Механизм E1
Как мы уже успели увидеть, этот механизм является двухстадийным, как и SN1-замещение. Еще одно их общее свойство – оба практически почти бесполезны, но если с реакцией SN1-замещения, как мы уже видели, еще можно сделать хоть что-то полезное, то к отщеплению E1 вполне точно применима меткая поговорка: “С паршивой овцы хоть шерсти клок”. Этим клоком шерсти является реакция кислотно-катализируемой дегидратации спиртов. А все остальное это E2. Мы увидим, что практически значимые реакции элиминирования, то есть те реакции, с помощью которых можно получать вещества в реальных колбах, почти всегда хорошо описываются механизмом E2. Более того, мы увидим, что под механизм E2 описывает очень широкий спектр реакций, потому что допускает возможность широкого варьирования структуры переходного состояния (пока не волнуйтесь, прочитав эту пафосную тарабарщину – скоро разберемся и поймем в чем дело), и уже механизмы E1 и E1cb перестают выглядеть как полноценные механизмы, а становятся чем-то более похожим на крайние разновидности механизма E2. Пока что посмотрим на оба механизма отдельно.
Механизм E1
Механизм E1 мы отлично знаем – успели уже его изучить и обсудить минимум два раза. Действительно, посмотрим на реакцию, описывающуюся механизмом E1 и прочитаем ее наоборот:
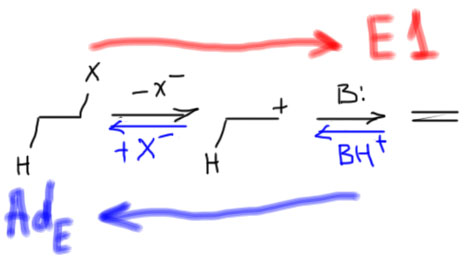
Вот так сюрприз! – это же просто электрофильное присоединение галогеноводорода к двойной связи. Да – в химии (и не только) действует так называемый принцип микрообратимости. Это очень простая идея. Выражаясь обыденным языком это значит, что если дорога из города Б в город А похожа на дорогу из города А в город Б, то это одна и та же дорога. Из этого следует много разных важных вещей.
- Раз реакция присоединения галогеноводорода обратима, и мы точно знаем, что она очень хорошо и почти количественно, и быстро идет в прямом направлении (это мы выяснили в теме Алкены), то обратная реакция, то есть как раз E1-отщепление, оказывается медленной и плохой – шанс на то, что она дойдет хотя бы до 5%-ного выхода, очень невелик. Равновесие смещено в сторону галогенпроизводного, а не олефина. Из этого правила есть исключения, но они очень редки и экзотичны. Нам они точно до лампочки.
- Карбокатион – продукт протонирования олефина. Напомню, что протонирование (перенос протона) – это понятие из теории кислот и оснований Бренстеда. Получается, что олефин – это основание Бренстеда, а карбокатион – его сопряженная кислота. Напомню, что кислотность кислоты и основность основания связаны одной константой равновесия – константой килотности (или основности, это одно и то же, взаимно обратные величины). Если основание сильно, то сопряженная кислота слаба, и наоборот. Простой вопрос: олефин – это сильное основание? Ответ очевиден: Нет, слабое, очень слабое, олефин можно протонировать только очень сильными кислотами (серной, толуолсульфоновой и т.п.). Значит, карбокатион – это тоже очень сильная кислота Бренстеда. А сильную кислоту легко депротонирует (то есть превращает карбокатион в олефин) любое основание, даже слабое. Очень сильную кислоту – вообще любое основание, даже совсем дохлое.
- И там, и там ключевой и единственный интермедиат – карбокатион. Чем стабильнее карбокатион, тем лучше. Это справедливо и для присоединения (отсюда правило Марковникова), и тем более для отщепления – про E1 имеет смысл говорить только тогда, когда образующийся карбокатион стабилизирован. Из обычных алкилгалогенидов – это третичные производные, те же самые, что соответствуют механизму замещения SN1.
Что следует из этих трех пунктов. Первое, чтобы попробовать увидеть E1-отщепление нужно взять что-нибудь типа трет-бутилбромида. Нужно растворить такой субстрат в растворителе, способствующем образованию ионов. Это мы уже знаем из SN1 – это полярные протонные растворители: спирты, карбоновые кислоты (муравьиная, уксусная), смеси воды с органическими растворителями. И терпеливо ждать, время от времени отбирая пробы и анализируя их с помощью доступного оборудование (спектров ЯМР, например, отслеживая соотношение винильных протонов образующегося олефина и метильных протонов исходного трет-бутилбромида с учетом их количества и при условии, что удалось их оттащить от метилов изобутилена). Через некоторое время вы получите смесь с переставшим изменяться соотношением исходного и продуктов (а там еще будет продукт сольволиза и его тоже придется как-то опознать в спектре). Установится равновесие. Карбокатион сам решит, во что ему превращаться, и повлиять на это мы в существенной степени не сможем, потому что мы вообще не очень хорошо умеем влиять на равновесия. Единственный ключ к этому – принцип Ле Шателье, и если мы сможем как-то удалять из реакционной смеси образующийся олефин (это не очень просто, так как хотя он и намного более летуч чем исходное и продукт сольволиза, но он отлично растворим в реакционной смеси) – то сможем и смещать равновесие в сторону продукта элиминирования. Две реакции всегда конкурируют – SN1-замещение и E1-элиминирование. Да, соотношение двух продуктов зависит от многих факторов – от конкретного растворителя (даже соотношения, если это смесь), уходящей группы, даже температуры. Все это можно прочитать в больших книгах, но нам это не очень интересно, потому что практически это применить нельзя. Иными словами, важно понять, что
- E1-элиминирование – механизм не самостоятельный, это часть общего механизма SN1-E1, имеющего общий интермедиат (карбокатион, или в более сложных рассуждениях, ионные пары карбокатиона), и два конкурирующих пути превращения превращения карбокатиона – реакцию с нуклеофилом и депротонирование (на самом деле конкурирующих пути три, так как образование карбокатиона из субстрата обратимо, и обратная реакция, то есть рекомбинация карбокатиона с уходящей группой – называется “внутренний возврат” тоже очень важна).
