Диены
Раздел посвящен в основном не всем диенам, а только сопряженным (1,3-диенам). Аллены (1,2-диены, они же кумулены) мы толком не обсуждаем, и никогда не используем активно, в задачах. Но не забывайте, что аллены появляются в ацетилен-алленовой перегруппировке, где играют очень важную роль.
Сопряженные диены мы рассматриваем серьезно. Главные вопросы здесь – структура, конкуренция 1,2 и 1,4-присоединения, реакция Дильса-Альдера. Обратите внимание, что на этом этапе изучения органики мы реально не можем синтезировать диены, просто не хватает знаний и инструментов. Все, что написано в программе про синтез диенов – общие сведения, которые невозможно применить в синтезе, за единственным маленьким исключением отщепления двух HBr от дибромидов. Только когда дойдем до карбонильных соединений получим метод синтеза диенов, тогда вернемся.
Программа по разделу
1. Диены: а) сопряжённые, б) аллены, в) несопряжённые. Длина сигма- С-С связи в сопряженных диенах. Сопряжение и УФ- спектры. S-цис– и S-транс– конформации.
2. 1,2- и 1,4-присоединение к сопряженным диенам. Энергетический профиль реакции. Кинетически и термодинамически контролируемые реакции. (Электрофилы, которые не дают 1,4-аддукты – RSCl, карбены, ВН3, эпоксидирование).
3. Реакция Дильса-Альдера как метод образования С-С связей и метод синтеза 6-ти членных циклов. Диены и диенофилы. Необходимая конформация нециклического диена. Механизм реакции (согласованное стереоспецифичное [2+4]-циклоприсоединение). Реакция с циклическими диенами (циклопентадиеном, циклогексадиеном). Экзо- и эндо- аддукты.
Реакции с ациклическими несиметрично-замещенными диенами и олефинами – региоселективность и так называемая “орто-пара-ориентация”.
4. Понятие о согласованных перициклических реакциях. Разрешенные и запрещенные по симметрии орбиталей процессы.
5. Понятие о еновой реакции. Аналогия между механизмом диенового синтеза и еновой реакции.
6. Аллены. Хиральность тетразамещенных алленов. Присоединение НBr к аллену.
7. Каучук.
МЕТОДЫ ПОЛУЧЕНИЯ СОПРЯЖЕННЫХ ДИЕНОВ.
1. А) БУТАДИЕН ИЗ КАРТОФЕЛЯ (Лебедев, 30-е гг.): Картофель→крахмал→спирт → бутандиол-1,3 → дегидратация в бутадиен-1,3. (Спирт на катализаторе Cr2O3/(ZnO)/Al2O3 при нагревании окисляется в ацетальдегид, последний конденсируется в альдоль и восстанавливается в бутандиол-1,3 (тема – альдольная конденсация).
Б) БУТАДИЕН ИЗ АЦЕТИЛЕНА И ФОРМАЛЬДЕГИДА по Фаворскому-Реппе.
В) ДЕГИДРИРОВАНИЕ С4 или С5 ФРАКЦИЙ, полученных при пиролизе нефтепродуктов с последующей экстрактивной ректификацией (напр. с ДМФА.)
2. ХЛОРОПРЕН Димеризация НС≡СН в СН2=СН-С≡СН, присоединение НСl (Cu2Cl2) по С≡С (старый способ).
3. ГИДРИРОВАНИЕ СОПРЯЖЕННЫХ ДИИНОВ (С≡С-С≡С) с P-2-Ni/H2, или с R2BH/потом AcOH. (СИН-гидрирование).
4. СИММЕТРИЧНЫЕ ДИЕНЫ. Присоединение к С≡С гидридов бора (Sia2BH) или алюминия (ДИБАЛ-Н) cпоследующей окислительной димеризацией с Cu2Cl2 или Сu2Br2.
5.КРОСС-СОЧЕТАНИЕ. RCH=CH-E (E= B, Sn, Zn) с Br-CH=CHR’ в присутствии Pd, или Niкатализаторов. (Тема – реакции, кат. Комплексами пер. металлов.)
Структура сопряженных диенов
Сопряжение в 1,3-диенах – довольно тонкий момент, испытывающий на прочность наши представления о сопряжении. Если они у нас есть, то все в порядке – не заблудимся. Итак, дано – две двойные связи рядом, то есть через одну простую. Вопрос- насколько сильно они взаимодействуют, и что из этого следует?
Ответ достаточно простой – это слабое взаимодействие, настолько слабое, что оно не препятствует вращению вокруг простой C-C связи. Энергия, которую приходится преодолевать при вращении вокруг этой связи (барьер вращения), составляет всего около 5 ккал/моль – это ненамного больше, чем энергетический барьер при вращении вокруг C-C связи в этане, где вовсе нет никакого сопряжения и весь барьер обусловлен очень слабым отталкиванием атомов водорода (или же отталкиванием слабых диполей C-H связей), и практически равен барьеру вращения вокруг центральной C-C связи в бутане. Это не удивительно. Мы уже договорились, что хорошее сопряжение требует, чтобы на концах цепи сопряжения были донор и акцептор. Если их нет, как в данном случае, то эту роль приходится брать на себя двойным связям. Граничные структуры при этом получаются очень невыгодные, вносящие очень малый вклад в реальную структуру диена. Обратите внимание, что граничные структуры B и B’ разные (это легко понять, если мысленно пометить любой из атомов углерода, например заменив на изотоп) и нарисовать нужно обе. Но так как их вклад мал, структура бутадиена вполне адекватно отображается обычной формулой без зарядов и с простой связью посредине.
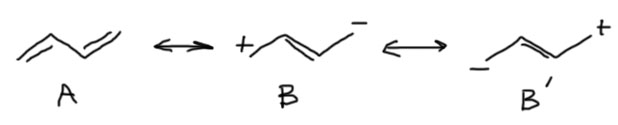
Так проявляется как-то реально сопряжение в молекулах сопряженных диенов, или это совсем не важно? Проявляется, и очень значительно. Даже слабые взаимодействия могут быть очень важными. Мы выяснили, что в молекуле диенов вращение вокруг простой связи практически свободно, а барьер мало чем отличается от барьеров в этане, бутане и других алканах. Но у алканов при вращении формы, соответствующие минимальной энергии – конформации – соответствуют такому расположению атомов, при котором они располагаются максимально далеко друг от друга, чтобы уменьшить отталкивание (заторможенные конформации), а заслоненные конформации соотвествуют максимумам энергии и избегаются.
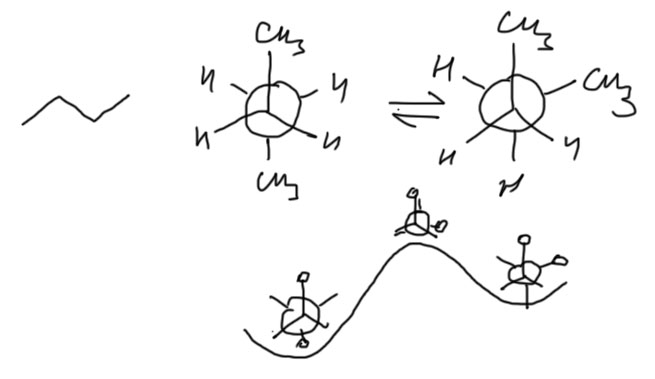 А в бутадиене все наоборот – минимумы соответствуют как раз заслоненным конформациям. Почему?
А в бутадиене все наоборот – минимумы соответствуют как раз заслоненным конформациям. Почему?
Мы уже обсуждали, что для взаимодействия π-орбиталей не обязательно, чтобы они были параллельны (находились в одной плоскости), а угол только ослабляет, но не уничтожает взаимодействие, лишь бы он не был равен 90º. Поэтому два фрагмента C=С относительно легко вращаются друг относительно друга, время от времени быстренько пролетая проклятый прямой угол. Так как взаимодействие максимально все-таки при параллельном расположении π-орбиталей, минимумы энергии соответствуют двум плоским формам 1,3-диена. Так как эти формы легко превращаются друг в друга при вращении вокруг простой C-C связи, эти формы являются не изомерами, а конформациями. Их называют s-транс и s-цис-конформацией. Буква s добавлена для того, чтобы это не путали с обозначениями настоящих диастереомеров, а взята она просто из слова сигма (sigma), так как речь идет о разном расположении фрагментов относительно сигма-связи. Почему нельзя было написать греческую букву σ непонятно, но скорее всего просто потому, что в докомпьютерную эпоху, когда это обозначение появилось, и тексты печатали на печатной машинке, букву σ приходилось вписывать от руки.
Поскольку конформации плоские, то в них атомы на концах простой связи находятся напротив друг друга – это именно заслоненные конформации. Но сопряжение стабилизирует их в большей степени чем отталкивание дестабилизирует. Итак, что такое по структуре 1,3-бутадиен или любой другой 1,3-диен? Ответ: это структура в двумя двойными и одной простой связью, вокруг которой происходит свободное вращение, обеспечивающее взаимопревращение двух плоских конформаций.
