Нуклеофильное ароматическое замещение
Внимание: эта страничка недоделана. Если честно, я про неё вообще забыл, но она есть и даже опубликована, следовательно, находится через поиск. Сейчас я ее прочитаю, чуток почищу, и куда-нибудь подлинкую. Тут всё правильно, только разработано не до конца, и местами оборвано на полуслове. Когда-нибудь доделаем.
Когда мы обсуждали нуклеофильное замещение в алифатическом ряду, механизмы SN2 и SN1, торжественно поклялись строго следить, чтобы углерод, на котором это замещение происходит, имел sp3-гибридизацию. Теперь настала пора разобраться, что это было – пустой запрет или что-то содержательное. Смысл того запрета был довольно прост: если мы разобрались в том, как происходит алифатическое нуклеофильное замещение, и как на практике проводятся те реакции, эти знания не помогли бы нам в ароматическом ряду. Бромбензол ни при каких обстоятельствах не будет реагировать с нуклеофилами в условиях, подходящих любому алифатическому бромпроизводному, ну хотя бы бромциклогексану, чтобы соблюсти некоторое внешнее сходство. Не будет бромбензол реагировать ни с, например, цианидом калия в ДМСО, ни с любым сильным нуклеофилом из 3-4 периодов. Вообще не будет. С другой стороны, тот же бромбензол вполне реагирует с теми нуклеофилами, за которым мы закрепили ярлык сильных оснований – с амидом натрия и литийорганическими соединениями, например. И результатом таких реакций, если судить по продуктами реакции будет именно продукты замещения, а не элиминирования – помните ведь, как страшно пугали вас элиминированием и призывали строго следить за основностью. Упустите основность, – говорили – не видать вам замещения, будет только элиминирование. А в ароматическом ряду все не так. Реакции или не идут, или идут, но по-другому. Попробуем разобраться.
Что здесь важно с самого начала.
- Механизмов больше, чем там. Даже в нашем сильно облегчённом курсе мы увидим не меньше трёх, а возможно и больше. И мы реально будем ими пользоваться. Один из основоположников этой химии, Джозеф Баннет, однажды попробовал посчитать механизмы ароматического нуклеофильного замещения и получил то ли 17, то ли 19. Он немного погорячился тогда, он вообще любил паясничать и изображать из себя лихого ковбоя с Дикого Запада, но факт остаётся фактом – не менее 10 основных уж точно, и еще множество вариантов.
- Заместители в ароматическом кольце играют очень важную роль. Именно они часто определяют механизм реакции.
- Особенно богатой химией нуклеофильного ароматического замещения обладают вовсе не производные бензола, а многочисленные гетероциклические соединения, у которых это один из основных видов реакционной способности – когда доберёмся до химии гетероциклов вновь вспомним про нуклеофильное ароматическое замещение.
- Уходящие группы не так важны, и при вдумчивом подходе в ароматическом замещении можно заместить то, что никогда или крайне редко работает в алифатическом, например, водород или фтор. Но в каждом случае придётся точно понять, как идёт реакция, и что ей нужно. С другой стороны, наша любимая уходящая группа из той химии, тозилат, в ароматической химии вообще бесполезна. Даже водород можно нуклеофильно заместить, а тозилат – нет. Можете хоть лопнуть от злости и вопить, что этого не может быть никогда, но это так. Мы еще вернёмся к этой курьёзной проблеме в химии фенолов во втором семестре.
- Нуклеофилы остаются нуклеофилами, и всё то, что мы про них узнали тогда, имеет значение и здесь, хотя акценты немного смещены. Соперничество нуклеофильности и основности в ароматическом замещении проявляется довольно необычным образом.
Если уходящая группы водород, комплексы принято называть комплексами Яновского. Это не совсем так хотя бы такой простой причине, что никакого Яновского на пути этих комплексов не было. Был чешский химик Ярослав Яновски (1850-1907), который первым наблюдал, что мета-динитробензол растворяется в растворах алкоголятов с образованием фиолетового окрашивания. Понятно, что в 1886 году никакой возможности разобраться, что стоит за этим окрашиванием, не было в принципе. Но, так или иначе, он действительно первым наблюдал интересное явление и сообщил об этом человечеству. За человечеством не заржавело – роль этого химика была признана, вот только по-русски лучше называть эти комплексы комплексами Яновски, а не Яновского. Достаточно нам своих знаменитых химиков, не стоит даже невольно тырить чужих. И не будем уподобляться австрийцам, хотя у них прав считать Яновски австрийским химиком чуть больше, потому в те времена всё это было Австро-Венгерской империей. Вообще, это еще одна поучительная история – хотите заниматься наукой, будьте наблюдательны и подмечайте всё необычное. Даже если сами не разберётесь, возможно, привлечёте внимание более поздних исследователей, и с высоты небес будете радоваться, что послужили причиной чей-то славы. Да и научная этика, как ни странно, работает неплохо. В последние полвека стало даже принято пересматривать историю всяких важных исследований, и нередко это приводит к тому, что ранее забытые имена достаются из пыльных полок библиотек, и начинают сиять заново.
Нуклеофильное замещение в активированных субстратах: основы
Сразу оговоримся: в этом разделе речь идёт только о производных бензола, ну и, возможно, нафталина. Гетероциклические соединения, вступающие в похожие реакции, но со своей очень яркой спецификой, здесь не рассматриваем.
Прямое нуклеофильное ароматическое замещение без дополнительных ухищрений – то есть просто взяв галогенпроизводное и нуклеофил, возможно подобрав подходящий растворитель и использовав обычные способы стимулирования реакций (нагревание, хорошее перемешивание и т.п.) – идёт только в случае, если в кольце есть акцепторные заместители, почти всегда -М-типа: нитро-группа, карбонильная группа и все ее разновидности, нитрил (циано), сульфоновая группа и т.п., одна или две или три, и обязательно в орто- и пара-положениях относительно уходящей группы.
Такие субстраты принято называть активированными, а нуклеофильное замещение в таких субстратах часто называют активированным нуклеофильным замещением. Мы так и будем это называть время от времени.
Итак, если у нас есть такой субстрат, и какой-нибудь нуклеофил, то с хорошей вероятностью они будут реагировать просто при смешивании в растворе и, возможно, при нагревании, но без дополнительных реагентов, катализаторов, и т.п. Это не значит, что не бывает таких случаев, когда добавление чего-нибудь такого не поможет реакции, это лишь значит, что в большинстве случаев это не нужно. Повторим еще раз – активирующее действие оказывают только акцепторные заместители в орто- и пара-положениях к уходящей группе!
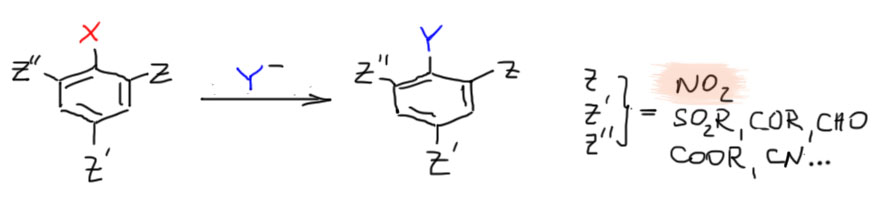
Хотя активирующими заместителями могут быть любые -M-группы, в активированном нуклеофильном замещении особую, потрясающую и ни с чем не сравнимую роль играет нитро-группа. Можно даже сказать, что в этом замещении есть нитро-группа, и есть всё остальное. Речь идёт об обычных группах, с которыми мы имеем дело в нашем курсе. Есть несколько более экзотических групп, сравнимых с нитро-группой по активирующему влиянию и даже превосходящих ее, но это необычные вещи, с которыми мы не столкнёмся. Для нас в активированном нуклеофильном замещении выдающаяся роль нитро-группы безальтернативна.
Очень важно следить, чтобы уходящая группа находилась в активированном положении. В других местах никакого замещения не будет, например: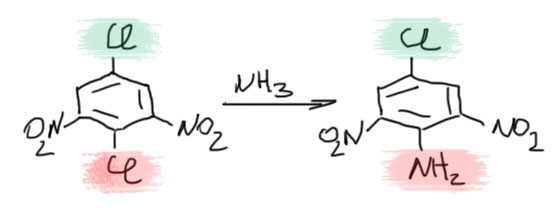
Активированные субстраты делятся на:
- те, у которых три нитро-группы в положениях 2,4,6 (такой остаток называется пикрилом), те у которых две нитро-группы в положениях 2 и 4. Эти соединения проявляют выдающуюся реакционную способность и реагируют практически с любыми нуклеофилами, даже с очень слабыми, и в очень мягких условиях. Косвенным свидетельством такого поведения является то, что если раствор любого такого соединения попадёт на руки, они желтеют и надолго, потому что в белках много нуклеофильных групп, и на них это тут же и сядет. Работайте в перчатках!
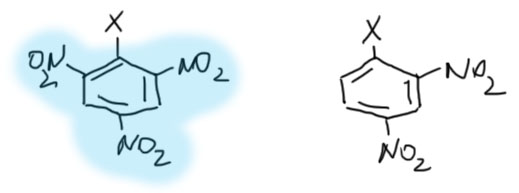
- все остальные – то есть те у которых одна нитро-группа, или до трёх групп другого вида. Такие субстраты реагируют с нуклеофилами намного медленнее, всегда нужно предпринять усилия – взять нуклеофил посильнее, использовать обычные способы повышения нуклеофильности, о которых мы говорили в SN2-замещении: спецрастворители типа ДМСО, ДМФА и т.п., межфазный перенос и т.п. Реакции часто проводят при нагревании, иногда очень сильном и продолжительном. Такие условия плохо сказываются на сохранности заместитей типа карбонилов, поэтому такие субстраты имеют гораздо более ограниченное применение в сравнении с динитро- и тринитро.
