Нуклеофильное ароматическое замещение
Когда мы обсуждали нуклеофильное замещение в алифатическом ряду, механизмы SN2 и SN1, торжественно поклялись строго следить, чтобы углерод, на котором это замещение происходит, имел sp3-гибридизацию. Теперь настала пора разобраться, что это было – пустой запрет или что-то содержательное. Смысл того запрета был довольно прост: если мы разобрались в том, как происходит алифатическое нуклеофильное замещение, и как на практике проводятся те реакции, эти знания не сильно помогли бы нам в ароматическом ряду. Бромбензол ни при каких обстоятельствах не будет реагировать с нуклеофилами в условиях, подходящих любому алифатическому бромпроизводному, ну хотя бы бромциклогексану, чтобы соблюсти некоторое внешнее сходство. Не будет бромбензол реагировать ни с, например, цианидом калия в ДМСО, ни с любым сильным нуклеофилом из 3-4 периодов, например, тиолятом, очень хорошим нуклеофилом, которому нет никаких проблем реагировать по SN2 даже не с самыми лучшими субстратами, такими, как вторичный алкилгалогенид. А бромбензол ни в этих условиях, ни в каких других (речь идёт о варьировании условий ркакции – растворителя, температуры, и т.п., но не добавления каких-то других реагентов) реагировать не будет. Вообще не будет. 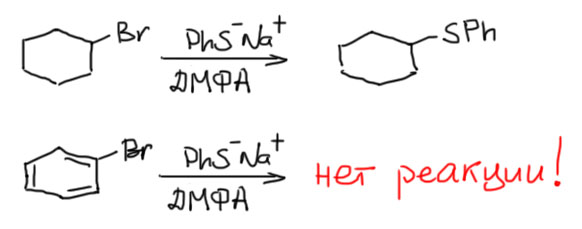 С другой стороны, тот же бромбензол вполне реагирует с теми нуклеофилами, за которым мы закрепили ярлык сильных оснований – с амидом натрия и литийорганическими соединениями, например. И результатом таких реакций, если судить по продуктами реакции будет именно продукты замещения, а не элиминирования – помните ведь, как страшно пугали вас элиминированием и призывали строго следить за основностью. Упустите основность, – говорили – не видать вам замещения, будет только элиминирование. А в ароматическом ряду все не так.
С другой стороны, тот же бромбензол вполне реагирует с теми нуклеофилами, за которым мы закрепили ярлык сильных оснований – с амидом натрия и литийорганическими соединениями, например. И результатом таких реакций, если судить по продуктами реакции будет именно продукты замещения, а не элиминирования – помните ведь, как страшно пугали вас элиминированием и призывали строго следить за основностью. Упустите основность, – говорили – не видать вам замещения, будет только элиминирование. А в ароматическом ряду все не так. 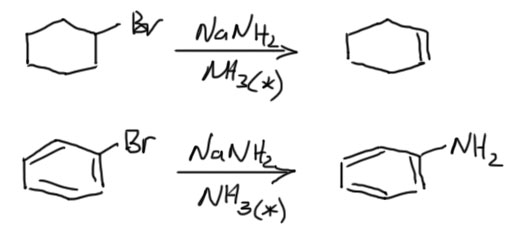 Реакции или не идут, или идут, но очень сильно по другому, как будто это другие реакции. Это типичный признак того, что в разных рядах действуют разные механизмы. Попробуем разобраться.
Реакции или не идут, или идут, но очень сильно по другому, как будто это другие реакции. Это типичный признак того, что в разных рядах действуют разные механизмы. Попробуем разобраться.
Что здесь важно с самого начала.
- Механизмов больше, чем там. Даже в нашем сильно облегчённом курсе мы увидим не меньше трёх, а возможно и больше. И мы реально будем ими пользоваться. Один из основоположников этой химии, Джозеф Баннет, однажды попробовал посчитать механизмы ароматического нуклеофильного замещения и получил то ли 17, то ли 19.
- В химии ароматического нуклеофильного замещения нет одного основного механизма, пригодного для большей части субстратов. Каждый механизм играет сво. специфическую роль для некоторого, достаточно узкого множества, и совершенно бесполезен для всего остального.
- Заместители в ароматическом кольце играют очень важную роль. Именно они часто определяют механизм реакции.
- Особенно богатой химией нуклеофильного ароматического замещения обладают вовсе не производные бензола, а многочисленные гетероциклические соединения, у которых это один из основных видов реакционной способности – когда доберёмся до химии гетероциклов вновь вспомним про нуклеофильное ароматическое замещение.
- Уходящие группы не так важны, и при вдумчивом подходе в ароматическом замещении можно заместить то, что никогда или крайне редко работает в алифатическом, например, водород или фтор. Но в каждом случае придётся точно понять, как идёт реакция, и что ей нужно. С другой стороны, наша любимая уходящая группа из той химии, тозилат, в ароматической химии практически совершенно бесполезна. Мы еще вернёмся к этой курьёзной проблеме в химии фенолов во втором семестре.
- Нуклеофилы остаются нуклеофилами, и всё то, что мы про них узнали в замещении на насыщенном атоме углерода, имеет значение и здесь, хотя акценты немного смещены и сильно зависят от конкретного механизма.
- Конкуренция нуклеофильности и основности в ароматическом замещении проявляется довольно необычным образом, и скорее является сотрудничеством, чем соперничеством.
Ароматическое нитрование – очень мощная и удобная реакция, с помощью которой ещё в 19 века наполучали много нитро-соединений, в том числе динитро- и тринитропроизводные замещённых бензолов. И быстро обнаружили, что такие соединения имеют необычные свойства, например, они растворяются в растоврах щелочей и алкоголятов, и очень легко обменивают галоген на другие полезные группы. Фактически, такие соединения ведут себя подобно насыщенным галогенпроизводным – продукты замещения получаются просто при смешивании галогенпроизводного и нуклеофила в растворе. Реакции идут настолько легко и чисто, что часто даже не требуют никаких специальных растворителей, нагревания и прочей подобной мороки. Одно из самых легкодоступных соединений этого типа, 2,4-динитрохлорбензол, например, очень быстро реагирует почти с любыми нуклеофилами. 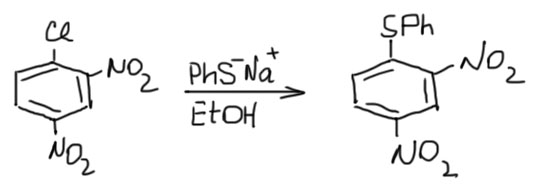
Замещение в таких активированных ароматических соединениях и является основной реакцией ароматического нуклеофильного замещения. Совершенно неудивительно поэтому, что и механизм этой реакции ароматического нуклеофильного замещения обозначается просто – SNAr – то есть буквально “замещение нуклеофильное ароматическое”. Содержательно, не правда ли? Вот с него и начнём.
Нуклеофильное замещение в активированных субстратах: основы
Прямое нуклеофильное ароматическое замещение без дополнительных ухищрений – то есть просто взяв галогенпроизводное и нуклеофил, возможно подобрав подходящий растворитель и использовав обычные способы стимулирования реакций (нагревание, хорошее перемешивание и т.п.) – идёт только в случае, если в кольце есть акцепторные заместители, почти всегда -М-типа: нитро-группа, карбонильная группа и все ее разновидности, нитрил (циано), сульфоновая группа и т.п., одна или две или три, и обязательно в орто- и пара-положениях относительно уходящей группы.
Такие субстраты принято называть активированными, а нуклеофильное замещение в таких субстратах часто называют активированным нуклеофильным замещением. Мы так и будем это называть время от времени.
Итак, если у нас есть такой субстрат, и какой-нибудь нуклеофил, то с хорошей вероятностью они будут реагировать просто при смешивании в растворе и, возможно, при нагревании, но без дополнительных реагентов, катализаторов, и т.п. Это не значит, что не бывает таких случаев, когда добавление чего-нибудь такого не поможет реакции, это лишь значит, что в большинстве случаев это не нужно. Повторим еще раз – активирующее действие оказывают только акцепторные заместители в орто- и пара-положениях к уходящей группе!
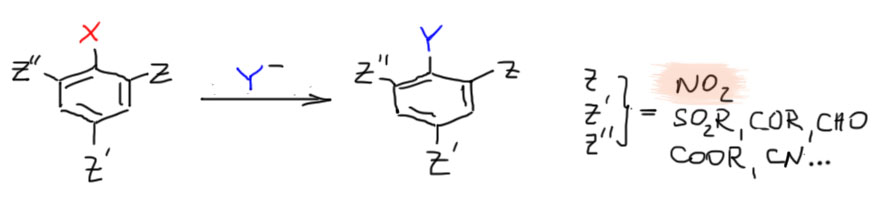
Хотя активирующими заместителями могут быть любые -M-группы, в активированном нуклеофильном замещении особую, потрясающую и ни с чем не сравнимую роль играет нитро-группа. Можно даже сказать, что в этом замещении есть нитро-группа, и есть всё остальное. Речь идёт об обычных группах, с которыми мы имеем дело в нашем курсе. Есть несколько более экзотических групп, сравнимых с нитро-группой по активирующему влиянию и даже превосходящих ее, но это необычные вещи, с которыми мы не столкнёмся. Для нас в активированном нуклеофильном замещении выдающаяся роль нитро-группы безальтернативна.