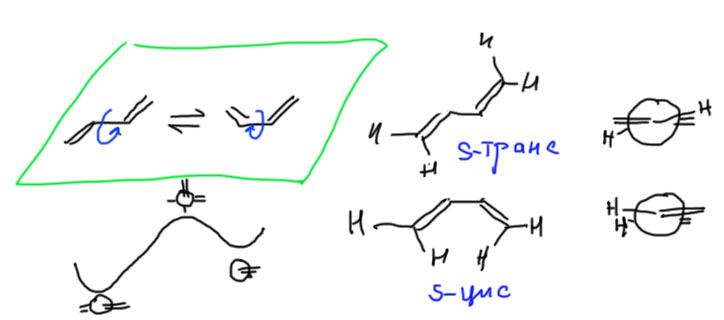
Конформации – очень важная вещь. Напомню, что если бы мы ничего не знали о конформациях, то нам было бы очень трудно работать со сложными молекулами с нежесткой структурой. Когда мы знаем, какие конформации есть у нежесткой молекулы, мы можем использовать эти конформации, как настоящие молекулы, и работать именно с ними, пренебрегая всеми промежуточными конфигурациями. В этом смысле, на вопрос, является ли молекула бутадиена плоской, мы можем с чистой совестью сказать, что да, является. Обе конформации бутадиена плоские, и других конформаций у бутадиена нет. Значит, нам нет никакого дела до всех других геометрических конфигураций этой молекулы. Нам плевать на эти формы. Мы никогда с ними не встретимся. Все эти формы, а понятно, что если одна конформация превращается в другую, то молекула просто не может не побывать в каждой из них, ведь это превращение – просто механическое вращение вокруг центральной связи, а не дискретные прыжки из одной в другую – нас тем не менее не волнуют, потому что молекула не задерживается в этих конфигурациях – это просто точки на непрерывном пути, а у точки нет времени жизни. Это то же самое, как химическая реакция – нас интересуют только исходные реагенты и продукты, и обычно не интересует, как точно происходит превращение. В тех случаях, когда мы хотим немного подробнее разобраться в том, как происходит химическая реакция, мы все равно не останавливаемся в каждой точке пути реакции, но смотрим еще на так называемое переходное состояние – конфигурацию, соответствующую перевалу из долины, где живут исходные, в долину, где будут жить продукты. У переходного состояния, точно так же как у любой точки на пути, тоже нет времени жизни – молекула в этом месте не задерживается, а продолжает движение, но положение переходного состояния на шкале энергии говорит нам об энергии активации, а следовательно о скорости реакции. Так же и с конформациями – мы можем рассматривать превращение конформаций, как химическую реакцию изомеризации, в данном случае s-транс формы в s-цис форму, а переходным состоянием будет перпендикулярная форма (конфигурация, не конформация). В этой перпендикулярной форме π-связи двух двойных связей строго перпендикулярны друг другу и вообще не взаимодействуют (правда, там возникают другие взаимодействия типа гиперконъюгации, стабилизирующие эту форму и делающие барьер превращения конформаций друг в друга таким удивительно невысоким, но это отдельная проблема). Именно эти несколько простых вещей и важны:
- то, что конформации бутадиена плоские и их всего две штуки,
- то, что конформация с противоположно-направленными двойными связями (s-транс) немного стабильнее, чем s-цис-конформация, причем важно и то, что стабильнее, и то, что стабильнее немного – это говорит нам о том, что при необходимости s-цис-конформация может участвовать в реакциях совершенно спокойно, и никаких проблем с этим не будет, потому что
- скорость взаимопревращения конформаций очень велика, так как барьер (он же энергия переходного состояния, он же энергия конфигурации с перпендикулярными двойными связями) невелик, естественно, при очень низких температурах это будет не так, но мы не делаем реакции с бутадиеном при температуре жидкого азота, поэтому нам это до лампочки.
Молекулу такого типа можно и даже нужно представлять, как равновесие конформаций, в данном случае s-цис и s-транс, в котором преобладает более выгодная s-транс форма, но так как разница энергий не очень велика, то и s-цис формы в равновесии немало. И если у нас есть реакция, которая выбирает из двух форм именно менее выгодную s-цис форму, то такая реакция, во-первых, пойдет, потому что такая форма в равновесии есть, и, во-вторых, имеет все шансы пройти до конца, до полного исчерпания диена в реакционной смеси, потому что по законам равновесия, по мере расходования s-цис формы и нарушения равновесия, равновесие будет смопроизвольно стараться восстановиться, превращая нереакционноспособную s-транс форму в s-цис, и так до полного исчерпания первой. Так идет, например, реакция Дильса-Альдера, о чем подробнее в соответствующем месте.
Кроме реакций, плоские конформации молекулы бутадиена и других сопряженных диенов проявляются в спектрах, особенно и в буквальном смысле очевидно – в спектрах электронного поглощения. В буквальном смысле потому что сопряжение приводит к сдвигу полос поглощения в видимую область спектра, и у молекул появляется настоящий цвет. Виноват, чушь сморозил, у молекул цвета быть никакого не может, цвет появляется у вещества, состоящего из этих молекул. У сопряженных диенов смщение полосы поглощения не настолько велико, чтобы заехать в видимую область, но при увеличении количества сопряженных двойных связей она смещается все ближе и ближе к видимой области с начиная с 5-6 связей хвостиком полосы начинает в нее заезжать, а вещество начинает слегка отдавать желтизной. Пока на этом остановимся.
1,2 и 1,4-присоединение
Одной из главных особенностей сопряженных диенов считается 1,4-присоединение электрофилов. Важно понять, что эта особенность никак не связана с сопряжением двойных связей в исходном диене, а является очевидным следствием реализации механизма электрофильного присоединения к алкенам, прежде всего потому что электрофил – любой электрофил – всегда атакует не сопряженную систему двойных связей целиком, а отдельные двойные связи, а образование продукта 1,4-привоединения всегда объясняется превращением образовавшегося в результате такой атаки промежуточного карбокатиона (или мостикового иона).
Посмотрим на 1,3-бутадиен как на самый обычный алкен, и присоединим к нему какой-нибудь электрофил, например, бром. Будем действовать так, как действовали с любыми другими алкенами. Так как мы уже неплохо разобрались с присоединением брома к двойной связи, сразу зададим вопрос, идет ли эта реакция через карбокатион, или через мостиковый ион. Обсуждая присоединение к одной двойной связи в разделе Алкены, мы пришди к выводу, что настоящий бромониевый ион образуется только из симметричных алкенов, а также, возможно, из алкенов, в которых карбокатионы с обеих сторон дестабилизированы. А если карбокатион стабилизирован, то образуется карбокатион или несимметричный мостиковый ион, смысл и структура которого не сильно отличаются от карбокатиона (подробнее в разделе Алкены, бромирование). Очевидно, что при присоединении к 1,3-диену мы можем ожидать образования сильно стабилизированного аллильного катиона, и мостиковый ион можно не рассматривать. Поэтому ограничимся карбокатионом, и прямо сразу его запишем.
Присоединим электрофил и посмотрим, какой из карбокатионов лучше. Так как две двойные связи одинаковы, присоединить можно только двумя способами. Разница между карбокатионами очевидна – один стабилизирован только индуктивным эффектом, другой – мезомерным. Аллильный катион, конечно, намного стабильнее. Сразу отметаем все остальное, и пишем этот карбокатионю Присоединение пойдет через него, а второй путь отправляется в мусорное ведро.
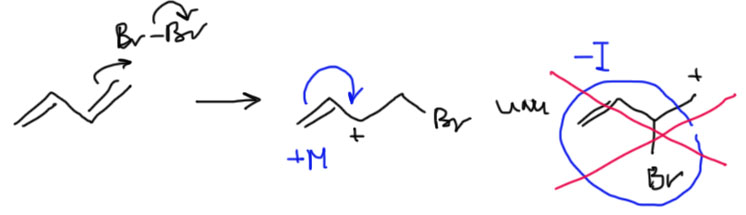
Распишем структуру карбокатиона граничными структурами и обратим внимание на то, что положительный заряд делокализован, фактически поровну, на двух атомах углерода. Когда к карбокатиону соберется присоединяться нуклеофил, к какому атому он пойдет? Видимо, можно ожидать образования смеси двух продуктов. Если мы пронумеруем атомы от углерода, на котором сел электрофил, получаются 1, 2 и 1,4-продукты. Так их и называют. Это немного тонкий момент, потому что с точки зрения номенклатуры и нумерации атомов в продукте, где приоритет имеет положение двойной связи, и нумеровать нужно от нее, и тога в продукте 1,2-присоединения электрофил оказывается на атомах 3 и 4. Если будете читать оригинальные статьи по этой теме, не удивляйтесь, что вместо 1,2-присоединения часто говорят о 3,4-присоединении. Но в учебной и обзорно-методической литературе это обозначение не закрепилось, и везде говорят про 1,2-присоединение.
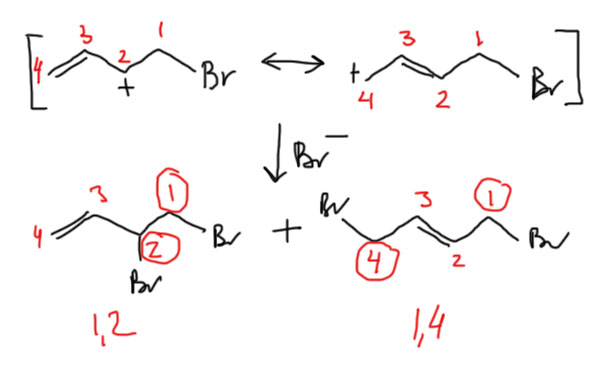
Итак, просто из общих соображений должны получаться оба продукта. И тогда, как обычно, органическая химия задается вопросом – всегда ли образуется смесь, или можно найти факторы, благоприятствующие образованию более-менее чистых продуктов, в идеале – каждого из типов. Можно ли сделать присоединение электрофила селективным?
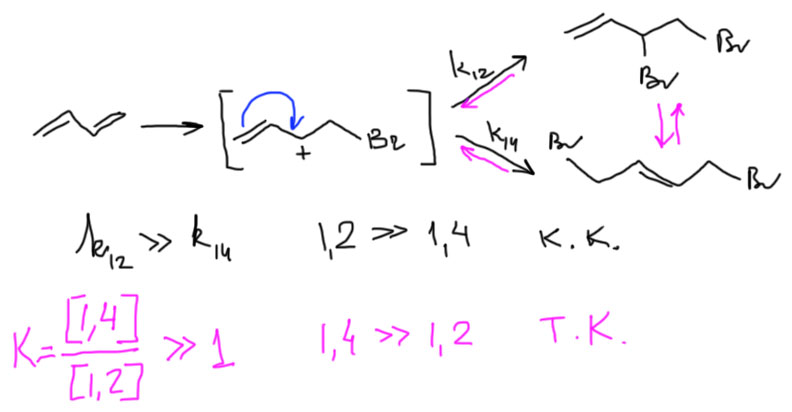
Ответ на этот вопрос даже для такой простой реакции как бромирование настолько непрост, что стал поводом для утверждения важнейшей концепции термодинамического и кинетического контроля, а это, в свою очередь, привело к возникновению удивительного мифа, попавшего во все учебники. Этот миф зажил своей жизнью, став реальностью. Это очень типичный пример того, о чем стали много говорить в самые последние годы уже в самом широком смысле: любой вздор и чистая выдумка могут овладеть массами, если в их распространение и продвижение будут вложены большие силы и средства.
