не 6, приходится посадить по одному на каждую из вырожденных связывающих. Это скверно, мы это уже обсуждали, поэтому цикл искажается, эти орбитали расходятся и электроны падают на нижнюю. Но – и это один из важнейших признаков антиароматических систем – сохраняется очень небольшой зазор между ВЗМО и НСМО. Чем меньше, тем хуже. Реально этот зазор определяется тем, насколько сильно может исказиться кольцо. Но это всегда небольшие величины 1-2-максимум 3 эВ. У ароматических молекул этот зазор всегда большой – от 4 эВ и более.
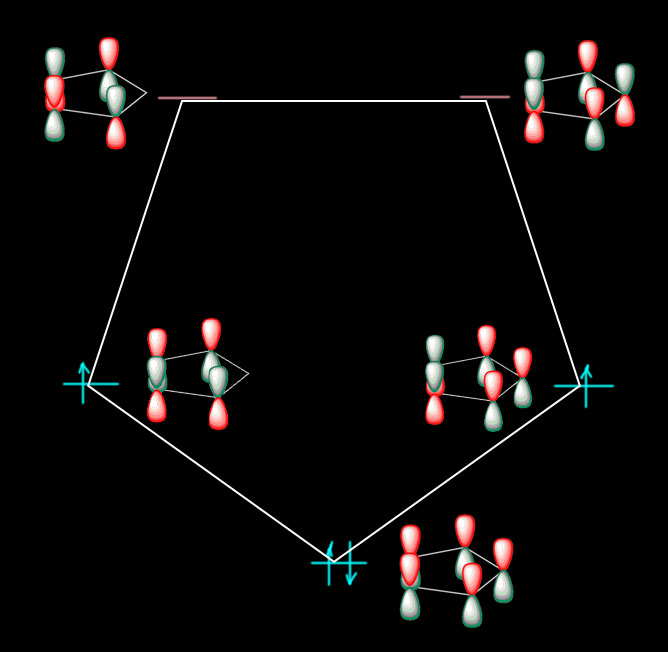
Смотрим расчетные орбитали. Их восемь по числу пи-электронов: два на двойной связи, два на азоте, по два на карбонилах. Мы как всегда наплюём на самые верхние разрыхляющие – они нафиг никому не нужны. Оставим шесть. Что видим? Первый сюрприз – ВЗМО не является пи-орбиталью, это сигма-орбиталь, в основном это неполеленные пары кислородов в плоскости кольца. Совершенно бездарная орбиталь, даром никому не нужна. Но вот она есть, придется признать. Эта орбиталь отвечает за нуклеофильность молекулы, и нам просто неинтересна, потому что никакой особой нуклеофильности мы не видим. Интересно, что если бы мы этим все же заинтересовались, такая ВЗМО значит, что азот кольца не является главным нуклеофильным центром. Ну так малеимид никто и не использует как нуклеофил. Но мы здесь напряжёмся, потому что мы понимаем, что малеимид похож на фталимид, а фталимид мы как раз алкилируем по азоту – это метод Габриэля. Но алкилируем мы анион, и если бы мы посчитали анион, наверняка вот та следуюшая и уже пи-МО с большим весом на азоте выехала бы вверх и стала бы ВЗМО, и молекула стала бы нуклеофилом. Проверять будем?
Более мы ничего интересного н видим. Все МО можео описать не из МО [5]-аннулена (сравниваем глазами, желающие могут разрисовать восьмёрками-гантельками), а тупо комбинируя МО двойной связи, двух карбонилов, и азота. Снизу полносимметричная комбинация всего, следующая это просто разность связывающих МО карбонилов, следующие две это то же самое, только с обратным знаком. По карбонилам хорошо видно связывание, никак не получится представить карбонилы с большой степенью поляризации, пониженным пи-связыванием. В общем, обычная молекула, хорошо соответствующая самой обычной структуре с карбонилами и двойной связью. По-моему, тема про антиароматичность закрыта. Причём, по-моему, гораздо безапелляционнее, чем история про тропон и трополон. Нет здесь никакой антиароматичности. Конечно, наверняка в литературе кто-то посчитал NICS и нашел там пару единиц, но это уже совершенно полная туфта, не стояшая внимания.