Очень важно следить, чтобы уходящая группа находилась в активированном положении. В других местах никакого замещения не будет, например: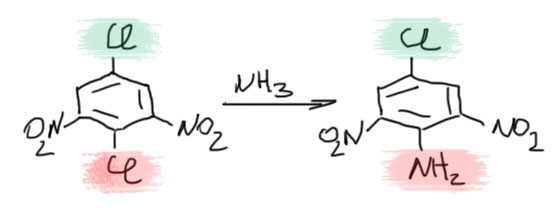
Активированные субстраты делятся на:
- те, у которых три нитро-группы в положениях 2,4,6 (такой остаток называется пикрилом), те у которых две нитро-группы в положениях 2 и 4. Эти соединения проявляют выдающуюся реакционную способность и реагируют практически с любыми нуклеофилами, даже с очень слабыми, и в очень мягких условиях. Косвенным свидетельством такого поведения является то, что если раствор любого такого соединения попадёт на руки, они желтеют и надолго, потому что в белках много нуклеофильных групп, и на них это тут же и сядет. Работайте в перчатках!
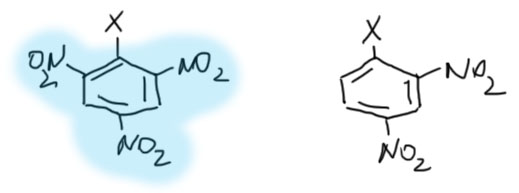
- все остальные – то есть те у которых одна нитро-группа, или до трёх групп другого вида. Такие субстраты реагируют с нуклеофилами намного медленнее, всегда нужно предпринять усилия – взять нуклеофил посильнее, использовать обычные способы повышения нуклеофильности, о которых мы говорили в SN2-замещении: спецрастворители типа ДМСО, ДМФА и т.п., межфазный перенос и т.п. Реакции часто проводят при нагревании, иногда очень сильном и продолжительном. Такие условия плохо сказываются на сохранности заместитей типа карбонилов, поэтому такие субстраты имеют гораздо более ограниченное применение в сравнении с динитро- и тринитро.
В качестве уходящих групп ограничимся галогенами. Активированное ароматическое нуклеофильное замещение имеет ярчайшую особенность, отличающую его от всех остальных видов замещения, как алифатического, так и ароматического – ряд галогенов выглядит совершенно необычно и вызывающе. Наилучшим галогеном является фтор, причём с огромным отрывом от всех остальных – часто разница в скоростях с идущим на втором месте хлором до 1000, а иногда и до 10000 раз! Это колоссальное преимущество, настолько большое, что остальные галогены по сравнению с фтором можно считать “мёртвыми” – нет никаких шансов, что они заместятся, если в молекуле есть активированный фтор. Это не так заметно для сильно-активированных динитро- и тринитро-производных, которые и так реагируют “со свистом” с любой уходящей группой, но для менее активированных субстратов типа моно-нитро-производных, фтор даёт возможность провести реакции замещения в более-менее нормальных условиях, в то время как остальные галогены замещаются с большим трудом и требуют очень серьезных усилий. И в тех условиях, когда в соединении есть разные галогены в активированных положениях, естественно, при такой разнице в реакционной способности замещается только фтор, например: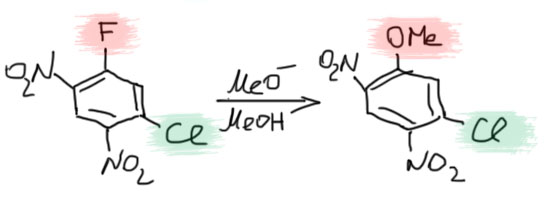
Остальные галогены в активированном положении тоже замещаются, причём способность к уходу хлора и брома приблизительно одинакова, а вот иод уходит хуже всех, часто на порядок-другой медленнее хлора или брома, а уж рядом со фтором иод совершенно неповоротлив. Как это непохоже на порядок уходящих групп в алифатическом нуклеофильном замещении, где всё наоборот – иод лучше всех, а фтор можно своротить только совместными усилиями всех нуклеофилов во Вселенной. Почему так происходит посмотрим ниже, разобрав механизм.
Механизм SNAr
Коротко
Механизм нуклеофильного ароматического замещения в активированных субстратах принято обозначать как SNAr, то есть просто как ароматическое нуклеофильное замещение. Несмотря на то, что применение этого механизма и реакций, подчиняющихся этому механизму ограничено только ароматическими соединениями, содержащими от одной до трёх акцепторных групп в орто- и пара-положениях относительно уходящей группы, именно этот механизм считается наиболее общим и хорошо исследованным. Фактически мы говорим, что раз нуклеофильное замещение без особых ухищрений и использования дополнительных, прямо не связанных с собственно реакцией замещения стадий, идёт только для таких субстратов, то нет смысла переживать по поводу того, что мезанизм и реакции неприменимы для более простых ароматических производных типа хорбензола.
Итак, у нас есть производное бензола с уходящей группой, обычно галогеном, и от одного до трёх заместителей, обладающих -M-эффектом в орто- и пара-положениях по отношению к уходящей группе. Сразу скажем, что заместителей таких много, но в подавляющем большинстве реальных реакций этого типа хотя бы один из них – нитро-группа. И есть нуклеофил. Нуклеофилом в этой реакции мы считаем то же самое, что мы уже использовали в SN2-замещении. По причинам ограничений, связанных как раз с природой активирующих заместителей, в этой реакции почти никогда не используют простые карбанионы (литий- и магнийорганические соединения, ацетилениды и пр.). Всё остальное годится.
Реакция нуклеофила с активированным ароматическим соединением происходит так:
- сначала нуклеофил присоединяется к тому углероду, на котором уходящая группа. Это медленная стадия, определяющая скорость всего процесса замещения. Эта стадия обратима, но когда уходящая группа галоген, про это можно не думать и считать стадию необратимой.
- скорость присоединения или, иными словами, реакционная способность субстрата зависит от числа и качества активирующих групп, а при одинаковом количестве и качестве заместителей – от электрофильности углерода, на котором происходит замещение. Электрофильность тем больше, чем больше электроотрицательность галогена. Поэтому скорость реакции наибольшая для фторпроизводных. Ряд уходящих групп выглядит очень необычно и не имеет прецедентов в дргих реакциях замещения: F >> Cl или Br > I. В реакциях этого типа фтор замещается намного легче других галогенов.
- присоединение нуклеофила даёт так называемый комплекс Мейзенгеймера, в котором атом, несущий уходящую группу и нуклеофил имеет sp3-гибридизацию и исключается из сопряжения. Минус, принесённый нуклеофилом, делокализуется по орто- и пара-положениям кольца, и дальше в мезомерные акцепторы в тех положениях, где они есть. Поскольку схема общая, мы это не показываем, а точнее, как это устроено можно посмотреть ниже в разделе Структура комплексов Джексона-Мейзенгеймера.
- Комплекс Мейзенгеймера быстро теряет уходящую группу, образуя продукт замещения. Замещение в целом необратимо (если X – галоген).
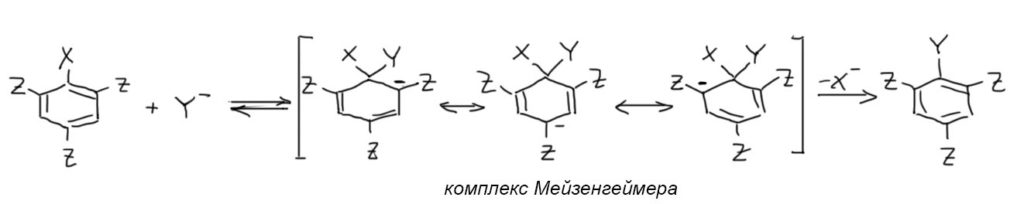
Несколько дополнительных пояснений:
- Чем больше активирующих заместителей (три лучше двух лучше одного) тем лучше. Нитро-группа лучше всех. Одна нитро-группа лучше двух карбонилов, карбоксилов, нитрилов и т.п. Две нитро-группы лучше трёх карбонилов и т.д. Три нитро-группы лучше всех.
- А если нитро-групп четыре? Нет, сказано ясно – только в орто- и пара-положениях, а таких максимум три. Если нитро-групп четыре, то одна из них будет не на месте. В этом случае нитро-группа становится уходящей и замещается точно так же, как галоген, причём очень легко. То же самое произойдёт, если нитро-групп две или три, но расположены они друг относительно друга не в мета-положениях (например, в орто-динитробензоле или 1,2,4-тринитробензоле). Это интересный случай, который мы когда-нибудь отдельно рассмотрим для особо любопытных.
- А если это не бензол, а нафталин? В нафталин ведь можно больше напихать нитро-групп. Да, но каждое кольцо более-менее автономно, и нитро-группы в том кольце, где нет уходящей группы, нас прямо не интересуют. А тогда никаких принципиальных отличий от бензола нет за одним исключением – если и уходящая группа и нитро-группа находятся в β-положениях (2-X-3-нитро), то такая нитро-группа не является активирующей – нарисуйте сами делокализацию в комплексе Мейзенгеймера, чтобы в этом убедиться.