Чтобы избежать недоразумений сразу скажу, что кинетический и термодинамический контроль – очень важное и прекрасно работающее поянтие, применяемое в десятках и сотнях реальных реакций. Здесь нет обмана.
И 1,2 и 1,4-присоединение к сопряженным диенам – совершенно точно установленный факт, проявляющийся в десятках и сотнях реальных реакций.
Мифом является другое – утверждение о том, что 1,2-аддукт (аддукт – это короткое название продукта присоединения) это продукт кинетического контроля, а 1,4-аддукт – это продукт термодинамического контроля. Но так учат? Да, ничего страшного, учат и хорошо. Лет тридцать назад на полном серьезе и до полного автоматизма реакций учили про три источника и три составные части марксизма. И где теперь эти источники и части?
Поэтому разобьем проблему на две части. В первой сделаем вид, что мы полностью верим в концепцию кинетического и термодинамического контроля в присоединении к диенам. На этой части стоит остановиться всем, кому нужно просто сдать экзамен и не париться (это, повторю в сотый раз, очень правильное отношение к делу, если ваша деятельность в будущем не будет связана с органической химией). Во второй же части я расскажу немногим, кому любопытно, как обстоят дела на самом деле.
Кинетический и термодинамический контроль.
Химия конкретная наука, иксов и игреков не любит, и все же иногда, ненадолго можно посмотреть на проблему в общем виде. Представим себе реакцию A и B с образованием двух продуктов. Это типичная проблема селективности. Соотношение продуктов будет соответствовать отношению скоростей, точнее, констант скоростей. Мы уже это обсуждали – относительные константы скоростей показывают нам селективность. Если отношение равно единице (что равнозначно утверждению, что относительные константы равны единицам), то реакция неселективна, если не равно, то реакция селективна (более или менее) и в ней образуется больше того продукта, относительная константа для которого больше. Это вообще издевательски банальная идея – больше того продукта, который образуется быстрее. Типа, золотую медаль получит спортсмен, который бежит быстрее других. А следующий тоже получит, но серебряную. И т.д. Надо же! Кто бы мог подумать!! А мы-то думали, что будет, как в старинной сказке про кролика и черепаху, где победила черепаха. Какая мощная наука, эта химия. Все, как в жизни, а не в сказке. Победил кролик! Да, и это называется кинетический контроль. Не зацикливайтесь на слове “контроль” – никто ничего не контролирует. В английском языке, а оттуда этот термин и идет, слово “контроль” не имеет пафосного смысла длинной руки верховного божества или правящей партии. Это слово просто означает, что речь идет про основной фактор, определяющий результат какого-нибудь сложного процесса. В данном случае результат реакции определяется ее скоростью, то есть кинетикой. Кинетика – это наука о скоростях химических реакций, поэтому понятия скорость и кинетика в данном случае полные синонимы, но от слова кинетика проще образовать прилагательное, не создав при этом ненужных ассоциаций с правилами дорожного движения.

Вообще, как несложно понять, большинство реакций, хоть органических, хоть неорганических, хоть каких-либо еще определяется их скоростью, то есть кинетикой. Это настолько банально, что может показаться издевательством – если есть несколько реакций, то если они идут одновременно, больше всего продукта получится из самой быстрой реакции. И надо понимать, что реакция, приводящая к нескольким продуктам – это, на самом деле, несколько одновременно идущих реакций. Мы уже сто раз имели дело с такими ситуациями. Вспомним хотя бы радикальное галогенирование алканов, где мы рассматривали относительную ракционную способность разных C-H связей, и пытались понять, как это связано с распределением прдуктов галогенирования. Это типичный случай кинетического контроля. И это действительно было бы банально, если бы не существовали реакции, результат которых определяется принципиально другим фактором.
А теперь представим себе, что реакции обратимы. Термодинамика ( а это и есть наука о равновесиях и обратимых процессах) говорит, что в этом случае можно дело представить так, что продукты обратимо превращаются друг в друга, и можно просто написать равновесие, а равновесие определяется константой равновесия. А константа равновесия определяется относительной стабильностью (в термодинамическом смысле) продуктов. В равновесии больше того вещества, которое стабильнее. Это более сложная идея, чем в кинетике, но тоже вполне немудреная – на нижней полке лучше чем на верхней, в случае чего падать ближе. Нужно только иметь в виду, что под стабильностью имеется в виду значение термодинамической функции энергии Гиббса, а не просто энергия образования. Но это преодолимая сложность.
Итак, у нас обратимая реакция дает два разных продукта. По обоим продуктам реакция обратима. Как и в случае скоростей, мы и здесь можем считать, что это две разных реакции, но в данном случае обратимых. Термодинамика хороша тем, что позволяет совсем не заморачиваться деталями. Важны только две вещи: установилось ли равновесие, и какова константа равновесия. Причем равновесие позволяет совсем все упростить, так один из основных законов равновесия говорит, что оно, равновесие, не зависит от пути его достижения. Поэтму, хотя продукты равновесных реакций не превращаются друг в друга напрямую, а только за счет обратимости реакций, в которых они образуются, но мы имеем полное право написать равновесие взаимопревращения продуктов, и дать этому равновесию константу равновесия. Один из самых ужасных выводов из этого состоит в том, что если представить себе образование тех же продуктов в какой-то другой реакции, результат термодинамического контроля будет ровно тот же самый (сразу оговоримся – условия должны быть сохранены, температура и растворитель в первую очередь, потому что константы равновесия зависят и от температуры, и от растворителя). И если есть сто разных реакций, дающих эти продукты, то все сто раз будет одно и то же. Это действительно страшный вывод – соотношение продуктов обратимой реакции вообще не зависит от самой реакции. Следовательно, продукты можно вообще взять не из реакции, а, например, купить на рынке, выпросить у кого-нибудь, выменять на коллекцию магнитиков, и даже, не попусти, Господи, искушение, похитить. Но если мы оставим эти продукты, они будут превращаться друг в друга, и когда равновесие установится, соотношение их будет вновь тем же (не забываем про температуру и растворитель – если мы хотим получить точно то же самое, нужно будет взять раствор с той же концентраций, что мы имели в реакциях, но обычно в несколько иных условиях, в другом, но похожем растворителе, и другой концентрацией, соотношение будет отличаться не сильно). Это может быть очень медленно, и даже, возможно, потребуется какой-нибудь катализатор (напомню, что катализатор не влияет на положение равновесия, но может дрматически ускорить его достижение), но смысл неизменен – в условиях термодинамического контроля, то есть, установившегося равновесия, соотношение продуктов зависит только от константы равновесия, или, что то же самое, от разницы стандартной свободной энергии Гиббса образования этих продуктов ΔGºf. А это еще откуда брать? Из статей, справочников, да и посчитать можно с довольно приличной точностью. В общем, это очень доступные данные, намного более доступные, чем константы скоростей.
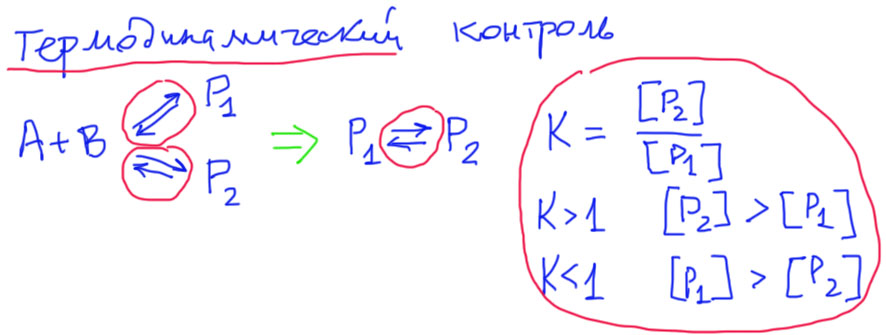
До этого момента все это – довольно банальная констатация того, что бывают скорости реакций, а бывают равновесия, и обращаться с ними нужно соответственно. По-настоящему интересным это становится тогда, когда эти два понятия сталкиваются в одной реакции. Ничего необычного в этом нет. Если есть обратимая реакция, дающая два (или три и т.д.) продукта, то их соотношение сначала зависит от соотношения констант скоростей (работает кинетический контроль), а когда равновесие установится, то от константы равновесия. И очень часто эти соотношения различаются. Даже можно сказать, что они должны различаться, потому что законы кинетики и равновесия очень сильно различаются. В этом случае мы говорим, что в первом случае соотношение продуктов соотвествует кинетическому контролю, во втором – термодинамическому.
А теперь самое главное – может ли в одной реакции быть и то и другое. И даже еще страшнее – могут ли эти контроли в таком случае тянуть в разные стороны? Если да, то мы получаем очень интересную ситуацию – разные продукты в разных условиях, и если мы поймем закономерности, то можем даже управлять направлением реакции и ее селективностью. Такая игра точно стоит свеч. Попробуем.
На первый вопрос ответить проще всего. Если равновесий нет, то нет и термодинамического контроля, только кинетический, и говорить не о чем, и вообще это неинтересно. А вот если равновесия есть, то можно еще вспомнить о том, что состояние равновесия редко достигается мнгновенно, в первую очередь потому, что сначала должен накопиться продукт реакции, скорость обратной реакции часто очень мала. Для установления равновесий могут потребоваться часы, а иногда и дни, недели, месяцы, годы. При повышении температуры все скорости (и прямой и обратной реакции) увеличиваются, и равновесие устанавливается быстрее. А пока равновесие не установилось, нет и термодинамического контроля, потому что этот контроль и означает, что распределение продуктов соответствует равновесию. Получаем такую картину: если реакции обратимые, то до установления равновесия продолжает работать кинетический контроль (естественно, чем ближе к равновесию, тем сильнее отклонения от чистого кинетичкского контроля).