В качестве уходящих групп ограничимся галогенами. Активированное ароматическое нуклеофильное замещение имеет ярчайшую особенность, отличающую его от всех остальных видов замещения, как алифатического, так и ароматического – ряд галогенов выглядит совершенно необычно и вызывающе. Наилучшим галогеном является фтор, причём с огромным отрывом от всех остальных – часто разница в скоростях с идущим на втором месте хлором до 1000, а иногда и до 10000 раз! Это колоссальное преимущество, настолько большое, что остальные галогены по сравнению с фтором можно считать “мёртвыми” – нет никаких шансов, что они заместятся, если в молекуле есть активированный фтор. Это не так заметно для сильно-активированных динитро- и тринитро-производных, которые и так реагируют “со свистом” с любой уходящей группой, но для менее активированных субстратов типа моно-нитро-производных, фтор даёт возможность провести реакции замещения в более-менее нормальных условиях, в то время как остальные галогены замещаются с большим трудом и требуют очень серьезных усилий. И в тех условиях, когда в соединении есть разные галогены в активированных положениях, естественно, при такой разнице в реакционной способности замещается только фтор, например: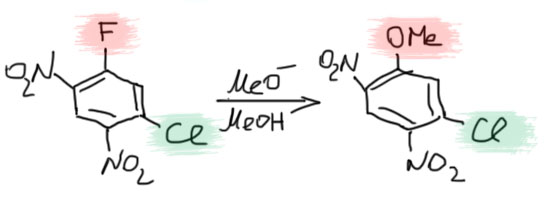
Остальные галогены в активированном положении тоже замещаются, причём способность к уходу хлора и брома приблизительно одинакова, а вот иод уходит хуже всех, часто на порядок-другой медленнее хлора или брома, а уж рядом со фтором иод совершенно неповоротлив. Как это непохоже на порядок уходящих групп в алифатическом нуклеофильном замещении, где всё наоборот – иод лучше всех, а фтор можно своротить только совместными усилиями всех нуклеофилов во Вселенной. Почему так происходит посмотрим ниже, разобрав механизм.
Механизм SNAr
Коротко
Механизм нуклеофильного ароматического замещения в активированных субстратах принято обозначать как SNAr, то есть просто как ароматическое нуклеофильное замещение. Несмотря на то, что применение этого механизма и реакций, подчиняющихся этому механизму ограничено только ароматическими соединениями, содержащими от одной до трёх акцепторных групп в орто- и пара-положениях относительно уходящей группы, именно этот механизм считается наиболее общим и хорошо исследованным. Фактически мы говорим, что раз нуклеофильное замещение без особых ухищрений и использования дополнительных, прямо не связанных с собственно реакцией замещения стадий, идёт только для таких субстратов, то нет смысла переживать по поводу того, что мезанизм и реакции неприменимы для более простых ароматических производных типа хорбензола.
Итак, у нас есть производное бензола с уходящей группой, обычно галогеном, и от одного до трёх заместителей, обладающих -M-эффектом в орто- и пара-положениях по отношению к уходящей группе. Сразу скажем, что заместителей таких много, но в подавляющем большинстве реальных реакций этого типа хотя бы один из них – нитро-группа. И есть нуклеофил. Нуклеофилом в этой реакции мы считаем то же самое, что мы уже использовали в SN2-замещении. По причинам ограничений, связанных как раз с природой активирующих заместителей, в этой реакции почти никогда не используют простые карбанионы (литий- и магнийорганические соединения, ацетилениды и пр.). Всё остальное годится.
Реакция нуклеофила с активированным ароматическим соединением происходит так:
- сначала нуклеофил присоединяется к тому углероду, на котором уходящая группа. Это медленная стадия, определяющая скорость всего процесса замещения. Эта стадия обратима, но когда уходящая группа галоген, про это можно не думать и считать стадию необратимой.
- скорость присоединения или, иными словами, реакционная способность субстрата зависит от числа и качества активирующих групп, а при одинаковом количестве и качестве заместителей – от электрофильности углерода, на котором происходит замещение. Электрофильность тем больше, чем больше электроотрицательность галогена. Поэтому скорость реакции наибольшая для фторпроизводных. Ряд уходящих групп выглядит очень необычно и не имеет прецедентов в дргих реакциях замещения: F >> Cl или Br > I. В реакциях этого типа фтор замещается намного легче других галогенов.
- присоединение нуклеофила даёт так называемый комплекс Мейзенгеймера, в котором атом, несущий уходящую группу и нуклеофил имеет sp3-гибридизацию и исключается из сопряжения. Минус, принесённый нуклеофилом, делокализуется по орто- и пара-положениям кольца, и дальше в мезомерные акцепторы в тех положениях, где они есть. Поскольку схема общая, мы это не показываем, а точнее, как это устроено можно посмотреть ниже в разделе Структура комплексов Джексона-Мейзенгеймера.
- Комплекс Мейзенгеймера быстро теряет уходящую группу, образуя продукт замещения. Замещение в целом необратимо (если X – галоген).
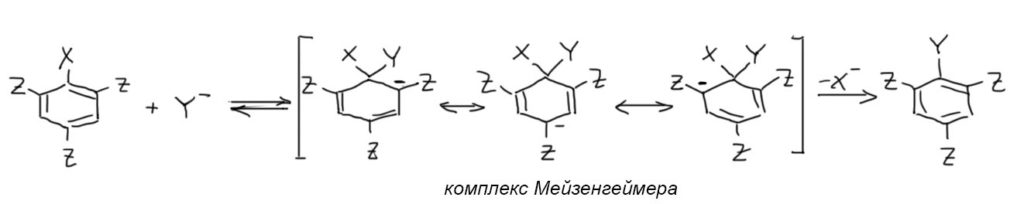
Несколько дополнительных пояснений:
- Чем больше активирующих заместителей (три лучше двух лучше одного) тем лучше. Нитро-группа лучше всех. Одна нитро-группа лучше двух карбонилов, карбоксилов, нитрилов и т.п. Две нитро-группы лучше трёх карбонилов и т.д. Три нитро-группы лучше всех.
- А если нитро-групп четыре? Нет, сказано ясно – только в орто- и пара-положениях, а таких максимум три. Если нитро-групп четыре, то одна из них будет не на месте. В этом случае нитро-группа становится уходящей и замещается точно так же, как галоген, причём очень легко. То же самое произойдёт, если нитро-групп две или три, но расположены они друг относительно друга не в мета-положениях (например, в орто-динитробензоле или 1,2,4-тринитробензоле). Это интересный случай, который мы когда-нибудь отдельно рассмотрим для особо любопытных.
- А если это не бензол, а нафталин? В нафталин ведь можно больше напихать нитро-групп. Да, но каждое кольцо более-менее автономно, и нитро-группы в том кольце, где нет уходящей группы, нас прямо не интересуют. А тогда никаких принципиальных отличий от бензола нет за одним исключением – если и уходящая группа и нитро-группа находятся в β-положениях (2-X-3-нитро), то такая нитро-группа не является активирующей – нарисуйте сами делокализацию в комплексе Мейзенгеймера, чтобы в этом убедиться.
Немного истории (можно пропустить)
Механизм реакции нуклеофильного замещения в активированных субстратах сформулировал Джозеф Баннет в начале 1950-х, но основную вещь в этом механизме – образование стабилизированных аддуктов нуклеофила и субстрата исследовали намного раньше и независимо друг от друга Чарльз Лоринг Джексон в США и Якоб Майзенхаймер в Германии.
Этот Джексон, вероятно, является не много не мало первым химиком-органиком в США. Это в наше время первенство США в науках, в том числе в химии, и в органической химии оспаривается только далёкими от науки и вообще от реальной жизни патриотическими мечтателями. Это первенство обеспечено наилучшей системой образования, востребованностью профессии, прекрасно оборудованными и оснащёнными исследовательскими лабораториями, великолепной логистикой, огромной и жёсткой конкуренцией, привлекательностью для всего остального мира и всем прочим. Доминирование учёных США в химии с каждым годом увеличивается, и ему никак не угрожают ни Великобритания, ни Евросоюз, ни Япония, ни Китай, ни, тем более, безнадёжно оставшие все остальные страны мира вместе взятые. А ведь когда-то всё было не так. В первые сто лет химии в ней доминировала Германия. Очень мощно химией занимались во Франции (этому мы обязаны тем, что странно называем некоторые важные радикалы не по исходному соединению, например, от бензола – фенилом, а от этилена винилом – это французские корни, во втором случае догадайтесь, какой). Много превосходных и даже великих химиков водилось и в Российской империи – посчитайте просто сколько русских именных реакций среди вечной классики – Бородин, Кучеров, Фаворский, Реформатский, Зелинский, Зайцев, Зимин, Кижнер, и т.п. Были великие органики в Швеции, Англии, Шотландии, Италии, Австрии… А вот в США в 19 веке, золотом веке классической органики, с органической химии была почти полная труба – никого и ничего, органической химии даже нигде не учили – единичным желающим приходилось отправляться в Европу. Понятно, что совсем долго так продолжаться не могло. Первым человеком, который получил серьезное образование (в Германии, естественно, у Гофмана, того самого, которого элиминирование, и много чего ещё) и поехал обратно на родину раскочегаривать первый американский очаг органической химии прямо в Гарварде, был этот самый Чарльз Лоринг Джексон. Раскочегарил, надо признать, знатно. Кроме комплексов, которые мы обсуждаем, Джексону принадлежит ещё один трюк, которым мы пользуемся – использование сульфирования для защиты ароматического кольца при жёстком нитровании – так до сих пор получают тринитрофенол, пикриновую кислоту.
В конце 19 века многие химики с удовольствием баловались динитро- и тринитро-производными бензола и его замещенных: галогенбензолов, фенола, анизола и т.п. Соединения были интересные, получались легко, да и перспективы применения таких соединений для любимой забавы человечества – истреблять друг друга, нарисовались очень быстро. Занятным совпадением можно считать то, что комплексы Мейзенгеймера были описаны в тот самый год, когда на арене войны дебютировал тротил, тринитротолуол и сразу завоевал признание военных. Многих необычайно занимали яркие окрашивания, наблюдаемые при действии на эти соединения щелочей, алкоголятов и других основний и нуклеофилов, хотя тогда и слов таких не было. Разные химики пытались разгадать причину образования этих окрасок, но обычно попадали пальцем в небо, так как не было в те времена ещё адекватных представлений о структуре. Первый исследователь, который догадался прямо на рубеже веков, что гидроксид- или метилат-ион просто присоединяются к атому углерода в кольце, причём получается нечто с хиноидной структурой, а это уже могда считалось причиной образования окраски, был как раз Джексон, и он собственно правильно нарисовал и структуру. Всего через год с небольшим, в 1902 Якоб Майзенхаймер (по старой русской традиции передачи немецких имён мы его называем Мейзенгеймером, что в наше время стало довольно забавно – что за геймер такой нарисовался в древности…) сделал просто фундаментальную работу, в которой на десятке примеров рассмотрел образующиеся комплексы. Мейзенгеймер не изучал замещение, он изучал интересные красиво окрашенные соединения, которые образуются из нитропроизводных при действии алкоголятов. Структуру он влял у Джексона, работу которого хорошо и обильно цитирует. Но он пошел дальше, от гипотез к доказательствам. Многие такие соединения совершенно устойчивы и выделяются в виде красных кристаллов, что позволяло применить к ним единственный медод исследования, который тогда существовал – элементный анализ, дающий брутто-формулу. Вот, как он нарисовал комплекс, полученный из тринитроанизола и метилата калия: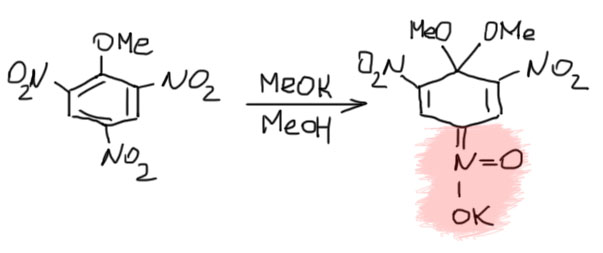 Как видим, очень точно угадал (именно угадал – не было способов установить структуру в те времена, только предположить) он структуру, имея экспериментально установленную брутто-формулу. Да, сейчас нам запрещают рисовать пять связей от азота, но до открытия электронной природы связей и правила октета оставалось еще 15 с лишним лет и вся Первая мировая война. Наиболее интересное и непосредственно связанное с механизмом нуклеофильного замещения наблюдение Мейзенгеймер сделал, когда показал, что из тринитроанизола и этилата калия образуется точно такой же комплекс, как из тринитрофенетола и метилата калия. И что при разложении таких комплексов образуется смесь исходных – в этом месте ое фактически установил не только факт замещения, но и его обратимость в том случае, когда уходящая группа и нуклеофил имеют одинаковую природу.