Немного истории (можно пропустить)
Механизм реакции нуклеофильного замещения в активированных субстратах сформулировал Джозеф Баннет в начале 1950-х, но основную вещь в этом механизме – образование стабилизированных аддуктов нуклеофила и субстрата исследовали намного раньше и независимо друг от друга Чарльз Лоринг Джексон в США и Якоб Майзенхаймер в Германии.
Этот Джексон, вероятно, является не много не мало первым химиком-органиком в США. Это в наше время первенство США в науках, в том числе в химии, и в органической химии оспаривается только далёкими от науки и вообще от реальной жизни патриотическими мечтателями. Это первенство обеспечено наилучшей системой образования, востребованностью профессии, прекрасно оборудованными и оснащёнными исследовательскими лабораториями, великолепной логистикой, огромной и жёсткой конкуренцией, привлекательностью для всего остального мира и всем прочим. Доминирование учёных США в химии с каждым годом увеличивается, и ему никак не угрожают ни Великобритания, ни Евросоюз, ни Япония, ни Китай, ни, тем более, безнадёжно оставшие все остальные страны мира вместе взятые. А ведь когда-то всё было не так. В первые сто лет химии в ней доминировала Германия. Очень мощно химией занимались во Франции (этому мы обязаны тем, что странно называем некоторые важные радикалы не по исходному соединению, например, от бензола – фенилом, а от этилена винилом – это французские корни, во втором случае догадайтесь, какой). Много превосходных и даже великих химиков водилось и в Российской империи – посчитайте просто сколько русских именных реакций среди вечной классики – Бородин, Кучеров, Фаворский, Реформатский, Зелинский, Зайцев, Зимин, Кижнер, и т.п. Были великие органики в Швеции, Англии, Шотландии, Италии, Австрии… А вот в США в 19 веке, золотом веке классической органики, с органической химии была почти полная труба – никого и ничего, органической химии даже нигде не учили – единичным желающим приходилось отправляться в Европу. Понятно, что совсем долго так продолжаться не могло. Первым человеком, который получил серьезное образование (в Германии, естественно, у Гофмана, того самого, которого элиминирование, и много чего ещё) и поехал обратно на родину раскочегаривать первый американский очаг органической химии прямо в Гарварде, был этот самый Чарльз Лоринг Джексон. Раскочегарил, надо признать, знатно. Кроме комплексов, которые мы обсуждаем, Джексону принадлежит ещё один трюк, которым мы пользуемся – использование сульфирования для защиты ароматического кольца при жёстком нитровании – так до сих пор получают тринитрофенол, пикриновую кислоту.
В конце 19 века многие химики с удовольствием баловались динитро- и тринитро-производными бензола и его замещенных: галогенбензолов, фенола, анизола и т.п. Соединения были интересные, получались легко, да и перспективы применения таких соединений для любимой забавы человечества – истреблять друг друга, нарисовались очень быстро. Занятным совпадением можно считать то, что комплексы Мейзенгеймера были описаны в тот самый год, когда на арене войны дебютировал тротил, тринитротолуол и сразу завоевал признание военных. Многих необычайно занимали яркие окрашивания, наблюдаемые при действии на эти соединения щелочей, алкоголятов и других основний и нуклеофилов, хотя тогда и слов таких не было. Разные химики пытались разгадать причину образования этих окрасок, но обычно попадали пальцем в небо, так как не было в те времена ещё адекватных представлений о структуре. Первый исследователь, который догадался прямо на рубеже веков, что гидроксид- или метилат-ион просто присоединяются к атому углерода в кольце, причём получается нечто с хиноидной структурой, а это уже могда считалось причиной образования окраски, был как раз Джексон, и он собственно правильно нарисовал и структуру. Всего через год с небольшим, в 1902 Якоб Майзенхаймер (по старой русской традиции передачи немецких имён мы его называем Мейзенгеймером, что в наше время стало довольно забавно – что за геймер такой нарисовался в древности…) сделал просто фундаментальную работу, в которой на десятке примеров рассмотрел образующиеся комплексы. Мейзенгеймер не изучал замещение, он изучал интересные красиво окрашенные соединения, которые образуются из нитропроизводных при действии алкоголятов. Структуру он влял у Джексона, работу которого хорошо и обильно цитирует. Но он пошел дальше, от гипотез к доказательствам. Многие такие соединения совершенно устойчивы и выделяются в виде красных кристаллов, что позволяло применить к ним единственный медод исследования, который тогда существовал – элементный анализ, дающий брутто-формулу. Вот, как он нарисовал комплекс, полученный из тринитроанизола и метилата калия: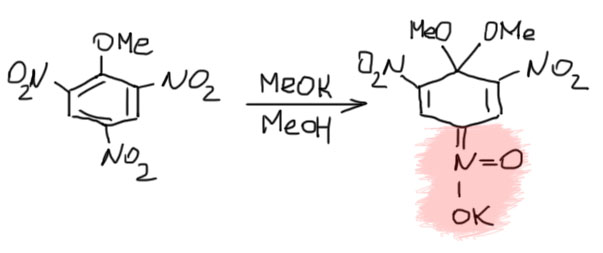 Как видим, очень точно угадал (именно угадал – не было способов установить структуру в те времена, только предположить) он структуру, имея экспериментально установленную брутто-формулу. Да, сейчас нам запрещают рисовать пять связей от азота, но до открытия электронной природы связей и правила октета оставалось еще 15 с лишним лет и вся Первая мировая война. Наиболее интересное и непосредственно связанное с механизмом нуклеофильного замещения наблюдение Мейзенгеймер сделал, когда показал, что из тринитроанизола и этилата калия образуется точно такой же комплекс, как из тринитрофенетола и метилата калия. И что при разложении таких комплексов образуется смесь исходных – в этом месте ое фактически установил не только факт замещения, но и его обратимость в том случае, когда уходящая группа и нуклеофил имеют одинаковую природу.
Как видим, очень точно угадал (именно угадал – не было способов установить структуру в те времена, только предположить) он структуру, имея экспериментально установленную брутто-формулу. Да, сейчас нам запрещают рисовать пять связей от азота, но до открытия электронной природы связей и правила октета оставалось еще 15 с лишним лет и вся Первая мировая война. Наиболее интересное и непосредственно связанное с механизмом нуклеофильного замещения наблюдение Мейзенгеймер сделал, когда показал, что из тринитроанизола и этилата калия образуется точно такой же комплекс, как из тринитрофенетола и метилата калия. И что при разложении таких комплексов образуется смесь исходных – в этом месте ое фактически установил не только факт замещения, но и его обратимость в том случае, когда уходящая группа и нуклеофил имеют одинаковую природу.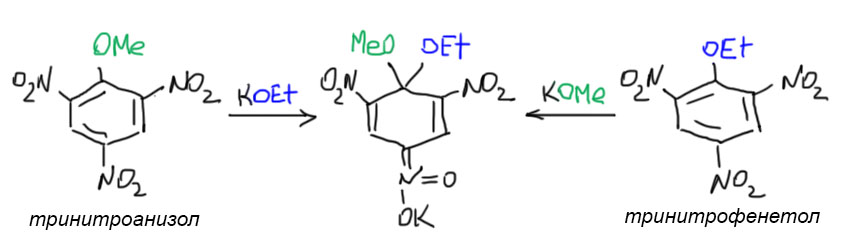
Гипотеза Джексона и Мейзенгеймера об устройстве соединений, образующихся при действии нуклеофила на активированное ароматическое соединение, оказалась верной. И хотя эти исследователи не изучали механизм – тогда не было даже мыслей про механизмы реакций – преложенные структуры можно считать самой первой в органической химии гипотезой об образовании промежуточных соединений (интермедиатов) в органических реакциях. Неудивительно, что когда появился собственно механизм 50 лет спустя, ключевые интермедиаты стали называть комплексами Мейзенгеймера или комплексами Джексона-Мейзенгеймера (второе намного справедливее, но встречается реже). Джексона забывали по очень простой причине – хотя он был первым и приоритете его несомненен и признавался самим Мейзенгеймером, Джексон опубликовал свою работу в никому не ведомом американском журнале, а Мейзенгеймер – в самом главном журнале золотого века органической химии – Анналах Юстуса Либиха, с чтения свежего номера которых начинал свой день всякий уважающий себя химик. То же происходит и наше время – печатаете свои труды чёрт знает где, не думайте, что их кто-нибудь заметит, даже если там будет что-то очень важное.