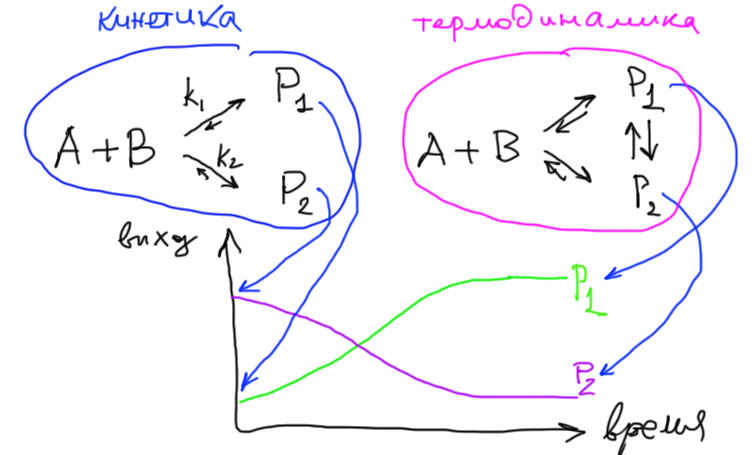
- кинетический контроль проявляется максимально при малом времени реакции и при более низких температурах
- термодинамический контроль вступает в силу после установления равновесия, это происходит быстрее при повышенных температурах, но произойдет и при низких, если дать реакционной смеси постоять подольше
Кинетический контроль не переходит в термодинамический скачком, и нет переключателя, который менял бы эти режимы по воле экспериментатора. Нужно понять, что в тех случаях, когда могут реализоваться оба эти режима – то есть, когда есть обратимая реакция, приводящая к двум или даже большему числу продуктов, – мы можем наблюдать за течением реакции и выходами продуктов (хроматографией или ЯМР, или ИК, или еще как-нибудь) и тогда увидим, что в начале процесса (в первых пробах) соотношение продуктов всегда соответствует кинетическому контролю, затем это соотношение начнет плавно меняться, с течением времени доедет до каких-то новых величин и практически станет постоянным на этом новом уровне – это и есть термодинамический контроль. Сколько потребуется времени для переходного периода заранее узнать нельзя – это зависит от констант скоростей прямой и обратной реакции, температуры и т.п.
Последний вопрос такой – почему кинетический и термодинамический контроль дают разные продукты? Ответ простой – в каждом случае нужно разбираться отдельно. Это не обязательно. В тех случаях, когда оба режима дают одни и те же продукты в близких количествах, это просто никому не интересно, и это никто даже не замечает. А вот когда продукты разные, и селективность меняется сильно, это вызывает интерес, попадает в статьи и учебники. Совсем яркий случай, хотя и весьма редкий, это когда продукты меняются полностью – кинетика дает один, а равновесие второй. Понятно, что для этого нужно соотношение констант скоростей в два порядка или больше, и константу равновесия в два порядка или больше. Это очень редкий, почти уникальный случай. Поэтому в реальной жизни сильно радоваться начинают, когда один из продуктов просто существенно преобладает, скажем соотношение 70:30 переходят в 20:80. Радость от долгожданной встречи с прекрасным в таких ситуациях бывает столь велика, что городу и миру горделиво сообщают о полном обращении селективности, позабыв про тридцать и двадцать процентов не тех продуктов в каждой из смесей. Ничего страшного: нет науки без обобщений и умолчания о несущественных деталях.
Как это работает в присоединении электрофилов к 1,3-диенам
В учебниках можно прочитать, что бром или HBr присоединяются к бутадиену с образованием 1,2-аддукта (продукты присоединения часто называют аддуктами) при 0ºC и 1,4-аддукта при 80ºС, что и является признаком кинетического и термодинамического контроля. И это настолько запоминается, что считается главным признаком кинетического и термодинамического контролей. Типа, если холодно, то кинетика, если тепло, то термодинамика. Термо- это же про тепло, в термосе горячий чай (или еще что-нибудь). Отлично запоминается!
Но это обман. Нас развели, как несмышленых младенцев. Подсунули яркую погремушку. Да так увлеклись, что многие разводилы и сами в это поверили. Если поверить в температуру, как главный фактор, определяющий 1,2- или 1,4-присоединение, то дальше в ход идут самые диковинные объяснения, как это работает. Я бы не стал это писать, если бы не выслушивал эти фантазии 20 лет на экзаменах.
Если мы разобрались в том, как работают эти контроли, то понимаем, что дело не в температурах, а в том, установилось или нет равновесие. При низкой температуре это происходит настолько медленно, что можно не обращать внимания на время реакции. Тем не менее, если мы, как это часто бывает в практикуме, провели бы реакцию при нуле (бром полностью израсходовался), и оставили бы смесь на неделю под тягой при комнатной температуре, то у удивлением обнаружили бы не кинетическое, а термодинамическое соотношение продуктов. И уж совсем неприятно то, что при попытке выделить и очистить 1,2-аддукт он имеет неприятную привычку медленно, но верно перегруппировываться в 1,4-аддукт.
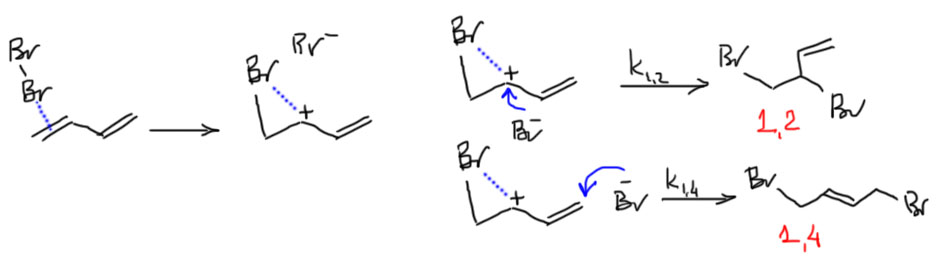
Но, во-первых, где же равновесие в этой реакции. Нам никто до сих пор не говорил, что бромирование олефинов обратимо. И правильно не говорил – бромирование обычных олефинов действительно необратимо. А вот диенов – это совсем другое дело. Напишем еще раз, как присоединяется бром к 1,3-диену, и обратим еще раз внимание, что карбокатион в этом случае хорошо стабилизирован донорным мезомерным эффектом – это катион аллильного типа. Когда катион стабилизирован, становится возможным его образование прямой диссоциацией (точнее, ионизацией) бромпроизводного. Мы еще вернемся к этой реакции довольно скоро в механизме SN1 нуклеофильного замещения. Ничего не поделаешь – в химии часто оказываются тесно связанными вещи, казалось бы весьма далекие друг от друга. Нарисуем теперь более точную схему добавив другим цветом равновесие, которое и включает термодинамический контроль.
Остались конкретные вопросы – почему 1,2-аддукт образуется быстрее 1,4-аддукта. Совсем однозначного ответа на это нет, но простое соображение помогает понять, почему так и должно быть и почему этот результат вполне ожидаем. Дело в том, что органические реакции проводят в органических растворителях. А органические растворители (хлороформ, эфир, ацетон, ТГФ и т.п.) обладают невысокой полярностью. Бромирование вообще обычно делают в неполярном CCl4. В таких растворителях катионы и анионы не могут отплыть далеко друг от друга, они держатся вместе (это называется ионная пара). Вот и получается, что бромид образуется рядом с местом присоединения, рядом с катионом, то есть гораздо ближе к положению 2. Если кажется, что это ерунда – молекула-то малюсенькая, подумаешь, велика работа проплыть несколько ангстрем! Не знаю, не пробовал, но не в этом дело. Дело в огромной реакционной способности карбокатионов, которые очень быстро реагируют с первым попавшимся нуклеофилом. У бромида просто мало шансов уплыть даже на пару ангстрем – он образуется вместе с карбокатионом и рядом с карбокатионом в виде ионной пары и немедленно с ним реагирует – это так и называется – коллапс ионной пары. 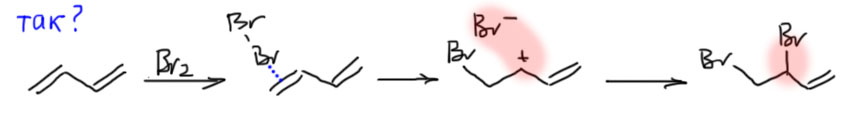 В этом объяснении все очевидно, и даже возникает вопрос – а 1,4-аддукт вообще как может получиться, если это так. Может быть, это всегда результат перегруппировки 1,2-аддукта? Нет, это проверяли и не раз – 1,4-аддукт все же образуется сразу, а при отрицательных температурах перегруппировка настолько медленна, что можно не принимать ее во внимание. К тому же есть сложность со стереохимией, мы ведь привыкли к анти-присоединению, а коллапс ионной пары неминуемо дал бы син-аддукт или только, или в смеси с анти. На бутадиене это правда не видно, но есть и другие данные. И как же тогда коллапс? Иногда коллапсирует, иногда не коллапсирует? Придется вспомнить, что в присоединении брома сначала образуется не карбокатион, а мостиковый ион. В данном случае, как мы уже договорились при обсуждении бромирования алкенов, мостиковый ион будет несимметричным – это карбокатион, стабилизированный донорно-акцепторным взаимодействием с неподеленными парами атома брома. Такой мостиковый ион все равно обладает очень высокой реакционной способностью по отношению к нуклеофилам, но реакция перестает выглядеть как коллапс ионной пары, а скорее как нуклеофильную атаку на электрофильный центр. И даже для образования 1,2-аддукта придется немного поплавать, чтобы подплыть с противоположной стороны, так как с этой приблизиться не дает мостиковый атом брома. Разница в атаке на второй и четвертый атом будет именно в невыгодности пространственного разделения зарядов в малополярной среде – физики назвали бы это работой против электростатического притяжения. Работать все любят поменьше, даже ионы. Но разница не так велика и бромид получает шанс уплыть и атаковать с аллильного положения, естественно с меньшей скоростью – в эту скорость входит с отрицательным знаком вклад скорости диффузии иона в малополярной среде. Уф, выглядит убедительно, хотя и малость смахивает на дрянной репортаж из жизни ионов. Причем это описание будет работать даже если мы узнаем, что реальная разница констант не так велика, но есть хотя бы небольшое преимущество 1,2-аддукта (напомню, что пока мы считаем, что оно большое).
