Енолизуемые карбонильные соединения
Важнейшей особенностью многих карбонильных соединений является возможность обратимого превращения в электронодонорные олефины с высокой реакционной способностью, могущие служить нуклеофилами в огромном количестве важнейших реакций. Именно это свойство делает карбонильные соединения (не все, но большинство) универсальными реагентами в органических реакциях. Получается, что в каждое такое карбонильное соединение встроена кнопка, переключающая электрофил в нуклеофил и обратно. Строго говоря, карбонильные соединения позволяют переключать себя несколькими способами, но именно тот, которому посвящена эта страница основной и самый мощный.
Для того, чтобы эта возможность была, необходимо, чтобы рядом с карбонильной группой был атом углерода, несущий хотя бы один атом водорода. Такой атом углерода может быть один (в альдегидах и многих других карбонильных соединениях) или два (в кетонах). Такие карбонильные соединения имеют две формы – кетонную (всегда так называется, вне зависимости от того, чем реально является карбонильное соединение – альдегидом, кетоном, карбоновой кислотой или ее производным и т.п.) и енольную.
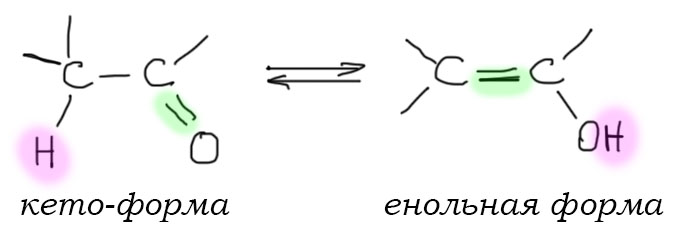
Сразу обращаем внимание на несколько важных вещей. Во-первых, это настоящее равновесие, а не мезомерия и граничные формы. Обе формы – настоящие молекулы, которые всегда существуют в реальности, но различаются по устойчивости в самом основном, термодинамическом смысле этого слова. Эти две формы связывает настоящее равновесие, которое принято называть кето-енольной таутомерией. Это старинный и довольно неудачный, но очень прочно укоренившийся термин, смысл которого можно даже не понимать, потому что главное здесь не то, как это называется и почему, а то, что это настоящее равновесие, которое мы очень легко можем понять как любое другое равновесие. Для интересующихся я где-нибудь внизу сделаю отдельный блок о том, что такое таутомерия.
Способность енолизуемых карбонильных соединений к обратимому образованию енолов имеет ряд чрезвычайно важных следствий.
- Способность к реакциям с электрофилами. Карбонильные соединения, как мы хорошо знаем, сами являются электрофилами, и поэтому вроде бы не должны реагировать с другими электрофилами, ведь одна из священных истин химии, которую мы каждый день повторяем, и состоит в том, что электрофилы не реагируют с электрофилами, а нуклеофилы не реагируют с нуклеофилами. Но енолизуемые карбонильные соединения реагируют с электрофилами, и именно потому что содержат в каких-то количествах нуклеофильный алкен – енол.
- Превращение кетона в енол происходит не само собой, а с помощью катализа кислотами или основаниями. Хорошо понимать эту особенность и как она работает чрезвычайно важно, потому что это объясняет не просто многое, а оень многое в поведении карбонильных соединений.
- Содержание енола в карбонильном соединении часто бывает очень мало, вполне достаточно для реакций с некоторыми электрофилами, но не достаточно для других реакций. При этом мы не можем целенаправленно превращать карбонильное соединение в соответствующий енол, потому что этот процесс управляется законами равновесия, и если равновесие смещено в сторону карбонильного соединения, то содержание енола в таком карбонильном соединении жестко ограничено очень малой константой равновесия. Не менее важно и то, что у нескоторых карбонильных соединений, например, несимметричных кетонов может быть не один енол, а два или даже более чем два разных, и мы никак не можем управлять их содержанием в равновесии – все диктуют константы равновесия. Но нам страшно повезло в том, что кроме енола существует много похожих на енол соединений, реакционная способность которых позволяет с успехом их использовать вместо енола, и отличающихся от енола тем, что их можно или получить как устойчивые реагенты, которые можно хранить и использовать по желанию, или использовать способы, которые позволяют получать их в реакционных смесях в значительно больших количествах и концентрациях чем енолы.
Кето-енольная таутомерия
Кето-енольная таутомерия. Кислотный и основный катализ.
В таутомерном превращении кетона в енол и обратно перемещается только один атом – водород, причем без электронов, то есть протон. Фокус состоит в том, что протон не умеет просто взять – и прыгнуть с одного атома на другой. Вернее, скажем так, умеет, но только в том случае, если эти два атома располагаются настолько рядом, что могут быть связаны водородной связью. Такие случаи в химии бывают, но кето-енольное превращение в простых карбонильных соединерях к ним не относится. Расстояние между углеродом и кислородом довольно значительно, водородной связи между ними нет. Кето и енол – две вполне самостоятельных молекулы, и для того, чтобы превратить одно в другое нужен посредник, который сначала возьмет протон, а затем отдаст его, или наоборот. Такой посредник называется переносчиком протона, и мы отлично знаем, что этот термин описывает хорошо знакомый нам класс веществ – кислоты и основания Бренстеда-Лоури. Переносчик протона, обеспечивающий превращение кетона в енол и обратно (напомню в очередной раз еще один из основных законов химии: прямую и обратную реакцию катализирует один и тот же катализатор по одному и тому же механизму). Посмотрим, как это делают кислоты.
Кислотный катализ таутомеризации
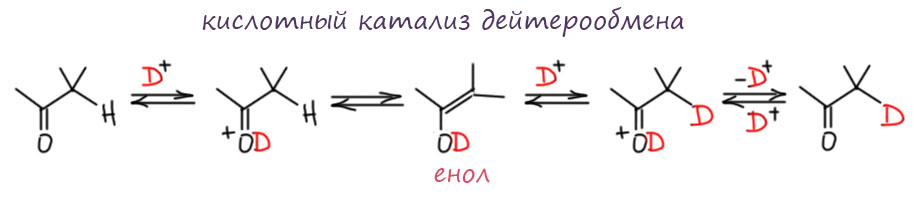
В химии много раз в разных местах повторяется одна и та же аргументация. Вот и в этот раз – хотим обсудить таутомерное превращение, а попадем прямиком в что-то похожее на E1-элиминирование. Действительно, протонирование карбонила по кислороду дает мезомерно-стабилизированный карбокатион, который по законам E1-элиминирования (в этом случае роль уходящей группы фактически играет π-связь карбонила C=O) отдает протон сопряженному основанию той кислоты, которая протонировала карбонил, и превращается в олефин, в этом случае олефином оказывается енол. Обратный процесс – взаимодействие очень реакционноспособного олефина енола по атому углерода с той же кислотой, причем получается тот же мезомерно стабилизированный карбокатион, и он отдает протон с атома кислорода сопряженному основанию протонирующей кислоты.
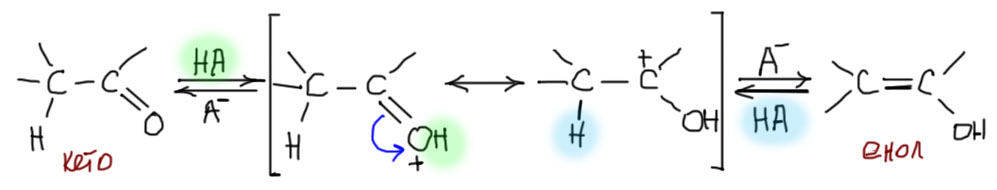
Обратим внимание и на катион в квадратных скобках – интермедиат между енолом и кето-формой. Формально это карбокатион, но реальная структура этой частицы ближе к форме протонированного по кислороду карбонила по очень простой причине – в этой форме все атомы имеют полную электронную оболочку, что всегда выгоднее, чем карбокатион с 6 электронами. Мы уже встречались с этим интермедиатом, например, когда рассматривали кислотный катализ реакции карбонильных соединений с нуклеофилами – протонирование кислорода увеличивает электрофильность карбонильного углерода. В этом одна из сложностей химии карбонильных соединений – одни и те же интермедиаты возникают в разных процессах, и тот же кислотный катализ активирует и присоединение к карбонильной группе, и кето-енольную таутомерию. Одно из последствий этого – кислотно-катализируемая альдольная конденсация.
При некоторой дополнительной дотошности можно усомниться в том, насколько действительно легко протонировать кетон или альдегид, ведь, наверное, это очень слабые основания, и кислота потребуется сильная. Да, это действительно так. Протонировать атом кислорода в карбонильной группе могут только действительно сильные кислоты, например, серная. Но, во-первых, для осуществления катализа нам не нужно, чтобы значительная часть или даже все карбонильное соединение в растворе было протонировано – достаточно только малой части, чтобы кето-енольное превращение стало возможно. Степень протонирования будет определять скорость этого процесса, скорость достижения равновесия. Меньше кислотность реакционной среды – меньше скорость, и все. И все? Нет, не совсем. Даже в присутствии слабых кислот, например, уксусной, равновесие устанавливается, хотя и гораздо медленнее, чем в присутствии сильной кислоты. Дело в том, что протонирование в полном смысле этого слова не обязательно. Достаточно образования водородной связи. Тогда при превращении кето-формы в енол получается что-то типа согласованного перемещения протона при посредничестве слабой кислоты – переносчика протона. Это даже можно изобразить внутримолекулярно, хотя это не обязательно, потому что можно использовать не одну, а две или более молекул переносчика. Обратим внимание на то, что в квадратных скобках изображены именно резонансные структуры, а не равновесие: в резонансных структурах все атомы, включая водороды, остаются на месте, смещается только электронная плотность.
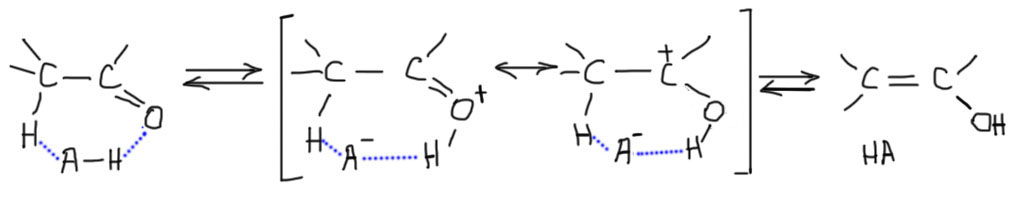
Такой кислотный катализ часто называют общим, потому что он работает всегда вне зависимости от способности кислоты к протонированию, и на него способен гораздо более широкий круг кислот. Скорость взаимопревращения кето-фомы и енола в этом случае зависит от общей концентрации кислоты, так же как и от ее константы кислотности. В химии карбонильных соединений этим видом катализа с удовольствием пользуются, прежде всего потому что этот тип катализа уменьшает вред от побочных реакций, в первую очередь альдольно-кротоновой самоконденсации, в которые часто вступают карбонильные соединения в присутствии сильных кислот.
Основный катализ таутомеризации
Пожалуйста, никогда не говорите “щелочной”, если не хотите прослыть незадачливыми гостями из далекого прошлого, когда серную кислоту называли купоросным маслом. Только оснóвный (не основнóй).
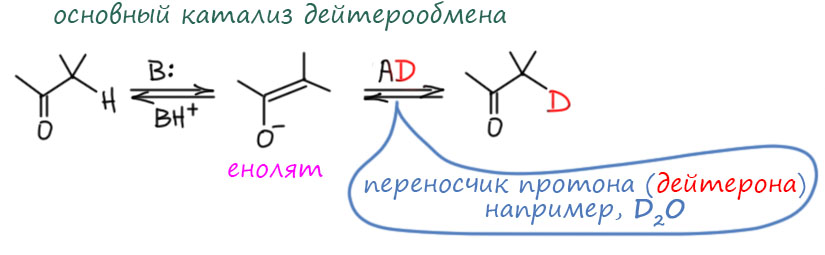
Суть основного катализа кето-енольного превращения – использование основания, которое может превращать как кето-форму, так и енол в сопряженное основание – енолят. Кето-форма – это CH-кислота, а енол – OH-кислота, просто спирт. Сопряженное основание у них одно, мезомерно-стабилизированный енолят, названный так потому что этот анион гораздо ближе соответствует форме с минусом на кислороде, гораздо более электроотрицательном атоме чем углерод. Так как перенос протона обратим, обратим и весь процесс, превращающий кето-форму в енол и обратно.
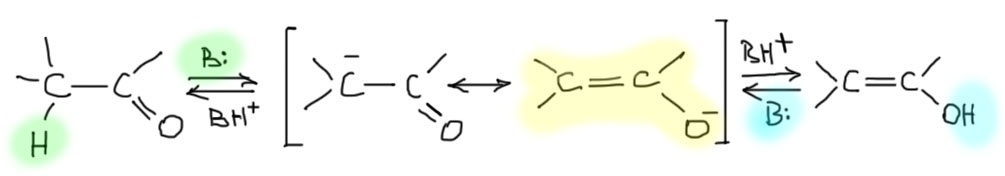
Какой силы должно быть основание, чтобы вызывать кето-енольное превращение? Здесь та же самая история, как и с кислотным катализом. От силы основания и его концентрации зависит скорость достижения равновесия, но даже более слабые основания по сравнению – по сравнению с чем? С кето-формой, конечно. Но кето-форма это не основание, а кислота. Отлично, именно так, потому что основность мы тоже измеряем константой кислотности сопряженной кислоты, то есть в данном случае как раз кето-формы. Это еще одна причина того, почему в химии принято характеризовать основания Бренстеда-Лоури не константой основности, а константой кислотности сопряженной кислоты. Получается невероятно удобно.
Итак, если мы посмотрим pK простых альдегидов и кетонов, то мы увидим величины порядка 24-26. Имеются в виду значения, полученные в полярном апротонном растворителе, но в реальности реакции альдегидов и кетонов с участием енолятов чаще делают в протонных растворителях типа спиртов или даже водных спиртов. Фокус в том, что мы не знаем надежной оценки pK альдегидов и кетонов в таких растворителях, но можем быть уверены, что это величины, существенно меньшие 24-26. Поэтому для обратимого депротонирования годятся простые основания типа алкоголятов или даже обычных щелочей. Очень часто останавливаются на таком компромиссе, как щелочь (LiOH, NaOH, KOH, Ca(OH)2 и т.п.), но в спиртовой среде (метанол, этанол), а не в водной. Основность гидроксид-аниона в спиртах несколько выше основности этого аниона в воде, а кислотность кето-форм альдегидов и кетонов, наоборот ниже чем в апротонной среде. К сожалению, мы не знаем надежных количественных оценок ни того, ни другого, потому что спирты – очень неприятные растворители для физико-химических измерений, но на качественном уровне понимаем, что разница не так велика, и основности гидроксид-иона в спиртах вполне достаточно для обратимого депротонирования кето-форм и катализа кето-енольной таутомеризации.
Соотношение кетонной и енольной форм
Если мы хорошо понимаем, что такое кислоты и основания Бренстеда-Лоури, а без этого в органической химии вообще никуда не проехать, то вопрос о соотношении кетонной и енольной форм у конкретных соединений поставит нас в тупик своей примитивностью. “Это же очевидно!” – воскликнем мы. Естественно, для того, чтобы ответить на этот вопрос количественно, нам потребовались бы надежные данные, но, как мы знаем, в химии обычно спрашивают не количественную оценку, а качественную, например, просят сравнить то и это, определить качественную тенденцию и т.п.
Если так, то у нас нет проблем. Кетон и енол – кислоты Бренстеда, причем очень забавные, потому что сопряженное основание у них одно и то же – енолят. Тогда все очевидно – протон будет в большей степени принадлежать более слабой кислоте, и чем больше разница в кислотности, тем больше будет разница в содержании кетонной и енольной форм – протон будет сидеть в той форме, кислотность которой меньше.

Получается даже нечто похожее на строгую формулу, ведь константа равновесия таутомерного превращения просто равна отношению констант кислотности енола и кето-формы. Но строгое применение этой формулы наталкивается на труднопреодолимые препятствия, потому что количественные величины этих констант редко известны. Но нам формула и не нужна, а нужно понять, какой формы больше, то есть больше или меньше единицы эта константа.
Вот откуда мы знаем кислотность енола и кето-формы? Строго говоря, как и почти все в органической химии – от фонаря. Того самого, под которым рекомендуется искать потерянные вещи, так как там светло и хотя бы что-то видно. Берем качественные оценки этих характеристик, используя разумные рассуждения и оценки. При этом мы можем рассуждать, например, так. Енол – это спирт, поэтому его кислотность не меньше, чем у обычного спирта типа этанола. Скорее всего даже немного больше, потому что все таки двойная связь сопряжена с неподеленной парой на кислороде, а это мы привыкли считать выгодным, стабилизирующим взаимодействием. А кето-форма, если мы говорим о простых альдегидах и кетонах, это CH-кислота с существенно более высоким pK. Разница между кислотностью енола и кето-формы может составлять от 5 до 10 порядков константы кислотности (от 5 до 10 единиц pK). Кето-форма – существенно более слабая кислота чем енол. Из этого непосредственно следует, что в простых альдегидах и кетонах кето-форма будет преобладать, и это полностью соответствует оценкам содержания енола в простых кетонах типа ацетона или циклогексанона в 0.0001 – 0.000001%.
Интересно, что если мы возьмем другие карбонильные соединения, то енольная форма довольно слабо чувствует изменения структуры, так как это всегда просто непредельный спирт, и кислотности енолов изменяются в довольно узких пределах. А вот CH-кислотность кето-формы гораздо более чувствительна к особенностям структуры и наличия стабилизирующих заместителей. Вот, например, скоро нам придется иметь дело с 1,3-дикетонами и похожими на них соединениями. В этих случаях CH-кислотность резко растет и запрыгивает уже сильно за пределы кислотности спиртов. Кето-форма становится более сильной кислотой чем енол. Результат – полное обращение соотношения кето/енол – именно енол становится преобладающей формой. Мы вернемся к этой проблеме в следующей теме.
Не путайте таутомерное равновесие кето-форма – енол и мезомерию енолята!!!
Как однажды сказал один из деятелей наполеоновской Франции по существенно менее важному поводу: “Это хуже чем преступление. Это ошибка.” Я бы даже сказал, что это не просто ошибка, а мать всех ошибок. Ведь действительно, как похоже:
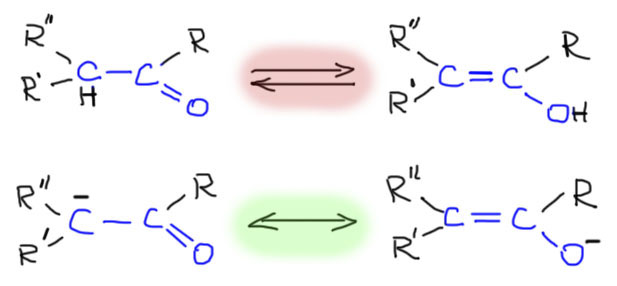
Но это две принципиально разные вещи. Извиняюсь за напоминание тривиальных вещей, но это действительно очень важно, потому что одна из основных причин ошибок – неряшливое, приблизительное отношение к основным понятиям. Поэтому напоминаю, что кето-форма и енол – две разные молекулы, которые могут превращаться друг в друга с большей или меньшей скоростью в присутствии кислотных или основных катализаторов, и связаны равновесием, которое изображается двойной стрелкой типа “туда-сюда”. Если умудриться получить чистые формы и тщательно исключить любые кислотные или основные примеси (придется, например, отказаться от стекла), а это сделать невероятно трудно, но не невозможно – кето-форма будет сохранять чистоту практически неограниченно, хотя енольная форма не сможет этого сделать, потому что сама обладает заметной бренстедовской кислотностью, а следовательно способна сама катализировать собственное превращение в кето-форму. В равновесных смесях мы можем отлично видеть каждую из форм разными спектроскопическими методами. Каждая из форм обладает своей и очень характерной реакционной способностью.
Енолят с конкретной структурой – одна молекула (ион – это тоже молекула). Структура енолята очень близка к правой граничной структуре, поэтому мы так его и называем, но теория мезомерии обязывает нас изображать и “кетонную” граничную структуру, связав обе обоюдоострой стрелкой, специально зарезервированной для этой теории, и не имеющей никаких других применений в химии. Эта стрелка означает то, что мы не имеем дело с реальными молекулами слева и справа от стрелки, но утверждаем, что реальная структура где-то между этими граничными случаями – не обязательно посредине, возможно даже, что очень близко к одной из них, но все равно для понимания поведения реальной молекулы надо иметь в виду, что каждая из граничных структур несет важную информацию. В случае енолята, например, хотя структура иона енолята очень близка к енолятной форме, но его поведение в реакциях говорит о делокализации электронной плотности. Наилучшим образом это можно было бы увидеть, разглядывая молекулярные орбитали енолята, но как-то у нас это не принято, и придется с этим смириться. Впрочем, немного пониже мы их все-таки порязглядываем.