Строение комплексов Джексона-Мейзенгеймера
Мы, конечно, не можем оставлять структуру комплексов в таком архаическом виде. Прерисуем ее по современным требованиям, заодно прояснив одну важную вещь. Во-первых, уберём катион и ограничимся анионной частью комплекса. Во-вторых, нарисуем делокализацию отрицательного заряда граничными структурами. Формально, отрицательный заряд распределён по трём положениям кольца, как на трёх верхних граничных структурах. Фактически, он уходит в нитро-группы, оказываясь на атоме кислорода, как на трёх нижних граничных структурах. Всего структур шесть, но граничные структуры с зарядом на углеродах кольца вообще не играют никакой роли в делокализации отрицательного заряда. Как мы уже разбирали в правилах рисования граничных структур, наилучшая ситуация – это когда на концах цепи сопряжения находится донор и акцептор – в этом случае никакие промежуточные структуры рисовать нет смысла – их вклад минимален, а можно нарисовать только зультат полного перераспределения заряда с донора на акцептор. Поскольку в случае тринитро-соединений акцептора три, реальную роль в делокализации заряда и будут играть три граничные структуры с минусами на кислородах.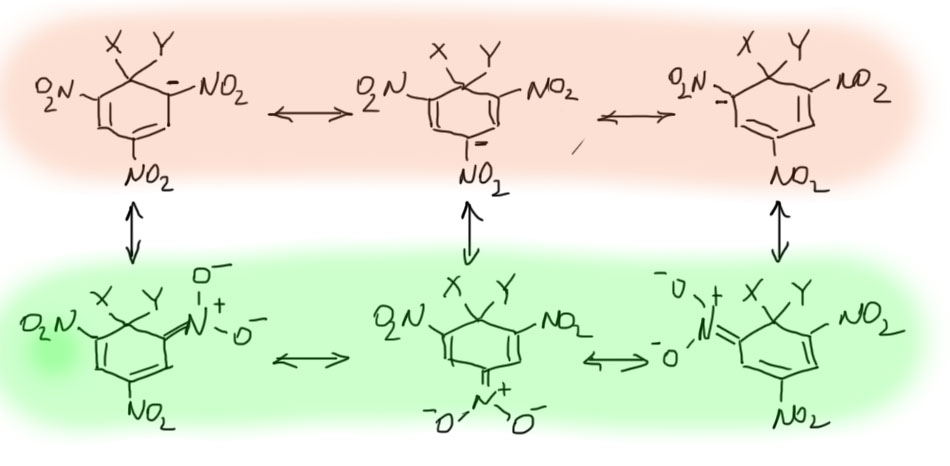
Всё это довольно очевидно, но в литературе, в том числе учебной, очень любят изображать делокализацию в комплексах Мейзенгеймера подковой внутри кольца с минусом посредине, в основном просто для простоты. Как часто бывает, и в этом случае простота оказывается хуже воровства. Время и усилия такое изображение, конечно, экономит, но невольно дезориентирует тех, кто пользуется таким изображением, особенно, если структура не была хорошо осмыслена. 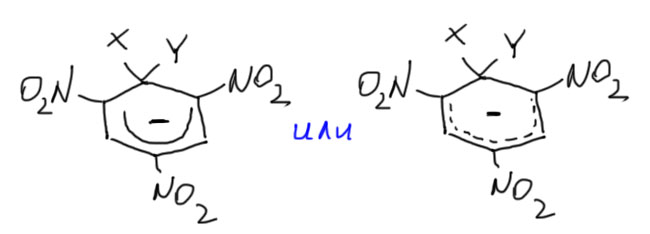 Нет, не в кольце делокализован минус. На углеродах минуса нет вообще, и если посчитать методами квантовой химии заряды на атомах в таких комплесах, на углеродах они останутся положительными, что неудивительно, ведь кольцо несёт аж три сильнейших мезомерных и индуктивных акцептора. Углероды кольца и в комплексе остаются электрофильными, и это хорошо известно, так как такие комплексы отлично присоединяют еще один нуклеофил, а в некоторых работах (Rochester, J.Chem.Soc., 1965, 2404) наблюдали даже аддукты с тремя нуклеофилами, что полностью использует акцепторный потенциал всех трёх нитро-групп. Это было бы невозможно, если бы в кольце действительно был минус, потому что нуклеофил с нуклеофилом не реагирует. Но минуса на углеродах нет, а аддукты, если к ним присмотреться, представляют собой ненасыщенные нитросоединения с нитро-группой на двойной связи. Это типичные электроноакцепторные олефины, котррые присоединяют нуклеофилы, как положено таким непредельным соединениям, и мы скоро увидим многочисленные примеры такой реакционной способности в других классах органических соединений (это называется присоединением по Михаэлю).
Нет, не в кольце делокализован минус. На углеродах минуса нет вообще, и если посчитать методами квантовой химии заряды на атомах в таких комплесах, на углеродах они останутся положительными, что неудивительно, ведь кольцо несёт аж три сильнейших мезомерных и индуктивных акцептора. Углероды кольца и в комплексе остаются электрофильными, и это хорошо известно, так как такие комплексы отлично присоединяют еще один нуклеофил, а в некоторых работах (Rochester, J.Chem.Soc., 1965, 2404) наблюдали даже аддукты с тремя нуклеофилами, что полностью использует акцепторный потенциал всех трёх нитро-групп. Это было бы невозможно, если бы в кольце действительно был минус, потому что нуклеофил с нуклеофилом не реагирует. Но минуса на углеродах нет, а аддукты, если к ним присмотреться, представляют собой ненасыщенные нитросоединения с нитро-группой на двойной связи. Это типичные электроноакцепторные олефины, котррые присоединяют нуклеофилы, как положено таким непредельным соединениям, и мы скоро увидим многочисленные примеры такой реакционной способности в других классах органических соединений (это называется присоединением по Михаэлю). 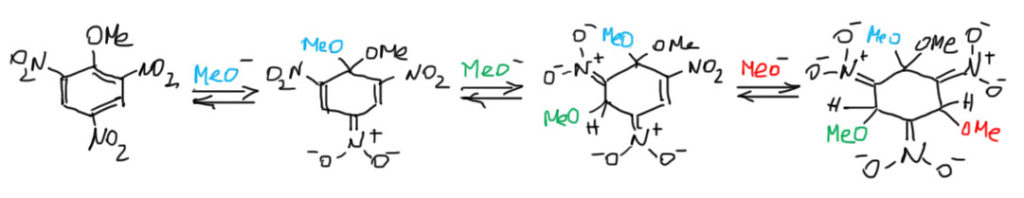
Я предлагаю избегать изображения делокализации “подковой” – не будем сбивать самих себя с толку, будем корректны и аккуратны. Это точно не тот случай, когда подкова приносит счастье. Химия любит уважительное отношение к структуре, потому что в химии нет ничего важнее структуры, и воздаёт сторицей тем, кто не гонится за краткостью ценой сути. Делокализацию же проще и точнее выражать любой из нормальных граничных структур с минусом на кислороде, если лень рисовать все три, нарисуйте любую, это уже будет точнее, чем подкова.
Механизм SNAr
Вернёмся к механизму. Нас интересует замещение, а не просто образование комплексов Джексона-Мейзенгеймера. Обратите внимание на то, что эти комплексы были описаны для активированных проматических соединений, не имеющих очевидной уходящей группы. Поэтому они вполне устойчивы, выделяются из растворов как устойчивые твёрдые соли, их можно потрогать руками (в перчатках), понюхать, положить в пузырёк, и так далее. Но если уходящая группа есть, например, галоген, комплексов наблюдать не получится, а вместо этого получается продукт замещения. Вот именно это обстоятельство, – комплексы не наблюдаются, и вызывало основные проблемы в том, чтобы разобраться, как идёт это замещение. К комплексам Мейзенгеймера все привыкли, их многие получали и исследовали, хорошо знали, что они очень интенсивно окрашены. А если взять не тринитроанизол, а тринитрохлорбензол или динитрохлорбензол, и тот же метилат, замещение идёт, а окрашивания нет. Это замещение пытались описать так же, как замещение у насыщенного атома углерода – согласованный механизм. В те времена многое еще не было понятно, поэтому эта гипотеза не была заведомо плохой. На бумаге всё выглядит вполне прилично – нуклеофил замещает уходящую группу в одностадийном процессе, как обычно в таких процессах рисуем переходное состояние: 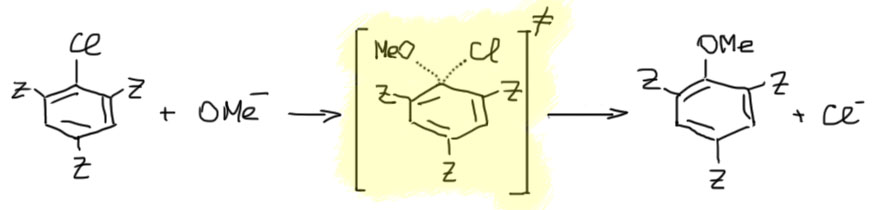
То, что такой механизм принципиально невозможен, и его нужно заменить другим, использовав давно известные комплексы Джексона-Мейзенгеймера, предположил в начале 1950-х молодой американский химик Джо Баннет, буквально за несколько лет создавший огромный раздел органической химии – нуклеофильное ароматическое замещение. Это был человек совершенно бешеной энергии и упорства, что сослужило органической химии не только хорошую, но и плохую услугу: ему мало было классического ароматического нуклеофильного замещения, он придумал еще и радикальное, и нам придётся его тоже изучать, хотя оно того не совсем стоит – но об этом ниже. Умер он всего несколько лет назад, только немного не дожив до своего столетия, в последние лет двадцать занимаясь вместо нуклеофильного замещения всякими шарлатанскими прожектами по уничтожению химического оружия. Один был особенно хорош – предполагалось собрать его всё в одном месте и шарахнуть атомной бомбой (заодно уничтожалась атомная бомба, числом одна).