В этом объяснении все очевидно, и даже возникает вопрос – а 1,4-аддукт вообще как может получиться, если это так. Может быть, это всегда результат перегруппировки 1,2-аддукта? Нет, это проверяли и не раз – 1,4-аддукт все же образуется сразу, а при отрицательных температурах перегруппировка настолько медленна, что можно не принимать ее во внимание. К тому же есть сложность со стереохимией, мы ведь привыкли к анти-присоединению, а коллапс ионной пары неминуемо дал бы син-аддукт или только, или в смеси с анти. На бутадиене это правда не видно, но есть и другие данные. И как же тогда коллапс? Иногда коллапсирует, иногда не коллапсирует? Придется вспомнить, что в присоединении брома сначала образуется не карбокатион, а мостиковый ион. В данном случае, как мы уже договорились при обсуждении бромирования алкенов, мостиковый ион будет несимметричным – это карбокатион, стабилизированный донорно-акцепторным взаимодействием с неподеленными парами атома брома. Такой мостиковый ион все равно обладает очень высокой реакционной способностью по отношению к нуклеофилам, но реакция перестает выглядеть как коллапс ионной пары, а скорее как нуклеофильную атаку на электрофильный центр. И даже для образования 1,2-аддукта придется немного поплавать, чтобы подплыть с противоположной стороны, так как с этой приблизиться не дает мостиковый атом брома. Разница в атаке на второй и четвертый атом будет именно в невыгодности пространственного разделения зарядов в малополярной среде – физики назвали бы это работой против электростатического притяжения. Работать все любят поменьше, даже ионы. Но разница не так велика и бромид получает шанс уплыть и атаковать с аллильного положения, естественно с меньшей скоростью – в эту скорость входит с отрицательным знаком вклад скорости диффузии иона в малополярной среде. Уф, выглядит убедительно, хотя и малость смахивает на дрянной репортаж из жизни ионов. Причем это описание будет работать даже если мы узнаем, что реальная разница констант не так велика, но есть хотя бы небольшое преимущество 1,2-аддукта (напомню, что пока мы считаем, что оно большое).
А почему в равновесии преобладает другой аддукт? А вот это мы знаем! Более замещенные олефины более устойчивы – надеюсь вы не забыли этот лозунг. Здесь это ровно то, что нужно. 1,4-аддукт – это дизамещенный олефин, а 1,2-аддукт – монозамещенный. Следовательно, 1,2-аддукт будет изомеризоваться в 1,4-аддукт, который и преобладает в реакционной смеси. Все сходится. По крайней мере, пока мы не задаем лишних вопросов (спойлер: если зададим, сходится перестанет, и об этом можно прочитать на следующей вкладке). Ой, забыл, мы же написали равновесие, определяющее возможность термодинамического контроля, идет через аллильный карбокатион, а теперь как-то незаметно стали использовать мостиковый ион. Это меняет картину равновесия? Нет не меняет, хотя и делает ее несколько менее привлекательной, потому что у нас больше нет очень выгодного аллильного катиона. Но несимметричный мостиковый ион тоже стабилизирован и взаимодействием с неподеленными парами атома брома, и взаимодействием с π-орбиталью двойной связи – это, к сожалению, уже не будет полноценная мезомерия, которую можно изобразить граничными структурами, но стрелочкой смещения электронной плотности ее показать можно. Кстати, это неплохо соответствует реальности – как мы, возможно, узнаем, из следующей вкладки, это равновесие не так уже легко и устанавливается. 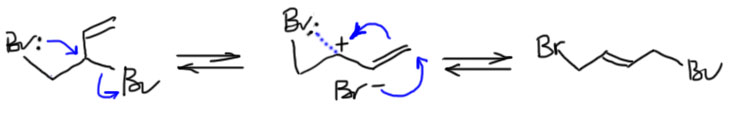
1,2 и 1,4-присоединение: а как на самом деле?
Строго факультативно! Не читайте это, если вы не собираетесь заниматься органической химией, чтобы не засорять мозги лишней информацией, уменьшающей ясность. А вот тем, кто собирается сделать органику профессией я бы порекомендовал ознакомится с этим типичным случаем, как желание красиво подать материал приводит к вопиющему насилию над реальной информацией.
Прежде чем начать разбираться, как обстоят дела на самом деле, сразу и категорично скажу, что картина бромирования бутадиена из учебников – при пониженной температуре 1,2- и кинетический контроль, а при 60 или 80 градусах 1,4- и термодинамический контроль, – просто выдумана. Это такая научная фантазия. Довольно старая, поэтому концов уже не найти, да мы и не собираемся заниматься криминалистикой. У всех таких фантазий, а их немало в химии, источник один – сначала кто-то рисует красивую теорию для учебника, потом со всей дури подгоняет к ней данные, потом это без конца переписывают, не удосужившись проверить, насколько это соответствует экспериментальным данным. Иногда это вполне безобидно и в рамках приличий (округлим числа, забудем про побочный продукт и т.п., возьмем один подходящий эксперимент, а про три другие забудем, и .т.п). Иногда беспардонно – просто рисуем данные так, как нам хотелось бы, чтобы они выглядели. В этом здорово помогает и обширнейшая научная литература, в которой часто можно найти один подходящий эксперимент, а на сто неподходящих внимания не обратить.
Если отжать суть дела, настоящая картина получается такая.
Бромирование бутадиена бромом в разных растворителях дает смесь 1,2- и 1,4-продуктов. Реакция в кинетически контролируемом режиме дает смесь с небольшим преобладанием 1,2-аддукта: обычное соотношение 1,2/1,4 = 60-65% к 30-34% (это составляет от 2 до 2.2), причем это соотношение не очень сильно зависит ни от температуры в диапазоне от -20 до +20ºC. Можно ли считать 1,2-аддукт продуктом кинетического контроля? Нет, нельзя. Продуктом кинетического контроля является именно смесь. Это значит одну простую вещь – константы скоростей 1,2- и 1,4-присоединения отличаются не очень сильно. Собственно, понятно, что они отличаются как раз в эти 2-2.2 раза. В принципе, это неплохая разница, и она очень хорошо соответствует описанию этой реакции на предыдущей странице – вот эти 2 раза и есть цена, которую приходится платить за путешествие бромида, чтобы атаковать мостиковый ион с аллильного положения. Качественно картина верна, количественно нет. Реакцию бромирования в режиме кинетического контроля нельза признать селективной. И совсем практический вывод – 1,2-аддукт получить бромированием бутадиена в ощутимых количествах почти невозможно – в смеси продуктов слишком много 1,4-аддукта, а разделение этой смеси почти нереально, если не прибегать к услугам дорогостоящей хроматографии – в процессе разделения будет происходить изомеризация в более устойчивый 1,4-аддукт.
Выше 20-25ºС (то есть комнатной температуры) бутадиен не бромируют – это трудновыполнимо практически, и приведет к изменению механизма реакции.
А как дела обстоят с термодинамическим контролем? Очень просто, если мы хорошо поняли что это такое – для этого не обязательно даже ставить реакцию. Действительно, нагревание кинетического продукта реакции (то есть смеси аддуктов бромирования в кинетическом режиме без разделения) до 60-100ºС в течение нескольких минут приводит к установлению равновесия, в котором преобладает 1,4-изомер (не менее 80%), который может быть легко выделен кристаллизацией, что приводит к смещению равновесия и образованию дополнительного количества 1,4-аддукта – так можно почти весь дибромид перевести в эту форму. Вывод простой: 1,4-аддукт можно с чистой совестью считать продуктом термодинамического контроля. Как и в случае любого другого продукта термодинамического контроля, для получения 1,4-аддукта не обязательно вести реакцию в режиме термодинамического контроля – проще просто погреть продукт реакции, полученный в неравновесных условиях. Можно было бы и реакцию провести – при 25ºС пришлось бы просто подождать неделю-другую до установления равновесия. Практический вывод: 1,4-аддукт получить бромированием бутадиена можно легко и с высоким выходом. Бромировать лучше при охлаждении, иначе придется решать проблему, как удержать бутадиен от выкипания. Нагревать можно при меньшей температуре, но тогда потребуется полчаса-час (например, при 77ºС – температуре кипения CCl4).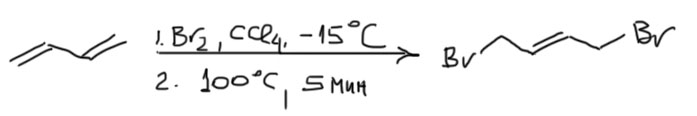
Желающие узнать подробности могут почитать дальше.
История вопроса с нудными подробностями
Как ни смешно, но самое близкое приближение к этой хрестоматийной картинке можно найти только в самой первой работе, где изучалась реакция бутадиена с бромом. Малоизвестный французский химик Гринер в лаборатории Шарля Фриделя (того самого, который во Фриделе-Крафтсе) в Сорбонне, исследовавший возможность превращения бутадиена в четырехатомный спирт эритрит, сообщил, что реакция бутадиена с бромом при -21ºС в хлороформе дает жидкий продукт. А если этот жидкий продукт погреть (не реакцию провести, а продукт реакции погреть!) при 100ºС то после охлаждения получается другой продукт, который закристаллизовывается (M.G.Griner, Compt.Rend. 1893, 117, 553). В те далекие времена статьи писали кое-как, и понять, что конкретно делалось и как анализировали наблюдения, почти невозможно.