Как видим, очень точно угадал (именно угадал – не было способов установить структуру в те времена, только предположить) он структуру, имея экспериментально установленную брутто-формулу. Да, сейчас нам запрещают рисовать пять связей от азота, но до открытия электронной природы связей и правила октета оставалось еще 15 с лишним лет и вся Первая мировая война. Наиболее интересное и непосредственно связанное с механизмом нуклеофильного замещения наблюдение Мейзенгеймер сделал, когда показал, что из тринитроанизола и этилата калия образуется точно такой же комплекс, как из тринитрофенетола и метилата калия. И что при разложении таких комплексов образуется смесь исходных – в этом месте ое фактически установил не только факт замещения, но и его обратимость в том случае, когда уходящая группа и нуклеофил имеют одинаковую природу.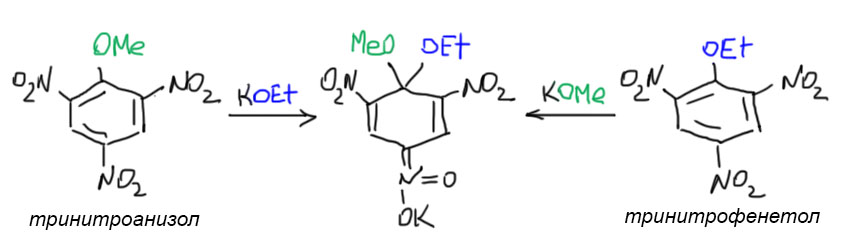
Гипотеза Джексона и Мейзенгеймера об устройстве соединений, образующихся при действии нуклеофила на активированное ароматическое соединение, оказалась верной. И хотя эти исследователи не изучали механизм – тогда не было даже мыслей про механизмы реакций – преложенные структуры можно считать самой первой в органической химии гипотезой об образовании промежуточных соединений (интермедиатов) в органических реакциях. Неудивительно, что когда появился собственно механизм 50 лет спустя, ключевые интермедиаты стали называть комплексами Мейзенгеймера или комплексами Джексона-Мейзенгеймера (второе намного справедливее, но встречается реже). Джексона забывали по очень простой причине – хотя он был первым и приоритете его несомненен и признавался самим Мейзенгеймером, Джексон опубликовал свою работу в никому не ведомом американском журнале, а Мейзенгеймер – в самом главном журнале золотого века органической химии – Анналах Юстуса Либиха, с чтения свежего номера которых начинал свой день всякий уважающий себя химик. То же происходит и наше время – печатаете свои труды чёрт знает где, не думайте, что их кто-нибудь заметит, даже если там будет что-то очень важное.
Строение комплексов Джексона-Мейзенгеймера
Мы, конечно, не можем оставлять структуру комплексов в таком архаическом виде. Прерисуем ее по современным требованиям, заодно прояснив одну важную вещь. Во-первых, уберём катион и ограничимся анионной частью комплекса. Во-вторых, нарисуем делокализацию отрицательного заряда граничными структурами. Формально, отрицательный заряд распределён по трём положениям кольца, как на трёх верхних граничных структурах. Фактически, он уходит в нитро-группы, оказываясь на атоме кислорода, как на трёх нижних граничных структурах. Всего структур шесть, но граничные структуры с зарядом на углеродах кольца вообще не играют никакой роли в делокализации отрицательного заряда. Как мы уже разбирали в правилах рисования граничных структур, наилучшая ситуация – это когда на концах цепи сопряжения находится донор и акцептор – в этом случае никакие промежуточные структуры рисовать нет смысла – их вклад минимален, а можно нарисовать только зультат полного перераспределения заряда с донора на акцептор. Поскольку в случае тринитро-соединений акцептора три, реальную роль в делокализации заряда и будут играть три граничные структуры с минусами на кислородах.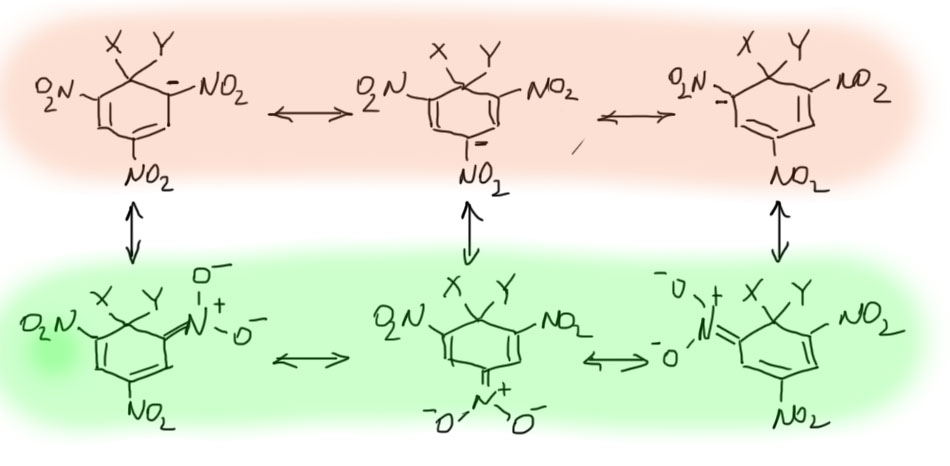
Всё это довольно очевидно, но в литературе, в том числе учебной, очень любят изображать делокализацию в комплексах Мейзенгеймера подковой внутри кольца с минусом посредине, в основном просто для простоты. Как часто бывает, и в этом случае простота оказывается хуже воровства. Время и усилия такое изображение, конечно, экономит, но невольно дезориентирует тех, кто пользуется таким изображением, особенно, если структура не была хорошо осмыслена. 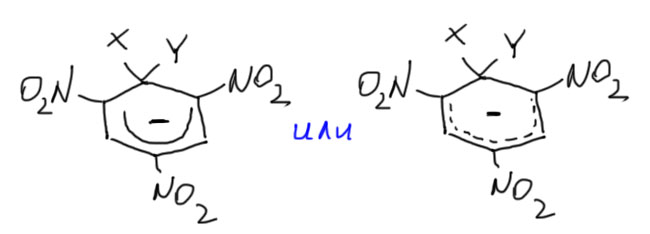 Нет, не в кольце делокализован минус. На углеродах минуса нет вообще, и если посчитать методами квантовой химии заряды на атомах в таких комплесах, на углеродах они останутся положительными, что неудивительно, ведь кольцо несёт аж три сильнейших мезомерных и индуктивных акцептора. Углероды кольца и в комплексе остаются электрофильными, и это хорошо известно, так как такие комплексы отлично присоединяют еще один нуклеофил, а в некоторых работах (Rochester, J.Chem.Soc., 1965, 2404) наблюдали даже аддукты с тремя нуклеофилами, что полностью использует акцепторный потенциал всех трёх нитро-групп. Это было бы невозможно, если бы в кольце действительно был минус, потому что нуклеофил с нуклеофилом не реагирует. Но минуса на углеродах нет, а аддукты, если к ним присмотреться, представляют собой ненасыщенные нитросоединения с нитро-группой на двойной связи. Это типичные электроноакцепторные олефины, котррые присоединяют нуклеофилы, как положено таким непредельным соединениям, и мы скоро увидим многочисленные примеры такой реакционной способности в других классах органических соединений (это называется присоединением по Михаэлю).
Нет, не в кольце делокализован минус. На углеродах минуса нет вообще, и если посчитать методами квантовой химии заряды на атомах в таких комплесах, на углеродах они останутся положительными, что неудивительно, ведь кольцо несёт аж три сильнейших мезомерных и индуктивных акцептора. Углероды кольца и в комплексе остаются электрофильными, и это хорошо известно, так как такие комплексы отлично присоединяют еще один нуклеофил, а в некоторых работах (Rochester, J.Chem.Soc., 1965, 2404) наблюдали даже аддукты с тремя нуклеофилами, что полностью использует акцепторный потенциал всех трёх нитро-групп. Это было бы невозможно, если бы в кольце действительно был минус, потому что нуклеофил с нуклеофилом не реагирует. Но минуса на углеродах нет, а аддукты, если к ним присмотреться, представляют собой ненасыщенные нитросоединения с нитро-группой на двойной связи. Это типичные электроноакцепторные олефины, котррые присоединяют нуклеофилы, как положено таким непредельным соединениям, и мы скоро увидим многочисленные примеры такой реакционной способности в других классах органических соединений (это называется присоединением по Михаэлю). 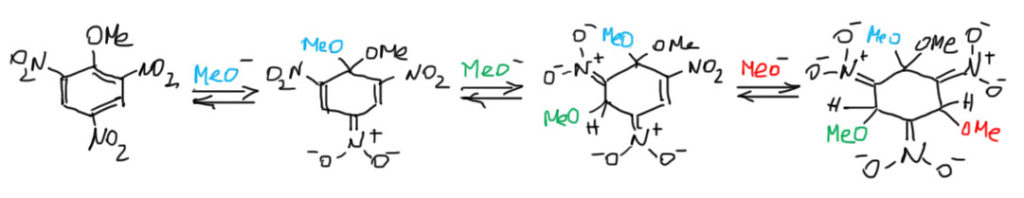
Я предлагаю избегать изображения делокализации “подковой” – не будем сбивать самих себя с толку, будем корректны и аккуратны. Это точно не тот случай, когда подкова приносит счастье. Химия любит уважительное отношение к структуре, потому что в химии нет ничего важнее структуры, и воздаёт сторицей тем, кто не гонится за краткостью ценой сути. Делокализацию же проще и точнее выражать любой из нормальных граничных структур с минусом на кислороде, если лень рисовать все три, нарисуйте любую, это уже будет точнее, чем подкова.