SN2 механизм невозможен в ароматическом ряду потому что это всегда атака нуклеофила на атом углерода с противоположной стороны от уходящей группы (атака с тыла). Возможность противоположного сценария – атаки с фронта пытались доказать многие, и экспериментально и теоретически, но ничего не вышло. Проблему эту можно просто объяснить так: в реакции участвует гибридная орбиталь связи углерода с уходящей группой, и p-орбиталь нуклеофила. Как и положено в химии, мы должны взять такие орбитали, чтобы в сумме на них было 2 электрона: логично эти 2 электрона отдать нуклеофилу на неподеленную пару как раз на p-орбитали, и тогда нам понадобится разрыхляющая пустая орбиталь связи с уходящей группой. Но σ-связи (связь с уходящей группой – σ-связь) устроены так, что у них связывающая и разрыхляющая орбиталь по форма одинаковы, они различаются только наличием узловой плоскости между атомами у разрыхляющей, и тем, что в связывающую орбиталь больший вес даёт атом уходящей группы, а в разрыхляющую – углерод. Теперь понятно, почему так удобна атака с тыла – там как раз есть задняя доля пустой гибридной орбитали с приличным весом, а вся электронная плотность связи на связывающей орбитали сидит ближе к уходящей группе и не мешает. А теперь попробуем наехать спереди: тут нас ждёт сильное отталкивание орбитали нуклеофила и электронной плотности на связывающей орбитали, обслуживающей связь с уходящей группой. До пустой разрыхляющей и не доберешься! Полный назад!! Атака с фронта отменяется – это не распивочная, а полицейский участок.
А с тыла у ароматики тоже нельзя – нет там тыла, там серединка кольца, надёжно прикрытого сверху и снизу шапками ароматической системы.
И тогда Баннет вспомнил про комплексы Джексона-Мейзенгеймера.
Но ведь при образовании комплекса тоже происходит атака нуклеофила с фронта – а мы ведь ее уже заклеймили! Как, почему, опять обман… Ничего подобного, атака, да не туда, не на ту орбиталь. Нуклеофилу уже нет никакого дела до связи углерода с уходящей группой – его мишенью стала разрыхляющая орбиталь ароматической системы. Нуклеофил выхватывает из частокола p-орбиталей ароматического кольца одну p-орбиталь, выворачивает её, превращая в гибридную орбиталь новой связи, при этом старая гибридная орбиталь связи с уходящей группой отгибается, отталкиваясь в сторону. Всё нормально и вполне реально. При этом, не нужно забывать, на ароматической системе оставалась ещё законная связывающая орбиталь ароматической системы, которая, в том ещё целом кольце, составляла полный комплект с ещё двумя π-орбиталями, несущими в сумме тот самый ароматический секстет, 6 электронов. Теперь мы из каждой орбитали этого набора выдернули одну p-орбиталь, использовав ее для связи с нуклеофилом. Но 6 электронов-то остались, мы ими не пользовались, электроны для новой связи целиком пришли от нуклеофила. Получается на сопряжённой системе из 5 атомов углерода шесть электронов – избыток. Вот тут и помогают акцепторные группы, которые буквально откачивают эту лишнюю плотность из сопряжённой 5-углеродной системы. Так, мы получаем орбитальное описание того же, о чём говорят нам граничные структуры в описании комплекса.
Но на комплексе мы не задерживаемся: у нас же есть уходящая группа, и острое желание восстановить, во что бы то ни стало, порушенную ароматическую систему. Выпихиваем уходящую группу, не пожалев отдать её пару электронов, а опустевшую орбиталь выпрямляем обратно и заполняем тем же электроном, который успешно пересидел эти нуклеофильно-электрофильные разборки на уютной акцепторной группе у кольца. Всё хорошо, что хорошо кончается, и единственным немного обиженным персонажем остается та самая акцепторная группа, которой электрон дали подержать, да и то ненадолго. Такова учесть всех жадин (жадина – исконно русский перевод иноземного слова акцептор).
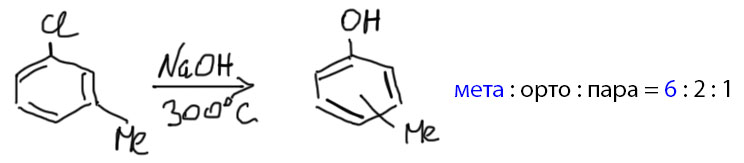
Активированное нуклеофильное замещение – очень хорошая реакция, с помощью которой можно получить много полезных и важных соединений. Но всё же она сильно ограничена. В органической химии, особенно такой, которая связана с промышленностью, очень востребованы самые простые производные бензола с гидроксильной группой (фенолы), амино-группой (анилины) и их производные. Как быстрее и проще сделать фенол или анилин из бензола. Первое, что приходит в голову – бензол хлорировать, а хлорбензол использовать для замещения. И этот способ действительно применяли, но это оказалось непросто, потому что реакция требует очень жёстких условий – нагревания под давлением до температуры выше 400ºС. Когда тот же способ применили для получения метилфенолов (эти смешные соединения называются крезолами, смешные они потому, что запах почти неотличим от неповторимого аромата свинарника – если вы никогда не бывали в свинарнике и не имеете шансов побывать, некоторое представление об аромате можно получить, посетив в зоопарке павильон с жирафами), обнаружили ещё один сюрприз. Гидролизу подвергали прямо смесь хлортолуолов, полученную хлорированием толуола. В такой смеси много орто-изомера, чуть меньше пара-изомера и немного мета-изомера (приблизительный состав орто:мета:пара – 62:8:30). А в продукте реакции, также требующей экстремально жёстких условий, тоже все три изомера – но преобладает мета-изомер (приблизительное соотношение 25:50:25).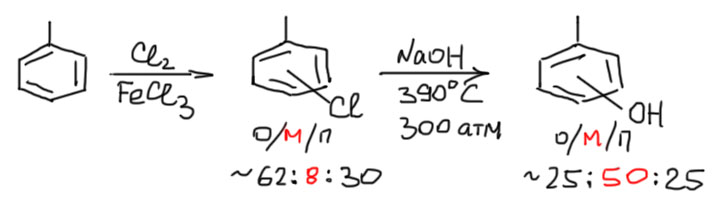
С анилинами похожая история: хлорбензол с аммиаком вообще не реагирует, но замещение можно сделать, использовав амиды щелочных металлов. Для промышленности такая реакция вообще смысла не имеет – слишком сложно и дорого. Но для науки о том, как идут органические реакции – это бесценные результаты, приведшие к открытию весьма необычного, почти невероятного механизма, которым мы и займёмся
Your Title Goes Here
Свернуть однако все же можно, и даже не одним способом, но всякий раз для этого потребуются ухищрения. Самый полезный метод использует, как это часто называют, грубую силу (brute force). Мы отлично знаем, что можно сделать, когда замещение не идет – тогда может идти элиминирование. Элиминирование в целом более универсально чем замещение, и если взять основание посильнее, то почти всегда есть шанс оторвать сначала протон из соседнего положения, если он там есть, конечно. Но что будет, если это сделать в ряду бензола? Должна получиться тройная связь. Но это невозможно – в циклах размером меньше 8 атомов настоящей тройной связи быть не может! Напомню, что такое настоящая тройная связь. Это связь между атомами в состоянии sp-гибридизации, что означает, что эти атомы и непосредственно связанные с ними лежат на прямой линии, и ничего с этим сделать нельзя. Если по какой-то причине эти 4 атома отклоняются от прямой, то в середине уже не настоящая тройная связь, а какое-то недоразумение и посмешище, готовое от стыда и позора провалиться под землю, – ну или быстренько с чем-нибудь прореагировать, если уж провалиться не получится. Вот поэтому настоящей тройной связи в небольших циклах (до 7-членных) быть не может – вы просто не сможете соединить прямой четырехуглеродный фрагмент в цикл, распялив между 1 и 4 атомами линейного фрагмента оставшиеся вам атомы – это такая геометрия нехитрая, обойти которую нельзя.
Тем не менее, если невозможна настоящая тройная связь, может быть возможна какая-нибудь другая, послабее. Посмотрим. От хлор- или бромбензола действительно элиминировать можно, и молекула, которая при этом получается не является ацетиленом, хотя ее рисуют с тройной связью, и называют бензином. Да, именно так, и всем плевать на страдания носителей русского языка, у которых этим словом обозначается одна из самых священных жидкостей – в других языках такой коллизии нет. Будем поэтому мучиться и вздрагивать – этот бензин на бензоколонке не продают, потому что он очень неустойчив и живет малые доли секунды. Обладая совершенно гигантской реакционной способностью, бензин (или в общем виде арин), всегда найдет во что превратиться, и за это особенно ценим химиками.