Через 35 лет за реакцию взялись уже серьезно. Торп с сотрудниками попробовали воспроизвести эксперименты Гринера (J.Chem.Soc., 1928, 729). К этому времени уже вполне сложился стандарт представления научной статьи с экспериментальной частью, тщательным описанием эксперимента и полным разделением описания работы и обсуждения результатов, позволяющее легко понять, где реальные данные, а где вымысел. В этом случае выяснили, что реакция бутадиена с бромом при -15ºС в хлороформе дает не один продукт, а смесь двух дибромпроизводных. Структуры были установлены озонолизом. В хлороформе эта смесь состояла из 63% 1,4-дибромида и 37% 1,2-дибромида. Ой! Что-то пошло не так. 1,4-продукт при отрицательной температуре?! Пока забудем. Там же было найдено, что соотношение 1,2/1,4 сильно зависит от растворителя, и, в совсем неполярном гексане соотношение было обратным – 62% 1,2-дибромида и 38% 1,4-дибромида. Если любую из этих смесей погреть при 100ºС соотношение изменяется да 80% 1,4-продукта и 20% 1,2-продукта. Делаем вывод, что в 1928 ясности не прибавилось, и картина сильно не похожа на ту, к которой мы привыкли.
В 1953 Мислоу однозначно определил, что 1,4-продукт имеет транс-конфигурацию. Цис-изомера в продуктах не обнаружено. В принципе, это понятно. Мы ожидаем, что 1,4-продукт – наиболее стабильный олефин из возможных продуктов бромирования. Мы уже знаем, что более замещенный олефин более устойчив, чем менее замещенный, но по стерическим прифинам это относится только к транс-изомеру. Цис-изомер стерически дестабилизирован относительно транс, и эта разница съедает выгоду от перемещения двойной связи внутрь цепи. Количественно это прояснили Хетч и сотр. (JACS, 1959, 81, 5943) – разница между 1,2-дибромидом и транс-1,4-дибромидом всего 1,4 ккал/моль. Ну и обычная разница между транс- и цис-дизамещенными алкенами те же 1 ккал/моль, с атомами брома будет побольше. Вернемся к разнице между 1,2- и транс-1,4. Не маловато ли – всего 1,4 ккал/моль? Нет, в самый раз. Разницу в свободных энергиях легко пересчитать в константу равновесия, разделив на RT в тех же единицах измерения, и возведя в эту степень с отрицательным знаком число e – будет как раз около 85:15 при 100ºС.
Итак, в одном разобрались. Транс-1,4-дибромид действительно термодинамически более устойчив, чем 1,2-дибромид, и при установлении равновесия при нагревании до 100ºС он будет преобладать в равновесной смеси. Это именно то, что и наблюдалось в упомянутых статьях, и в нескольких других. 15% 1,2-дибромида в равновесной смеси можно пренебречь. Получается, что, по крайней мере, в этом отношении учебники не врут? Не совсем так – многие учебники зачем-то рассказывают, что 1,4-дибромид получается при проведении реакции бромирования при 80 или 100ºС. Но реакцию бромирования бутадиена никто никогда не делал при этой температуре, прежде всего потому что это очень непросто сделать – бутадиен это газ (т.кип. -4ºС) и чтобы сделать реакцию при повышенной температуре нужно городить очень сложное устройство для работы под давлением, причем потребуется очень быстрое смешение реагентов, так как реакция бромирования бутадиена очень быстра уже при отрицательной температуре, а очень быстрые реакции часто дают странные результаты, потому что реагенты не успевают равномерно распределяться по объему растворителя и в месте падения капли реагента образуется большой его избыток и получаются не те продукты. Но это-то как раз очень небольшое прегрешение, ведь мы уже договорились, что в термодинамическом контроде необязательно проводить саму реакцию, достаточно показать, как состав продуктов изменяется при достижении равновесия. При нагревании до 80-100ºС равновесие устанавливается за несколько минут, а вот при комнатной температуре намного медленнее, и потребуется не менее недели для достижения такого же состава равновесной смеси. Нет, не такого же – так как RT при комнатной температуре меньше, то и константа равновесия будет больше, и 1,4-продукта будет больше 90%, но это небольшой приз за долгое ожидание. Тем более, что и так уже найдено, что можно поступить проще просто за счет кристаллизации и смщения равновесия – так мы можем получить почти чистый 1,4-дибромид с почти количественным выходом.
А вот с кинетическим контролем все не так весело. В той же статье Хетча и сотр. измерили состав при бромировании при отрицательной температуре в нескольких растворителях, и получили везде почти одно и то же – смесь 1,2 и 1,4 почти один к одному. Противоречие? Что-то типа того. После в нескольких статьях это еще раз измеряли, и получали несколько разные результаты. Разгадку этой загадке дали Хизли и Тейлор (J.Org.Chem., 1969, 34, 2779), и вроде бы им удалось поставить точку в этой истории. По крайней мере до июня 2019 года опровержения или существенного уточнения этой работы не появилось, так что можно считать, что это и есть окончательное описание реакции. Фокус в том, что бром может присоединяться не только электрофильно, но и свободнорадикально. Вот так: 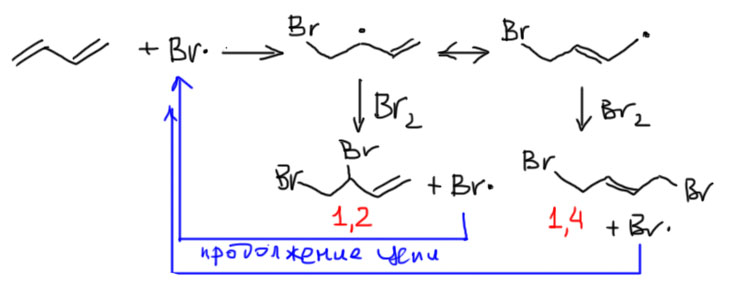
Получается та же смесь 1,2- и 1,4-присоединения, но с преобладанием 1,4-продукта – около 80%. Довольно хорошо понятно почему. Радикал, образовавшийся после присоединения атома брома тоже делокализован аллильньным способом, но для образования продукта радикальный центр забирает атом брома из молекулы брома. Просто по стерическим причинам это более вероятно с менее затрудненного концевого атома углерода. И здесь не работает соображение о том, что до дальнего положения нужно доплыть, так как реакция идет не с ионом, а с молекулярным бромом равновероятно распределенном в объеме растворителя. Образующийся атом брома возвращается в цепь. Иными словами, в свободнорадикальном присоединении кинетический контроль дает то же самое, что и термодинамический (напомню, что термодинамический контроль не зависит ни от механизма, ни от реакции, он всегда один и тот же). Это случайность и не это главное. Важно то, что там установили, что инициирование свободнорадикальной реакции происходит очень легко, и нужно тщательно исключать все возможности, в том числе свет, примеси, а также использовать небольшую концентрацию, так как есть гипотетическая возможность инициирования свободнорадикальной реакции через образования комплекса олефина с бромом. Во всех предыдущих работах это не делали, и странные несовпадения отношения 1,2/1,4 объясняются прото вкладом свободнорадикального бромирования. Но если тщательно исключить свободнорадикальное присоединение, то уходят и все аномалии в непостоянстве выходов и соотношения 1,2/1,4. Результат такой – чистое электрофильное присоединения брома к бутадиену в условиях строгого кинетического контроля дает смесь 1,2 и 1,4-продукта с небольшим преобладанием 1,2-аддукта: соотношение 1,2/1,4 в разных условиях, в том числе разных равстоврителях, и температурах в диапазоне от -15 до +25ºС очень хорошо ложится в узкий диапазон между 2 и 2.2.
Очень похожая ситуация наблюдается в хлорировании бутадиена. Хлорирование еще больше чем бромирование склонно переезжать на свободнорадикальный механизм. Но в темноте и в присутствии ингибиторов свободнорадикальной реакции (кислорода обычного) хлорирование дает смесь 1,2- и 1,4-аддуктов практически 1:1 (J.Org.Chem., 1966, 31, 4167). И в отличие от бромирования, в хлорировании нет равновесия (хлорид – более плохая уходящая группа чем бромид) и 1,2-аддукт вполне устойчив – смесь можно разделить. Надо сказать, что если мы приняли объяснение, почему при кинетическом контроле в бромировании получается больше 1,2-аддукта, то хлорирование будет для нас большим испытанием. По тем же рассуждениям здесь 1,2-аддукта должно быть больше, а не меньше, хотя бы потому что в хлорировании мостиковые ионы больше похожи на настоящий карбокатион, и коллапс ионной пары был бы более вероятен. Но нет. Почему? Можете попробовать сами что-нибудь придумать. В этом вся органическая химия – удачное объяснение, найденное для одной реакции непостижимым образом проваливается для похожей реакции. 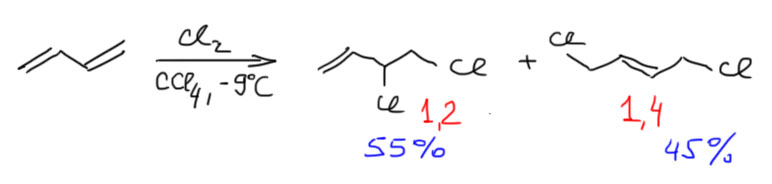
Прямое иодирование бутадиена никто не делал, но реакции с хлористы и бромистым иодом изучали (J.Chem.Soc., Perkin Trans II, 1991, 1271). И вновь при кинетическом контроле нет большого преимущества в 1,2-аддукте. Здесь, на самом деле, предсказать результат было бы труднее всего, потому что электрофильное иодирование идет через мостиковый ион, наиболее сильно напоминающий иодониевый ион, и тогда он должен будет раскрываться нуклеофилами с аллильной перегруппировкой и без нее. Оставим это в покое, так как это очень сложная проблема, в которой так никто до сих пор не разобрался. 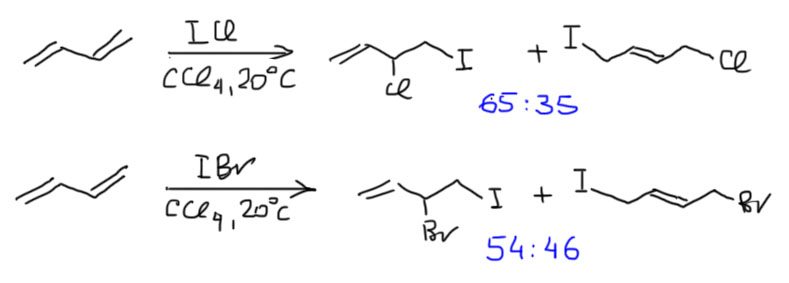
Так или иначе, мы видим что из четырех реакций электрофильного галогенирования в режиме кинетического контроля всегда получается смесь 1,2- и 1.4-аддуктов или вообще без преимущества или с небольшим преимуществом 1,2-аддукта. Ни в одном случае 1,2-аддукт нельзя назвать главным продуктом кинетического контроля.