Механизм SNAr
Вернёмся к механизму. Нас интересует замещение, а не просто образование комплексов Джексона-Мейзенгеймера. Обратите внимание на то, что эти комплексы были описаны для активированных проматических соединений, не имеющих очевидной уходящей группы. Поэтому они вполне устойчивы, выделяются из растворов как устойчивые твёрдые соли, их можно потрогать руками (в перчатках), понюхать, положить в пузырёк, и так далее. Но если уходящая группа есть, например, галоген, комплексов наблюдать не получится, а вместо этого получается продукт замещения. Вот именно это обстоятельство, – комплексы не наблюдаются, и вызывало основные проблемы в том, чтобы разобраться, как идёт это замещение. К комплексам Мейзенгеймера все привыкли, их многие получали и исследовали, хорошо знали, что они очень интенсивно окрашены. А если взять не тринитроанизол, а тринитрохлорбензол или динитрохлорбензол, и тот же метилат, замещение идёт, а окрашивания нет. Это замещение пытались описать так же, как замещение у насыщенного атома углерода – согласованный механизм. В те времена многое еще не было понятно, поэтому эта гипотеза не была заведомо плохой. На бумаге всё выглядит вполне прилично – нуклеофил замещает уходящую группу в одностадийном процессе, как обычно в таких процессах рисуем переходное состояние: 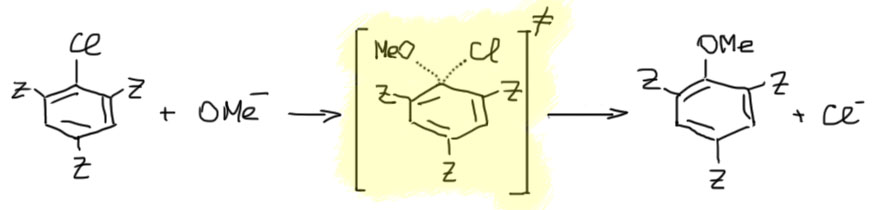
То, что такой механизм принципиально невозможен, и его нужно заменить другим, использовав давно известные комплексы Джексона-Мейзенгеймера, предположил в начале 1950-х молодой американский химик Джо Баннет, буквально за несколько лет создавший огромный раздел органической химии – нуклеофильное ароматическое замещение. Это был человек совершенно бешеной энергии и упорства, что сослужило органической химии не только хорошую, но и плохую услугу: ему мало было классического ароматического нуклеофильного замещения, он придумал еще и радикальное, и нам придётся его тоже изучать, хотя оно того не совсем стоит – но об этом ниже. Умер он всего несколько лет назад, только немного не дожив до своего столетия, в последние лет двадцать занимаясь вместо нуклеофильного замещения всякими шарлатанскими прожектами по уничтожению химического оружия. Один был особенно хорош – предполагалось собрать его всё в одном месте и шарахнуть атомной бомбой (заодно уничтожалась атомная бомба, числом одна).
SN2 механизм невозможен в ароматическом ряду потому что это всегда атака нуклеофила на атом углерода с противоположной стороны от уходящей группы (атака с тыла). Возможность противоположного сценария – атаки с фронта пытались доказать многие, и экспериментально и теоретически, но ничего не вышло. Проблему эту можно просто объяснить так: в реакции участвует гибридная орбиталь связи углерода с уходящей группой, и p-орбиталь нуклеофила. Как и положено в химии, мы должны взять такие орбитали, чтобы в сумме на них было 2 электрона: логично эти 2 электрона отдать нуклеофилу на неподеленную пару как раз на p-орбитали, и тогда нам понадобится разрыхляющая пустая орбиталь связи с уходящей группой. Но σ-связи (связь с уходящей группой – σ-связь) устроены так, что у них связывающая и разрыхляющая орбиталь по форма одинаковы, они различаются только наличием узловой плоскости между атомами у разрыхляющей, и тем, что в связывающую орбиталь больший вес даёт атом уходящей группы, а в разрыхляющую – углерод. Теперь понятно, почему так удобна атака с тыла – там как раз есть задняя доля пустой гибридной орбитали с приличным весом, а вся электронная плотность связи на связывающей орбитали сидит ближе к уходящей группе и не мешает. А теперь попробуем наехать спереди: тут нас ждёт сильное отталкивание орбитали нуклеофила и электронной плотности на связывающей орбитали, обслуживающей связь с уходящей группой. До пустой разрыхляющей и не доберешься! Полный назад!! Атака с фронта отменяется – это не распивочная, а полицейский участок.
А с тыла у ароматики тоже нельзя – нет там тыла, там серединка кольца, надёжно прикрытого сверху и снизу шапками ароматической системы.
И тогда Баннет вспомнил про комплексы Джексона-Мейзенгеймера.
Но ведь при образовании комплекса тоже происходит атака нуклеофила с фронта – а мы ведь ее уже заклеймили! Как, почему, опять обман… Ничего подобного, атака, да не туда, не на ту орбиталь. Нуклеофилу уже нет никакого дела до связи углерода с уходящей группой – его мишенью стала разрыхляющая орбиталь ароматической системы. Нуклеофил выхватывает из частокола p-орбиталей ароматического кольца одну p-орбиталь, выворачивает её, превращая в гибридную орбиталь новой связи, при этом старая гибридная орбиталь связи с уходящей группой отгибается, отталкиваясь в сторону. Всё нормально и вполне реально. При этом, не нужно забывать, на ароматической системе оставалась ещё законная связывающая орбиталь ароматической системы, которая, в том ещё целом кольце, составляла полный комплект с ещё двумя π-орбиталями, несущими в сумме тот самый ароматический секстет, 6 электронов. Теперь мы из каждой орбитали этого набора выдернули одну p-орбиталь, использовав ее для связи с нуклеофилом. Но 6 электронов-то остались, мы ими не пользовались, электроны для новой связи целиком пришли от нуклеофила. Получается на сопряжённой системе из 5 атомов углерода шесть электронов – избыток. Вот тут и помогают акцепторные группы, которые буквально откачивают эту лишнюю плотность из сопряжённой 5-углеродной системы. Так, мы получаем орбитальное описание того же, о чём говорят нам граничные структуры в описании комплекса.
Но на комплексе мы не задерживаемся: у нас же есть уходящая группа, и острое желание восстановить, во что бы то ни стало, порушенную ароматическую систему. Выпихиваем уходящую группу, не пожалев отдать её пару электронов, а опустевшую орбиталь выпрямляем обратно и заполняем тем же лектроном, который успешно пересидел эти нуклеофильно-электрофильные разборки на уютной акцепторной группе у кольца. Всё хорошо, что хорошо кончается, и единственным немного обиженным персонажем остается та самая акцепторная группа, которой электрон дали подержать, да и то ненадолго. Такова учесть всех жадин (жадина – исконно русский перевод иноземного слова акцептор).
Ариновый механизм
Если мы имеем дело с неактивированным производным бензола, в котором есть атом галогена и либо вообще нет заместителей, или есть донорные или слабо-акцепторные заместители, то в обычных условиях такие соединения не вступают в реакцию нуклеофильного замещения, остаются инертными к действию нуклеофилов в полном соответствии с тем, что мы обсуждали в теме алифатическое нуклеофильное замещение – говорили же тогда, что если галоген висит на sp2-гибридном углероде, то с места его не свернешь, гиблое дело.
Свернуть однако все же можно, и даже не одним способом, но всякий раз для этого потребуются ухищрения. Самый полезный метод использует, как это часто называют, грубую силу (brute force). Мы отлично знаем, что можно сделать, когда замещение не идет – тогда может идти элиминирование. Элиминирование в целом более универсально чем замещение, и если взять основание посильнее, то почти всегда есть шанс оторвать сначала протон из соседнего положения, если он там есть, конечно. Но что будет, если это сделать в ряду бензола? Должна получиться тройная связь. Но это невозможно – в циклах размером меньше 8 атомов настоящей тройной связи быть не может! Напомню, что такое настоящая тройная связь. Это связь между атомами в состоянии sp-гибридизации, что означает, что эти атомы и непосредственно связанные с ними лежат на прямой линии, и ничего с этим сделать нельзя. Если по какой-то причине эти 4 атома отклоняются от прямой, то в середине уже не настоящая тройная связь, а какое-то недоразумение и посмешище, готовое от стыда и позора провалиться под землю, – ну или быстренько с чем-нибудь прореагировать, если уж провалиться не получится. Вот поэтому настоящей тройной связи в небольших циклах (до 7-членных) быть не может – вы просто не сможете соединить прямой четырехуглеродный фрагмент в цикл, распялив между 1 и 4 атомами линейного фрагмента оставшиеся вам атомы – это такая геометрия нехитрая, обойти которую нельзя.
Тем не менее, если невозможна настоящая тройная связь, может быть возможна какая-нибудь другая, послабее. Посмотрим. От хлор- или бромбензола действительно элиминировать можно, и молекула, которая при этом получается не является ацетиленом, хотя ее рисуют с тройной связью, и называют бензином. Да, именно так, и всем плевать на страдания носителей русского языка, у которых этим словом обозначается одна из самых священных жидкостей – в других языках такой коллизии нет. Будем поэтому мучиться и вздрагивать – этот бензин на бензоколонке не продают, потому что он очень неустойчив и живет малые доли секунды. Обладая совершенно гигантской реакционной способностью, бензин (или в общем виде арин), всегда найдет во что превратиться, и за это особенно ценим химиками.