Существует ли виниловый спирт и другие простые енолы?
Простое правило гласит, что непредельные спирты с гидроксильной группой у двойной связи неустойчивы и самопроизвольно перегруппировываются в карбонильные соединения. Правилу этому очень много лет (оно основано на работах 1877 и 1880 года), в нашей стране оно известно как правило Эльтекова-Эрленмейера, а в остальном мире просто как правило Эрленмейра, и относится оно к большой группе эмпирических правил, унаследованных нами от описательной науки 19 века, когда еще не было никаких представлений о химической связи, электронах и прочих важных вещах, а выводы делались на основе интуитивного обобщения накопленного, ещё не очень обширного экспериментального мтериала. Польза от этих правил для истории органической химии была несомненно, поэтому мы и поминаем их до сих пор, в основном просто чтобы показать, что мы соблюдаем приличия, и не занимаемся каждые 50 лет выбрасыванием с корабля всего, что на нём накопилось. Но правила необходимо переосмыслять на основании и нового материала, и новых понятий и теорий. То же и с правилом Эльтекова-Эрленмейера. Оно не абсолютно, а всего лишь выражает хорошо нам уже известное наблюдение, что для простых енолизуемых карбонильных соединений равновесие енолизации сдвинутов сторону кето-форм. И более того, постольку мы теперь знаем намного больше, то утверждаем, что для превращения виниловых спиртов в карбонильные соединения обязателен посредник, переносчик протона, кислота или основание Бренстеда-Лоури, и что после установления равновесия некоторое количество такого непредельного спирта, или енола, остается и может быть обнаружено с помощью высокочувствительных физико-химических методов, и даже просто по реакционной способности. Но значит ли это, что простые енолы в принципе можно получить как самостоятельные молекулы и даже вещества, и как-то наблюдать за их жизнью. Существуют ли самые простые енолы, например, енол ацетальдегида – виниловый спирт – или енол ацетона – пропен-2-ол?
Да, безусловно существуют, и за последние полвека в литературе описаны многочисленные успешные попытки получения и того, и другого. И вполне доказанным можно считать то, что эти соединения вполне устойчивы, если из системы исключены любые кислоты и основания Бренстеда-Лоури. А вот если они присутствуют, хотя бы в следовых количествах, то простые енолы быстро перегруппировываются в ацетальдегид или ацетон. Получить простые енолы можно многими способами. Самый очевидный – быстрое подкисление енолята в растворе сильной кислотой – в этом случае перенос протона происходит чрезвычайно быстро, и образующийся енол вполне удается увидеть с помощью какой-нибудь быстрой спектроскопии (к сожалению, к таким методам не относится ЯМР – для регистрации спектра ЯМР по принципиальным причинам требуется значительное время, скажем очень приблизительно, что это время измеряется секундами, в то время как таутомерное превращение простых енолов в среде, где есть переносчики протонов, а без них подкислить ничего нельзя, – измеряется долями миллионных долей секунды). Но есть много очень шустрых спектроскопических методов, и с их помощью виниловый и пропениловый спирты были не только обнаружены, но и измерены кинетические параметры перегруппировки. Метод оказался хорош, и если бы удалось добавить настолько точное количество кислоты, чтобы каждая молекула енолята превратилась бы в енол – то можно было бы попробовать получить чистый енол в отсутствие катализаторов. Увы, это за пределами возможностей эксперимента – ничтожный избыток кислоты создаст кислотный катализ, а ничтожный недостаток – основный, ведь непрореагировавший енолят – очень сильное основание, и будет сам катализировать превращение енола в кето-форму.
Исключить сильные кислоты и основания удавалось более инструментальными методами, например, вакуумным пиролизом (очень быстрым нагреванием разреженного пара) этиленгликоля – при этом происходит термическое элиминирование воды. В такой системе в парах при пониженном давлении виниловый спирт устойчив минутами и даже часами. Но и там есть вода, и хотя в парах вероятность встречи молекулы винилового спирта и молекулы воды невелика, а вода это слабая кислота Бренстеда-Лоури, и вполне способна вызывать более медленное, но неотвратимое превращение енола в кето-форму. Сконденсировать эти пары и получить жидкий виниловый спирт тоже идея спорная, потому что сам виниловый спирт является слабой кислотой, более слабой чем вода, но все равно способной катализировать превращение других молекул винилового спирта в ацетальдегид.
Но в высоком вакууме, где молекул мало и вероятность их встречи пренебрежимо мала, виниловый спирт и другие простые енолы, скорее всего, как говорят, бесконечно устойчивы – то есть не превращаются с измеримой скоростью в свои таутомеры. Одно из надежных доказательств этого найдено ни много ни мало – в космосе. Еще в 2001 году было обнаружено, что в нашей родной галактике в межзвездном пространстве полно винилового спирта, Полно, естественно, в космическом смысле – от одной молекулы до другой пришлось бы пару сотен лет пешком идти. Вот и хорошо, но молекулы-то есть, существуют там тоже в космической шкале времени, возможно, найдутся и такие, которые динозавров наших из своего далека видели (не придирайтесь к скорости света, у нас не очень большая галактика, плюс-минус 50-100 тысяч лет это не принципиально). И не превращаются в ацетальдегид (давно бы превратились, если бы это происходило). Обязательно кто-нибудь заметит – да ведь там чертовски холодно, мало ли какая молекула может выжить при температуре жидкого гелия. Верно, но здесь весь вопрос в том, сколько выжить. Таутомеризация – это просто перемещение протона, и если бы этот процесс мог происходить без посредников, то низкая температура ему бы не помешала.
И все это вместе представляет собой чрезвычайно поучительную историю – как бы ни была неустойчива или реакционноспособна молекула, но если нет условий для реализации механизма ее превращения в другие молекулы, например, катализатора, то она будет ждать своего часа столько, сколько захочет ей предоставить слепой случай, то есть, возможно, вечно. И скоро мы увидим другие, уже совершенно земные примеры неограниченно устойчивых енолов, которые подтверждают это правило без космического экстремизма.
Реакции карбонильных соединений, происходящие за счет енола
Содержание енолов в простых альдегидах и кетонах – величины порядка от одной тысячной до одной десятимиллионной части процента. И что – мы будем обсуждать такую безделицу?! Какой смысл обсуждать такие ничтожные примеси? Ведь если мы возьмем ацетальдегид, ацетон или циклогексанон, даже очень чистый, из хорошей реактивной лавки типа Мерк-Сигма-Олдрич, обслуживающей весь мир первосортными реактивами, то будем на банках иметь чистоту 97-98%. А что все остальное? Вода, и еще много всякой дряни и ерунды. И такой чистоты обычно хватает для большинства работ. Если раскошелимся, можем купить и высокочистые реактивы – 99%, даже 99,9%, – и все равно 0.1% примесей. А енола – в сто, тысяча, и даже больше раз меньше чем этих никому не интересных примесей. Почему нас не волнуют эти анонимные примеси, но волнует нечто в тысячи раз более ничтожное?
Прежде всего потому что у енола есть два свойства, которые делают ничтожную примесь критически важной.
- У енола другая реакционная способность – это чрезвычайно реакционноспособный олефин, который охотно и очень быстро реагирует со многими электрофилами;
- если включается кето-енольное равновесие, то на смену прореагировавшему енолу получается новый, он тоже реагирует, и так далее – работает принцип Ле Шателье, равновесие смещается в ту сторону, которая куда-то девается, и нужно все время производить новый енол, иначе может обидеться константа равновесия, обнаружив, что кто-то стырил ее числитель, и она превратилась в бесконечно малую. И так может прореагировать весь альдегид или кетон – через енол. Получается, что енол – это такая дверка потайная, мизерная, не видная даже в микроскоп, но через которую умудряется полностью перебраться в другое место енолизуемое карбонильное соединение. Но чтобы это произошло, должно включиться кето-енольное равновесие, то есть должен найтись рядом и благородно подставить плечо кислотный или основный катализатор. Если этого не произойдет, то наличный енол прореагирует и дело встанет, и мы даже этого не заметим, потому что мало кто интересуется реакциями с выходом в тысячную долю процента.
Посмотрим поподробнее на некоторые важные реакции, в которых енолизуемое карбонильное соединение реагирует через енол.
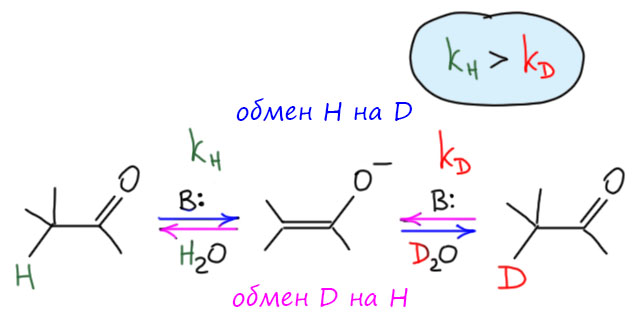
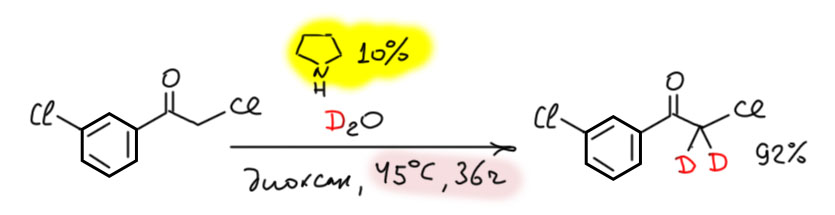
Начнём мы с очень специфической, но очень важной реакции – дейтерообмена. В этой реакции фактически ничего не происходит, не изменяются ни скелет, ни какие-то группы – можно даже и не заметить, что что-то происходит. Но среди изотопов элементов пара протий-дейтерий занимает совершенно особенное и совершенно уникальное место – это единственный пример изотопного обмена с устойчивым, нерадиоактивным изотопом, который приводит к серьёзному изменению свойств, настолько серьёзному, что это часто можно видеть “невооруженным глазом” и не требуются прецизионные измерения свойств или скоростей реакций. Поэтому дейтерообмен очень важен и не только в исследованиях, но и с практической точки зрения. Разберемся в нем поэтому попобробнее, впрочем, как всегда, можно ограничиться основами (на первой вкладке), а подробности – для любопытных, факультативно, на следующих вкладках.
Дейтерообмен. Самое важное.
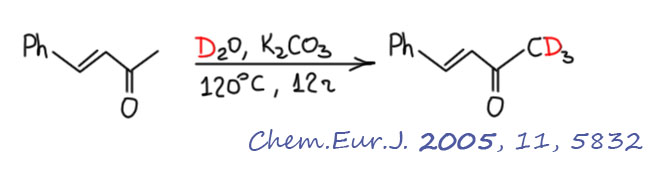
Первое интересное следствие обратимого образования енола – дейтерообмен α-протонов. Он происходит в том случае, если карбонильное соединение растворить в протонодонорной среде, в которой обменивающиеся протоны заменены на дейтерий, и в присутствии каталитического количества кислоты или основания. Обмениваются только протоны на соседних с карбонилом атомах углерода и только на насыщенных, например:
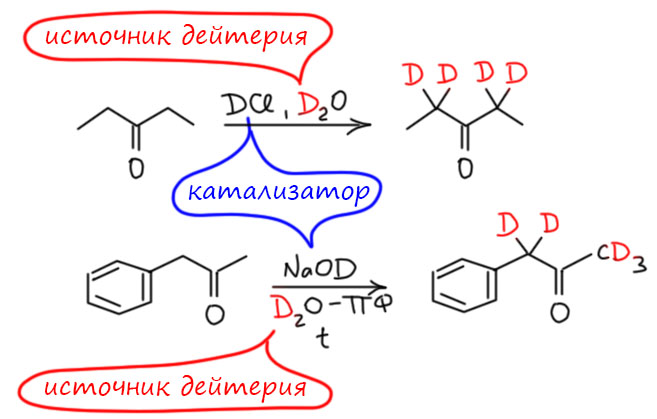
Обратите на это внимание – протоны (дейтероны) идут не от кислоты или основания, а от среды. Кислота или основание – катализаторы, их может быть очень мало, и они могут быть даже самые обычные, хотя если есть под рукой дейтерированные, то и хорошо – это чуть-чуть ускорит дело, а почему, скоро разберемся. Средой обычно служит тяжелая вода или ее смесь с апротонным растворителем (только не берите ацетон!!!). Но можно, в принципе, взять спирты с дейтерированным гидроксилом (остальные протоны вполне могут быть обычными), или уксусную кислоту или что-то подобное, причем и в том и в том дейтерием должен быть только кислый гидрон.
См дейтерообмен идет по очевидной причине, хотя для кислотных и основных условий придется написать немного разные схемы. Если немного подумать, то механизм дейтерообмена это то е самое, что механизм кислотного и основного катализа кето-енольной таутомерии, только в среде, являющейся донором не протонов, а дейтеронов. В кислотном катализе енол будет протонироваться кислотой, переносящей протон (дейтерон) от протонодонорной среды, например, воды. В основных условиях, протонироваться будет уже енолят в равновесии сопряженной кислотой основания, опять таки, переносящей протон (дейтерон) от среды.
А теперь внимание! Это очень жесткий тест на то, понимаем мы или нет, что такое равновесие. Равновесие – это одновременно и страшная, почти непреодолимая сила, и одновременно чрезвычайно грубый инструмент Природы. Для работы с равновесием Природа выдала нам единственный инструмент – принцип Ле Шателье, и другого нет и не предвидится. Это обстоятельство просто бесит, но придется смириться. И не забывать неустанно прославлять Анри Луи Ле Шателье, который снабдил нас таким мощныи и единственным инструментом на все времена. Говорят, почти то же самое сформулировал одновременно Вант Гофф, но тот столько всего сформулировал, что не убудет, если здесь отдаст кусочек славы другому. В этом, может быть, есть глубокий смысл – если захотите что-нибудь сформулировать важное, не жадничайте, и формулируйте понемногу, давайте людям возможность переварить сформулированное. Люди будут вам благодарны и понесут ваше имя в вечность. А то бывают такие персонажи – так любят формулировать, что уже если начнут, никак не могут остановиться, всё формулируют и формулируют. Люди обычно с подозрением к таким относятся и немедленно забывают всё наформулированное захлёбом.
Так вот, если у нас в кетоне или альдегиде несколько α-протонов (в ацетальдегиде и ацетофеноне три, в циклогексане 4, в ацетоне аж 6, и больше не бывает), то мы ни при каких обстоятельствах и ни в каких обратимых условиях не сможем обменять только часть из них, например, один. Не поможет ни недостаток или избыток чего бы то ни было, ни температура, ни время реакции, ни заклинания, ни проклятия, ни помощь потусторонних сил, ничего. Скорости и константы равновесия для недейтерированных и дейтерированных субстратов отличаются, но не сильно и этим можно пренебречь (здесь есть нюансы и ниже мы их немного разберём), считая вероятность обмена просто равной вероятности встретить молекулы с разным числом атомов дейтерия, а это просто их соотношение в растворе. Например, если у нас обмениваемых протонов два, а мы хотим один и возьмем один эквивалент источника дейтерия (полмоля тяжелой воды на моль кетона) то в самом начале процесса у нас будет обмениваться один протон, но как только монодейтерированный продукт накопится хотя бы в количестве 10%, вероятность обмена второго протона станет вполне существенной, округленно 0.1 и начнет накапливаться дидейтерированный продукт, и далее все пойдет со все более сближающимися скоростями (или вероятностями, что в данном случае одно и то же).
Более того, даже если мы получим каким-нибудь целенаправленным синтезом частично дейтерированный кетон или альдегид, скажем, монодейтероацетон (почему бы и нет – вы сами без труда напишете такие синтезы, например, восстановление формальдегида дейтерированным алюмогидридом лития даст вам монодейтерометанол, который вы без труда превратите несколькими способами в монодейтероацетон), то при выделении этого продукта из реакционной смеси вам придется тщательно избегать присутствия кислот и оснований, и источника протонов, например, воды. Если вы вспомните, как обычно заканчиваются органические реакции и как из реакционной смеси добывается продукт, то вы поймете, что действовать придется с полным включением сознания, обдумывая каждый шаг прежде чем реально его делать. Иначе весь ваш с таким трудом введенный дейтерий слетит и размажется – по-серьезному это называется рандомизацией метки, то есть приведением изотопного соотношения в соответствие с неумолимыми законами теории вероятности.
Единственно, что мы можем сделать, это обменять все α-протоны на дейтерий. И это не совсем просто, если мы хотим достигнуть хорошей степени обмена. Сразу поймем, что 100%-ного обмена в одной реакционной смеси достичь невозможно, так как протоны, выходящие из объекта обмена включаются в общее равновесие, и не дают ему прийти к полной замене протона на дейтерон. Скажем, если у нас два протона и мы взяли 100-кратный избыток воды (то есть соотношение D:H = 100:1), то обменять больше 99% у нас не получится. И это отличный результат – редко нужно больше. Но если нужно 99.9, то нам потребуется 1000-кратный избыток тяжелой воды. И хотя тяжелая вода не самый дорогой на свете реактив, но даже 100-кратный избыток может остановить многих, а 1000-кратный. Такой несуразный избыток, скорее всего, остановил бы всех желающих даже в некоторых странах Персидского залива, где деньги, выделяемые на науку, измеряются порядками числа Авогадро, и уже точно ни в какие ворота не лезет в странах, где соответствующие фонды имеют порядок постоянной Планка.
Средство от этого хорошо известно вообще в любых областях, где имеют дело с равновесием. В экстракции, хроматографии, ректификации, делении изотопов и т.д., и т.п. всегда делают одно и то же – то, что не удается достичь одним равновесием, получается, если процесс достижения равновесия последовательно повторить несколько, иногда много раз. Так и здесь. Первый раз берем 10-кратный избыток тяжелой воды. Получим что-то около 10% остаточных протонов, или дейтериевую чистоту в 85-90%. Выделяем из реакционной смеси кетон с таким содержанием, и повторно запускаем с тем же количеством тяжелой воды. В этот раз по отношению к остаточным протонам избыток источника дейтерия получится уже почти 100-кратным, соотвественно и результатом будет остаточное содержание протонов сильно ниже 1%. Выделяем, пускаем третий раз в тех же условиях. По отношению у остатоным протонам избыток дейтерия станет уже ближе в 1000-кратному, результат будет соотвествующий, и это всего за три раза с суммарным избытком всего 30-кратным. Более того, мы сможем еще сэкономить, если будем повторять процесс с новыми порциями кетона, тогда оставшуюся тяжелую воду с 3 стадии, содержащую небольшое количество обычных протонов, используем на второй в следующий раз, а со второй – на первой. Вот как-то так приблизительно и делают.
Зачем? Например, для того чтобы делать растворители для ЯМР. Все современные приборы (современные = последние лет 40) требуют использования дейтерированных растворителей не только для того, чтобы протоны растворителя не мешались, но и для автоматической настройки параметров прибора. Все известные растворители имеют дейтерированные версии, которые можно купить. Но стоят они все очень по-разному. Есть более доступные (дейтерохлороформ, гексадейтероацетон, гексадейтеродиметилсульфоксид, тяжелая вода), а есть очень дорогие (дейтерированные ТГФ, дихлорметан, диметилформамид и т.п.). Разница в цене определяется очень просто – все, что можно получить прямым дейтерообменом, приблизительно так, как мы это только что разобрали, стоят, условно говоря, по цене дейтерия, потому что сам процесс довольно прост и эффективен, а первичный источник дейтерия, тяжелая вода, производится атомной промышленностью в неограниченных количествах. А те, что нельзя получить прямым дейтерообменом, а требуется многостадийный синтез с использованием меченых реагентов, дороги, потому что приходится оплачивать сложную препаративную работу.
И когда в следующий раз будете делать образец для ЯМР, растворяя свое вещество в дейтерохлороформе или дейтероацетоне, обратите внимание на этикетку растворителя, где всегда указаны остаточные протоны. И обратите внимание на то, что 100%-но чистые дейтерированные растворители почти никогда не используются, хотя есть в продаже, но очень дороги потому что прямым дейтерообменом можно получить какую угодно чистоту, хоть 99,99999%, но потребуется на это очень много повторов равновесия обмена.