Итак, во-первых, бензин – это не ацетилен, не алкин, в нем нет настоящей тройной связи между углеродами в состоянии sp-гибридизации. Чтобы понять, что это достаточно просто подумать, как он получается. У галогенбензола с двух соседних атомов уходит протон (а электронная пара связи остается на месте), и галогенид, уносящий пару с собой. При этом, все это происходит в плоскости кольца, то есть на месте остаются гибридные орбитали углерода, ранее отвечавшие за эти связи. Сравним это с тем, что происходит, если мы элиминируем от галогеналкена – нечто похожее, но на месте бывших связей C-H и C-X появились p-орбитали (так как весь фрагмент распрямился, связи выстроились в линию), перекрывание которых дает полноценную π-связь – образуется настоящая тройная связь. В бензоле уже такое быть не может – потому что связи кольца имеют углы 120º и не собираются от этого отказываться, и в линию вытянуться не могут, а без этого нет sp-гибридизации. Гибридизация углерода остается sp2, и вместо p-орбиталей мы имеем гибридные орбитали под углом, что не дает им эффективно перекрываться и образовывать полноценную связь. Поэтому бензин – это не алкин, хоть именно так его и положено изображать, просто потому что это удобно. Но реально это устроено скорее как нечто среднее между двумя граничными структурами, в которых эти гибридные орбитали попеременно несут пару и дырку (минус и плюс, карбанион и карбокатион). Одна из этих граничных структур просто отражает, как образовался бензин – от соседних атомов ушли две уходящие группы, оставив на месте как раз пару и дырку. Поскольку в бензине два атома углерода одинаковы, законы квантовой науки не дают нам возможности сказать, на каком из углеродов что конкретно находится – после ухода протона и галогенида бензин “забывает”, что и где у него было до ограбления. Это “забывает” мы и изображаем в виде граничных структур. Или, чтобы не мучиться, в виде такого фейкового алкина с тройной связью. Главное, чтобы у нас память оказалась не так коротка, как у бензина, и мы помнили и что случилось, и что это значит. Итак, принимаем, что в бензине есть связь, обслуживаемая перекрыванием гибридных орбиталей в сумме несущих пару электронов (мы уже много раз убеждались, что это важный признак химической связи – именно пара электронов в общем пользовании двух, а иногда и более атомов), но эта связь слаба, что делает такую молекулу короткоживущей и невероятно реакционноспособной.
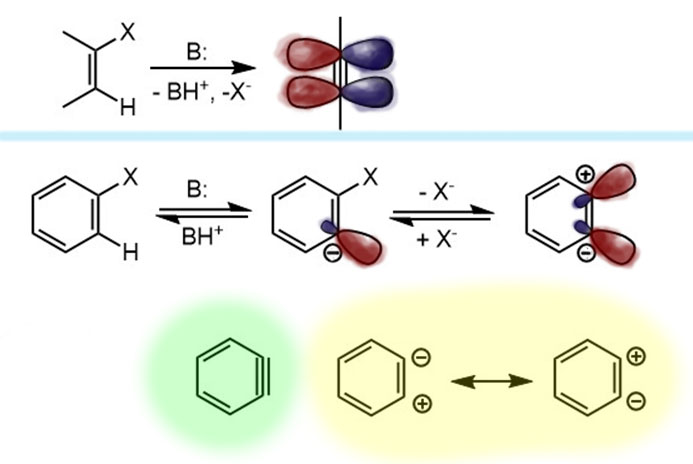
У самого бензина два углерода, образующие “тройную связь” совершенно одинаковы, и это означает, что вклад двух граничных структур совершенно одинаков, и на атомах реально нет зарядов. Но все может быть иначе, если в молекуле бензина есть заместитель рядом с этой “тройной связью”. В этом случае углероды становятся разными, различимыми, и законы квантовой науки более не заставляют нас ничего забывать. А так как у заместителей бывают донорные или акцепторные эффекты, граничные структуры становятся неравноценными. Например, если заместитель акцепторный (речь идет об индуктивном эффекте, так как мезомерный в такой конфигурации не действует), то граничная структура с плюсом рядом с заместителем дестабилизируется и понижает свой вклад за счет второй – это означает, что ближний углерод становится немного карбанионным, а дальний карбокатионным. Эта поляризация “тройной связи” имеет интересные последствия, которые мы обсудим чуть дальше.
Теперь можно нарисовать полный механизм нуклеофильного замещения с участием бензина (арина). Сначала для галогенбензола без других заместителей.
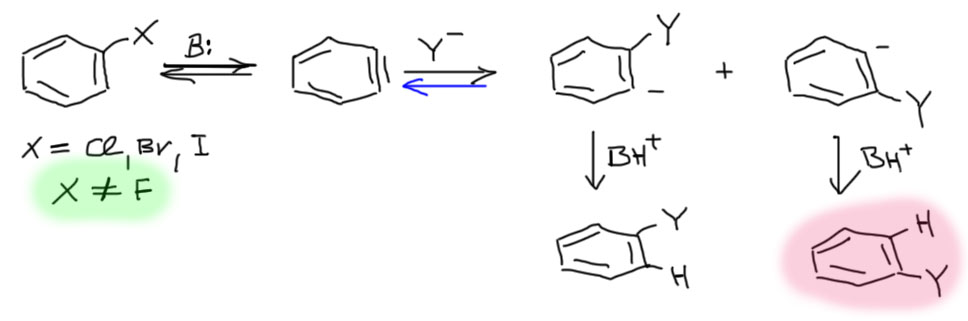
При отщеплении протона от галогенбензола (фтор не годится(примеры таких реакций в литературе найти можно, но они редкие и довольно спорные)) и быстрого ухода галогенида (фторид просто не уходит и поэтому фтор не годится) образуется промежуточный бензин, который немедленно присоединяет к своей “тройной связи” нуклеофил из реакционной смеси. Поскольку углерода “тройной связи” неразличимы, то нуклеофил с равной вероятностью присоединяется либо к одному, либо к другому. При этом образуется карбанион. Стадия присоединения нуклеофила обычно необратима, но в тех случаях, когда она бывает обратима, случаются интересные реакции (например, бешеная пляска галогенов, посмотрите одну из задач внизу и разберитесь сами, как это может быть). Карбанионы эти обладают огромной основностью и немеделенно находят протон, хотя бы в сопряженной кислоте основания, которое отрывало протон от исходного галогенбензола. Из-за равновероятного присоединения получается два продукта, которые можно различить, использовав изотопные метки в исходном галогенбензоле. В одном из продуктов нуклеофил садится туда, где была уходящая группа – это нормальное замещение. В другом – нуклеофил садится не туда, где была уходящая группа. Этот сенсационный результат невозможно объяснить по другому, и именно он стал ключевым доказательством этого механизма, который без этого доказательства так и остался бы безумной гипотезой. Путь, ведущий к этому продукту получил особое название кине-замещения.
Основные сведения
Ароматические галогенпроизводные, не содержащие активирующих групп в орто- и пара-положениях, открывающих для замещения механизм SNAr, который мы уже рассмотрели, могут реагировать с нуклеофилами, являющимися по совместительству очень сильными основаниями. Типичный пример такого нуклеофила – амид-ион, существующий в виде амидов щелочных металлов, обычно в растворе жидкого аммиака (больше эти амиды ни в чём просто не растворяются, а для этой реакции совершенно критично, чтобы она шла в растворе, а не гетерогенно, на поверхности).
Реакции идут по многостадийному механизму, включающему сначала β-элиминирование, скорее всего, по механизму E1cb – сначала отрываем протон, потом уходит галогенид. Образуется весьма своеобразное и очень неусточивое соединение – арин. Структуру этого соединения рисуют с тройной связью, хотя в небольших циклах настоящей тройной связи быть не может. Вместо настоящей π-связи, как у ацетилена, в арине слабая и очень реакционноспособная связь. Эта связь быстро присоединяет нуклеофилы с образованием нового карбаниона. Этот карбанион забирает протон у аммиака (это не совсем точно, желающие более корректного описания найдут его на более подробных вкладках ниже).
Доказательство Робертса
Идея о том, что в таком замещении участвует своеобразный ацетилен, бензин или арин, приходила в голову многим в конце 1940х – начале 1950х, но как это доказать? Было вполне понятно, что это очень неустойчмвый интермедиат, шансов выделить его и как-то зафиксировать не было. Косвенные доказательства были, но косвенные они та то и косвенные, что при желании их можно интерпретировать и другим способом. Методов исследования в те времена было ещё не очень много. Но кое-что важное появилось и было использовано одним из легендарных химиков 20 века, Джоном Робертсом для почти неоспоримого доказательства учатия арина в реакции нуклеофильного замещения в присутствии очень сильного основания.