Но все меняется, если в бромировании попробовать внешний нуклеофил. Например, если проводить бромирование в метаноле, то нуклеофилом в основном будет сам растворитель – его много, и ему не нужно никуда плавать. И что бы мы ожидали? Понятия не имею. В таких случаях сначала лучше посмотреть на эксперимент, а не позориться предсказаниями. Пальцем в небо попасть – дело нехитрое.
Образуется (V.Heasley, Chamberlain, J.Org.Chem., 1970, 35, 539), как и можно ожидать, в основном продукт присоединение брома и метанола, а обычный продукт дибромирования только как побочный продукт, и что характерно с уже знакомым нам соотношением 1,2/1,4 около двух. Но основной продукт почти целиком 1,2-аддукт, с очень небольшим количеством 1,4-аддукта.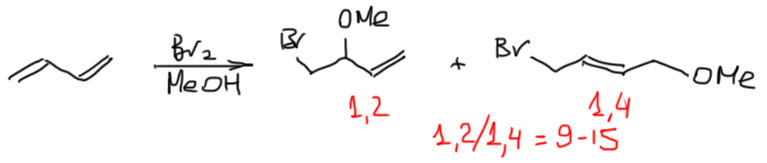
Нужно сразу сказать, что в этом случае обратимость исключена – что метокси-группа, что метанол – очень плохие уходящие группы (подробнее об этом в теме Нуклеофильное замещение). Оба продукта вполне стабильны и никуда не перегруппировываются. 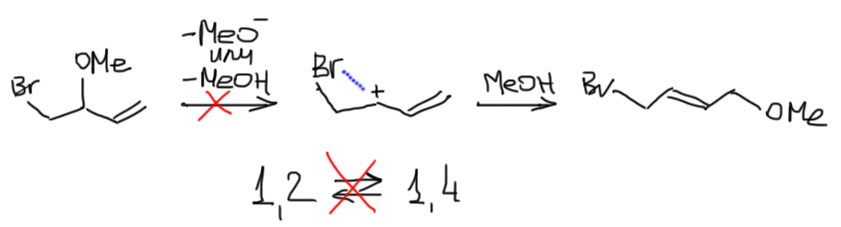
Значит только кинетический контроль, и основной продукт 1,2. Наконец-то. Если не в просто бромировании, то хоть в бромировании с чужим нуклеофилом! Но проблема в том, что это существенно другая ситуация, и объяснение, почему 1,2 здесь должно быть другим. В обычном бромировании нуклеофил образовывался из электрофильного реагента, брома, в непосредственной близости от положения 2, и с этим была связана вся аргументация, хотя и оказавшаяся в конце концов не такой уж безупречной. Здесь – метанол, растворитель, он везде, как и положено равстворителю – любая молекула в растворе окружена множеством молекул растворителя (говорят даже “сольватной оболочкой”, хотя это, скорее, метафора). Другое объяснение искали как раз в связи с несимметричным мостиковым ионом. В этом ионе на углероде 2 предполагается положительный заряд, но так как это не настоящий свободный карбокатион, в нем нет мезомерной делокализации. Нуклеофил может атаковать такую частицу со стороны крайнего углерода (в Нуклеофильном замещении увидим, что именно так и ведут себя аллильные субстраты), но поскольку метанол – нуклеофил слабый, то ему гораздо легче атаковать более положительный углерод-2.
Реакция бутадиена с другими электрофилами
Реакция Дильса-Альдера

Обратим внимание на то, что с точки зрения диена происходит 1,4-присоединение. В целом эта реакция представляет собой присоединение, только непонятно, олефин присоединяется к диену или наоборот. Не будем морочить себе голову такими вопросами, а просто назовем эту реакцию циклоприсоединением и будем считать реагенты равноправными. Поскольку всяких циклоприсоединений много, уточним, что присоединяется фрагмент из двух атомов к фрагменту из четырех – это [2+4]-циклоприсоединение.
Мы уже привыкли, что если есть реакция, тем более важная, то сейчас начнется нудное обсуждение механизма. Не тут-то было! Мы бы с удовольствием, но у реакции Дильса-Альдера нет механизма. Просто есть исходные вещества и продукт, в процессе исходные превращаются в продукт, а посредине ничего нет. Чтобы хоть что-то написать, изображают стрелками смещение электронных пар, причем написать можно хоть в одну сторону, хоть в другую – это одно и то же. Обратим внимание, что перемещаются три пары, принадлежавшие π-связям, и образуются три новые связи – две простые и одна π-связь.
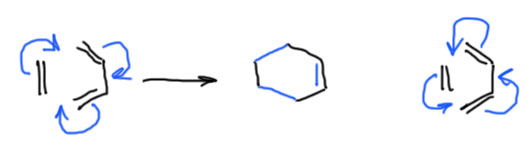
Перемещаются эти три пары одновременно (для знатоков квантовой физики такое утверждение – невероятный вздор, но мы люди попроще и не заморачиваемся, у нас это отлично работает). Нам это важно, потому что, если бы пары перемещались бы не одновременно, то по дороге образовались бы всякие катионы и анионы, а исследователям этой реакции вроде бы совершенно ясно, что ничего подобного там, как правило, нет. Итак, имеем ситуацию, когда по дороге от реагентов до продуктов ничего конкретного нет – это одностадийная реакция. Такие реакции в химии чрезвычайно важны, они носят немного странное название согласованных. Вероятно, предполагается, что задумав реакцию, олефин и диен отправляются в вышестоящую инстанцию с планом превращения, согласовывают его, получают кучу подписей и печатей, после чего немедленно вступают. На самом деле, это просто перевод на русский английского термина concerted, у которого нет таких лишних смыслов. Английское слово вызывает в памяти слово концерт, то есть дружные совместные действия – в этом и смысл термина, – некоторое количество молекул, фрагментов, атомов, связей вместе что-то совершают, и получается, как будто чудом и сразу, довольно непростой и красивый результат.
Если от балабольства перейти к делу, то отсутствие каких-либо интермедиатов (катионов, анионов, радикалов, еще чего-нибудь) по пути от исходных к продуктам имеет интересное следствие – сама реакция почти ни от чего не зависит, ни от растворителей, ни, строго говоря, от заместителей в молекулах диена и олефина. Но – и это важно – в олефине должны быть акцепторные заместители, и посильнее, желательно мезомерного типа. Какие и сколько – не важно, но должны быть. Олефины, способные вступать в реакцию Дильса-Альдера, называются диенофилы. Реакционная способность, при этом, ни от количества, ни от конкретного типа не зависит – мы знаем, что они, скорее всего, реагируют, но не знаем, кто быстрее, кто медленнее.
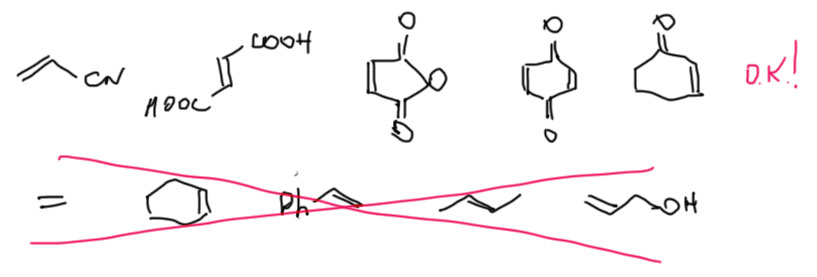
К диенам требования даже мягче. Желательно, чтобы там были донорные заместители, но вполне сносно реагирует и сам бутадиен, и некоторые его производные с акцепторными заместителями. Но вот что точно должно быть – диеновая система должна иметь возможность принять s-цис-конформацию, или быть жестко встроенной в цикл, где такая геометрия закреплена. Чем жестче закреплена такая конфигурация, тем лучше, поэтому всякие циклические диены обычно более активны, чем нециклические. Еще сильно влияет на реакционную способность диена всякое напряжение и дестабилизация, но мы об этом пока ничего не знаем, оставим.
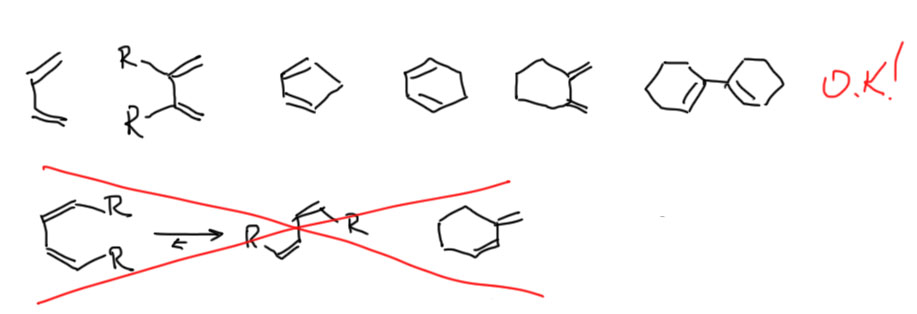
В остальном все просто. Нарисовать реакцию, исходя из готовых диена и диенофила, очень просто. Просто и догадаться от структуре диена и диенофила, если дан аддукт. Где двойная связь, там был диен. Просто пилим, как показано, и внимательно рассматриваем, что получилось. Если все нормально, то есть олефин похож на диенофил, а в диене нет ничего, противодействующего правильному расположению двойных связей, утверждаем. Если что-то не так, значит где-то ошибка.