Безусловно, есть не только равновесные, термодинамические способы дейтерообмена, но и кинетические – когда используется какая-то конкретная необратимая реакция с источником дейтерия, например, реакция нестабилизированной литий или магнийорганики с дейтерированной кислотой. Или гидрирование дейтерием. Или восстановление карбонильной группы алюмодейтеридом лития. Что-то такое можно придумать и для частичной замены протонов на дейтерий в енолизуемых положениях. Только не забудьте, что продукт такой реакции придется выделять из реакционной смеси – вам придется найти такой способ, который не приведет к включению равновесного обмена и размыванию уже введенной дейтериевой метки. В химии альдегидов и кетонов это очень непростая задача.
Насколько легко идет дейтерообмен
Тезис первый: дейтерообмен – реакция весьма небыстрая, но из этого не следует, что можно сделать ее ступенчатой, сначала один дейтерий, потом второй и так далее. Не получится – всегда будут смеси, от начала реакции до конца. Поэтому дейтерообмен всегда делают в расчете на полное дейтерирование – обмен всех енолизуемых протонов на дейтерии. Для этого используют большой избыток (50-100 кратный) источника дейтерия, почти всегда это тяжелая вода, самый дешевый источник дейтерия.
Тезис второй: хоть дейтерообмен – реакция небыстрая, но это когда надо получить дейтерированное производное. А когда есть возможность испортить уже готовое, нужно не зевать, если у вас есть дейтерированный кетон, вы его купили или с большими трудами получили, то ни в коем случае не нужно забывать его в протонной среде (в протонных растворителях: воде, спиртах, карбоновых кислотах и т.п.) в присутствии оснований или кислот, любых – Бренстеда-Лоури или Льюиса. Вы очень быстро получите испорченное дейтерированное соединение – количество обычных протонов станет таким, что возможно, задуманный эксперимент, для которого могло быть нужно иметь большое содержание дейтерия, придется отложить. Если вы уже имели дело с приготовлением образцов для ЯМР, то могли заметить, что такие растворители как дейтероацетон или дейтеродиметилсульфоксид (в сульфоксиде дейтерообмен происходит по похожим законам, хотя катализ годится только основный), когда отрываете новую баночку, такие все очень чистенькие, и вы видите только очень маленький и очень характерный сигнальчик остаточных протонов, который даже полезен, потому что по нему удобно проверять шкалу химических сдвигов. Но когда баночка уже старая, рестворителя в ней уже меньше половины, ее часто используют и не всегда хорошо закрывают крышку, а даже если и хорошо, то имеют обыкновение держать баночку открытой, пока не спеша готовят образец – то мы уже, во первых, видим немаленький широкий сигнал воды, а во-вторых, сигнал остаточных протонов становится уже сосем не сигнальчиком, а так хорошо возвышается над сигналами собственно образца. Это следствие того, что обычная вода, которую успели нахватать растворители, а все сухие растворители по определению гигроскопичны, вступает в изотопный обмен и обратно вытесняет дейтерии. Этот процесс катализируется просто стеклом баночки, да и кроме того, за месяцы даже и без катализа равновесие все равно берет своё. Чтобы этого избежать и расторители не портились, во первых, стараются максимально сократить “время открытой крышки”, и, во-вторых, сразу после открывания свежей баночки добавляют немного свежеактивированных молекулярных сит.
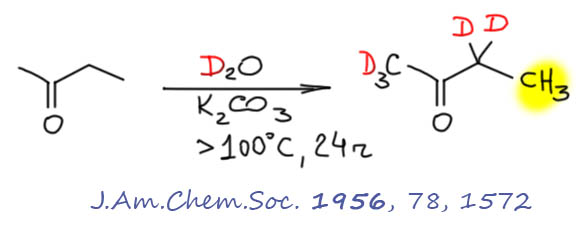
Хотя дейтерообмен идет и в присутствии кислот, и в присутствии основний, но, во-первых, всё же по-разному, и во-вторых, есть большая разница между сильными и слабыми основаниями. При выполнении дейтерообмена еще очень важно помнить, что те же катализаторы вызывают и другие реакции, в первую очередь альдольно-кротоновую самоконденсацию. Это, во-первых, делает практически невозможным дейтерообмен в альдегидах – они очень быстро самоконденсируются, и большая часть уйдет в продукты конденсации. Поэтому дейтерированные альдегиды, если понадобятся, лучше получать не дейтерооменом, а синтезом. Далее, даже у кетонов сильные кислоты и основания скорее вызовую самоконденсацию, чем слабые, поэтому в препаративной практике чаще применяют именно слабые основания, и намного чаще, чем слабые кислоты, с которыми процессы идут слишком медленно. Самая популярная методика использует карбонаты калия или натрия, в тяжелой воде, чистой, или с добавлением смешивающегося с водой органического растворителя типа диоксана или ТГФ. Реакция идет весьма медленно, и для установленя равновесия требуется многочасовое кипячение реакционной смеси, например:
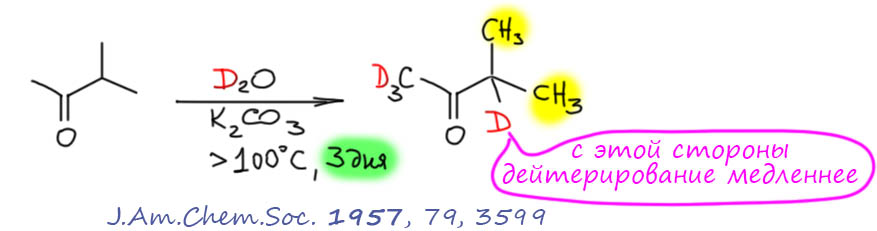
Еще медленнее идут реакции с разветвленными кетонами, например, содержащими вторичные алкилы. Вроде бы, как мы обсуждали, у таких кетонов более устойчивые енолы, но не надо забывать, что енолизация в присутствии оснований зависит от кислотности протонов, а в разветвленных кетонах кислотность ниже (донорный эффект алкилов) и основание медленнее отрывает протон с этой стороны. Только не думайте, что вы сможете, пользуясь разницей в скорости энолизации, успеть загнать дейтерии только с незамещенной стороны – получите неполное дейтерирование, смесь, никому не нужную. И не надо забывать, что и здесь и предыдущем примере эту процедуру повторяют несколько раз, каждый раз выделяя продукт (но без очистки) и загружая в новый цикл дейтерообмена, так что суммарное время реакций исчисляется неделями. С продукта каждый раз делают ЯМР и определяют количество оставшихся протонов (это очень просто сделать, если в соединении остаются недейтерированные группы, тогда ответ дает относительный интеграл остаточных протонов и недейтерированной группы)
Более сильные основния тоже иногда используют, это быстрее, но нужно строго следить, чтобы реакция не зашла в самоконденсацию слишком глубоко. Впрочем, в работах, где используют дейтерированные соединения, обычно решают какие-то сложные и дорогие задачи, денег обычно у таких исследователей много, тяжелой воды не жалеют, и за выходами не гонятся – если успеет сконденсироваться и осмолиться 80% исходного, то никто не парится. Если попадутся когда-нибудь статьи с такойхимией, не пугайтесь выходов дейтерированного продукта процентов в 15 – это как раз и означает, что пока сутками кипятили, большая часть успела накрыться.
Что делают с тяжелой водой из-под дейтерообмена? Собирают и отправляют на регенерацию, если есть такая служба. В промышленности очистка тяжелой воды от обычной проблемы не представляет, но гораздо дешевле очистить от примеси обычной воды “испачканную” тяжелую воду, чем делать ее заново. Если объёмы небольшие, но дейтерообменом занимаются часто, то есть смысл в лаборатории собирать тяжелую воду с отдельных стадий дейтерообмена, и заново использовать на ранних стадиях. Если, например, дейтерирование делают за пять повторов, а так бывает довольно часто, если нужно содержание дейтерия выше 99%, то на первой стадии можно использовать 90%-ную воду практически с тем же результатом, как и намного более дорогую свежую 99%-ную. Но на последующих стадиях качество тяжелой воды увеличивают.
Кислотный катализ тоже используют, обычно берут дейтерированную соляную, иногда серную кислоту. Если дейтерированной солянки под руками нет, а есть только тяжёлая вода, можно взять немного любого быстро гидролизующегося соединения, типа безводного хлористого алюминия или триметилсилилхлорида. Вот пример полного дейтерирования, заметно, что условия помягче, но в общем не так уж радикально:
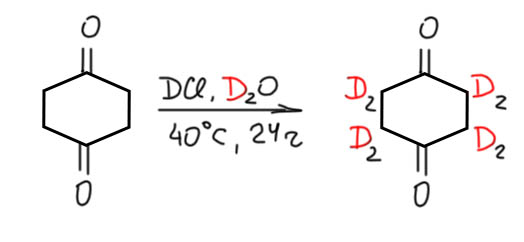
И вот дикетон с большим расстоянием между карбонилами, где есть неенолизуемые положения. Поскольку соединение крупное, в воде нерастворимо, то приходится добавлять органический растворитель, весьма немудреный, но это тоже делает условия более мягкими:
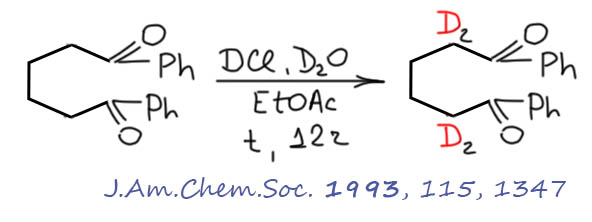
Зависит ли скорость дейтерообмена от CH-кислотности
Конечно зависит. Особенно сильно это проявляется в реакциях катализируемых основаниями, так как скорость-определяющей стадией всегда является отщепление протона. Мы уже видели это в сильном замедлении дейтерирования кетона с вторичной алкильной группой. Можно даже пойти дальше и поискать что-нибудь с кислотностью меньше, чем у карбонильных соединений. Среди карбонильных соединений мы такого не найдём, но можем взять, например, терминальные ацетилены – это рК немного меньше 30. Мы уже не будем делать дейтерирование в условиях равновесного обмена, но вполне могли бы, взяв основание посильнее, например, трет-бутилат калия в трет-бутаноле t-BuOD (в трет-бутиле дейтерии не нужны). Проблема в том, что мы вызовем ацетилен-алленовую перегруппировку и получим дейтерированную смесь. Нужна она нам? Скорее всего нет, нам не нужны смеси. Поэтому, когда мы хотим запихать дейтерий в терминальное положение ацетиленов, мы количественно отщепляем протон, переводим ацетилен в литиевое, натриевое, или магниевое производное, и после аккуратно гасим его тяжелой водой.
Но если наоборот увеличивать кислотность, мы получим значительное ускорение дейтерообмена. Увеличить кислотность у карбонильных соединений проще простого – навесить еще один акцептор на енолизуемю группу. Мы получим 1,3-дикетоны, β-кетоэфиры, и т.п. – все это великолепные CH-кислоты с повышенной кислотностью и рК в районе 10-12 единиц, для депротонирвоания которых можно применять самые простые основания типа щелочей, карбонатов, третичных аминов и т.п., а анионы которых расторяются в воде и не подвергаются ощутимому гидролизу. Мы очень серьёзно займёмся этими соединениями в следующем семестре, а сейчас убедимся, чт дейтерообмен в таких соединенияъ, особенно в присуствии основных катализаторов очень быстр. Вот здесь, наконец, мы получаем возможность частичного дейтерирования. Вот, например, типичное такое соединение, ацетилацетон. Во-первых, мы сразу должны сказать, что такие соединения в основном сущесвуют в виде стабильных енолов, дополнительно стабилизированных внутримолекулярной водородной связью.
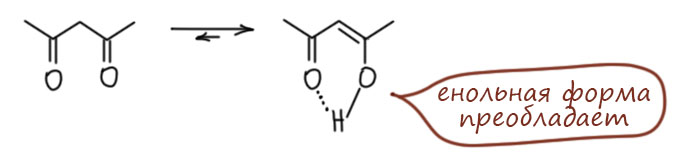
Поэтому для дейтерообмена этих кислых протонов ничего кроме самое воды не нужно. Но малейшие количества кислот или оснований делают этот обмен совсем быстрым.
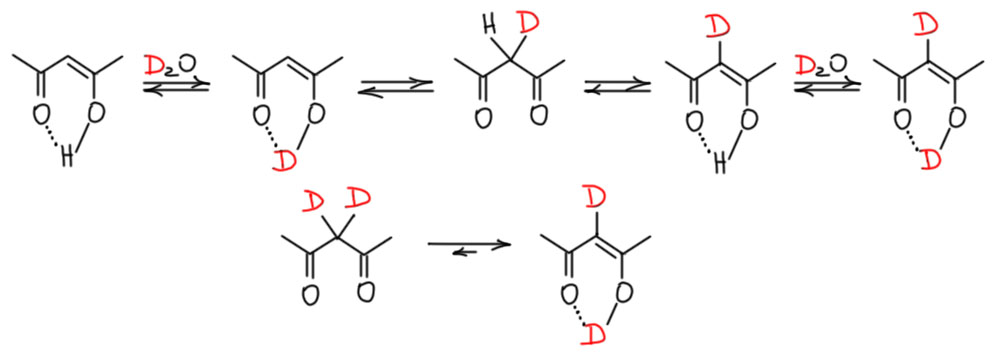
Если нужно заменить все альфа-протоны, то придется действовать так же как с обычными кетонами – долго кипятить в присутствии карбоната калия или что-то из этой серии.
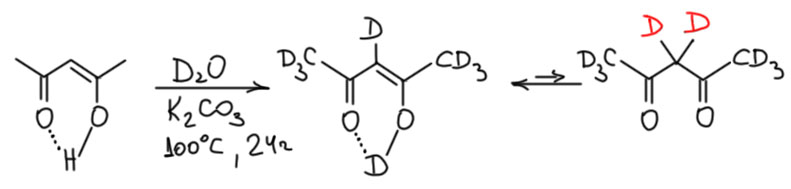
Надо только не забывать, что при выделении полностью дейтерированного соединения такого типа придется строго исключить не только обычную воду, но и все другие протонные растворители, напрмер, спирты. И апротонные растворители придется проверить на содержание в них воды. Потому что иначе два кислых дейтерия слетят в мгновение. Но, с другой стороны так можно получть соединение, дейтерированное только по метилам.
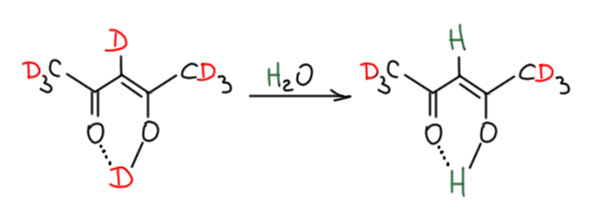
Обменять шило на мыло не то же самое, что обменять мыло на шило
Когда мы обсуждаем дейтерообмен, всегда говорим, что обычный водород обменивается на дейтерий обратимо, то есть в условиях изотопного обмена идут все возможные реакции, меняющие протон на дейтерон и дейтерон на протон. В этом случае результат реакции будет определяться простой теорией вероятности, так как мы неявно считаем оба процесса равновероятными, и в каждой стадии этого процесса должны просто прикинуть относительные количества атомов каждого сорта и из этого делать прогноз.
Но это не совсем верно, а точнее, иногда верно, иногда нет. Чаще неверно. Почему? Потому что если мы учтем механизм дейтерообмена, то должны будем учитывать и первичный кинетический изотопный эффект (который чаще называют просто изотопным эффектом, потому что это самый распротраненный тип изотопного эффекта из большого разнообразия возможных). Дело в том, что если мы измерим скорости двух реакций, отличающихся только заменой обычного водорода на дейтерий в тех местах молекулы, которые непосредственно участвуют в реакции (должны разрываться или образовываться связи с атомами водорода или дейтерия), то эти скорости могут отличаться, иногда даже довольно сильно. Могут и не отличаться или отличаться очень слабо – тогда говорят об отсутствии изотопного эффекта. Могут отличаться, и по теории этот эффект может достигать почти 7-кратного, причем скорость реакции с дейтерированным субстратом всегда медленнее скорости реакции с недейтерированным субстратом. Не будем разбираться в теории этого явления, просто заметим, что это довольно очевидно, если подумать, что реакции, в которых рвутся или образуются связи, это такие колебания. Атомы в молекулах колеблются, самые важные колебания это так называемые валентные колебания – когда связь работает как пружинка – стягивается и растягивается. Когда связь рвется – это колебание, в котором связь растягивается, не расчитав своих сил, и лопается. Скорость реакции, в которой участвует такое движение довольно естественно сравнивать с частотой колебания, чем больше частота, тем быстрее колебания, тем больше скорость реакции. А частота колебания прямо зависит от массы частиц на концах пружинки – чем больше массы, тем медленнее колебания, это следствие из обычного закона Гука, да и просто из здравого смысла – большую массу труднее сдвинуть с места, а тем более труднее этой массой дёргать туда-сюда. Когда мы говорим о связях и реакциях, естественно такое грубое приближение имеет шанс хорошо работать только если связи и частицы на концах не отличаются ничем кроме массы. И вот изотопное замещение как раз этому и соотвествует – с химической точки зрения (валентных возможностей, электронной структуры, орбиталей, всего такого) изотопы идентичны. Но масса у них разная. И среди всех изотопов всех элементов два имеют самые большие различия – массы отличаются аж в два раза (конечно, есть еше обычный водород и тритий, тритиевые изотопные эффекты хорошо известны и иногда очень велики, но желающих измерять их немного, тритий довольно короткоживущий изотоп, и работать с ним не столько опасно, сколько дорого и неудобно, да и информации будет ненамного больше, чем с дейтерием – если вы научились измерять дейтериевые изотопные эффекты, зачем вам тритиевые, они просто будут в соответствующее число раз больше и всё, небольшая экономия на точности измерения при огромных затратах на материалы).
Итак, дейтериевый изотопный эффект в дейтерообмене – есть или нет? На это не так просто ответить, потому что мы должны точно знать скоростьопределяющую стадию и степень участия в ней атомов водорода и дейтерия. Даже в такой простой с виду реакции как изотопный обмен участвует несколько стадий, и кинетика всего процесса может быть непростой. Тем не менее, по крайней мере в реакции, катализируемой основаниями, скорость-определяющей стадией почти наверняка является отщепление гидрона (напоминаю, что это общий термин для протона и дейтерона), и тогда мы можем ожидать большого изотопного эффекта. Действительно, в литературе можно найти примеры таких измерений и убедиться, что изотопный эффект достигает максимально возможного – почти семикратной разницы.
То есть, если поставить эксперимент, в котором атомов дейтерия и водорода будет строго поровну, например, в реакции того же ацетона взять не 100-кратный избыток тяжелой воды, а всего навсего три моля воды на моль ацетона, то результатом будет не равное распределение изотопов по молекулам ацетона и воды (в среднем три там, три сям), а существенно большая степень дейтерирования. Дейтерий как будто стремиться залезть в ацетон. Конечно, семикратная разница в скорости не эквивалентна тупому стократному избытку, но все же это немало. И в обратном смысле, если у нас уже есть дейтерированный ацетон, а мы его подвергаем обмену с обычной водой, обмен будет, качество ацетона ухудшится, но далеко не так сильно, как можно было бы ожидать. Дейтерированные молекулы держат дейтерий сильнее, чем недейтерированные обычный водород.
Это явление хорошо известно. Явление иногда называют фракционированием изотопов. В чем явление? В том, что в среде, содержащей некоторое количество дейтерия, дейтерообмен приводит к обогащению дейтерием. Например, если у нас есть обычная вода с малым содержанием дейтерия, скажем 0.1%, и мы там пополощем ацетон или ацетилацетон, и потом выделим и определим в нем содержание дейтерия, то оно будет заметно отличаться в большую сторону от 0.1% (с поправкой на эквиваленты). Ну и чем больше дейтерия в среде, тем относително больше его будет в продуктах обмена. И тот же эффект усугубляет токсичный эффект тяжелой воды в организме – обогащение дейтерием биомолекул приводит к изменению скоростей биохимических процессов, а для организма такие фокусы крайне нежелательны – там все очень хорошо подогнано друг к другу. Но тот же эффект приводит к тому, что в организмах живых существ скорости биохимических реакций с участием дейтерированных соединений могут быть существенно меньше, чем скорости обычных субстратов. Замедление метаболизма стали использовать при разработке лекарственных препаратов совершенно целенаправлено, и с этим связан новый интерес к методам дейтерирования, о чём я немного расскажу на последней вкладке.