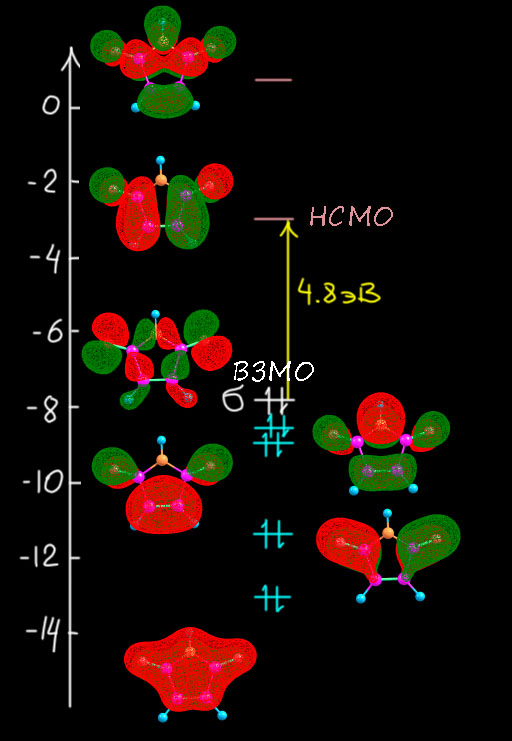
А есть ли более надёжные примеры активации за счет антиароматичности?
Да, конечно, и то, что мы зарубили (гипотетически! – никогда не забывайте, что в естественных науках не бывает строгих доказательств, типа, как в математике – и любое высказывание в физике, химии, биологии это гипотеза, которую нужно всячески пробовать на вшивость экспериментальной работой) малеимид как пример соединения, явно повышенная реакционная способность которого якобы объясняется вкладом антиароматической граничной структуры. Мы сказали – нет, это не так. И вклад настолько мал, что себя не проявляет. И причина, скорее всего, проще – просто угловое напряжение. Но это не значит, что в других случаях это явление – активация молекулы за счет существенного вклада антиароматической структуры – не может проявиться. Таких примеров полно на самом деле, и это очень инетересная область. Подробно мы в этом копаться пока не будем, но одну работу, довольно свежую, разберём, хотя бы потому что в ней сошлись два героя этой странички, и уже нам близкий и родной тропон и развенчанный из антиароматиков малеимид (Lucas J. Karas, Adam T. Campbell, Igor V. Alabugin, Judy I. Wu Antiaromaticity Gain Activates Tropone and Nonbenzenoid Aromatics as Normal-Electron-Demand Diels–Alder Dienes Org. Lett. 2020, 22, 7083–7087). Джуди Ву и её коллеги из Техаса и Флориды задались целью вовлечь тропон … в реакцию Дильса-Альдера. Исследователи предположили, что тропон не торопится участвовать в этой реакции из-за его ароматичности. Нам такое объяснение конечно же теперь не очень понравится, потому что мы знаем, что к ароматичности тропон имеет не самое большое отношение, а многим настоящим ароматическим соединениям ароматичность совсем не мешает быть и диенами и диенофилами, конечно, не самыми шустрыми, но всё же. И сами же авторы делают презабавнейший ляп, заметив, что тропон всё же можно понудить к непривычной для него реакции в условиях кислотного катализа. Это здорово, только мы знаем, что координация по кислороду тропона как раз и увеличивает ароматический характер и даже делает его вполне ясно выраженным. И что же – это больше не мешает реакции Дильса-Альдера, хоть и специфической, в варианте акцепторный диен – донорный олефин (это очень коряво называется реакцией Дильса-Альдера с обращёнными электронными требованиями, reverse electron-demand Diels-Alder reaction). Что-то не сходится. Видимо, низкая реакционная способность тропона в циклоприсоединении вызвана не ароматичностью, а обычным сопряжением, делающим кольцо плоским, так что при развитии реакции долно произойти немаленькое искажение, что всегда дает более высокую энергию активации, а следовательно медленную реакцию. Ладно, не важно. Давайте посмотрим на замысел самой статьи. Он интересен. И состоит в том, чтобы превратить тропон в производное, в котором экзоциклическая двойная связь будет поляризована в обратную сторону, что и должно дать вклат антиароматической граничной структуры и дестабилизацию, а за ней – и ускорение реакции Дильса-Альдера. Ожидаемая реакция [4+2]-циклоприсоединения должна выглядеть так. С самим тропоном (X=O) она не идёт. Рабочая гипотеза авторов статьи (не наша!) в том, что ароматический характер кольца, обеспечиваемый поляризацией связи в сторону кислорода, делает невыгодным нарушение ароматичности при уиклоприсоединении.