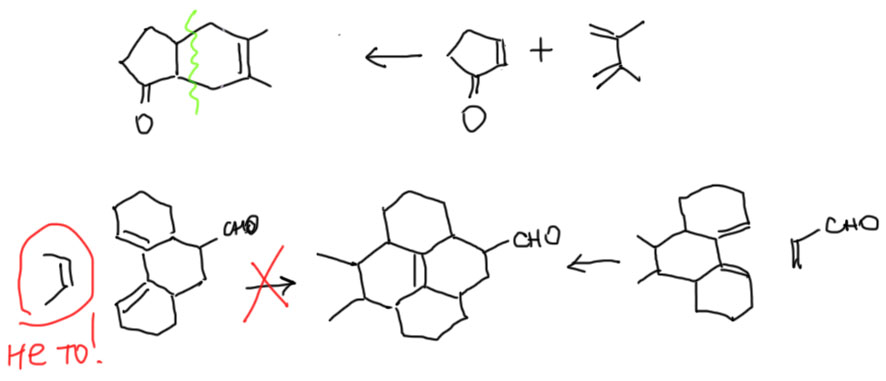
Реакция Дильса-Альдера, эндо/экзо-селективность
Это правильно, и если это вас устраивает, свободно пользуйтесь, и никто вам дурного слова не скажет. Но, может быть, душа ваша жаждет настоящих подвигов. Тогда поучимся рисовать это ближе к реальности. Оборвем пока заместители и посмотрим как выглядит это кольцо с мостиком. Посмотрим сбоку, со стороны двойной связи – и увидим, как в стереохимии принято говорить, ванну. Мостик станет такой ручкой сверху. Вообще это больше похоже на корзинку, а не на ванну, но принято говорить именно про ванну (по-английски, это даже не ванна, а лодочка, boat – сами решите, у кого воображение лучше). Но на виде сбоку обычно не останавливаются, а смотрят на эту ванну-корзинку-лодочку со стороны углерода с тремя связями. Получают такой вид, на котором хорошо просматриваются боковые связи, что очень удобно когда там придется рисовать заместители со своей стереохимией, а это делать приходится. Оба других вида для этого не годятся. Поучимся получше рисовать такую проекцию. Это делается очень легко, просто следите за тем, чтобы противолежащие связи имели приблизительно равную длину, а боковые еще и были визуально параллельны (при этом их наклон никого не волнует). Мостик сверху рисуем сверху, не гнем его набок – он ни в чем не виноват. Тренируйтесь. После десятого раза само будет рисоваться, одним росчерком.
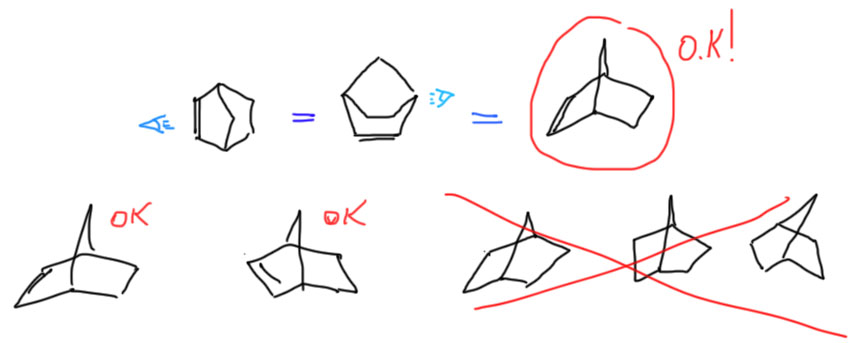
Теперь добавим заместители. Здесь нас ожидает маленькая проблема – их можно нарисовать двумя способами.
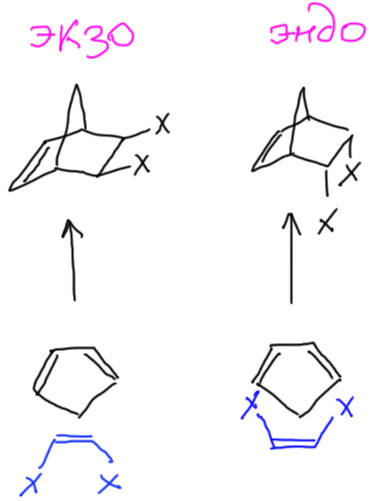 Эти два продукта отражают два возможных способа сближения диена и диенофила. Если бы диен был нециклический, то разницы бы не было. Мостик задает два направления относительно себя – заместители наружу от мостика, и замесители внутрь от мостика. Поэтому продукты называют экзо (наружу) и эндо (внутрь). Очень важным является то, что в эндо-продукте заместители при сближении диена и диенофила оказываются рядом с образующейся двойной связью. А при экзо-сближении они максимально удалены друг от друга. Именно это и определяет результат большинства реакций Дильса-Альдера – образуется эндо-аддукт или совсем как единственный, или в смеси с экзо, но с явно выраженной эндо-селективностью. Можно просто это запомнить, и не задаваться вопросом почему. Если все же задаться таким вопросом, тогда мы замечаем, что вновь говорим о реакции, которая дает или может дать два разных, но изомерных продукта. Возникает вопрос селективности, а теперь мы еще и знаем про кинетический и термодинамический контроль. Эндо-продукт – это что? Это продукт, который получается быстрее экзо-продукта, потому что эндо-сближение реагентов выгоднее экзо-сближения именно потому, что при этом возникает стабилизирующее взаимодействие образующейся двойной связи (пока замнем, что это за взаимодействие и отчего оно стабилизирующее). Итак, эндо-продукт – это продукт кинетического контроля. А экзо-продукт неужели термодинамический? Да, только большинство реакций Дильса-Альдера в условиях их проведения необратимы, хотя есть исключения.
Эти два продукта отражают два возможных способа сближения диена и диенофила. Если бы диен был нециклический, то разницы бы не было. Мостик задает два направления относительно себя – заместители наружу от мостика, и замесители внутрь от мостика. Поэтому продукты называют экзо (наружу) и эндо (внутрь). Очень важным является то, что в эндо-продукте заместители при сближении диена и диенофила оказываются рядом с образующейся двойной связью. А при экзо-сближении они максимально удалены друг от друга. Именно это и определяет результат большинства реакций Дильса-Альдера – образуется эндо-аддукт или совсем как единственный, или в смеси с экзо, но с явно выраженной эндо-селективностью. Можно просто это запомнить, и не задаваться вопросом почему. Если все же задаться таким вопросом, тогда мы замечаем, что вновь говорим о реакции, которая дает или может дать два разных, но изомерных продукта. Возникает вопрос селективности, а теперь мы еще и знаем про кинетический и термодинамический контроль. Эндо-продукт – это что? Это продукт, который получается быстрее экзо-продукта, потому что эндо-сближение реагентов выгоднее экзо-сближения именно потому, что при этом возникает стабилизирующее взаимодействие образующейся двойной связи (пока замнем, что это за взаимодействие и отчего оно стабилизирующее). Итак, эндо-продукт – это продукт кинетического контроля. А экзо-продукт неужели термодинамический? Да, только большинство реакций Дильса-Альдера в условиях их проведения необратимы, хотя есть исключения.
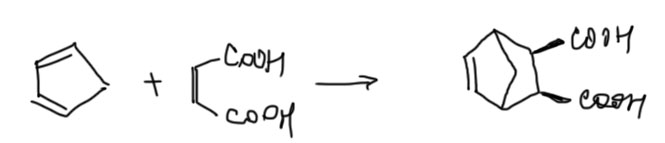
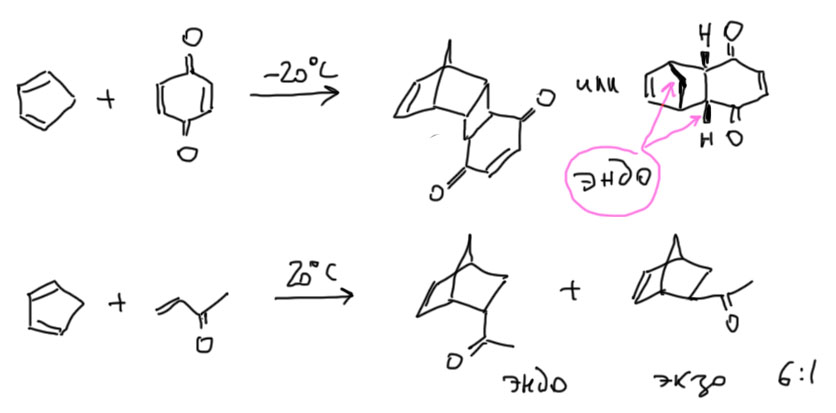
Условия реакций Дильса-Альдера просты – нагревание смеси диена и диенофила часто без растворителя или в каком-нибудь простом растворителе. И все – если реакция идет, через некоторое время охлаждай и выгружай продукт. Если не идет, нужно попробовать нагреть посильнее (иногда и до 200º). Если и так не идет, признаем поражение, потому что за редкими исключениями ускорять реакции Дильса-Альдера нечем. С очень активными диенами типа того же циклопентадиена реакция часто идет при комнатной температуре или даже при охлаждении. Вот несколько примеров (заодно посмотрите, как корректно обозначать эндо-продукт на самой простой проекции для тех, кто не хочет учиться рисовать):
Задачи и упражнения к коллоквиуму по теме диены
1,2 и 1,4-присоединение
Если мы все поняли про кинетический и термодинамический контроль, попробуем объяснить такой экспериментальный факт. При бромировании 1,3-диенов в присутствии избытка воды образуются, как и положено, продукты присоединения брома и воды (посмотрите еще раз присоединение к простым алкенам, если забыли). Так вот, при этом никогда не наблюдается 1,4-присоединения, только 1,2. Что это может значить? Напишите механизм присоединения брома и воды к 1,3-бутадиену, и постарайтесь понять, куда мог запропаститься термодинамический контроль.
Реакция Дильса-Альдера