Практическим следствием этого эффекта является то, что особенно в современных методах дейтерообмена стали экономить на дейтерии – используют меньшие избытки и очень часто стали ограничиваться однократным обменом без повторов, и все равно достигать очень высоких степеней дейтерирования. В таких местах обычно задают вопрос – а раньше-то куда смотрели? Да просто в современной химии стали ценить экономные методы, и стали на это обращать внимание. А раньше на это закрывали глаза – подумаешь, лишний литр тяжелой воды.
Дейтерообмен в непредельных кетонах
Еноны (и енали) для нас почти исключительно связаны с электронодефицитной двойной связью, реакцией Михаэля и амбидентной электрофильностью. Но у таких соединениях может быть и кето-енольная таутомерия, и все что с этим связано. И здесь всё намного сложнее, чем в обычных енолизуемых кетонах и альдегидах, потому что возникает множество возможностей для делокализации, которые здорово смешивают карты, и делают ситуацию крайне запутанной. Строго говоря, систематически это, кажется, до сих пор никто так и не исследовал, но частные случаи применения разбросаны по литературе. В синтезе эта особенность непредельных карбонильных соединений применяется очень часто, но каждый раз поведение конкретного соединения приходится исследовать, потому что заранее предсказать основное направление енолизации бывает непросто.
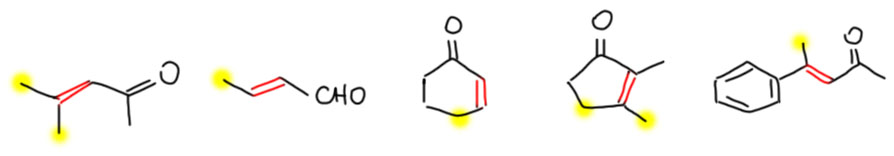
Возьмем самый простой фрагмент, где это может проявиться. Для этого нам понадобится карбонил, сопряженная с ним двойная связь, и еще один атом углерода хотя бы с одним атомом водорода. Таких фрагментов полно в непредельных альдегидах и кетонах. Вот несколько примеров, жёлтеньким помечены те группы, где находятся атомы водорода, которые могут быть оторваны основаниями разумной силы, но только такие, которые отделены от карбонила двойной связью. В некоторых приведенных ниже молекулах есть и обычные енолизуемые положения, которые хорошо видны и я не стал их помечать. Мы обсудим взаимоотношения таких положений ниже.
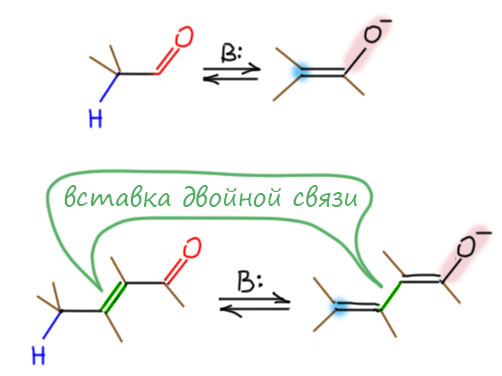
Вставка двойной связи между карбонильной группой и СН-группой, которая может быть депротонирована, приводит к тому, что взаимодействие сохраняется – двойная свзь работает как проводник сопряжения. Мы подробно разбирали это свойство двойной связи, когда разбирали сопряжение и мезомерию. Сам по себе этот приём часто называют винилогией – по аналогии с гомологией, извините, но здесь вставка винильного фрагмента, как часто называют два углерода, связанных двойной связью. Точнее было бы назвать его винилиденом, так как вставка с двумя связями (двухвалентный радикал), но это лишняя суета. Тем более, что сам теримин “винилогия” давно уже вышел из моды и употребляется довольно редко – мы и без этого термина видим суть дела.
Сразу заметим, что у соединений с тройной связью тоже есть нечто подобное, но там это еще сложнее, поэтому оставим их пока в стороне.
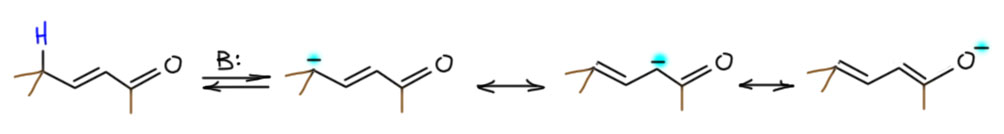
Итак, посмотрим на депротонирование такой системы еще раз, только поточнее. Снятие протона дает мезомерно стабилизированный карбанион, но здесь уже не две, а три граничные структуры. В этом месте еще раз вспомним мезомерию. Сопряжение между карбанионом и карбонилом – типичнейшее взаимодействие донор-акцептор. Мы пришли к выводу, что в таких случаях основная граничная структура соответствует полному смещению электронной плотности (пары электронов) от донора к акцептору. В обычном енолизуемом альдегиде или кетоне это енолят. В винилогичном енолизуемом альдегиде или кетоне тоже енолят, только точнее диенолят – двойных связи две и они сопряжены. Вон он в конце мезомерии. А в середине еще одна структура, в которой смещение неполное, до карбонила оно вообще ещё не дошло, роль акцептора сыграла та самая вставленная двойная связь. Мы знаем, что это не очень важная граничная структура. Фокус в том, что она, тем не менее, есть, и отрицателный заряд частично делокализован на этот средний углерод. В химии реальных енолизуемых енонов и еналей эта средняя граничная структура проявляется очень шизофреническим способом, то проявляется, то не проявляется. Сейчас мы это увидим на примерах.
Представим себе теперь дейтерообмен в присутствии оснований. Сразу обратим внимание на одно обстоятельство – непредельные кетоны и особенно альдегиды чрезвычайно реакционноспособны, при попытке их равновесно депротонировать образующиеся еноляты будут охотно присоединяться по Михаэлю к исходному енону (еналю), что приведет к олигомеризации, образованию смесей олигомеров, которое проще назвать осмолением. Енали просто обречены. Некоторые еноны, особенно разветвленные, циклические, бициклические в таких условиях олигомеризуются не так быстро и для них можно наблюдать дейтерообмен, почти всегда с не очень большим выходом – это то, что успевает обменяться до олигомеризации. Поэтому реальных результатов не очень много. Простые, хорошо нам известные еноны типа метилфинилкетона, окиси мезитила, пент-3-ен-2-она и т.п. осмоляются и нормально дейтерировать их невозможно – если понядобятся дейтерированные производные таких соединений, придётся их синтезировать, а не получать дейтерообменом.
В качестве примера исчерпывающего дейтерирования (дейтерий влез во все места, куда мог добраться) приведу дейтерообмен изофорона (изофорон – один из продуктов альдольной самоконденсации ацетона, подробно об этом можно прочитать на странице про альдольную конденсацию) в обычных условиях осноно-катализируемого дейтерообмена (J.Org.Chem. 1973, 38, 880). Дейтерий действительно влез во все места, затронутые енолизацией. И ещё в одно. Как он там оказался?
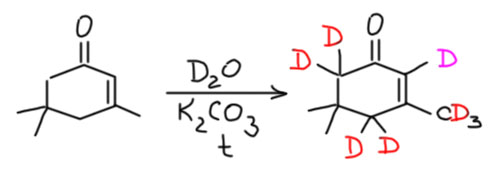
Дейтерообмен идёт через все три возможных енолята, которые находятся в равновесии, при это нас не интересует их относительное содержание в реакионной смеси и величины констант равновесий, потому что реакцию ведет в равновесных условиях так долго, как нужно для максимально возможной степени дейтерирования в условиях большого избытка источника дейтеронов. А это более необыное место, при двойной связи совершенно очевидно связано со структурой двух винилогичных енолятов, и как мы уже видели, отрицательный заряд частично делокализован и на этот протон. Я уверен в том, что если бы за ходом дейтерирования следили, а это можно отлично делать с помощью ЯМР, например, по относительному уменьшению сигналов протонов в процессе дейтерирования, а в этом соединении есть отличные метильные группы, которые не обмениваются и сохраняют обычные протоны – то этот протон обменивался медленнее всех остальных. Увы, никто этого не делал. Так или иначе, мы видим, что в енонах могут обмениваться все протоны, задействованные в образовании енолятов.
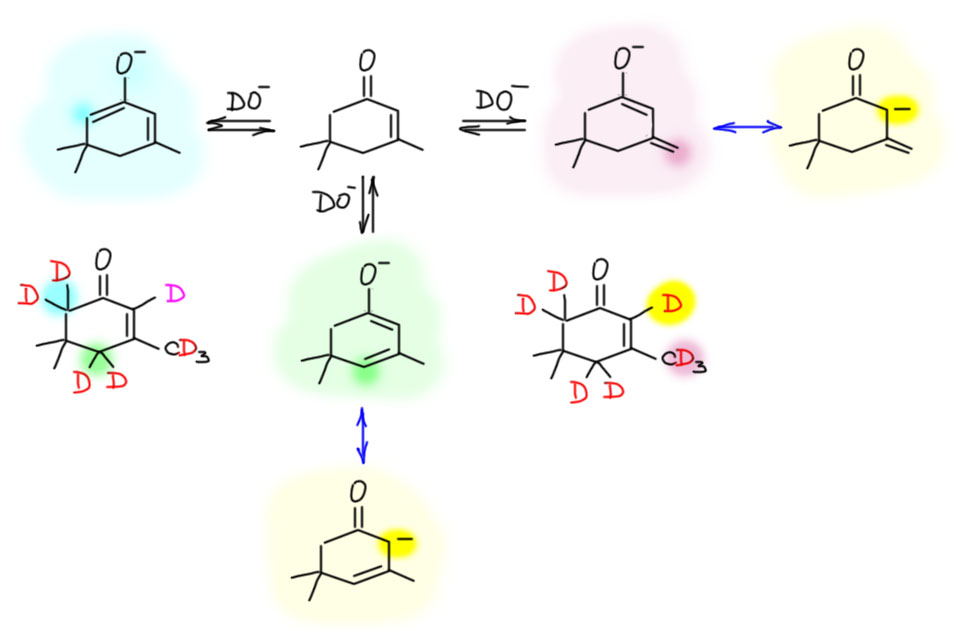
А вот в условиях кислотного катализа получается нечто другое. Циклопентенон, например, дал только обмен в сторону без двойной связи. Из этого мы можем сделать вывод о том, что второй диенол либо не хочет образовываться, либо настолько неустойчив, что реакция в эту сторону приводит не к дейтерообмену, а к образованию олигомеров или осмолению, а дейтерий обнаруживается только в том, что можно выделить из реакции. С этим вполне согласуется то, что из 8 грамм исходного циклопентенонона выделили только 0.5 грамм дейтерированного производного, а всё остальное куда-то делось (J.Mol.Spectroscopy, 1973, 48, 266). Реакцию, впрочем, делали спектроскописты, а не настоящие органики, чем, видимо и объясняется более чем странный результат. Кислотно-катализируемая енолизация может дать два разных енола, точнее, диенола. Эти диенолы изомерны, двойный связи в них сопряжены (а в пятичленном кольце по другому не получится). Обратимая кето-енольная таутомерия в присутствии источника дейтерона, тяжёлой воды, могла бы дать два разных продукта дейтерообмена, но получается только один. Можете сами на досуге поразмышлять, почему так получается. Возможно, эти диенолы сильно различаются по устойчивости – в одном из них мезомерный донор, гидроксил, находится на крайнем атоме диеновой системы, во втором – на внутреннем. Это точно довольно сильно влияет на электронную структуру. В дополнение здесь есть еще одна сложность. В химии циклопентадиенов хорошо известны перегруппировка – миграция двойных связей, которую проще представлять как миграцию насыщенного атома по кольцу. Такие миграции могут быть как катализируемыми (совершенно очевидно, как это может катализировать основание – опять, можете сами это нарисовать), так и спонтанными, потому что есть такое явление как сигматропная перегруппировка. Здесь я это оставлю, потому что это долгий разговор и мы его когда-нибудь разберём. Пока просто зафиксируем такой факт, немного странный, но интересный – образуется только один продукт дейтерообмера.
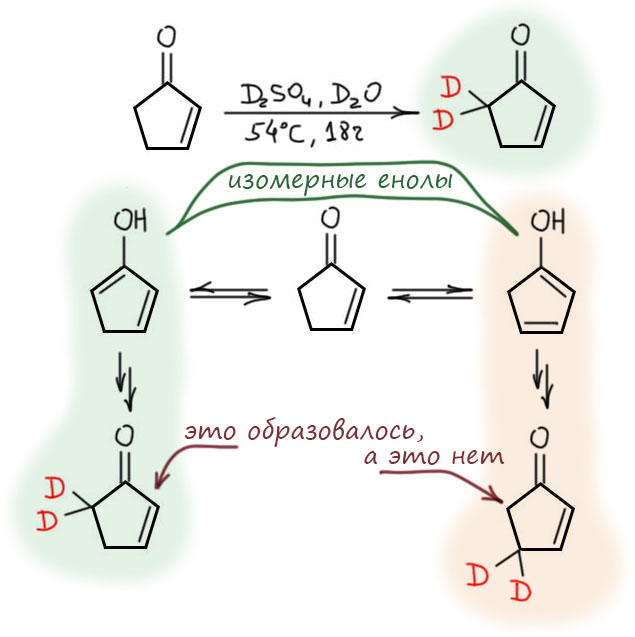
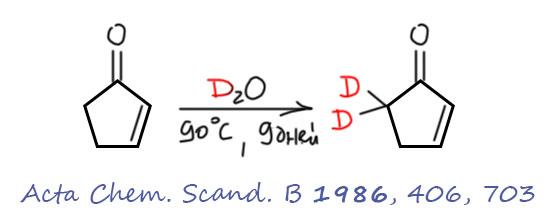
Но то, что в циклопентеноне два протона со стороны без двойной связи обмениваются легче подтверждает другая работа, в которой енон просто тупо грели в тяжелой воде 9 (девять!) суток, то есть очевидно воспользовались очень медленной кето-енольной таутомерии без катализа (вода, в принципе, тоже катализатор кето-енольной таутомерии, но очень слабый)
Ещё более странно идет дейтерообмен в циклогексеноне, настолько странно, что я пока не буду это рассматривать. Можете сами посмотреть, что получилось в статье Spectrochimica Acta, 1962, 18, 697. То ли опять спектроскописты, а не органики намутили, то ли там что-то очень интересное, но не очень понятное.
Ну и в соединениях попроще, в енонах с ароматическими кольцами, легкодоступных продуктах кротоновой конденсации ацетона и бензальдегидов, легко обмениваются енолизуемые протоны, и дейтерий не лезет в сторону двойной связи. Но его там никто и не ждёт. Дейтерирование, впрочем, идёт невесело.
Ещё немного вернемся к этой интересной теме, когда будем рассматривать новые методы дейтерирования, которые позволили вовлечь в дейтерообмен более широкий круг непредельных кетонов.
Новые методы дейтерообмена
В новом столетии много в органической химии стали делать заново. Одна из причин нового интереса к старым проблемам – поиск более надежных и экономных решений. Старые методы дейтерообмена, как мы уже видели, это обычно многочасовое, а иногда и многодневное кипячение карбонильного соединения в присутсвии немаленьких количеств кислот или оснований. Эта процедура повторяется от трех до пяти раз, чтобы добиться высокой степени дейтерообмена. Немудрено, что при этом значительная, а часто и большая часть кетона просто осмоляется, подвергаясь множественным альдольным конденсациям. Современные методы ищут более мягких условий. И находят. Разберем несколько свежих работ, в которых предлагают новые методы и посмотрим, какие подходы стали использовать для того чтобы решать в общем-то вполне несложные задачи. Работ таких в последние 10-15 лет появилось не менее десятка. Разберем несколько типичных.
Сильная кислота образуется in situ.
Работа таиландских исследователей (P. Kittakoop et al Org. Biomol. Chem., 2021, 19, 7390) предлагает весьма остроумное решение. Обратим внимание, как расширилась география химии. Лет тридцать-сорок назад серьёзной химией занимались от силы в полутора десятках стран. Сейчас работы высокого уровня идут из всех частей мира, даже из таких мест, которые никак ни с какой серьёзной наукой еще недавно вообще не ассоцировались.
Дейтерообмен катализируется сильными кислотами, и мы видели много таких примеров из старой химии, но сильная кислота вызывает и кротоновую самоконденсацию и много других плохих вещей. При этом сильная кислота – катализатор сильный, и нужно ее очень немного. Но если кислоту специально добавлять, мало добавить очень непросто. Если добавлять что-то легко гидролизующееся, все равно это происходит почти моментально. Таиландские исследователи решили соединить две вещи. Если взять галогенпроизводное, легко подвергающееся сольволизу, то в такой смеси будет постоянно образовываться сильная кислота, концентрация будет близка к постоянной, потому что при малой концентрации скорость сольволиза будет приблизительно постоянной. Взяли разные аллилбромиды – как мы отлично значем, очень хорошие субстраты и для SN1 и для SN2, и нам в общем-то всё равно по какому механизму идет этот сольволиз, нам важно что при этом образуется небольшая конценрация HBr. Лучше всего работал изопренилбромид, это вот такой аллильный бромид, очень легкодоступный.
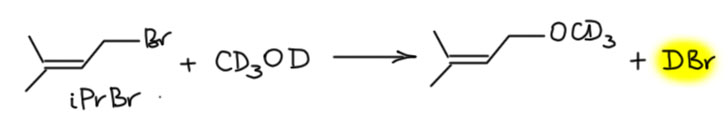
Так и делали дейтерообмен. В качестве источника дейтерия использовали дейтерометанол, не вполне понятно только зачем там дейтерий в метиле, но, видимо, монодейтерометанол не так просто найти. Растворяли в нём то, что нужно дейтерировать и немного 1-3% изопренилбромида, и ставили перемешиваться в теплое место. В Таиланде тепло, и такое место найти было нетрудно. У них комнатной температурой считают 28º, а может и ещё побольше. Так или иначе, реакция в таком теплом месте занимала 16 часов, и была однократной. Степень дейтерообмена составляла не менее 90% при почти количественном возврате – сколько положиди, столько и выделили, то есть в этих условиях не происходит олигомеризация и осмоление, всё чистенько. Дейтерировали и обычные енолизуемые кетоны, не будем на это обращать внимание, тут всё понятно. В статье много интересных кетонов, в том числе непредельных, природных терпеновых кетонов из душистых масел. Вот, например, три штуки: карвон, пиперитон, пулегон. И опять видим странность. Два последник кетона добросовестно дейтерируются во все места, куда дотягиваются енолы. Но первый, карвон, почему-то не дейтерируется в очевидное положение винилогичного енола, отмечено зелененьким. Кетон рядом, пиперитон, очень похожий по строению, честно дейтерируется во все доступные места. Почему? – не знаю.