Итак, во-первых, бензин – это не ацетилен, не алкин, в нем нет настоящей тройной связи между углеродами в состоянии sp-гибридизации. Чтобы понять, что это достаточно просто подумать, как он получается. У галогенбензола с двух соседних атомов уходит протон (а электронная пара связи остается на месте), и галогенид, уносящий пару с собой. При этом, все это происходит в плоскости кольца, то есть на месте остаются гибридные орбитали углерода, ранее отвечавшие за эти связи. Сравним это с тем, что происходит, если мы элиминируем от галогеналкена – нечто похожее, но на месте бывших связей C-H и C-X появились p-орбитали (так как весь фрагмент распрямился, связи выстроились в линию), перекрывание которых дает полноценную π-связь – образуется настоящая тройная связь. В бензоле уже такое быть не может – потому что связи кольца имеют углы 120º и не собираются от этого отказываться, и в линию вытянуться не могут, а без этого нет sp-гибридизации. Гибридизация углерода остается sp2, и вместо p-орбиталей мы имеем гибридные орбитали под углом, что не дает им эффективно перекрываться и образовывать полноценную связь. Поэтому бензин – это не алкин, хоть именно так его и положено изображать, просто потому что это удобно. Но реально это устроено скорее как нечто среднее между двумя граничными структурами, в которых эти гибридные орбитали попеременно несут пару и дырку (минус и плюс, карбанион и карбокатион). Одна из этих граничных структур просто отражает, как образовался бензин – от соседних атомов ушли две уходящие группы, оставив на месте как раз пару и дырку. Поскольку в бензине два атома углерода одинаковы, законы квантовой науки не дают нам возможности сказать, на каком из углеродов что конкретно находится – после ухода протона и галогенида бензин “забывает”, что и где у него было до ограбления. Это “забывает” мы и изображаем в виде граничных структур. Или, чтобы не мучиться, в виде такого фейкового алкина с тройной связью. Главное, чтобы у нас память оказалась не так коротка, как у бензина, и мы помнили и что случилось, и что это значит. Итак, принимаем, что в бензине есть связь, обслуживаемая перекрыванием гибридных орбиталей в сумме несущих пару электронов (мы уже много раз убеждались, что это важный признак химической связи – именно пара электронов в общем пользовании двух, а иногда и более атомов), но эта связь слаба, что делает такую молекулу короткоживущей и невероятно реакционноспособной.
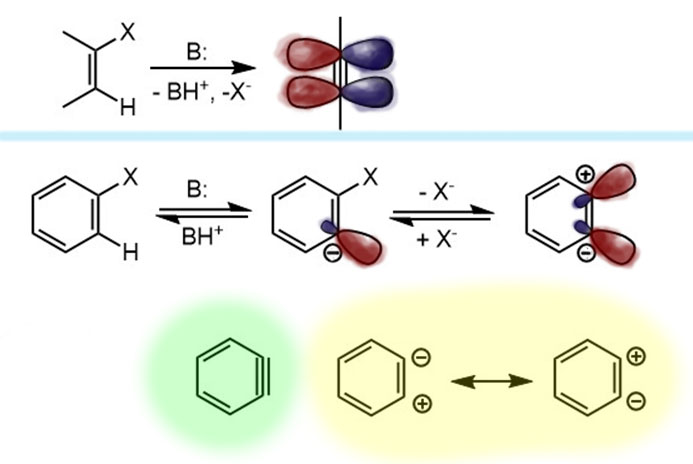
У самого бензина два углерода, образующие “тройную связь” совершенно одинаковы, и это означает, что вклад двух граничных структур совершенно одинаков, и на атомах реально нет зарядов. Но все может быть иначе, если в молекуле бензина есть заместитель рядом с этой “тройной связью”. В этом случае углероды становятся разными, различимыми, и законы квантовой науки более не заставляют нас ничего забывать. А так как у заместителей бывают донорные или акцепторные эффекты, граничные структуры становятся неравноценными. Например, если заместитель акцепторный (речь идет об индуктивном эффекте, так как мезомерный в такой конфигурации не действует), то граничная структура с плюсом рядом с заместителем дестабилизируется и понижает свой вклад за счет второй – это означает, что ближний углерод становится немного карбанионным, а дальний карбокатионным. Эта поляризация “тройной связи” имеет интересные последствия, которые мы обсудим чуть дальше.
Теперь можно нарисовать полный механизм нуклеофильного замещения с участием бензина (арина). Сначала для галогенбензола без других заместителей.
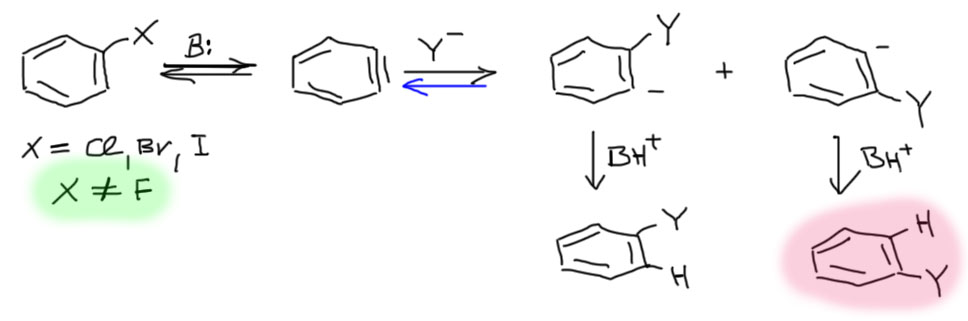
При отщеплении протона от галогенбензола (фтор не годится(примеры таких реакций в литературе найти можно, но они редкие и довольно спорные)) и быстрого ухода галогенида (фторид просто не уходит и поэтому фтор не годится) образуется промежуточный бензин, который немедленно присоединяет к своей “тройной связи” нуклеофил из реакционной смеси. Поскольку углерода “тройной связи” неразличимы, то нуклеофил с равной вероятностью присоединяется либо к одному, либо к другому. При этом образуется карбанион. Стадия присоединения нуклеофила обычно необратима, но в тех случаях, когда она бывает обратима, случаются интересные реакции (например, бешеная пляска галогенов, посмотрите одну из задач внизу и разберитесь сами, как это может быть). Карбанионы эти обладают огромной основностью и немеделенно находят протон, хотя бы в сопряженной кислоте основания, которое отрывало протон от исходного галогенбензола. Из-за равновероятного присоединения получается два продукта, которые можно различить, использовав изотопные метки в исходном галогенбензоле. В одном из продуктов нуклеофил садится туда, где была уходящая группа – это нормальное замещение. В другом – нуклеофил садится не туда, где была уходящая группа. Этот сенсационный результат невозможно объяснить по другому, и именно он стал ключевым доказательством этого механизма, который без этого доказательства так и остался бы безумной гипотезой. Путь, ведущий к этому продукту получил особое название кине-замещения.
Как делают реакции по ариновому механизму
С ариновым механизмом замещения разобрались, теперь посмотрим на то, как это делают. Уже ясно, что нужно сильное основание и нуклеофил. Как мы знаем из алифатического нуклеофильного замещения, нуклеофилы иногда бывают сильными основаниями, и там это было почти проклятием, потому что не давало провести замещение, но здесь это стало благословением, потому что нам ровно это и нужно. Основания требуются довольно сильные: амиды щелочных металлов, простые литийорганические соединения, трет-бутилат в ДМСО. Амиды щелочных металлов чаще всего берут в растворе жидкого аммиака, в основном потому что эти ионные соединения с очень прочной кристаллической решеткой малорастворимы в обычных растворителях. Самый простой вариант – берем два моля основания-нуклеофила на моль галогенбензола. Пара примеров:
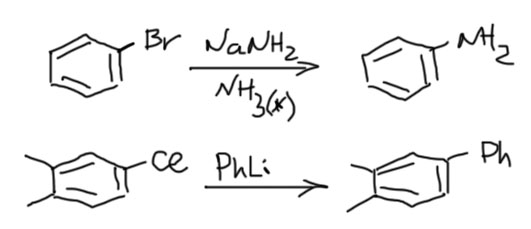
Так можно получать самые разные амины, вначале получив из амина его соль с щелочным металлом (такие соли называют амидами, и нужно быть очень внимательным, чтобы не путать эти амиды с амидами карбоновых кислот).
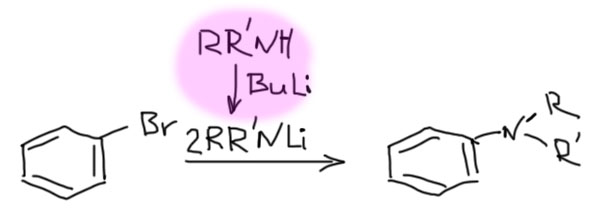
Иногда можно разделить основание и нуклеофил, первый использовать для генерации арина, второй для дела. Таких реакций довольно много, но планировать их трудно, и нам стоит избегать этот вариант, просто потому что у нас не хватит знаний корректно предсказать результат таких реакций. Посто для примера приведу образование сульфида. В этом случае все более-менее просто, потому что основность сульфида недостаточна для депротонирования, а нуклеофильность у него ого-го какая! А у трет-бутилата все наоборот. Из этой реакции мы можем сделать весьма важный вывод: несмотря на очень высокую реакционную способность аринов, они неплохо разбираются в нуклеофилах и выбирают те, что получше. В этом смысле они сильно и в лучшую сторону отличаются от совсем неразборчивых карбокатионов в SN1-замещении.

Все это совершенно замечательно, и кажется, что цены нет такому потрясающему методу. В современном синтезе эту реакцию не очень любят из-за низкой экономичности – один моль основания-нуклеофила теряется впустую (да, это серьезный аргумент). Нам это не очень важно. Но стоит нам ввести заместители в бензольное кольцо, как начинаются проблемы с селективностью, а это уже проблема тяжелая и, мы поклялись ею не пренебрегать. Как мы видели в механизме реакции, арин присоединяет нуклеофил к двум разным углеродам, и получается или нормальный продукт, или продукт кине-замещения, а чаще всего их смесь. Как мы видели в структуре арина на углеродах “тройной связи” может быть больше карбокатионного, или больше карбанионного характера, в зависимости от заместителей. В реальности, если заместитель находится в пара- или мета-положении, его эффект на поляризацию “тройной связи” не очень велик, и почти всегда образуются смеси, и нам точно нет смысла разбираться в том, чего там может быть больше и когда, да и не только нам, а и в большом органическом синтезе вся эта суета очень не нравится – не любят синтетики эту реакцию, грязная она. Вот, например, что получается из мета-изомера в типичной реакции с ариновым механизмом.
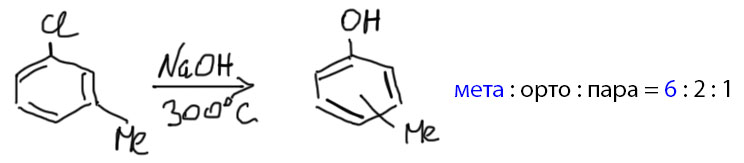
Высокая температура нужна, чтобы заставить щелочь – недостаточно сильное основание – реагировать по этому механизму. А что, температура так сильно влияет на основность??? Да, но только в том случае, когда отщепление протона необратимо, и в дело вступают законы кинетики (помните заклинание: При повышении температуры на десять градусов скорость химической реакции увеличивается в 2-4 раза – повторять натощак каждый день десять раз, выходные и праздники можно пропустить). На равновесия температура влияет, но далеко не так сильно. В случае аринового механизма отщепление протона необратимо, потому что за ним немедленно следует отщепление галогенида и реакция арина. И это делает возможным раскочегарить примитивную щелочь на ариновый механизм. Этим активно пользовались в допотопной промышленности для синтеза фенолов. После потопа изобрели более изящные методы, о которых мы поговорим в своем месте.