Рабочая гипотеза состояла в том, что нужно изменить направление поляризации в сторону от заместителя, так чтобы на кольце граничная структура имела минус, а значит вклад антиароматического состояния. Вроде бы, если бы нас попросили придумать субстрат для такого эксперимента, мы бы взяли хорошо известный гептафульвен с донорным заместителем на экзо-углероде (мы это всё обсуждали в самом начале этой страницы). Но авторы придумали другое. Они решили поэксплуатировать альфа-эффект – отталкивание электронных пар. Если взять гидразон – преположили они – то отталкивание пары дальнего азота сместит пару внутреннего в сторону кольца. Но это конечно, будет слабый эффект, а чтоы сделать его сильным, надо внешнюю группу депротонировать. А чтобы депротонировать было легче, повесить дуда обычный акцептор, типа сульфонильной группы. Сказано – нет, не сделано – нарисовано:
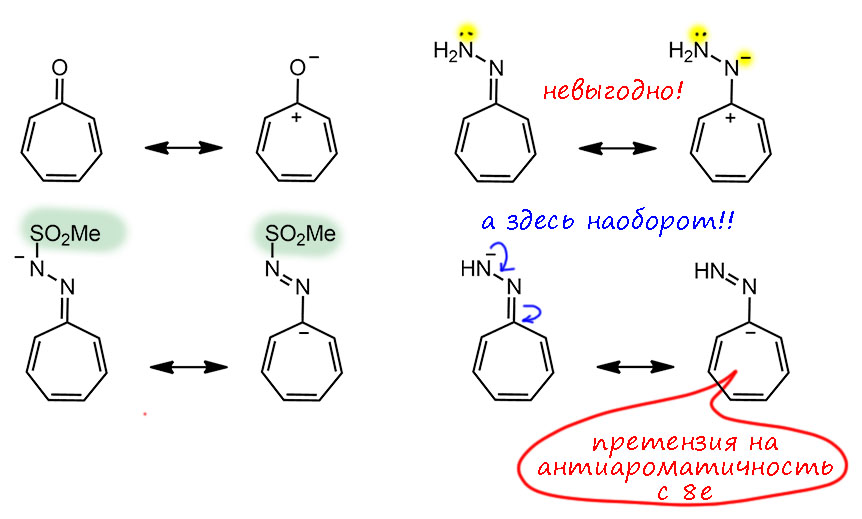
Так, выглядит занятно, хотя мы, конечно, малость сомневаемся в том, что семичленный цикл, получив немного антиароматической дестабилизации, останется плоским, а не выгнется, сломав циклическую делокализацию. Имея избыток электронов, это можно сделать через регибридизацию. Но ладно, давайте посмотрим, что у них получилось в эксперименте. Ой, а у них нет эксперимента. Как нет, такого не может быть, это же химия, а не чистописание. Глазам своим не верим, тем более что текст написан в настоящем времени без намёков на гипотетичность ситуации. Смотрим всё два раза, лезем в дополнительные материалы – да, нет никакого эксперимента. одни расчёты, расчетное моделирование путей реакций, из которого следует то, что они и хотели нам сказать – что у реакций с анионами гидразонов барьеры активации (они же энергии переходных состояний) ниже, чем для реакции тропона и тем более нейтрального гидразона. Правда тоже с приключениями. Одна из двух реакций оказывается не согласованной, а стадийной. И это чертовски важно, сейчас разберём почему.
Почему мне кажется, что это исследование, говоря мягко, не достигает поставленной цели, и нам, к сожалению, не даёт то, что мы хотели найти – пример повышенной реакционной спсобности молекулы, у которой нет настоящей антиароматичности, но есть граничная структура, обладающая признаком ароматичности. Потому что, увы, как бы ни были хороши современные расчеты, которые дают нам отличные результаты в том, что касается молекулярных свойств (структуры, многих спектров, дипольного момента, много чего ещё), остаются крайне приблизительными и малонадежными во всем, что касается вещества – совокупностей огромного числа молекул, подчиняющихся законам статистики – и это в первую очередь, скорости реакций. Если у вас есть эксперимент, то расчет может вам помочь уточнить механизм, увидеть тенденции и так далее. Но просто ставить реакции на компьютерном чипе еще очень сильно рано – ценность таких результатов невелика. А здесь мы вообще имеем дело сразу с довольно сомнительной гипотезой, и с ее проверкой только расчетом, но это не проверка, а подгонка под заранее известный результат. Почему я так уверенно это пишу? Разве мы не должны руководствоваться предположением о добросовестности исследвоателей? Конечно должны, и что самое занятное, я эту добросовестность под сомнение и не ставлю. Проблема только в том, что когда вы делаете эксперимент (добросовестно), то результатом управляет Природа – вы не можете изменить пути Природы. Что она вам даст, то и даст – вот и объясняйте, что получили. А когда вы делаете расчетное моделирвание, то в ваших руках необозримое разнообразие всяких трюков – выбор базиса, функционала, алгоритма построения пути реакции, критериев сходимости, чёрт знает чего ещё – и дело не в том, что вы сознательно подгоняете результат, а в том. что вы ищете метод расчета. который вам даст результат, и этот процесс исключительно субъективен. Рано еще делать химию, как говорят, в кремнии (in silico), а не в стекле (in vitro).