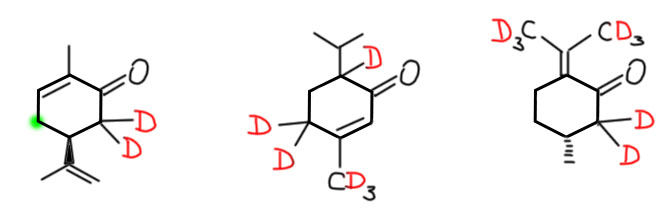
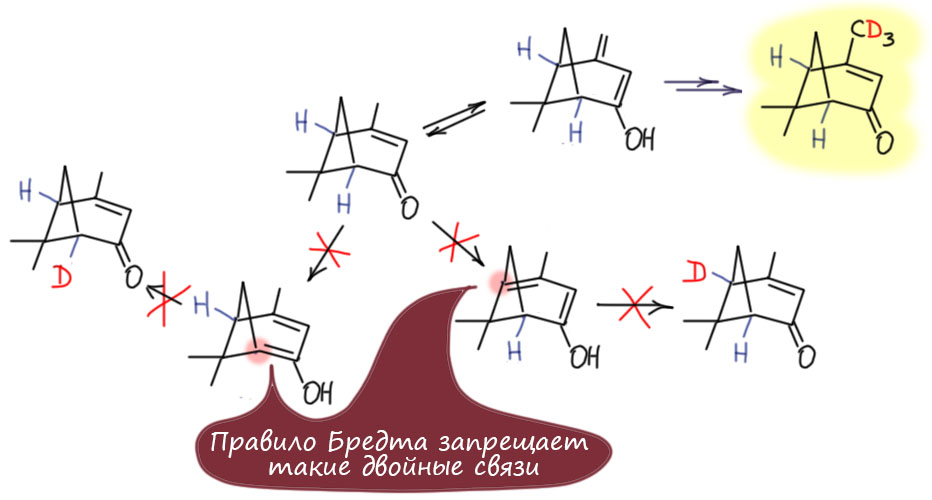
А вот особенно поучительный случай, причем со всех сторон. Бициклический кетон вербенон содежит все радости жизни – и енолизацию в обычную и винилогичную сторону. И протоны в голове мостиков. Дейтерирование затрагивает единственное место – в метильной группе на конце винилогичного енона, соответствующий енол легко рисуется и не имеет никаких проблем. А вот два других енола образоваться не могут. Причина этого железна – правило Бредта строго запрещает двойную связь в голове моста, если мост соотвествует небольшим циклам. “В голове моста” означает – перед вами бициклическая система (трициклическая, тетрациклическая и т.п.), в таких системах всегда есть такие перемычки, которые соединяют стороны более крупного цикла. Здесь, например, есть шестичленный цикл и перемычка между тремя атомами. Перемычка короткая, из одного атома. Такие перемычки и называют мостами или мостиками. Двойная связь во главе – означает – упёрлась в мостик, в один из атомов, на которых висит мостик. Мостик образует небольшой цикл – здесь четырехчленный. Это невозможно. Немецкий химик Юлиус Бредт угадал это в 1902 году по обобщению экспериментального материала по природным бициклическим соединениям, и это совершенно гениальная идея. Когда позже разобрались со структурой двойной связи и стереохимией, поняли что это исключительно важный закон химии – нельзя насиловать тригональный атом углерода, уводя его далеко от плоской треугольной конфигурации. Сейчас мы уже можем не просто эмпирически применять это правило, а узнавать молекулярную геометрию и точно видеть, когда насилие над двойной связью еще возможно и когда еще нет. Но если цикл четырехчленный шансов точно нет, такие двойные связи невозможны, енолы невозможны, и дейтерирование не идёт. Водороды во главе моста нельзя оторвать и основанием – еноляты точно так же невозможны. Поэтому мы и не имеем дейтерирования в эти два положения. И во всех подобных случаях будем иметь ту же проблему. Самое интересное, что таиландские авторы этой статьи, видимо, не знают про правило Бредта, потому что они удивляются, отчего у них нет дейтерия в этих положениях. Ничего удивительного, никакой химии в Таиланде еще 20 лет назад не было. Так быстро все не дается. Со временем разберутся. А правило Бредта – исключительно интересная вещь, когда-нибудь мы до него добереся серьёзнее.
Органокатализ
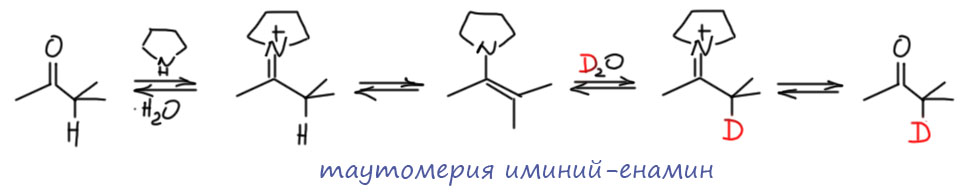
Модный органокатализ, за который только что дали нобелевскую премию, естественно не мог остаться в стороне, тем более что львиная доля достижений органокатализа – это про химию карбонильных соединений. Собственно, именно с органокаталитической работы китайских исследователей, Чэнь Юаньвэя с сотрудниками, и началось новое наступление на дейтеорообмен (M. Zhan, T. Zhang, H. Huang, Y. Xie, Y. Chen, J. Labelled Compd.
Radiopharm. 2014, 57, 533). Эта первая работа была очень проста, и использовала самое первое достижение органокатализа – использование вторичных аминов для активации енолизуемых карбонильных соединений через образование in situ енамина в равновесии с солью иминия. И как только все предыдущие поколения этого не заметили! Вот в этом и состоит достижение нынешних нобелевских лауреатов, Б.Листа и Д.Макмиллана, что они обратили внимание на такой общедоступный слой химии и его нераскрытый потенциал. Чэнь с сотрудниками просто добавили пирролидин. Очаровательна формулировка – “смесь тяжёлой воды и безводного ТГФ”; понятно, что люди просто не ходят разбавлять дейтерий остаточной легкой водой из растворителя, но от кривой ухмылки удержаться невозможно – всё же хорошо видно, что китайцы совсем недавно стали заниматься химией, и хотя уже достигли невероятных результатов и впечатляющего уровня, вот такие остаточные симптомы продолжают выдавать неофитов. Так или иначе, реакция удивительно быстра – за 3-6 часов при комнатной температуре и без повтра достигается очень высокая степень дейтерообмена, хотя в некоторых случаях требуется более жеские условия. Ассортимент кетонов очень большой. Некоторые результаты вызывают легкое удивление, например:
Так и хочется спросить – неужели такой активный галоген рядом с карбонилом никак не реагирует с нуклеофилом, тем более что условия для конкретно этой реакции уже не такие уж и щадящие, а время реакции оставляет возможности даже для медленных процессов. Но в науке есть жесткое правило, восходящее к знаменитой сентенции Понтия Пилата “что я написал, то написал” – иными словами, если у вас есть вопросы, то это ваши проблемы. Но дальше мы встречаем другие странные вещи, например, легкое дейтерирование не кетона, а сложного эфира (и еще амида), и это уж совсем настораживает. Производные кислот, безусловно, карбонильные соединения, и енолизация им не чужда, вот только происходит это намного труднее, да и в придачу, непросто было бы избежать переамидирования. Про карбоновые кислоты, их производные и особенные свойства мы скоро отдельно поговорим, а тут пока отметим, что у китайской химии есть некоторые особенности, которые надо учитывать. Учитывать просто – принимать к сведению, но относится с легким предубеждением. Скорее всего, описанное явление имеет место, но как минимум часть примеров для статьи нарисована “от фонаря”. Предполагаемые механизм реакции предполагает образование енамина, енамин-ининиевую таутомерию и обмен в этом процессе.
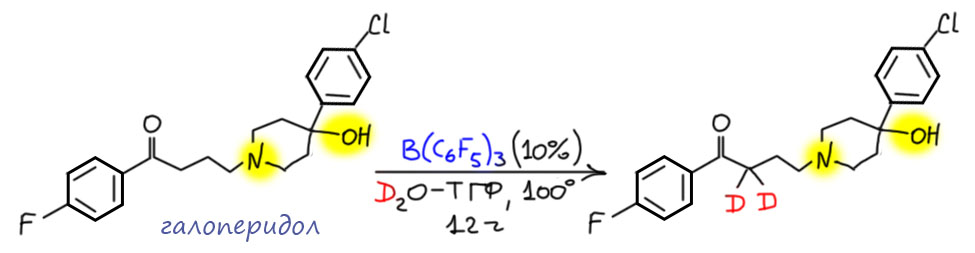
Специальные кислоты Льюиса
Кислоты Льюиса отлично катализируют кето-енольную таутомерию, но на надо еще доставить дейтероны от переносчика дейтерия типа тяжелой воды или дейтерометанола. И обычные, известные нам кислоты Льюиса типа хлористого алюминия или цинка или трехфтористого бора этой водой просто прогидролизуются, выделится сильная протонная кислота и будет опять катализировать альдольную конденсацию и другие безобразия. А нам нужна современная, чистая химия, выходы за 90% и никаких побочных. В современной химии есть такие кислоты Льюиса. Очень популярная штука, например, это особо электронодефицитный боран с тремя пентафторфенильными заместителями. Бор ведь в боранах и сам по себе секстетный, а на нем еще три солидных акцептора. Но кроме того, эти заместители, расположенные пропеллером вокруг атома бора, создают немаленькое стерическое препятствие, и такая кислота Льюиса сочетает большую жадность к основаниям с весьма большой избирательностью, потому что крупное основание просто не может подойти к атому бора. Такое свойство создает хорошую селективность, кислота Льюиса такого типа не пристает ко всем атомам с неподеленной парой, что создает немало совершенно необычных ситуаций. На таких кислтах Льюиса, например, делают такую необычную вещь как так называемые фрустрированные ионные пары, необычайно модные в современной химии, но об этом как-нибудь в другом месте. Здесь же приведу работу японского исследователя Масаюки Васы, работающего впрочем в Штатах (M.Vasa et al Adv. Synth. Catal. 2019, 362, 360-364) и сотрудников. В работе не занимались простыми кетонами, но дейтерировали известные лекарственные препараты, поотому что, оказывается, в современной науке о лекарствах весьма активно применяют дейтерированные аналоги известных препаратов. Дейтерирование изменяет кинетику метаболизма, а это иногда очень полезно. Мы уже обсуждали кинетический изотопный эффект – вот это именно про него, скорости реакций замедляются, если на месте протона оказывается дейтерий. Организмы живых существ обычно стараются избавиться от чужеродных соединений, быстро превращая их во что-нибудь, что можно быстро вышвырнуть из организма. Это правильно, но одновременно сокращает время действия лекарственных веществ, и несколько замедлить скорость метаболизма оказывается неплохой идеей. Поэтому в современной химии все бросились все дейтерировать. В данной работе дейтерировали кетоны, и среди известных лекарственных препаратов кетонов немало. Например, это печально известный галоперидол, препарат для успокоения во время острых психозов, очень популярный у всяких спецслужб – в увеличенных дозах превращает человека в овощ, на время, но с очень неприятными побочными эффектами. Галоперидолом, например, “лечили” советских диссидентов, потому что любую критику советской власти считали проявлением психического заболевания, которое цинично назвали “вялотекущей шизофренией” – человека с такими идеями направляли в психушку, где накачивали галоперидолом. Некоторые не выдерживали такого “лечения” и признавались во всех грехах, которые на них вешали гебисты. Впрочем, тот же препарат в нормальных дозах весьма полезный препарат, которым снимают всякие болезненные состояния, психозы, и прочие нервные расстройства. Галоперидол – кетон, содержащий немало других функциональных групп, но в присутствии этой кислоты Льюиса довольно гладко дейтерируется. Если бы взяли кислоту Льюиса попроще, она тут же уцепилась бы за амин и снесла бы ко всем чертям третичную спиртовую группу. Результатом будет тот же галоперидол, но с увеличенным сроком действия.
Как перекачать дейтерий из одного кетона в другой
Все рассмотренные новые методы дейтерирования в принципе ничего принципиально нового не содердажи – это был обмен через катализ таутомерии кислотами или основаниями, а вся новизна состояла в изобретательности в выборе катализаторов или их генерации in situ. Источниками дейтерия всегда являлась дейтерированная протонная кислота, обычно просто тяжелая вода. Методы получились вполе успешные, но некоторые важные ограничесния они не снимают – в протонной среде всё равно не исключены побочные реакции, хоть и сильно снижено их влияние. Но вот совсем новый метод, предложенный российскими исследователями, В. Ананиковым и сотрудниками из ИОХа (K.I.Galkin, E.G. Gordeev, V.P.Ananikov Adv. Synth. Catal. 2021, 363, 1368– 1378) выполняет более изощренный фокус – пересадку дейтерия из апротонного источника дейтерия, обычного дейтерированного растворителя дейтероацетона, и в ходе процесса никаких других источников протона(дейтерона) не видно. Дейтерии перелетают из дейтероацетона в енолизуемые кетоны или сложные эфиры или лактамы. Кто же их перетаскивает? Придется признать, что никаких других кандидатов на этот благородный труд кроме енола не видно. Концентрация енола ничтожна, а тут еще получается должны встретиться два енола – кажется, что вероятность такого события должна быть пренебрежимо мала. Но перенос происходит, и с вполне серьёзной скоростью, значит сомнения побоку – в химии все диктует эксперимент: если реакция идёт, бесполезно говорить, что этого не может быть, надо смириться и искать объяснение. Надо сказать, что в статье предложен ну очень изощренный механизм, в котором перенос дейтерона с енола на енол происходит согласованно в очень хитро собранном реакционном комплексе. Я к таким механизмам отношусь без энтузиазма – слишком сложно, слишком отрицательной должна быть энтропия активации. Да и надо сказать, что авторы настолько усложнили описание предполагаемого механизма, что я вообще ничего не понял, но это мои проблемы. Современные исследователи любят строить изощренные концепции – это помогает хорошо позиционировать исследование; таковы нынче правила игры и нужно ими владеть, чтобы уметь обращать внимание на свои работы. Во всём, конечно, желательно блюсти разумную меру, но это дело вкуса. Поэтому я не буду тут пересказывать гипотезу, высказанную в статье. Захотите, сами посмотрите. Разберем только очевидные вещи.
Главное действующее лицо работы – еще одна невероятно модная штука в последнее десятилетие – производные имидазола, из которых что только не делают – и ионные жидкости, и новые лиганды, и стабильные нуклеофильные карбены, и органокатализаторы и много чего еще. Вся эта химия восходит к совершенно гениальной старой работе Рональда Бреслоу, которую я подробно разобрал на страничке про последнюю нобелевскую премию. Некоторые пятичленные азотные гетероциклы в форме ониевых солей можно легко депротонировать и получить то, что в современной химии называют NHC-карбенами. В статье использовали один из самых популярных тких гетероциклов, который обычно сокращают как BMIM, и в виде солей с инертными противоионами используют как самую популярную ионную жидкость. При депротонировании она дает нейтральную частицу, которую можно рассматривать и как карбанион илидного типа (рядом плюс и минус) и как карбен. Депротонируют ее в работе равновесно и гетерогенно лежащим на дне карбонатом натрия или калия.
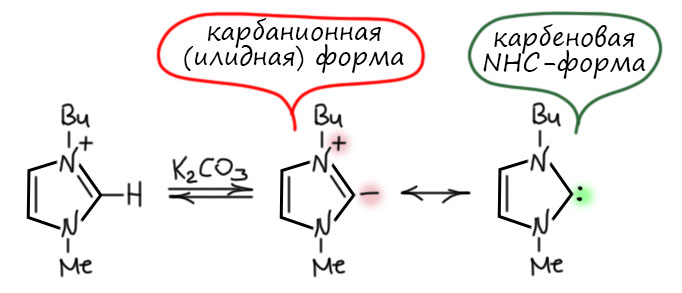
Важно то, что эта штука одновременно и нуклеофил, и основание. Поэтому она естественно может катализировать кето-енольную таутомерию дейтероацетона. И таким образом вытаскивать дейтерий на кислород, то есть в положение, готовое к обмену.
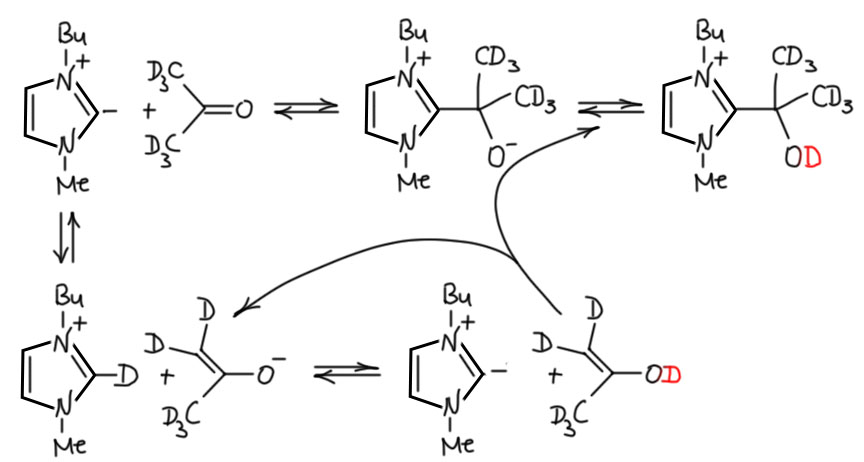
Дальше можете написать сами – тот же карбанион-карбен с тем же успехом может катализирвоать кето-енольную таутомерию кетона, который надо дейтерировать. И в енольной форме происходит обмен протона на дейтерон, а общее направление задается тем, что дейтероацетон берется, как всегда в таких процессах, в изрядном избытке. Впрочем, здесь как раз и есть одна загвоздка, которая, возможно, объясняет, как может быть перенос гидрона (протона, дейтерона) между двумя енолами, каждый из которых находится в ничтожной равновесной концентрации. В системе обязательно должен быть еще и противоион типа хлорида. И вот именно он, возможно, и таскает дейтерии от одного енода к другому. На возмущенный возглас: Этого не может быть, потому что HCl – сильная кислота, а у нас тут карбонат на дне! Ответим, что это она в водном растворе сильная, а в апротонной среде и в малой концентрации никакая она не сильная, и очень даже вполне может быть искомым переносчиком. Ну, пусть авторы сами со своей системой разбираются, как она работает, но она работает и, судя по описанию, должна быть довольно удобна особенно для маленьких количеств. В качестве примера приведу только дейтерообмен в окиси мезитила, которая не выдерживает обычных методов дейтерирования, а здесь обменивает все без исключения протоны, а почему, я уже разобрал на одной из вкладок выше.
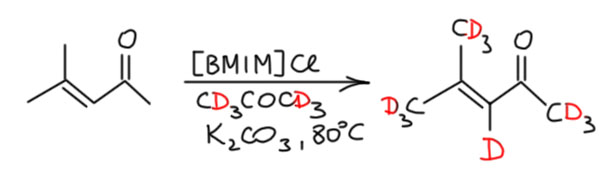
На этом, пожалуй, хватит. В литературе последних лет вы найдете еще несколько интересных методов дейтерообмена и не только в кетонах, но и в ароматических соединениях и гетероциклах. Эта тема становится актуальной как раз из-за того, что дейтерированные соединения стали использовать не только в теоретических исследованиях, но у них нашлись практические применения особенно в разработке лекарственных препаратов, и сразу понадобились современные, селективные и мягкие методы, и пошли в ход все современные штучки.
и всё остальное
Рацемизация
Если у нас есть альдегид или кетон (или любое другое карбонильное соединение), которое имеет рядом с карбонильной группой асимметрический атом углерода и хотя бы один водород на нем, и имеем это соединение в виде одного из энантиомеров, то нам придется соблюдать очень строгие меры предосторожности, иначе мы будем терять оптическую чистоту, пока не получим рацемическую смесь.
Происходит это по очевидной причине – при образовании енола асимметрический атом углерода становится sp2-гибридным, плоским, и обратное присоединение протона при превращении енола в кето-форму происходит с равной вероятностью снизу и сверху этой плоскости (это высказывание являеся строгим только если в молекуле нет других стереогенных центров или элементов, а обратное перемещение протона не происходит от сопряженной кислоты хирального основания – в таких случаях вероятности снизу и сверху не равны, и рацемизация все равно происходит, но может быть неполной; в нашем курсе мы с такими случаями не встретимся). В результате, какой бы энантиомер мы не взяли, в конце концов они превратятся в рацемат. Поэтому этот процесс называется рацемизацией.
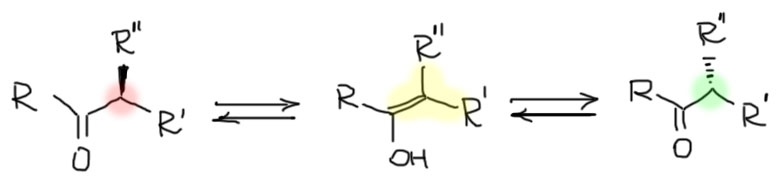
Для рацемизации нужен кислотный или основный катализ, который может прийти, откуда не ждали, например, от стенок стеклянной банки (стекло – силикат натрия, обладает весьма приличной основностью), или от силикагеля (силикагель – весьма приличная кислота в основном по причине наличия на поверхности гидроксильных, так называемых силанольных групп) при попытке очистить соединение хроматографией. Поэтому и получить и сохранять оптически активные альдегиды и кетоны очень непросто. Но не невозможно, что в успехом демонстрирует нам Природа, так как, например, все углеводы, включая глюкозу и фруктозу (но не сахарозу), являются энантиомерно чистыми альдегидами или кетонами и минимум один из стереогенных центров в этих соединениях находится на атоме углерода, участвующем в енолизации. Почему это так, мы разберемся, когда дойдем до углеводов.
Еще один пример оптически чистых карбонильных соединений, имеющих енолизуемый стереогенный центр – природные аминокислоты, из которых состоят белки и пептиды. Это не альдегиды и кетоны, поэтому сильно вперед забегать не будем, просто отметим, что в химии аминокислот тщательно избегают реакций, в условия которых входит действие сильных оснований в протонодонорной среде. Именно потому. что как огня боятся рацемизации, катализируемой основанием. Почему в этом случае не боятся кислотно-катализируемой рацемизации, тоже разберемся в своем месте. А для простых альдегидов и кетонов и кислоты, и основания одинаково опасны.