- О, ужас! Картина еще сложнее – механизм E1 не только часть общего механизма SN1-E1, но все это вместе – часть еще более общего механизма электрофильного присоединения к двойной связи. То есть мы получаем общий процесс AdE2-SN1-E1 и все эти направления равновесны: в равновесии находятся, например, олефин, бромпроизводное, продукт его сольволиза, и HBr. Подбор условий реакции может несколько упростить картину, например, если мы возьмем растворитель, неспособный к сольволизу (апротонный и ненуклеофильный, типа какого-нибудь хлоруглеводорода), то отпадет SN1 и в равновесии останется только олефин, продукт присоединения и HBr. Именно это мы имели, например, когда присоединяли HBr к сопряженным диенам, наблюдая последствия установления или неустановления равновесия – термодинамический и кинетический контроль, 1,4 против 1,2-присоединения.
- E1-элиминирование из галогенпроизводных или тозилатов не используется для получения олефинов
- Субстраты для SN1-E1, в первую очередь, третичные (вспомните еще раз Определитель реакций) при действии сильных оснований охотно подвергаются селективному элиминированию, но по механизму E2. Эта реакция отлично используется для получения олефинов. например, 2-метилпропен отлично получается при действии спиртовой щелочи на трет-бутилхлорид, но это не E1, а E2-отщепление.
Полезный случай E1-элиминирования – дегидратация спиртов
Механизм E1 не совсем бесполезен. Именно этим механизмом объясняется хорошо известная древняя реакция – дегидратация спиртов, по крайней мере третичных и вторичных. Дегидратация первичных спиртов в жестких условиях также дает олефины, но мы не можем предполагать образование первичных карбокатионов – все известные данные категорически против этого, первичные катионы слишком нестабильны и обладают настолько высокой энергией, что реакция, приводящая к таким частицам неизбежно имела бы огромную энергию активации, и следовательно ничтожную скорость. Оставим пока этот спорный момент, и посмотрим, как идет дегидратация типичного третичного спирта. Реакция идет в присутствии каталитического количества сильной кислоты, серной или фосфорной (по первой ступени фосфорную кислоту можно считать сильной), которую мы условно обозначаем протоном. Когда мы пишем -H+ мы имеем в виду перенос протона от протонированной частицы, являющейся сильной кислотой Бренстеда, на любое имеющееся в смеси основание Бренстеда, например воду или анион кислоты (бисульфат, дигидрофосфат, и т.п.).
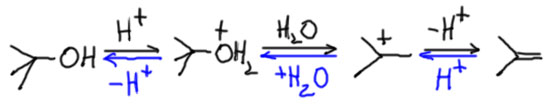
Видим очень простую вещь, гидроксильная группа протонируется, и плохая уходящая группа -OH превращается в хорошую – нейтральную молекулу воды. Далее все типично для E1 – карбокатион, с удовольствием отдающий протон. Почему же дегидратация спирта – вполне полезная реакция, с помощью которой отлично можно получить олефин, а E1-элиминирование из трет-бутилхлорида или бромида – бесполезная трата времени. Посмотрим еще раз на схему процесса. Видим, что все стадии обратимы, а следовательно превращение спирта в олефин обратимо. Мы легко узнаем обратную реакцию – это кислотно катализируемая гидратация олефина – электрофильное присоединение протона и воды к олефину. Здесь ничего нового нет, мы уже выяснили, что элиминирование и электрофильное присоединение – это одна и та же обратимая реакция. Это очевидное обстоятельство поможет нам не тратить много времени на обсуждение дегидратации, так как на странице про алкены мы уже довольно подробно обсудили реакцию гидратации.
Гидратация и дегидратация – одна и та же реакция с одним механизмом, и единственное, что позволяет пустить этот процесс по необходимости в одну или другую сторону – применения принципа Ле Шателье.
Но здесь нам помогают физические свойства веществ. Спирты всегда менее летучи чем соответствующие олефины – и из-за большей молярной массы (более тяжелых молекул), и из-за водородных связей в жидкости. Поэтому мы можем, добавив сильную кислоту к спирту, просто нагреть и отогнать олефин, присоединив к реакционной колбе насадку с нисходящим холодильником. Саму реакцию часто просто ведут в приборе для перегонки. В отгоне будет в основном олефин. Из-за непрерывного удаления образующегося олефина равновесие по принципу Ле Шателье смещается в сторону олефина, и реакция проходит с хорошим выходом и вполне удобно.