Полное его имя – Джон Домбровски Робертс, что вместе с годом рождения, 1918, намекает на то, что его родители, скорее всего, тоже прибыли в США в той самой волне эмиграции из Российской империи, что уже дала нам Брауна и Хараша, и даст еще много кого. А может не из Российской, а из Австро-Венгерской, мало чем отличавшейся в те времена по отношению к меньшинствам. Робертс прожил огромную жизнь, всего двух лет не дожив до столетия, и даже немного пережив Баннета. Пример этих двух ясно показывает, что большие успехи в исследовании ароматического нуклеофильного замещения весьма способствуют долголетию. Нам этот Робертс очень хорошо известен по учебнику Робертс-Касерио. Этот учебник до сих пор в ходу и нисколько не устарел, входит в число рекомендованных, хотя сейчас выглядит достаточно непритязательно, мало кому нравится и почти никем не используется – там, говорят, нет ничего. Но в своё время, в начале 1970-х он произвёл настоящую революцию. Я очень хорошо это помню, потому что поступил на химфак в год выхода этой великолепной книги, чёрной такой, красивой и стильной, с большими красными цифрами томов. Все учебники органики до этого – подробные, для кого увлекательные, для большинства смертельно занудные описания классов органических соединений и их важных представителей, перечисление конкретных реакций с выходами, температурами, временем, внешним видом, запахом, иногда даже вкусом и всеми прочими подробностями. Всё это полагалось запоминать, и кто помнил больше всех конкретных реакций и веществ со всеми этими деталями, тот и был знатоком органической химии. До 1960-х это вполне неплохо работало, так как так и было – реакций, приводящих к тому или другому важному органическому соединению было одна-две, и всё это вполне хорошо запоминалось, часто даже до подробностей методики. Но как раз в это время органическую химию хорошенько пришпорили неугомонные люди типа этого Робертса, и она понеслась, как бешеная, вскачь с головокружительным ускорением. Реакций становилось всё больше, методы стали размножаться как кролики, новых веществ – лавина, и старый способ изучения органической химии превратился в изнурительное упражнение памяти, при этом довольно бессмысленное, потому что один запомнил одни реакции, другой другие, третий третьи, и что из этого важно, что верно, кто лучше знает органическую химию, и что это вообще значит – знать органическую химию? Выучить наизусть целиком на языках оригинала Ветхий Завет, Илиаду, Евгения Онегина и Поминки по Финнегану – не проще ли? Вот в это время и выходит Робертс с новым учебником, где описаний веществ почти нет совсем, конкретных реакций – на пальцах пересчитать, а ведь весьма толстая книга, в русском переводе, сделанном, кстати, на нашей кафедре, – 2 тома свёрстанных очень рыхлой модерновой вёрсткой с кучей картинок, в основном спектров. И всё обсуждение крутится только вокруг важнейших механизмов, основных понятий химии, структуры и спектроскопии, и иллюстрируется типичными примерами, кейсами, как это принято в Америке. Робертс сказал нам. – Мы не будем больше запоминать все эти реакции и вещества. – Вот вам основные инструменты, разберитесь в них получше, а дальше думайте и применяйте их самостоятельно. – Сами разбирайтесь, короче, а я пошёл. После Робертса-Касерио способ изучения и преподавания органической химии очень быстро изменился, и стал тем, что мы и знаем – к реакциям и веществам через механизм, структуру и спектр. Сложновато для тех, кто привык заучивать, но хотя бы запоминать выходы больше не надо.
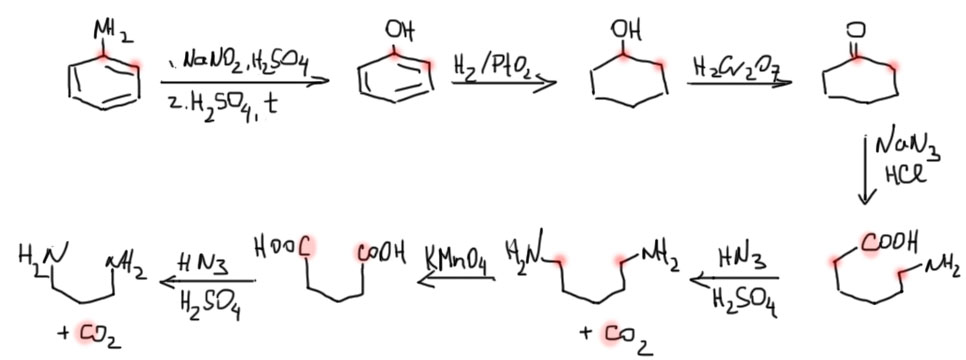
Джон Робертс использовал меченый по одному углероду радиоактивным изотопом 14С анилин, купленный готовым у одной из компаний, возникших вокруг только что зародившейся ядерной отрасли. Анилин был превращён с помощью реакции Зандмейера через диазотирование (2-й семестр) в иод- или бромбензол, меченый строго по углероду под галогеном. Этот меченый субстрат был подвергнут замещению амидом калия в жидком аммиаке, в типичных условиях, в которых наблюдали кине-замещение. Меченый углерод пометим красненькой точкой. Если ариновый механизм действует, должно получится два сорта молекул анилина, и поскольку арин строго симметричен. Изотоп никак не влияет на реакционную способность в пределах точности эксперимента и даже сильно за ее пределами. Есть такое явление – изотопный эффект на константу скорости, но для изотопов, различающихся по массе так, как изотопы углерода 12С и 14С этот эффект очень мал, тем более, что никто не собирается делать и использовать в качестве метки 100%-ный изотоп – вполне достаточно уже небольшого обогащения относительно природного фона. Этот изотоп есть в природном углероде, потому что он непрерывно образуется в атмосфере из изотопа азота, поэтому его содержание в природной углекислоте постоянно и очень мало, и из неё он распределяется по другим углеродсодержащим молекулам, на чём и основан радиоуглеродный анализ. Природный изотоп будет размазан по всем углеродам любой органической молекулы, в том числе и анилина, использованного в эксперименте. Чтобы использовать изотоп как метку, приходится использовать искусственно обогащённые исходные вещества, долей процента обогащения достаточно, так как содержание природного изотопа имеет порядок 10-10 %. Поэтому мы можем спокойно считать, что количество молекул меченого анилина каждого сорта относится как 1:1. 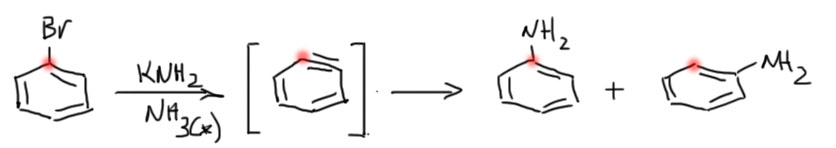
Чтобы доказать, что метка расползлась на два положения в образовавшемся анилине Робертсу и его сотрудникам пришлось проделать длиннющую и трудоёмкую цепочку превращений, разделяя атомы углерода на те, которые остаются в колбе, и те, которые улетают из неё в виде газа (CO2). Чтолбы этого достичь, причём количественно, пришлось проделать много реакций, выбирая такие, которые идут легко и количественно, и позволяют легко измерять радиоактивность (углерод-14 имеет β-активность, то есть просто стреляет электронами, это совсем не опасно и не требует особого оборудования, только аккуратной работы). Без потери смысла можем вместо двух сортов меченого анилина показывать одну с расползшейся меткой. Если вам что-то покажется странным, просто повторите цепочку с каждым из сортов меченого анилина, и проследите судьбу метки в каждом случае, не забывая, что каждая такая цепочка следит за половиной исходной полной активности. Диазотированием получили фенол, фенол прогидрировали в циклогексанол, который окислили в циклогексанон. Дальше Робертс активно использует перегруппировку Шмидта, с которой мы тоже разберёмся во 2 семестре. Сначала метка разползается уже на три углерода. Потом следуют два раунда вышибания углеродов в виде CO2. Если бы кине-замещения не было, вся активность целиком вылетела бы в первом раунде, а все оставшиеся вещества стали бы нерадиоактивными. Но нет – вторая порция CO2 имеет такую же активность (в пределах точности), как и первая. А такой результат согласуется только с механизмом с участием арина.
Эта работа Робертса и сотрудников навсегда останется классикой теоретической органической химии, примером того, как изобретательный ум может решить сложную проблему, используя те инструменты, которые можно найти в его время. Каждое время рождает свои инструменты. Робертсу, как ни странно, помогла послевоенная гонка вооружений между СССР и США – в обеих сверхдержавах лихорадочно работали над ядерным оружием, и это привело, в качестве побочного эффекта, к появлению целой индустрии радиоактивных изотопов. Радиоактивные метки очень понравились исследователям в 1950-е, и их стали использовать направо и налево, чтобы следить за приключениями реагентов и интермедиатов в реакциях, быстро и надёжно измерять скорости и равновесия. Но как видно из работы Робертса, изотоп сам не решает проблем, и приходится тщательно планировать и выполнять исключительно сложный и трудоёмкий ксперимент.