Бромирование
Если у нас есть енол, то этот высокореакционноспособный олефин будет легко взаимодействовать с такими замечательными электрофилами как галогены, например, бром. Реакция будет развиваться точно так же, как реакция любого алкена с бромом – сначала присоединение электрофильной части с образованием самого стабильного карбокатиона из двух возможных. В данном случае получится нечто особенное. Более выгодно присоединение по α-атому углерода, при этом получается мезомерно-стабилизированный катион, с которым мы уже встречались и не раз. Структура этого катиона ближе к второй граничной струтктуре с плюсом на кислороде, но зато и с полностью заполненными валентными оболочками у всех атомов. Поэтому этот катион – не что иное, как протонированный кетон, α-бромкетон. Это и есть продукт присоединения брома к енолу. Обратим внимание, что второй продукт реакции – HBr, сильная кислота.
Теперь представим себе, что мы прибавляем бром к чистому кетону или альдегиду, или к раствору в каком-нибудь чистом апротонном растворителе. Тогда будет любопытный эффект – моментально прореагирует только тот енол, который содержится в карбонильном соединении согласно равновесию. И все – реакция остановится: енола больше нет, а просто кетон или альдегид в кето-форме с галогеном не реагирует – это электрофил, а не нуклеофил, а электрофил с электрофилом не реагирует.
Возникает возможность, которую поняли очень давно, как только появилась идея о кето-енольном равновесии. А почему бы так не попробовать измерить содержание енола. Ведь это просто титрование. Надо взять побольше кетона или альдегида, и аккуратно титровать раствором брома в инертном растворителе, используя окраску брома как индикатор – первая же капля лишнего окрасит раствор в хорошо видный желтый цвет. Так и делали. В старых учебниках можно найти цифры, которые получались этим методом – в ацетоне и циклогексаноне намерили до 1% енола. Эти цифры на полном серьезе обсуждались еще не так давно, и из них делались глубокомысленные выводы по поводу разных эффектов. Не исключено, что на свете до сих пор существуют люди, верящие в такие цифры содержания енолов.
Очевидно, что это сильно завышенные значения. Если учесть реальное содержание енола в этих кетонах, то завышение в тысячи и десятки тысяч раз. Мышь перепутали со слоном. Даже не мышь, а таракана.
Причина ошибки очевидна. При ручном титровании, как бы ни был искусен экспериментатор, время добавления капли, перемешивания и фиксации цвета глазом довольно велико. И за это время часть кето-формы успевает перейти в енол, тем более что выделяющаяся кислота HBr является катализатором процесса. Ну и дальше получается некоторое случайное сочетание скорости реакции экспериментатора и скорости кето-енольного превращения. Очевидно, что кето-енольное превращение обошлось с теми экспериментаторами довольно жестоко.

Позже методику изменили – стали использовать хлорид иода вместо брома для титрования Задание: напишите реакцию ацетона с хлоридом иода.
Только не ищите в самой этой реакции причин, почему измерение получилось точнее – это зависело от более-менее случайной совокупности факторов. Но вот еще позже от ручного титрования перешли к электрохимическому. Не будем рассматривать это в деталях, но просто для общего образования отметим, что в этом случае титрующий реагент получают редокс-реакцией на одном из электродов, реакция присоединения происходит в очень малом межэлектродном пространстве, а еще один электрод используют для точного и высокочувствительного измерения концентрации этого реагента в растворе, что позволяет фиксировать точку эквивалентности очень точно и с огромной скоростью, и тут уже скорость кето-енольного превращения оказывается ни на что не влияющим поползновением улитки. И вот так приблизились, во-первых, к результатам, которые неплохо воспроизводились, и во-вторых, к таким, которые мы и сегодня считаем достаточно точно отражающими реальность. Мало, очень мало енолов в простых карбонильных соединениях.
Теперь попробуем понять, как нужно делать реакцию бромирования енолизуемого кетона или альдегида, чтобы получить продукт бромирования. Сразу замечу, что в альдегидами это не так чисто и удобно, как с кетонами, из-за побочных реакций, в частности из-за окисления альдегидной группы, но в первом приближении можно не обращать на это внимание. Возьмем, например, ацетон, и попробуем его пробромировать, добавив эквивалент брома. Увидим любопытный эффект. Сначала получится раствор брома в ацетоне и он довольно долго будет стоять безо всяких изменений. И в какой-то момент вдруг моментально вскипит и обесцветится.
Это очень интересное явление, называемое автокатализом. Автокатализ очень часто встречается и в химии и в биологии (то есть тоже в химии), и этот слкчай дает прекрасную простую модель, которая показывает, как это работает. Фокус в том, что у нас есть реакция, которая производит катализатор для себя самой. И в начале у нас нет катализатора. Мы смешиваем реагенты, но они не реагируют, потому что нет катализатора. Смесь может сидеть так без дела неопределенно долго – этот период называют или индукционным или латентным в зависимости от того, что там в этой тиши происходит или наоборот не происходит. Могут быть два варианта: 1) там понемногу идет реакция за счет наличия очень малого количества реакционноспособной формы и начинает накапливаться катализатор; 2) там происходит что-то вообще случайное, и первый катализатор возникнет там волей случая – ветром надует, из космоса прилетит, муха что-нибудь уронит. У нас очевидно, первый случай – есть очень мало енола и он мгновенно реагирует, образовав ничтожное количество кислоты-катализатора. Поскольку концентрация катализатора мала, он катализирует кето-енольное превращение, но очень медленно. Но катализатор-то никуда не девается, он накапливается. Скорость реакции понемногу становится все быстрее и быстрее, катализатор накапливается все быстрее, но до поры до времени мы этого не замечаем. С каждым процентом превращения все больше катализатора, все быстрее реакция. Такие системы в самых разных областях науки и техники имеют еще одно название – система с положительной обратной связью – то есть процесс сам порождает причину своего ускорения, усиления, и т.п., работает с ускорением, с саморазгоном. В какой-то момент скорость становится огромной – это может быть вскипание, выброс, даже взрыв. К счастью, в простых химических реакциях есть и естественный ограничитель этого – просто заканчивается реагент – наступает насыщение, полное превращение, торможение – больше нечему реагировать.
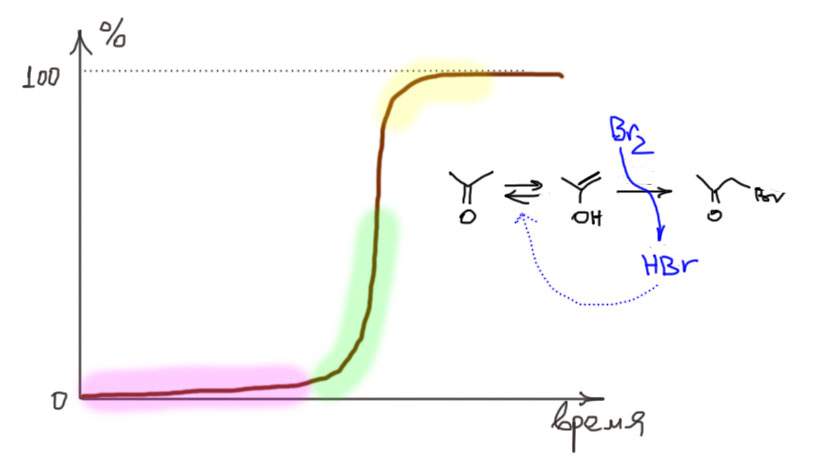
Такая постановка эксперимента дает интересный и поучительный эффект, но когда цель эксперимента не позабавиться, а получить конкретный продукт с хорошим выходом, она не годится – нормальные химики не любят реакций, которые сначала не идут, а потом вылетают в окно вместе с плачущим экпериментатором (плачущим – потому что α-бромкетоны почти всегда являются слезоточивыми веществами). Но тот же механизм отлично подсказывает нам, как сделать так, чтобы никто никуда не вылетел, а реакция шла “быстро и с высоким выходом” (это популярнейший мем органического синтеза). Нужно обеспечить постоянную и приличную скорость кето-енольного превращения, сразу взяв подходящий катализатор в более-менее постоянной концентрации. И тогда нам останется просто прикапывать бром с такой скоростью, чтобы он расходовался. Прекрасно, таким катализатором может быть, например, достаточно слабая уксусная кислота, которую можно еще и взять в качестве растворителя – тогда и концентрация будет постоянной. Так и делают – посмотрите на странице про методы в соответствующей вкладке.
Галоформная реакция
Если бромирование идет через енол, то как идет галоформная реакция – ведь это почти то же самое. Енолизуемый кетон, правда не любой, а только метилкетон в присутствии галогена (хлора, брома, иода), взятого в избытке, но минимум в трехкратном количестве, и обязательно в присутствии водной щелочи, дает необычные продукты, расщепляясь в галоформ и соль кислоты на 1 углерод короче. При этом, нужно различать препаративную галоформную реакцию, когда мы ее делаем для того чтобы превратить кетон в кислоту (см. на странице методы), и качественную галоформную реакцию, которую в старину, когда реакции ставили при свечах и под пение псалмов, использовали как пробу на наличие некоторых веществ в моче вьючных верблюдов. Качественную галоформную реакцию дают не только метилкетоны и ацетальдегид, но и соединения, которые могут окисляться в метилкетоны или ацетальдегид галогеном в щелочной среде, например, спирты типа этилового и изопропилового. После изобретения всяких спектроскопий эта реакция даром никому стала не нужна, а препаративно ее использовать нельзя, потому что щелочной галоген, на самом деле, очень плохой окислитель, слабый и неселективный. Для качественного теста это не важно – окислится 5%, выпадет галоформ, – тест положительный, зачет. Но для препаративных целей выход в 5% с параллельной возможностью этим галогеном еще куда-нибудь заехать совершенно неприемлем. Поэтому лучше забыть про качественную галоформную реакцию, как про предание старины глубокой.
А вот препаративная галоформная реакция – вполне полезная реакция, у которой нет хорошей (то есть простой, дешевой и эффективной) замены даже в самой наисовременной химии. В отличие от бромирования, которое идет в условиях кислотного катализа или автокатализа, галоформная реакция идет в условиях основного катализа. Так, но мы недавно разобрались в том, что катализ не влияет на равновесие, в том числе на соотношение енолов, если их два, как в несимметричных кетонах, енолизуемых в обе стороны. И как же тогда получается, что при бромировании получается более замещенный бромкетон, а в галоформной реакции – продукт из менее замещенных бромкетонов.
Поскольку равновесие обмануть нельзя, значит в галоформной реакции нет равновесия. Реакция идет в условиях основного катализа, и, как мы знаем, в этом случае енолизация протекает через посредство енолята. Енолят по свойствам вполне аналогичен енолу – это высокодонорный нуклеофильный олефин, но только еще более нуклеофильный, на порядки более нуклеофильный. Когда доберемся в следующем семестре до фенолов, увидим прямую аналогию, и то, настолько более реакционноспособен фенолят (это такой енолят в ряду бензола) чем фенол (енол в ряду бензола) по отношению к электрофилам. Разница огромная и качественная. Возвращаемся к енолизации через енолят, и представляем себе, что будет, если енолят образуется в присутствии галогена, а галоформную реакцию именно так и делают – галоген кладут сразу. Енолят немедленно прореагирует с галогеном, и это необратимая реакция. Результат – кето-енольное равновесие не достигается, и енолят немедленно перехватывается галогеном (в английском языке используют более образное слово scavenge – буквально, “подбирать мусор” – увидел, бумажка валяется, подобрал, больше не валяется; так же и галоген – увидел, енолят валяется, подобрал, больше не валяется). До енола и равновесия мы просто не добираемся. А протон быстрее отщепляется от менее замещенного положения, поэтому мы и называем этот енолят кинетическим – образующимся быстрее. Дальше еще проще, у галогенкетона кислотность протона на углероде с галогеном повышена, и отщепляется именно он, – а третий тем более. Тригалогенирование происходит очень быстро, каждая следующая стадия быстрее предыдущей. Итак, в отличие от галогенирования кетонов в условиях кислотного катализа, подчиняющегося законам термодинамического контроля, галоформная реакция – типичный случай контроля кинетического. Обратим еще внимание, что при присоединении электрофила к еноляту вместо обычного карбокатиона, или даже хотя бы оксониевого катиона, сразу получается нормальная молекула – кетон, потому что ожидаемый карбокатион просто соответствует граничной структуре с разделенными зарядами самой карбонильной группы. Понтяно, что такой путь просто не может не быть чрезвычайно выгодным и быстрым.
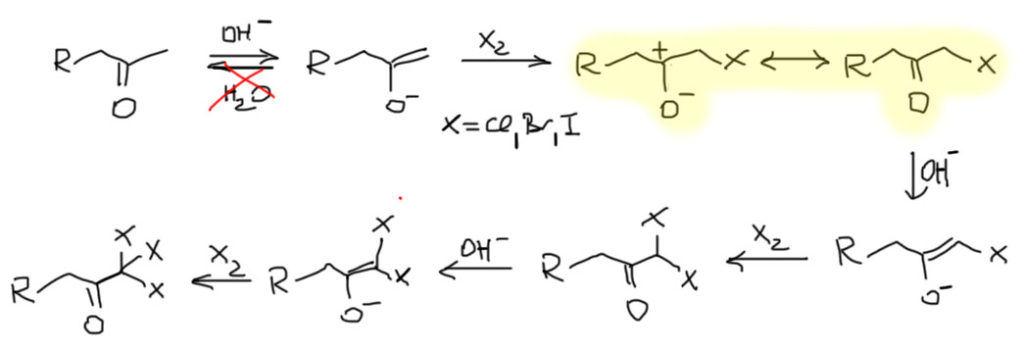
Стоит сказать, что хотя мы пишем чистое галогенирование по метилу даже в несимметричных кетонах, у которых с другой стороны есть CH2-группа, реально такой селективности достигнуть трудно. Поэтому галоформную реакцию гораздо чаще применяют к метилкетонам, у которых нет такого соблазна, и с другой стороны или ароматическое кольцо, или двойная связь, или что-то третичное, неенолизуемое. Гидроксид все же очень маленький и недостаточно сильный как основание, чтобы давать чистое депротонирование по менее замещенной стороне, то есть результат, который мы получаем (и то не на 100%!) при генерации енолята действием очень сильного и громоздкого LDA.
Последняя стадия галоформной реакции довольно интересна, потому что мы в ней встречаем старого знакомого в новой роли, и убеждаемся, что поведение реакционноспособных частиц очень сильно зависит от условий, в которых они находятся. После образования тригалогенкетона карбонильная группа в этом соединении становится чрезвычайно электрофильной – тригалометильная группа является сильным индективным акцептором. Поэтому это соединение быстро присоединяет нуклеофил, гидроксид-ион. Обратимо образуется тетраэдрический интермедиат, у которого, казалось бы, нет никаких возможностей во что-то превратиться дальше. Но такая возможность есть, и это отщепление тригалометильного карбаниона, неплохо стабилизированного индуктивным эффектом трех галогенов, что делает его достаточно приличной уходящей группой. В SN2-химии мы не видели такой уходящей группы (на самом деле и там такое бывает, но это очень большая редкость), но в химии карбонила закономерности ухода от тетраэдрического интермедиата не такие жесткие, как в SN2, и уйти может почти любой анион, который хоть немного делокализован и стабилизирован. Мы еще увидим огромное множество таких примеров. Но главная проблема не в этом, а в том, что мы уже однажды видели трихлометильный анион, и узнали про него, что он быстро и необратимо распадается на дихлоркарбен и хлорид – вспомните реакцию алкенов с хлороформом и крепкой щелочью в межфазных условиях, которая приводит к образованию дихлорциклопропанов. Почему же здесь такого не происходит? Да просто потому, что тригалометильный анион в галоформной реакции выходит в водную среду с невысокой основностью (в галоформной реакции не используют больших избытков щелочи) и немедленно протонируется, ведь это все же очень сильное основание, карбанион, стабилизированный всего-навсего индуктивными акцепторами. Образование сопряженной кислоты тригалометильного аниона, то есть галоформа HCX3 происходит в этих условиях быстрее, чем α-элиминирование хлорида. А галоформ, как только образуется уходит в отдельную фазу, или жидкую (хлороформ или бромоформ), или кристаллическую (иодоформ) и покидает поле битвы.
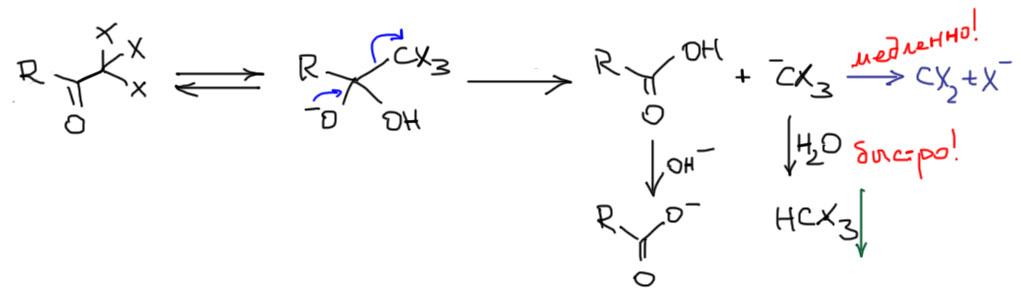
Тут стоит заметить еще одну занятную вещь. Иногда подмечают, что после ухода тригалометильного аниона из тетраэдрического интермедиата формально образуется карбоновая кислота, куда более сильная кислота чем вода, и, дескать, именно она протонирует тригалометильный анион по праву более сильной кислоты. Это замечание столь же глубокомысленно, сколь и бессмысленно, потому что культурное обращение с кислотностью и основностью по Бренстеду-Лоури требует признания, что источником протона в протонодонорной среде, например, в воде, всегда является сама среда и ее основные компоненты – сопряженные кислоты и основания (в воде это вода и гидроксид, или вода и гидроксоний), а прямой передачей протона можно пренебречь. Другое дело, если бы среда была апротонной, и там нет альтернативы прямому переносу протона от кислоты к основанию, но тогда и реакция пошла бы не так. Это замечание выглядит довольно неприличным занудством, но уверяю вас, вы много приобрете, если научитесь корректно работать с кислотными и основными протонодонорными средами. Игра стоит свеч.
Нитрозирование
Понять, что делает реакция нитрозирования енолизуемых карбонильных соединений в нашем, в общем-то вводном курсе органической химии, довольно трудно. Это очень частная реакция, не имеющая широкого применения. Если нам повезет, мы возможно встретимся с этой реакцией во 2 семестре, если решим разобрать исторически очень важный, но в наше время малополезный метод синтеза пирролов по Кнорру, где эта реакция используется как источник α-аминокетонов.
В этой реакции используют только енолизуемые кетоны. Альдегиды легко окисляются в условиях нитрозирования, потому что нитрозо-соединения – это окислители, работающие по механизму гидридного переноса, а это один из основных механизмов окисления альдегидной группы (второй – свободнорадикальный).
Нитрозированием вообще называется введение в органические молекулы нитрозо-группы NO. Это очень странная группа с очень необычной химией, и мы коснемся ее только мимоходом. Источником нитрозо-группы в органических молекулах обычно является электрофильное нитрозирование – реакция органических нуклеофилов с нитрозоний-катионом NO+ или ковалентными нитрозо-соединениями с уходящими группами типа нитрозилхлорида NOCl, нитрозилбромида NOBr, азотистого ангидрида ON-O-NO и др. Все эти молекулы элементарно образуются при действии на нитриты сильных кислот – анион кислоты становится уходящей группой в нитрозосоединении, а если анион не имеет заметной нуклеофильности, как например, анион серной кислоты, то получается азотистый ангидрид.
Если в этот момент рядом находятся енолизуемые кетоны, то любая из этих молекул легко реагирует как электрофил. Все эти частицы и даже сам нитрозоний-катион – довольно слабые электрофилы, например, они не реагируют с большинством ароматических соединений кроме самых реакционноспособных типа фенолов и анилинов. Во 2 семестре мы еще посмотрим, что получается там с нитрозированием. В любом случае, это неплохая аналогия: фенолы и енолы это очень близкие по структуре и реакционной способности соединения.
Нитрозирование енолов происходит при действии любого из электрофильных нитрозо-соединений. Логично предположить, что самый электрофильный из них сам нитрозоний-катионом NO+ и он-то и реагирует, и так было бы проще всего. Увы, не все так просто. Такие катионы (какие такие? – маленькие, с локализованным зарядом) живут только в сильнокислой среде с малой нуклеофильностью. Мы видели такие условия в очень похожей на нитрозирование реакции нитрования, где работает нитроний-катион, и там нам нужна была концентрированная серная кислота или даже что-то еще более кислотное. Но для нитрозирования используют гораздо более простые условия – берут кетон, нитрит натрия или эфиры азотистой кислоты и прикапывают разбавленную соляную кислоту. В таких условиях нитрозоний-катион не живет, а образуются ковалентные нитрозо-соединения, в данном случае NOCl. Если кетон несимметричный, то условия реакции – обратимое образование енола – диктуют участие более замещенного енола и образование более замещенного продукта.