А вот пример ариновой реакции с пара-замещенным. А продукт основной – мета, увы, далеко не чистый, и пара-изомера тоже немало.
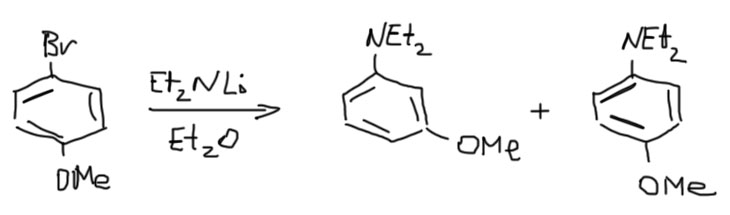
Ну и зачем нам такая химия? К счастью в некоторых частных случаях все получается гораздо чище и определеннее. Обсудим их подробнее на отдельной вкладке: Кине-замещение.
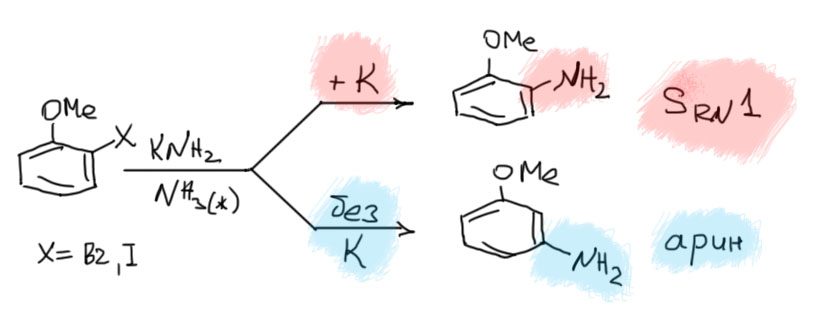
SRN1 - основная информация
В строгой формулировке механизма электрон нужен только для инициирования реакции, дальше она должна происходить как цепной процесс. Простая аналогия с реакциями свободнорадикального замещения, с которыми мы много разбирались в хими алканов, говорит, что радикалы куда-то деваются (происходит случайный обрыв цепи), поэтому инициирование нужно постоянно во время реакции, и инициатор расходуется в значительных количествах, иногда даже эквимольно по реагирующему субстрату.
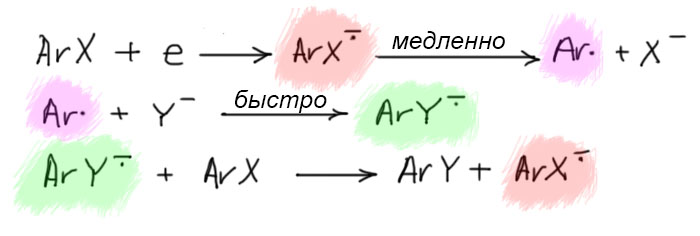
Итак, берем галогенпроизводное бензола (замещенный фенил называют арилом). Добавляем электрон. Образуется анион-радикал. Он быстро распадается на арильный радикал и галогенид-ион. Самая интересная и необычная часть механизма – арильный радикал взаимодействует с нуклеофилом, образуя анион-радикал продукта замещения. Этот анион-радикал быстро передает электрон исходному галогенпроизводному, образуя продукт замещения, и всё начинается сначала – так развивается цепь. 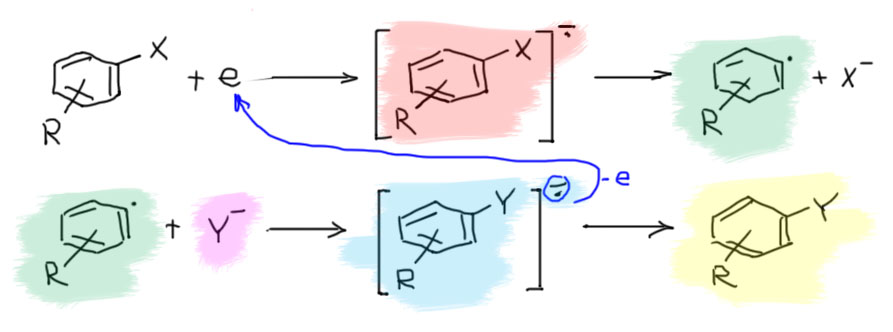
Нарисовать такой механизм относительно легко, сделать реально намного труднее. Во-первых, очень трудно удержать арильный радикал, а это очень активный радикал, от обычных радикальных фокусов, – отрыва атома водорода от растворителя. Поэтому для этой реакции выбор растворителя совершенно критичен, и в большинстве случвев это жидкий аммиак. Это довольно полярный растворитель, что важно, потому что в неполярных растворителях не происходит перенос электрона с образованием заряженных частиц, которые необходимо сольватировать и оттаскивать друг от друга. И от аммиака почти невозможно отодрать атом водорода – связь NH прочнее большинcтва связей CH.
Реакции SRN1 замещения почти всегда проводят в жидком аммиаке
Выбор такого растворителя подсказывает и выбор источника электронов. Как мы уже знаем, в растворе жидкого аммиака легко получить просто сами электроны – раствор электронов. Такие растворы тёмно-синего, почти чёрного цвета образуются при растворении щелочных металлов в жидком аммиаке. Это почти идеальный восстановитель – просто передаёт электроны восстанавливаемому веществу. В этой реакции чаще всего используют раствор калия, а не натрия или лития. Если интересно почему, почитаете более подробное описание этой реакции. Если нет – проще запомнить, что калий и жидкий аммиак – своеобразные опознавательные признаки этой реакции.
Применение реакции SRN1 очень непросто, так как в ней есть множество сложностей, явно выходящих за пределы того, что можно обсуждать на 3 курсе. А попала она к нам по очень простой причине – как своеобразный довесок к ариновому механизму. Собственно именно так её и обнаружил сам Баннет. Подробнее это будет в другом месте для желающих, а здесь просто скажем, что если взять o-галогенанизол или другой похожий субстрат и дать ему прореагировать в жидком аммиаке с амидом калия, то мы получим, как и ожидаем, ариновый механизм и кине-замещение. А если к этой реакционной смеси добавить немного калия, то результат драматически изменится и основным продуктом станет о-анизидин, потому что механизм переключится на SRN1,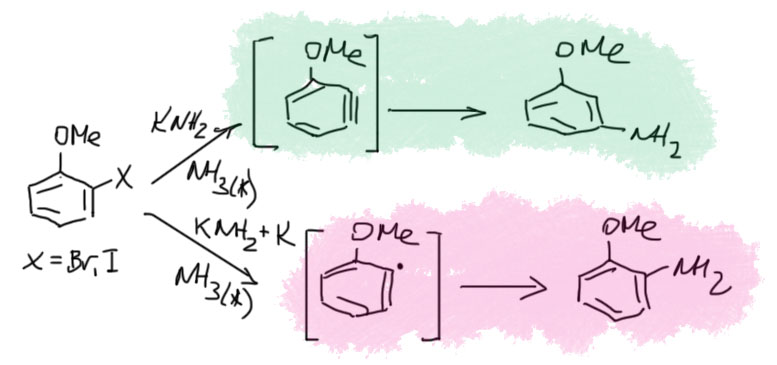
в механизме SRN1 нуклеофил всегда встаёт на место уходящей группы
SRN1 - механизм, которого нет (в том виде, как его рисуют)
Всё, что написано на этой вкладке и дальше строго факультативно, только для любопытных
Разберёмся, как работает механизм SRN1, вернее, как он должен был бы работать, если бы не некоторые труднопреодолимые проблемы.
Возьмём ещё раз обычный бромбензол, потому что пока нам не очень интересны заместители в кольце – в этой реакции они действительно играют очень скромную роль. Как устроить в бромбензоле нуклеофильное замещение с помощью очень сильного основания мы более-менее разобрались. Но есть как минимум ещё один способ.
Чтобы устроить замещение нужно порвать связь C-Br и потом подсунуть в образовавшееся место нуклеофил. Как разорвать связь углерод-галоген хорошо известно – нужно ее восстановить, дать ей электроны. Хорошо и давно известно, что это приводит к дегалогенированию, образованию бензола и бромид-иона. Если отвлечься от восстановителя, можно просто записать полуреакцию: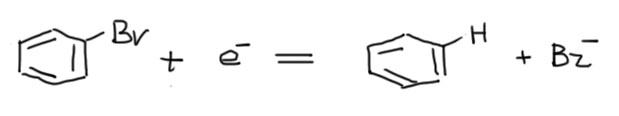
Стоп. Тут что-то не сходится. А водород откуда? И вообще, мы хотели нуклеофильным замещением заняться, а это зачем?