Если бы Робертс не торопился вписать своё имя в историю химии, то его вписал бы кто-нибудь другой, а подождал всего несколько лет, доказательство могло бы быть получено намного проще. В начале 1960-х в самых продвинутых лабораториях США появился новый метод исследования – спектроскопия ЯМР. Робертс был одним из тех, кто сразу понял мощь этого метода и принялся энергично его использовать для органической химии. И если у нас есть спектроскопия 13С ЯМР, получить доказательство участия арина можно гораздо быстрее. Правда для этого всё равно понадобится меченый галогенбензол, но в качестве метки использовался бы стабильный изотоп 13С. Как бы знаем, это природный изотоп, в природном углероде его около 1%, и на этом содержании основывается спектроскопия 13С ЯМР. В самом распространенном исполнении этой спектроскопии (широкополосная развязка от протонов) каждый атом углерода в этой спектроскопии даёт узкую резонансную линию с некоей средней интенсивностью, конкретная величина которой практически случайна и ни о чём не говорит, за одним исключением – атомы углерода, не связанные с протонами (четвертичные атомы углерода) имею совсем маленькую интенсивность. Теперь сделаем достаточно простую вещь – возьмём бром или иодбензол, у которого углерод под галогеном обогащён изотопом 13С – достаточно и 5% (чем больше степень обогащения, тем дороже, и нет смысла платить больше за то, что можно получить дешевле), это всё равно будет в пять раз больше, чем природное содержание. Если мы зарегистрируем спектр 13С ЯМР обычным методом с такогго бромбензола, получим такой же спектр, как у необогащённого, с одним исключением – интенсивность сигнала четвертичного углерода будет необычно велика, мы не сможем этого не заметить. Ну и теперь просто сделаем реакцию замещения с тем же амидом калия и жидким аммиаком, а с образовавшегося анилина тоже сделаем спектр. И сразу увидим очень ясно, что две резонансные линии – от первого и второго углерода необычно интенсивны. Всё, доказательство получено, не нужно ничего долго и нудно разрушать, ловить CO2 и всё остальное. Если покажется мало, я арсенале ЯМР есть много способов уточнить спектральную информацию, точно доказав, какой атом с камим связан и как. Робертс понял это очень хорошо и одним из первых, и поэтому и сделался из автора одного из самых знаменитых экспериментов в доказательстве механизмов органической химии с использованием радиоактивной метки в одного из тех, кто буквально втащил ЯМР в органическую химию, сделав этот метод совершенно необходимым инструментом и убедив в этом всех остальных. Сейчас это кажется совершенно очевидным, но в те годы ЯМР для большинства выглядел так, как сейчас для нас Большой адронный коллайдер – предствьте себе, что какой-то странный фрик советует вам ваше вещество из курсовой или диплома засунуть в один из детекторов БАК, ну там, договориться с знакомым, который там работает, найти дырочку в которую это можно туда засунуть прямо на путь протонов, и внимательно рассмотреть бозон Хиггса, который может быть оттуда вылетит… Что вы скажете? Ну, что-то такое, уклончивое… Тогдашние ЯМР-спектрометры выглядели не намного менее убедительно, чем сейчас детекторы коллайдера – огромные магниты, шкафы с электроникой, мотки проводов и прочего. Делалось это штучно, стоило это космических денег. Совсем не похоже на то, что можно просто взять и пристроить к обычной органической лаботратории. Но очень быстро стало незаменимым.
Your Title Goes Here
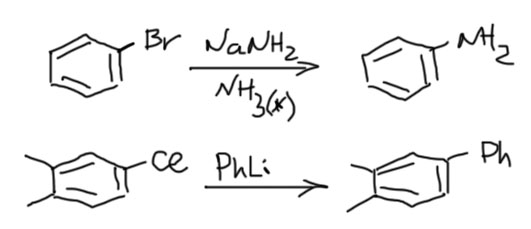
Так можно получать самые разные амины, вначале получив из амина его соль с щелочным металлом (такие соли называют амидами, и нужно быть очень внимательным, чтобы не путать эти амиды с амидами карбоновых кислот).
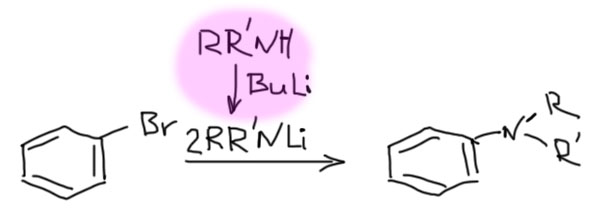
Иногда можно разделить основание и нуклеофил, первый использовать для генерации арина, второй для дела. Таких реакций довольно много, но планировать их трудно, и нам стоит избегать этот вариант, просто потому что у нас не хватит знаний корректно предсказать результат таких реакций. Посто для примера приведу образование сульфида. В этом случае все более-менее просто, потому что основность сульфида недостаточна для депротонирования, а нуклеофильность у него ого-го какая! А у трет-бутилата все наоборот. Из этой реакции мы можем сделать весьма важный вывод: несмотря на очень высокую реакционную способность аринов, они неплохо разбираются в нуклеофилах и выбирают те, что получше. В этом смысле они сильно и в лучшую сторону отличаются от совсем неразборчивых карбокатионов в SN1-замещении.

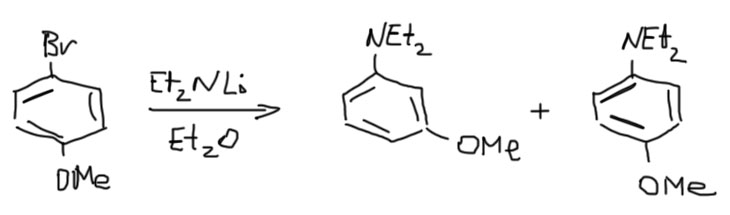
Все это совершенно замечательно, и кажется, что цены нет такому потрясающему методу. В современном синтезе эту реакцию не очень любят из-за низкой экономичности – один моль основания-нуклеофила теряется впустую (да, это серьезный аргумент). Нам это не очень важно. Но стоит нам ввести заместители в бензольное кольцо, как начинаются проблемы с селективностью, а это уже проблема тяжелая и, мы поклялись ею не пренебрегать. Как мы видели в механизме реакции, арин присоединяет нуклеофил к двум разным углеродам, и получается или нормальный продукт, или продукт кине-замещения, а чаще всего их смесь. Как мы видели в структуре арина на углеродах “тройной связи” может быть больше карбокатионного, или больше карбанионного характера, в зависимости от заместителей. В реальности, если заместитель находится в пара- или мета-положении, его эффект на поляризацию “тройной связи” не очень велик, и почти всегда образуются смеси, и нам точно нет смысла разбираться в том, чего там может быть больше и когда, да и не только нам, а и в большом органическом синтезе вся эта суета очень не нравится – не любят синтетики эту реакцию, грязная она. Вот, например, что получается из мета-изомера в типичной реакции с ариновым механизмом.
Высокая температура нужна, чтобы заставить щелочь – недостаточно сильное основание – реагировать по этому механизму. А что, температура так сильно влияет на основность??? Да, но только в том случае, когда отщепление протона необратимо, и в дело вступают законы кинетики (помните заклинание: При повышении температуры на десять градусов скорость химической реакции увеличивается в 2-4 раза – повторять натощак каждый день десять раз, выходные и праздники можно пропустить). На равновесия температура влияет, но далеко не так сильно. В случае аринового механизма отщепление протона необратимо, потому что за ним немедленно следует отщепление галогенида и реакция арина. И это делает возможным раскочегарить примитивную щелочь на ариновый механизм. Этим активно пользовались в допотопной промышленности для синтеза фенолов. После потопа изобрели более изящные методы, о которых мы поговорим в своем месте.
А вот пример ариновой реакции с пара-замещенным. А продукт основной – мета, увы, далеко не чистый, и пара-изомера тоже немало.
Ну и зачем нам такая химия? К счастью в некоторых частных случаях все получается гораздо чище и определеннее. Обсудим их подробнее на отдельной вкладке: Кине-замещение.
Реакции нуклеофильного замещения с участием аринов очень интересны и дают возможность делать разные очень непростые вещи, которые почти невозможно делать по-другому – то же кине-замещение, например. Только для одной цели она почти бесполезна – для простого и банального нуклеофильного замещения, когда вы просто хотите взять что-то простое, например, пара-бромтолуол, и получить просто продукт, в котором нуклеофил сидит там, где был галоген. Не тут-то было – получите смесь. Ариновый механизм почти всегда даёт смеси, иногда с преобладанием кине-продукта, иногда с преобладанием нормального продукта, но почти никогда не даёт чистый продукт номального замещения. 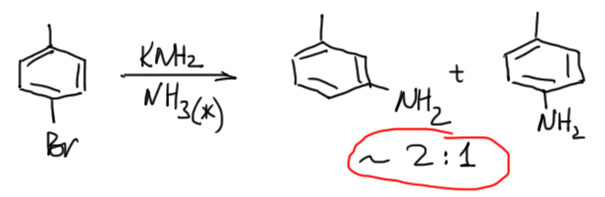
Вторым недостатком аринового замещения является его неэкономность – на один входящий нуклеофил требуется два эквивалента этого самого нуклеофила, так как второй эквивалент работает как основание и теряется. В современной органической химии этот аргумент – экономичность реакции – стал очень часто определяющим. И даже без отсылки к современным заморочкам, иногда явно демагогическим, вы сами можете столкнуться в этой проблемой, если захотите в качестве нуклеофила использовать не банальный амид, а что-нибудь сильно посложнее, например, амин какой-нибудь оптически чистый и бешено дорогой. И тогда вам будет жалко не то что лишнего эквивалента, а молекулы лишней.
Решение этой проблемы нашёл тот же Баннет, скорее всего, случайно. Он исследовал реакцию галогенпроизводных замещённых бензолов с амидом калия во вполне ариновых условиях. Амид калия для таких реакций получают растворением калия в жидком аммиаке, причём сначала получается раствор сольватированных электронов, к которому добавляют немного соли железа, действующей как катализатор превращения в амид калия. Раствор при этом обесцвечивается. Если не дождаться полного обесцвечивания и добавить галогенпроизводное, то конкурентно пойдёт совсем другая реакция без участия арина, и получится в основном продукт без кине-замещения. Амида калия для этой реакции, теоретически, нужен ровно один эквивалент, а электроны, то есть некоторый избыток металлического калия над тем, что нужен для получения амида, теоретически, можно взять в каталитическом количестве. Это звучит по-настоящему прикольно – реакция, катализируемая электронами. Так ли это и что это за реакция посмотрим на следующих вкладках.
Your Title Goes Here
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Your Title Goes Here
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.