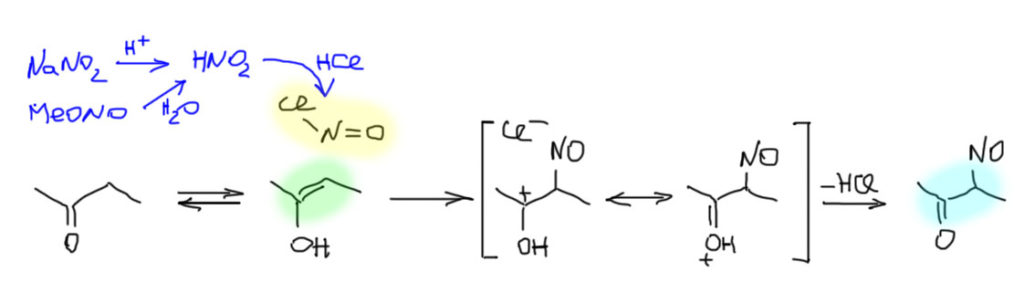
Образующийся нитрозокетон – очень интересная штука, потому что он очень похож на 1,3-дикетоны, которыми мы еще займемся, и точно также должен быть склонен к таутомерии, причем можно нарисовать два разных таутомера. Сразу заметим, что оставаться в нитрозо-кето-форме, так как нарисовано на схеме реакции, у него нет никаких шансов. Нитрозо-группа – явно очень сильный акцептор, и индуктивный, и мезомерный. Она очень похожа на карбонильную, но имеет в составе более электроотрицательный азот вместо углерода. Поэтому атом водорода на углероде между двух сильных акцепторов обречен иметь немаленькую кислотность, pK такой CH-кислоты не может быть выше 10, а скорее всего и существенно ниже. Поэтому в равновесии такой формы не может быть много (посмотрите еще раз обсуждение соотношения таутомеров на вкладке выше). Но перебраться он может как на кислород нитрозо-группы (нитрозо-оксимная таутомерия), так и на кислород карбонила (обычная кето-енольная таутомерия). Выбрать между этими двумя формами непросто, и на основании общих соображений не получится. Один фактор учесть можно – водородную связь внутри молекулы с образованием шестичленного цикла – это всегда очень выгодно, и ложится увесистой гирькой на чашку весов имеющего такую особенность таутомера. В оксимной форме образование такой связи очевидно и легко, в альтернативной нитрозоенольной форме могут быть еще и два диастереомера – (E) и (Z), и водородную связь может иметь только один из них, следовательно это уж точно не проще чем в оксимной форме. Так или иначе именно оксимная форма преобладает.
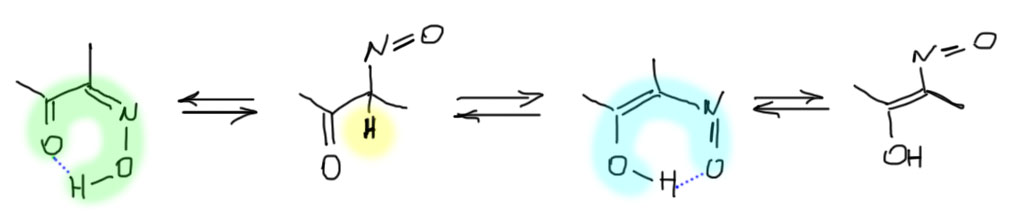
Результат интересный – получается, что нитрозирование енолизуемых кетонов дает моно-оксим дикетона. В принципе, его можно было бы гидролизовать в 1,2-дикетон, но есть более надежный способ синтеза этих соединений. С другой стороны, можно добавить гидроксиламин и получить полный оксим дикетона, а это очень интересные соединения. Из метилэтилкетона при этом получается знаменитое вещество – диоксим диацетила, хорошо известный под названием диметилглиоксим (гли – от глиоксаля, так исторически называется простейший диальдегид, этандиаль) он же реактив Чугаева.
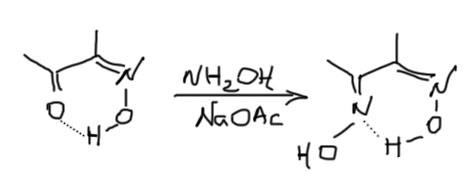
Реактив Чугаева – совершенно потрясающе интересный хелатный лиганд. Особенно хорошо известен его комплекс с никелем, имеющий кроваво- красный цвет. Цвет хорош, но еще красивее структура комплекса, который образован двумя моноанионами лиганда, образующими координационную сферу металла таким образом, что один лиганд дополнительно сцеплен с другим за счет очень прочных водородных связей, и такая структурра образуется из-за очень точной пространственной подгонки обоих лигандов друг под друга, или, как это часто называют, самокомплиментарности. Хотя водородные связи обычно считают межмолекулярными, но здесь они реализованы в одной молекуле комплекса, фактически связывая два отдельных лиганда в макроциклический общий лиганд.
Реакция с диоксидом селена
Окисление диоксидом селена носит название реакции Райли (Riley) в честь первооткрывателя. Довольно полезная реакция, но не очень популярная, потому что диоксид селена редко можно найти в органической лаборатории, и нужно заранее озаботиться покупкой этого реактива, а это довольно серьезный аргумент – обычно, когда нужно что-то сделать, ищут реактивы у себя на полках и спрашивают у соседей. Ничего не найдя, бросают замысел, или выбирают другую дорогу. К тому же с селеном связано много подсознательных страхов – считается, что соединения селена имеют отвратительный запах и очень токсичны. Некоторая правда в этом есть, хотя это не относится к оксидам селена и их производным, да и сам селен – вполне безобидное вещество.
Диоксид селена и селенистая кислота – аналоги сернистого ангидрида и сернистой кислоты, но свойства и реакционная способность у этих близких родственников драматически различаются. Производные черырехвалентной серы – скорее нуклеофилы, а сам сернистый ангидрид – удивительно малореакционноспособное вещество, из-за чего его часто применяют в жидком виде как растворитель, который сам никому и ничему не мешает. А производные четырехвалентного селена обладают весьма богатой и разнообразной реакционной способностью, в первую очередь это вполне неплохие электрофилы, но еще они умеют играть в сложные игры согласованных реакций, участвуя в циклических переходных состояниях с одновременным перемещанием электронов и связей. Диоксид селена – совершенно уникальный окислитель, осуществляющий превращения, которые другими способами потребовали бы многостадийных цепочек. У диоксида селена есть два любимых реакционных центра – сильнодонорные двойные связи и аллильные атомы водорода, – которые он ловко выцепляет из очень сложных структур, и не обращая внимание ни на что другое, превращает в очень полезные продукты. Такие свойства очень нравятся синтетикам. Окислители вообще обычно довольно грубые и бесцеремонные ребята, и каждый раз в тех случаях когда нужно что-то селективно и аккуратно окислить в сложной молекуле, всегда приходится основательно напрягаться, чтобы не окислить что-то лишнее или не переокислить то, что нужно, потому что почти любой окислитель втайне или откровенно мечтает окислить любое органическое вещество до угольного ангидрида и воды.
Диокид селена же не таков – механизмы его действия очень специфичны и аккуратны, они развиваются в основном не как окислительные (а окисление – это обычно банальный грабеж, состоящий в том, чтобы оторвать и присвоить электроны или атомы какие-нибудь вместе с их электронами), а как обычные реакции замещения, присоединения, согласованной еновой реакции, и изменение степени окисления наступает как формальный результат образования и разрыва связей.
Кроме того, это еще и один из первых настоящих реагентов в органическом синтезе в современном смысле этого понятия. Реакция была открыта аж в 1932 году, то есть в то странное время, когда органики обнаружили, что они занимаются этим делом уже приблизительно сто лет, и за это время ничего не поняли, хотя уже перепробовали все доступные им вещества и открыли все возможные реакции между ними, и им остро нужно, во-первых, наконец перестать просто смешивать все подряд и задуматься о том, как эти реакции идут (из чего тут же родилась теория органической химии, то есть наука о механизмах), а во-вторых, выйти из органических лабораторий и зайти к соседям неорганикам в поисках новых реактивов. О, это был непростой шаг. До этого (а многие верят в это до сих пор) органики были совершенно уверены в том, что их химия совершенно особенная, главная, единственная, заслуживающая названия “химия”. Обратите внимание на тот забавный факт, что у неорганической химии вообще нет названия – это просто не органическая химия, все остальное; то есть есть на свете только одна настоящая химия со своим собственным названием. Это удивительно, это все равно, как если бы, например, в языке не было бы слов для каких-то вполне важных вещей, например, вместо “мужчина” говорили бы “неженщина”, а вместо взрослый человек – “неребенок”… И ведь и смысл какой-то в этом можно было бы легко найти (возможно, даже есть такие языки на свете, где это именно так, или как-то похоже, но я в этом не уверен). Но вот с неорганической химией так и получилось, и никто не против. Они, коварные неорганики, просто догадывались, что разок попробовав что-то откровенно неорганическое, органики увлекутся и сами того не заметив, погрузятся в совершенно неисчерпаемые запасы неорганических соединений, и что из этих робких опытов вырастут современный синтез и катализ, и химия координационных соединений, и химия материалов, и что все это ускоренными темпами начнет вытеснять классическую органическую химию на обочину прогресса. В современных журналах, как бы они ни назывались, вы уже с изрядным трудом сможете понять, о какой химии там говорят, но точно не об органической. Это поучительная история, которая показывает, что пафосное название не всегда гарантирует блестящее будущее.
Вернемся к окислению енолизуемых карбонильных соединений диоксидом селена. За уже почти сто скоро как сто лет никто не удосужился изучить механизм этой реакции, но это не помешало целым двум мощным нобелевским лауреатам, Элайасу Кори и Барри Шарплесу сцепиться по поводу этого механизма. С тех пор в литературе попеременно встречается то один, то другой механизм, без попыток выяснить, который из них правильный. Мне более разумным кажется механизм Шарплеса, к тому же он хоть как-то обоснован. Он довольно прост, хотя и содержит пару не очень внятных стадий. Но начинается все просто реакцией электрофильного диоксида селена с равновесным енолом, и, как всегда в таких случаях, если кетон несимметричный, то преобладает более замещенный енол. Отдельный вопрос заключается в том, как обеспечивается катализ кето-енольного превращения в реакции диоксида селена, которую часто проводят в инертном растворителе типа диоксана. Ответ, видимо, простой – сам диоксида селена, видимо, всегда содержит примесь селенистой кислоты, обладающей вполне достаточной кислотностью для того чтобы обеспечивать приемлемую скорость кето-енольного превращения. Итак, присоединяем диоксид селена к двойной связи енола и получаем, после обычного перемещения протона, некий интермедиат, у которого рядом с карбонилом остаток селенистой кислоты (это называется кетоселениновая кислота). Тут мы замечаем уже знакомый нам атом водорода с повышенной кислотностью между двумя акцепторами, и видим неплохую перспективу ухода этого протона с образованием нуклеофильного центра. Но вот проблема – это-то нам как раз и не нужно – нам нужно окислять этот атом, а не восстанавливать, а минус на атоме – признак восстановления. Как быть? Спасение приходит откуда не ждали, потому что дальше происходит нечто, что иногда называют перегруппировкой Пуммерера (до собственно перегруппировки дело не доходит, но некоторая аналогия, заключающаяся в неожиданном изменении реакционной способности атома углерода с обычной нуклеофильной енольно-енолятного типа, на электрофильную из-за того, что между этим атомом и селеном возникает нечто похожее на двойную связь за счет ухода от селена уходящей группы (в этом случае воды). Вот это ровно то, что нам нужно, так как атом углерода в одночасье сделавшийся из нуклеофила электрофилом, спокойно присоединяет воду, а селеновый остаток, который незаметно для себя уже восстановился от Se(4+) до Se(2+), немедленно разваливается на металлический селен и воду. Схема не лишена признаков легкого мошенничества, но таковы многие органические механизмы, и пока никто не возражает, сойдет.
И еще много чего, например, кислотно-катализируемая альдольная конденсация и т.п.
И немало других реакций карбонильных соединений зависят от того, что в енолизуемых карбонильных соединениях содержится небольшое количество енола. Этот енол, выражаясь немного посовременнее, обслуживает важный канал превращения карбонильного соединения в продукты разнообразныз реакций, в основном через электрофильное присоединение к енолу.
Упомянем только еще одну реакцию – кислотно-катализируемую альдольную конденсацию. Почему только кислотно-катализируемую? Потому что более распространенная альдольная конденсация, катализируемая основанием, идет не через енол, а через енолят, а это немного другая история, которой мы займемся отдельно.
В кислотно катализируемой альдольной конденсации нуклеофилом (метиленовой компонентой) является равновесный енол, но что является электрофилом. Иногда, увы слишком часто, говорят, что само карбонильное соединение. Это безусловно не так, потому что, если бы это было так, енолизуемые карбонильные соединения превращались бы в альдоли просто при стоянии. Но ничено подобного не происходит – и ацетальдегид и ацетон не превращаются в свои альдоли. Ацетон вообще стоит годами в закрытой таре без видимых изменений, а ацетальдегид медленно полимеризуется, но не в альдоль и не через альдоль.
Придется вспомнить, что карбонильная группа тоже изменяет реакционную способность в условиях кислотного катализа – увеличивает электрофильность или за счет протонирования по кислороду (сильный эффект, требует присутствия сильных кислот), или за счет образования водородной связи по атому кислорода (слабый эффект, для этого достаточно слабых кислот, желательно в избытке, но в альдольной конденсации этот тип кислотного катализа почти не работает). Возможен еще катализ не протонными кислотами, а кислотами Льюиса типа безводного хлорида цинка, но мы не будем трогать этот путь, так как там очень трудно разобраться в каше возможных путей и продуктов. Просто пока заметим, что этот путь является основным, если в качестве метиленовой компоненты берут не енол (еще раз повторю – енол взять нельзя, можно только смиренно дождаться милостей от никак не зависящего от вас равновесия), а стабильные и полученные заранее эфиры енолов. Обсудим это отдельно.
Итак, в кислотно катализируемой альдольной конденсации равновесный енол реагирует с протонированным карбонильным соединением в присутствии сильной кислоты. Кислотно-катализируемая альдольная конденсация почти не применяется в современной химии потому что она очень неселективна, ее почти невозможно выполнить в перекрестном варианте между разными карбонильными соединениями даже в тех случаях, конда нормально работает ненаправленная конденсация, катализируемая основаниями. В кислотно-катализируемой конденсации образуются смеси продуктов, и она почти никогда не останавливается на простом альдоле или продукте его дегидратации, а идет дальше. Иногда этим даже можно воспользоваться, как в самоконденсации ацетона и других метилкетонов в 1,3,5-тризамещенные бензолы, но в большинстве случаев получается просто неопределенная смесь.
Некоторое минимальное применение кислотно-катализируемая конденсация находит для получения продуктов самоконденсации простых альдегидов и кетонов, при этом почти всегда получается продукт дегидратации. Например, имено так можно получить собственно кротоновый альдегид и окись мезитила (старинное название для продукта дегидратации самоальдоля из ацетона). Распишем реакцию на примере кротонового альдегида. Во-первых, обратим внимание на весьма примечательный факт: обе реагирующие частицы – электрофил и нуклеофил, карбонильная и метиленовая компоненты, протонированный карбонил и енол – образуются в результате действия одного и того же катализа фактически в одном и том же равновесии. Кислотный катализ умудряется добывать сразу оба типа реакционной способности, в отличие от основного катализа, который активирует только нуклеофильность. К сожалению, в химии такая доблесть не всегда ценится по достоинству: вот если бы на каждую молекулу енола образовывалась бы ровно одна протонированная карбонильная группа, и они тут же и количественно бы, и необратимо бы реагировали, и продукт этой реакции тут же бы куда-нибудь девался бы, например сам собой складывался бы в чистую баночку с карсивой этикеткой – то цены бы не было такой промысловатости. Увы, в реальности получается каша из десятков сцепленных друг с другом равновесий, все реагируют со всеми, и добром такой пир реакций не заканчивается. Тем не менее, наши далекие предки потратили много времени и сил на то, чтобы отработать методики таких простых реакций – температуру, концентрации, время, растворители, порядки смешения, скорость смешения и др. и пр. – и для таких реакций можно найти старые работающие методики, которые придется воспроизводить буквально и как можно ближе к тексту. Это занудно, но в целом дешево и работает, но по методике, скажем, для ацетальдегида не получится сконденсировать даже его ближайший аналог, для которого придется искать свою методику.
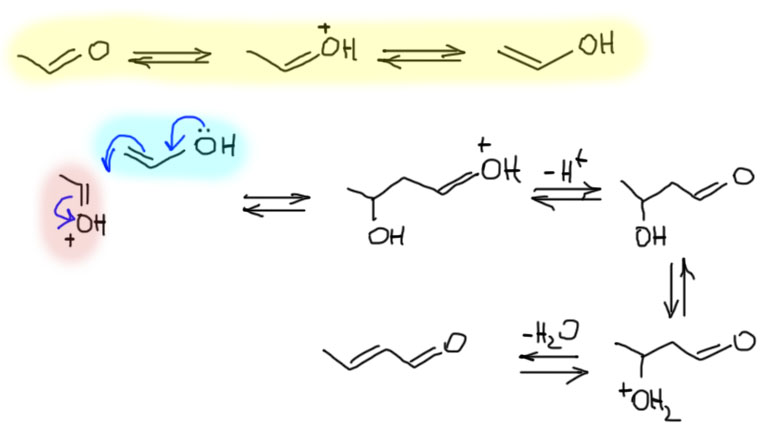
А что такое вообще таутомерия
Строго факультативно. Не читайте этот раздел, если не хотите забивать голову лишними сведениями, сеющими сомнения.
Таутомерия – это очень важная разновидность изомерии, в которой изомеры (таутомеры) связаны реальным равновесием, а превращение одного таутомера в другой происходит за счет переноса протона (такая таутомерия называется прототропной) или реже других атомов, например, металлов (тогда это металлотропная таутомерия) между атомами, связанными чередующимися простыми и кратными связями с одновременным перераспределением этих связей. Наиболее часто такой процесс происходит в трехатомных фрагментах (тогда таутомерия называется триадной), но бывает и таутомерия в пятиатомных, семиатомных и т.п. фрагментах, но это все довольно большие редкости, поэтому сразу ограничим себя только самой важной и невероятно часто встречающейся триадной таутомерией, в которой протон перемещается между крайними атомами трехатомного фрагмента. Примеров такой таутомерии многие десятки, вот хотя бы три примера (неиспользованные валентности не показаны) – кето-енольная, нитрозо-оксимная, имин-енаминная.
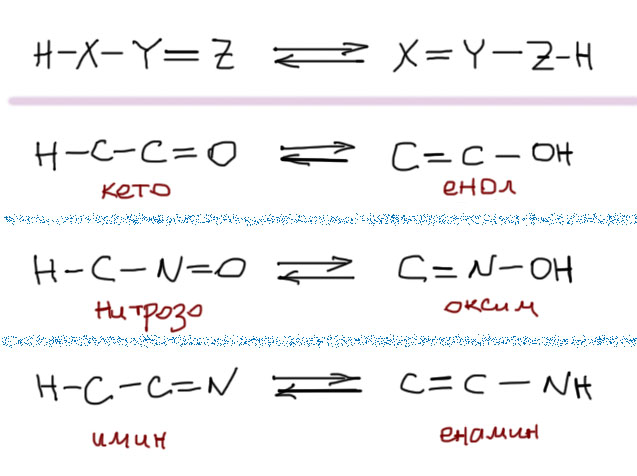
Некоторым читателям может прийти в голову другая аналогия – а как связана триадная таутомерия и аллильная перегруппировка, с которой мы тоже неплохо знакомы, хотя бы в такой форме как взаимопревращение продуктов 1,2- и 1,4-присоединения электрофилов к сопряженным диенам. Некоторым может даже прийти в голову такая вещь, как сигматропные перегруппировки, в частности чрезвычайно похожая на обычную таутомерию [1,3]-сигматропная перегрупировка.