Хорошо, хватит брюзжать, может тут все хорошо получилось. Нет, и мы это видим сами, но оказывается только что было опубликовано опровержение этой работы группой голландских исследователей (Eveline H. Tiekink, Pascal Vermeeren, Trevor A. Hamlin Not antiaromaticity gain, but increased asynchronicity enhances the Diels–Alder reactivity of tropone Chem. Commun., 2023, 59, 3703). Вообще в химии опровержения – невероятная редкость. Химию трудно опровергать, если только речь не идет об откровенной ошибке. Тем более трудно опровергать расчетную химию. Но ведь написали люди такую статью. Может они эксперимент сделали и у них ни черта не получилось? Нет, и эти все делают расчетами. Берут очень модный современный метод моделирования барьеров активации. То есть один расчёт опровергают другим. Битва на мониторах. Выглядит это опровержение поэтому не менее странно, чем то, что опровергают. А мы даже не будем в этом копаться, потому что это опровержение оказывается воспроизводит то, что уже было найдено в опровергаемой статье. Реакция с анионами гидразона тропона оказалась не согласованной, а стадийной. Что это значит и почему это ставит крест на любых спекуляциях про антиароматичность?
Дело в том, что механизм реакции Дильса-Альдера иногда бывает довольно прост. В тех случаях когда в олефине есть сильная акцепторная группа, а в диене сильная донорная (просто доноры и акцепторы в диене и диенофиле есть всегда, но здесь речь идет именно о сильных донорах и акцепторах мезомерного типа, которые умеют серьёзно делокализовать плюс и минус), то вместо согласованного механизма с шестичленным переходным состоянием, в котором 6 электронов более-менее одновременно перемещаются, созлавая и разрушая связи, может быть просто присоединение донора к акцептору со смещением плотности и образованим интермедиата с разделёнными зарядами (цвиттер-иона), который затем закрывает цикл просто рекомбинацией карбокатион-карбанион.
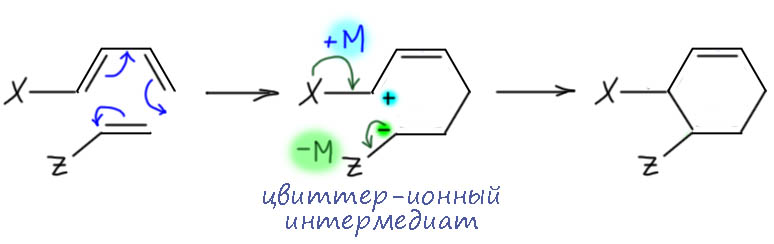
Как только в реакции Дильса-Альдера есть подозрение на такой механизм вместо обычного согласованного, к чёрту летят орбитальные соображения, правила Вудварда-Хоффмана, антиароматичность и всё такое, потому что в дело встпают куда более простые факторы – влияние заместителей, способность к делокализации, стабилизация карбокатионов и карбанионов. Время жизни цвиттер-иона может быть разным. Иногда это почти то же переходное состояние, только сильно асинхронное – некоторые связи еще даже не начали образовываться, а другие уже порвались. Иногда это прямо настоящий интермедиат, который долго плавает и думает, н пора ли ему уже наконец образовать последнюю связь и что ему за это будет. Это все зависит и от мощности стабилизирующих заместителей, и от стерики, и от (наконец) полярности растворителя, и от погоды и международной обстановки. Важно то, что всякие умные рассуждения про антиароматичность в исходном уже не работают, птому что его, исходного, уже нет. Да и стабилизация зарядов обычно более мощный энергетический фактор, чем какая-то потешная дестабилизация исходного за счет одной граничной структуры.
И вот, уже сами авторы первой статьи в своей статье признают, что с самым интересным из субстратов, анионом гидразона, механизм переключается в стадийный. И потому на это упирают и авторы статьи-опровержения, что довольно глупо выглядит – зачем опровергать то, что и так уже прозвучало. “Опровергатели” могли заметить, что авторы первой статьи недооценили этот фактор, ну и хорошо – только, видимо, без громкого заявления об “опровержении” их не пустили бы со статьёй в некогда самый престижный химический журнал Chemical Communications, ныне растерявший почти весь свой престиж и ставший хорошим, но одним из. Ладно, чёрт с ним, проще забыть об этом “опровержении” тем более что авторы там усердно ломятся в открытую дверь – ну, им там надо протестировать одну модную теорию, ну и пусть себе забавляются. И просто нарисовать стадийный механизм реакции сопряжённого основания тропонового гидразона с малеимидом. Карбонил малеимида отлично служит мезомерным акцептором, а депротонированный гидразон – донором. Здесь даже ещё выгоднее получается, потому что анионный характер донора в данном случае уничожает заряд со стороны диена, поэтому такое действительно кажется вполне возможным.