Попробуем разобраться, как вообще идут такие реакции. Мы уже обсуждали и не раз, что происходит, если молекуле дать лишний электрон. Сначала должен получиться анион-радикал – электрон приклеивается к самой низшей свободной орбитали. Ароматические соединения мы уже так восстанавливали в реакции Бёрча, и помним, что электрон клеится к ароматической π-системе, и дальше происходят важные процессы перераспределения электронной плотности, из присутствующего в растворе этанола берется протон, и происходит восстановление. Источник протона мы таким образом нашли – это растворитель или что-то, что к нему добавили. Но – если в ароматической молекуле есть еще и галоген, картина “приклеивания” электрона сильно изменяется. В галогенбензолах низшая свободная орбиталь – это не орбиталь π-системы, а разрыхляющая орбиталь связи C-галоген. Получается, что после добавления электрона на этой связи оказывается три электрона: раньше было два, как положено любой ковалентной связи, а стало три. Это приводит к ослаблению этой связи и её лёгкому разрыву, причём из трёх электронов два берёт себе галоген, а один – углерод (догадайтесь почему). 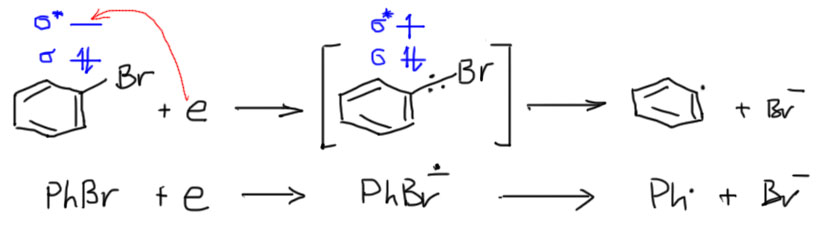
Первое уточнение. В механизме всегда рисуют анион-радикал галогенбензола, который распадается на арильный радикал и галогенид. Распад считался медленной стадией механизма, и именно поэтому его назвали SRN1 по аналогии с SN1 – в обоих механизмах медленной стдией считается распад субстрата, с единственной разницей, что в SN1 это самопроизвольный процесс, а в SRN1 распад вызывается одноэлектронным восстановлением. Уточнение состоит в том, что как показали многочисленные исследования, в первую очередь виднейшего французского электрохимика Жана-Мишеля Савеана (J.M.Saveant), электрон рвёт связь моментально, и никакого анион-радикала поймать невозможно: связь C-галоген в галогенбензоле, как положено, колеблется, сжимается-разжимается – и как только к ней прилипает электрон она тут же разжимается на всю катушку, отфутболив галогенид-ион куда подальше. Поскольку реакция происходит в жидкости, где жуткая теснота, далеко он не улетит, но достаточно, чтобы связь считалась полностью разорванной. Тем не менее, если это учесть, то медленной стадией становится не распад анион-радикала, а присоединение электрона к галогенпроизводному. Но это ничего не меняет в сути реакции, потому что это присоединение немедленно приводит к разрыву связи. И – сильно меняет суть механизма. Проблема в том, что скорость-определяющей стадией становится не мономолекулярный распад анион-радикала (а именно это и обозначает цифра 1 в названии механизма), а вполне себе бимолекулярная реакция переноса электрона. У такой реакции совершенно другие закономерности, в частности, приходится озадачиваться концентрацией восстановителя. Пренебречь этим невозможно, хотя бы потому что в реальной реакции идут два конкурирующих процесса – восстановительное замещение и ариновое замещение, и так и есть на самом деле. Если мы хотим, чтобы шла в основном или даже исключительно реакция восстановительного (радикального) замещения, приходится брать концентрацию восстановителя побольше – до эквимольной или даже избыток, а тогда механизм перестаёт быть цепным, а это, вроде как, было в определении механизма. Иными словами, это другой механизм, точно не SRN1. А что тогда? Вот в том-то и дело, что на этот вопрос невозможно ответить, потому что это зависит от источника электронов – об этом на отдельной вкладке. Пока поедем дальше.
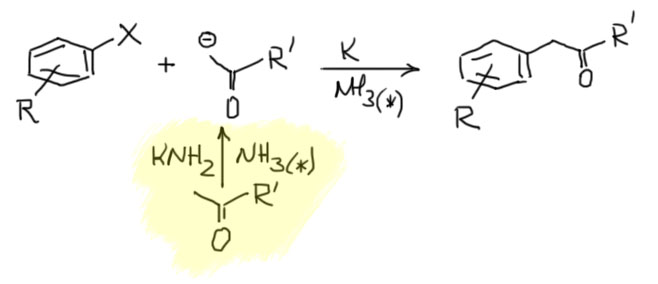
Итак, получили радикал. Обычно радикалы стараются оторвать атом водорода у какой-нибудь зазевавшейся молекулы, но в жидком аммиаке оторвать водород не получается – связь NH прочнее связей CH (на всякий случай напомню, что прочность связей определяется по распаду на радикалы, а не на ионы). Сдвоится с таким же, образовав дифенил? Для этого нужно, чтобы радикалы накопились, и их встреча стала вероятной. Но они не накопятся, потому что, оказывается, радикалы любят реагировать с нуклеофилами, причём со многими нуклеофилами арильные радикалы реагируют с огромной скоростью. Это быстрая стадия. В такой реакции должен получиться анион-радикал ожидаемого продукта замещения. 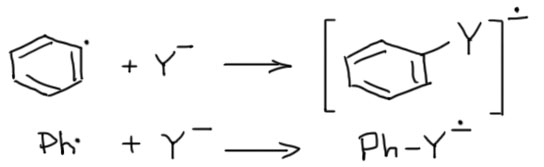
Так. А как это так получается, что один анион-радикал моментально распадается, а другой почему-то тут же получается, да еще и с огромной скоростью. Похоже на очередное жульничество. Ничего подобного. Все нормально. Судьба анион-радикала зависит от того, куда попадает лишний электрон – какова самая нижняя свободная орбиталь. Только у галогенбензолов она сидит на связи C-галоген. У большинства других производных бензола это или π-орбиталь ароматической системы, или вообще она где-то на заместителях. Радикал потому так быстро с нуклеофилом и реагирует, что новая связь образуется вполне нормальная, двухэлектронная, а лишний электрон прямо в процессе образования связи откочёвывает на новое место несения службы. С анион-радикалами на ароматической системе мы вполне уже познакомились в реакции Бёрча. А почему здесь тогда не получается такой же продукт, как в Бёрче? Потому что протонировать некому, нет здесь добавки спирта. А если добавить? А тогда арильный радикал погибнет, оторвав у него водород, и не доживёт до встречи с нукелофилом. Значит, анион-радикал образуется и пока не понятно, что он будет делать дальше. Перерисуем тогда эту стадию так, чтобы это показать, использовав нелюбимую нами гайку для простоты, чтобы показать, что анион-радикал сидит на ароматической системе. 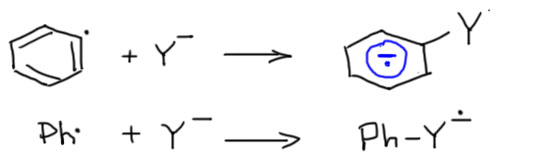
В конце надо придумать, куда девать лишний электрон. Это тяжёлый вопрос. Баннет, написавший механизм, решил, что этот анион-радикал просто передаёт электрон исходному галогенбензолу. И это не фантазии, а достаточно разумная гипотеза, основанная, во-первых, на некотором знании электрохимии, которая способна измерить восстановительные потенциалы и галогенбензола и продукта реакции, и во многих случаях действительно показать, что первые восстанавливаются легче вторых, а значит анион-радикалы вторых действительно могут передать электрон галогенбензолам. Во вторых, в самой первой работе (JACS, 1970, 92, 7464) Ким и Баннет увидели, что реакцию можно осуществить, использовав меньше чем эквивалент калия, то есть на полное восстановление электронов не хватало, и это подтверждало идею, что электроны используются повторно. Они специально взяли орто-галогенанизол, а не бромбензол, чтобы видеть по сохранению положения вошедшего заместителя, что механизм сменился с аринового на новый, восстановительно-радикальный. 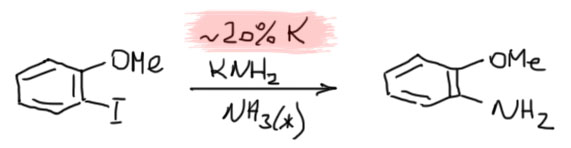
Восстановитель фактически играет роль катализатора. А вся реакция имеет признаки радикального цепного процесса. Запишем последнюю стадию механизма.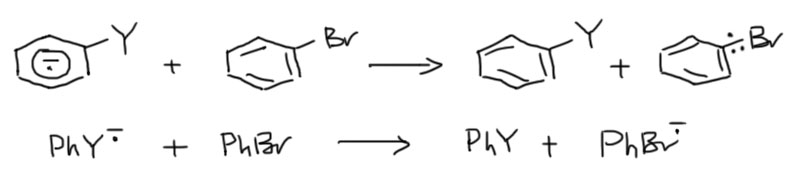
Всё сходится. И даже экспериментальное доказательство есть. Механизм готов, и вроде, за маленьким изменением, соответствует придуманному Баннетом.
Можно нести ложку дёгтя. Пока одну. Потом принесу ещё.
Это экспериментальное доказательство одно-единственное, да и то кривое. Выход правильного изомера в нём всего 67%, кроме него получается много второго изомера, то есть ариновый путь тоже работает наряду с восстановительным. Во всех остальных реакциях, описанных славными основоположниками, калия или эквивалентное количество, или даже избытки, причём совершенно произвольные. Видимо, дело было так – ставят новую реакцию, отрежут ломтик калия на глазок – шасть в колбу, а когда реакция уже прошла, каким-то образом всё же узнавали массу ломтика (возможно, взвешивали оставшийся кусок или даже определяли массу остатка в колбе после испарения аммиака, и по разнице с другими реагентами рассчитывали) и заносили в таблицу. И только один раз у них калия получилось существенно меньше эквивалента – тот самый единственный раз. Как это понимать? Да чёрт его знает! Сейчас бы такую статью завернули из редакции, тем более джакса, с ехидными вопросами и рекомендацией отправить ее в корзину или переделать с начала до конца. А тогда и джакс был пожиже, и стандарты написания статей помягче, особенно, когда их представляли такие авторитеты, как тот же Джо Баннет. Не спрашивать же его, отчего у него никак соотношения реагентов не сходятся, когда такой крутой новый механизм вырисовывается. Но оставим Баннета в покое, тем более что и сам он уже оставил наш мир. И подумаем, так куда же девается лишний электрон. Да чёрт его знает! Куда-то девается, он маленький такой, не уследишь, говорят он вообще на одном месте никогда не сидит, а шастает, как ненормальный. Или даже не шастает, а просто присутствует везде, где только можно. Может он сам как-то из колбы вылез… Если немного серьезнее, то в химии таких фокусов тьма тьмущая. Нарисуем красивый механизм, так, в общих чертах его подтвердим, а там хоть трава не расти. Но дав волю снобскому брюзжанию, заметим одну действительно важную вещь, восславив то, как когда-то делалась наука. Вот ведь честные были люди в то время! Богатыри, не мы. Садились писать статью и писали всё, как было, до мельчайших деталей и явных несуразностей, соотношения реагентов, выходы, даже если маленькие, так и писали, каждый эксперимент по описанию можно повторить. Не сходятся полученные данные с нарисованным механизмом – как есть, так и есть – кому будет дело разбирайтесь, критикуйте, переделывайте, ищите свои объяснения. Экспериментальные данные и наблюдения для настоящего учёного в естественных науках намного важнее интерпретации и теорий, их нельзя ни подгонять, ни искажать, если данные не вписываются в теорию, это проблема теории, а не данных. Но делать так можно только тогда, когда на 100% уверен в том, что делал, и в том, что это описано точно и без искажений. Это высочайший стандарт работы, и в те времена это действительно было так, поэтому те результаты и эксперименты до сих пор составляют основу нашей химии. Попробуйте-ка теперь в современных статьях найти выходы меньше 95%, и понять, что и как было сделано… А плата за это великолепие проста, как гвоздь – 99% современных статей не находят ни продолжения, ни применения, как будто и не было их никогда.