Как ответить на этот вопрос? Увы, есть проблема. Понятие таутомерии не поддается формальному определению. Это частая ситуация в химии и вообще в науках, когда одна часть исследователей любит все обобщать и везде искать аналогии, а другая безуспешно стремиться к строгости формулировок, стараясь наоборот каждое определение делать максимально точным. Эти партии никогда не могут договориться, а поскольку в химии и вообще в науках (кроме марксизма-ленинизма) нет верховных начальников или жрецов, то все эти споры обычно ничем не кончаются. В химии своеобразным законодательным правом обладает уже знакомый нам ИЮПАК. По крайней мере, с номенклатурой органических, неорганических и координационных соединений и некоторыми другими вещами у этой международной организации что-то получилось – в научных статьях используют именно ее. ИЮПАК попробовал определить и наиболее важные термины химии и издал Золотую Книгу (IUPAC Golden Book, по цвету обложки вполне в геральдическом смысле, она не золотая, а всего лишь желтая, но именно так – золотом – называется этот цвет в гербах и флагах), или полностью – Компендиум Химической Терминологии. Золотую книгу можно с полными основаниями назвать эпическим провалом – о ее существовании мало кто знает, и почти никто ей не пользуется, но другого авторитетного источника терминологии все равно нет. Книга не смогла дать строгого определения и таутомерии, и это и есть надежный признак того, что такого определения дать и не получится. При этом, данное Золотой книгой определение не лишено простоты и изящества, что не удивительно, так как над определениями этой книги работали очень серьезные ученые. Согласно Золотой книге таутомерией является взаимопревращение изомеров, происходящее за счет перенос какого либо атома или остатка (G) с одновременной перестройкой связей, причем этот процесс должен а) происходить легко (readily) – и никто не объясняет, что значит “легко”; б) перемещающаяся группа или атом в процессе становится либо электрофугом (то есть перемещается, не забрав с собой электронную пару бывшей связи), либо нуклеофугом (то есть перемещается с парой электронов).
 Скажем ИЮПАК спасибо, потому что под это определение полностью подпадают и аллильные перегруппировки, в том числе и превращение продуктов 1,2-присоединения электрофилов к сопряженным диенам в продукты 1,4-присоединения, именно потому что определение Золотой книги допускает перемещение нуклеофуга, в частности, бромид-аниона вместо обычного для традиционного понимания таутомерии электрофуга типа протона. Тем не менее, вы практически никогда не встретите применения термина таутомерия к аллильным перегруппировкам. Этот пример показывает, насколько нестрогой является общепринятая химическая терминология, и как важно понимать суть происходящих процессов, а не уповать на механическое запоминание названий и правил.
Скажем ИЮПАК спасибо, потому что под это определение полностью подпадают и аллильные перегруппировки, в том числе и превращение продуктов 1,2-присоединения электрофилов к сопряженным диенам в продукты 1,4-присоединения, именно потому что определение Золотой книги допускает перемещение нуклеофуга, в частности, бромид-аниона вместо обычного для традиционного понимания таутомерии электрофуга типа протона. Тем не менее, вы практически никогда не встретите применения термина таутомерия к аллильным перегруппировкам. Этот пример показывает, насколько нестрогой является общепринятая химическая терминология, и как важно понимать суть происходящих процессов, а не уповать на механическое запоминание названий и правил.
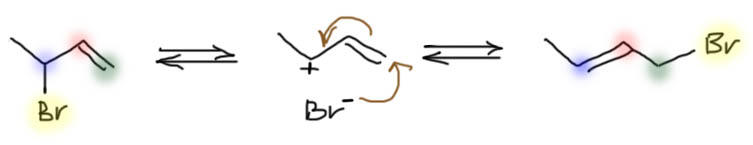
Остроумие определения Золотой книги заключается в том, что средней группой Y может быть все, что угодно – это не обязательно один атом. Поэтому, под это определение отлично подходят и таутомерии в более длинных системах – пентадные и т.п., и даже так называемая кольчато-цепная таутомерия, очень популярная в химии углеводов. Кольчато-цепная таутомерия – это просто внутримолекулярное образование полуацеталя из кетона или альдегида, в молекулах которых есть гидроксильная группа на расстоянии, обеспечивающем образование 5- или 6-членного цикла. Таутомерией это называли просто по традиции, и правильнее всего было бы перестать использовать это двусмысленное и бесполезное название. Но вот ведь как получается – это равновесие отлично подходит под определение Золотой книги, если мы представим под группой Y просто часть цепи. Для удобства разметим все теми же цветами. Получается вполне аналогично, особенно, если мы учтем, что раз Y это в этот раз не просто атом, а целая цепь, то и новая связь с фрагментом X не обязана исходить с того же атома, а может быть как раз замыканием цикла.
Енолят-анионы
Раздел в работе. Пока только несколько важных вещей.
Енолят – это сопряженное основание енолизуемого карбонильного соединения. Обе таутомерные формы, енол и кето-форма, дают один и тот же енолят. Несимметричные кетоны, имеющие α-атомы углерода с обоих сторон от карбонильной группы, могут давать два разных енолята, и с этим связана проблема региоселективности в реакциях, идущих через еноляты – могут образоваться два разных продукта, а если еще учесть стереохимию, то даже больше. Стереохимию мы учитывать не будем.
Енолят - амбидентный нуклеофил и может реагировать как по атому углерода, так и по атому кислорода
Главная проблема, которая может и даже должна возникнуть при обсуждении реакций енолята, – почему он реагирует по атому углерода. Источник противоречия прост: с одной стороны мы весьма категорично говорим, что структура енолята почти точно отображается именно енолятной формой с отрицательным зарядом на кислороде. Следовательно, отрицательный заряд в еноляте и находится на атоме кислорода. Ну и что? Но разве нуклеофильность не связана именно с отрицательным зарядом на атоме? разве мы не утверждали, когда обсуждали нуклеофильность, что анионы – более сильные нуклеофилы, чем нейтральные молекулы?
Нет, конечно. Анионы – более сильные нуклеофилы только по сравнению с нейтральными молекулами, являющимися сопряженными кислотами этих анионов. Гидроксид-ион сильнее воды, а ацетат – уксусной кислоты, мы уже не говорим о разнице в нуклеофильности метильного карбаниона и метана. Да, и енолят – более сильный нукелофил чем сам енол, и это точно верное утверждение. Но мы не можем сравнивать два разных нуклеофила или, как в еноляте, два разных нуклеофильных центра только на основании заряда на атомах. Нет такой закономерности в представлениях о нуклеофильности, просто нет и все, а если вам кажется, что есть, то, уверяю вас, вы ее сами выдумали и сами же в нее и поверили.
А как же быть, какая есть закономерность? Вот мы точно в разделе о нуклеофильности связывали нуклеофильность и основность для элементов 2 периода, то есть как раз тех самых C, N, O. Больше основность (больше pK) – больше нуклеофильность. Это качественная тенденция, не дающая численных оценок, а работающая в терминах больше-меньше. Если разница большая, то и оценка получается довольно надежная. Да, вот это уже неплохо, ведь мы только что обсуждали, что само явление таутомерии связано с различием CH- и OH-кислотности кето-формы и енола, а это вполне можно интерпретировать так, что енолят простых карбонильных соединений – существенно более сильное основание Бренстеда-Лоури по атому углерода (этому кислотно-основному равновесию соответствует CH-кислотность кето-формы), чем по атому кислорода (этому равновесию соответствует OH-кислотность енола). Вроде работает.
Но некоторое ощущение неудовлетворености от такого объяснения не проходит. Если чуть-чуть забежать вперед, то там окажется, что еноляты 1,3-дикетонов и кетоэфиров типа ацетоуксусного эфира, тоже в основном работают как C-нуклеофилы, хотя там как раз кислотность кето-формы очень сильно повышена, и порядок основности по углероду и по кислороду совсем иной. Строго говоря, разница в основностях там не очень велика, и закономерность основность-нуклеофильность просто перестает работать, именно потому что это очень грубая, качественная закономерность.
Второе объяснение очень часто применяют к амбидентным нуклеофилам, а енолят – типичный амбидентный нуклеофил. Это объяснение использует принцип жестких и мягких кислот и оснований (ЖМКО), перетолковывая его так, что понятия кислот и оснований отображается на понятия электрофилов и нуклеофилов. Подробнее мы это учение разберем где-нибудь в другом месте, но пока примем его без критики и доказательств. Оно до сих пор очень популярно и меет множество сторонников, которые искренне считают его доброкачественной теорией. Для амбидентных нуклеофилов этот подход работает хорошо и просто. В амбидентных нуклеофилах всегда есть два нуклеофильных центра. Один мы объявляем жестким, а другой мягким. Жестким будет тот, на котором в основном сидит отрицательный заряд и который связан с более электроотрицательным атомом, расположенном повыще и правеее в периодической таблице. В еноляте это кислород, а, например, в другом знаменитом амбидентном нуклеофиле, цианид-ионе, это азот. Второй центр объявляем мягким. И в еноляте, и в цианиде – это углерод. Далее говорим следующее: с нейтральными электрофилами (мы их считаем мягкими) амбидентный нуклеофил предпочитает реагировать по мягкому нуклеофильному центру. Нейтральные электрофилы это например, алкилгалогениды и тозилаты из SN2-замещения, эпоксиды оттуда же, карбонильная группа альдегидов и кетонов. Все эти электрофилы реагируют с енолят-анионом по атому углерода, мягкому нуклеофильному центру. С другой стороны, есть и жесткие электрофилы – это электрофилы с настоящим положительным зарядом, электрофилы-катионы, например, сам протон (енолят всегда протонируется более сильными кислотами по кислороду, образуя енол – ай-яй-яй! караул!! только что же рассказывали, что енолят более сильное основание по углероду, а протонируется он, оказывается, по кислороду – совсем заврались! – Нет. Здесь, на самом деле, нет никаких противоречий, и ключевое слово – протонируется более сильными кислотами, когда важнее не разница в основностях, а скорость переноса протона, и мы опять призываем не путать равновесия и кинетику, ведь енол все равно перегруппируется в равновесную кето-форму). И еще сюда же относятся электрофилы с электрофильными центрами на более электроположительных атомах, имеющих огромное сродство (образующих очень прочные связи) с кислородом, например, кремния. Именно поэтому мы легко получаем кремниевые эфиры енолов реакцией енолятов с галогенпроизводными силанов.
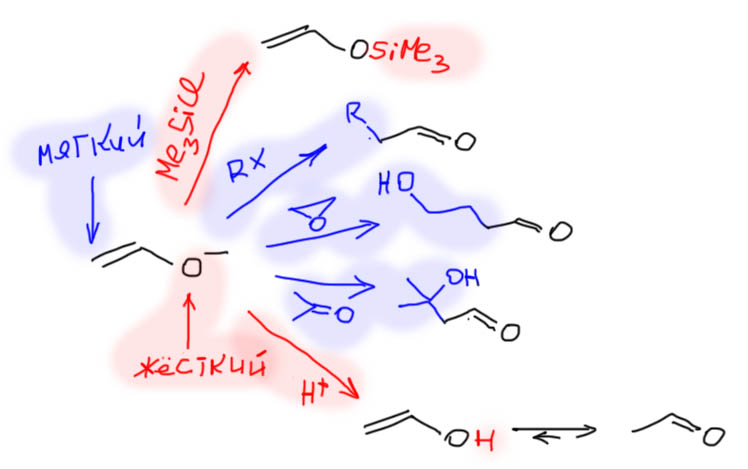
Итак, ЖМКО работает. И можно на этом остановиться, если только не задавать вопроса, а почему. Это больше похоже не на теорию, а на банальное обобщение экспериментального материала. Это так и есть, но чтобы под это почти религиозное учение подложить что-то, напоминающее настоящую науку, не обойтись без квантовой химии, с которой мы совсем не в ладах. Иными словами, дальше можно не читать. Объяснения с точки зрения ЖМКО всегда принимаются, и на экзаменах, и даже в научной литературе.
Но можно и почитать. Самое популярное квантово-химическое описание того, как реагируют молекулы, использует теорию молекулярных орбиталей, и в ней сосредоточивает внимание не на всех орбиталях, а только на так называемых граничных, особенно двух из них – самой последней по энергии (считая снизу вверх) занятой – высшей занятой (ВЗМО), и самой первой незанятой – низшей свободной (НСМО). Когда говорят о реакции нуклеофил-электрофил, то довольно очевидно, что для нуклеофила мы берем его ВЗМО (нуклеофильность определяется парой электронов, которая пойдет на образование новой связи), а для электрофила – его НСМО (пустую орбиталь, ведь и электрофил всегда предоставляет пустую орбиталь для образования новой связи). Дальше, мы должны откуда-то взять эти орбитали (посчитать или просто оценить на глазок, если мы знаем, как это делается), и в этом случае направление реакции будет соответствовать максимальному взаимодействию этих граничных орбиталей, в свою очередь соответствующему взаимодействию между теми атомами, на которых в этих орбиталях вклады соответствующих атомов максимальны (молекулярные орбитали строятся из атомных, и каждый атом дает какой-то вклад, какие-то атомы больший, какие-то меньший или совсем незначительный). Займемся этим для реакции енолята. В этом случае, нам потребуется ВЗМО енолята, который даст нам представление о том, где находится больший вклад в эту орбиталь – на кислороде или на углероде. Электрофилом и его НСМО нам заниматься не придется, потому что там никаких сюрпризов быть не может, если у электрофила один центр, то на нем и нужная орбиталь.
Как прикинуть орбитали енолята? Довольно просто, если мы представляем себе орбитали очень похожего на енолят аллил-аниона. Нам это даже вроде бы нужно – знать, как выглядят орбитали аллильной системы. Речь идет не о всех орбиталях, а только о π-орбиталях сопряженной системы, по разным причинам именно сопряженная система дает ВЗМО, что довольно разумно и понятно, ведь именно сопряженная система определяет большинство свойств. У аллильной системы три π-орбитали (три атома, у каждого по одной p-орбитали, π-орбитали делаются из p-орбиталей, количество молекулярных орбиталей равно количеству атомных, использованных для их изготовления). Снизу вверх: связывающая, несвязывающая, разрыхляющая. Число узлов (узловых плоскостей, на рисунке красный пунктирчик), как всегда увеличивается снизу вверх – ноль, одна, две. Узел – это место где встречаются доли орбиталей с разным знаком (разным цветом на картинке). Нарисовали, заполняем электронами – у аниона 4 электрона, два от двойной связи, два от аниона. Итого – ВЗМО аллильного аниона – это вторая (несвязывающая – но нам это неважно, нас интересует не энергия орбитали, а ее форма, вклады атомов) МО.
Рядом рисуем очень схематически орбитали енолята. Енолят отличается от аллильного аниона тем, что вместо одного углерода кислород, более электроотрицательный элемент. В остальном енолят и аллил-анион фактически одно и то же, такие частицы называют изоэлектронными (количество и распределение электронов одинаково). Это значит, что и орбитали в первом приближении одинаковы – у них так же распределены узловые поверхности и доли атомных орбиталей глядят в те же стороны. С одной важной разницей – аллил симметричен, а енолят нет, и там где у аллила одинаковые по размеру атомные орбитали, у енолята будут разные. Замена атома на более электроотрицательный в форме орбиталей отражается довольно просто – в аналогичных орбиталях атомы как будто меняются вкладами: более электроотрицательный элемент берет больше в нижележащих орбиталях, а менее электрооотрицательный за это получает компенсацию в более высоколежащей. Получается вес кислорода увеличивается в самой первой орбитали, а вес углерода во второй. Но это и есть ВЗМО! Вот её-то нам и надо, именно она съела нашу морковь, в смысле электрофил!!
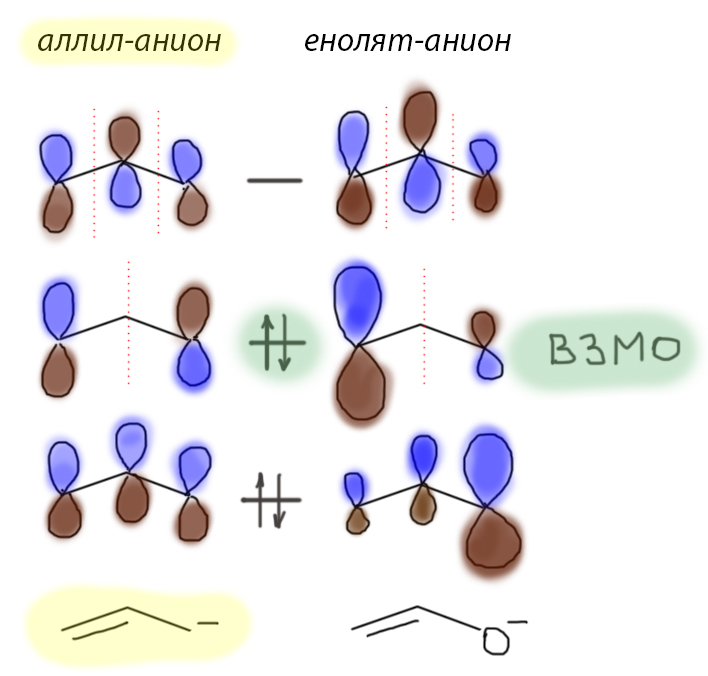
Такой подход позволяет понять, почему два реагента реагируют именно по таким атомам, а не другим. Реагенты стараются сблизиться и образовать связи так, чтобы были максимальными орбитальные взаимодействия, среди которых считаются самыми главными взаимодействия ВЗМО-НСМО. Такой способ взаимодействия называют орбитальным контролем. И тогда, если немного подумать и сопоставить, именно орбитальный контроль соответствует взаимодействию мягкий-мягкий в ЖМКО. Фактически получается, что представление об орбитальном контроле это объяснение тому, как работает концепция ЖМКО. Мы даже можем сказать, что эта теория примиряет нас с теорией ЖМКО, которая сама по себе весьма голословна, но с подкладкой из орбитального контроля становится как будто бы обоснованной, по крайней мере в части мягкое-к-мягкому.
А тогда что такое взаимодействие жесткий-жесткий из ЖМКО? Это называется обычно зарядовым контролем, то есть таким направлением реакции, в которой максимальными делаются не орбитальные взаимодействия, а просто взаимное притяжение мест с максимальными зарядами. В протонировании енолята сильной кислотой, например, положительный протон предпочитает атом с наибольшим отрицательным зарядом – кислород. Но и заряды на атомах тоже берутся из квантово-химических расчетов и представлений, только в определении заряда участвуют не граничные орбитали, а все вообще орбитали, образованные атомными орбиталями валентной оболочки.
Вот как-то так приблизительно и принято объяснять реакции в последние лет 40-50 после первых работ Роберта Вудварда и Роалда Хоффмана в середине 1960-х. Это не единственный способ и не самый современный, но точно самый популярный, отмеченный Нобелевской премией 1981 года, сочетающий достаточно солидное научное обоснование, и вполне доступную нормальным людям наглядность.
Почему алкилируют енамины, а не еноляты
Вообще-то, если поискать в литературе, то реакции алкилирования енолятов вполне нередко попадаются. Да и в учебниках они упоминаются, часто вообще без комментариев. В реальных синтезах из литературы это почти всегда очень специфические случаи, а идея запоминать частные случаи в органической химии – плохая идея, слишком много в органике частных случаев, которые нельзя обобщить. Мы предпочитаем использовать более надежные методы, которые можно использовать для многих похожих реакций. И в случае выбора – енолят или енамин – в качестве нуклеофила для алкилирования, выбирать мы будем енамин.
Главная проблема при алкилировании енолятов простая – алкилирование сразу дает готовое карбонильное соединение, альдегид или кетон, а поскольку реакция алкилирования не очень быстрая, то в реакционной смеси одновременно оказываются и карбонильное соединение и непрореагировавший енолят. И они вступают – не могут не вступать – в реакцию альдольной конденсации алкилированного карбонильного соединения (в качестве карбонильной компоненты) и еще не прореагировавшего енолята (в качестве метиленовой компоненты). Результатом будут невысокие выходы продуктов алкилирования и образование смеси побочных продуктов.
Эта проблема особенно важна при использовании енолятов альдегидов: из-за очень высокой реакционной способности альдегидов альдольная конденсация пойдет с большой скоростью, и побочная реакция станет основной.
В отличие от енолятов, енамины при алкилировании непосредственно в реакции дают соли иминия, которые не реагируют с непрореагировавшим енамином (могли бы, но не хватает реакционной способности), поэтому алкилирование протекает намного более гладко, и всячески поэтому рекомендуется, хотя, как мы знаем, и у него есть проблемы – невысокая нуклеофильность енамина делает реакцию алкилирования медленной, если только не используются очень активные SN2-субстраты.