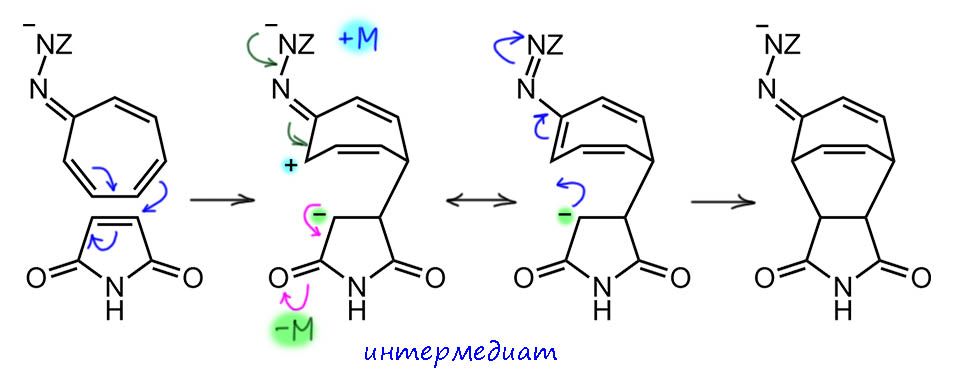
Чтро из этого следует? Во-первых, это поучительный пример того, как исследователи выбрали неудачную модель для того, чтобы найти подтверждение своей теории. Теория, напомню, состояла в том, что антиароматический характер тропонового диена с таким заместителем, смещающим плотность в сторону кольца, будет активировать олекулу к циклоприсоединению. Но, увы, сами же и нашли более простое объяснение эффекта – изменение механизма с согласованного на стадийный, причём работать должен тот же эффект – донорный. Это очень скверно, когда один фаактор работает в пользу двух альтернатив. Авторам так хочется, чтобы подтвердилась их теория что они все равно настаивают на том, что именно антиароматичность причина. Увы, мы всё время забываем, чему учил нас средневековый мыслитель-монах брат Вильгельм Оккамский (в просторечьи Оккам) – перефразируя его принцип, скажем так – из двух объяснений всегда выбирайте более простое и привычное, если у вас не очень веских причин выбирать мене привычное и новое. В данном случае точно нет, о чем свидетельствует и сама работа и ее “опровержение” – обе они обнаружили стадийность механизма. И увы, и то и другое еще и не вполне полноценные исследования, так как вообще не делают эксперимент, а здесь он мог бы вообще сломать все ожидания, так как депротонированный гидразон – отличный нуклеофил, а здесь для нуклеофила есть и другая работа – например, присоединяться по Михаэлю азотом к малеимиду.
В заключение обсуждения этой странной, но поучительной истории, замечу, что это всё равно хорошая наука. Хорошей её делает добросовестность изложения – мы именно из самой работы понимаем, что в ней не так, потому что всё, что нужно, описано и изложено исчерпывающе – никто не пытается как-то манипулировать данными и что-то скрывать. Это говорит о том, как важна научная добросовестность – ведь сделать не совсем удачное исследование не страшно; страшно сделать лживое исследование и быть пойманным на этом коллегами или собственной совестью. Когда исследование добросовестно, то даже из не совсем удачного замысла и реализации можно узнать немало полезного, и, что самое главное, этому можно доверять. Добросовестным исследование делают две вещи – собственно исследователи, которые работают в серьёзной науке и не собираются заниматься чепухой, их профессиональная репутация и положение в научном сообществе. И второе – качество журнала, в котором они публикуются. Так уже повелось, что издания Американского химического общества (ACS), Королевского химического общества (RCS) и издательского дома Wiley, собравшего все серьезные континентальные европейские журналы и унаследовавшего знаменитые немецкие издания с двухвековой безпречной историей – известны высоким уровнем публикаций, неукоснительным соблюдением всех процедур публикации и высоким авторитетом. Материалу, опубликованному в изданиях этих издателей, обычно можно верить – ну а дальше никто не мешает прочитанное интерпретировать по-своему. Увы, в журналах других издательских групп, а их нынче море, можно найти всё что угодно в любой пропорции.