SRN1: откуда берется электрон?
Чтобы сделать реакцию по этому механизму нужно иметь галогенбензол и нуклеофил и ещё источник электронов. И найти подходящий – очень непростая задача.
Самая простая идея – использовать то же, что в реакции Бёрча – раствор сольватированного электрона, получаемый растворением щелочных металлов в жидком аммиаке. Только спирт добавлять ни в коем случае не нужно – в присутствии спирта арильный радикал немедленно схватит атом водорода из спирта и вы получите просто восстановление галогенбензола до бензола, то чего и началась вся эта история.
Росси и Баннет ровно так и поступили. Но – очень странным способом: они взяли не натрий или литий, которые обычно и берут, чтобы сделать тёмно-синий раствор электронов в аммиаке, а – калий. Понять, зачем они это сделали, решительно невозможно; они сами хранят от этом полное молчание, взяли и всё, почему нет, что нам, калия жалко… Это тем более странно, что щелочной металл сначала растворяют в аммиаке, после чего, казалось бы, вообще не должно быть важно, какой был щелочной металл, он всё равно в этот момент уже стал безобидным катионом, до которого никому нет никакого дела. Более того, нет никаких оснований считать, что калий растворяется в аммиаке намного лучше и быстрее натрия или лития: мы уже выяснили, что движущей силой такого растворения является сольватация катиона металла, а катион калия сольватируется слабее меньших по размеру и обладающих существенной льюисовой кислотностью катионов натрия и лития. Но – взяли калий и всё, так это и осталось.
Но это же было 50 лет назад! Неужели с тех пор не появилось десятков исследований со сравнением всех щелочных металлов в этой важнейшей реакции??? Нет, не появилось. И реакция эта не важнейшая. Строго говоря, она даром никому не нужна. Изучал ее всю дорогу один человек, Роберто Росси, аспирант Баннета из Аргентины, открывший эту реакцию в своей диссертации. И ставший практически единственным её исследователем, необычайно дотошным и плодовитым, написавшим многие десятки огромных статей, обзоров и даже книгу. Он до сих пор этим занимается – 50 лет одной реакцией! Воздух в Аргентине хороший, как ясно говорит название столицы этой страны, Буэнос Айрес, еда тоже, живут там долго даже несмотря на чрезвычайно дрянную политику и экономику, так что еще лет двадцать SRN1 замещение будет кому исследовать. А почему такая реакция попала к нам в программу 3 курса? Просто потому что на нашей кафедре в 70-80-е годы прошлого века очень много занимались нуклеофильным ароматическим замещением, приезжали сюда и Баннет, и Росси, и очень увлекательно и темпераментно рассказали про свою реакцию, она очень здесь всем понравилась, и так и попала в программу. Её очень удобно сравнивать с ариновым замещением – там есть кине-замещение, а здесь нет; такие фокусы в химии очень любят. Вот с тех пор мы до сих пор и изучаем реакцию, про которую в остальном мире мало кто знает. Почему – немного в другом месте, когда мы попробуем выяснить ее реальные возможности.
Ровно поэтому и получилась эта страннейшая вещь – когда говорят про замещение SRN1, всегда всплыват именно калий, и многим кажется, что именно в калии есть какая-то большая и очень серьмяжная правда. Ничего подобного. Просто так показалось лучше Роберто Росси, а кроме него никто этим не заинтересовался. И как любая вещь, интересная только одному или очень маленькому количеству людей, она, мягко говоря, со странностями. Нет никакого смысла волноваться по этому поводу – вам никогда ее реально, в колбе использовать не придётся, а на бумаге – какая разница, калий или натрий.
Фотохимическая реакция
Когда мы рассматриваем рекции SRN1-типа, всё время Реакция в присутствии раствора электронов в жидком аммиаке в реальной жизни не является самой
SRN1: механизма нет, а реакция есть
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
SRN1 замещение: достоинства и проблемы
Лучше было бы наоборот – проблем у этой реакции намного больше чем достоинств. Но и достоинства есть.
- Самая главная проблема SRN1 реакций – они довольно сложны экспериментально. Жидкий аммиак – не такая сложная вещь, баллоны ярко-жёлтого цвета с аммиаком не так сложно найти, в баллонах он именно жидкий, и с помощью нехитрого приспособления прямо выливается оттуда в жидком виде. Работать с жидким аммиаком тоже не намного сложнее, чем с обычными растворителями. Нужет просто обратный холодильник с охлаждающей смесью, и тогда аммиак просто потихоньку кипит в колбе и стекает обратно. Реакцию проводят при кипячении с обратным холодильником, только это температура -33ºС. Чтобы работать с металлическим калием, амиак прямо из баллона не подойдёт, его придётся перегнать, пропустив через осушающие колонки с чем-нибудь страшным типа натрия на стекловолокне. А тем, кто решает использовать фотохимический вариант, придётся искать очень непростое оборудование, которое позволит облучать ультрафиолетом содержимое сосуда с жидким аммиаком. Ничего страшного во всём этом нет, и те, кто много работают с жидким аммиаком, считают всё это пустяками, и оборудование найти можно. Но для тех, кто работает в синтезе и применяет методы по мере надобности, чтобы достичь желаемой цели, всё это покажется нежелательной морокой. Если бы альтернативы методам SRN1 не было, всё это не имело бы значения. Но альтернатива есть. Самые плохие новости для SRN1 реакций – они не являются уникальным способом синтеза – то, что можно получить с их помощью, можно получить и другими методами. Особенно жестокую конкуренцию SRN1 составляют современные реакции с использованием катализа комплексами переходных металлов.
- Реакции SRN1 далеко не так селективны, как их иногда изображают. Очевидная проблема – восстановление галогенпроизводного – замена галогена на водород. Хотя мы сказали, что арильный радикал не может оторвать атом водорода от аммиака, в присутствии сильного восстановителя радикалы довольно легко восстанавливаются до карбанионов – мы активно это используем и в восстановлении по Бёрчу и в гидрировании ацетиленов – а арильный карбанион является очень сильным основанием, и обратимо депротонирует аммиак – pK у ароматических протонов и протонов аммиака очень близки.
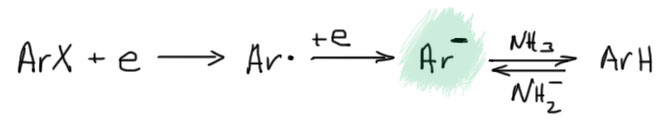 Атом водорода иногда можно оторвать и у нуклеофила, если он посложнее. Радикалы вступают и в другие бесполезные реакции. Любимые нами реакции с амидом калия в присутствии калия почти всегда идут как конкурирующие реакции через арин и арильный радикал, и некоторая доля кине-замещения почти всегда присутствует.
Атом водорода иногда можно оторвать и у нуклеофила, если он посложнее. Радикалы вступают и в другие бесполезные реакции. Любимые нами реакции с амидом калия в присутствии калия почти всегда идут как конкурирующие реакции через арин и арильный радикал, и некоторая доля кине-замещения почти всегда присутствует. - Далеко не все нуклеофилы годятся для реакций SRN1-типа. Не годится гидроксид – гидроксиды щелочных металлов просто нерастворимы в аммиаке, а даже если бы и были, то образующийся фенолят стал бы конкурирующим нуклеофилом. Не годятся почти любые алкоксилаты (RO–) кроме фенолята (PhO–). Плохо работает такой важный нуклеофил как цианид-ион. Плохо работают азотные нуклеофилы: и даже про наш любимый амид калия мы теперь знаем, что это не так гладко. Сопряжённые основания других аминов (алкиламиды, диалкиламиды калия или других щелочных металлов) тоже не годятся по совершенно той же причине, что и алкоксиды – арильный радикал легко отрывает у них атом водорода с образованием анион-радикалов иминов. Анилины, впрочем, годятся, точно так же как феноляты, но реальные выходы в этом случае невелики и реакция не привлекла внимания синтетиков.
- Мало заместителей в арил галогениде совместимы с условиями восстановительной радикальной реакции. Не годятся, например, мезомерные акцепторы типа карбонила или карбоксила – такие заместители подменяют низшую свободную орбиталь своей, и лишний электрон вместо связи C-галоген клеится к таким группам, что приводит совсем к другим реакциям, в которых галоген остаётся, где был. Также не годится всякая ненасыщенность – она в условиях свободнорадикальной реакции норовит заполимеризоваться. Остаётся не так много – алкилы, алкокси, третичные амины, фтор. Это не тот набор заместителей, которые мы ждём от хорошей и гибкой реакции замещения. Впрочем, у аринового механизма приблизительно такие же ограничения.
- замещаемые галогены фактически ограничены бромом и иодом, причём безусловно лидирует последний, но это и самый дорогой и неудобный (прежде всего огромной атомной массой) из галогенов. И хлор и даже фтор проявляют некоторую реакционную способность в таких реакциях, но замещение идёт очень медленно (особенно с фтором), что открывает возможности для успешной конкуренции со стороны всяких побочных реакций.
Но всё не так плохо. Надо сказать, что реакции этого типа всё же довольно интересны и небесполезны. У них есть несколько важных преимуществ.
- для реакции годятся много интересных нуклеофилов, прежде всего делокализованных карбанионов типа енолятов – подробнее на страничке про методы. Очень интересно применение таких методов для нуклеофилов производных серы, фосфора, элементов группы фосфора, даже некоторых металлов, но всё это далеко за пределами нашего курса.
- в качестве субстратов годятся не только галогенпроизводные, но и некоторые другие соединения, в том числе весьма экзотические. Это одна из немногих реакций, в которых допустимы даже кислородные уходящие группы типа феноксидов.
- очень ценное качество, даже ценнейшее – практически полная нечувствительность к стерике. Арильные радикалы так легко реагируют с нуклеофилами, что этому не могут помешать даже заместители рядом с реакционным центром.
- использование жидкого аммиака как растворителя безусловно отталкивает, но если эти трудности преодолеть, то этот растворитель может легко стать любимым, даже любимейшим – он весьма дешев, а главное – очень легко удаляется после реакции – просто перестают подливать охлаждающую жидкость в обратный холодильник и он аккуратно и медленно сам выкипит без остатка – была бы хорошая тяга. Сами же реакции в жидком аммиаке очень эффектны. Справившись с этой химией, любой экспериментатор становится выше ростом, а взгляд его/её наполняется шестым и седьмым чувствами советского человека – чувством законной гордости и глубокого морального удовлетворения. Экспериментатор начинает понимать, что теперь сам чёрт ему/ей не брат, и нет таких реакций, которые бы ей/ему не покорились.